

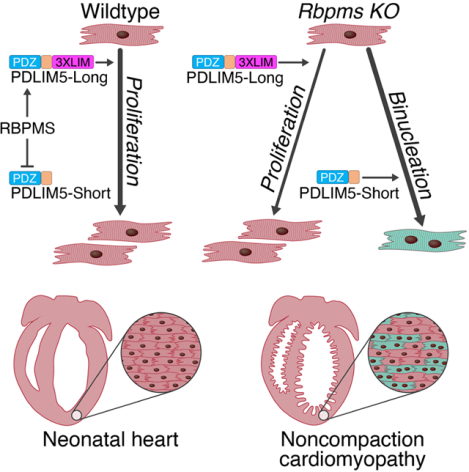
Summary
Noncompaction cardiomyopathy is a common congenital cardiac disorder associated with abnormal ventricular cardiomyocyte trabeculation and impaired pump function. The genetic basis and underlying mechanisms of this disorder remain elusive. We show that genetic deletion of RNA binding protein with multiple splicing (Rbpms), an uncharacterized RNA binding factor, causes perinatal lethality in mice due to congenital cardiovascular defects. Loss of Rbpms causes premature onset of cardiomyocyte binucleation and cell cycle arrest during development. Human iPSC-derived cardiomyocytes with RBPMS gene deletion have a similar blockade to cytokinesis. Sequencing analysis revealed that RBPMS plays a role in RNA splicing and influences RNAs involved in cytoskeletal signaling pathways. We found that RBPMS mediates isoform switching of the heart-enriched LIM domain protein Pdlim5. Loss of Rbpms leads to abnormal accumulation of Pdlim5-short isoforms, disrupting cardiomyocyte cytokinesis. Our findings connect premature cardiomyocyte binucleation to noncompaction cardiomyopathy and highlight the role of Rbpms in this process.
Keywords: Noncompaction cardiomyopathy, hypertrabeculation, patent ductus arteriosus, cardiomyocyte binucleation, RNA binding protein, alternative splicing
Graphical Abstract
eTOC Blurb
Gan et al. demonstrate the roles of an uncharacterized RNA splicing factor Rbpms in mouse heart development. Rbpms regulates cytokinesis of embryonic cardiomyocytes, while absence of Rbpms causes cytokinesis failure, premature binucleation, and noncompaction cardiomyopathy.
Introduction
Cardiac function requires second-to-second contractility of the ventricular chambers, which are lined by finger-like projections of the myocardium, referred to as trabeculae. During mouse heart development, the myocardial layer protrudes into the ventricular lumen at embryonic day 9.5 (E9.5) and forms trabecular ridges. Trabeculae extend radially to form a trabecular network, and the bases of the trabeculae thicken and merge to form the compact myocardium beginning at approximately E11.5. Remodeling and compaction of the trabeculae require rapid cardiomyocyte proliferation and are complete at E14.5 (Samsa et al., 2013). Disruption of these processes causes noncompacted myocardium, a potentially fatal disorder characterized by excessive trabeculation and hypoplastic ventricular walls with reduced cardiac contractility (Sedmera et al., 2000). Persistence of noncompacted myocardium postnatally can cause heart failure, arrhythmias, stroke, thromboembolism and sudden death (Almeida and Pinto, 2013, Jefferies et al., 2015). Noncompacted myocardium can exist as an independent disorder, such as left ventricular non-compaction (LVNC), which is the third most common primary congenital cardiomyopathy in children, after dilated cardiomyopathy (DCM) and hypertrophic cardiomyopathy (HCM) (Nugent et al., 2003). Noncompacted myocardium is also associated with many other disorders such as Barth syndrome, Emery-Dreifuss muscular dystrophy, and myotubular myopathy (Weiford et al., 2004). Cardiomyocyte proliferation defects are a major cause of myocardium noncompaction (Wilsbacher and McNally, 2016).
Embryonic murine cardiomyocytes are highly proliferative, mononuclear and diploid, whereas postnatal cardiomyocytes exit the cell cycle soon after birth, and become binucleated and polyploid (Gan et al., 2020a), coinciding with the loss of cardiac regenerative capacity (Porrello et al., 2011, Soonpaa et al., 1996). Recent studies revealed that although most adult murine cardiomyocytes are binucleated, the residual mononuclear diploid cardiomyocytes are proliferative and contribute to the limited regenerative capacity of the adult heart (Patterson et al., 2017). Cardiomyocytes become polyploid when DNA replication in S phase of the cell cycle occurs without the completion of karyokinesis or cytokinesis. Disruption of karyokinesis or cytokinesis can cause cell cycle arrest in the mitotic phase and polyploidization thereafter, which is common in postnatal mammalian cardiomyocytes (Gan et al., 2020a). Premature cardiomyocyte polyploidization has been observed in mice and Drosophila with cardiac developmental defects, such as DCM, tetralogy of Fallot, and hypertrophy (Stopp et al., 2017, Liu et al., 2019, Yu et al., 2013). However, it is unknown how premature cardiomyocyte polyploidization influences heart development.
RNA binding proteins (RBPs), such as RBM20 and RBM24 (Blech-Hermoni and Ladd, 2013, Guo et al., 2012, Yang et al., 2014), have been shown to play essential roles in heart development. RNA binding protein with multiple splicing (RBPMS), also known as hermes in frog and chick, is highly expressed in the heart and contains a conserved RNA recognition motif (RRM) (Gerber et al., 1999, Gerber et al., 2002, Akerberg et al., 2019), but its potential functions in mammalian heart development have not been explored (Akerberg et al., 2019). In this study, we show that Rbpms is required for normal heart development in mice and its absence results in premature cardiomyocyte binucleation and noncompaction cardiomyopathy. Among its various targets, RBPMS mediates splicing of the PD-LIM domain protein Pdlim5, a sarcomeric scaffolding protein, thus governing the appropriate timing of cardiomyocyte binucleation in embryonic hearts. The absence of Rbpms leads to accumulation of Pdlim5-short variants, which directly inhibit cardiomyocyte cytokinesis, thereby diminishing cardiomyocyte number and heart growth, causing noncompaction cardiomyopathy. These findings provide new insights into the molecular basis of heart development and congenital heart disease.
Results
Genetic deletion of Rbpms causes perinatal lethality and cardiovascular abnormalities.
Rbpms is highly expressed in the ventricles of the adult mouse heart (Figure 1A) and in hearts of other species, including chicken and Xenopus (Gerber et al., 1999, Gerber et al., 2002), suggesting an evolutionarily conserved role in the heart. During development, Rbpms is expressed in the heart as early as E12.5 and thereafter (Figure 1B). Rbpms mRNA is also detected in smooth muscle-enriched organs, such as aorta, pulmonary artery, intestine, bladder, and esophagus at E15.5 (Figure 1B). We observed high Rbpms expression in both endocardium and myocardium of E12.5 hearts, but decreased Rbpms expression in the endocardium of E15.5 hearts (Figure S1A).
Figure 1. Perinatal lethality and noncompaction cardiomyopathy of Rbpms KO mice.
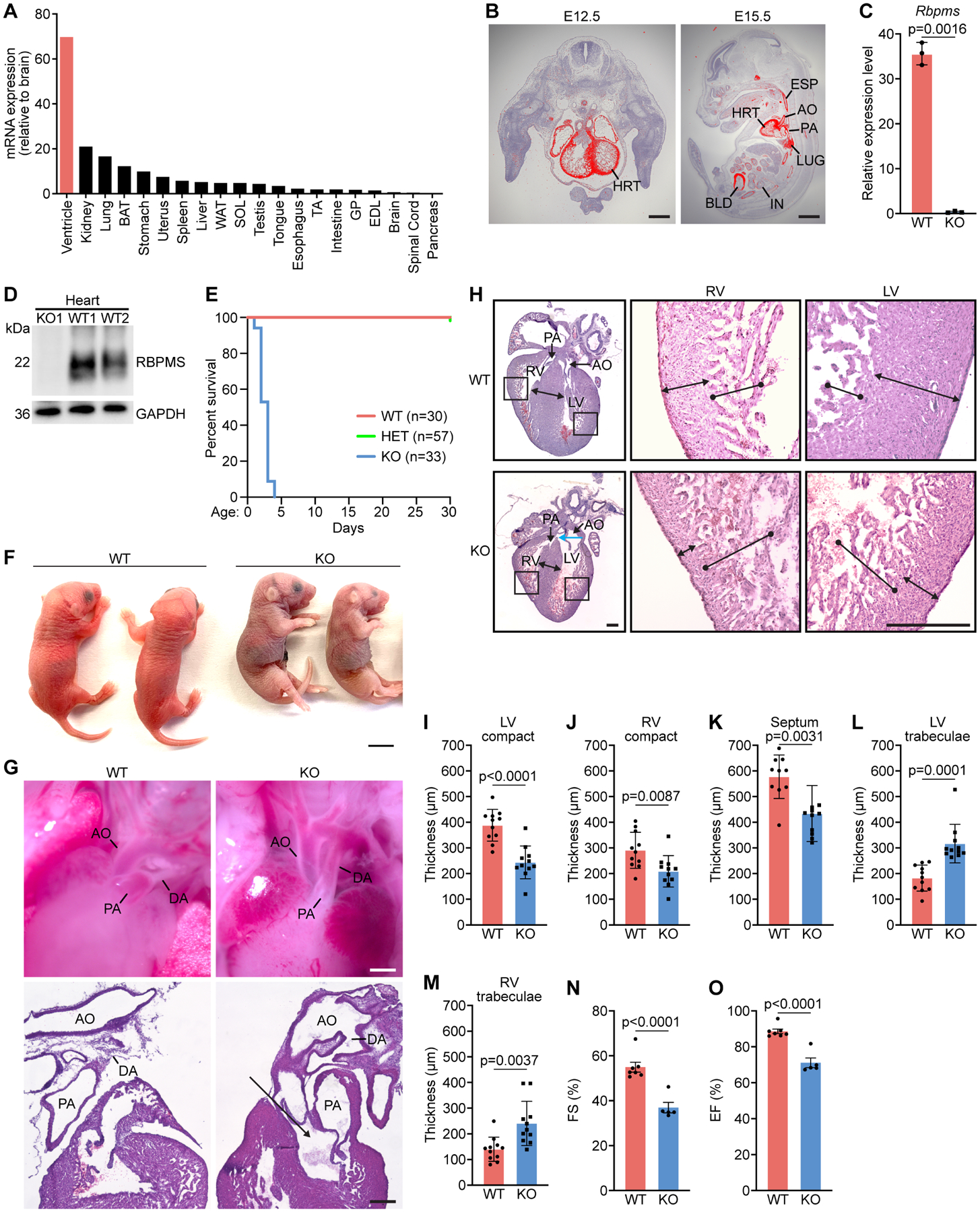
A. Relative expression levels of Rbpms mRNA in adult mouse tissues as measured by qRT-PCR. B. In situ hybridization showing cardiac and smooth muscle expression of Rbpms mRNA at the indicated embryonic time points. HRT, heart; AO: aorta; PA: pulmonary artery; IN, intestine; LUG, lung; BLD, bladder; ESP, esophagus. Scale bars (left): 500 μm, (right): 2 mm. C. Expression level of Rbpms mRNA in P1 WT and KO hearts, measured by RNA-seq (n = 3 for WT, and 3 for KO). D. Western blot analysis showing loss of RBPMS protein in P1 hearts of Rbpms KO mice. GAPDH is a loading control. E. Survival curve of Rbpms KO mice. F. Representative image of Rbpms WT and KO pups at P2. Scale bar: 5 mm. G. Whole mount (top panel) and H&E sagittal sections showing the ductus arteriosus (DA), pulmonary artery (PA), aorta artery (AO) in WT and KO pups 6 h after birth. Black arrow indicates double Outlet Right Ventricle (DORV) in KO heart. Scale bars (top): 500 μm, (bottom): 200 μm. H. H&E-stained coronal sections of representative hearts from P1 WT and KO pups fixed in diastole. Magnified images show left and right ventricular regions. Blue arrow in KO heart section indicates overriding aorta. Scale bar: 300 μm. I-M. Measurements of left ventricle (LV) compact myocardium, right ventricle (RV) compact myocardium, septal, LV trabecular and RV trabecular zone thicknesses in coronal sections at the level of papillary muscle roots (n = 11 for WT and KO). N, O. Fractional shortening (FS%) and ejection fraction (EF%) of WT and KO mouse hearts between P1–P3 (n = 6 for WT, and 5 for KO). All data are presented as mean ± SEM.
To explore the function of Rbpms, we generated Rbpms knockout (KO) mice using CRISPR/Cas9-mediated genome editing. The murine Rbpms gene spans seven exons. We deleted the first exon using two single-guide RNAs (sgRNAs) (Figure S1B), eliminating the translation start site (TSS) of the open reading frame (ORF), thus disrupting Rbpms expression. Loss of Rbpms mRNA and protein in KO mice were confirmed by RNA sequencing and western blot analysis, respectively, in the heart at postnatal day 1 (P1) (Figures 1C and 1D).
Rbpms KO mice were born at Mendelian ratios from heterozygous intercrosses (Figure S1C) and were readily identifiable by their smaller body size at P1 (Figure S1D). Homozygous mutant mice failed to thrive after birth, as evidenced by the failure to gain weight, and all KO pups died by P4 (Figures S1E and 1E). The KO pups were cyanotic before death (Figure 1F), suggesting poor oxygenation. Consistent with this observation, in all KO pups (7 out of 7 pups) the ductus arteriosus of the heart failed to close after birth, a condition termed patent ductus arteriosus (PDA) (Figure 1G). The ductus arteriosus provides an arterial connection between the pulmonary artery and the descending aorta during fetal life. It is permanently closed soon after birth to separate the pulmonary and systemic circulations. PDA, in which the ductus fails to close, causes juvenile lethality in mice (Turgeon and Meloche, 2009). Necropsies on dead pups revealed severe and broad hemorrhages in pulmonary alveoli in KO lungs (Figures S1F and S1G), a likely consequence of PDA. We also analyzed lung morphology of KO pups immediately after birth, and did not observe any obvious defects (Figure S1H), suggesting that Rbpms deletion does not affect lung function. Although Rbpms is also expressed in other tissues, such as kidney and intestine (Figure 1A), we did not detect morphological abnormalities in these tissues in KO pups (Figures S1I and S1J), and the dead pups all contained milk in their stomachs (Figure S1K). Taken together, these findings infer that KO pups died primarily, if not solely, because of cardiovascular developmental defects, including PDA.
Hypertrabeculation and noncompaction cardiomyopathy of Rbpms KO hearts.
Histological analysis revealed hyper-trabeculated ventricles with thinner compact myocardium without fibrosis in hearts of KO mice compared to WT mice at P1 (Figures 1H and S1L). At the level of the papillary muscle, the thickness of the ventricular compact zone and interventricular septum were reduced, whereas trabecular thickness were increased in KO hearts (Figures 1I–1M). In both left and right ventricles, the trabecular-to-compact myocardium thickness ratios were significantly higher in KO mice than WT mice at P1 (Figures S1M and S1N). We also observed ventricular septal defects (VSDs) and double outlet right ventricle (DORV) in histological sections of KO hearts with a high penetrance (7 out of 12 pups at P1) (Figures 1G, 1H and S1O). The hyper-trabeculation and reduced thickness of the compact myocardium of the left and right ventricles are hallmarks of noncompaction cardiomyopathy. KO hearts showed significantly reduced contractile function, as revealed by decreased fractional shortening (FS) and ejection fraction (EF) measured by echocardiography between P1 to P3 before death (Figures 1N and 1O). Rbpms heterozygous mice were viable and showed no discernable abnormalities but displayed subtle reductions in cardiac FS and ventricular wall thickness as revealed by echocardiographic analysis at 4 months of age (Figures S1P and S1Q, Supplemental Table 1), indicative of modest cardiac functional defects caused by Rbpms haploinsufficiency.
Absence of Rbpms disrupts cardiomyocyte cytokinesis and causes premature binucleation.
Abnormalities in cardiomyocyte proliferation are associated with noncompaction cardiomyopathy (Kodo et al., 2016, Chen et al., 2009, Arndt et al., 2013, Luxan et al., 2013). Therefore, we analyzed cardiomyocyte proliferation by immunofluorescence staining with markers for different stages of the cell cycle in P1 hearts. We first performed immunofluorescence staining for Ki67, which is expressed throughout the cell cycle (Hutchins et al., 2010). Cardiomyocyte nuclei were detected by pericentriolar material 1 (PCM1) immunofluorescence staining, and cardiomyocytes were stained with cardiac troponin I (cTnI) (Bergmann et al., 2009). No difference was observed in the frequency of Ki67 positive cardiomyocyte nuclei between WT and KO hearts at P1 (Figures 2A, 2B and S2A), indicating that Rbpms does not affect overall cell cycle activity of cardiomyocytes. We next assessed phospho-histone H3 (pH3), a marker for mitosis. KO hearts showed a significant decrease in the frequency of pH3-positive cardiomyocytes compared with WT littermates (Figures 2C, 2D and S2B), suggesting a G2-M phase transition defect in KO hearts at P1. Given that Rbpms is also expressed in non-cardiomyocyte lineages (Figure 1B), we assessed the proliferation rate of non-cardiomyocytes in the heart that are pH3-positive and PCM1-negative, and did not see any difference between WT and KO hearts (Figure S2C). We also analyzed the proliferation rate of atrial cardiomyocytes, and did not observe any difference between WT and KO hearts (Figures S2D and S2E), suggesting that Rbpms deletion causes proliferation defects mainly in ventricular cardiomyocytes. Intriguingly, we found that KO pups had a significantly higher percentage of pH3 positive nuclei in aorta and pulmonary arteries (Figures S2F and S2G), suggesting that Rbpms deletion promotes cell proliferation in arteries, in contrast to the phenotype in ventricular cardiomyocytes.
Figure 2. Cytokinesis defects of P1 Rbpms KO mouse cardiomyocytes.
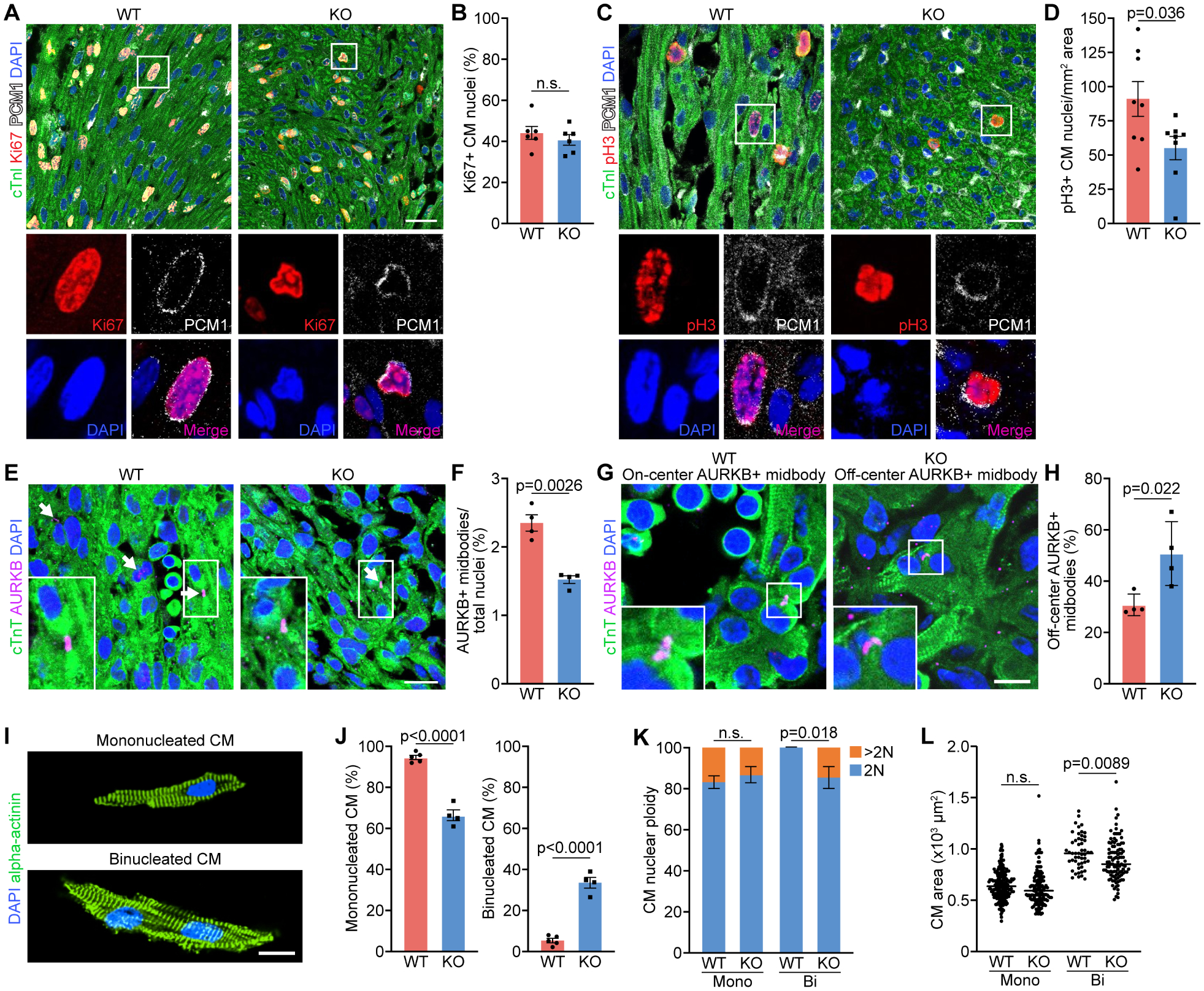
A, B. Immunofluorescence staining for Ki67, cardiac troponin I (cTnI), PCM1 and DAPI on coronal sections of WT and KO myocardium at P1 and quantification of percentage of Ki67+ cardiomyocyte (CM) nuclei over total nuclei (n = 6 for WT and KO). Scale bar: 30 μm. C, D. Immunofluorescence staining for phosphorylated histone H3 (pH3), cTnI, PCM1 and DAPI of WT and KO myocardium at P1, and quantification of number of pH3+ CM nuclei per mm2 area. (n = 8 for WT and KO). Scale bar: 30 μm. E, F. Immunofluorescence staining for aurora B kinase (AURKB), cTnT and DAPI of WT and KO hearts at P1 and quantification of AURKB-positive midbody frequency (n = 4 for WT and KO). Scale bar: 25 μm. White arrows indicate AURKB-positive midbodies between nuclei. G. Representative immunofluorescence staining images of on-center and off-center AURKB-positive midbodies in P1 WT and KO heart sections. Scale bar: 10 μm. H. Percentage of off-center AURKB-positive midbodies in WT and KO hearts at P1 (n = 4 for WT and KO). I. Representative immunofluorescence images of P1 mononucleated and binucleated CMs stained for alpha-actinin. Scale bar: 20 μm. J. Percentage of mononucleated and binucleated CMs of P1 WT and KO hearts (n = 5 for WT, and 4 for KO). K. Quantification of the nuclear ploidy of the mononucleated (mono) and binucleated (Bi) CMs in P1 WT and KO hearts (n = 5 for WT mice, and 4 for KO mice, average 100–200 cardiomyocytes per mouse). n.s., not significant. L. Areas of individual mononucleated and binucleated CMs (n = 5 for WT mice, and 4 for KO mice, average 100–200 cardiomyocytes per mouse). n.s., not significant. All data are presented as mean ± SEM.
To determine the percentage of cardiomyocytes completing the cell cycle, we assessed the cytokinesis marker aurora B kinase (AURKB), which is localized in the central mitotic spindle and cleavage furrow (Krenn and Musacchio, 2015). We found that KO hearts had a significant decrease in the percentage of AURKB-positive midbodies compared to WT (Figures 2E, 2F, S2H and S2I), suggesting a cytokinesis defect in KO cardiomyocytes. A recent report showed that on-center positioned AURKB in the midbody predicts successful cytokinesis, while off-center AURKB-positive midbodies correspond to incomplete cytokinesis (Hesse et al., 2018). In this regard, we observed both on-center and off-center AURKB-positive midbodies in sections of hearts at P1 (Figure 2G). Interestingly, the KO hearts showed a significantly higher percentage of off-center AURKB-positive midbodies than WT controls, and reduced on-center AURKB-positive midbodies (Figures 2H and S2J), further confirming the cytokinesis failure of KO cardiomyocytes.
Cytokinesis defects cause binucleation of cardiomyocytes (Gan et al., 2020). We therefore analyzed individual cardiomyocytes isolated from WT and KO hearts at P1. As expected, the percentage of binucleated cardiomyocytes was 6-fold higher in KO hearts than WT hearts, with a concomitant decrease in the percentage of mononucleated cardiomyocytes, indicating that the cytokinesis defects of KO cardiomyocytes result in increased binucleation (Figures 2I and 2J). To further study karyokinesis, we performed ploidy analysis of the mononucleated and binucleated cardiomyocyte nuclei (Gan et al., 2019, Gan et al., 2020b). In WT hearts, 83% of the mononucleated cardiomyocytes contain diploid nuclei (2N), while 17% are polyploid (>2N). The polyploid nuclei in mononucleated cardiomyocytes represent those undergoing DNA synthesis before karyokinesis. All the binucleated cardiomyocyte nuclei in P1 WT hearts are diploid (2N). There was no change in ploidy levels of KO mononucleated cardiomyocytes (Figure 2K). Intriguingly, there was a significant increase in polyploid (>2N) nuclei in KO binucleated cardiomyocytes, suggesting continuous DNA synthesis without karyokinesis (Figures 2K and S2K). This result is consistent with the normal DNA synthesis detected by Ki67 staining in KO hearts (Figure 2B).
The mean cell size of mononucleated cardiomyocytes in P1 KO hearts was comparable to WT littermates, whereas the binucleated KO cardiomyocytes were slightly smaller than WT (Figure 2L). To exclude the possibility that the noncompaction cardiomyopathy in KO hearts was caused by abnormal cardiomyocyte apoptosis, we performed TUNEL assay, and observed no difference between KO and WT hearts (Figures S2L and S2M). In summary, the noncompaction cardiomyopathy in Rbpms KO mice correlated with defects in cardiomyocyte cytokinesis.
Noncompaction cardiomyopathy in Rbpms KO mice is a developmental defect caused by premature cardiomyocyte binucleation.
To determine the onset of noncompaction cardiomyopathy observed at P1, we analyzed WT and KO embryonic hearts from E12.5 to E18.5 by immunofluorescence staining for cardiac troponin T (cTnT). At E12.5, no obvious morphological differences were observed between KO and WT hearts (Figure 3A). From E14.5 onwards, the KO hearts began to show excessive trabeculation and significant decreases in the thickness of the LV and RV compact zones and septum thickness measured at the level of the papillary muscle compared to WT hearts (Figures 3B–3D), suggesting that the noncompaction cardiomyopathy is a developmental defect that manifests before E16.5. We also observed VSDs and overriding aorta in KO hearts at E16.5 and E18.5 (Figure 3A, indicated by yellow arrows).
Figure 3. Noncompaction cardiomyopathy in Rbpms KO mice is a developmental defect.
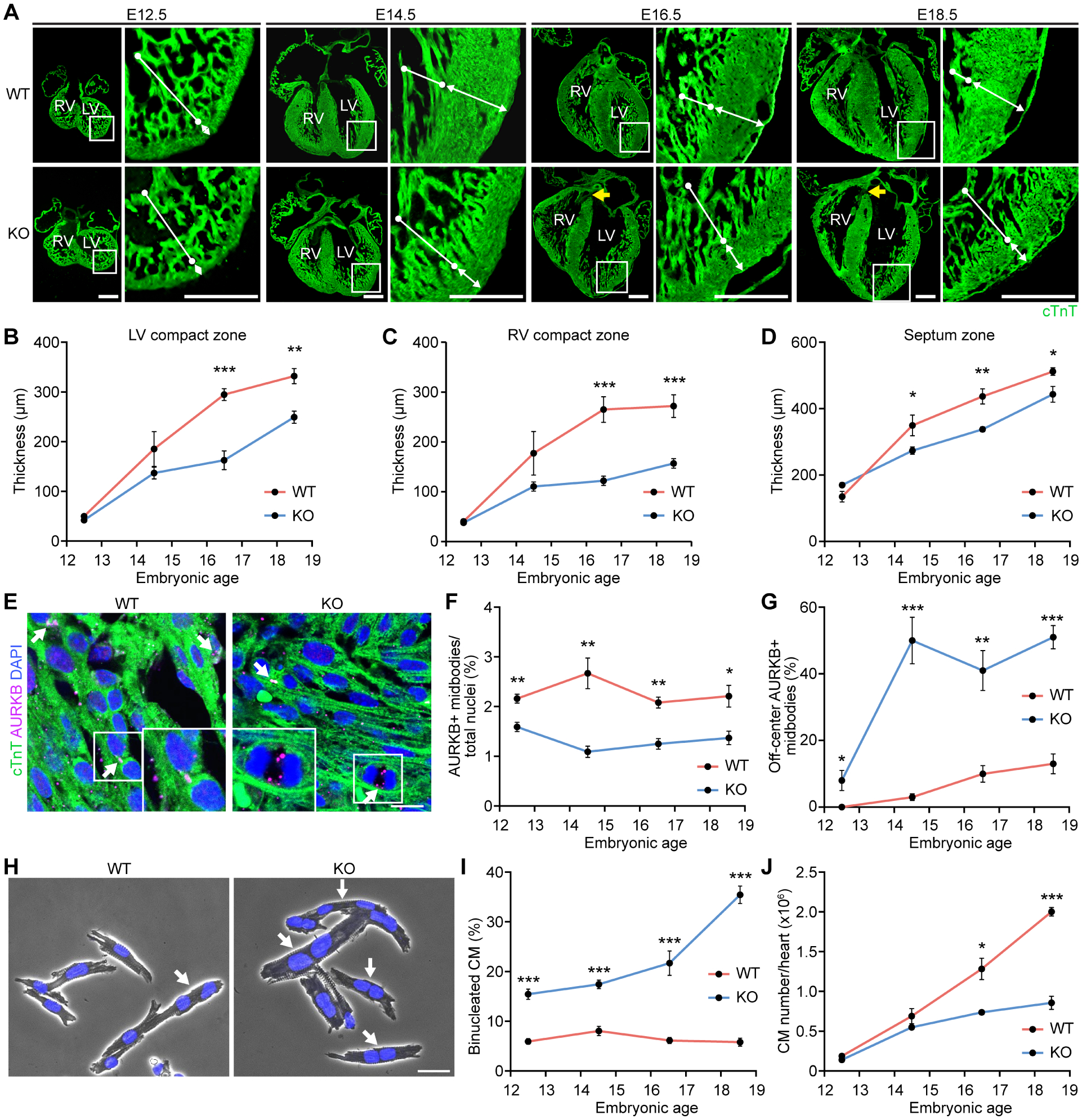
A. Representative immunofluorescence images of coronal heart sections at indicated embryonic time points stained for cTnT. White bars with round head indicate LV trabeculae, and white bars with arrowhead indicate LV compact zone. Yellow arrows in KO heart sections indicate overriding aorta. Scale bar: 400 μm. B-D. Quantification of thickness of LV compact zone, RV compact zone and septum zone in embryonic hearts (n = 5–7 for WT and KO at each time point). E. Representative immunofluorescence images of WT and KO hearts at E16. 5 stained for AURKB and cTnT. Scale bar: 25 μm. White arrows indicate AURKB-positive midbodies between nuclei. F. Quantification of AURKB-positive midbody frequency in embryonic hearts (n = 5–7 for WT and KO at each time point). G. Quantification of off-center AURKB-positive midbody percentage in embryonic hearts (n = 4–6 for WT and KO at each time point). H. Representative phase contrast microscopy images of isolated cardiomyocytes from E18.5 WT and KO hearts stained with DAPI. Scale bar: 30 μm. White arrows indicate binucleated cardiomyocytes. I. Quantification of binucleated cardiomyocytes in embryonic hearts (n = 3–6 for WT and KO mice at each time point, average 100–200 cardiomyocytes per mouse). J. Total cardiomyocyte number in hearts at the indicated embryonic time points (n = 5–7 for WT and KO at each time point). *p<0.05, **p<0.01; ***p<0.001; n.s., not significant. All data are presented as mean ± SEM.
We next analyzed cardiomyocyte cell cycle and cytokinesis activities across developmental stages. Consistent with the results from P1 hearts, no difference was observed in the percentage of Ki67-positive or pH3-positive cardiomyocyte nuclei between WT and KO hearts, except for a decrease in pH3 staining in KO hearts at E18.5 (Figures S3A and S3B), suggesting that the cardiomyocyte G2-M transition defect manifested only in late embryogenesis. Interestingly, at all embryonic stages, KO hearts showed substantially lower frequencies of AURKB-positive midbodies (Figures 3E and 3F), while the off-center AURKB-positive midbody frequencies were noticeably higher in KO hearts across the time course (Figure 3G), suggesting that cytokinesis defects of Rbpms KO cardiomyocytes manifest as early as E12.5. There was a 3-fold increase in the precent of binucleated cardiomyocytes in KO hearts compared to WT hearts at E12.5, consistent with the cytokinesis defect (Figures 3H and 3I).
The frequency of cardiomyocyte binucleation in KO hearts increased during embryonic development (Figure 3I). In contrast, the binucleation frequency in WT cardiomyocytes remained low across embryonic stages, consistent with previous observations (Soonpaa et al., 1996) (Figure 3I). By E18.5, 35% of the KO cardiomyocytes were binucleated compared with only 5.8% of WT cardiomyocytes (Figure 3I), suggesting that the binucleated cardiomyocytes begin to accumulate in Rbpms KO hearts at E12.5.
Cardiomyocyte cytokinesis failure was previously shown to lead to an increase in binucleated cardiomyocytes and a decrease in cardiomyocyte number (Stopp et al., 2017, Liu et al., 2019). To determine cardiomyocyte numbers at different developmental stages, we performed stereological analysis of heart sections and calculated total cardiomyocyte number based on the total pericentriolar material 1 positive (PCM1+) cardiomyocyte nuclei number and the mono/binucleated cardiomyocyte ratio at each developmental stage. Rbpms KO mice had significantly fewer cardiomyocytes than WT mice starting from E16.5 (Figure 3J). Since a fraction of Rbpms KO pups also developed VSDs, we checked whether the cardiomyocyte proliferation defects influence the emergence of VSDs. We compared cardiomyocyte proliferation of the KO hearts with VSDs with those without VSDs, and observed no significant difference in cardiomyocyte mitosis or cytokinesis between VSD and non-VSD KO animals at E18.5 and P1 (Figures S3C and S3D).
To exclude the possibility that the reduced cardiomyocyte number in KO hearts is due to increased apoptosis, we performed TUNEL assay on heart sections and observed no difference in cardiomyocyte apoptosis at any time point (Figure S3E). Therefore, we conclude that the cardiomyocyte cytokinesis defects in KO hearts during development resulted in a decrease in cardiomyocyte number, leading to noncompaction cardiomyopathy.
Deletion of RBPMS in human iPSC-derived cardiomyocytes causes cytokinesis defects.
Human and mouse RBPMS proteins share 99% homology in amino acid sequences, suggesting a conserved function (Figure S4A). To explore the function of RBPMS in human cardiomyocytes, we generated an isogenic RBPMS knockout (KO) human induced pluripotent stem cell (hiPSC) line by CRISPR/Cas9 genome editing. We designed a sgRNA to target the second exon of RBPMS and selected a clone with a biallelic 1 base-pair insertion in the middle of exon 2 (Figure S4B). The +1 bp insertion caused a frameshift of the RBPMS ORF and introduced a premature stop codon, thus disrupting RBPMS protein expression. RBPMS-KO hiPSCs were differentiated into cardiomyocytes, indicating that deletion of RBPMS does not affect cardiogenesis in hiPSCs. We confirmed the loss of RBPMS protein in the KO line by western blot analysis and immunofluorescence staining (Figures S4C and S4D). We next assessed whether the RBPMS-KO hiPSC-cardiomyocytes recapitulated the cytokinesis defect in Rbpms KO mice. On day 22 of differentiation, we observed a notably higher percentage of binucleated RBPMS-KO hiPSC-cardiomyocytes than WT cardiomyocytes (Figure 4A). Time course analysis revealed a higher percentage of binucleated RBPMS-KO cardiomyocytes than WT cardiomyocytes starting from day 21, and the difference became more pronounced over time (Figure 4B). Moreover, the growth rate of RBPMS-KO hiPSC-cardiomyocytes was substantially lower than that of WT cardiomyocytes (Figure 4C). These results indicate that in hiPSC-derived cardiomyocytes, loss of RBPMS caused increased cardiomyocyte binucleation and delayed cardiomyocyte growth, resembling the phenotypes seen in KO mouse hearts.
Figure 4. Cytokinesis defects in RBPMS-KO hiPSC-derived cardiomyocytes.
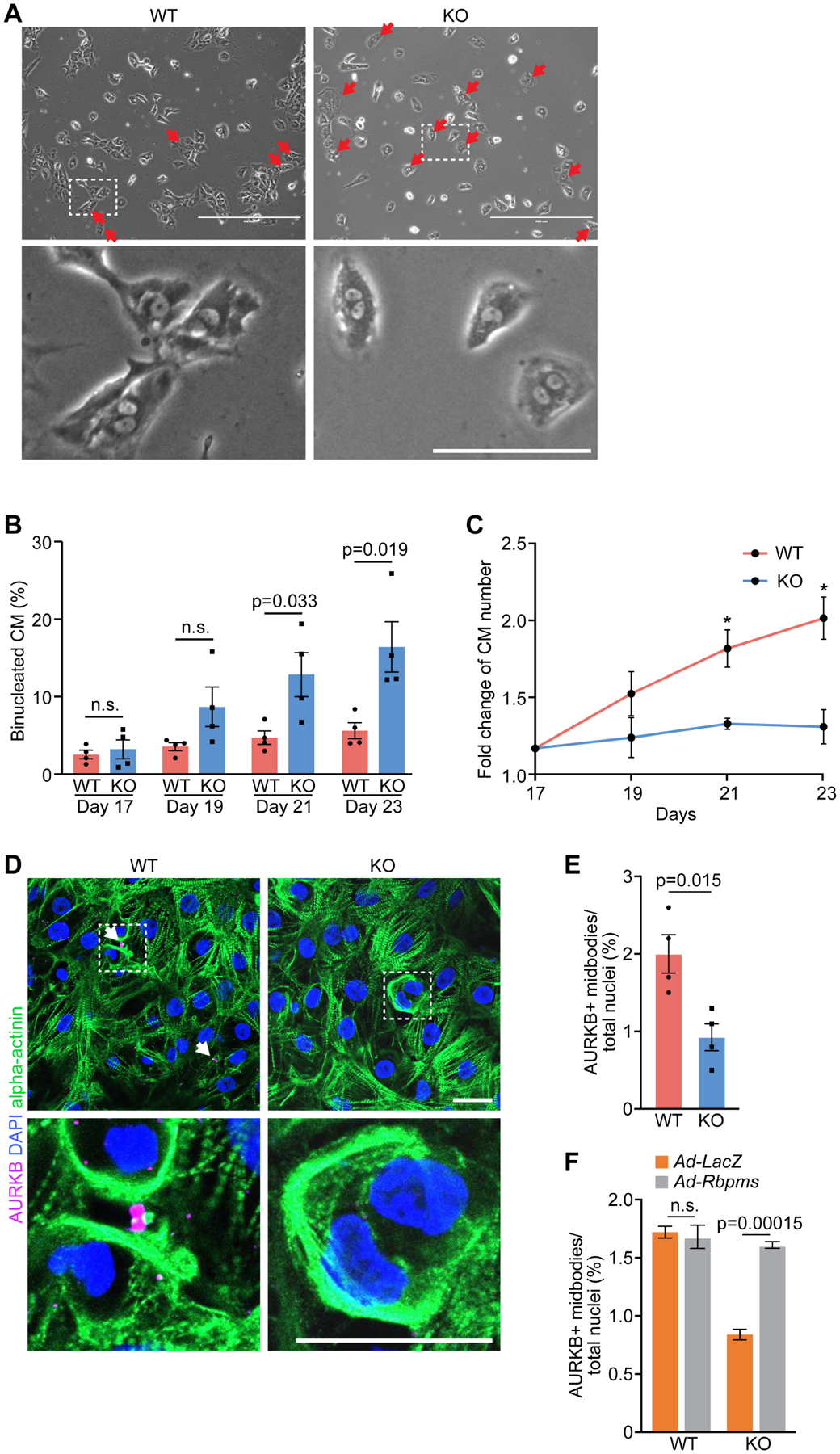
A. Phase contrast microscopy of WT and RBPMS-KO hiPSC-cardiomyocytes. Red arrows indicate binucleated hiPSC-cardiomyocytes. Scale bars (top): 500 μm, (bottom): 100 μm. B. Percentage of binucleated WT and RBPMS-KO hiPSC-cardiomyocytes at indicated time points (n = 3 for WT groups, and 4 for KO groups, average 200 cardiomyocytes per group). n.s., not significant. C. Fold change of cardiomyocytes number in WT and RBPMS-KO cardiomyocytes at indicated time points. D. Immunofluorescent staining for AURKB and alpha-actinin in WT and RBPMS-KO hiPSC-cardiomyocytes. White arrows indicate AURKB+ midbodies in between nuclei. Scale bar: 25 μm. E. Quantification of AURKB-positive midbody frequencies in WT and RBPMS-KO hiPSC-cardiomyocytes (n = 4 for WT and KO groups, average 300 cardiomyocytes per group). F. Quantification of AURKB-positive midbody frequencies in WT and RBPMS-KO hiPSC-cardiomyocytes infected with Ad-LacZ and Ad-Rbpms (n = 4 for WT groups, and 3 for KO groups, average 300 cardiomyocytes per group). All data are presented as mean ± SEM.
To determine whether the increased binucleation of RBPMS-KO hiPSC-cardiomyocytes was also caused by cytokinesis failure, we performed immunofluorescence staining for AURKB. RBPMS-KO hiPSC-cardiomyocytes showed a noticeably reduced frequency of AURKB+ midbodies compared to WT (Figures 4D and 4E). Furthermore, overexpression of murine Rbpms in KO hiPSCs-cardiomyocytes rescued the number of AURKB+ midbodies to the level of WT hiPSCs-cardiomyocytes (Figures 4F, S4E–S4G). Interestingly, Rbpms overexpression in WT hiPSC-cardiomyocytes did not further promote cytokinesis (Figure 4F). These results confirmed that the absence of RBPMS in hiPSC-cardiomyocytes directly caused cytokinesis defects and binucleation.
Rbpms regulates alternative splicing events in the heart.
RBPMS is classified as an RNA binding protein, but little is known about its functions in RNA processing or other cellular functions. To elucidate the molecular functions of Rbpms in the heart, we performed paired-end RNA-sequencing (RNA-seq) on ventricles of Rbpms KO and WT mice at P1, and identified 99 upregulated genes and 327 downregulated genes in KO samples compared with WT littermate controls (Figure 5A). Gene ontology analysis revealed that cytoskeleton-related pathways, such as cilium movement and organization, microtubule-based movement, and microtubule bundle formation, were the most downregulated biological pathways in KO hearts (Figure 5B). These data suggest that Rbpms may contribute to cardiomyocyte cytoskeleton-associated biological processes. Cell division is a process regulated by cytoskeleton reorganization, including the formation of the mitotic spindle for chromosome segregation and the contractile ring for cytokinesis (Ali et al., 2020). Thus, RNA-seq analysis data corroborate the role of Rbpms in regulating cardiomyocyte cytokinesis.
Figure 5. Paired-end RNA sequencing reveals Pdlim5 as a splicing target of RBPMS.
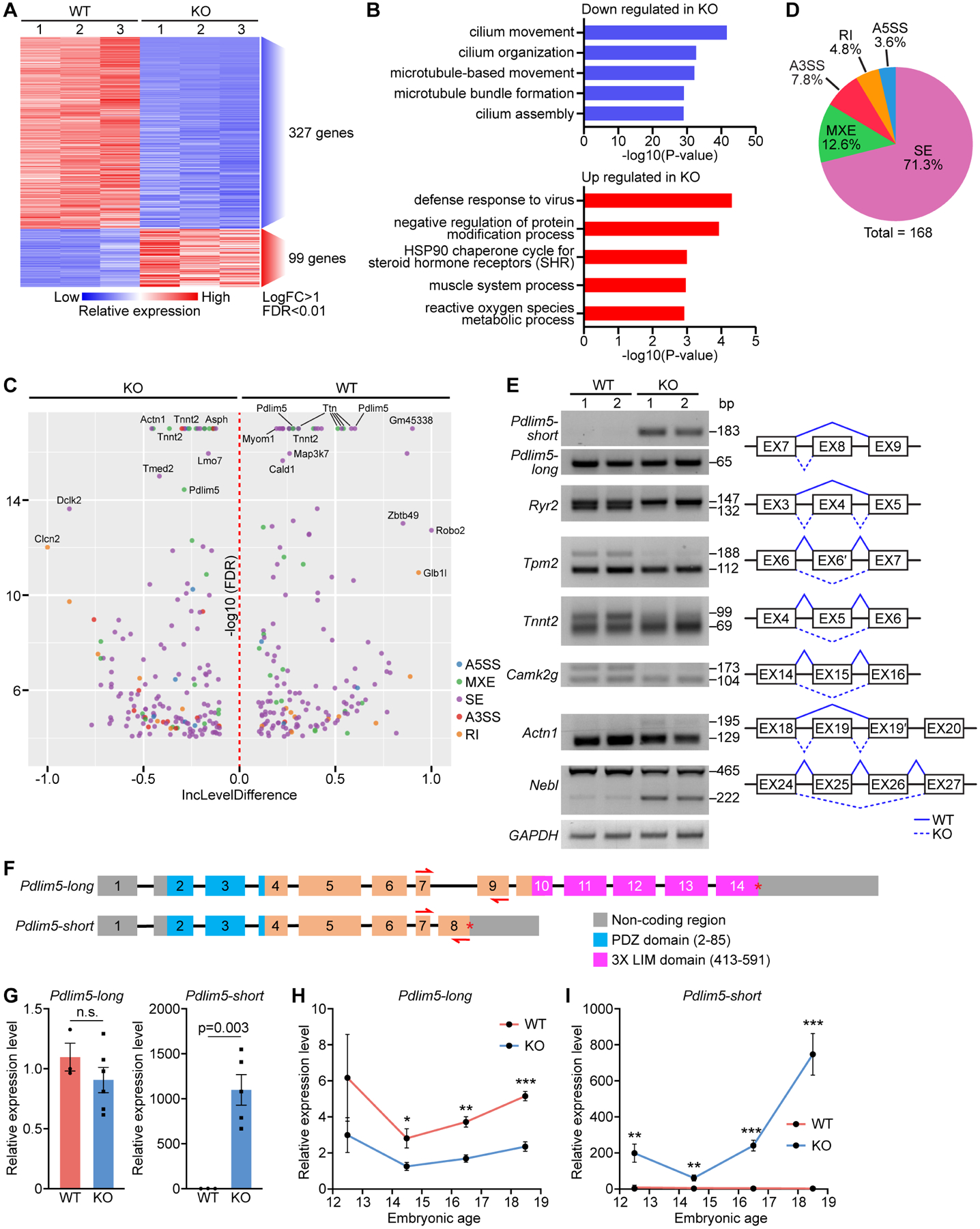
A. Heatmap of differentially expressed genes between WT and KO P1 heart ventricles identified by RNA-seq. B. Top GO terms for down and upregulated genes in KO samples. C. Volcano plot showing the inclusion (Inc) level differences (WT Inclevel – KO Inclevel, X axis) and False Discovery Rates (FDR) for differential alternative splicing events (ASEs) between WT and KO P1 heart ventricles. Y axis is presented as −log10(FDR). D. Pie chart showing the percentage of different ASEs in Rbpms KO mice (FDR<0.01). SE: skipped exon, MXE: mutually exclusive exon, RI: retained intron, A3SS: alternative 3’ splice site, and A5SS: alternative 5’ splice site. E. Representative RT-PCR confirmation of abnormal splicing events in KO heart, and schematic diagram of SE events based on rMATs analysis. Solid lines indicate normal splicing events, and dashed lines indicate abnormal events in KO hearts. F. Schematic of murine Pdlim5 long and short isoform gene structures. Boxes represent exons, different functional domains are labelled with different colors. Red arrows indicate the locations of qRT-PCR primers for determining long and short isoforms expression. G. qRT-PCR of the relative expression of Pdlim5 long and short isoforms in P1 WT and KO heart (n = 3 for WT, and 6 for KO). H, I. qRT-PCR of relative expression levels of Pdlim5-long and short isoforms in WT and KO hearts at different embryonic time points (n = 4–6 for WT or KO). *p<0.05, **p<0.01; ***p<0.001; n.s., not significant. All data are presented as mean ± SEM.
A previous report demonstrated that RBPMS acts as a smooth muscle RNA splicing factor (Nakagaki-Silva et al., 2019). To determine whether RBPMS is also involved in RNA splicing in mouse hearts, we performed replicate multivariate analysis of transcript splicing (rMATS) on the paired-end RNA-seq dataset to identify alternative splicing events in KO and WT hearts (Shen et al., 2014). We identified 168 differential alternative splicing events (ASEs) in KO hearts (Figure 5C). The most common ASEs were skipped exons (SEs), representing 71.3% of the total ASEs, followed by mutually exclusive exons (MXEs, 12.6%), alternative 3’ splice sites (A3SSs, 7.8%), retained introns (RIs, 4.8%), and alternative 5’ splice sites (A5SSs, 3.6%) (Figure 5D), suggesting that RBPMS is involved in differential alternative splicing events. The nuclear localization of RBPMS in hiPSC-cardiomyocytes (Figure S4D) supports its role as an RNA splicing regulator.
We validated multiple alternative splicing events of the top ranked genes by RT-PCR, including Pdlim5, Ryr2, Tpm2, Tnnt2, Camk2g, Actn1, Nebl (Figure 5E). Many of these target genes are involved in cardiac myofibrillogenesis and sarcomere structure. Mutations in sarcomeric genes account for approximately 50% of LVNC patients (Hirono et al. 2020). We observed that those genes that are known to associate with LVNC in humans are dysregulated in Rbpms KO hearts, such as Myh7, Myh6, Prdm16 and Dsp (Figures S5A to S5D), indicating a potentially shared pathological pathway of noncompaction cardiomyopathy between mice and humans.
Rbpms mediates long and short isoform switching of Pdlim5 transcripts.
Among the top-ranked alternatively spliced genes, we focused on PDZ and LIM domain protein 5 (Pdlim5), which was reported to influence cytoskeletal reorganization during cell division (Cheng et al., 2010, Huang et al., 2020). PDLIM5 protein contains a PDZ domain at its N-terminus and three LIM domains at its C-terminus (Yamazaki et al., 2010). The primary Pdlim5 transcripts contain several splice variants, clustered into two main groups: Pdlim5-long transcripts and Pdlim5-short transcripts (Figure 5F). The long and short variants differ in the inclusion of exon 8, which contains a stop codon and is spliced out in the long variants (Figure 5F). Pdlim5-long transcripts encode the LIM-containing isoforms, while Pdlim5-short transcripts encode the LIM-less isoforms. Expression of the long and short transcripts is dynamically regulated during heart development (Yamazaki et al., 2010). Pdlim5-long transcripts are highly expressed at E14.5, and expression gradually decreases after birth (Figure S5E). In contrast, expression of the short transcript is low at E14.5 and increases dramatically starting from P1 (Figure S5F). Interestingly, the temporal switching of the long and short transcripts coincides with the onset of cardiomyocyte binucleation (Soonpaa et al., 1996).
The frequency of Pdlim5 exon 8 exclusion (skipped exon, SE) was substantially higher in WT hearts than in KO hearts (Figure 5C), which was validated by RT-PCR analysis (Figure 5E). We further quantified the level of the short transcript by qRT-PCR. Using primer pairs that can specifically recognize the Pdlim5 long and short variants, respectively (Figures 5F and 5G), we found that expression of the Pdlim5-short isoforms in KO samples was >1,000-fold higher than in WT hearts at P1, while no obvious difference was observed in the expression level of the Pdlim5-long isoforms (Figure 5G). Total Pdlim5 transcript levels were slightly higher in KO samples (Figure S5G). We further confirmed that the increased expression of the Pdlim5-short transcripts leads to increased expression of the short LIM-less PDLIM5 protein, as detected by western blot analysis (Figure S5H).
Next, we measured the relative expression of the Pdlim5 long and short variants in KO and WT hearts during embryonic development by qRT-PCR. We found that expression of Pdlim5-long variants was slightly lower in KO hearts across the time course (Figure 5H). In contrast, expression of the Pdlim5-short variants was higher in KO hearts compared to WT controls from E12.5 and the difference between KO and WT increased through development (Figure 5I), coinciding with the increased cardiomyocyte binucleation in KO hearts during embryogenesis (Figure 3I). These findings confirmed that deletion of Rbpms caused an increased accumulation of Pdlim5-short transcripts in embryonic hearts.
The Pdlim5-short variant inhibits cardiomyocyte cytokinesis.
Human PDLIM5 long and short isoforms differ by the inclusion of exon 9 (equivalent to exon 8 of mouse Pdlim5) (Figure S6A). To determine whether PDLIM5 is also a splicing target of RBPMS in hiPSC-cardiomyocytes, we performed qRT-PCR analysis with primer pairs recognizing human PDLIM5 long and short variants, respectively (Figure S6A). In RBPMS-KO hiPSC-cardiomyocytes, PDLIM5-short transcripts were expressed at 50-fold higher levels than in WT cardiomyocytes, while no difference was observed in the expression of the long transcripts between KO and WT cardiomyocytes (Figure 6A). To determine whether the absence of RBPMS directly causes an increase in PDLIM5-short transcripts in KO cardiomyocytes, we used an adenovirus construct to overexpress murine Rbpms in KO hiPSC-cardiomyocytes. qRT-PCR demonstrated that Rbpms overexpression in KO iPSC-cardiomyocytes dramatically reduced the level of PDLIM5-short transcripts but had no effect on the expression of PDLIM5-long transcripts (Figure S6B), confirming that RBPMS inhibits the expression of PDLIM5-short transcripts.
Figure 6. Pdlim5 long and short variants regulate cytokinesis in hiPSC-cardiomyocytes.
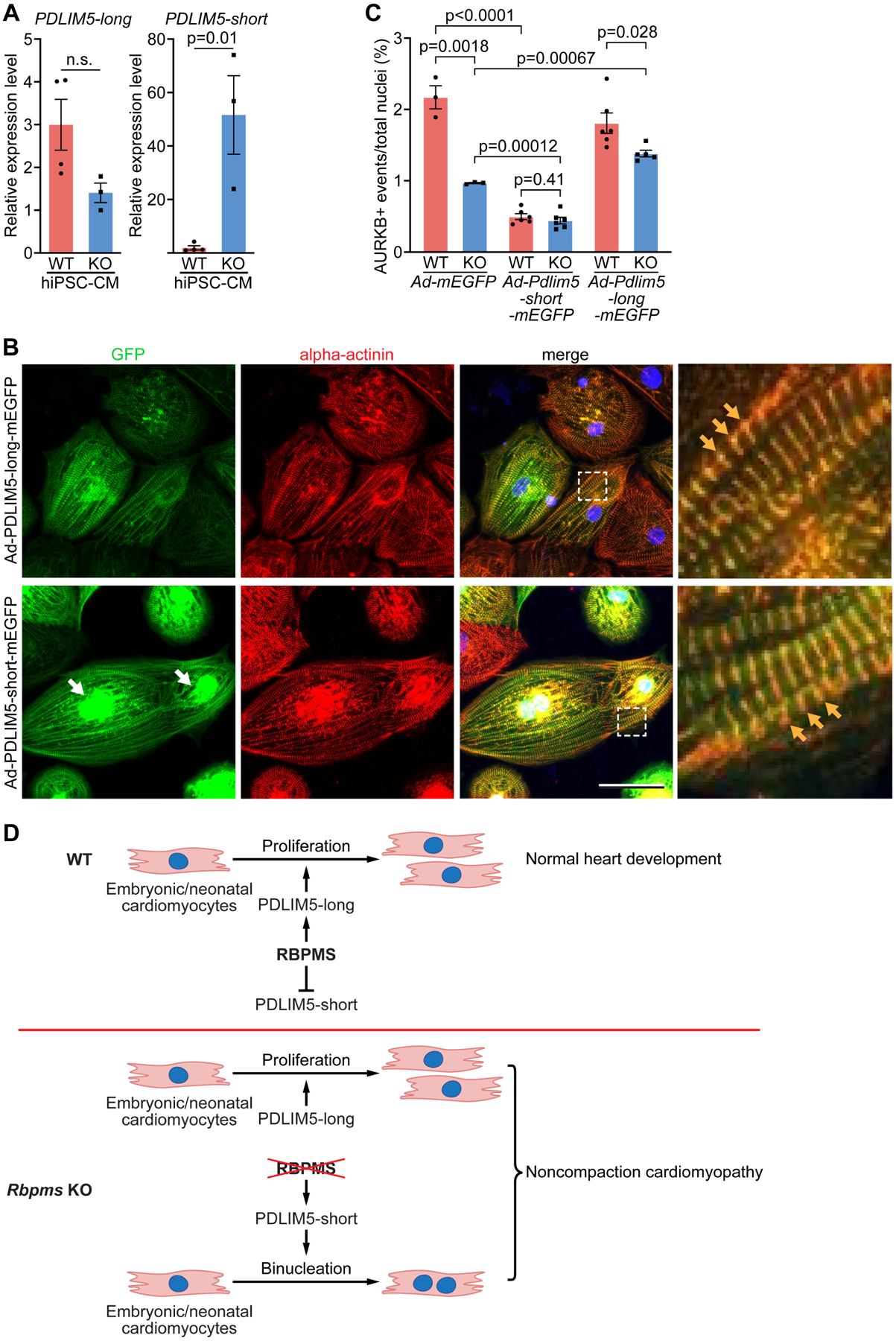
A. Quantification of relative expression levels of PDLIM5 long and short isoforms in WT and KO hiPSC-cardiomyocytes by qRT-PCR (n = 4 for WT groups, and 3 for KO groups). B. hiPSC-cardiomyocytes were infected with Ad-Pdlim5-long-mEGFP and Ad-Pdlim5-short-mEGFP and immunostained for GFP and alpha-actinin. White arrows indicate the accumulation of Pdlim5-short variants surrounding nuclei. Yellow arrows indicate Pdlim5 long and short isoforms colocalizing with alpha-actinin at Z-discs. Scale bar: 50 μm C. Quantification of AURKB-positive midbody frequency in WT and KO hiPSC-cardiomyocytes infected with Ad-mEGFP, Ad-Pdlim5-long-mEGFP and Ad-Pdlim5-short-mEGFP (n = 4–6 for WT or KO groups, average 300 cardiomyocytes per group). D. Schematic diagrams showing the function of RBPMS in cardiomyocyte proliferation and WT heart development (top), and loss of Rbpms causes cardiomyocyte binucleation and noncompaction cardiomyopathy (bottom). All data are presented as mean ± SEM.
The temporal correlation between the Pdlim5 long to short isoform switch and cardiomyocyte binucleation after birth strongly implicates Pdlim5 in cardiomyocyte binucleation. To further test this hypothesis, we infected WT and KO hiPSC-cardiomyocytes with adenoviruses overexpressing murine Pdlim5-long and short isoforms, respectively (Figures S6C and S6D). In hiPSC-cardiomyocytes, PDLIM5 long and short proteins both colocalized with sarcomeric alpha-actinin at the Z-disc of sarcomeres (Figure 6B). Overexpression of the Pdlim5-short variant in WT hiPSC-cardiomyocytes inhibited cytokinesis activity, as shown by the reduced AURKB-positive midbodies from 2.2% to 0.5% (Figures 6C and S6E). Overexpression of Pdlim5-short variant in KO hiPSC-cardiomyocytes further reduced cytokinesis activity from 1.0 % to 0.44% (Figures 6C and S6E), suggesting a dosage-dependent effect of the Pdlim5-short variant in inhibiting cytokinesis (Figure 6C). Meanwhile, overexpression of the Pdlim5-long variant in RBPMS-KO hiPSC-cardiomyocytes enhanced cardiomyocyte cytokinesis activity from 1.0% to 1.4%, but had no effect on WT hiPSC-cardiomyocytes (Figure 6C). Taken together, these results confirmed that the Pdlim5-short variant can directly inhibit cardiomyocyte cytokinesis, and the overexpression of Pdlim5-long variant can partially rescue the cardiomyocyte cytokinesis defects of RBPMS-KO hiPSC-cardiomyocytes. Interestingly, we observed that the PDLIM5-short variant aggregated together with alpha-actinin surrounding the nuclei of hiPSC-cardiomyocytes, in addition to its Z-disc localization, while the PDLIM5-long variant was barely seen around nuclei (Figure 6B). It has been demonstrated that the increased accumulation of alpha-actinin and actin filaments causes cardiomyocyte cytokinesis failure (Mukhina et al., 2007). It is likely that the PDLIM5-short variants entangle with alpha-actinin and other sarcomeric proteins surrounding cardiomyocyte nuclei, disturbing cytokinesis and causing cell binucleation.
Discussion
A major conclusion from this work is that premature cardiomyocyte binucleation during heart development impairs cardiomyocyte proliferation and causes noncompaction cardiomyopathy. We show that a cardiac RNA binding protein, RBPMS, maintains cardiomyocyte cytokinesis during embryogenesis. RBPMS functions, at least in part, by regulating alternative splicing of Pdlim5 in the heart. RBPMS represses the Pdlim5 short isoforms by splicing out exon 8 of Pdlim5 to maintain expression of the Pdlim5-long variants in embryonic cardiomyocytes. The absence of Rbpms in mice and hiPSC-derived cardiomyocytes leads to the accumulation of Pdlim5-short variants, which directly inhibit cardiomyocyte cytokinesis and cause cardiomyocyte binucleation from E12.5 onward. The premature onset of cardiomyocyte binucleation leads to the reduction of cardiomyocyte number increase during embryonic heart development, which then causes noncompaction cardiomyopathy (Figure 6D).
Cardiomyocyte polyploidization influences heart development.
Heart formation depends on active cardiomyocyte proliferation and a robust increase in cardiomyocyte number to sustain cardiac pump function. More than 95% of embryonic cardiomyocytes are mononuclear, diploid and highly proliferative, whereas most adult cardiomyocytes exit the cell cycle and become binucleated (polyploid) (Soonpaa et al., 1996). Cardiomyocyte polyploidization is a finely tuned process, which occurs between P4 and P10 in mice and is coincident with the loss of heart regeneration capacity (Porrello et al., 2011). Previous studies have revealed that disruption of cardiomyocyte polyploidization can alter cardiomyocyte proliferation and heart regeneration capacity thereafter (Patterson et al., 2017, Gonzalez-Rosa et al., 2018, Hirose et al., 2019). However, why mammalian cardiomyocyte polyploidization occurs only after birth, and how polyploidization contributes to postnatal heart development remain unknown. We show that premature onset of cardiomyocyte binucleation before E12.5 leads to precocious cell cycle exit, limiting the increase in cardiomyocyte number during embryogenesis. The reduction in myocardial growth and trabecular remodeling results in postnatal noncompaction cardiomyopathy. Interestingly, besides noncompaction cardiomyopathy, premature cardiomyocyte polyploidization was also observed in other types of cardiac developmental defects, like DCM, hypertrophy, and tetralogy of Fallot (Stopp et al., 2017, Liu et al., 2019, Yu et al., 2013). Taken together, our findings reveal a potential mechanism whereby premature cardiomyocyte cell cycle exit caused by polyploidization might underlie certain congenital heart defects. This may explain why most mammalian cardiomyocytes become polyploid only after birth when the heart is structurally fully developed. Further elucidation of the molecular mechanisms governing cardiomyocyte proliferation and polyploidization may lead to therapeutic interventions for congenital heart diseases and adult heart regeneration.
RBPMS regulates cardiomyocyte cytokinesis and heart development.
RBPs regulate multiple stages of heart development, including heart tube formation, cardiomyocyte differentiation and maturation, myocardial trabeculation and compaction (Blech-Hermoni and Ladd, 2013). Mutations in RBPs have been shown to cause severe cardiac defects. For example, Rbm20 mutations cause DCM (Guo et al., 2012), loss of Rbm24 causes ventricular-septal defects, reduced trabeculation and compaction, and atrial dilation (Yang et al., 2014), and RBFox1 deficiency leads to HCM (Gao et al., 2016). RBPMS is highly expressed throughout cardiac development, which involves active cardiomyocyte proliferation and dramatic changes in cytoskeleton reorganization, sarcomere formation and maturation. In this study, we found that global Rbpms knockout in mice caused neonatal lethality, noncompaction cardiomyopathy and PDA, highlighting the essential role of Rbpms in heart development. Patients with LVNC do not display juvenile lethality (Vaidya et al., 2021). Therefore, the neonatal lethality of Rbpms KO may be caused by other abnormalities of embryogenesis such as vascular development. Indeed, we observed PDA and an increase in arterial cell proliferation in the tunica media in KO pups. Ductus arteriosus closure requires the phenotypic switching of vascular smooth muscle cells from a proliferative state to a contractile state shortly after birth, and failure of vascular smooth muscle cells to contract results in PDA (Huang et al., 2008, Feng et al., 2010). RBPMS has been shown to facilitate the transition of vascular smooth muscle cells to a contractile state, while loss of Rbpms retains cells in the proliferative state (Nakagaki-Silva et al., 2019). This is consistent with our finding of increased arterial cell proliferation in KO pups. It would be interesting to explore how RBPMS inhibits vascular smooth muscle cell proliferation, while promoting cardiomyocyte proliferation.
Our analysis also revealed that RBPMS regulates mRNA alternative splicing in the heart, and primarily influences genes associated with cytoskeletal pathways. RBPMS has also been shown to play key roles in regulating cytoskeleton-related targets in smooth muscle cells (Nakagaki-Silva et al., 2019). In addition to cytoskeletal pathways, it is worth noting that many other aberrant splicing events are found in Rbpms KO hearts, as observed for Ryr2, Tpm2, and Tnnt2 transcripts, suggesting that RBPMS may also regulate cardiomyocyte contractility and calcium fluctuation. Analysis of other alternative splicing events influenced by Rbpms may reveal additional functions of Rbpms in the heart. Rbpms shares 72% sequence identity with its homolog Rbpms2 (Akerberg et al., 2019). It would be intriguing to investigate whether Rbpms and Rbpms2 function synergistically in the heart.
Pdlim5 alternative splicing regulates cardiomyocyte binucleation during heart development.
PDLIM5 is a cytoskeletal protein highly expressed in heart and skeletal muscle (Nakagawa et al., 2000). Cardiac-specific Pdlim5-knockout mice display cardiomyocyte contractile defects and DCM (Cheng et al., 2010). PDLIM5 interacts with multiple sarcomeric components, protein kinases and transcription factors involved in cell proliferation and cardiomyocyte physiology (Huang et al., 2020). The long and short isoforms of PDLIM5 differ in the inclusion of the 3XLIM domain at the carboxyl terminus. The long-to-short Pdlim5 isoform switch has been observed in heart development, however, the biological significance of this switch remains elusive (Yamazaki et al., 2010). Our study revealed that Pdlim5-long variants are required for maintaining regular cardiomyocyte cytokinesis in the embryonic heart, while misexpression of Pdlim5-short variants causes cytokinesis defects in postnatal cardiomyocytes. The mechanism whereby Pdlim5-short affects cardiomyocyte cytokinesis is unclear. We speculate that as a postnatal isoform of Pdlim5, the short variant may facilitate cardiomyocyte contraction synergistically with other sarcomeric proteins, thus inhibiting cytoskeletal reorganization during cell division. RBPMS functions in the embryonic heart to prevent the generation of short variants. Pdlim5-short variants act as antagonists of Pdlim5-long variants in postnatal mouse hearts (Yamazaki et al., 2010), which is consistent with our observation that overexpression of Pdlim5-short variants in hiPSC-cardiomyocytes inhibited cytokinesis, while overexpression of Pdlim5-long variants in Rbpms-KO hiPSC-cardiomyocytes partially rescued the cytokinesis defects (Figures 6C). Other RBPs have also been reported to mediate Pdlim5 mRNA splicing, including RBM20, RBM24 (Ito et al., 2016), RBFox2 (Wei et al., 2015), and QKI (Chen et al., 2021). Our findings revealed that RBPMS mediates the splicing of exon 8 and maintains Pdlim5-long expression. It is intriguing that RBPMS plays this role in the embryonic heart, given that Rbpms expression is generally constant during development. One possibility is that RBPMS might splice Pdlim5 mRNA together with stage-specific cofactors in embryonic hearts. Another possibility is that the increased expression of other RBPs during heart development competes with RBPMS in mediating Pdlim5 mRNA splicing, as indicated by the splicing of Pdlim5 occurring postnatally by several other RBPs (Ito et al., 2016, Wei et al., 2015, Chen et al., 2021). As revealed in our rMATS analysis, RBPMS is also involved in many other alternative splicing events that may contribute to the abnormalities in Rbpms KO mice.
In summary, our findings provide new insights into the mechanisms whereby an RNA binding protein (RBPMS) and one of its key splicing targets (Pdlim5) regulate cardiomyocyte proliferation and heart development. It will be of interest to determine the potential involvement of Rbpms and its targets in various forms of human congenital heart disease.
Limitations of the study
In this study, we created and maintained Rbpms knockout (KO) mice on a B6C3F1 and C57BL/6 mixed background, and the strain background heterogeneity may influence the manifestation of the Rbpms KO phenotype. Another limitation is that we analyzed both male and female pups but did not separate the genders, because all Rbpms KO pups died before P4. Further work will be necessary to address possible gender specific effects of Rbpms in heart development.
STAR Methods
Resource Availability.
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Eric Olson (Eric.Olson@UTSouthwestern.edu).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact upon request.
Data and Code Availability
All sequencing data have been deposited in the Gene Expression Omnibus under accession number: GSE182949, and is available to the public.
The code generated during this study are available from the Lead Contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
Experimental model and subjects details
Experimental Animals
Animal work described in this manuscript has been approved and conducted under the oversight of the UT Southwestern Institutional Animal Care and Use Committee. Neonatal Sprague-Dawley rats (Envigo) were used to isolate neonatal rat ventricular cardiomyocytes (NRVMs). Animals were housed in a 12 h light/dark cycle in a temperature-controlled room in the Animal Research Center of UT Southwestern, with ad libitum access to water and food. The ages of mice are indicated in the figure legends or methods. Sex was not determined for neonatal pups in the study.
Cell lines and cell culture
All cells were cultured at 37°C with 5% CO2. NRVMs were isolated from 1- or 2-day-old Sprague-Dawley rats with the Isolation System for Neonatal Rat/Mouse Cardiomyocytes (Cellutron, nc-6031) according to the manufacturer’s instructions. NRVMs were plated at a density of 3 × 105 cells/well to gelatin-coated 6-well plates and were maintained in DMEM/M199 medium (3:1, with 3% FBS), and penicillin-streptomycin for 48 h before adenoviral infection. 48 h after adenoviral infection, NRVMs were imaged with Keyence BZX700 microscope (10X objective) for fluorescence to evaluate infection efficiency, and cell lysates were collected using RIPA buffer (Sigma-Aldrich, R0278). HC01–5, male human iPSCs were cultured in mTeSR™1 media (Stemcell Technologies) and passaged approximately every 6 days (1:16 to 1:24 split ratio depending on the needs) by Versene Solution (Cat. no. 15040066). Dissociated human iPSCs were seeded onto Matrigel (hESC-qualified, Corning 354277)-coated cell culture plates and treated with 10 μM ROCK inhibitor (Y-27632) for 24 h after passaging.
Method Details
Generation of Rbpms KO mice
CRISPR Cas9 single guide (sg) RNAs flanking exon 1 of the Rbpms gene were selected from CRISPR 10K Genome Browser Track, cloned into pX458 (Addgene, #48138), transfected into N2a cells, FACS sorted, and cutting efficiency was assessed by T7E1 assay as per the provider’s instructions (New England BioLabs #E3321).
#1 Rbpms-sgRNA1 5’-accccgtggacctaggcgtc-3’
#2 Rbpms-sgRNA2 5’-aacttttaacaccgaaaggg-3’
Cas9 mRNA and Rbpms sgRNAs (#1 and #2) were injected into the pronucleus and cytoplasm of zygotes. For zygote production, C57BL/6 female mice were treated for superovulation and mated to C57BL/6 stud males. Zygotes were isolated, transferred to M16 and M2 medium, injected with Cas9 mRNA and sgRNA, and cultured in M16 medium for 1 h at 37 °C. Injected zygotes were transferred into the oviducts of pseudo-pregnant ICR female mice.
Tail genomic DNA was extracted from F0 mice and used for genomic analysis with PCR primers that amplify the targeted region. Primers 1 and 2 amplify fragments of different size in WT (1,678bp) and KO (991bp) mice. Primers 1 and 3 only amplify the WT allele (1,012bp).
#1 Rbpms-F0 5’-GCCTAGAGAGCGATAGCGG-3’
#2 Rbpms-R0 5’-CACCACAGCCGCAAATTGTT-3’
#3 Rbpms-R2 5’-cctcagtacCTCCTCCTCCTGAA-3’
Mosaic mice were mated to C57BL6 mice and a mouse line with a 690-bp deletion was selected for further characterization.
Mouse echocardiography
Cardiac function was evaluated by two-dimensional transthoracic echocardiography on conscious mice at P1 or 4 months of age using a VisualSonics Vevo2100 imaging system as described previously (Wang et al., 2019). All measurements were performed by an experienced operator blinded to the study.
Quantitative Real Time PCR Analysis
Total RNA was extracted from mouse heart ventricular tissues or iPSC-derived cardiomyocytes using Trizol (Thermo Fisher Scientific, 15596026) and reverse transcribed using iScript Reverse Transcription Supermix (Bio-Rad, 1708840) with random primers. The Quantitative Polymerase Chain Reactions (qPCR) were assembled using KAPA SYBR Fast qPCR Master Mix (KAPA, KK4605). Assays were performed using a 7900HT Fast Real-Time PCR machine (Applied Biosystems). Expression values were normalized to GAPDH or 18s rRNA and were represented as fold change. The following oligonucleotides were ordered from Integrated DNA Technologies to measure transcript abundance, and primer sequences are in Supplementary Table 2.
Single mouse cardiomyocyte isolation
Neonatal or embryonic mouse ventricular tissues were fixed in 4% paraformaldehyde (PFA) or formalin for 1–2 days followed by incubation in 50% w/v potassium hydroxide solution for 30 min to 1 h, depending on the tissue size. After a brief wash with PBS, tissues were gently teased apart in PBS to release single cardiomyocytes. The cardiomyocyte suspension was filtered through a 250-μm nylon mesh to remove cell clusters and enrich single isolated cardiomyocytes.
Cardiomyocyte ploidy and size
Cardiomyocyte ploidy was analyzed following previous protocols (Gan et al., 2019, Gan et al., 2020b). Briefly, isolated cardiomyocytes were stained for mouse anti-alpha-actinin antibody (1:500, Sigma-Aldrich A7811) with an Alexa Fluor 488 secondary antibody (1:500, ThermoFisher, A11001) and 4’,6-diamidino-2-phenylindole (DAPI, 1:1,000) using standard procedures. Cardiomyocytes were then spread on slides and coverslipped with ProLong Diamond Antifade Mountant (ThermoFisher, P36962). The number of nuclei per cardiomyocyte was quantified using photographs taken at a uniform setting for all cell preparations on a Keyence BZ-X700 microscope (10X objective). Five photographs were taken from different regions of each slide, and at least 200 cells were counted per heart. An unpaired, two-tailed Student t-test was used to assess statistical significance when only two groups were compared. Quantification of cardiomyocyte size was performed with ImageJ software, and freehand selection tool was used to outline the cellular boundary of cardiomyocytes and the area was measured. At least 200 cells were analyzed per heart.
To evaluate the ploidy of cardiomyocyte nuclei, using ImageJ software, nuclei in photographs were identified and outlined with a standard threshold requirement for all samples, and DAPI fluorescence intensity of each nucleus automatically quantified by ImageJ. The median value of DAPI fluorescence intensity of binucleated cardiomyocytes nuclei was used as a diploid nucleus standard and given a value of 1. All other nuclear fluorescence signals were normalized to this value. Nuclei were assigned as being diploid if their intensity value was within the 0.5–1.5 range (indicated by a red box in some figures), tetraploid for values 1.5–2.5, and octaploid for values>3. Approximately, 300 nuclei were analyzed per heart.
Plasmids and cloning
The pSpCas9(BB)-2A-GFP (PX458) plasmid contained the human codon optimized SpCas9 gene with 2A-EGFP. pSpCas9(BB)-2A-GFP (PX458) was a gift from F. Zhang (Addgene, #48138). Cloning of sgRNA sequence into PX458 vector was done using BbsI site. Cloning of Rbpms, Pdlim5-long-mEGFP, Pdlim5-short-mEGFP and mEGFP sequences into Adeno-X Adenoviral System 3 were achieved by In-Fusion® HD Cloning Plus kits (Takara, 638920) according to manufacturer’s instructions (Clontech, 632269, 632268). Rbpms gene sequence was amplified from mouse cDNA library. Pdlim5-long and Pdlim5-short gene sequences were synthesized as gBlocks from Integrated DNA Technologies IDT. The sequences of the constructs will be available upon request.
Stereological estimation of ventricular cardiomyocyte number
Cardiomyocyte number quantification by stereological analysis based on heart sections was adapted from a published protocol (Sampaio-Pinto et al., 2021). Briefly, P1 mouse hearts were rinsed in cardioplegia solution (PBS, 4M KCl), and fixed in 4% formaldehyde for 2–3 days at 4°C. Atria and vessels were trimmed off, and ventricles were cryopreserved in 10% sucrose/PBS and 18% sucrose/PBS solution sequentially. Tissues were embedded in optimal cutting temperature (OCT) Compound (Fisher Healthcare, 23730571), and cryosectioned at 20μm intervals. Consecutive sections were placed in the same position on different slides, numbered from 1 to 10. After each series of 10 consecutive sections, the same procedure was repeated until the heart was fully sectioned from anterior to posterior. Each slide contained 7 or 8 sections, representing consecutively different layers of heart. One section from each heart sample was used for immunofluorescence staining with anti-cTnT antibody (Abcam, ab8295, 1:500), anti-PCM1 antibody (Sigma-Aldrich, HPA023374, 1:500) and DAPI. The images of each whole heart ventricular section were captured by a Keyence BZX700 microscope with a 4x objective. The area of each heart ventricular section was calculated by using ImageJ software, and the volume of each section was determined as the area multiplied by the thickness of the section (20 μm). The estimated heart ventricular volume was calculated by summing up volumes of each heart section on the same slide and multiplied by the total slide number (10). To assess cardiomyocyte nucleus density on heart sections, five 160 μm × 80 μm × 20 μm (length × width × thickness) regions on heart sections different were photographed by using a Zeiss LSM 800 confocal microscope with automatic Z-stacking (1-μm interval). Cardiomyocyte nucleus (PCM1+) number was calculated manually by using ImageJ software. Cardiomyocyte nucleus density of each heart sample was the average density of cardiomyocyte nuclei of five different imaged regions. Total ventricular cardiomyocyte nucleus number was calculated by multiplying estimated heart ventricular volume with cardiomyocyte nucleus density. In P1 mouse hearts, the number of cardiomyocytes with more than two nuclei was very rare and negligible. Thus, the total number of cardiomyocytes was calculated as follows: number of cardiomyocytes = total number of cardiomyocyte nuclei / (Mono% + 2 × Bi%).
Differentiation of human iPSCs to cardiomyocytes
To differentiate human iPSCs into cardiomyocytes, cells were cultured in mTeSR™1 media until they reached 80 to 90% confluency, and cells were cultured in CDM3-C media, consisting of RPMI 1640 (Life Technologies, 11875), 500 μg/mL Oryza sativa-derived recombinant human albumin (A0237, Sigma-Aldrich), and 213 μg/mL L-ascorbic acid 2-phosphate supplemented with 10 μM CHIR-99021 (Selleckchem, S2924) for two days. Cells were then cultured in CDM3-C media, supplemented with 2 μM WNT-C59 (Selleckchem, S7037) for 2 days. Cells were cultured in BASAL media (RPMI-1640 with B27 Supplement (Thermo Fisher Scientific, 17504044)) for 6 days, and media was changed every 2 days. Cardiomyocytes were selected by culturing in SELECTIVE media (RPMI-1640, no glucose (Gibco, 11879–020) with B27 Supplement (Thermo Fisher Scientific, 17504044)) for 6 days. Then, purified cardiomyocytes were dissociated using TrypLE Express Enzyme (Gibco, 12604021) and replated at 1×105 cells per well in a 6-well dish.
Generation of RBPMS KO hiPSC line
HC01–5, male human iPSCs were cultured in mTeSR™1 media to reach 95% confluency. 1 h before nucleofection, iPSCs were treated with 10 μM ROCK inhibitor (Y-27632) and dissociated into single cells using Accutase (Innovative Cell Technologies Inc., NC9839010). iPSCs (1 × 106) were mixed with 5 μg of PX458-sgRNA-RBPMS-2A-GFP plasmid (sgRNA-RBPMS: 5’-GTCCGGACCCTATTTGTCAG-3’) and nucleofected using the P3 Primary Cell 4D Nucleofector X Kit (Lonza, V4XP-3024) according to the manufacturer’s protocol. After nucleofection, iPSCs were cultured in mTeSR™1 media supplemented with 10 μM ROCK inhibitor for 24 h and changed to mTeSR™1 media the next day. Two days after nucleofection, media were changed into mTeSR™1 media supplemented with 10 μM ROCK inhibitor and Primocin (100 μg/ml) (InvivoGen, ant-pm-05) 1 h before fluorescence-activated cell sorting (FACS). GFP(+) and (−) cells were sorted by FACS and seeded back into culture dishes. Single clones derived from GFP(+) iPSCs were picked and sequenced.
HiPSC-cardiomyocyte cell number and ploidy analysis
HiPSC-cardiomyocytes were passaged onto Matrigel-coated 12-well-dishes at a density of 2 × 104 cells/well and maintained in BASAL media for 2–3 days until cardiomyocytes recovered autonomous beating. Three separate regions (the same size as a 20X view field) were marked on the bottom of each well, where there were more than 70% single cardiomyocytes. Phase contrast microscopy of each marked region was performed on a Keyence BZX700 microscope with a 20X objective. Imaging was performed every two days on each marked region, until cardiomyocytes started to overlap with each other. Cardiomyocyte number in each marked region and number of nuclei in each cardiomyocyte were quantified manually using ImageJ software, and roughly 300 cardiomyocytes were analyzed for each sample.
Adenovirus generation and hiPSC-cardiomyocyte infection assay
Adenoviruses for Rbpms, Pdlim5-long-mEGFP, Pdlim5-short-mEGFP and mEGFP were generated using the Adeno-X Adenoviral System 3 (Clontech, 632269, 632268). Packaging of adenoviruses was performed as previously described (Wang et al., 2019) by transfecting Adeno-X 293 cells (Clontech, 632271) with corresponding Adenoviral vectors. Primary lysates were used to re-infect Adeno-X 293 cells to generate higher-titer viruses.
For hiPSC-derived cardiomyocytes infection, cardiomyocytes were passaged onto Matrigel-coated 12-well-dishes at a density of 1 × 105 cells/well and maintained in BASAL media for 4–6 days. 2 h before adenovirus infection, media was changed to fresh BASAL media. 1–2 μL of the respective adenovirus with roughly equal viral titer was transduced into hiPSC-cardiomyocytes per well of 12-well-dishes and cultured for 48 h. Cells were then fixed with 4% PFA at room temperature for 15 min and permeabilized with 0.3% Triton X-100 in PBS for 10 min. To access cardiomyocyte cytokinesis activity, cells were immunostained for anti-alpha-actinin antibody (Sigma-Aldrich, A7811, 1:500), anti-Aurora B kinase antibody (Abcam, ab2254, 1:200) and DAPI. Imaging was performed on a Keyence BZX700 microscope. Five fields at 10X magnification were captured for each well. Quantification was carried out by counting the ratio of Aurora B+ events/total cardiomyocyte nuclei on immunofluorescent staining images. Only Aurora B+ midbodies localized between two nuclei in a dividing cell were quantified as an Aurora B+ and positive cytokinesis event. Roughly 2,000 cardiomyocyte nuclei were analyzed for each sample.
Histology, immunochemistry and TUNEL assay
Animals were euthanized by isoflurane anesthesia followed by cervical dislocation and removal of hearts. As previously described (Gan et al., 2021), freshly isolated hearts were immersed in room temperature 0.2M KCl to arrest in diastole. Hearts were then fixed in 4% PFA in PBS at 4°C with gentle agitation on a rocker for 2–3 days, cryopreserved in 10% sucrose/PBS and 18% sucrose/PBS solution sequentially, embedded in O.C.T. Compound (Fisher Healthcare, 23730571), and cryosectioned at 10-μm intervals. For H&E staining, heart sections were stained by hematoxylin and eosin. For antigen retrieval, either 1 mM EDTA with 0.05% Tween 20 in boiling water or epitope retrieval solution (IHC World) in a steamer (IHCTek Epitope Retrieval Streamer Set) were used. For immunofluorescence staining, cryosections were washed by PBS twice and air-dried for 20 min at room temperature and fixed with 4% PFA for 20 min. Section slides were then washed twice with PBS and permeabilized with 0.3% Triton X-100 (Fisher Scientific, BP151–500) in PBS (PBST) for 10 min. For hiPSC-cardiomyocytes, cells were cultured on 35 mm Glass bottom dish (Cellvis, D35-20-1.5H), fixed with 4% PFA for 10 min and permeabilized in PBST for 15 min. Sections or cells were then blocked in 10% goat serum (Sigma, G9023)/3% BSA/0.025% Triton X-100/PBS blocking solution for 1 h and stained with the indicated primary antibodies prepared in blocking solution at 4°C overnight using the following dilutions: cTnT (Abcam, ab8295, 1:500), pH3 (Cell Signaling Technology, 9701S, 1:250), Ki67 (Abcam, ab15580, 1:500), Aurora B kinase (Abcam, ab2254, 1:200), PCM1 (Santa Cruz Biotechnology, sc-398365, 1:100), Recombinant Alexa Fluor® 488 Anti-Cardiac Troponin I antibody (Abcam, ab196384, 1:250), RBPMS (Santa Cruz Biotechnology, sc-293285, 1:200). Samples were subsequently washed with PBS three times (5 min for each wash) and incubated with corresponding secondary antibodies conjugated to Alexa Fluor 488, 555 or 647 (Invitrogen) prepared in blocking solution with Hoechst (Thermo Scientific, 62249, 1:1,000) at room temperature for 3 h. After secondary antibody incubation, samples were washed three times with PBS, then mounted with Prolong Diamond Antifade Mountant (Thermo Fisher Scientific, P36962). Images were obtained using a Zeiss LSM 800 confocal microscope and analyzed in ImageJ software. TUNEL assay was performed using Click-iT Plus TUNEL Assay for In Situ Apoptosis Detection kit (Thermo Fisher Scientific, C10619) following manufacturer’s protocol.
RNA in situ hybridization
Probe DNA sequences were synthesized as gBlocks from IDT and cloned into pCRII-D-TOPO vector using TOPO TA Cloning kit (Invitrogen, 450640). 35S-labelled UTP (Perkin Elmer, NEG039C001MC) were used in the in vitro transcription reaction to synthesize 35S-labelled probes, using MAXIscript SP6/T7 Transcription kit (Invitrogen, AM1322). Antisense and sense probes were transcribed with T7 and SP6 polymerases, respectively. Radio-isotopic in situ hybridization was performed as previously described. Probe sequences (antisense) are available upon request.
Western blot analysis
Tissues were snap frozen in liquid nitrogen then homogenized with an OMNI TH homogenizer in RIPA buffer (Sigma-Aldrich, R0278) supplemented with 1x Complete protease inhibitor mixture (Roche, 04693159001) on ice. For cultured hiPSC-cardiomyocytes, media was aspirated, and cells were washed twice with 1ml per well of 6-well-dish of ice-cold PBS, then lysed by the addition of 200μl ice-cold RIPA buffer with protease inhibitor and homogenized by pipetting. Tissue and cell lysates were centrifuged at 14,000 RPM for 10 min at 4°C to remove insoluble material. Immunoblotting was performed by standard protocols with 5–50μg lysate per well. Primary antibodies used were rabbit anti-RBPMS (1:1,000, Invitrogen, PA5-31231), mouse anti-GAPDH (1:1,000, GeneTex, GT239), mouse anti-PDLIM5 (1:1,000, Millipore Sigma, WH0010611M1), rabbit anti-GFP (1:1,000, Invitrogen A-11122). HRP-coupled goat anti-mouse antibody or goat anti-rabbit antibody (Bio-Rad Laboratories, 1706516 and 1706515) at room temperature for 2 h were used and the blot was developed using Western Blotting Luminol Reagent (Santa Cruz, sc-2048).
Paired-end RNA-Sequencing (RNA-seq) and alternative splicing analysis
RNA from heart tissue was extracted using TRIzol reagents (1 mL per 50–100 mg of tissue) according to manufacturer’s protocol. RNA-seq was performed as previously described (Wang et al., 2019). Stranded mRNA-Seq libraries were generated using KAPA mRNA HyperPrep Kit (Roche, KK8581) following manufacturer’s protocol. Sequencing was performed on an Illumina Nextseq 500 system for 2×75bp paired-end sequencing. The bulk RNA-seq data have been deposited in GEO Database under accession number: GSE182949.
Quality control of RNA-Seq data was performed using FastQC Tool (Version 0.11.4). Sequencing reads were aligned to mouse GRCm38 (mm10) reference genome using HiSAT2 (v2.0.4) with default settings and --rna-strandness FR (Kim et al., 2015). Aligned reads were counted using featureCounts (v1.6.0) per gene ID (Liao et al., 2014). Differential gene expression analysis was performed with the R package edgeR (v3.30.3) using the GLM approach (Robinson et al., 2010). Genes with more than 1 CPM (Count Per Million) in at least three samples were considered as expressed and were used for calculating normalization factor. Cutoff values of absolute fold change greater than 2.0 and false discovery rate less than 0.01 were used to define differentially expressed genes. Normalized gene CPM values were used to calculate RPKM (Reads Per Kilobase per Million mapped reads) values, which were then used for heatmap plotting. Gene ontology analysis was performed using Metascape (Zhou et al., 2019). Alternative splicing events were analyzed using rMATS (v4.0.2), and events with |IncLevelDifference| > 0.1 and FDR < 0.05 were considered as significant (Shen et al., 2014). Sequences for the RT-PCR primers used to confirm alternative splicing are provided in Supplemental Table 2. Full rMATS results have been uploaded to GEO under the accession number GSE182949. An interactive version of the volcano plot (Figure 5C) is available at: https://zwang0715.github.io/Gan_et_al_rMATS/
Quantification and statistical analysis
Statistical analyses were performed using GraphPad Prism 9 (GraphPad Software, Inc.) using a 2-tailed unpaired t test, with P < 0.05 considered significant unless otherwise indicated. All data are displayed as mean ± SEM unless otherwise indicated.
Supplementary Material
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse Anti-Alpha-actinin Antibody | Sigma | Cat#A7811 |
| Mouse Anti-Cardiac Troponin T Antibody | Abcam | Cat#ab8295 |
| Rabbit Anti-PCM1 Antibody | Millipore Sigma | Cat#HPA023370 |
| Rabbit Anti-Aurora B Antibody | Abcam | Cat#ab2254 |
| Rabbit Phospho-Histone H3 (Ser10) Antibody | Cell Signaling Technology | Cat#9701 |
| Rabbit Anti-Ki67 Antibody | Abcam | Cat#ab15580 |
| Mouse Anti-PCM1 Antibody | Santa Cruz Biotechnology | Cat#sc-398365 |
| Mouse Anti-RBPMS Antibody | Santa Cruz Biotechnology | Cat#sc-293285 |
| Mouse Anti-RBPMS Antibody | Thermo Fisher Scientific | Cat#PA5-31231 |
| Mouse Anti-GAPDH Antibody | GeneTex | Cat#GTX627408 |
| Mouse Anti-PDLIM5 Antibody | Millipore Sigma | Cat#WH0010611M1 |
| Rabbit Anti-GFP Polyclonal Antibody | Thermo Fisher Scientific | Cat#A-11122 |
| Goat Anti-Rabbit IgG (H + L)-HRP Conjugate | Bio-Rad | Cat#1706515 |
| Goat Anti-Mouse IgG (H + L)-HRP Conjugate | Bio-Rad | Cat#1706516 |
| Rabbit Recombinant Alexa Fluor® 488 Anti-Cardiac Troponin I Antibody | Abcam | Cat#ab196384 |
| Goat Anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat#A11001 |
| Goat Anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen | Cat#A21422 |
| Goat Anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat#A21235 |
| Goat Anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | Cat#A21428 |
| Goat Anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen | Cat#A21235 |
| Goat Anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | Cat#A21244 |
| Bacterial and virus strains | ||
| Stellar Competent Cells | Takara | Cat#636766 |
| Ad-Rbpms | This paper | N/A |
| Ad-mEGFP | This paper | N/A |
| Ad-Pdlim5-long-mEGFP | This paper | N/A |
| Ad-Pdlim5-short-mEGFP | This paper | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Albumin human | Millipore Sigma | Cat#A0237 |
| CHIR-99021 (CT99021) HCl | Selleckchem | Cat#S2924 |
| WNT-C59 | Selleckchem | Cat#S7037 |
| Primocin® | InvivoGen | Cat#ant-pm-05 |
| Y-27632 RHO/ROCK pathway inhibitor | Stemcell Technologies | Cat#72302 |
| 35S-labelled UTP | Perkin Elmer | Cat#NEG039C001 MC |
| Critical commercial assays | ||
| Neomyt Kit | Cellutron | Cat#nc-6031 |
| Dulbecco’s Modified Eagle’s Medium - high glucose | Sigma | Cat#D5796 |
| Medium 199, Earle’s Salts | GIBCO | Cat#11150-059 |
| Fetal Bovine Serum | Gemini Bio Products | Cat#100-106 |
| Penicillin-Streptomycin | Sigma | Cat#P0781 |
| RIPA Buffer | Sigma | Cat#R0278 |
| mTeSR™1 | ||
| cGMP, feeder-free maintenance medium for human ES and iPS cells | Stemcell Technologies | Cat#85850 |
| Versene Solution | Thermo Fisher Scientific | Cat#15040066 |
| Matrigel® hESC-Qualified Matrix | Corning | Cat#354277 |
| T7 Endonuclease I | New England Biolabs | Cat#M0302 |
| TRIzol™ Reagent | Thermo Fisher Scientific | Cat#15596026 |
| iScript™ Reverse Transcription Supermix | Bio-Rad | Cat#1708840 |
| KAPA SYBR FAST qPCR Kits | Roche | Cat#07959435001 |
| Paraformaldehyde, 16% w/v aq. soln | Fisher Scientific | Cat#AA433689M |
| Hoechst 33342 Solution (20 mM) | Thermo Fisher Scientific | Cat#62249 |
| ProLong™ Diamond Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat#P36962 |
| Adeno-X Adenoviral System 3 | Clontech | Cat#632267 |
| In-Fusion HD Cloning Plus Kits | Takara | Cat#638920 |
| Tissue-Plus™ O.C.T. Compound | Fisher Scientific | Cat#23730571 |
| RPMI 1640 Medium | Thermo Fisher Scientific | Cat#11875 |
| B-27™ Supplement (50X), serum free | Thermo Fisher Scientific | Cat#17504044 |
| RPMI 1640 Medium, no glucose | Thermo Fisher Scientific | Cat#11879-020 |
| TrypLE™ Express Enzyme (1X) | Gibco | Cat#12604021 |
| Accutase | Innovative Cell Technologies | Cat#NC9839010 |
| P3 Primary Cell 4D-NucleofectorTM X Kit L | Lonza | Cat#V4XP-3024 |
| Triton™ X-100 (Electrophoresis) | Fisher Scientific | Cat#BP151-100 |
| Click-iT™ Plus TUNEL Assay for In Situ Apoptosis Detection, Alexa Fluor™ 647 dye | Thermo Fisher Scientific | Cat#C10619 |
| TOPO™ TA Cloning™ Kit | Thermo Fisher Scientific | Cat#450640 |
| MAXIscript™ SP6/T7 Transcription Kit | Thermo Fisher Scientific | Cat#AM1322 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Roche | Cat#04693159001 |
| Western Blotting Luminol Reagent | Santa Cruz Biotechnology | Cat#sc-2048 |
| KAPA mRNA HyperPrep Kit | Roche | Cat#08098123702 |
| Deposited data | ||
| Paired-end RNA Seq data | This paper | GEO: GSE182949 |
| Experimental models: Cell lines | ||
| Adeno-X 293 | Clontech | Cat#632271 |
| Neuro 2a | Millipore Sigma | Cat#89121404 |
| HC01-5 human iPSC | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Charles River | N/A |
| Rat: Sprague Dawley | Envigo | N/A |
| Oligonucleotides | ||
| Primers for qRT-PCR and splicing event verification, see Supplementary Table 2 | This paper | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP (PX458) | Addgene | Cat#48138 |
| pCRII-D-TOPO | This paper | |
| Software and algorithms | ||
| Adobe Illustrator 2021 | Adobe | N/A |
| BioRender | BioRender | https://biorender.com/ |
| Fiji/ImageJ | NIH | https://imagej.nih.gov/ij/ |
| FastQC Tool (Version 0.11.4) | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| HiSAT2 (v2.0.4) | Kim et al., 2015 | http://daehwankimlab.github.io/hisat2/ |
| featureCounts (v1.6.0) | Liao et al., 2014 | http://subread.sourceforge.net/ |
| R package edgeR (v3.30.3) | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Metascape | Zhou et al., 2019 | http://metascape.org/ |
| rMATS (v4.0.2) | Shen et al., 2014 | http://rnaseq-mats.sourceforge.net/rmats4.0.2/user_guide.htm |
| GraphPad Prism 9 | GraphPad Software Inc | N/A |
| NCBI Primer Blast | NCBI | RRID:SCR_004870 |
| Integrated DNA Technologies (IDT) PrimerQuest software | Integrated DNA technologies (IDT) | https://www.idtdna.com/primerquest |
| Other | ||
Highlights.
Rbpms regulates cardiomyocyte cytokinesis and binucleation in embryonic hearts
Absence of Rbpms causes myocardium noncompaction during development
RBPMS mediates alternative mRNA splicing in the heart
Pdlim5 regulates cardiomyocyte cytokinesis and is a key splicing target of RBPMS
Acknowledgements
We thank Cristina Rodriguez-Caycedo for maintaining the iPSC facility and providing technical instruction. We thank Dr. Andres Ramirez-Martinez for advice on the Rbpms gene knockout strategy, and John McAnally for performing microinjections for generating Rbpms KO mice. We thank Jose Cabrera for graphics. We thank Dr. Wei Tan for performing mouse echocardiography analysis, and the Molecular Histopathology Core managed by John Shelton for help with histology, Drs. Jian Xu and Yoon Jung Kim from the Next Generation Sequencing Core Facility at Children’s Research Institute for performing the Illumina sequencing, and Stephen Johnson for help with high-performance computing. We thank Dr. Miao Cui and other members of the Olson laboratory for helpful discussions. We are grateful to Dr. Henry M. Sucov (Medical University of South Carolina) for advice on cardiac histology. This work was supported by grants from the NIH (AR-067294, HL-130253, HL138426, and HD-087351), the Fondation Leducq Transatlantic Networks of Excellence in Cardiovascular Research, and the Robert A. Welch Foundation (Grant 1-0025 to E.N.O.). P.G. was supported by a postdoctoral fellowship from the American Heart Association (825635). Z.W. was supported by a predoctoral fellowship from the American Heart Association and the Harry S. Moss Heart Trust (19PRE34380436).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Competing interests
E.N.O. is on the editorial board of Developmental Cell. The authors declare no other competing interests.
References
- AKERBERG AA, BURNS CE & BURNS CG 2019. Exploring the Activities of RBPMS Proteins in Myocardial Biology. Pediatr Cardiol, 40, 1410–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALI H, BRAGA L & GIACCA M 2020. Cardiac regeneration and remodelling of the cardiomyocyte cytoarchitecture. FEBS J, 287, 417–438. [DOI] [PubMed] [Google Scholar]
- ALMEIDA AG & PINTO FJ 2013. Non-compaction cardiomyopathy. Heart, 99, 1535–42. [DOI] [PubMed] [Google Scholar]
- ARNDT AK, SCHAFER S, DRENCKHAHN JD, SABEH MK, PLOVIE ER, CALIEBE A, KLOPOCKI E, MUSSO G, WERDICH AA, KALWA H, HEINIG M, PADERA RF, WASSILEW K, BLUHM J, HARNACK C, MARTITZ J, BARTON PJ, GREUTMANN M, BERGER F, HUBNER N, SIEBERT R, KRAMER HH, COOK SA, MACRAE CA & KLAASSEN S 2013. Fine mapping of the 1p36 deletion syndrome identifies mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet, 93, 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERGMANN O, BHARDWAJ RD, BERNARD S, ZDUNEK S, BARNABE-HEIDER F, WALSH S, ZUPICICH J, ALKASS K, BUCHHOLZ BA, DRUID H, JOVINGE S & FRISEN J 2009. Evidence for cardiomyocyte renewal in humans. Science, 324, 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLECH-HERMONI Y & LADD AN 2013. RNA binding proteins in the regulation of heart development. Int J Biochem Cell Biol, 45, 2467–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN Q, CHEN H, ZHENG D, KUANG C, FANG H, ZOU B, ZHU W, BU G, JIN T, WANG Z, ZHANG X, CHEN J, FIELD LJ, RUBART M, SHOU W & CHEN Y 2009. Smad7 is required for the development and function of the heart. J Biol Chem, 284, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN X, LIU Y, XU C, BA L, LIU Z, LI X, HUANG J, SIMPSON E, GAO H, CAO D, SHENG W, QI H, JI H, SANDERSON M, CAI CL, LI X, YANG L, NA J, YAMAMURA K, LIU Y, HUANG G, SHOU W & SUN N 2021. QKI is a critical premRNA alternative splicing regulator of cardiac myofibrillogenesis and contractile function. Nat Commun, 12, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHENG H, KIMURA K, PETER AK, CUI L, OUYANG K, SHEN T, LIU Y, GU Y, DALTON ND, EVANS SM, KNOWLTON KU, PETERSON KL & CHEN J 2010. Loss of enigma homolog protein results in dilated cardiomyopathy. Circ Res, 107, 348–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FENG X, KREBS LT & GRIDLEY T 2010. Patent ductus arteriosus in mice with smooth muscle-specific Jag1 deletion. Development, 137, 4191–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAN P, BAICU C, WATANABE H, WANG K, TAO G, JUDGE DP, ZILE MR, MAKITA T, MUKHERJEE R & SUCOV HM 2021. The prevalent I686T human variant and loss-of-function mutations in the cardiomyocyte-specific kinase gene TNNI3K cause adverse contractility and concentric remodeling in mice. Hum Mol Genet, 29, 3504–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAN P, PATTERSON M & SUCOV HM 2020a. Cardiomyocyte Polyploidy and Implications for Heart Regeneration. Annu Rev Physiol, 82, 45–61. [DOI] [PubMed] [Google Scholar]
- GAN P, PATTERSON M, VELASQUEZ A, WANG K, TIAN D, WINDLE JJ, TAO G, JUDGE DP, MAKITA T, PARK TJ & SUCOV HM 2019. Tnni3k alleles influence ventricular mononuclear diploid cardiomyocyte frequency. PLoS Genet, 15, e1008354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAN P, PATTERSON M, WATANABE H, WANG K, EDMONDS RA, REINHOLDT LG & SUCOV HM 2020b. Allelic variants between mouse substrains BALB/cJ and BALB/cByJ influence mononuclear cardiomyocyte composition and cardiomyocyte nuclear ploidy. Sci Rep, 10, 7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO C, REN S, LEE JH, QIU J, CHAPSKI DJ, RAU CD, ZHOU Y, ABDELLATIF M, NAKANO A, VONDRISKA TM, XIAO X, FU XD, CHEN JN & WANG Y 2016. RBFox1-mediated RNA splicing regulates cardiac hypertrophy and heart failure. J Clin Invest, 126, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GERBER WV, VOKES SA, ZEARFOSS NR & KRIEG PA 2002. A role for the RNA-binding protein, hermes, in the regulation of heart development. Dev Biol, 247, 116–26. [DOI] [PubMed] [Google Scholar]
- GERBER WV, YATSKIEVYCH TA, ANTIN PB, CORREIA KM, CONLON RA & KRIEG PA 1999. The RNA-binding protein gene, hermes, is expressed at high levels in the developing heart. Mech Dev, 80, 77–86. [DOI] [PubMed] [Google Scholar]
- GONZALEZ-ROSA JM, SHARPE M, FIELD D, SOONPAA MH, FIELD LJ, BURNS CE & BURNS CG 2018. Myocardial Polyploidization Creates a Barrier to Heart Regeneration in Zebrafish. Dev Cell, 44, 433–446 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUO W, SCHAFER S, GREASER ML, RADKE MH, LISS M, GOVINDARAJAN T, MAATZ H, SCHULZ H, LI S, PARRISH AM, DAUKSAITE V, VAKEEL P, KLAASSEN S, GERULL B, THIERFELDER L, REGITZ-ZAGROSEK V, HACKER TA, SAUPE KW, DEC GW, ELLINOR PT, MACRAE CA, SPALLEK B, FISCHER R, PERROT A, OZCELIK C, SAAR K, HUBNER N & GOTTHARDT M 2012. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med, 18, 766–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HESSE M, DOENGI M, BECKER A, KIMURA K, VOELTZ N, STEIN V & FLEISCHMANN BK 2018. Midbody Positioning and Distance Between Daughter Nuclei Enable Unequivocal Identification of Cardiomyocyte Cell Division in Mice. Circ Res, 123, 1039–1052. [DOI] [PubMed] [Google Scholar]
- HIROSE K, PAYUMO AY, CUTIE S, HOANG A, ZHANG H, GUYOT R, LUNN D, BIGLEY RB, YU H, WANG J, SMITH M, GILLETT E, MUROY SE, SCHMID T, WILSON E, FIELD KA, REEDER DM, MADEN M, YARTSEV MM, WOLFGANG MJ, GRUTZNER F, SCANLAN TS, SZWEDA LI, BUFFENSTEIN R, HU G, FLAMANT F, OLGIN JE & HUANG GN 2019. Evidence for hormonal control of heart regenerative capacity during endothermy acquisition. Science, 364, 184–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG J, CHENG L, LI J, CHEN M, ZHOU D, LU MM, PROWELLER A, EPSTEIN JA & PARMACEK MS 2008. Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. J Clin Invest, 118, 515–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG X, QU R, OUYANG J, ZHONG S & DAI J 2020. An Overview of the Cytoskeleton-Associated Role of PDLIM5. Front Physiol, 11, 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUTCHINS JR, TOYODA Y, HEGEMANN B, POSER I, HERICHE JK, SYKORA MM, AUGSBURG M, HUDECZ O, BUSCHHORN BA, BULKESCHER J, CONRAD C, COMARTIN D, SCHLEIFFER A, SAROV M, POZNIAKOVSKY A, SLABICKI MM, SCHLOISSNIG S, STEINMACHER I, LEUSCHNER M, SSYKOR A, LAWO S, PELLETIER L, STARK H, NASMYTH K, ELLENBERG J, DURBIN R, BUCHHOLZ F, MECHTLER K, HYMAN AA & PETERS JM 2010. Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science, 328, 593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ITO J, IIJIMA M, YOSHIMOTO N, NIIMI T, KURODA S & MATURANA AD 2016. RBM20 and RBM24 cooperatively promote the expression of short enh splice variants. FEBS Lett, 590, 2262–74. [DOI] [PubMed] [Google Scholar]
- JEFFERIES JL, WILKINSON JD, SLEEPER LA, COLAN SD, LU M, PAHL E, KANTOR PF, EVERITT MD, WEBBER SA, KAUFMAN BD, LAMOUR JM, CANTER CE, HSU DT, ADDONIZIO LJ, LIPSHULTZ SE, TOWBIN JA & PEDIATRIC CARDIOMYOPATHY REGISTRY I. 2015. Cardiomyopathy Phenotypes and Outcomes for Children With Left Ventricular Myocardial Noncompaction: Results From the Pediatric Cardiomyopathy Registry. J Card Fail, 21, 877–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM D, LANGMEAD B & SALZBERG SL 2015. HISAT: a fast spliced aligner with low memory requirements. Nat Methods, 12, 357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KODO K, ONG SG, JAHANBANI F, TERMGLINCHAN V, HIRONO K, INANLOORAHATLOO K, EBERT AD, SHUKLA P, ABILEZ OJ, CHURKO JM, KARAKIKES I, JUNG G, ICHIDA F, WU SM, SNYDER MP, BERNSTEIN D & WU JC 2016. iPSC-derived cardiomyocytes reveal abnormal TGF-beta signalling in left ventricular non-compaction cardiomyopathy. Nat Cell Biol, 18, 1031–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRENN V & MUSACCHIO A 2015. The Aurora B Kinase in Chromosome Bi-Orientation and Spindle Checkpoint Signaling. Front Oncol, 5, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIAO Y, SMYTH GK & SHI W 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–30. [DOI] [PubMed] [Google Scholar]
- LIU H, ZHANG CH, AMMANAMANCHI N, SURESH S, LEWARCHIK C, RAO K, UYS GM, HAN L, ABRIAL M, YIMLAMAI D, GANAPATHY B, GUILLERMIER C, CHEN N, KHALADKAR M, SPAETHLING J, EBERWINE JH, KIM J, WALSH S, CHOUDHURY S, LITTLE K, FRANCIS K, SHARMA M, VIEGAS M, BAIS A, KOSTKA D, DING J, BAR-JOSEPH Z, WU Y, YECHOOR V, MOULIK M, JOHNSON J, WEINBERG J, REYES-MUGICA M, STEINHAUSER ML & KUHN B 2019. Control of cytokinesis by beta-adrenergic receptors indicates an approach for regulating cardiomyocyte endowment. Sci Transl Med, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUXAN G, CASANOVA JC, MARTINEZ-POVEDA B, PRADOS B, D’AMATO G, MACGROGAN D, GONZALEZ-RAJAL A, DOBARRO D, TORROJA C, MARTINEZ F, IZQUIERDO-GARCIA JL, FERNANDEZ-FRIERA L, SABATER-MOLINA M, KONG YY, PIZARRO G, IBANEZ B, MEDRANO C, GARCIA-PAVIA P, GIMENO JR, MONSERRAT L, JIMENEZ-BORREGUERO LJ & DE LA POMPA JL 2013. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med, 19, 193–201. [DOI] [PubMed] [Google Scholar]
- MUKHINA S, WANG YL & MURATA-HORI M 2007. Alpha-actinin is required for tightly regulated remodeling of the actin cortical network during cytokinesis. Dev Cell, 13, 554–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAKAGAKI-SILVA EE, GOODING C, LLORIAN M, JACOB AG, RICHARDS F, BUCKROYD A, SINHA S & SMITH CW 2019. Identification of RBPMS as a mammalian smooth muscle master splicing regulator via proximity of its gene with super-enhancers. Elife, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAKAGAWA N, HOSHIJIMA M, OYASU M, SAITO N, TANIZAWA K & KURODA S 2000. ENH, containing PDZ and LIM domains, heart/skeletal muscle-specific protein, associates with cytoskeletal proteins through the PDZ domain. Biochem Biophys Res Commun, 272, 505–12. [DOI] [PubMed] [Google Scholar]
- NUGENT AW, DAUBENEY PE, CHONDROS P, CARLIN JB, CHEUNG M, WILKINSON LC, DAVIS AM, KAHLER SG, CHOW CW, WILKINSON JL, WEINTRAUB RG & NATIONAL AUSTRALIAN CHILDHOOD CARDIOMYOPATHY, S. 2003. The epidemiology of childhood cardiomyopathy in Australia. N Engl J Med, 348, 1639–46. [DOI] [PubMed] [Google Scholar]
- PATTERSON M, BARSKE L, VAN HANDEL B, RAU CD, GAN P, SHARMA A, PARIKH S, DENHOLTZ M, HUANG Y, YAMAGUCHI Y, SHEN H, ALLAYEE H, CRUMP JG, FORCE TI, LIEN CL, MAKITA T, LUSIS AJ, KUMAR SR & SUCOV HM 2017. Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet, 49, 1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PORRELLO ER, MAHMOUD AI, SIMPSON E, HILL JA, RICHARDSON JA, OLSON EN & SADEK HA 2011. Transient regenerative potential of the neonatal mouse heart. Science, 331, 1078–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBINSON MD, MCCARTHY DJ & SMYTH GK 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAMPAIO-PINTO V, SILVA ED, LAUNDOS TL, DA COSTA MARTINS P, PINTO-DO OP & NASCIMENTO DS 2021. Stereological estimation of cardiomyocyte number and proliferation. Methods, 190, 55–62. [DOI] [PubMed] [Google Scholar]
- SAMSA LA, YANG B & LIU J 2013. Embryonic cardiac chamber maturation: Trabeculation, conduction, and cardiomyocyte proliferation. Am J Med Genet C Semin Med Genet, 163C, 157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEDMERA D, PEXIEDER T, VUILLEMIN M, THOMPSON RP & ANDERSON RH 2000. Developmental patterning of the myocardium. Anat Rec, 258, 319–37. [DOI] [PubMed] [Google Scholar]
- SHEN S, PARK JW, LU ZX, LIN L, HENRY MD, WU YN, ZHOU Q & XING Y 2014. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A, 111, E5593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SOONPAA MH, KIM KK, PAJAK L, FRANKLIN M & FIELD LJ 1996. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol, 271, H2183–9. [DOI] [PubMed] [Google Scholar]
- STOPP S, GRUNDL M, FACKLER M, MALKMUS J, LEONE M, NAUMANN R, FRANTZ S, WOLF E, VON EYSS B, ENGEL FB & GAUBATZ S 2017. Deletion of Gas2l3 in mice leads to specific defects in cardiomyocyte cytokinesis during development. Proc Natl Acad Sci U S A, 114, 8029–8034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURGEON B & MELOCHE S 2009. Interpreting neonatal lethal phenotypes in mouse mutants: insights into gene function and human diseases. Physiol Rev, 89, 1–26. [DOI] [PubMed] [Google Scholar]
- VAIDYA VR, LYLE M, MIRANDA WR, FARWATI M, ISATH A, PATLOLLA SH, HODGE DO, ASIRVATHAM SJ, KAPA S, DESHMUKH AJ, FOLEY TA, MICHELENA HI, CONNOLLY HM & MELDUNI RM 2021. Long-Term Survival of Patients With Left Ventricular Noncompaction. J Am Heart Assoc, 10, e015563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG Z, CUI M, SHAH AM, YE W, TAN W, MIN YL, BOTTEN GA, SHELTON JM, LIU N, BASSEL-DUBY R & OLSON EN 2019. Mechanistic basis of neonatal heart regeneration revealed by transcriptome and histone modification profiling. Proc Natl Acad Sci U S A, 116, 18455–18465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEI C, QIU J, ZHOU Y, XUE Y, HU J, OUYANG K, BANERJEE I, ZHANG C, CHEN B, LI H, CHEN J, SONG LS & FU XD 2015. Repression of the Central Splicing Regulator RBFox2 Is Functionally Linked to Pressure Overload-Induced Heart Failure. Cell Rep, 10, 1521–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEIFORD BC, SUBBARAO VD & MULHERN KM 2004. Noncompaction of the ventricular myocardium. Circulation, 109, 2965–71. [DOI] [PubMed] [Google Scholar]
- WILSBACHER L & MCNALLY EM 2016. Genetics of Cardiac Developmental Disorders: Cardiomyocyte Proliferation and Growth and Relevance to Heart Failure. Annu Rev Pathol, 11, 395–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMAZAKI T, WALCHLI S, FUJITA T, RYSER S, HOSHIJIMA M, SCHLEGEL W, KURODA S & MATURANA AD 2010. Splice variants of enigma homolog, differentially expressed during heart development, promote or prevent hypertrophy. Cardiovasc Res, 86, 374–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG J, HUNG LH, LICHT T, KOSTIN S, LOOSO M, KHRAMEEVA E, BINDEREIF A, SCHNEIDER A & BRAUN T 2014. RBM24 is a major regulator of muscle-specific alternative splicing. Dev Cell, 31, 87–99. [DOI] [PubMed] [Google Scholar]
- YU L, DANIELS J, GLASER AE & WOLF MJ 2013. Raf-mediated cardiac hypertrophy in adult Drosophila. Dis Model Mech, 6, 964–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU Y, ZHOU B, PACHE L, CHANG M, KHODABAKHSHI AH, TANASEICHUK O, BENNER C & CHANDA SK 2019. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun, 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data have been deposited in the Gene Expression Omnibus under accession number: GSE182949, and is available to the public.
The code generated during this study are available from the Lead Contact upon request.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.