

Abstract
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a serious, complex, and highly debilitating long-term illness. People with ME/CFS are typically unable to carry out their routine activities. Key hallmarks of the disease are neurological and gastrointestinal impairments accompanied by pervasive malaise that is exacerbated after physical and/or mental activity. Currently, there is no validated cure of biomarker signature for this illness. Impaired tryptophan (TRYP) metabolism is thought to play significant role in the pathobiology of ME/CFS. TRYP is an important precursor for serotonin and the essential pyridine nucleotide nicotinamide adenine dinucleotide (NAD+). TRYP has been associated with the development of some parts of the brain responsible for behavioural functions. The main catabolic route for TRYP is the kynurenine pathway (KP). The KP produces NAD+ and several neuroactive metabolites with neuroprotective (i.e., kynurenic acid (KYNA)) and neurotoxic (i.e., quinolinic acid (QUIN)) activities. Hyperactivation of the KP, whether compensatory or a driving mechanism of degeneration can limit the availability of NAD+ and exacerbate the symptoms of ME/CFS. This review discusses the potential association of altered KP metabolism in ME/CFS. The review also evaluates the role of the patient’s gut microbiota on TRYP availability and KP activation. We propose that strategies aimed at raising the levels of NAD+ (e.g., using nicotinamide mononucleotide and nicotinamide riboside) may be a promising intervention to overcome symptoms of fatigue and to improve the quality of life in patients with ME/CFS. Future clinical trials should further assess the potential benefits of NAD+ supplements for reducing some of the clinical features of ME/CFS.
Keywords: Kynurenine pathway, tryptophan, NAD+, myalgic encephalomyelitis/chronic fatigue syndrome, gut microbiota
1.Introduction
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a complex multisystem, long-term, disabling disorder. The key hallmark of ME/CFS is debilitating fatigue that is present for at least 6 months and is not ameliorated by rest. [1]. According to the Institute of Medicine (IOM) report, 83,000 to 2.5 million individuals suffer from ME/CFS in the United States with an estimated annual economic burden of 17-24 billion USD dollars [2]. It is estimated that the prevalence of ME/CFS to be 0.4-1% of the Australians, implying up to 250,000 people affected [3]. This disease is characterized by complicated symptoms associated with impairment in cognition, immune system, autonomous functions, and the endocrine system (Fig. 1). Individuals with ME/CFS suffer overwhelming fatigue, dizziness, and sleep disturbances [4]. The illness can affect all races and ages with women at three to four times higher risk than men [5-7]. Many patients are not diagnosed rapidly since there is no specific laboratory diagnostic markers or definitive tests available in the clinic. Efforts have been made to address this issue by unravelling the pathobiology of ME/CFS at both the molecular and clinical levels.
Figure 1.

Common symptoms and significant features of ME/CFS patients.
The aetiology of ME/CFS is also poorly understood. The main issue is that ME/CFS is usually diagnosed in the individuals when it is already at its severe stage [5]. About 50-80% of patients show sudden, persistent flu-like symptoms. ME/CFS is commonly occurred following bacterial or viral infections. These infections may render the immune system dysfunctional, initiating ME/CFS that is followed by multi-systemic impairments that may be long-lasting [8, 9]. We speculate that a higher number of COVID-19 patients may increase the reported cases of ME/CFS, as observed following the SARS epidemic [10].
Concerning the pathophysiology of ME/CFS, many studies reported evidence of serotonergic system dysfunction, chronic viral infection, and abnormalities in immune response (i.e., immune activation and chronic inflammation) and cellular bioenergetics [11]. Chronic neuroinflammation near the paraventricular nucleus of the hypothalamus is an important factor in ME/CFS [12]. Evidence from advanced molecular techniques has showed the increased cytokine production, immune activation and inflammation which are associated with deficits in cellular energy metabolism and mitochondrial function. Immune activation can increase flux of tryptophan (TRYP) metabolism via the kynurenine pathway (KP) through realising proinflammatory cytokines that induce the activity of indoleamine 2,3-dioxygenase 1 (IDO-1), the primary enzyme in the KP [13]. Up to 90% of TRYP is catabolized through the KP into neuroactive metabolites and nicotinamide adenine dinucleotide (NAD+) [14-17]. Metabolites of TRYP catabolism from the KP have been associated with inflammation, immune response, and neurological disorders. It is worth mentioning that any increase in KP activity will shunt away TRYP from serotonin pathways. Downregulation of the serotonin pathway may contribute to emergence of ME/CFS symptoms such as chronic fatigue, depression, sleep problems, and headaches.
The present study is a comprehensive review that i) discusses the possible association of KP with ME/CFS; ii) highlights the role of patient’s gut microbiota in the availability of TRYP; iii) emphasizes the significance of KP metabolites and their relation to disease symptoms, and iv) evaluates evidence for the role of raising levels of NAD+ precursors to attenuate disease symptoms. By disseminating this information, this review aims to provide researchers in this field a promising theragnostic concept and bringing us closer to developing a biosignature for ME/CFS.
2.Epidemiology of ME/CFS
The exact prevalence of ME/CFS remains controversial; however, a large number of studies reported a significant prevalence of ME/CFS among adults [6, 18-20]. This controversy is due to several factors ranging from differences in the laboratory methodologies and the type of population surveyed for definition and determination of the disease [21-25]. Therefore, a common diagnostic criterion in ME/CFS studies is essential but lacking. Care should be taken to exclude individuals that suffer from known pathological diseases as ME/CFS patients.
According to the Office of the Privacy Commissioner of Canada (OPC), ~27% of adults in the United Kingdom suffer from chronic fatigue which accounts for the prevalence rate of 13.4% in that population [26, 27]. A study conducted in England revealed an incidence rate of 4.7% (prevalence rate of 0.2%) of CFS among the 143,000 individuals (age, 18-64 years) [28]. Another study in the US estimated that over 6% of people experience significant fatigue that last more than 14 days [29]. Based on the current Centers for Disease Control and Prevention (CDC) statistics, more than one million individuals suffer ME/CFS in the US. According to a four-year study [30] in Wichita, Kansas, the prevalence of CFS was 235 per 100,000 persons, and was four times more common in women than men. Lawrie et al. (1997) [31] reported that the annual incidence and the prevalence of fatigue syndrome in Edinburgh were 370 per 100,000 and 740 per 100,000 individuals, respectively. While definition and classification of the disease vary and can affect the prevalence reports, it has been estimated that about 0.4-1% of Australians suffer ME/CFS [3]. A Norwegian study identified two peaks in age prevalence of ME/CFS. The first one appeared between 10 to 19 years and the other one was observed between 30 to 39 years [32]. Overall, it has been estimated that there are about 17-24 million people with ME/CFS around the world (Fig. 2) with women at four-time higher risk [7, 33].
Figure 2.
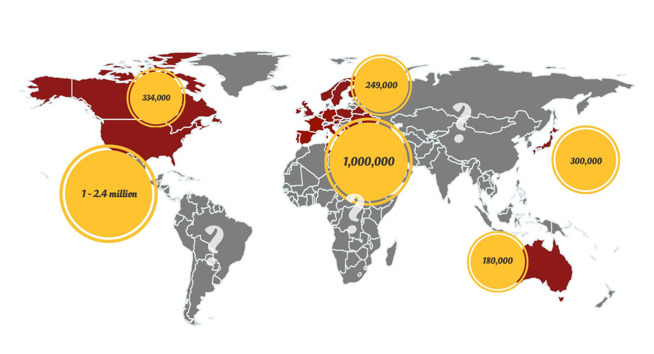
Worldwide population affected by ME/CFS (https://www.emerge.org.au/).
Approximately, a quarter of patients are totally disabled with an estimated annual cost of $20,000 per individual [34]. Due to complex symptomology, prognosis of patients is challenging. In most of the cases, an improvement of the symptoms is more common than full recovery. [35]. Patients with severe ME/CFS usually die from underlying health complications such as cardiovascular diseases and cancer [36-38].
3.Aetiology and pathophysiology of ME/CFS
Several clinical investigations have suggested infections, genetics, physical trauma, stress, and the environment as risk factors for the development of ME/CFS [39-43]. Patients with ME/CFS have several pathophysiological abnormalities that affect various organ systems. However, it is not clear whether these abnormalities occur after ME/CFS or are the initiator of the illness. The possible immune system abnormalities in ME/CFS could include higher production of pro-inflammatory cytokines (i.e.,IL-4, IL-5, IL-6, IL-12), immunosuppression due to dysfunctional natural killer cells (NK), decreased function of T cells (e.g. CD8+ and CD4+), and increased production of autoantibodies (e.g., rheumatic factor, anti-thyroid) [25, 44-46]. The severity of ME/CFS may be correlated with poor functioning NK cells. NK cells play key roles in primary recognition and destruction of hijacked cells by viruses. It has been reported in several cases that the susceptibility to viral infections caused by Epstein-Barr virus, cytomegalovirus, and human herpesviruses type 1, 6 and 7 could be increased in ME/CFS due to low-functioning NK cells [39, 47-49]. These infections may, in turn, cause inflammation and/or substantially limit NAD+ production. For example, human herpesvirus type 1 infection can trigger degradation of poly(ADP-ribose) glycohydrolase and activation of the NAD-dependent DNA nick sensor poly(ADP-ribose) polymerase (PARP). The latter enzyme consumes NAD+ when repairing damages to double strand DNA [50].
Chronic inflammation and oxidative stress in ME/CFS patients may also be triggered by increased production of pro-inflammatory cytokines [51, 52], reduced levels of antioxidant compounds (e.g., coenzyme Q10) and antioxidant enzymes [53], increased oxidative and nitrosative stress (IO&NS) [54, 55], damage to important macromolecules (e.g., DNA, proteins, fatty acids) [56, 57], mitochondrial dysfunction[58], and overactivation of apoptotic pathways [27, 59]. The increase in the levels of pro-inflammatory cytokines during ME/CFS can explain the observed fatigue and flu-like symptoms. However, the most accepted hypothesis for inflammation in ME/CFS is of infectious and particularly viral origin, as a compensatory mechanism to induce anti-viral immune responses and a systemic inflammation [39, 47].
Up to 60% of ME/CFS patients have high titre of autoantibodies [60]. It was indicated that the ME/CFS patients had an IgM-associated immune response against some normally not immunogenic degraded membrane lipids produced by oxidative and nitrosative stress-induced peroxidation and S-farnesyl-l-cysteine [59]. It was suggested that chronic immune system activation and inflammation, probably due to an infection, could cause these autoimmune complications in ME/CFS patients [46, 59]. Increased production of pro-inflammatory cytokines, for example, IL-1 and tumour necrosis factor-α (TNFα), and elevated levels of nuclear factor-κB further deteriorate the autoimmune condition. The number of effector B cells and autoreactive T cells increase in response to abnormal cytokine production [4, 61, 62]. In contrast, the population of NK cells decrease, triggering an imbalance in homeostasis and enhancing the survival of effector T cells [63]. Viral infections, usually observed in ME/CFS patients, can initiate autoimmunity by employing molecular mimicry. Moreover, bacterial translocation which is also seen in these patients can induce autoimmunity and inflammation [64, 65]. Autoimmunity can also be mediated by mitochondrial dysfunction and decreased level of NAD+ and ATP that interfere with apoptosis and necrosis [60, 66]. Individuals with ME/CFS have been also identified to possess autoantibodies against neurotransmitters and important components in the central nervous system (CNS) including serotonin, dopamine, gangliosides, muscarinic receptors, and the 5-hydroxytryptamine receptor 1A receptor. These abnormalities coul explain the presence of nervous system-associated symptoms such as chronic fatigue, cognitive impairment, and sleep disturbances [27, 67].
4.Tryptophan metabolism and the kynurenine pathway
TRYP is an essential amino acid which is a precursor for various bioactive molecules, the most noteworthy of which is serotonin. However, a small proportion of TRYP is converted into serotonin and more than 90% of that is metabolised to kynurenine (KYN), several downstream neuroactive metabolites, and de novo synthesis of NAD+. The KP is strongly induced by pro-inflammatory cytokines such as INFγ [14, 15]. Two important enzymes including tryptophan dioxygenase (TDO) and IDO-1 catalyse the first rate-limiting step of the KP that is the conversion of TRYP to KYN. TDO is mainly expressed in the liver while IDO-1 is present in many cells such as macrophages and brain cells [14, 16, 68]. Under normal physiological conditions, KYN is mainly converted to 3-hydroxykynurenine (3-HK) by the catalytic activity of the enzyme kynurenine monooxygenase (KMO). In the next steps, 3-HK is metabolized to 3-hydroxyanthranilic acid (3HAA), quinolinic acid (QUIN), and finally NAD+. The remaining KYN is also converted into kynurenic acid (KYNA) by kynurenine aminotransferase (KAT) isozymes. KYNA is known as a neuroprotective metabolite antagonising the excitotoxic potential of QUIN (Fig. 3).
Figure 3.

The kynurenine pathway and the NAD+ salvage pathway. IDO: Indoleamine-pyrrole 2,3-dioxygenase; TDO: Tryptophan 2,3-dioxygenase; KATs: Kynurenine aminotransferase; KMO: Kynurenine 3-monooxygenase; 3HAO: 3-hydroxyanthranilate oxidase; QPRT: Quinolinate phosphoribosyl transferase; ACMSD: ACMS decarboxylase.
4.1 Alteration of tryptophan metabolism in ME/CFS
4.1.1 Depression and mood disorders
Several studies have provided evidence that overactivation of IDO-1 and subsequent TRYP depletion may be associated with depression and other mood disorders [69-72]. Mood disorders are prevalent complications in ME/CFS patients [59, 73, 74]. IDO-1 is extensively expressed in different human tissues including the kidney, brain, lungs, dendritic cells, and macrophages. It is induced by IFN-γ, TNFα, IL-1β, amyloid β, and lipopolysaccharide [14, 15]. It is well-documented that mood disorders are linked with cell-mediated immune system activation, which is accompanied by increased inflammation, production of IFN-γ, IL-1β, and TNFα [69, 75]. Theses pro-inflammatory cytokines can strongly activate the KP. Overactivation of the KP diverts TRYP from the serotonin biosynthesis pathway, thus depleting serotonin levels while stimulating overproduction of KP metabolites. It should be noted that KYN has depressogenic properties, whereas KYNA is a neuroprotective metabolite. Therefore, the ratio of KYN to KYNA is a significant indicator of KAT activity and neurotoxic potential. Accordingly, Maes et al. (2011) [76] reported that depressive symptoms, particularly somatization, is directly associated with increased levels of serum KP metabolites as specified by the KYN:KYNA and KYN:TRYP ratios, and negatively relates to levels of serum TRYP in serum [76].
The reduced conversion of KYN into KYNA can be due to glial depressing factors that downregulate the activity of KAT-I [77, 78]. This phenomenon can lead to excessive levels of KYN and QUIN. Alternatively, the increase in KYN:KYNA ratio could be justified by the effect of microglia on astrocytes during inflammation. Chronic inflammation, which is widely observed in ME/CFS patients, activates both microglia and astrocytes [79]. These cells are considered as the major sites of TRYP catabolism [80]. Astrocytes are the main KYNA-producing cells whereas microglia are the main QUIN producers [81, 82]. Microglia activation induces KP in the brain and produces pro-inflammatory mediators [83]. The overproduction of QUIN by activated microglia, in turn, decreases KYNA levels by supressing astrocytes. High microglial populations and low astrocyte titre was reported in depressive individuals [84-86]. It has been suggested that microglia are activated in large parts of the brains of ME/CFS patients. Accordingly, Nakatomi et al (2014) [87] reported an increase in the binding potential values of 11C-(R)-PK11195, a ligand for a translocator protein that is expressed by activated microglia, in patients with ME/CFS compared to the healthy controls.
Microglial activation can also trigger upregulation of serotonin transporter (5-HTT) in astrocytes viareleasing IL-1β, decreasing the levels of extracellular serotonin leading to depression. The interaction between IL-1β released from microglia and astrocytic 5-HTT is well-known, proved by employing IL-1 receptor antagonists. IL-1β can directly induce the upregulation of 5-HTT in primary cultured rat astrocytes [88]. This pro-inflammatory cytokine has also been detected in significantly high concentrations in ME/CFS patients, which is produced by activated microglia [89].
4.1.2QUIN production and NAD+ metabolism in ME/CFS
Besides astrocyte suppression, overproduction of QUIN induces oxidative stress, neuroinflammation, mitochondrial dysfunction and cell death. Activated microglia and infiltrating macrophages are the main source of QUIN production in the brain. Like glutamate, QUIN is an agonist of the N-methyl-D-aspartate receptor (NMDAR); therefore, its accumulation can induce excitotoxicity in neurons and astrocytes. A potent mechanism of QUIN neurotoxicity is through lipid peroxidation. More specifically, QUIN can form a complex with iron that mediates the formation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) and cause oxidative damage to lipids, proteins, and nucleic acids. Many studies have reported increased levels of oxidative stress in ME/CFS patients, promoting lipid peroxidation and protein carbonyl formation [43, 90, 91]. Oxidative and nitrosative stress induce damage to endogenous epitopes (e.g., residue components of lipid peroxidation), and trigger secondary IgM and IgG mediated autoimmune responses against neoepitopes [92, 93]. QUIN could also increase the activity of inducible nitric oxide synthase (iNOS) and nitric oxide (NO) production, which contributes to the activation of immune cells in the brain. ME/CFS patients have significantly higher iNOS activity, compared to healthy people [92, 94]. Consequently, increased NO production is also expected in these patients [95].
Alternatively, excessive QUIN production in ME/CFS patients could negatively impacted NAD+ metabolism. TRYP degradation represents the de novo route of NAD+ production, and de novo NAD+ synthesis increases in chronic inflammatory conditions. NAD+ levels increase concomitant with increases in QUIN up to levels where saturation of the enzyme quinolinic acid phosphoribosyltransferase (QPRT) occurs. The cytotoxic effects of QUIN have been reported at higher levels. NAD+ plays an important role in the cellular respiratory chain and some other cellular processes (e.g., calcium homeostasis, apoptosis, ageing, DNA repair, immunogenicity, transcriptional regulation) [96-99]. Several studies have demonstrated that QUIN-induced oxidative stress could overactivate poly (ADP-ribose) polymerase (PARP), an enzyme that repairs damaged DNA following exposure to oxidative insult [98, 100, 101]. PARP overactivation depletes NAD+ and ATP, and significantly contributes to mitochondrial dysfunction, cell energy loss, and overproduction of ROS and RNS e.g., NO and superoxide, which are observed in ME/CFS.
Studies using animal models that reflect some of the clinical features of ME/CFS have indicated a strong association between decreased expression of mitochondrial complexes, changes in mitochondrial morphology, and ultimately mitochondrial dysfunction with fatigue-like performance, high pain sensitivity, and depression [102-104]. A recent Sequential Window Acquisition of All Theoretical Mass Spectra (SWATH-MS) analysis of ME/CFS peripheral blood mononuclear cell proteome identified a distinct decline in ATP and energy production in ME/CFS and an upregulation of Complex V in the mitochondrial respiratory chain, suggestive of increased ROS production [105]. Deficiencies in the natural coenzyme Q10 (CoQ10) and NADH, the reduced form of NAD+ have been reported in patients with ME/CFS [106]. Optimal levels of CoQ10 and NADH are required for mitochondrial oxidative phosphorylation and ATP production.
4.1.3Gut microbiota and ME/CFS
The human microbiota is a dynamic community comprising more than 10,000 different microbes including bacteria, archaea, viruses, fungi, and protozoa. Naturally, the microbiota is commensal or symbiotic flora which have been co-evolved with their host and play important roles in various physiological process such as immune responses and nutrition [14, 107]. More specifically, the human gastrointestinal (GI) microbiota contains a complex microbial ecosystem which strongly maintain homeostasis of the GI tract. It has been suggested that dysbiosis of the GI microbial population could be associated with ME/CFS [108-110]. Giloteaux et al [109] reported that GI microbial diversity in ME/CFS patients was lower, compared to healthy controls. The study also found higher levels of some inflammatory biomarkers including bacterial LPS, CD14, and LPS-binding protein in the blood samples of ME/CFS patients. For example, IL-1β, IL-6, IL-8, and TNFα are some of the pro-inflammatory cytokines that were increased during ME/CF [109]. Another study reported that up to 92% of ME/CFS patients suffer from Irritable Bowel Syndrome (IBS) [111].
TRYP can be metabolized either directly or indirectly by gut microbiota to several compounds such as serotonin, kYN, indolyl compounds, and tryptamine, which play important roles in gut-brain axis communication [14, 112, 113]. More specifically, in silico analyses have indicated that some GI bacterial species (e.g. phyla Bacteroides, Firmicutes, Fusobacteria, Actinobacteria, Proteobacteria) can catabolize TRYP in the GI system and produce neuroactive metabolites including KYN, KYNA, and QUIN [14, 114]. Compared to other GI bacteria, genera Burkholderia, Ralstonia, Klebsiella, and Citrobacterhave a higher potential to catabolize TRYP to neuroactive compounds [114]. Microbial indole and indole-derived compounds (e.g., indole-3-aldehyde, indole-3-acetic acid, indole-3-propionic acid, indole-3-acetaldehyde) could regulate GI homeostasis as well as IDO-1 expression. These indolyl compounds play important roles in GI as signalling molecules, transferring information among the GI, innate and adaptive immune system, various immune cells such as NK cells, T-cells, and macrophages [15, 115] through binding to the aryl hydrocarbon receptor (AhR) [14, 15]. Indoles and their derivatives are known as natural ligands of AhR, a ligand-activated transcription factor which mediates the communication between the host and microbiota [116]. The ligand-AhR complex is translocated into the nucleus and bound to AhR nuclear translocator (ARNT) protein to form a heterodimer structure inducing specific genes containing a sequence named aryl hydrocarbon response elements (AhREs) [116]. The AhR-ARNT complex can also regulate the expression of IL-6 in macrophages. IL-6 subsequently induces the expression of IDO-1 through JAK/STAT signalling [117], increasing the levels of TRYP metabolites and enhancing TRYP depletion [14]. Increasing the level of IL-6 in ME/CFS patients has been reported in several studies [89, 116, 118] and plays a role in development of ME/CFS symptoms. Therefore, the gut microbiota can potentially affect the level of IL-6 and inflammation in ME/CFS patients through consuming TRYP.
As mentioned earlier, it has been reported that intestinal microbial biodiversity decreases in ME/CFS patients [108, 109, 119, 120]. Decreased gut microbial diversity could affect the levels of circulating TRYP as well as KYN metabolism in the peripheral and central nervous systems [14, 116]. Frémont et al [116] reported significant differences in gut microbial composition in ME/CFS patients and healthy individuals, with increased number of genus Alistipes within Bacteroidetes phylum. Another study conducted by Lupo et al [121] reported increased abundance in genera Bacteroides and a reduction in Firmicutes (except Lactonifactor) population in ME/CFS patients. Several species of genus Bacteroides (e.g. B. thetaiotaomicron, B. ovatus, B. eggerthii, B. fragilis) have been recognized to produce indole and its derivatives from TRYP catabolism in the gut [116]. It could be hypothesized that increased abundance of genus Bacteroides in the GI of ME/CFS patients may enhance TRYP metabolism and indole production leading to TRYP depletion. The reduction in Firmicutes population [108] can decrease the production of short chain fatty acids (SCFAs) in ME/CFS patients’ guts. Butyrate, a SCFA with anti-inflammatory activity, decreases IDO-1 transcription through inhibition of histone deacetylase and decreasing the expression of signal transducer and activator of transcription 1 (STAT1) leading to inhibition of IFN-γ dependent STAT1 phosphorylation and finally decreasing the STAT1-driven transcriptional activity of IDO-1 [122]. Consequently, IDO-1 could be more active in ME/CFS patients, compared to the healthy individuals, due to lack of butyrate-producing bacteria.
5. NAD+ as a therapeutic target in ME/CFS
The activity of pro-inflammatory cytokines is the driving force for increased TRYP catabolism through the KP. Therefore, strategies aimed at suppressing the activity of pro-inflammatory cytokines, for example, synthesis of IFN-γ in the peripheral IL-6 in the CNS can counteract TRYP depletion. However, TRYP depletion also represents an important mechanism to starve pathogens and tumour cells and facilitate immune tolerance [123]. Moreover, therapeutic strategies aimed at inhibiting the KP can have a dramatic effect on de novo NAD+ synthesis [124].
Apart from the KP, NAD+ can also be produced from the salvage pathway using the precursors nicotinic acid (NA), nicotinamide (NAM), nicotinamide mononucleotide (NMN) and nicotinamide riboside (NR) [125]. However, it should be noted that NA therapy induces some unwanted negative adverse effects including significant skin flushing in most individuals below therapeutic doses, which limits its widespread clinical use [125]. NAM is the primary form of vitamin B3 found in meat and is synthesized as a by-product of enzymatic degradation of NAD+ by PARPs, Sirtuins and NAD+ glycohydrolases (e.g CD38) [125]. NAM aids recovery from depression and mood disorders by modulating monoamine-neurotransmitter synthesis and degradation, potent antioxidant effects and increasing intracellular NAD+ levels [126]. Although supplementation with NAM raises NAD+ without causing flushing, it is unlikely to be an ideal NAD+ supplement due to its enzyme inhibiting (e.g., PARPs, sirtuins, CD38), methyl depleting and hepatotoxic potential [125].
NMN can be produced endogenously from NR [125]. Animal studies have shown that NMN can ameliorate degeneration and improved age-related cognitive decline [127-131]. However, the intracellular uptake of NMN is unclear. Recently, one study reported that NMN is dephosphorylated to NR and then internalization by the solute carrier family 12 member 8 (Slc12a8) [132]. However, another report did not find enough evidence to support Slc12a8 as the ‘reclusive’ NMN transporter [133].
NR is a precursor that can be naturally obtained by regular consumption of cow milk [125]. NR is converted to NAD+ via the catalytic activity of NRK1 and NRK2. There also exists an NR kinase independent pathway which converts NR to NAM which is then salvaged back to NAD+ [125]. NR has been shown to prevent cognitive decline and ameliorate brain attenuate brain degeneration [126], and correct social deficits and anxiety disorders in animal models [134]. This suggests that NR may have beneficial effects in ME/CFS. Oral uptake of NR has been reported to increase NAD+ concentrations in whole blood and tissue in humans. NR is safe and more orally bioavailable than other NAD+ precursors [125]. Unlike other niacin supplements, NR does not cause flushing. However, there are no clinical trials using NAD+ precursors, particularly NMN and NR in ME/CFS. Further clinical evidence is necessary for the use of NMN and NR as NAD+ supplements for managing and preventing ME/CFS.
Mitochondrial dysfunction, which limits NAD+ and ATP levels, is the central agent for energy production in patients with ME/CFS. Supplementation with CoQ10 (200 mg/day) and NADH (20 mg/day) can enhance cellular bioenergetics and reduce fatigue and lipoperoxides, and improve biochemical parameters (e.g. CoQ10, ATP, citrate synthase and NAD+:NADH redox ratio, in humans) [135]. One study showed a significant reduction in anxiety and maximum heart rate in a Spanish CFS cohort [136]. Another study demonstrated a significant reduction in symptoms following treatment with NADH (10 mg/day), compared to placebo [137]. Oral supplementation with NADH was more effective in terms of symptom reduction than conventional nutritional supplements and psychotherapy over a 24-month period [138]. Oral NADH has been shown to be safe and well tolerated in humans [135-138]. However, results are conflicting for CoQ10. One study showed that supplementation with CoQ10 (50 mg/day) and NADH (5 mg/day) significantly reduced maximal heart rate during cycle ergometer tests. Fatigue perception was also reduced, although no improvements were reported for pain and sleep [139]. However, larger controlled trials are needed to confirm these findings.
6. Conlusions
ME/CFS is a poorly understood disorder with unknown aetiology and no established diagnostic criteria. During ME/CFS, NAD+ level significantly decrease, leaving the patient with severe fatigue. This drop can be largely explained by 1) the overactivation of IDO-1 shunts the available TRYP into KP, increasing KP metabolites, depleting serotonin, and causing mood disorders and other related neurological symptoms; 2) QUIN, at pathophysiological concentrations can trigger oxidative stress and apoptosis viaexcitotoxicity in different cells including neurons and astrocytes. The lower population of astrocytes, in turn, is translated into lower KYNA production, further enhancing neurological symptoms; 3) The available NAD+ and ATP are depleted due to overactivation of PARP, following QUIN-induced oxidative damages to DNA. This phenomenon significantly contributes to cell energy loss and leads to mitochondria dysfunctional; 4) Mitochondrial dysfunctional further promotes the production of ROS and RNS leading to oxidative stress and reduced function; and 5) The accumulation of QUIN can have positive feedback control on all the mechanisms explained above by triggering chronic inflammation. This chronic inflammation is due to QUIN-induced damage to macromolecules, ROS and RNS-induced damage to endogenous epitopes, and secondary IgM and IgG mediated autoimmune responses against neoepitopes. Alteration to the gut microbiota may further contribute to ME/CFS through increased production of indole and its derivatives and modified availability of circulatory TRYP. Reduction of microbial biodiversity in GI of ME/CFS patients may increase inflammation and IDO-1 activity leading to increased TRYP catabolism and depletion.
As well, raising intracellular NAD+ levels can improve the quality of life by improving neurological function, promoting energy production, and lowering fatigue. NAD+ precursors such as NR and NMN have been suggested as potential treatments to correct anxiety disorders, fatigue, and social deficits in animal models. However, there are no clinical trials using NAD+ precursors to alleviate the clinical features of ME/CFS and further clinical assessment are needed for the use of these precursors for increasing the level of NAD+ in ME/CFS patients. Supplements using NADH and CoQ10 are safe and well-tolerated and may be added to conventional CFS therapy subject to recommendation from a general practitioner.
Footnotes
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- [1].Blomberg J, Gottfries CG, Elfaitouri A, Rizwan M, Rosén A (2018). Infection elicited autoimmunity and myalgic encephalomyelitis/chronic fatigue syndrome: an explanatory model. Front Immunol, 9:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Clayton EW (2015). Beyond myalgic encephalomyelitis/chronic fatigue syndrome: an IOM report on redefining an illness. JAMA, 313:1101-1102. [DOI] [PubMed] [Google Scholar]
- [3].EmergeAustralia. 2019. What is ME/CFS? https://www.emerge.org.au/what-is-mecfs.
- [4].Sotzny F, Blanco J, Capelli E, Castro-Marrero J, Steiner S, Murovska M, et al. (2018). Myalgic encephalomyelitis/chronic fatigue syndrome-evidence for an autoimmune disease. Autoimmun Rev, 17:601-609. [DOI] [PubMed] [Google Scholar]
- [5].Rowe PC, Underhill RA, Friedman KJ, Gurwitt A, Medow MS, Schwartz MS, et al. (2017). Myalgic encephalomyelitis/chronic fatigue syndrome diagnosis and management in young people: a primer. Front Pediatr, 5:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Słomko J, Newton JL, Kujawski S, Tafil-Klawe M, Klawe J, Staines D, et al. (2019). Prevalence and characteristics of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME) in Poland: A cross-sectional study. BMJ Open, 9:e023955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Castro-Marrero J, Faro M, Aliste L, Sáez-Francàs N, Calvo N, Martínez-Martínez A, et al. (2017). Comorbidity in chronic fatigue syndrome/myalgic encephalomyelitis: a nationwide population-based cohort study. Psychosomatics, 58:533-543. [DOI] [PubMed] [Google Scholar]
- [8].Bested AC, Marshall LM (2015). Review of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome: an evidence-based approach to diagnosis and management by clinicians. Rev Environ Health, 30:223-249. [DOI] [PubMed] [Google Scholar]
- [9].Underhill R (2015). Myalgic encephalomyelitis, chronic fatigue syndrome: an infectious disease. Med Hypotheses, 85:765-773. [DOI] [PubMed] [Google Scholar]
- [10].Poenaru S, Abdallah SJ, Corrales-Medina V, Cowan J (2021). COVID-19 and post-infectious myalgic encephalomyelitis/chronic fatigue syndrome: a narrative review. Ther Adv Infect Dis, 8:20499361211009385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Noda M, Ifuku M, Hossain M, Katafuchi T (2018). Glial activation and expression of the serotonin transporter in chronic fatigue syndrome. Frontiers Psychiatry, 9:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mackay A, Tate WP (2018). A compromised paraventricular nucleus within a dysfunctional hypothalamus: a novel neuroinflammatory paradigm for ME/CFS. Int J Immunopathol Pharmacol, 32:2058738418812342. [Google Scholar]
- [13].Braidy N, Grant R (2017). Kynurenine pathway metabolism and neuroinflammatory disease. Neural Regen Res, 12:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dehhaghi M, Kazemi Shariat Panahi H, Guillemin GJ (2019). Microorganisms, tryptophan metabolism, and kynurenine pathway: A complex interconnected loop influencing human health status. Int J Tryptophan Res, 12:1178646919852996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Dehhaghi M, Panahi HKS, Heng B, Guillemin GJ (2020). The gut microbiota, kynurenine pathway, and immune system interaction in the development of brain cancer. Front Cell Dev Biol, 8:562812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Guillemin GJ, Smythe G, Takikawa O, Brew BJ (2005). Expression of indoleamine 2, 3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia, 49:15-23. [DOI] [PubMed] [Google Scholar]
- [17].Braidy N, Berg J, Clement J, Khorshidi F, Poljak A, Jayasena T, et al. (2019). Role of nicotinamide adenine dinucleotide and related precursors as therapeutic targets for age-related degenerative diseases: rationale, biochemistry, pharmacokinetics, and outcomes. Antioxid Redox Signal, 30:251-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jason L, Mirin A (2021). Updating the National Academy of Medicine ME/CFS prevalence and economic impact figures to account for population growth and inflation. Fatigue: Biomed. Health Behav, 1-5. [Google Scholar]
- [19].Lim EJ, Ahn YC, Jang ES, Lee SW, Lee SH, Son CG (2020). Systematic review and meta-analysis of the prevalence of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME). J Transl Med, 18:1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Jason LA, Benton MC, Valentine L, Johnson A, Torres-Harding S (2008). The economic impact of ME/CFS: individual and societal costs. Dyn Med, 7:1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Afari N, Buchwald D (2003). Chronic fatigue syndrome: a review. Am J Psychiatry, 160:221-236. [DOI] [PubMed] [Google Scholar]
- [22].Hornig M, Montoya JG, Klimas NG, Levine S, Felsenstein D, Bateman L, et al. (2015). Distinct plasma immune signatures in ME/CFS are present early in the course of illness. Sci Adv, 1:e1400121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].VanElzakker MB, Brumfield SA, Lara Mejia PS (2019). Neuroinflammation and cytokines in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): a critical review of research methods. Front Neurol, 9:1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Haney E, Smith MB, McDonagh M, Pappas M, Daeges M, Wasson N, et al. (2015). Diagnostic methods for myalgic encephalomyelitis/chronic fatigue syndrome: a systematic review for a National Institutes of Health Pathways to Prevention Workshop. Ann Int Med, 162:834-840. [DOI] [PubMed] [Google Scholar]
- [25].Rivas JL, Palencia T, Fernández G, García M (2018). Association of T and NK cell phenotype with the diagnosis of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Front Immunol, 9:1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Skapinakis P, Lewis G, Meltzer H (2000). Clarifying the relationship between unexplained chronic fatigue and psychiatric morbidity: results from a community survey in Great Britain. Int Rev Psychiatry, 157:1492-1498. [DOI] [PubMed] [Google Scholar]
- [27].Cortes Rivera M, Mastronardi C, Silva-Aldana CT, Arcos-Burgos M, Lidbury BA (2019). Myalgic encephalomyelitis/chronic fatigue syndrome: a comprehensive review. Diagnostics, 9:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nacul LC, Lacerda EM, Pheby D, Campion P, Molokhia M, Fayyaz S, et al. (2011). Prevalence of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) in three regions of England: a repeated cross-sectional study in primary care. BMC Med, 9:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nijrolder I, van der Windt DA, Twisk JW, van der Horst HE (2010). Fatigue in primary care: longitudinal associations with pain. Pain, 150:351-357. [DOI] [PubMed] [Google Scholar]
- [30].Reyes M, Nisenbaum R, Hoaglin DC, Unger ER, Emmons C, Randall B, et al. (2003). Prevalence and incidence of chronic fatigue syndrome in Wichita, Kansas. Archives of internal medicine, 163:1530-1536. [DOI] [PubMed] [Google Scholar]
- [31].Lawrie S, Manders D, Geddes J, Pelosi A (1997). A population-based incidence study of chronic fatigue. Psychol Med, 27:343-353. [DOI] [PubMed] [Google Scholar]
- [32].Mensah FKF, Bansal AS, Ford B, Cambridge G (2017). Chronic fatigue syndrome and the immune system: where are we now? Neurophysiologie Clinique/Clinical Neurophysiol, 47:131-138. [DOI] [PubMed] [Google Scholar]
- [33].CDC. 2018. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. https://www.cdc.gov/me-cfs/index.html
- [34].Reynolds KJ, Vernon SD, Bouchery E, Reeves WC (2004). The economic impact of chronic fatigue syndrome. Cost Eff Resour Alloc, 2:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Griffith JP, Zarrouf FA (2008). A systematic review of chronic fatigue syndrome: don't assume it's depression. Prim Care Companion J Clin Psychiatry, 10:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jason LA, Corradi K, Gress S, Williams S, Torres-Harding S (2006). Causes of death among patients with chronic fatigue syndrome. Health Care Women Int, 27:615-626. [DOI] [PubMed] [Google Scholar]
- [37].McManimen SL, Devendorf AR, Brown AA, Moore BC, Moore JH, Jason LA (2016). Mortality in patients with myalgic encephalomyelitis and chronic fatigue syndrome. Fatigue, 4(4): 195-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dimmock ME, Mirin AA, Jason LA (2016). Estimating the disease burden of ME/CFS in the United States and its relation to research funding. J Med Ther, 1:1-7. [Google Scholar]
- [39].Rasa S, Nora-Krukle Z, Henning N, Eliassen E, Shikova E, Harrer T, et al. (2018). Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). J Trans Med, 16:1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Chia J, Chia A, Voeller M, Lee T, Chang R (2010). Acute enterovirus infection followed by myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and viral persistence. J Clinic Pathol, 63:165-168. [DOI] [PubMed] [Google Scholar]
- [41].Dibble JJ, McGrath SJ, Ponting CP (2020). Genetic risk factors of ME/CFS: a critical review. Hum Mol Genet, 29:R117-R124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Nacul L, O'Boyle S, Palla L, Nacul FE, Mudie K, Kingdon CC, et al. (2020). How Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) Progresses: The Natural History of ME/CFS. Front Neurol, 11:826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Morris G, Maes M (2014). Oxidative and nitrosative stress and immune-inflammatory pathways in patients with myalgic encephalomyelitis (ME)/chronic fatigue syndrome (CFS). Curr Neuropharmacol, 12:168-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].CDC. 2018. Etiology and Pathophysiology. https://www.cdc.gov/me-cfs/healthcare-providers/presentation-clinical-course/etiology-pathophysiology.html.
- [45].Montoya JG, Holmes TH, Anderson JN, Maecker HT, Rosenberg-Hasson Y, Valencia IJ, et al. (2017). Cytokine signature associated with disease severity in chronic fatigue syndrome patients. Proc Natl Acad Sci U S A, 114:E7150-E7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lorusso L, Mikhaylova SV, Capelli E, Ferrari D, Ngonga GK, Ricevuti G (2009). Immunological aspects of chronic fatigue syndrome. Autoimmu Rev, 8:287-291. [DOI] [PubMed] [Google Scholar]
- [47].Chapenko S, Krumina A, Logina I, Rasa S, Chistjakovs M, Sultanova A, et al. (2012). Association of active human herpesvirus-6,-7 and parvovirus b19 infection with clinical outcomes in patients with myalgic encephalomyelitis/chronic fatigue syndrome. Adv Virol, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Agliari E, Barra A, Vidal KG, Guerra F (2012). Can persistent Epstein-Barr virus infection induce chronic fatigue syndrome as a Pavlov reflex of the immune response? J Biol Dyn, 6:740-762. [DOI] [PubMed] [Google Scholar]
- [49].Brenu E, Hardcastle S, Atkinson G, van Driel M, Kreijkamp-Kaspers S, Ashton K, et al. (2013). Natural killer cells in patients with severe chronic fatigue syndrome. Auto Immun Highlights, 4:69-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Grady SL, Hwang J, Vastag L, Rabinowitz JD, Shenk T (2012). Herpes simplex virus 1 infection activates poly (ADP-ribose) polymerase and triggers the degradation of poly (ADP-ribose) glycohydrolase. J Virol, 86:8259-8268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Milrad SF, Hall DL, Jutagir DR, Lattie EG, Ironson GH, Wohlgemuth W, et al. (2017). Poor sleep quality is associated with greater circulating pro-inflammatory cytokines and severity and frequency of chronic fatigue syndrome/myalgic encephalomyelitis (CFS/ME) symptoms in women. J Neuroimmunol, 303:43-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Morris G, Maes M (2012). Increased nuclear factor-κB and loss of p53 are key mechanisms in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Med Hypotheses, 79:607-613. [DOI] [PubMed] [Google Scholar]
- [53].Maes M, Mihaylova I, Kubera M, Uytterhoeven M, Vrydags N, Bosmans E (2009). Coenzyme Q10 deficiency in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is related to fatigue, autonomic and neurocognitive symptoms and is another risk factor explaining the early mortality in ME/CFS due to cardiovascular disorder. Neuro Endocrinol Lett, 30:470-476. [PubMed] [Google Scholar]
- [54].Maes M, Twisk F (2009). Why myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) may kill you: disorders in the inflammatory and oxidative and nitrosative stress (IO&NS) pathways may explain cardiovascular disorders in ME/CFS. Neuroendocrinol Lett, 30:677-693. [PubMed] [Google Scholar]
- [55].Maes M, Kubera M, Stoyanova K, Leunis JC (2021). The Reification of the Clinical Diagnosis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) as an Immune and Oxidative Stress Disorder: Construction of a Data-Driven Nomothethic Network and Exposure of ME/CFS Subgroups. Curr Top Med Chem, PMID: 34315375. [DOI] [PubMed] [Google Scholar]
- [56].Maes M, Mihaylova I, Kubera M, Uytterhoeven M, Vrydags N, Bosmans E (2009). Increased 8-hydroxy-deoxyguanosine, a marker of oxidative damage to DNA, in major depression and myalgic encephalomyelitis/chronic fatigue syndrome. Neuroendocrinol Lett, 30:715. [PubMed] [Google Scholar]
- [57].Glassford JA (2017). The neuroinflammatory etiopathology of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Front Physiol, 8:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Booth NE, Myhill S, McLaren-Howard J (2012). Mitochondrial dysfunction and the pathophysiology of myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Int J Clin Exp Med, 5:208. [PMC free article] [PubMed] [Google Scholar]
- [59].Maes M, Mihaylova I, Kubera M, Leunis JC, Twisk FN, Geffard M (2012). IgM-mediated autoimmune responses directed against anchorage epitopes are greater in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) than in major depression. Metab Brain Dis, 27:415-423. [DOI] [PubMed] [Google Scholar]
- [60].Morris G, Berk M, Galecki P, Maes M (2014). The emerging role of autoimmunity in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Mol Neurobiol, 49:741-756. [DOI] [PubMed] [Google Scholar]
- [61].Thorarinsdottir K, Camponeschi A, Cavallini N, Grimsholm O, Jacobsson L, Gjertsson I, et al. (2016). CD21-/low B cells in human blood are memory cells. Clin Exp Immunol, 185:252-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Klimas NG, Salvato FR, Morgan R, Fletcher MA (1990). Immunologic abnormalities in chronic fatigue syndrome. J Clin Microbiol, 28:1403-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Brenu EW, Van Driel ML, Staines DR, Ashton KJ, Hardcastle SL, Keane J, et al. (2012). Longitudinal investigation of natural killer cells and cytokines in chronic fatigue syndrome/myalgic encephalomyelitis. J Translation Med, 10:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Maes M, Ringel K, Kubera M, Anderson G, Morris G, Galecki P, et al. (2013). In myalgic encephalomyelitis/chronic fatigue syndrome, increased autoimmune activity against 5-HT is associated with immuno-inflammatory pathways and bacterial translocation. J Affect Disord, 150:223-230. [DOI] [PubMed] [Google Scholar]
- [65].Maes M, Leunis JC, Geffard M, Berk M (2014). Evidence for the existence of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS) with and without abdominal discomfort (irritable bowel) syndrome. Neuroendocrinol Lett, 35:445-453. [PubMed] [Google Scholar]
- [66].Myhill S, Booth NE, McLaren-Howard J (2013). Targeting mitochondrial dysfunction in the treatment of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS)-a clinical audit. Int J Clin Exp Med, 6:1. [PMC free article] [PubMed] [Google Scholar]
- [67].Loebel M, Grabowski P, Heidecke H, Bauer S, Hanitsch LG, Wittke K, et al. (2016). Antibodies to β adrenergic and muscarinic cholinergic receptors in patients with Chronic Fatigue Syndrome. Brain Bhav Immu, 52:32-39. [DOI] [PubMed] [Google Scholar]
- [68].Chen Y, Guillemin GJ (2009). Kynurenine pathway metabolites in humans: disease and healthy states. Int J Tryp Res, 2: IJTR. S2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Maes M, Leonard B, Myint A, Kubera M, Verkerk R (2011). The new ‘5-HT’hypothesis of depression: cell-mediated immune activation induces indoleamine 2, 3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog Neuropsychopharmacol Biol Psychiatry, 35:702-721. [DOI] [PubMed] [Google Scholar]
- [70].Maes M, Mihaylova I, Ruyter MD, Kubera M, Bosmans E (2007). The immune effects of TRYCATs (tryptophan catabolites along the IDO pathway): Relevance for depression--And other conditions characterized by tryptophan depletion induced by inflammation. Neuroendocrinol Lett, 28:826-831. [PubMed] [Google Scholar]
- [71].Wigner P, Czarny P, Galecki P, Su KP, Sliwinski T (2018). The molecular aspects of oxidative & nitrosative stress and the tryptophan catabolites pathway (TRYCATs) as potential causes of depression. Psychiatry Res, 262:566-574. [DOI] [PubMed] [Google Scholar]
- [72].Kanchanatawan B, Sirivichayakul S, Carvalho AF, Anderson G, Galecki P, Maes M (2018). Depressive, anxiety and hypomanic symptoms in schizophrenia may be driven by tryptophan catabolite (TRYCAT) patterning of IgA and IgM responses directed to TRYCATs. Prog Neuropsychopharmacol Biol Psychiatr, 80:205-216. [DOI] [PubMed] [Google Scholar]
- [73].Crawley E, Hunt L, Stallard P (2009). Anxiety in children with CFS/ME. Eur Child Adolesc Psychiatry, 18:683-689. [DOI] [PubMed] [Google Scholar]
- [74].Bould H, Lewis G, Emond A, Crawley E (2011). Depression and anxiety in children with CFS/ME: cause or effect? Arch Dis Childhood, 96:211-214. [DOI] [PubMed] [Google Scholar]
- [75].Leonard B, Maes M (2012). Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci Biobehav Rev, 36:764-785. [DOI] [PubMed] [Google Scholar]
- [76].Maes M, Galecki P, Verkerk R, Rief W (2011). Somatization, but not depression, is characterized by disorders in the tryptophan catabolite (TRYCAT) pathway, indicating increased indoleamine 2, 3-dioxygenase and lowered kynurenine aminotransferase activity. Neuroendocrinol Lett, 32:264-273. [PubMed] [Google Scholar]
- [77].Dantzer R, O’Connor JC, Lawson MA, Kelley KW (2011). Inflammation-associated depression: from serotonin to kynurenine. Psychoneuroendocrinol, 36:426-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Baran H, Kepplinger B, Draxler M (2010). Endogenous kynurenine aminotransferases inhibitor is proposed to act as “Glia Depressing Factor”(GDF). Int J Trypt Res, 3:IJTR. S3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Guillemin GJ, Smith DG, Smythe GA, Armati PJ, Brew GJ.2003. Expression of the kynurenine pathway enzymes in human microglia and macrophages. In Developments in Tryptophan and Serotonin Metabolism: Springer. 105-112. [DOI] [PubMed] [Google Scholar]
- [80].Grant RS, Naif H, Espinosa M, Kapoor V (2000). IDO induction in IFN-γ activated astroglia: a role in improving cell viability during oxidative stress. Redox Rep, 5:101-104. [DOI] [PubMed] [Google Scholar]
- [81].Guillemin GJ, Kerr SJ, Smythe GA, Smith DG, Kapoor V, Armati PJ, et al. (2001). Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection. J Neurochem, 78:842-853. [DOI] [PubMed] [Google Scholar]
- [82].Guillemin GJ (2012). Quinolinic acid, the inescapable neurotoxin. FEBS, 279:1356-1365. [DOI] [PubMed] [Google Scholar]
- [83].Tao X, Yan M, Wang L, Zhou Y, Wang Z, Xia T, et al. (2020). Homeostasis imbalance of microglia and astrocytes leads to alteration in the metabolites of the kynurenine pathway in LPS-induced depressive-like mice. Int J Mol Sci, 21:1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Busse M, Busse S, Myint AM, Gos T, Dobrowolny H, Müller UJ, et al. (2015). Decreased quinolinic acid in the hippocampus of depressive patients: evidence for local anti-inflammatory and neuroprotective responses? Eur Arch Psychiatry Clin Neurosci, 265:321-329. [DOI] [PubMed] [Google Scholar]
- [85].Steiner J, Walter M, Gos T, Guillemin G, Bernstein H, Sarnyai Z, et al. (2011). Severe depression is associated with increased quinolinic acid immunoreactivity in the dorsal and ventral anterior cingulum: further evidence for an immune modulation of glutamatergic neurotransmission. J Neuroinflammation, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Stone T, Behan W, MacDonald M, Darlington L (2000). Possible mediation of quinolinic acid-induced hippocampal damage by reactive oxygen species. Amino Acids, 19:275-281. [DOI] [PubMed] [Google Scholar]
- [87].Nakatomi Y, Mizuno K, Ishii A, Wada Y, Tanaka M, Tazawa S, et al. (2014). Neuroinflammation in patients with chronic fatigue syndrome/myalgic encephalomyelitis: an 11C-(R)-PK11195 PET study. J Nuc Med, 55:945-950. [DOI] [PubMed] [Google Scholar]
- [88].Ifuku M, Hossain SM, Noda M, Katafuchi T (2014). Induction of interleukin-1β by activated microglia is a prerequisite for immunologically induced fatigue. Eur J Neurosci, 40:3253-3263. [DOI] [PubMed] [Google Scholar]
- [89].Maes M, Twisk FN, Kubera M, Ringel K (2012). Evidence for inflammation and activation of cell-mediated immunity in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): increased interleukin-1, tumor necrosis factor-α, PMN-elastase, lysozyme and neopterin. J Affect Disord, 136:933-939. [DOI] [PubMed] [Google Scholar]
- [90].Maes M, Kubera M, Uytterhoeven M, Vrydags N, Bosmans E (2011). Increased plasma peroxides as a marker of oxidative stress in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Med Sci Monit, 17:SC11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Singh A, Naidu PS, Gupta S, Kulkarni SK (2002). Effect of natural and synthetic antioxidants in a mouse model of chronic fatigue syndrome. J Med Food, 5:211-220. [DOI] [PubMed] [Google Scholar]
- [92].Maes M, Mihaylova I, De Ruyter M (2006). Lower serum zinc in Chronic Fatigue Syndrome (CFS): relationships to immune dysfunctions and relevance for the oxidative stress status in CFS. J Affect Disord, 90:141-147. [DOI] [PubMed] [Google Scholar]
- [93].Geffard M, Bodet D, Martinet Y, Dabadie M (2002). Detection of the specific IgM and IgA circulating in sera of multiple sclerosis patients: interest and perspectives. Immuno-analyse & biologie sp écialisé, 17:302-310. [Google Scholar]
- [94].Maes M, Mihaylova I, Kubera M, Bosmans E (2007). Not in the mind but in the cell: increased production of cyclo-oxygenase-2 and inducible NO synthase in chronic fatigue syndrome. Neuroendocrinol Lett, 28:463-469. [PubMed] [Google Scholar]
- [95].Maes M, Twisk FN, Johnson C (2012). Myalgic encephalomyelitis (ME), chronic fatigue syndrome (CFS), and chronic fatigue (CF) are distinguished accurately: results of supervised learning techniques applied on clinical and inflammatory data. Psychiatry Res, 200:754-760. [DOI] [PubMed] [Google Scholar]
- [96].Ying W (2008). NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal, 10:179-206. [DOI] [PubMed] [Google Scholar]
- [97].Kim MY, Zhang T, Kraus WL (2005). Poly (ADP-ribosyl) ation by PARP-1:PAR-laying'NAD+ into a nuclear signal. Genes Dev, 19:1951-1967. [DOI] [PubMed] [Google Scholar]
- [98].Dehhaghi M, Tan V, Heng B, Braidy N, Mohammadipanah F, Guillemin GJ (2019). Neuroprotective effect of myxobacterial extracts on quinolinic acid-induced toxicity in primary human neurons. Neurotox Res, 35:281-290. [DOI] [PubMed] [Google Scholar]
- [99].Dehhaghi M, Tan V, Heng B, Mohammadipanah F, Guillemin GJ (2019). Protective effects of myxobacterial extracts on hydrogen peroxide-induced toxicity on human primary astrocytes. Neurosci, 399:1-11. [DOI] [PubMed] [Google Scholar]
- [100].Dehhaghi M, Kazemi Shariat Panahi H, Braidy N, Guillemin GJ (2020). Herpetosiphon secondary metabolites inhibit amyloid-β toxicity in human primary astrocytes. J Alzheimers Dis.:1-11. [DOI] [PubMed] [Google Scholar]
- [101].Braidy N, Grant R, Adams S, Guillemin GJ (2010). Neuroprotective effects of naturally occurring polyphenols on quinolinic acid-induced excitotoxicity in human neurons. FEBS, 277:368-382. [DOI] [PubMed] [Google Scholar]
- [102].Surapaneni DK, Adapa SRSS, Preeti K, Teja GR, Veeraragavan M, Krishnamurthy S (2012). Shilajit attenuates behavioral symptoms of chronic fatigue syndrome by modulating the hypothalamic-pituitary-adrenal axis and mitochondrial bioenergetics in rats. J Ethnopharmacol, 143:91-99. [DOI] [PubMed] [Google Scholar]
- [103].Wang J, Sun C, Zheng Y, Pan H, Zhou Y, Fan Y (2014). The effective mechanism of the polysaccharides from Panax ginseng on chronic fatigue syndrome. Arch Pharmacal Res, 37:530-538. [DOI] [PubMed] [Google Scholar]
- [104].Vichaya EG, Vermeer DW, Christian DL, Molkentine JM, Mason KA, Lee JH, et al. (2017). Neuroimmune mechanisms of behavioral alterations in a syngeneic murine model of human papilloma virus-related head and neck cancer. Psychoneuroendocrinol, 79:59-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Sweetman E, Kleffmann T, Edgar C, de Lange M, Vallings R, Tate W (2020). A SWATH-MS analysis of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome peripheral blood mononuclear cell proteomes reveals mitochondrial dysfunction. J Transl Med, 18:1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Castro-Marrero J, Cordero MD, Sáez-Francas N, Jimenez-Gutierrez C, Aguilar-Montilla FJ, Aliste L, et al.2013. Could mitochondrial dysfunction be a differentiating marker between chronic fatigue syndrome and fibromyalgia? : Mary Ann Liebert, Inc. 140 Huguenot Street, 3rd Floor New Rochelle, NY 10801 USA. [DOI] [PubMed] [Google Scholar]
- [107].Dehhaghi M, Kazemi Shariat Panahi H, Guillemin GJ (2018). Microorganisms’ footprint in neurodegenerative diseases. Front Cell Neurosci, 12:466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Frémont M, Coomans D, Massart S, De Meirleir K (2013). High-throughput 16S rRNA gene sequencing reveals alterations of intestinal microbiota in myalgic encephalomyelitis/chronic fatigue syndrome patients. Anaerobe, 22:50-56. [DOI] [PubMed] [Google Scholar]
- [109].Giloteaux L, Goodrich JK, Walters WA, Levine SM, Ley RE, Hanson MR (2016). Reduced diversity and altered composition of the gut microbiome in individuals with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome, 4:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Navaneetharaja N, Griffiths V, Wileman T, Carding SR (2016). A role for the intestinal microbiota and virome in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS)? J Clin Med, 5:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Loohuis LMO, Mangul S, Ori AP, Jospin G, Koslicki D, Yang HT, et al. (2018). Transcriptome analysis in whole blood reveals increased microbial diversity in schizophrenia. Transl Psychiatry, 8:1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Kennedy PJ, Cryan JF, Dinan TG, Clarke G (2017). Kynurenine pathway metabolism and the microbiota-gut-brain axis. Neuropharmacol, 112:399-412. [DOI] [PubMed] [Google Scholar]
- [113].Agus A, Planchais J, Sokol H (2018). Gut microbiota regulation of tryptophan metabolism in health and disease. Cell Host Microbe, 23:716-724. [DOI] [PubMed] [Google Scholar]
- [114].Kaur H, Bose C, Mande SS (2019). Tryptophan metabolism by gut microbiome and gut-brain-axis: an in silico analysis. Front Neurosci, 13:1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Cheong JE, Sun L (2018). Targeting the IDO1/TDO2-KYN-AhR pathway for cancer immunotherapy-challenges and opportunities. Trends Pharmacol Sci, 39:307-325. [DOI] [PubMed] [Google Scholar]
- [116].Anderson G, Reiter RJ (2020). Melatonin: roles in influenza, Covid-19, and other viral infections. Rev Med Virol, 30:e2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Kim H, Chen L, Lim G, Sung B, Wang S, McCabe MF, et al. (2012). Brain indoleamine 2, 3-dioxygenase contributes to the comorbidity of pain and depression. J Clin Invest, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Domingo JC, Cordobilla B, Ferrer R, Giralt M, Alegre-Martín J, Castro-Marrero J (2021). Are Circulating Fibroblast Growth Factor 21 and N-Terminal Prohormone of Brain Natriuretic Peptide Promising Novel Biomarkers in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome? Antioxid Redox Signal, 34(18):1420-1427. [DOI] [PubMed] [Google Scholar]
- [119].Shukla SK, Cook D, Meyer J, Vernon SD, Le T, Clevidence D, et al. (2015). Changes in gut and plasma microbiome following exercise challenge in myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). PLoS One, 10:e0145453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Nagy-Szakal D, Williams BL, Mishra N, Che X, Lee B, Bateman L, et al. (2017). Fecal metagenomic profiles in subgroups of patients with myalgic encephalomyelitis/chronic fatigue syndrome. Microbiome, 5:1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Lupo GFD, Rocchetti G, Lucini L, Lorusso L, Manara E, Bertelli M, et al. (2021). Potential role of microbiome in Chronic Fatigue Syndrome/Myalgic Encephalomyelits (CFS/ME). Sci Rep, 11:1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Gao K, Mu CL, Farzi A, Zhu WY (2020). Tryptophan metabolism: a link between the gut microbiota and brain. Adv Nut, 11:709-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Moffett JR, Namboodiri MA (2003). Tryptophan and the immune response. Immunol Cell Biol, 81:247-265. [DOI] [PubMed] [Google Scholar]
- [124].Braidy N, Guillemin GJ, Grant R (2011). Effects of kynurenine pathway inhibition on NAD+ metabolism and cell viability in human primary astrocytes and neurons. Int J TrypRes, 4:IJTR. S7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Braidy N, Berg J, Clement J, Khorshidi F, Poljak A, Jayasena T, et al. (2019). Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxid Redox Signal, 30:251-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Song SB, Park JS, Chung GJ, Lee IH, Hwang ES (2019). Diverse therapeutic efficacies and more diverse mechanisms of nicotinamide. Metabolomics, 15:1-28. [DOI] [PubMed] [Google Scholar]
- [127].Hosseini L, Farokhi-Sisakht F, Badalzadeh R, Khabbaz A, Mahmoudi J, Sadigh-Eteghad S (2019). Nicotinamide Mononucleotide and Melatonin Alleviate Aging-induced Cognitive Impairment via Modulation of Mitochondrial Function and Apoptosis in the Prefrontal Cortex and Hippocampus. Neurosci, 423:29-37. [DOI] [PubMed] [Google Scholar]
- [128].Johnson S, Wozniak DF, Imai S (2018). CA1 Nampt knockdown recapitulates hippocampal cognitive phenotypes in old mice which nicotinamide mononucleotide improves. NPJ Aging Mech Dis, 4:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Kiss T, Balasubramanian P, Valcarcel-Ares MN, Tarantini S, Yabluchanskiy A, Csipo T, et al. (2019). Nicotinamide mononucleotide (NMN) treatment attenuates oxidative stress and rescues angiogenic capacity in aged cerebromicrovascular endothelial cells: a potential mechanism for the prevention of vascular cognitive impairment. Gerosci, 41:619-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Tarantini S, Valcarcel-Ares MN, Toth P, Yabluchanskiy A, Tucsek Z, Kiss T, et al. (2019). Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol, 24:101192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Wang X, Hu X, Zhang L, Xu X, Sakurai T (2020). Nicotinamide mononucleotide administration after sever hypoglycemia improves neuronal survival and cognitive function in rats. Brain Res Bull, 160:98-106. [DOI] [PubMed] [Google Scholar]
- [132].Grozio A, Mills KF, Yoshino J, Bruzzone S, Sociali G, Tokizane K, et al. (2019). Slc12a8 is a nicotinamide mononucleotide transporter. Nat Metab, 1:47-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Schmidt MS, Brenner C (2019). Absence of evidence that Slc12a8 encodes a nicoinamide mononucleotide transporter. Nat Metab, 1:660-661. [DOI] [PubMed] [Google Scholar]
- [134].Gerasimenko M, Cherepanov SM, Furuhara K, Lopatina O, Salmina AB, Shabalova AA, et al. (2020). Nicotinamide riboside supplementation corrects deficits in oxytocin, sociability and anxiety of CD157 mutants in a mouse model of autism spectrum disorder. Sci Rep 10:1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Castro-Marrero J, Cordero MD, Segundo MJ, Sáez-Francas N, Calvo N, Román-Malo L, et al.2015. Does oral coenzyme Q10 plus NADH supplementation improve fatigue and biochemical parameters in chronic fatigue syndrome? : Mary Ann Liebert, Inc. 140 Huguenot Street, 3rd Floor New Rochelle, NY 10801 USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Forsyth LM, Preuss HG, MacDowell AL, Chiazze L Jr, Birkmayer GD, Bellanti JA (1999). Therapeutic effects of oral NADH on the symptoms of patients with chronic fatigue syndrome. Ann Allergy Asthma Immunol, 82:185-191. [DOI] [PubMed] [Google Scholar]
- [137].Santaella ML, Font I, Disdier OM (2004). Comparison of oral nicotinamide adenine dinucleotide (NADH) versus conventional therapy for chronic fatigue syndrome. P R Health Sci J, 23. [PubMed] [Google Scholar]
- [138].Birkmayer W, Birkmayer G, Vrecko K, Mlekusch W, Paletta B, Ott E (1989). The coenzyme nicotinamide adenine dinucleotide (NADH) improves the disability of parkinsonian patients. Journal of Neural Transmission-Parkinson's Disease and Dementia Section, 1:297-302. [DOI] [PubMed] [Google Scholar]
- [139].Castro-Marrero J, Sáez-Francàs N, Segundo MJ, Calvo N, Faro M, Aliste L, et al. (2016). Effect of coenzyme Q10 plus nicotinamide adenine dinucleotide supplementation on maximum heart rate after exercise testing in chronic fatigue syndrome-A randomized, controlled, double-blind trial. Clin Nutr, 35:826-834. [DOI] [PubMed] [Google Scholar]