

Abstract
p53 is a vital transcriptional protein implicated in regulating diverse cellular processes, including cell cycle arrest, DNA repair, mitochondrial metabolism, redox homeostasis, autophagy, senescence, and apoptosis. Recent studies have revealed that p53 levels and activity are substantially increased in affected neurons in cellular and animal models of Parkinson's disease (PD) as well as in the brains of PD patients. p53 activation in response to neurodegenerative stress is closely associated with the degeneration of dopaminergic neurons accompanied by mitochondrial dysfunction, reactive oxygen species (ROS) production, abnormal protein aggregation, and impairment of autophagy, and these pathogenic events have been implicated in the pathogenesis of PD. Pathogenic p53 integrates diverse cellular stresses and activate these downstream events to induce the degeneration of dopaminergic neurons; thus, it plays a crucial role in the pathogenesis of PD and appears to be a potential target for the treatment of the disease. We reviewed the current knowledge concerning p53-dependent neurodegeneration to better understand the underlying mechanisms and provide possible strategies for PD treatment by targeting p53.
1. Introduction
Parkinson's disease (PD) is a common neurodegenerative disorder caused by the selective and progressive loss of dopaminergic neurons in the substantia nigra (SN) of the midbrain and depletion of dopamine neurotransmitter in the striatum [1]. The etiology responsible for the progressive degeneration of dopaminergic neurons remains unclear. However, multiple pathogenic events, including mitochondrial dysfunction, oxidative stress, abnormal protein aggregation, and impairment of mitophagy, have been documented to be mechanistically linked to the pathogenesis of PD [2–5]. p53 is known to be an essential apoptotic inducer and becomes activated in response to diverse cellular stresses. Pathogenic p53 integrates the cellular stresses to trigger the death of different cell types, including dopaminergic neurons [6, 7]. Studies in cellular models of PD have demonstrated that p53 levels and activity are substantially increased, and these changes are closely associated with dopaminergic neuron death in neurodegenerative conditions. The high levels of p53 were also observed in the brains of PD patients as well as PD animal models, supporting the link between p53 activation and the degeneration of dopaminergic neurons in PD [8]. The activation of p53 induces neurodegeneration through diverse cell death pathways, including mitochondrial dysfunction, mitochondrial Ca2+ overloading, reactive oxygen species (ROS) production, abnormal protein aggregation, and impairment of mitophagy [9, 10]. p53 brings together diverse pathogenic signals to initiate downstream pathogenic events and consequent neurodegeneration; thus, it plays a central role in the pathogenesis of PD and provides a potential target for therapeutic intervention of the disease. This article reviewed the involvement of apoptotic mediator p53 in pathogenic events associated with the loss of dopaminergic neurons and the underlying mechanisms responsible for p53-mediated neurodegeneration in PD.
2. Property of p53
p53 is a transcriptional protein encoded by the TP53 gene. It was initially described as a tumor suppressor. However, later studies revealed that p53 is a multifunctional protein involved in regulating numerous cellular processes by activating diverse downstream signal cascades [11–15]. Structurally, p53 contains five highly conserved domains: a central DNA-binding domain, an N-terminal transactivation domain, a proline-rich region, a tetramerization domain, and a C-terminal basic domain, which are associated with the transcriptional or posttranscriptional regulating function of p53 [16]. The DNA-binding domain is a primary functional domain of p53 that recognizes and binds to specific DNA sequences in target genes, triggering the transcription of sets of genes with diverse biological functions [17–19]. The C-terminal basic domain stabilizes the formation of p53-DNA complexes by inducing the conformational changes in the core DNA-binding domain. It is also a specific site for p53 posttranslational modifications including phosphorylation, acetylation, ubiquitination, methylation, SUMOylation, and neddylation, and these structural changes are closely associated with p53 stability and functional activity [20–25]. The N-terminal domain and the proline-rich region are correlated with p53 transcriptional activation, while the oligomerization domain contributes to the stability of p53-DNA complexes, thereby promoting p53 transcriptional function. Normally, p53 is an unstable protein that is continuously degraded by proteasomes. The murine double minute-2 (Mdm2) protein is known to be a major negative regulator that targets p53 for proteasomal degradation and inhibits its subcellular translocation by ubiquitinating p53 [26]. Phosphorylation of p53 at Thr377 and Ser378 decreases its acetylation and activity and facilitates its ubiquitination and degradation, while Ser15, Thr18, or Ser20 phosphorylation increases its ability to counteract ubiquitin-mediated protein degradation, promotes C-terminal acetylation and nuclear transport, and enhances its DNA binding and transcriptional activity [27–30]. Phosphorylation at Ser46 of cytoplasmic p53 activates its conformational change and mitochondrial translocation [31, 32]. p53 is a response gene that regulates the transactivation of many target genes involved in diverse biological processes. p53 activation-associated degeneration of dopaminergic neurons has been reported to be closely associated with the development of PD [10, 33].
3. p53 with Mitochondria
Mitochondria are multifunctional subcellular organelles that are essential for numerous cellular functions, including generation of cellular energy, intracellular Ca2+ homeostasis, ROS production, and activation of intrinsic cell death pathways [34–36]. Mitochondrial dysfunction has been implicated in a series of diverse diseases including PD and has been reported to be a central event in PD pathogenesis [37]. Activation of p53-mediated mitochondrial apoptotic changes and the subsequent cell death of dopaminergic neurons have been underlined in neurodegeneration [38]. Experimental and clinical studies have demonstrated that the levels and activity of p53 are highly increased in PD cellular and animal models as well as in the brains of PD patients, and these changes are closely associated with the dysfunction of mitochondria and the cell death of dopaminergic neurons [8]. p53 activation has a profound influence on mitochondrial integrity and function through transcription-dependent mechanisms and transcription-independent actions.
3.1. p53 and Mitochondrial ROS Production
Oxidative stress is a pathogenic condition resulting from an imbalance between ROS production and cellular enzymatic and nonenzymatic antioxidative defenses. Oxidative damage to dopaminergic neurons has been considered as an essential pathogenic factor in the development of PD [39]. This is supported by the findings that the brain tissues of PD patients express high levels of oxidative products, including lipid peroxidation product 4-hydroxyl-2-nonenal (HNE), carbonyl modifications of soluble proteins, and DNA and RNA oxidation products 8-hydroxy-deoxyguanosine and 8-hydroxyguanosine [40–43]. Oxidative damage of dopaminergic neurons has also been observed in PD animal and cellular models, supporting the correlation of oxidative stress with the degeneration of dopaminergic neurons in PD [44–46]. Mitochondria are a primary intracellular source of ROS production in the electron transport chain (ETC) of oxidative phosphorylation. Respiratory chain complexes I and III are the major sites of ROS generation in mitochondria [47–49]. During oxidative phosphorylation, the respiratory chain complexes transfer electrons to oxygen, mainly producing superoxide radicals and subsequently hydrogen peroxide (H2O2) and hydroxyl radicals [49, 50]. This production of ROS can be detoxified by cellular defense systems, including mitochondrial superoxide dismutase, manganese superoxide dismutase (MnSOD), glutathione peroxidase, catalase, and glutathione (GSH) [51–53]. When the balance of ROS production and antioxidant defense is perturbed, ROS accumulate and result in oxidative damage to the target cells. Cellular redox homeostasis is tightly regulated by p53 through transcription and modification of pro-oxidant and antioxidant protein [54, 55]. Various forms of cellular stress activate p53 to inhibit ROS generation and promote cell repair or to increase cellular oxidative damage and induce senescence or apoptosis under conditions of severe, irreversible stress [12]. Numerous studies have revealed that the levels and activity of p53 are substantially increased in various neurodegenerative conditions, accompanied by oxidative damage of macromolecule proteins and DNA [56–58]. Overexpression of p53 transactivates a series of pro-oxidative genes, including p53-inducible gene 3 (PIG3), p66shc, and proline oxidase gene associated with ROS production [54, 59–62]. PIGs activation, for example, causes oxidative damage of target cells through increased ROS production via NADPH-quinone oxidoreductase and inhibition of ROS scavenging by catalase [63, 64]. p53 affects mitochondrial respiratory activity by regulating the synthesis of cytochrome c oxidase 2 (SCO2). SCO2 is a nuclear DNA-encoding subunit, which is essential for regulating the cytochrome c oxidase (COX) complex, the major site of oxygen utilization in eukaryotic cells. p53 transactivates the expression of SCO2 by binding its promoter in nuclear DNA, resulting in ROS production [65]. Moreover, p53 following cellular stress induces the expression of proapoptotic proteins including B-cell lymphoma 2 (Bcl-2)-associated X-protein (Bax), p53 upregulated modulator of apoptosis (PUMA), and nicotinamide adenine dinucleotide phosphate oxidase activator (NOXA), which disturb mitochondrial function resulting in upregulation of ROS generation [66]. In addition, cytosolic p53 decreases the ubiquitin-mediated degradation of α-synuclein protein [67]. α-Synuclein targets mitochondria to induce profound mitochondrial alterations, including collapse of transmembrane potential, impairment of respiratory chain complexes, disturbance of mitochondrial Ca2+ homeostasis, and, finally, ROS production and oxidative stress [68–70]. Accumulation of p53 in the mitochondrial matrix binds and inactivates MnSOD, a critical mitochondrial enzyme, involved in cellular defense against oxidative stress by scavenging ROS [71]. p53 overexpression also impairs mitochondrial morphology, resulting in decreased mitochondrial Ca2+ transients, followed by ROS production [72]. (Figure 1). The mitochondrial ETC is a primary cellular target of ROS-induced oxidative stress, and oxidative damage leads to further inhibition of the ETC and excessive ROS production [73]. Thus, a vicious pathogenic cycle develops between the defects in ETC and ROS generation, which may be critical in the progressive loss of dopaminergic neurons and the development of PD [74]. p53 plays an essential role in these processes and provides a potential target for therapeutic intervention.
Figure 1.
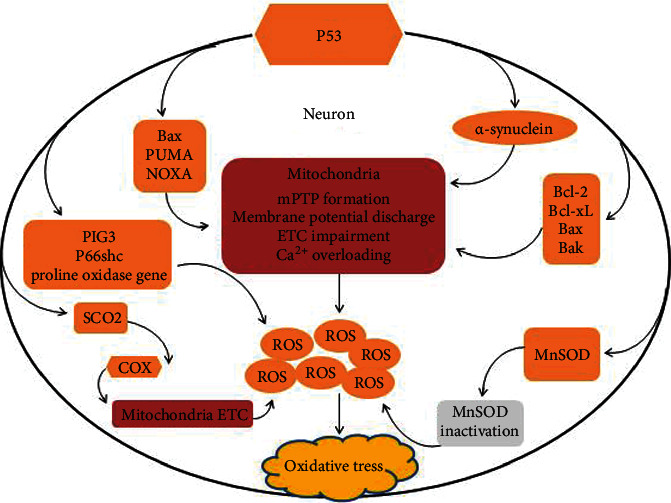
p53 activation in ROS production under irreversible stress conditions. p53 regulates cellular redox homeostasis through transcriptional action and modified expression of pro-oxidant and antioxidant proteins. p53 activation induces the expression of many proapoptotic proteins, such as Bax, PUMA, and NOXA, and facilitates Bax/Bak-mediated permeabilization of the outer mitochondrial membrane leading to the discharge of the membrane potential and ETC impairment followed by excessive ROS production. p53 also activates pro-oxidative genes including PIG3, proline oxidase, and p66shc to induce elevated levels of oxidative stress. The pro-oxidative activities of p53 also include the activation of expression of SCO2 gene, which is essential for regulating COX complex, the major site of oxygen utilization in the eukaryotic cells. The accumulation of cytosolic p53 protects α-synuclein from ubiquitin-mediated degradation inducing ROS generation and oxidative stress. α-Synuclein, in respone to cellular stress specifically targets mitochondria causing their profound alterations, including collapse of transmembrane potential, impairment of respiratory chain complexes, disturbance of mitochondrial Ca2+ homeostasis, and subsequent excessive ROS production. Accumulation of p53 in the mitochondrial matrix also contributes to oxidative damage to target cells by binding and inactivating the antioxidant MnSOD.
The NS dopaminergic neurons are vulnerable to oxidative stress. Increased iron levels have been detected in the SN of PD patients compared to healthy controls [75]. Iron promotes the generation of highly reactive oxygen species, resulting in further oxidative damage. DNA oxidative damage in vulnerable dopaminergic neurons is a hallmark of PD [76, 77]. Proliferating cell nuclear antigen (PCNA) is an essential protein that protects DNA from oxidative damage by regulating a wide range of enzymes and regulatory proteins [78, 79]. p53 is an upstream regulator of PCNA, and high concentration of p53 reduces the expression levels of PCNA by inhibiting its promoter, which diminishes its ability to protect DNA from oxidative damage [80,81]. Consistent with these reports, our previous studies in MPP+-induced neuronal PC12 cells suggested that PCNA downregulation caused by p53 activation contributed to the DNA oxidative damage in dopaminergic neurons [74]. This evidence supports the conclusion that p53 functioning as a converging signal for the generation of ROS plays a crucial role in PD pathogenesis.
3.2. p53 and Mitochondrial mPTP
p53 in response to cellular stress undergoes posttranscriptional modifications that increase its stabilization and subcellular translocation [29]. Nuclear translocated p53 binds to specific response sequences in the target genes and induces the expression of many proapoptotic proteins, such as Bax, PUMA, NOXA [82–84]. These proteins are essential for forming the mitochondrial permeability transition pore (mPTP) and inducing mitochondria-mediated intrinsic cell death under pathological conditions [38, 74]. Bax and Bcl-2 antagonist/killer (Bak) are proapoptotic proteins involved in mPTP formation. The antiapoptotic Bcl-2 family proteins Bcl-2 and B-cell lymphoma-extra large (Bcl-xL) combine with Bak to counter their proapoptotic function under normal conditions. Activation of p53 following cellular stress interacts with Bcl-2/Bcl-xL and releases Bax/Bak to open mPTP, leading to the release of cytochrome c from the mitochondria into the cytosol [85]. Mitochondrial translocation of p53 can directly bind Bax/Bak to disrupt the protein complex and activate the intrinsic apoptotic pathway [86,87].
p53 transcriptionally activates the proapoptotic protein PUMA [88]. Activation of PUMA binds all of the antiapoptotic BCL-2 members and facilitates Bax/Bak-mediated permeabilization of the outer mitochondrial membrane (OMM), resulting in the release of cytochrome c and activation of the caspase cascade [14]. PUMA also induces the release of cytosolic p53 from BCL-xL to activate Bax and Bak [89].
In addition, p53 induces the expression of the apoptotic regulating factor NOXA, which facilitates the opening of mPTP and release of cytochrome c to trigger cell death [90]. Besides OMM permeabilization, p53 mitochondrial translocation also induces the opening of the permeability transition pore in the inner mitochondrial membrane (IMM) by activating the translocation of cyclophilin D (CypD) from the mitochondrial matrix to the IMM. The translocated CypD interacts with the IMM protein adenine nucleotide translocator (ANT) to induce its morphological changes and subsequent formation of the ANT channel [91]. The permeabilization of outer mitochondrial membranes together with the channel formed by ANT in inner mitochondrial membranes constitutes a tunnel-like structure that causes the release of apoptotic mediators from the mitochondria into the cytosol to trigger caspase activation and eventual cell death (Figure 2). p53 has been implicated in the regulation of mitochondrial Ca2+ homeostasis in numerous ways. Nuclear p53 transrepresses the expression of Pten-induced kinase 1(PINK1) through binding and inactivating its promotor [9]. PINK1 physiologically regulates calcium efflux from the mitochondria via the ion exchanger, and its deficiency causes impaired Ca2+ efflux resulting in mitochondrial Ca2+ overloading [92]. Mitochondrial translocation of p53 reduces mitochondrial Ca2+ transients and facilitates Ca2+ release into the mitochondrial matrix [72]. Ca2+ is an essential ion for the activation of numerous mitochondrial enzymes that are necessary for mitochondrial metabolism [93]. Mitochondrial Ca2+ overloading has profound consequences for the cell, including defective synthesis of adenosine triphosphate (ATP), the collapse of the mitochondrial transmembrane potential, ROS production, and activation of mitochondrial mediated cell death [94]. Ca2+ overloading and excessive ROS production, in turn, facilitate the mPTP opening by inducing the translocation of the mitochondrial matrix CypD to the inner membrane and activating the mPTP regulator ANT [95]. Thus, p53 overexpression and subcellular translocation play a crucial role in mitochondrial apoptotic changes and subsequent neurodegeneration.
Figure 2.
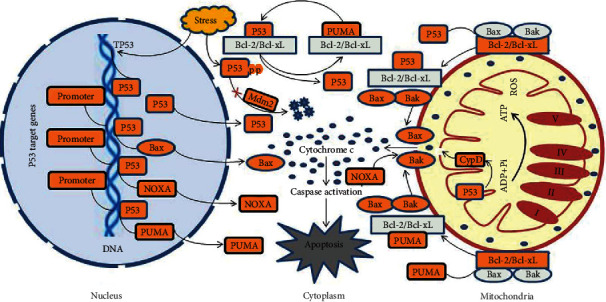
Association of p53 activation with mitochondria-mediated cell death. p53 is a response factor and becomes activated in response to cellular stress. Activation of p53 transactivates the expression of many proapoptotic proteins, including Bax, PUMA, and NOXA, which induces the opening of mPTP and initiates the mitochondria-mediated intrinsic cell death. p53 in response to cellular stress undergoes posttranscriptional modifications to control its activity and subcellular localization and to resist proteasomal degradation by Mdm2. Mitochondrial Bax and Bak are essential proapoptotic proteins that are combined with the antiapoptotic Bcl2 protein under normal conditions. Activation of p53 interacts with Bcl-2/Bcl-xL and releases Bax/Bak to opening mPTP, leading to the release of cytochrome c from the mitochondria into the cytosol and subsequent caspase activation. p53 transcriptionally activates proapoptotic protein PUMA to induce mPTP formation. PUMA contributes to the release of p53 from Bcl-xL complex into cytosol, resulting in p53-dependent Bax/Bak activation and subsequent permeabilization in the outer mitochondrial membrane. PUMA also directly interacts with proapoptotic proteins Bax and Bak to open mPTP and release cytochrome c. NOXA is a p53 target protein that impairs mitochondria via mPTP triggering intrinsic cell death. Mitochondrial p53 also interacts directly with CypD to open mPTP and trigger apoptosis.
4. p53 with Autophagy and Protein Aggregation
Neurodegenerative disorders are characterized by the accumulation of abnormal protein and damaged mitochondria that are associated with dysregulation of either proteasomal and/or autophagic quality control systems [96]. p53 has been known to be a key regulator in autophagic response, and activation following neurodegenerative stress leads to autophagic failure and subsequent neurodegeneration [97].
4.1. p53 and Autophagy
Autophagy is a major intracellular process for the elimination of deleterious proteins and damaged mitochondria; dysfunctional autophagy has been linked to the pathogenesis of numerous neurodegenerative disorders, including Alzheimer's disease (AD), Huntington's disease (HD), amyotrophic lateral sclerosis (ALS), and PD [96, 98]. p53 has been increasingly recognized to be a key autophagic regulator that functions primarily through transcriptional effects on a wide range of downstream target genes, as well as regulation of the mTOR pathway in a transcription-dependent manner [99, 100].The differential regulation of autophagy by p53 following cellular stress is dependent on its subcellular localization, targeting genes, and stress conditions. The accumulation of p53 in the cytosol has been suggested to inhibit autophagic clearance of abnormally aggregated proteins in pathogenic conditions [67, 101, 102]. p53-associated dysfunction of autophagy is increasingly considered as a potential mechanism responsible for the degeneration of dopaminergic neurons in PD pathogenesis [9, 67, 103]. Neurodegenerative conditions induce high levels of p53 that are closely associated with the abnormal accumulation of α-synuclein and dysfunctional mitophagy [9].
4.2. p53 and α-Synuclein Aggregation
Neuropathologically, PD is characterized by the presence of protein inclusions termed Lewy bodies (LBs) in the vulnerable neurons of the SN [104]. The synaptic protein α-synuclein has been identified as the primary component of LBs [105, 106]. α-Synuclein is an intracellular protein normally localized in the presynaptic terminals, and aggregation and dimer formation of α-synuclein are caused by dysfunctional cellular proteostasis [107–109]. Aberrant α-synuclein accumulation and formation of LBs in dopaminergic neurons have implicated the neurodegeneration [107]. Protein aggregation disrupts cellular function, leading to the activation of cell death signals and subsequent neuron injury and death [107]. p53 is a stress response gene involved in the regulation of autophagy via diverse pathways [101, 110].
Chaperone and cochaperone systems are essential for protein folding or refolding and degradation of aggregated protein; thus, they prevent the cytotoxicity caused by aberrant protein accumulation [111, 112]. p53 regulates the functional activity of HSP70 and HSP90 chaperone and cochaperone systems in neurodegenerative conditions [67, 113]. Studies in PD cellular and animal models have shown that p53 activation increases the aggregation of α-synuclein in vulnerable neurons through inhibiting HSP70-mediated protein folding activity, accompanied by BAG5 protein overexpression [113]. BAG5 is an important stress-induced backup nucleotide exchange factor of HSP70 associated with the protein activation. High levels of BAG5, however, inhibit the folding activity of the HSP70 chaperone, resulting in dysfunction of protein folding and refolding and subsequent abnormal protein aggregation. BAG5 expression is transcriptionally regulated by the p53 gene under stress conditions [113]. The gene silence of p53 causes a substantial decrease in BAG5 mRNA and protein levels in the stressed cells. Mechanism studies reveal that p53 can directly bind to the promoter and activate BAG5 transcription, leading to elevated levels of BAG5 under irreversible stress conditions [113]. p53 activation induces overexpression of BAG5 to inhibit the protein folding activity of HSP70, leading to the aggregation and accumulation of α-synuclein and subsequently cell toxicity and death.
c-Abl is a critical tyrosine kinase associated with the accumulation of pathogenic α-synuclein and neurodegeneration in PD [114–116]. c-Ab1 is activated in response to cellular stress, including oxidative stress and DNA damage [67]. Activation of c-Ab1 directly phosphorylates α-synuclein or decreases its autophagic degradation [116, 117]. Pharmacological inhibition of c-Ab1 has been shown to reduce α-synuclein levels or its aggregation via the activation of autophagy in PD cellular and animal models [115]. Several lines of evidence have suggested that c-Abl-dependent inhibition of autophagy also involves p53 activation and p53-dependent mTOR signal pathway [67]. c-Abl directly phosphorylates Mdm2, decreasing its ligase activity [118]. Mdm2 is a key E3 ligase that ubiquitinates p53 for proteasomal degradation and prevents p53 transcription by binding to its N-terminal domain [119]. Decreased levels and activity of Mdm2 cause the accumulation of p53 under stress conditions [120]. Studies in PD cellular and animal models have demonstrated that pharmacological inhibition of p53 can block α-synuclein aggregation and autophagy defects caused by c-Ab1 activation. These results support the conclusion that c-Ab1 mediates the accumulation and aggregation of α-synuclein, which at least in part occurs through the p53-dependent pathway under neurodegenerative conditions.
5. p53 and Mitophagy
Mitophagy is a protective mechanism for mitochondria to maintain their homeostasis through clearance of damaged mitochondria or fission-fragmented mitochondria via lysosomal degradation [121]. This protective function is crucial for neuronal cells due to the sensitivity of neurons to toxic aggregation. Mitophagy impairment causes the accumulation of defective mitochondria resulting in toxicity to the vulnerable neurons and eventually neuronal degeneration, and this cell death pathway has been underlined in the pathogenesis of neurodegenerative disorders, including PD [122, 123]. PINK1 and Parkin have been suggested to play a crucial role in the process of mitophagy [121]. PINK1 is a serine/threonine kinase possessing a mitochondrial targeting sequence, which allows the kinase to enter into the mitochondria and translocate to the IMM. The mitochondrial translocated PINK1 is normally cleaved and inactivated by the IMM protease presenilin-associated rhomboid-like protein (PARL) and subsequently degraded through the N-end rule pathways, resulting in low levels of PINK1 in the healthy mitochondria [124, 125]. However, mitochondrial depolarization inhibits PINK1 translocation to the IMM and subsequent degradation by PARL, which contribute to the accumulation of PINK1 on the OMM and the subsequent recruitment of Parkin from the cytoplasm into the damaged mitochondria. Parkin is an E3 ubiquitin ligase that ubiquitinates mitochondrial membrane proteins to trigger the elimination of defective mitochondria by lysosomes. The PINK1/Parkin-mediated mitophagy is crucial for mitochondrial quality control and to clean damaged mitochondria. This functional activity of PINK1/Parkin can be disturbed by p53 activation, leading to impaired mitophagy. p53 transrepresses the expression of PINK1 under normal as well as pathogenic conditions. This is supported by the finding that pharmacological phosphorylation of p53 leads to the decreased expression of PINK1 in SH-SY5Y neuroblastoma cells and inhibition of p53 activity increases both PINK1 protein expression and mRNA levels in the cell treated with pifithrin-α (PFT), a well-known p53 inhibitor. p53 adenoviral overexpression in mouse striatal neurons causes the decrease in PINK1 and mRNA levels, while depletion of endogenous p53 promotes its expression and mRNA levels, supporting p53 as a transcriptional inhibitor of PINK1 transcription [9]. p53 also directly interacts with Parkin to inhibit its translocation to the damaged mitochondria, resulting in the impairment of mitophagy [126]. Parkin is shown to repress the transcription of p53, which in turn transactivates the expression of Parkin [127, 128]. This interplay could increase the expression of PINK1 since its transcription is tightly controlled by p53 and p53 repression by Parkin results in PINK1 transactivation. The interplay among p53, PINK1, and Parkin creates an intricate regulating network for elimination of defective mitochondria by mitophagy, while overexpression of p53 during neurodegenerative stress decreases PINK1 levels and inactivates mitophagic activity of Parkin, resulting in impairment of mitophagy and consequent neurodegeneration.
6. Conclusion and Future Perspectives
p53 is a multifunctional protein that regulates numerous diverse cellular processes through transcription-dependent mechanisms and transcription-independent actions. p53-dependent neuronal death has been mechanistically linked to the pathogenesis of many neurodegenerative disorders including PD. Activation of p53 in response to neurodegenerative stress facilitates mitochondrial dysfunction, oxidative stress, aberrant protein aggregation, and autophagy impairment. These are central events associated with the degeneration of dopaminergic neurons and fundamental processes in the pathogenesis of PD. p53 plays a significant role in neurodegeneration through the integration of various neurodegenerative signals triggering neuronal death, making it a potential target for the treatment of PD. Strategies to inhibit the high levels and activity of p53 could inhibit the progression of pathological changes and alleviate the progressive degeneration of dopaminergic neurons in PD. In particular, Mdm2 binds to the transactivation domain of p53, inhibits its transcriptional activity, and mediates p53 ubiquitination and degradation via proteasomes. Pharmacological stimulation of Mdm2 has been shown to decrease p53 activity and levels and promote neuronal survival under neurodegenerative conditions. Therefore, Mdm2 appears to be a potential therapeutic target that could be used in the development of novel neuroprotective strategies for PD. In conclusion, p53-dependent therapeutic intervention is needed.
Acknowledgments
This study was supported by Technology Research of the Education Department of Jilin Province, China (grant no. JJKH20190653 KJ) to Da-Wei Li.
Abbreviations
- PD:
Parkinson's disease
- ROS:
Reactive oxygen species
- SN:
Substantia nigra
- Mdm2:
Murine double minute-2
- Bax:
Bcl-2-associated X-protein
- PUMA:
p53-upregulated modulator of apoptosis
- NOXA:
Nicotinamide adenine dinucleotide phosphate oxidase activator
- Bak:
BCL-2 antagonist/killer
- mPTP:
Mitochondrial permeability transition pore
- Bcl-2:
B-cell lymphoma 2
- CypD:
Cyclophilin D
- ATP:
Adenosine triphosphate
- HNE:
4-Hydroxyl-2-nonenal
- H2O2:
Hydrogen peroxide
- MnSOD:
Manganese superoxide dismutase
- SCO2:
Cytochrome c oxidase 2
- COX:
Cytochrome c oxidase
- ETC:
Electron transport chain
- PCNA:
Proliferating cell nuclear antigen
- LBs:
Lewy bodies
- 6-OHDA:
6-Hydroxydopamine
- PINK1:
Pten-induced kinase 1
- Bcl-xL:
B-cell lymphoma-extra large.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Authors' Contributions
Qiang Luo and Wei Sun have contributed equally.
References
- 1.Raza C., Anjum R., Shakeel N. U. A. Parkinson’s disease: mechanisms, translational models and management strategies. Life Sciences . 2019;226:77–90. doi: 10.1016/j.lfs.2019.03.057. [DOI] [PubMed] [Google Scholar]
- 2.Borsche M., Pereira S. L., Klein C., Grunewald A. Mitochondria and parkinson’s disease: clinical, molecular, and translational aspects. Journal of Parkinson’s Disease . 2021;11(1):45–60. doi: 10.3233/jpd-201981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Trist B. G., Hare D. J., Double K. L. Oxidative stress in the aging substantia nigra and the etiology of Parkinson’s disease. Aging Cell . 2019;18(6) doi: 10.1111/acel.13031.e13031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu J., Wu M., Yue Z. Autophagy and Parkinson’s disease. Autophagy: Biology and Diseases . 2020;1207:21–51. doi: 10.1007/978-981-15-4272-5_2. [DOI] [PubMed] [Google Scholar]
- 5.Rocha E. M., De Miranda B., Sanders L. H. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiology of Disease . 2018;109(Pt B):249–257. doi: 10.1016/j.nbd.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Wawryk-Gawda E., Chylinska-Wrzos P., Lis-Sochocka M., et al. P53 protein in proliferation, repair and apoptosis of cells. Protoplasma . 2014;251(3):525–533. doi: 10.1007/s00709-013-0548-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haronikova L., Olivares-Illana V., Wang L., Karakostis K., Chen S., Fahraeus R. The p53 mRNA: an integral part of the cellular stress response. Nucleic Acids Research . 2019;47(7):3257–3271. doi: 10.1093/nar/gkz124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mogi M., Kondo T., Mizuno Y., Nagatsu T. P53 protein, interferon-γ, and NF-κB levels are elevated in the parkinsonian brain. Neuroscience Letters . 2007;414(1):94–97. doi: 10.1016/j.neulet.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Goiran T., Duplan E., Rouland L., et al. Nuclear p53-mediated repression of autophagy involves PINK1 transcriptional down-regulation. Cell Death & Differentiation . 2018;25(5):873–884. doi: 10.1038/s41418-017-0016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dai C. Q., Luo T. T., Luo S. C., et al. P53 and mitochondrial dysfunction: novel insight of neurodegenerative diseases. Journal of Bioenergetics and Biomembranes . 2016;48(4):337–347. doi: 10.1007/s10863-016-9669-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beckerman R., Prives C. Transcriptional regulation by p53. Cold Spring Harbor Perspectives in Biology . 2010;2(8) doi: 10.1101/cshperspect.a000935.a000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu X., Fan L., Lu C., Yin S., Hu H. Functional role of p53 in the regulation of chemical-induced oxidative stress. Oxidative Medicine and Cellular Longevity . 2020;2020:1–10. doi: 10.1155/2020/6039769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vaseva A. V., Marchenko N. D., Ji K., Tsirka S. E., Holzmann S., Moll U. M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell . 2012;149(7):1536–1548. doi: 10.1016/j.cell.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li M. The role of P53 up-regulated modulator of apoptosis (PUMA) in ovarian development, cardiovascular and neurodegenerative diseases. Apoptosis . 2021;26(5-6):235–247. doi: 10.1007/s10495-021-01667-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Talebi M., Kakouri E., Farkhondeh T., Pourbagher-Shahri A. M., Tarantilis P. A., Samarghandian S. Tantalizing role of p53 molecular pathways and its coherent medications in neurodegenerative diseases. International Journal of Biological Macromolecules . 2021;172:93–103. doi: 10.1016/j.ijbiomac.2021.01.042. [DOI] [PubMed] [Google Scholar]
- 16.Joerger A. C., Fersht A. R. Structural biology of the tumor suppressor p53. Annual Review of Biochemistry . 2008;77(1):557–582. doi: 10.1146/annurev.biochem.77.060806.091238. [DOI] [PubMed] [Google Scholar]
- 17.Tebaldi T., Zaccara S., Alessandrini F., Bisio A., Ciribilli Y., Inga A. Whole-genome cartography of p53 response elements ranked on transactivation potential. BMC Genomics . 2015;16(1):p. 464. doi: 10.1186/s12864-015-1643-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitayner M., Rozenberg H., Kessler N., et al. Structural basis of DNA recognition by p53 tetramers. Molecules and Cells . 2006;22(6):741–753. doi: 10.1016/j.molcel.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 19.Lane D., Levine A. P53 Research: the past thirty years and the next thirty years. Cold Spring Harbor Perspectives in Biology . 2010;2(12) doi: 10.1101/cshperspect.a000893.a000893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bell S., Klein C., Muller L., Hansen S., Buchner J. P53 contains large unstructured regions in its native state. Journal of Molecular Biology . 2002;322(5):917–927. doi: 10.1016/s0022-2836(02)00848-3. [DOI] [PubMed] [Google Scholar]
- 21.Toledo F., Wahl G. M. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nature Reviews Cancer . 2006;6(12):909–923. doi: 10.1038/nrc2012. [DOI] [PubMed] [Google Scholar]
- 22.Lavin M. F., Gueven N. The complexity of p53 stabilization and activation. Cell Death & Differentiation . 2006;13(6):941–950. doi: 10.1038/sj.cdd.4401925. [DOI] [PubMed] [Google Scholar]
- 23.Bode A. M., Dong Z. Post-translational modification of p53 in tumorigenesis. Nature Reviews Cancer . 2004;4(10):793–805. doi: 10.1038/nrc1455. [DOI] [PubMed] [Google Scholar]
- 24.Feng L., Lin T., Uranishi H., Gu W., Xu Y. Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Molecular and Cellular Biology . 2005;25(13):5389–5395. doi: 10.1128/mcb.25.13.5389-5395.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu B., Zhu W. G. Surf the post-translational modification network of p53 regulation. International Journal of Biological Sciences . 2012;8(5):672–684. doi: 10.7150/ijbs.4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks C. L., Gu W. Dynamics in the p53-Mdm2 ubiquitination pathway. Cell Cycle . 2004;3(7):893–897. doi: 10.4161/cc.3.7.997. [DOI] [PubMed] [Google Scholar]
- 27.Saito S., Yamaguchi H., Higashimoto Y., et al. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. Journal of Biological Chemistry . 2003;278(39):37536–37544. doi: 10.1074/jbc.m305135200. [DOI] [PubMed] [Google Scholar]
- 28.Sakaguchi K., Saito S., Higashimoto Y., Roy S., Anderson C. W., Appella E. Damage-mediated phosphorylation of human p53 threonine 18 through a cascade mediated by a casein 1-like kinase. Journal of Biological Chemistry . 2000;275(13):9278–9283. doi: 10.1074/jbc.275.13.9278. [DOI] [PubMed] [Google Scholar]
- 29.Shieh S. Y., Ikeda M., Taya Y., Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell . 1997;91(3):325–334. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 30.Chernov M. V., Bean L. J., Lerner N., Stark G. R. Regulation of ubiquitination and degradation of p53 in unstressed cells through C-terminal phosphorylation. Journal of Biological Chemistry . 2001;276(34):31819–31824. doi: 10.1074/jbc.m103170200. [DOI] [PubMed] [Google Scholar]
- 31.D’Orazi G., Cecchinelli B., Bruno T., et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nature Cell Biology . 2002;4(1):11–19. doi: 10.1038/ncb714. [DOI] [PubMed] [Google Scholar]
- 32.Hofmann T. G., Moller A., Sirma H., et al. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nature Cell Biology . 2002;4(1):1–10. doi: 10.1038/ncb715. [DOI] [PubMed] [Google Scholar]
- 33.Schapira A. H., Jenner P. Etiology and pathogenesis of Parkinson’s disease. Movement Disorders . 2011;26(6):1049–1055. doi: 10.1002/mds.23732. [DOI] [PubMed] [Google Scholar]
- 34.Martin L. J. Biology of mitochondria in neurodegenerative diseases. Progress in Molecular Biology and Translational Science . 2012;107:355–415. doi: 10.1016/b978-0-12-385883-2.00005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trancikova A., Tsika E., Moore D. J. Mitochondrial dysfunction in genetic animal models of Parkinson’s disease. Antioxidants and Redox Signaling . 2012;16(9):896–919. doi: 10.1089/ars.2011.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malpartida A. B., Williamson M., Narendra D. P., Wade-Martins R., Ryan B. J. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: from mechanism to therapy. Trends in Biochemical Sciences . 2021;46(4):329–343. doi: 10.1016/j.tibs.2020.11.007. [DOI] [PubMed] [Google Scholar]
- 37.Xu S., Zhang X., Liu C., et al. Role of mitochondria in neurodegenerative diseases: from an epigenetic perspective. Frontiers in Cell and Developmental Biology . 2021;9:p. 688789. doi: 10.3389/fcell.2021.688789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang D. B., Kinoshita C., Kinoshita Y., Morrison R. S. P53 and mitochondrial function in neurons. Biochimica et Biophysica Acta - Molecular Basis of Disease . 2014;1842(8):1186–1197. doi: 10.1016/j.bbadis.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dias V., Junn E., Mouradian M. M. The role of oxidative stress in Parkinson’s disease. Journal of Parkinson’s Disease . 2013;3(4):461–491. doi: 10.3233/jpd-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoritaka A., Hattori N., Uchida K., Tanaka M., Stadtman E. R., Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proceedings of the National Academy of Sciences of the United States of America . 1996;93(7):2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Floor E., Wetzel M. G. Increased protein oxidation in human substantia nigra pars compacta in comparison with basal ganglia and prefrontal cortex measured with an improved dinitrophenylhydrazine assay. Journal of Neurochemistry . 2002;70(1):268–275. doi: 10.1046/j.1471-4159.1998.70010268.x. [DOI] [PubMed] [Google Scholar]
- 42.Alam Z. I., Jenner A., Daniel S. E., et al. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. Journal of Neurochemistry . 2002;69(3):1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- 43.Isobe C., Abe T., Terayama Y. Levels of reduced and oxidized coenzyme Q-10 and 8-hydroxy-2’-deoxyguanosine in the cerebrospinal fluid of patients with living Parkinson’s disease demonstrate that mitochondrial oxidative damage and/or oxidative DNA damage contributes to the neurodegenerative process. Neuroscience Letters . 2010;469(1):159–163. doi: 10.1016/j.neulet.2009.11.065. [DOI] [PubMed] [Google Scholar]
- 44.Yang C., Mo Y., Xu E., et al. Astragaloside IV ameliorates motor deficits and dopaminergic neuron degeneration via inhibiting neuroinflammation and oxidative stress in a Parkinson’s disease mouse model. International Immunopharmacology . 2019;75 doi: 10.1016/j.intimp.2019.05.036.105651 [DOI] [PubMed] [Google Scholar]
- 45.Li X., Zhang J., Rong H., Zhang X., Dong M. Ferulic acid ameliorates MPP(+)/MPTP-Induced oxidative stress via ERK1/2-dependent Nrf2 activation: translational implications for Parkinson disease treatment. Molecular Neurobiology . 2020;57(7):2981–2995. doi: 10.1007/s12035-020-01934-1. [DOI] [PubMed] [Google Scholar]
- 46.Villavicencio Tejo F., Quintanilla R. A. Contribution of the Nrf2 pathway on oxidative damage and mitochondrial failure in Parkinson and Alzheimer’s disease. Antioxidants . 2021;10(7):p. 1069. doi: 10.3390/antiox10071069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kussmaul L., Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proceedings of the National Academy of Sciences of the United States of America . 2006;103(20):7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kudin A. P., Debska-Vielhaber G., Kunz W. S. Characterization of superoxide production sites in isolated rat brain and skeletal muscle mitochondria. Biomedicine & Pharmacotherapy . 2005;59(4):163–168. doi: 10.1016/j.biopha.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 49.Kudin A. P., Bimpong-Buta N. Y. B., Vielhaber S., Elger C. E., Kunz W. S. Characterization of superoxide-producing sites in isolated brain mitochondria. Journal of Biological Chemistry . 2004;279(6):4127–4135. doi: 10.1074/jbc.m310341200. [DOI] [PubMed] [Google Scholar]
- 50.Brand M. D., Affourtit C., Esteves T. C., et al. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radical Biology and Medicine . 2004;37(6):755–767. doi: 10.1016/j.freeradbiomed.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 51.Antunes F., Han D., Cadenas E. Relative contributions of heart mitochondria glutathione peroxidase and catalase to H(2)O(2) detoxification in in vivo conditions. Free Radical Biology and Medicine . 2002;33(9):1260–1267. doi: 10.1016/s0891-5849(02)01016-x. [DOI] [PubMed] [Google Scholar]
- 52.Rush J. D., Koppenol W. H. Oxidizing intermediates in the reaction of ferrous EDTA with hydrogen peroxide. Reactions with organic molecules and ferrocytochrome c. Journal of Biological Chemistry . 1986;261(15):6730–6733. doi: 10.1016/s0021-9258(19)62677-3. [DOI] [PubMed] [Google Scholar]
- 53.Giustarini D., Colombo G., Garavaglia M. L., et al. Assessment of glutathione/glutathione disulphide ratio and S-glutathionylated proteins in human blood, solid tissues, and cultured cells. Free Radical Biology and Medicine . 2017;112:360–375. doi: 10.1016/j.freeradbiomed.2017.08.008. [DOI] [PubMed] [Google Scholar]
- 54.Yuan Y., Wang H., Wu Y., et al. P53 contributes to cisplatin induced renal oxidative damage via regulating P66shc and MnSOD. Cellular Physiology and Biochemistry . 2015;37(4):1240–1256. doi: 10.1159/000430247. [DOI] [PubMed] [Google Scholar]
- 55.Niwa-Kawakita M., Ferhi O., Soilihi H., Le Bras M., Lallemand-Breitenbach V., de The H. PML is a ROS sensor activating p53 upon oxidative stress. Journal of Experimental Medicine . 2017;214(11):3197–3206. doi: 10.1084/jem.20160301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chatoo W., Abdouh M., Bernier G. P53 pro-oxidant activity in the central nervous system: implication in aging and neurodegenerative diseases. Antioxidants and Redox Signaling . 2011;15(6):1729–1737. doi: 10.1089/ars.2010.3610. [DOI] [PubMed] [Google Scholar]
- 57.Smith J. A., Park S., Krause J. S., Banik N. L. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochemistry International . 2013;62(5):764–775. doi: 10.1016/j.neuint.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Erekat N. S., Al-Jarrah M. D. Association of Parkinson disease induction with cardiac upregulation of apoptotic mediators P53 and active caspase-3: an immunohistochemistry study. Medical Science Monitor Basic Research . 2018;24:120–126. doi: 10.12659/msmbr.910307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jin H., Yin S., Song X., Zhang E., Fan L., Hu H. P53 activation contributes to patulin-induced nephrotoxicity via modulation of reactive oxygen species generation. Scientific Reports . 2016;6(1):p. 24455. doi: 10.1038/srep24455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Italiano D., Lena A. M., Melino G., Candi E. Identification of NCF2/p67phox as a novel p53 target gene. Cell Cycle . 2012;11(24):4589–4596. doi: 10.4161/cc.22853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Natalicchio A., Tortosa F., Labarbuta R., et al. The p66(Shc) redox adaptor protein is induced by saturated fatty acids and mediates lipotoxicity-induced apoptosis in pancreatic beta cells. Diabetologia . 2015;58(6):1260–1271. doi: 10.1007/s00125-015-3563-2. [DOI] [PubMed] [Google Scholar]
- 62.Donald S. P., Sun X. Y., Hu C. A., et al. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Research . 2001;61(5):1810–1815. [PubMed] [Google Scholar]
- 63.Asher G., Lotem J., Cohen B., Sachs L., Shaul Y. Regulation of p53 stability and p53-dependent apoptosis by NADH quinone oxidoreductase 1. Proceedings of the National Academy of Sciences of the United States of America . 2001;98(3):1188–1193. doi: 10.1073/pnas.98.3.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang M. Y., Kim H. B., Piao C., et al. The critical role of catalase in prooxidant and antioxidant function of p53. Cell Death & Differentiation . 2013;20(1):117–129. doi: 10.1038/cdd.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matoba S., Kang J. G., Patino W. D., et al. p53 regulates mitochondrial respiration. Science . 2006;312(5780):1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 66.Jazvinscak Jembrek M., Orsolic N., Mandic L., Sadzak A., Segota S. Anti-oxidative, anti-inflammatory and anti-apoptotic effects of flavonols: targeting Nrf2, NF-κB and p53 pathways in neurodegeneration. Antioxidants . 2021;10(10):p. 1628. doi: 10.3390/antiox10101628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Karim M. R., Liao E. E., Kim J., et al. α-Synucleinopathy associated c-Abl activation causes p53-dependent autophagy impairment. Molecular Neurodegeneration . 2020;15(1):p. 27. doi: 10.1186/s13024-020-00364-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ganjam G. K., Bolte K., Matschke L. A., et al. Mitochondrial damage by alpha-synuclein causes cell death in human dopaminergic neurons. Cell Death & Disease . 2019;10(11):p. 865. doi: 10.1038/s41419-019-2091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vicario M., Cieri D., Brini M., Cali T. The close encounter between alpha-synuclein and mitochondria. Frontiers in Neuroscience . 2018;12:p. 388. doi: 10.3389/fnins.2018.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Culmsee C., Landshamer S. Molecular insights into mechanisms of the cell death program: role in the progression of neurodegenerative disorders. Current Alzheimer Research . 2006;3(4):269–283. doi: 10.2174/156720506778249461. [DOI] [PubMed] [Google Scholar]
- 71.Pani G., Galeotti T. Role of MnSOD and p66shc in mitochondrial response to p53. Antioxidants and Redox Signaling . 2011;15(6):1715–1727. doi: 10.1089/ars.2010.3499. [DOI] [PubMed] [Google Scholar]
- 72.Ottolini D., Cali T., Negro A., Brini M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Human Molecular Genetics . 2013;22(11):2152–2168. doi: 10.1093/hmg/ddt068. [DOI] [PubMed] [Google Scholar]
- 73.Camara A. K., Lesnefsky E. J., Stowe D. F. Potential therapeutic benefits of strategies directed to mitochondria. Antioxidants and Redox Signaling . 2010;13(3):279–347. doi: 10.1089/ars.2009.2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li D. W., Li G. R., Zhang B. L., Feng J. J., Zhao H. Damage to dopaminergic neurons is mediated by proliferating cell nuclear antigen through the p53 pathway under conditions of oxidative stress in a cell model of Parkinson’s disease. International Journal of Molecular Medicine . 2016;37(2):429–435. doi: 10.3892/ijmm.2015.2430. [DOI] [PubMed] [Google Scholar]
- 75.Sian-Hulsmann J., Mandel S., Youdim M. B. H., Riederer P. The relevance of iron in the pathogenesis of Parkinson’s disease. Journal of Neurochemistry . 2011;118(6):939–957. doi: 10.1111/j.1471-4159.2010.07132.x. [DOI] [PubMed] [Google Scholar]
- 76.Henchcliffe C., Beal M. F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nature Clinical Practice Neurology . 2008;4(11):600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- 77.Yan M. H., Wang X., Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radical Biology and Medicine . 2013;62:90–101. doi: 10.1016/j.freeradbiomed.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koundrioukoff S., Jonsson Z. O., Hasan S., et al. A direct interaction between proliferating cell nuclear antigen (PCNA) and Cdk2 targets PCNA-interacting proteins for phosphorylation. Journal of Biological Chemistry . 2000;275(30):22882–22887. doi: 10.1074/jbc.m001850200. [DOI] [PubMed] [Google Scholar]
- 79.Waga S., Hannon G. J., Beach D., Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature . 1994;369(6481):574–578. doi: 10.1038/369574a0. [DOI] [PubMed] [Google Scholar]
- 80.Morris G. F., Bischoff J. R., Mathews M. B. Transcriptional activation of the human proliferating-cell nuclear antigen promoter by p53. Proceedings of the National Academy of Sciences of the United States of America . 1996;93(2):895–899. doi: 10.1073/pnas.93.2.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shivakumar C. V., Brown D. R., Deb S., Deb S. P. Wild-type human p53 transactivates the human proliferating cell nuclear antigen promoter. Molecular and Cellular Biology . 1995;15(12):6785–6793. doi: 10.1128/mcb.15.12.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jeffers J. R., Parganas E., Lee Y., et al. Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell . 2003;4(4):321–328. doi: 10.1016/s1535-6108(03)00244-7. [DOI] [PubMed] [Google Scholar]
- 83.Shibue T., Takeda K., Oda E., et al. Integral role of Noxa in p53-mediated apoptotic response. Genes & Development . 2003;17(18):2233–2238. doi: 10.1101/gad.1103603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Toshiyuki M., Reed J. C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell . 1995;80(2):293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 85.Blandino G., Valenti F., Sacconi A., Di Agostino S. Wild type- and mutant p53 proteins in mitochondrial dysfunction: emerging insights in cancer disease. Seminars in Cell & Developmental Biology . 2020;98:105–117. doi: 10.1016/j.semcdb.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 86.Chipuk J. E., Kuwana T., Bouchier-Hayes L., et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science . 2004;303(5660):1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 87.Leu J. I. J., Dumont P., Hafey M., Murphy M. E., George D. L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nature Cell Biology . 2004;6(5):443–450. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- 88.Hikisz P., Kilianska Z. M., Puma PUMA, a critical mediator of cell death--one decade on from its discovery. Cellular and Molecular Biology Letters . 2012;17(4):646–669. doi: 10.2478/s11658-012-0032-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chipuk J. E., Bouchier-Hayes L., Kuwana T., Newmeyer D. D., Green D. R. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science . 2005;309(5741):1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 90.Seo Y. W., Shin J. N., Ko K. H., et al. The molecular mechanism of Noxa-induced mitochondrial dysfunction in p53-mediated cell death. Journal of Biological Chemistry . 2003;278(48):48292–48299. doi: 10.1074/jbc.m308785200. [DOI] [PubMed] [Google Scholar]
- 91.Yang H., Li R., Zhang L., et al. P53-cyclophilin D mediates renal tubular cell apoptosis in ischemia-reperfusion-induced acute kidney injury. American Journal of Physiology - Renal Physiology . 2019;317(5):F1311–F1317. doi: 10.1152/ajprenal.00072.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gandhi S., Wood-Kaczmar A., Yao Z., et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Molecular Cell . 2009;33(5):627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dubois C., Prevarskaya N., Vanden Abeele F. The calcium-signaling toolkit: updates needed. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research . 2016;1863(6):1337–1343. doi: 10.1016/j.bbamcr.2015.11.033. [DOI] [PubMed] [Google Scholar]
- 94.Liu X. L., Wang Y. D., Yu X. M., Li D. W., Li G. R. Mitochondria-mediated damage to dopaminergic neurons in Parkinson’s disease (Review) International Journal of Molecular Medicine . 2018;41(2):615–623. doi: 10.3892/ijmm.2017.3255. [DOI] [PubMed] [Google Scholar]
- 95.Halestrap A. P., Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Current Medicinal Chemistry . 2003;10(16):1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- 96.Chu C. T. Mechanisms of selective autophagy and mitophagy: implications for neurodegenerative diseases. Neurobiology of Disease . 2019;122:23–34. doi: 10.1016/j.nbd.2018.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ho D. H., Seol W., Son I. Upregulation of the p53-p21 pathway by G2019S LRRK2 contributes to the cellular senescence and accumulation of alpha-synuclein. Cell Cycle . 2019;18(4):467–475. doi: 10.1080/15384101.2019.1577666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guo F., Liu X., Cai H., Le W. Autophagy in neurodegenerative diseases: pathogenesis and therapy. Brain Pathology . 2018;28(1):3–13. doi: 10.1111/bpa.12545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ventura A., Kirsch D. G., McLaughlin M. E., et al. Restoration of p53 function leads to tumour regression in vivo. Nature . 2007;445(7128):661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 100.Mrakovcic M., Frohlich L. F. P53-Mediated molecular control of autophagy in tumor cells. Biomolecules . 2018;8(2):p. 14. doi: 10.3390/biom8020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tasdemir E., Maiuri M. C., Galluzzi L., et al. Regulation of autophagy by cytoplasmic p53. Nature Cell Biology . 2008;10(6):676–687. doi: 10.1038/ncb1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tasdemir E., Maiuri M. C., Morselli E., et al. A dual role of p53 in the control of autophagy. Autophagy . 2008;4(6):810–814. doi: 10.4161/auto.6486. [DOI] [PubMed] [Google Scholar]
- 103.Desplats P., Spencer B., Crews L., et al. α-Synuclein induces alterations in adult neurogenesis in Parkinson disease models via p53-mediated repression of Notch1. Journal of Biological Chemistry . 2012;287(38):31691–31702. doi: 10.1074/jbc.m112.354522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Spillantini M. G., Schmidt M. L., Lee V. M. Y., Trojanowski J. Q., Jakes R., Goedert M. α-Synuclein in Lewy bodies. Nature . 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 105.Grazia Spillantini M., Anthony Crowther R., Jakes R., Cairns N. J., Lantos P. L., Goedert M. Filamentous alpha-synuclein inclusions link multiple system atrophy with parkinson’s disease and dementia with lewy bodies. Neuroscience Letters . 1998;251(3):205–208. doi: 10.1016/s0304-3940(98)00504-7. [DOI] [PubMed] [Google Scholar]
- 106.Baba M., Nakajo S., Tu P. H., et al. Aggregation of alpha-synuclein in lewy bodies of sporadic parkinson’s disease and dementia with lewy bodies. American Journal Of Pathology . 1998;152(4):879–884. [PMC free article] [PubMed] [Google Scholar]
- 107.Wong Y. C., Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nature Medicine . 2017;23(2):1–13. doi: 10.1038/nm.4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen T., Li J., Chao D., et al. δ-Opioid receptor activation reduces α-synuclein overexpression and oligomer formation induced by MPP+ and/or hypoxia. Experimental Neurology . 2014;255:127–136. doi: 10.1016/j.expneurol.2014.02.022. [DOI] [PubMed] [Google Scholar]
- 109.Kurtishi A., Rosen B., Patil K. S., Alves G. W., Moller S. G. Cellular proteostasis in neurodegeneration. Molecular Neurobiology . 2019;56(5):3676–3689. doi: 10.1007/s12035-018-1334-z. [DOI] [PubMed] [Google Scholar]
- 110.Suzuki N., Johmura Y., Wang T. W., et al. TP53/p53-FBXO22-TFEB controls basal autophagy to govern hormesis. Autophagy . 2021;17(11):3776–3793. doi: 10.1080/15548627.2021.1897961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zininga T., Shonhai A. Small molecule inhibitors targeting the heat shock protein system of human obligate Protozoan parasites. International Journal of Molecular Sciences . 2019;20(23):p. 5930. doi: 10.3390/ijms20235930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sousa R. Structural mechanisms of chaperone mediated protein disaggregation. Frontiers in Molecular Biosciences . 2014;1:p. 12. doi: 10.3389/fmolb.2014.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chen H. Y., Lin C. H., Teng S. C. Stress-induced p53 drives BAG5 cochaperone expression to control α-synuclein aggregation in Parkinson's disease. Aging . 2020;12(20):20702–20727. doi: 10.18632/aging.103998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lonskaya I., Hebron M. L., Desforges N. M., Franjie A., Moussa C. E. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Molecular Medicine . 2013;5(8):1247–1262. doi: 10.1002/emmm.201302771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hebron M. L., Lonskaya I., Moussa C. E. H. Tyrosine kinase inhibition facilitates autophagic SNCA/α-synuclein clearance. Autophagy . 2013;9(8):1249–1250. doi: 10.4161/auto.25368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Brahmachari S., Ge P., Lee S. H., et al. Activation of tyrosine kinase c-Abl contributes to alpha-synuclein-induced neurodegeneration. Journal of Clinical Investigation . 2016;126(8):2970–2988. doi: 10.1172/jci85456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mahul A. l, Fauvet B., Gysbers A., et al. c-Abl phosphorylates alpha-synuclein and regulates its degradation: implication for alpha-synuclein clearance and contribution to the pathogenesis of parkinson’s disease. Proceedings of the Qatar Foundation Annual Research Conference Proceedings; 2014; Doha, Qatar. pp. 2858–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Goldberg Z., Vogt Sionov R., Berger M., et al. Tyrosine phosphorylation of Mdm2 by c-Abl: implications for p53 regulation. The EMBO Journal . 2002;21(14):3715–3727. doi: 10.1093/emboj/cdf384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Levav-Cohen Y., Goldberg Z., Zuckerman V., Grossman T., Haupt S., Haupt Y. C-Abl as a modulator of p53. Biochemical and Biophysical Research Communications . 2005;331(3):737–749. doi: 10.1016/j.bbrc.2005.03.152. [DOI] [PubMed] [Google Scholar]
- 120.Marine J. C., Lozano G. Mdm2-mediated ubiquitylation: p53 and beyond. Cell Death & Differentiation . 2010;17(1):93–102. doi: 10.1038/cdd.2009.68. [DOI] [PubMed] [Google Scholar]
- 121.Liu J., Liu W., Li R., Yang H. Mitophagy in Parkinson’s disease: from pathogenesis to treatment. Cells . 2019;8(7):p. 712. doi: 10.3390/cells8070712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dagda R. K., Cherra S. J., 3rd, Kulich S. M., Tandon A., Park D., Chu C. T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. Journal of Biological Chemistry . 2009;284(20):13843–13855. doi: 10.1074/jbc.m808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Osellame L. D., Duchen M. R. Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy . 2013;9(10):1633–1635. doi: 10.4161/auto.25878. [DOI] [PubMed] [Google Scholar]
- 124.Harper J. W., Ordureau A., Heo J. M. Building and decoding ubiquitin chains for mitophagy. Nature Reviews Molecular Cell Biology . 2018;19(2):93–108. doi: 10.1038/nrm.2017.129. [DOI] [PubMed] [Google Scholar]
- 125.Yamano K., Youle R. J. PINK1 is degraded through the N-end rule pathway. Autophagy . 2013;9(11):1758–1769. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hoshino A., Mita Y., Okawa Y., et al. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nature Communications . 2013;4(1):p. 2308. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- 127.da Costa C. A., Sunyach C., Giaime E., et al. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nature Cell Biology . 2009;11(11):1370–1375. doi: 10.1038/ncb1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang C., Lin M., Wu R., et al. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proceedings of the National Academy of Sciences of the United States of America . 2011;108(39):16259–16264. doi: 10.1073/pnas.1113884108. [DOI] [PMC free article] [PubMed] [Google Scholar]