

Abstract
Lung infections caused by Gram-positive Staphylococcus aureus and co-infections caused by S. aureus and Gram-negative Pseudomonas aeruginosa are challenging to treat, especially with the rise in the number of antibiotic-resistant strains of these pathogens. Bacteriophage (phage) are bacteria-specific viruses that can infect and lyse bacteria, providing a potentially effective therapy for bacterial infections. However, the development of bacteriophage therapy is impeded by limited suitable biomaterials that can facilitate effective delivery of phage to the lung. Here, we demonstrate the ability of porous microparticles engineered from poly(lactic-co-glycolic acid) (PLGA), a biodegradable polyester, to effectively deliver phage to the lung. The phage-loaded microparticles (phage-MPs) display potent antimicrobial efficacy against various strains of S. aureus in vitro and in vivo, and arrest the growth of a clinical isolate of S. aureus in the presence of sputum supernatant obtained from cystic fibrosis patients. Moreover, phage-MPs efficiently mitigate in vitro co-cultures of S. aureus and P. aeruginosa and display excellent cytocompatibility with human lung epithelial cells. Therefore, phage-MPs represents a promising therapy to treat bacterial lung infection.
Keywords: antimicrobial biomaterials, phage therapy, poly(lactic-co-glycolic acid), bacterial infections, Cystic Fibrosis
Graphical Abstract
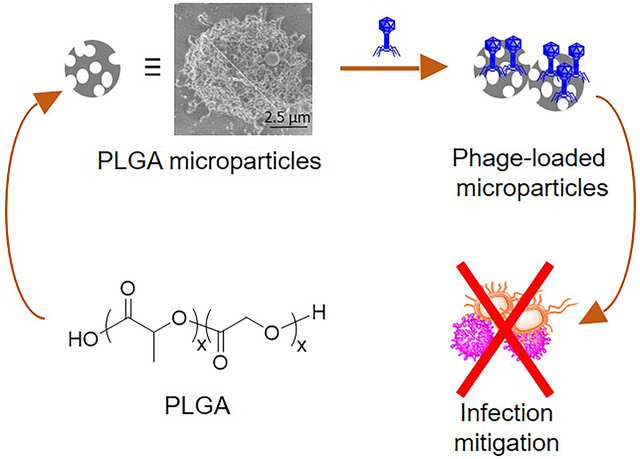
Porous, poly(lactic-co-glycolic) acid microparticles to facilitate the delivery of bacteriophages to the lungs were engineered. Bacteriophage-loaded microparticles (phage-MP) prepared under mild conditions mitigate S. aureus lung infection and inhibit the growth of S. aureus and P. aeruginosa co-cultures. This work demonstrates the promising potential of phage-MP therapy as an effective alternative to antibiotic regimens to treat bacterial lung infections.
INTRODUCTION
The rise of multi-drug resistant bacteria has rendered the treatment of bacterial infections very challenging, especially those caused by Staphylococcus aureus and Pseudomonas aeruginosa.[1] Infections caused by resistant strains of Gram-positive and Gram-negative bacteria are responsible for infections in bone,[2] wounds,[3] and lungs.[4] Notably, lung infections associated with pneumonia or cystic fibrosis (CF), a genetic disorder manifested by decreased mucociliary clearance and neutrophilic inflammation, often results in accelerated decline in lung function and premature death due to respiratory failure.[5],[6] Thus, alternative antibacterial therapies need to be developed to combat such infections that are unresponsive to standard-of-care antibiotics. Bacteriophage (phage) therapy is a viable strategy to mitigate such infections because of its ability to kill bacteria within a biofilm as well as its efficacy to inhibit infections caused by antibiotic-resistant strains of bacteria.[7] Indeed, phage therapy has been shown to be safe and effective in the treatment of infections that were non-responsive to antibiotic treatment.[8] Phages are viruses specific against particular bacterial strains, and thus can be used for targeted treatment of specific infections with minimal detrimental effect on the commensal flora of the host.[9]
Whereas phage therapy has been explored as an alternative to antibiotics to treat challenging infections,[10] the ability to achieve targeted delivery of high titers of phage to the lung while preserving the morphology and functionality of phage is crucial in the development of phage therapy. The discouraging results of a recent clinical trial on evaluation of efficacy of phage cocktail to treat burn wounds highlights the need for a biomaterial-based platform that can improve the retention of phage at the site of infection and provide with a desirable release profile.[11] Pulmonary delivery of phage as a liquid formulation with the use of nebulizers or intratracheal instillation results in reduction of phage titers upon delivery, with mode of delivery such as intratracheal instillation limited to animal models.[12] Although different phage carriers have been explored,[13] the conditions required to prepare the phage-carrier formulation often result in the loss of phage activity. Thus, there is a need for biomaterial-based phage formulations that can be prepared under mild conditions to maintain full phage activity and provide efficient mitigation of bacterial infection.[14] We previously showed that microparticles (MPs) engineered from poly(lactic-co-glycolic acid) (PLGA) and loaded with phage mitigate lung infections caused by Gram-negative P. aeruginosa.[15] However, MP-based phage therapy has not been developed for treatment of lung infection caused by Gram-positive S. aureus, which exhibits differences in sensitivity to antibiotics and immune responses as compared to Gram-negative bacteria due to the differences in peptidoglycan layer thickness and composition, and presence of an outer membrane.[16] Moreover, there is a need for the development of phage therapy to treat challenging co-cultures of S. aureus and P. aeruginosa.
Herein, we engineered phage-loaded PLGA MP formulations to deliver one or more phage active against S. aureus and S. aureus with P. aeruginosa. Phage-MP formulations were prepared under mild conditions to prevent loss of phage activity. The development of a MP-based phage approach allows for the development of a dry powder formulation with potential benefits of ease of handling, administration, and improved patient compliance over a conventional strategy of delivery of phage as a liquid formulation. We demonstrate efficient loading of phage on PLGA MPs and the efficacy of this formulation to mitigate S. aureus and P. aeruginosa co-cultures in vitro, and S. aureus in a murine model of acute lung infection.
RESULTS AND DISCUSSION
Preparation of phage-MP formulation
Porous PLGA MPs were prepared by a double emulsion method (water/oil/water) and homogenization process (Figure 1A). Particle shape, porosity, and particle size were characterized using dynamic light scattering (DLS) and scanning electron microscopy (SEM). The optimized fabrication process yielded highly porous microparticles of 5.7 ± 0.4 μm in size (Figure 1B and 1C). Whereas particles with a range of 1–5 micron are ideal for pulmonary delivery,[17] porous and larger-sized particles were prepared on purpose to lower their phagocytosis by alveolar macrophages,[18] while retaining their desirable aerodynamic properties required for lung delivery, because of the porous nature of the MPs.[19] Bacteriophage K, a phage active against S. aureus, was obtained from the Centers for Disease Control and Prevention (US CDC), amplified on the host bacteria (S. aureus 19685), and purified using fast protein liquid chromatography (FPLC). This process yielded a stock of phage K at high titers (109 to 1010 plaque forming units per mL or PFU/mL). Phage K was physically adsorbed on the surface of the microparticles by incubating MPs in phage solution. The choice of loading phage on the surface of the MP instead of encapsulating them within enables preparation of the formulation while avoiding exposure of phage to the relatively harsh conditions used to fabricate the MPs. Loading efficiency was quantified by dissolving phage-MPs in chloroform followed by extracting the free phage in an aqueous layer, which was serially diluted and plated on an agar-overlay assay containing the host bacteria (loading efficiency of 106 to 107 phage/mg of MPs). Stability of phage in chloroform was evaluated, and no reduction in phage titers were observed upon exposure in the time course of this assay (Figure S-1). Dynamic light scattering analysis of phage-loaded MPs showed reduction in the particle size as compared to unloaded MPs and appearance of a new signal (around 100 nm) corresponding to phage released from the MP during sample preparation for the measurement (Figure S-2A–C). This signal was present in the stock of free phage that was used for the loading process. The release profile of phage K loaded on the microparticles showed an initial burst released of the phage followed by sustained release over a period of 12 hours (Figure S-3).
Figure 1.
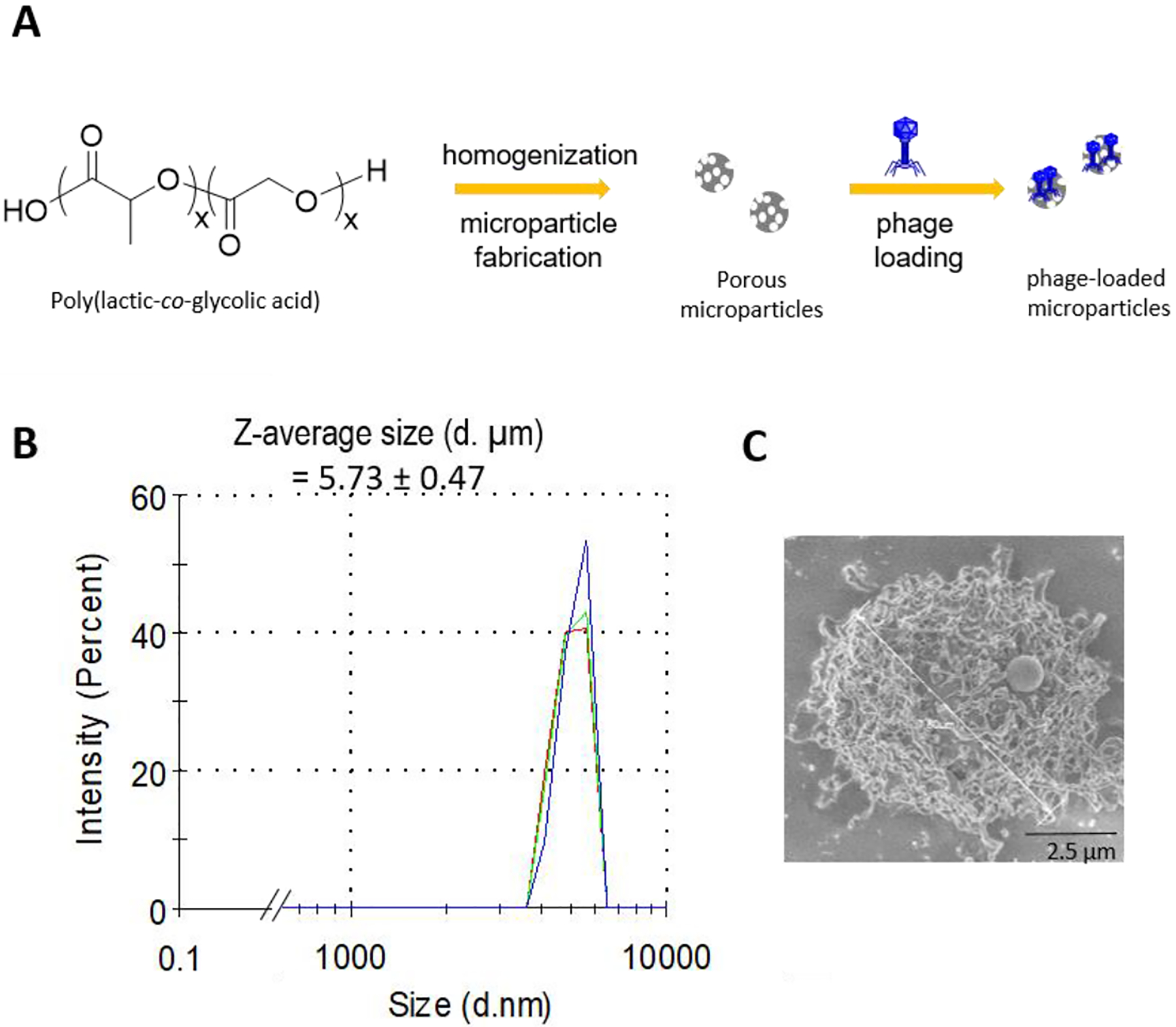
PLGA MP synthesis and characterization. A, Schematic of porous PLGA MP fabrication and bacteriophage loading. B, Dynamic light scattering profile of MPs (3 traces shown in different colors shows three separate runs on the same sample). C, Scanning electron microscopic image of porous MPs.
Phage-MPs kill S. aureus in vitro
Killing efficacy of phage K-loaded MPs was evaluated against four strains of S. aureus. Each strain was incubated in Lysogeny broth (LB) media in the presence of phage K-MPs or unloaded control MPs, and the bacterial counts were enumerated at the end of the assay by plating the mixture on agar plates. As shown in Figure 2A–D, treatment of bacteria with phage K-MPs reduced viable bacteria below the detection limits of the assay for all strains. In contrast, in the absence of phage K, all four strains of S. aureus were able to grow rapidly even in presence of control unloaded MPs (P < 0.0001). To evaluate the ability of phage K-MPs to mitigate co-cultures caused by multiple strains of S. aureus, a mixture of all four strains of S. aureus was grown in the presence of phage K-MPs or control unloaded MPs. The robustness of the phage K-MPs is demonstrated by complete clearance of all the strains of S. aureus in the co-culture following treatment (P < 0.0001 vs. unloaded MPs, Figure 2E).
Figure 2.
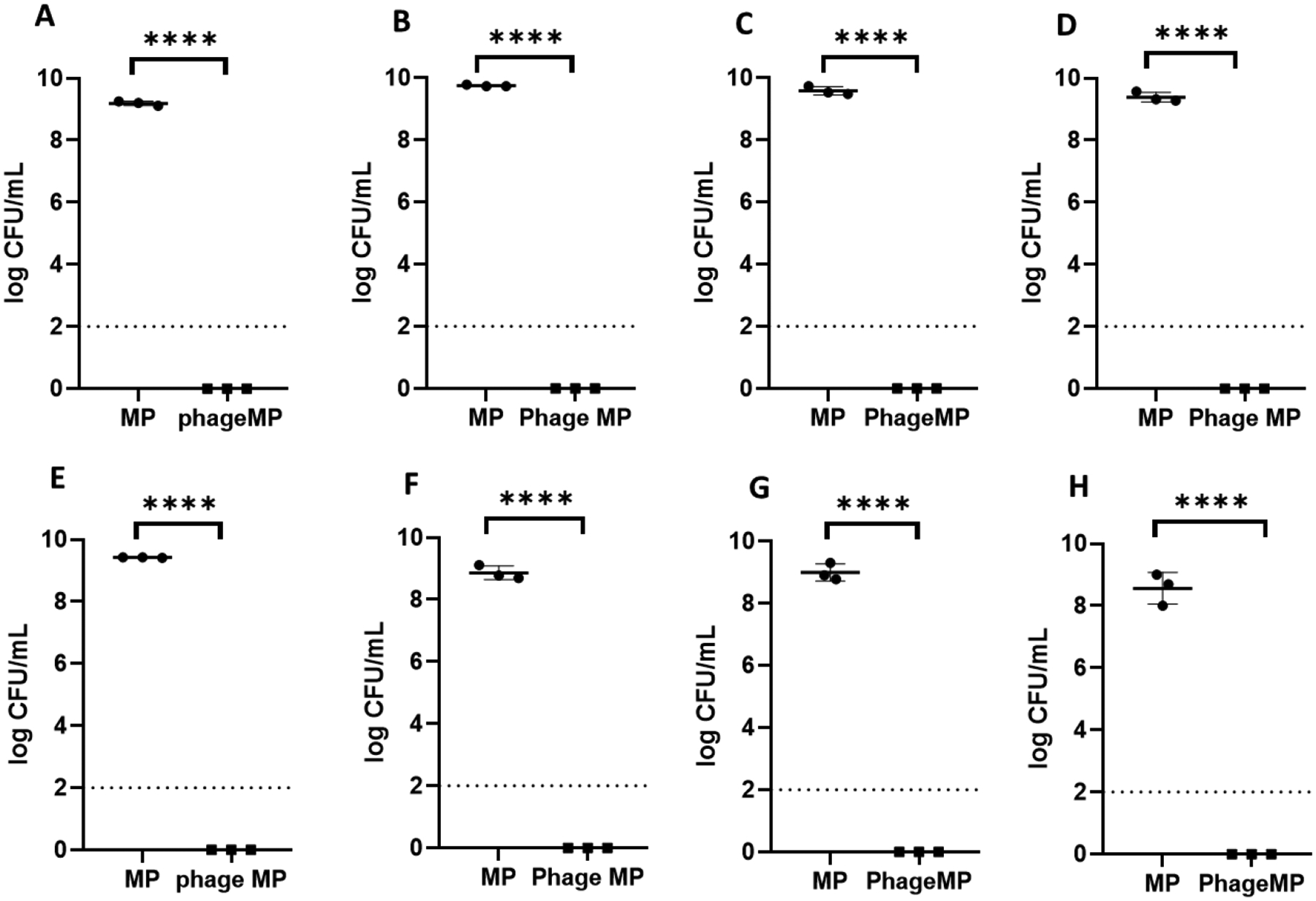
Efficacy of phage K-MPs against different strains of S. aureus. A, Xen29. B, SA 46106A. C, SA 46106B. D. SA 19685. E, mixture of four strains of S. aureus (SA Xen29 + SA 46106A + SA 46106B + SA 19685). F, UAMS-1, a clinical isolate of S. aureus, G Methicillin-resistant S. aureus and H, mixture of MRSA and UAMS-1. 100 μL bacterial suspension (1×107 CFU/mL) in LB was treated with MPs or phage K-MPs (2 mg/mL) for 24 hours with intermittent shaking. The suspensions were serially diluted, plated on agar plates, incubated at 37 °C, followed by enumeration of bacterial colonies after 16–20 hours. Dashed line represents limit of detection.****P < 0.0001 via unpaired two-tailed t test.
Phage-MPs kill clinical isolates and antibiotic-resistant strains of S. aureus
Whereas the robust mitigation of growth of different strains for S. aureus is encouraging, its efficacy against antibiotic-resistant strains and clinically isolated strains with a propensity to form biofilms must be evaluated as a step in the development of phage-MP therapy as an alternative to antibiotics. We evaluated the ability of phage K-MPs to clear a clinical isolate and methicillin-resistant S. aureus strain (MRSA). UAMS-1 is a clinical isolate from an osteomyelitis patient and a known biofilm former.[20] Incubation of phage K-MPs with UAMS-1 showed complete clearance of live bacteria while incubation with control unloaded MP showed high bacterial counts (Figure 2F). A dose-response study showed that a 10-fold lower dose of phage K-MPs was effective in eliminating live bacteria. As expected, further reduction in the dose of phage K-MPs resulted in loss of antibacterial activity (Figure S-4). Phage K-MP was also effective against MRSA and complete clearance was observed upon treatment of phage K-MPs with MRSA alone (Figure 2G), or co-culture of MRSA and UAMS-1, Figure 2H.
Phage-MPs mitigate co-culture of S. aureus and P. aeruginosa in vitro
An advantage of biomaterial-based delivery is that it can be used for co-delivery of multiple agents. To combat co-cultures of P. aeruginosa and S. aureus, we engineered the MP platform to co-deliver two or more phage that are active against one of the two bacteria. Phage 14, a phage active against PAO1 strain of P. aeruginosa, and phage K, a phage active against S. aureus, were loaded onto MPs simultaneously by incubating MPs in a mixed solution of the two phage (ratio of phage by volume = 1:1). Successful loading of both phage on the MPs was evaluated using an agar over-lay assay (phage K: 8 ×107 PFU/mg; phage 14: 4×107 PFU/mg). The ability of this formulation to mitigate co-culture of PAO1-GFP (P. aeruginosa) and UAMS-1 (S. aureus) was evaluated by measuring the change in the optical density of the bacterial co-suspension incubated with phage-loaded or control (no phage) MPs. Optical density of the bacterial suspension is proportional to the concentration of live bacteria in the suspension (Figure S-5). MPs co-loaded with phage 14 and phage K successfully reduced the optical density of the mixture of PAO1-GFP and UAMS-1 (P < 0.0001 compared to unloaded microparticles, P < 0.0001 vs. t=0 Figure 3A). Enumeration of the bacterial counts post treatment with the phage-loaded MPs showed orders of magnitude reduction in the bacterial counts (Figure 3B). To advance the therapy further, we evaluated the ability of phage MPs to inhibit co-culture of UAMS-1 and PA103, a strain of P. aeruginosa derived from the sputum of a patient. MPs co-loaded with phage K and two phage active against PA103 (phage 22 and phage E2005C) at a ratio of phage K:phage 22: E2005C = 2:1:1, (loading efficiency = phage K: 1 ×107 PFU/mg; phage E2005C + phage22: 2.5×107 PFU/mg) successfully inhibited the increase in the optical density of the mixture of the two bacteria (P < 0.0001 vs unloaded MPs, Figure 3C) and showed significant reduction in the counts of live bacteria as compared to the control unloaded MPs (Figure 3D).
Figure 3.
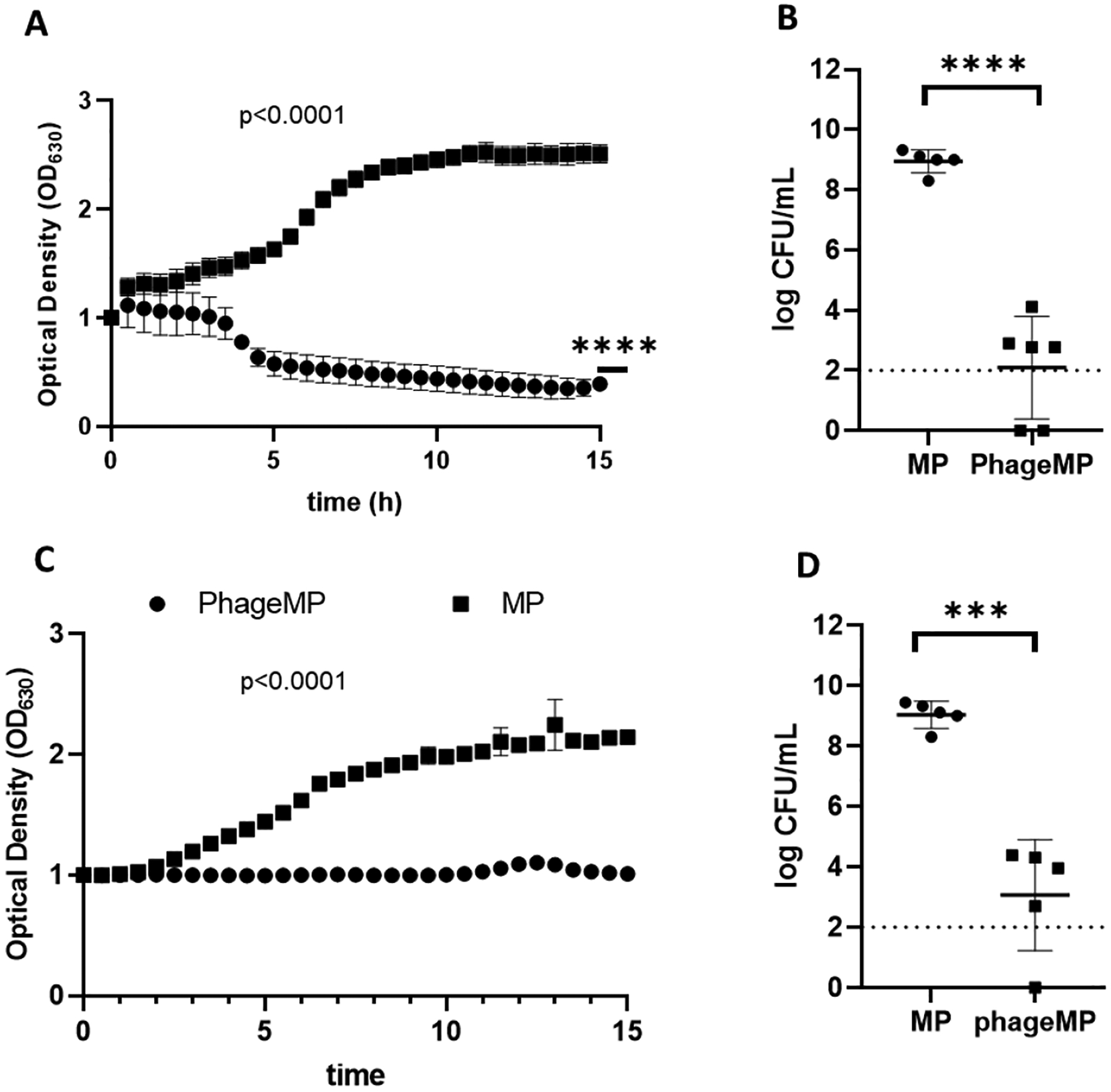
Efficacy of MPs co-loaded with phage active against S. aureus and P. aeruginosa against: A, B, mixture of PAO1-GFP and UAMS-1; and C, D, UAMS-1 and PA103. 50 μL of UAMS-1 (1×107 CFU/mL) in LB and 50 μL of PAO1-GFP (7×106 CFU/mL) or PA103 (1×107 CFU/mL) was treated with MP or phage-MP (2 mg/mL). For A and C, Normalized optical density was measured at 630 nm over a period of 15 hours. For B and D, the suspensions were serially diluted, plated on agar plates, and incubated at 37 °C, followed by enumeration of bacterial colonies after 16–20 hours. Dashed line represents limit of detection. Two-way ANOVA with Sidak’s test for multiple comparison. Two-way ANOVA with Dunnett’s multiple comparison test relative to time 0. ****P < 0.0001, ***P < 0.001 via unpaired two-tailed t test, N = 3–6.
Because the susceptibility of bacteria to antibacterial agents in a co-culture can be different compared to monocultures,[21] we performed a study to confirm that delivery of both phage K and phage 14 is necessary to inhibit mixture of UAMS-1 and PAO1-GFP. The two bacterial species were incubated with individual phage as well as a mixture of the two phage. Although each phage was able to inhibit growth caused by its target bacteria species (Figure S-6A and S-6B), neither was able to completely arrest the growth of the bacterial mixture (Figure S-6C and S-6D). In contrast, co-culture growth was significantly limited in the presence of both phage (Figure S-6E). This result demonstrates the need for both phage to target co-culture of UAMS-1 and PAO1-GFP. Similarly, exposure of mixture of UAMS-1 and PA103 to i) phage K (active against UAMS-1), ii) a mixture of phage 22 and phage E2005C (active against PA103), or iii) mixture of phage K, phage 22 and phage E2005C shows that phage against both S. aureus and P. aeruginosa are required to mitigate co-culture of UAMS-1 and PA103 (Figure S-7).
Phage-MPs kill a clinical isolate of S. aureus in CF sputum
To develop the phage therapy to treat bacterial infections in CF patients, we evaluated the efficacy of the phage-MP to mitigate S. aureus in presence of CF sputum. CF sputum contains mucin glycoproteins, enzymes, and other products that could interfere with phage activity.[22] Sputum supernatant samples from CF patients were obtained from the CF Biospecimen Repository at the Children’s Healthcare of Atlanta and Emory University CF Discovery Core (Emory IRB00042577) (see Table S-1 for details). UAMS-1 was incubated with LB and CF sputum supernatant (LB: sputum = 4:1 by volume) for 5 hours followed by addition of phage K-MPs. As shown in Figure 4A, phage K-MPs successfully prevented an increase in the optical density of UAMS-1 in contrast to unloaded MPs, which did not deter the growth of the bacteria (P < 0.0001). After establishing the efficacy of phage K-MPs to mitigate bacterial growth in sputum supernatant, we evaluated their efficacy to eliminate UAMS-1 culture in viscous CF sputum. Artificial CF sputum that recapitulates sputum conditions found in CF patients[23] was prepared and was used to grow UAMS-1 in presence of phage K-MPs or unloaded MPs. Enumeration of the bacterial counts post treatment showed elimination of live bacteria by phage K-MPs (Figure 4B). These results support the development of an alternative, phage-based strategy for the treatment of bacterial infections in CF patients.
Figure 4.
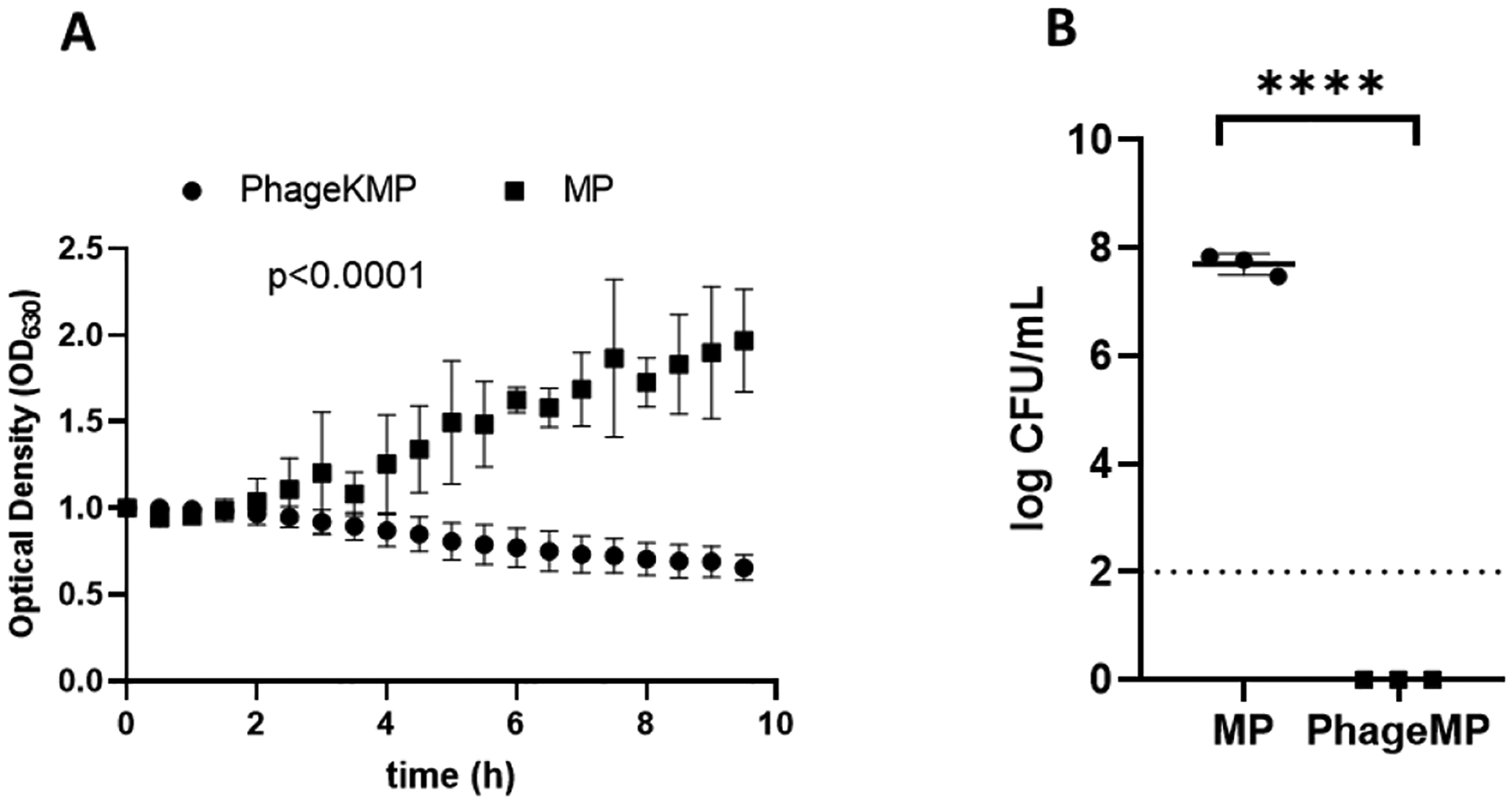
Efficacy of phage K-MPs against UAMS-1 in presence of CF sputum supernatant and viscous artificial sputum. A, Normalized optical density of UAMS-1 suspension in a mixture of LB and sputum supernatant was measured at 630 nm over a period of 10 hours in presence of phage K-MPs or unloaded MPs. B, Enumeration of bacterial counts post treatment of phage K-MPs or MPs with UAMS-1 grown in artificial CF sputum. A mixture of 90 μL bacterial suspension (1×107 CFU/mL) in artificial CF sputum medium and 10 μL LB was treated with MPs or phage K-MPs (2 mg/mL) for 24 hours. The suspensions were serially diluted, plated on agar plates, and incubated at 37 °C, followed by enumeration of bacterial colonies after 16–20 hours. Dashed line represents limit of detection. Two way ANOVA with Sidak test for multiple comparison. ****P < 0.0001 via unpaired two-tailed t test, N = 3.
MPs facilitate efficient delivery of phage to the lungs of mice
An advantage of development of MP-based formulation is the ability to lyophilize phage-MP to prepare a dry powder formulation, which has potential benefits of longer shelf life, ease of transportation, improved patient compliance while providing an efficient delivery of phage to deep lung tissue.[15] The improvement in the ability of MPs to deliver phage to the lungs as a dry powder was evaluated by the endotracheal delivery of phage K-MP formulation prepared by mixing lyophilized phage-MPs with lactose at a ratio of 1:9 by weight, Figure 5A. Lactose was added to improve the handling of the formulation. For comparison, a free phage formulation was prepared by mixing an equivalent dose of phage K with lactose (no MPs) followed by lyophilization. Approximately, a dose of 7.5 × 106 PFU/mouse of Phage-MP formulation or the free phage control formulation were delivered using endotracheal delivery. The dose of the free phage formulation was matched to that of phage-MP such that all mice were delivered the same amount of lactose by weight. The lungs of the mice were isolated post-delivery and analyzed for bacteriophage counts. Recovery of high titer of phage K from the lungs (trachea and intubation tube not included in the analysis) confirms successful lung delivery of phage K-MPs (Figure 5B). In contrast, delivery of phage K without the MP carrier did not lead to any detectable recovery of phage in the lungs. While previous studies have shown successful delivery of free phage to the lungs, it should be noted that those studies delivered phage as a liquid formulation and at a significantly higher dose of phage.[24],[25] In this study, free phage was delivered as a dry powder and at a low concentration to match the dose of phage-MP formulation and the lack of detection of free phage is consistent with our previous study.[15]
Figure 5.
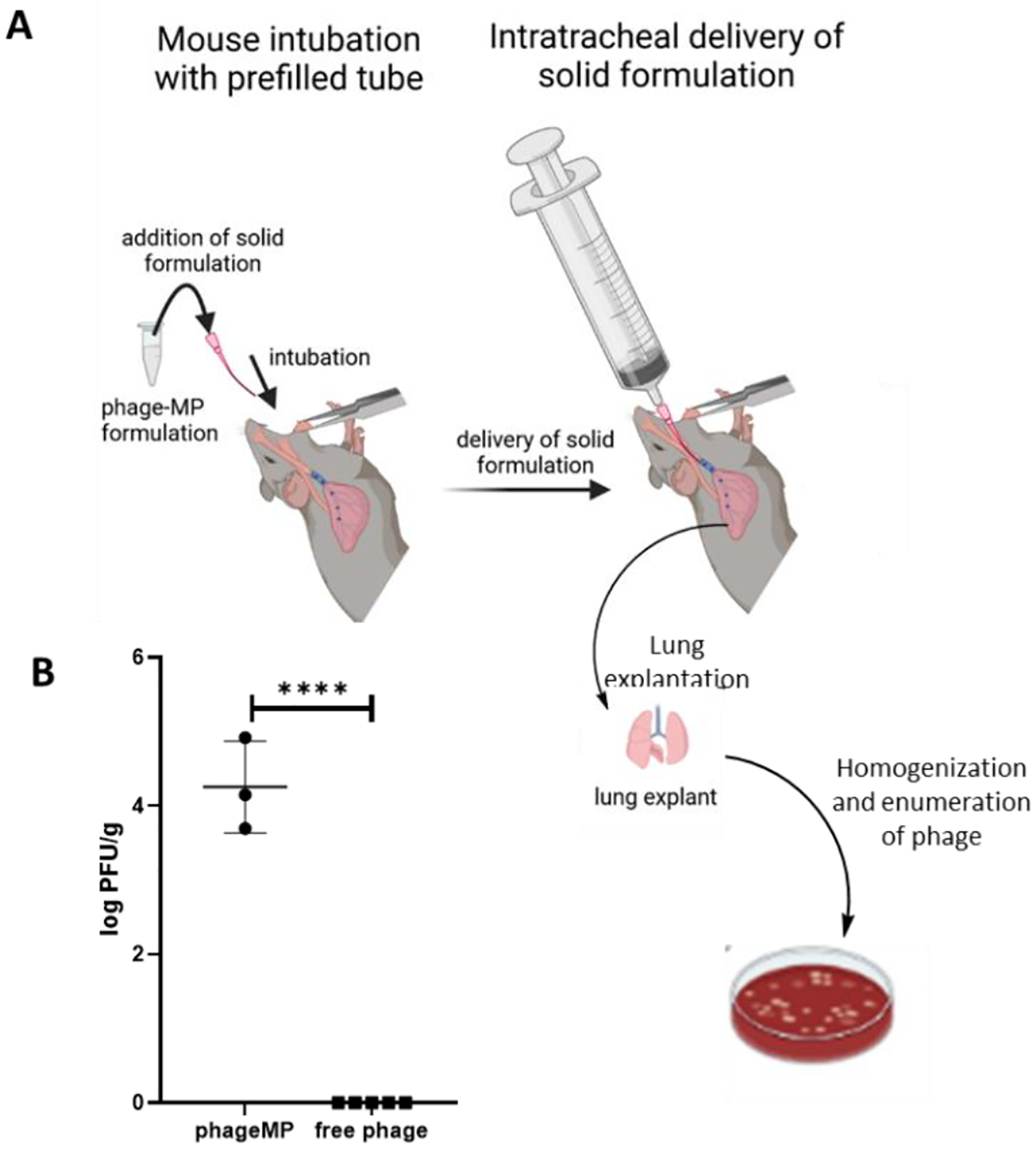
Delivery of phageK-MPs as a dry powder formulation in presence or absence of MPs to the lungs of mice. A, Schematic of endotracheal delivery of the dry powder formulation (Created with BioRender.com). B, Phage recovered in the lungs post-delivery of phage-MPs or free phage. Dry powder formulation was prepared by mixing lyophilized phage K-MPs with respiratory grade lactose (1:9 by weight). Free phage formulation consisted of phage K and lactose (no MPs). A dose of 7.5 × 106 PFU/mouse was delivered to each mouse. ****P < 0.0001 via unpaired two-tailed t test, N = 3–5.
Phage-MPs mitigate acute S. aureus lung infection in mice
The ability of phage-MPs to mitigate S. aureus lung infection was evaluated by delivering phage-MPs and JE-2 strain of S. aureus to the lungs of mice via the endotracheal route (Figure 6A). Loading the formulation directly into the top of the endotracheal tube precludes the need for the use of a dry powder insufflator. JE-2 is a strain derived from methicillin-resistant S. aureus.[26] Prior to the treatment study, a bacterial dosing study was performed to identify a dose of JE-2 that results in a sustained lung infection in mice over 24 hours. While initial studies on the delivery of phage-MPs bearing a single phage (phage K) did not lead to mitigation of infection (Figure S-9), a formulation prepared by loading five different phage that infect this bacterial strain (phage K + phage 110 + phage 134 + phage 135 + phage 136) on MPs led to statistically significant mitigation of infection (P < 0.0001, compared to JE-2, and P <0.001 compared JE-2 +MP, Figure 6B). The lack of difference between bacterial counts between the JE-2 and JE-2 + MP groups indicates that MPs by themselves do not impact bacterial numbers. Moreover, the phage-MP formulation outperformed a formulation consisting of a mixture of free phage preparations (phage K + phage 110 + phage 134 + phage 135 + phage 136) and lactose (no MPs), highlighting the advantage in the use of MP-based carrier system. We posit that multiple phage were needed for efficacy due to differences in infectivity and virulence towards the bacteria, which leads to improved efficacy when used as a mixture. Moreover, by lowering the concentration of individual phage in a phage mixture, the potential for development of resistance is reduced.
Figure 6.
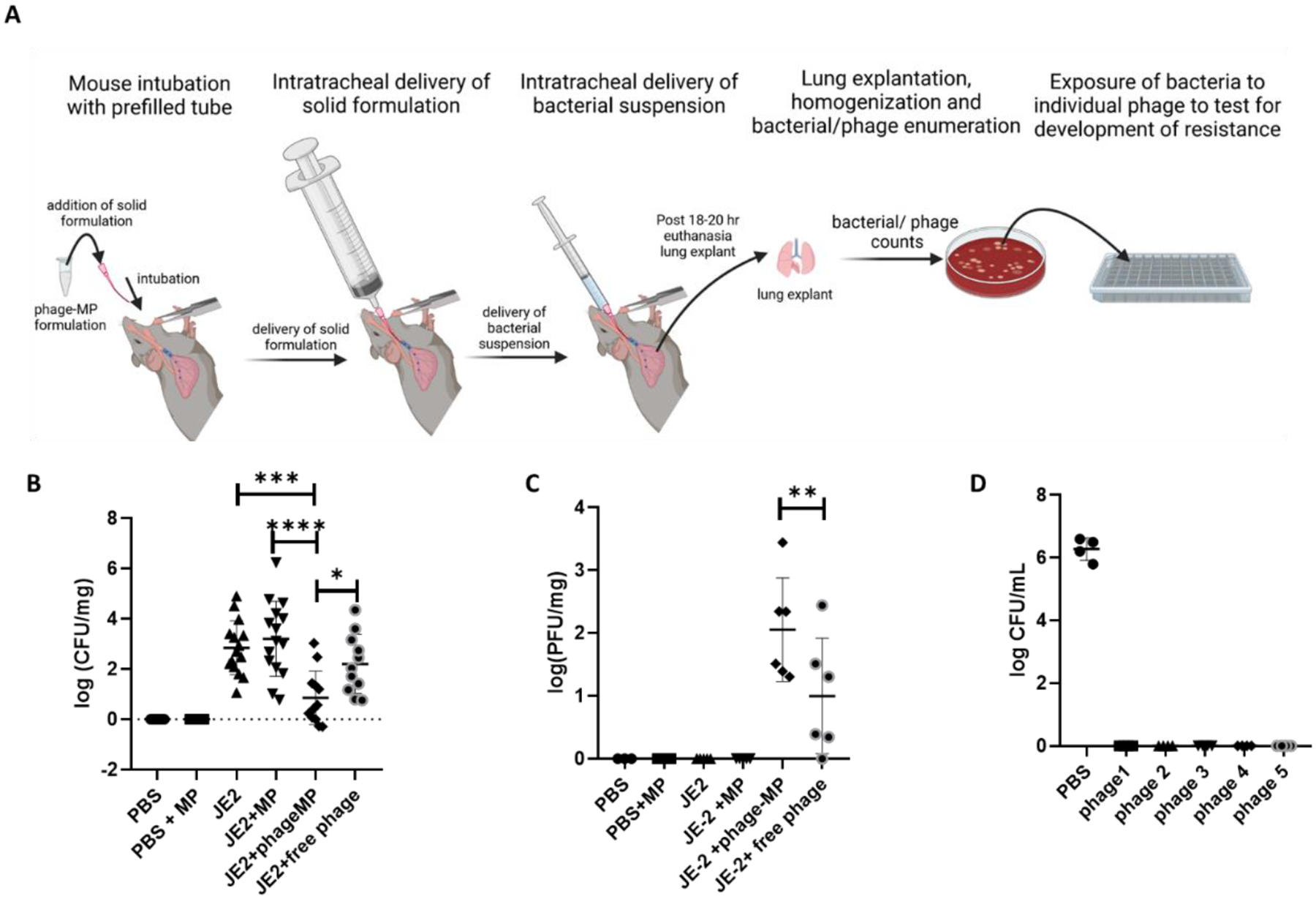
Efficacy of phage-MPs to mitigate acute S. aureus lung infection. A, Schematic overview of animal procedure (Created with BioRender.com). Phage-MPs were prepared by loading a mixture of five phage (phage K + phage 110 + phage 134 + phage 135 + phage 136) active against S. aureus strain JE-2. The dry powder formulation was prepared by lyophilization of phage-MPs followed by mixing with lactose at 1:1 ratio. B, Bacterial counts recovered from the lungs of mice at 18–20 hours post-treatment. Input dose of JE-2 = 1 × 108 CFU/mouse; input phage dose = 1 × 108 PFU/mouse. N = 7–15, data pooled from three separate studies. C, Phage recovered from the lungs of mice. N = 3–6. D, Evaluation of sensitivity of JE-2 colonies recovered from four different mice treated with phage-MPs to individual phage, without MPs. N = 4. One-way ANOVA with Holm-Sidak’s multiple comparisons. ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05
Mice that received phage-MPs displayed higher phage counts in the lungs as compared to mice that received free phage (Figure 6C). No detectable phage were recovered from the lungs of mice that received free phage in absence of infection (Figure 5B); this result is not unexpected as the presence of bacteria is necessary for phage replication (Figure 6C). While the ability of phage-MP to mitigate the infection is very promising, this study demonstrates the efficacy of phage-MP formulation for prophylactic treatment against S. aureus. Additional studies need to be performed to evaluate the efficacy of phage-MP to treat chronic lung infections. However, such studies are often impeded by challenges in the development of a mouse model that display chronic S. aureus lung infection.
Bacterial susceptibility to phage post phage-MP therapy
To evaluate the susceptibility of bacteria to re-exposure of phage, bacterial colonies isolated from mice treated with phage-MPs were grown in LB media in the presence of individual components of phage mixture. As a control, the colonies were exposed to PBS to confirm their ability to grow. The continued susceptibility of JE-2 to phage is demonstrated by their complete killing upon exposure to individual phage components (Figure 6D) as well as by the lack of increase in the optical density of the suspension (Figure S-10). While the susceptibility of bacteria recovered from mice treated with phage-MPs to phage re-exposure are encouraging, additional studies should be performed to probe for development of anti-phage antibodies and resistance over multiple dosing of phage form.
Phage-MPs exhibit acceptable cytocompatibility with mammalian cells
Finally, the metabolic activity of human lung epithelial cells (NuLi cells)[27] in the presence of phage K-MPs and MPs co-loaded with phage K, phage 14 and phage 22 and phage E2005C was evaluated following procedure described in the experimental section. The cells were exposed to three different concentrations of the phage-loaded MPs, including a concentration of 2 mg/mL that was used for the antibacterial assays, and incubated over a period of 24 hours. Human epithelial cells exposed to phage K-MPs (Figure 7A) or co-loaded with phage K, phage 14, phage 22, and phage E2005C (Figure 7B) maintained high metabolic activity over a period of 24 hours which was statistically equivalent to the metabolic activity of the control cells exposed to media alone. These results display that the phage-MP preparations were cytocompatible with these airway epithelial cells.
Figure 7.
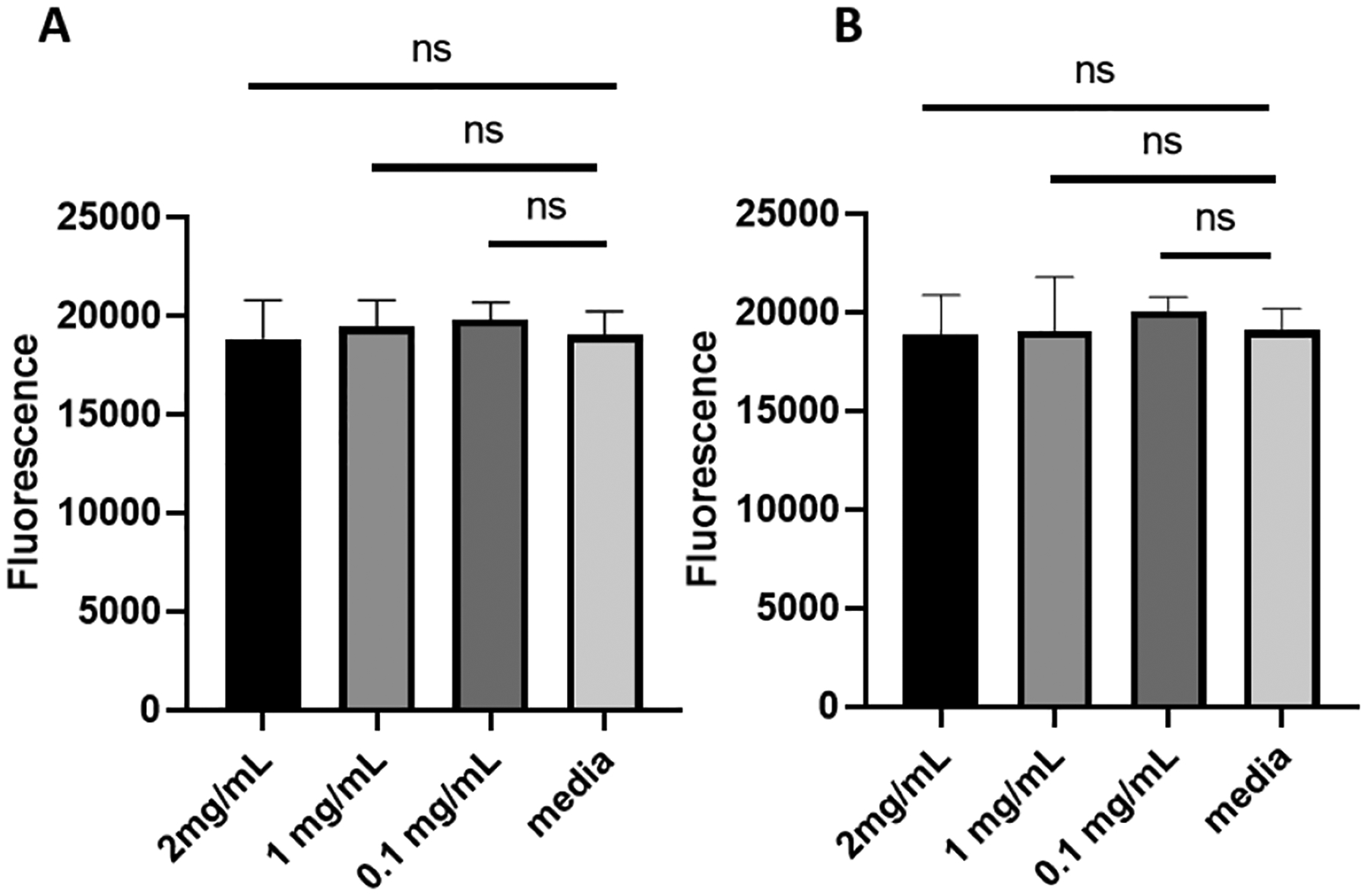
Metabolic activity assay of human lung epithelial cells. NuLi cells were incubated for 24 hours with three different concentrations of: A, phage K-MPs, B, phage K+14+22+E2005C-loaded MPs. 10% Alamar blue reagent in growth media was added to each group, incubated at 37 °C for 3–4 hours and fluorescence (λex = 560 nm and λem = 590 nm) was measured. One-way ANOVA with Tukey post-hoc test. Measurements (n=6–8/group, mean ± SD) were taken from two separate runs. ns=not significant..
CONCLUSION
We demonstrate that phage-loaded MPs exhibit potent antimicrobial efficacy against different strains of S. aureus, including clinical isolates, biofilm formers, and methicillin-resistant strains. Phage-MPs show robust mitigation of S. aureus in an acute lung infection mouse model, and arrest the growth of S. aureus in viscous artificial sputum and in presence of supernatant from CF sputum samples obtained from patients. In addition, the phage-MPs did not show any gross toxicity towards human lung epithelial cells. The MP platform allowed for simultaneous delivery of multiple phage active against different bacterial species, including inhibiting the growth of Gram-positive S. aureus and Gram-negative P. aeruginosa co-cultures. These results demonstrate the potential of phage-MP therapy as an effective alternative to antibiotic regimens to treat S. aureus lung infections, and provide encouraging preliminary results to treat polymicrobial bacterial infections.
Experimental methods
Synthesis of Poly(lactic-co-glycolic) acid (PLGA) MPs:
MPs were prepared following procedures described elsewhere.[15] Briefly, 200 mg of PLGA (Resomer® RG 503 H, Sigma; acid terminated, lactide:glycolide 50:50, Mw 24,000–38,000) was dissolved in 10 mL of methylene chloride (CH2Cl2). In a separate vial, 1% polyvinyl alcohol (PVA, Mw = 31000–50000, Sigma Aldrich) solution in DI water was prepared. Ammonium bicarbonate (Sigma Aldrich) solution (40 mg/mL) was prepared and quickly added to PLGA solution. The mixture was immediately homogenized at 4000 rpm for 2 min using PRO Scientific Benchtop Homogenizer D-Series. For preparation of MP of different particle size, different homogenization speeds were carried out. The homogenized mixture was added to 50 mL of PVA solution, and the mixture was again homogenized for 2 min. The homogenized mixture was added to 100 mL of PVA solution and stirred for 4–6 hours at room temperature in an open beaker to facilitate evaporation of CH2Cl2. The microparticles were then washed thrice with DI water and lyophilized for 24 hours. Microparticles were characterized by dynamic light scattering (Malvern Zetasizer Nano series) and scanning electron microscopy (Hitachi SU8010 SEM).
Preparation of bacteriophage stocks:
Bacteriophage (phage K, phage 14, phage 22, and phage E2005C) were prepared from samples generously provided by Dr. Rodney Donlan, Biofilm Laboratory, Centers for Disease Control and Prevention, Atlanta, GA, USA. Purified stocks of phage 110, phage 134, phage 135, phage 136 were obtained from the Naval Medical Research Center. Bacteriophage were grown on a lawn of host bacteria (Table S2) by overnight incubation on TSA plates at 37 °C. Individual plaques of the phage were isolated, suspended in PBS and the suspension was plated on the center of a lawn of host bacteria on a TSA plate and again incubated overnight for 37 °C. Next, the phage plaques were scraped using a cell scraper and grown in 1 L Trypticase soy broth growth media supplemented with the host bacteria for 4–8 hours at 37 °C on an orbital shaker. Chloroform was added to the mixture to kill the bacteria and phage were collected by aqueous extractions. The aqueous layer was then purified using fast protein liquid chromatography.[28]
Preparation of phage-loaded MPs:
PLGA MPs were washed thrice sequentially in ethanol, water, and PBS. After the final wash PBS was removed, the microparticles were suspended in a solution of phage and kept on a rocker for 4 hours at room temperature. When loading more than one phage, a mixture of phage was prepared, and the MPs were suspended in the phage mixture for 4 hours (ratio of phage against S. aureus to P. aeruginosa = 1:1). Next, the mixture was centrifuged at 500g for 5 minutes, and the collected MPs were washed in PBS. Phage-loaded MPs were suspended in PBS, and the suspension was incubated with different strains of S. aureus or a mixture of S. aureus and P. aeruginosa.
Measurement of phage loading on MPs:
Qualitative evaluation of phage loading on MPs was performed by plating 10 μL of phage-MP suspension at the center of an agar overlay containing the host bacteria on a TSA plate followed by overnight incubation at 37 °C. Quantification of phage loading on MPs was performed by addition of 100 μL of chloroform to 100 μL of phage-MP suspension to dissolve PLGA MPs. The aqueous layer of the biphasic mixture was collected, serially diluted, and plated on an agar overlay containing the host bacteria on a TSA plate followed by overnight incubation at 37 °C.
Release assay.
Phage-MPs were suspended in PBS at a concentration of 10 mg/mL and kept on a rocker at 37 °C. At each time point, the suspension was centrifuged at 500g for five minutes and the supernatant was collected. The supernatant was serially diluted and plated on an agar overlay containing host bacteria, and plaque forming units (PFU) were quantified after incubation of the plates at 37 °C overnight. The remaining MPs were resuspended in PBS and an equal volume of chloroform was added to dissolve the MPs. The aqueous layer of the biphasic mixture was collected, serially diluted, and plated on an agar overlay of the host bacteria and quantified after incubation of the plates at 37 °C overnight.
Antimicrobial assay:
Frozen glycerol stocks of bacteria were cultured on TSA plates overnight at 37 °C. Individual bacterial suspensions were prepared by swabbing a few colonies of the bacteria in LB media to reach a bacterial concentration of 1 × 107 CFU/mL. For assay utilizing a single strain of S. aureus, 100 μL of the bacterial suspension in LB was added to 100 μL of phage-MP suspension in PBS or 100 μL of unloaded MPs in PBS so that the final concentration of phage-MPs (or MPs) was 2 mg/mL. In the dose response study, 100 μL of phage-MPs, diluted to different concentrations, were incubated with 100 μL of the bacterial suspension. Absorbance was measured using BioTek PowerWave XS at 37 °C for 15 hours with intermittent shaking for three seconds every minute. For assay utilizing two or more strains of S. aureus, a bacterial mixture was prepared by mixing suspensions of the strains at equal ratio by volume. In a 96 well-plate, 100 uL of the bacterial mixture was added to 100 μL of phage-MPs suspension in PBS or 100 μL of unloaded MPs in PBS so that the final concentration of phage-MPs (or MPs) was 2 mg/mL. Absorbance was measured at 37 °C for 15 hours with intermittent shaking for three seconds every minute. For the co-culture assay, bacterial stock suspensions of S. aureus and P. aeruginosa were prepared separately by inoculating a few colonies of the bacteria in LB media. In a 96 well-plate, 50 μL each of the two bacterial stock suspensions in LB were added to 100 μL of phage-MPs suspension in PBS or 100 μL of unloaded MPs in PBS so that the final concentration of phage-MPs (or MPs) was 2 mg/mL. To demonstrate that phage active against both S. aureus and P. aeruginosa are necessary to mitigate the growth of the co-culture, 50 μL each of S. aureus and P. aeruginosa suspensions were added to 100 μL of: i) phage active against S. aureus, ii) phage active against P. aeruginosa, iii) a solution of phage active against both S. aureus and P. aeruginosa prepared at equal ratio by volume, and iv) PBS. The absorbance was measured at 37 °C for 10–15 hours. To evaluate antibacterial efficacy of phage-MPs in CF sputum supernatant (obtained from the CF Biospecimen Repository, Emory University) a bacterial stock solution was prepared by inoculating a few colonies of the bacteria in LB media. 40 μL of the bacteria in LB was added to 10 μL of CF sputum supernatant and incubated for 4–5 hours. 50 μL of phage-MPs or control MPs was added to each well and optical density was measured for 10 hours at 37 °C, with intermittent shaking for three seconds every minute. Data were normalized to time zero. Antimicrobial assay in artificial CF sputum (prepared as described elsewhere)[23] was performed by incubating 90 μL of artificial sputum and 10 μL of LB containing approximately 1 × 107 CFU/mL bacteria. 100 μL of phage-MP or control MP (final concentration 2 mg/mL) was added to the bacterial suspension. After 24 hours, the mixture was serially diluted, plated on TSA plates and incubated at 37 °C. Bacterial colonies were counted after overnight incubation.
Enumeration of bacterial colony forming units:
Post measurement of optical density, the mixture in each well of the 96-well plate were serially diluted, plated on TSA plates, and incubated at 37 °C. Bacterial colonies were counted after overnight incubation of the plates.
Metabolic activity assay:
Metabolic activity was measured using Alamar blue reagent, a calorimetric assay to quantify the proliferation of the cells.[29] Approximately 1×104 NuLi cells[27] (ATCC® CRL-4011™) in growth media (PneumaCult™) were plated in the wells of a 48-well plate at 37 °C. After 24 hours, the media was removed and replaced by 300 μL of phage-MPs suspended in media at different concentrations (2 mg/mL, 1 mg/mL and 0.1 mg/mL). Fresh media was added to the control wells. Cells were incubated for 24 hours at 37 °C. After 24 hours, media was removed from each well and 300 μL of 10% Alamar blue reagent in growth media was added to each well. The well-plate was incubated at 37 °C for 3–4 hours. Fluorescence (λex = 560 nm and λem = 590 nm) was measured using a BioTek Synergy H4 microplate reader.
Determination of correlation of optical density of bacterial suspension to bacterial concentration (CFU/mL):
Bacterial stock suspensions were prepared at different optical density measured at 590 nm. These stock suspensions were serially diluted, and the aliquots were plated on a TSA plate. The TSA plates were incubated at 37 °C for 16–20 hours and the colonies were enumerated to calculate CFU/mL at each optical density of the bacterial suspension.
Animal studies
All animal procedures were conducted according to the guidelines of the Emory University Institutional Animal Care and Use Committee (IACUC), under approved protocol number DAR-PROTO201700349-N. The study was carried out in strict accordance with established guidelines and policies at Emory University School of Medicine, and recommendations in the Guide for Care and Use of Laboratory Animals, as well as local, state, and federal laws. C57BL/6 female mice (12–14 weeks old) were obtained from Jackson Laboratories, Bar Harbor-ME were anesthetized by intraperitoneal injection of 0.2 ml of a mixture of ketamine (6.7 mg/ml) and xylazine (1.3 mg/ml). Anesthetized mice were placed on a 45°-angled platform held by upper incisors.
Endotracheal delivery of phage-MP to the lungs:
Dry powder formulation consisting of a mixture of phage K-MP and lactose (1:9) ratio by weight was prepared. Control consisted of free phage with lactose (no MP). All mice received a formulation with same amount of lactose by weight. The mice were intubated using 18-G catheter filled with 10 mg of the MP formulation and connected to a 3 mL syringe. The air in the syringe was pushed through catheter to deliver the dry powder formulation to the lungs. After the procedure, the mice were woken-up under heat lamp, euthanized in 30 minutes and the lungs were collected. The collected lungs were homogenized in PBS using 24 Fast Prep homogenizer, serially diluted in PBS and plated on an agar overlay containing the host bacteria on a TSA plate followed by overnight incubation of the plate at 37 °C for phage enumeration.
In vivo antibacterial study:
A dry powder formulation consisting of a mixture of phage-MPs (phage mixture is an equal volume mixture of phage K + phage 110 + phage 134 + phage 135 + phage 136; phage = 1 × 108 PFU/mouse) and lactose at a ratio of 1:1 by weight was prepared. Controls consisted of unloaded MPs mixed with lactose (1:1 ratio) and free phage mixed with lactose (no MPs). The mice were intubated using 18-G catheter filled with 10 mg of the MP formulation and connected to a 3 mL syringe. The air in the syringe was pushed through catheter to deliver the dry powder formulation to the lungs. Immediately after the delivery of the dry powder formulation, bacterial suspension (25 μL corresponding to 1× 108 CFU/mouse) was delivered via the same route. After the procedure, the mice were monitored closely until fully recovered. Mice were euthanized at 18–20 hours post infection using cervical dislocation under isoflurane anesthesia, and the whole lungs were collected aseptically. The collected lungs were homogenized in PBS using 24 Fast Prep homogenizer, serially diluted in PBS. For bacterial enumeration, lung homogenates were plated on mannitol-salt agar, tryptic soy agar (TSA) containing an overlay of the host bacteria (JE-2) for phage enumeration. The plates were incubated at 37 °C for 16–20 hours and bacterial and phage counts were recorded.
Evaluation of sensitivity of bacteria to re-exposure of phage:
Bacteria collected from the lungs of mice exposed to JE-2 and phage-MPs were added to LB media. 100 μL aliquots of the bacterial suspension were added to different wells of a 96 well plate. 50 μL of individual phage or PBS was added to well and the optical density of the suspension was measured at 630 nm for 10 hours. Next, the samples from each well were serially diluted, serial dilutions added to agar plates and the plates were incubated at 37 °C. Bacterial colonies were counted after overnight incubation of the plates.
Statistical analysis:
Statistical comparisons were performed using GraphPad Prism 7.0. Normal distribution was determined using Shapiro-Wilk test. Two-group comparisons were conducted using unpaired two-tailed t test. One-way ANOVA with Tukey’s post-hoc test was performed for in vitro metabolic activity assay. One-way ANOVA with Holm-Sidak’s multiple comparisons test was performed to measure efficacy of phage-MP formulation in vivo. Two-way ANOVA and Sidak’s post-hoc analysis was performed to detect differences between the optical density of bacteria in presence of phage-MPs and control MPs. Two-way ANOVA with Dunnett’s test was used to evaluate statistical difference in reference to t=0 in the antibacterial assay. A P-value of <0.05 was deemed significant. All results were reported as mean ± standard deviation, unless otherwise noted.
Supplementary Material
Acknowledgement.
We thank Dr. Rodney Donlan, Biofilm Laboratory, Centers for Disease Control and Prevention, Atlanta, GA, USA and Naval Medical Research Center for providing bacteriophage samples. CF human subject samples were provided by the Cystic Fibrosis Biospecimen Repository at the Children’s Healthcare of Atlanta and Emory University CF Discovery Core. Mice were obtained from the Animal Models Core of the Center for CF and Airways Disease Research, Children’s Healthcare of Atlanta and Emory University. We acknowledge the core facilities at the Parker H. Petit Institute for Bioengineering and Bioscience at the Georgia Institute of Technology for the use of their shared equipment, services, and expertise. We appreciate Dr. Thomas and Dr. Roy at the Georgia Institute of Technology for providing access to the DLS and the homogenizer. This work was supported by grants from Cystic Fibrosis Foundation CFF GARCIA17G0 and National Institutes of Health (NIH R01 AR062920) to AJG.
Footnotes
Supporting information
The supporting information is available from the Wiley Online Library or from the authors.
Conflict of Interest
The authors declare no conflict of interest
Contributor Information
Pranav P. Kalelkar, Woodruff School of Mechanical Engineering and Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, 315 Ferst Dr. NW, Atlanta, GA 30332, USA
Dina A. Moustafa, Department of Pediatrics and Children’s Healthcare of Atlanta, Center for Cystic Fibrosis and Airway Diseases Research, Emory University School of Medicine, 1510 Clifton Road NE, Atlanta, GA 30322
Milan Riddick, Wallace H. Coulter Department of Biomedical Engineering and Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, 315 Ferst Dr. NW, Atlanta, GA 30332, USA.
Joanna B. Goldberg, Department of Pediatrics and Children’s Healthcare of Atlanta, Center for Cystic Fibrosis and Airway Diseases Research, Emory University School of Medicine, 1510 Clifton Road NE, Atlanta, GA 30322
Nael A. McCarty, Department of Pediatrics and Children’s Healthcare of Atlanta, Center for Cystic Fibrosis and Airways Disease Research, Emory University School of Medicine, 2015 Uppergate Drive, Atlanta, GA 30322, USA
Andrés J. García, Woodruff School of Mechanical Engineering and Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, 315 Ferst Dr. NW, Atlanta, GA 30332, USA
References
- [1].a) Sprenger M, Fukuda K, Science 2016, 351, 1263; [DOI] [PubMed] [Google Scholar]; b) Edelstein MV, Skleenova EN, Shevchenko OV, D’Souza JW, Tapalski DV, Azizov IS, Sukhorukova MV, Pavlukov RA, Kozlov RS, Toleman MA, Walsh TR, Lancet Infect. Dis 2013, 13, 867; [DOI] [PubMed] [Google Scholar]; c) Caplin JD, García AJ, Acta Biomater. 2019, 93, 2; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Costerton JW, Stewart PS, Greenberg EP, Science 1999, 284, 1318; [DOI] [PubMed] [Google Scholar]; e) Koo H, Allan RN, Howlin RP, Stoodley P, Hall-Stoodley L, Nat. Rev. Microbiol 2017, 15, 740; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Beaudoin T, Yau YCW, Stapleton PJ, Gong Y, Wang PW, Guttman DS, Waters V, npj Biofilms and Microbiomes 2017, 3, 25; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Briaud P, Camus L, Bastien S, Doléans-Jordheim A, Vandenesch F, Moreau K, Sci. Rep 2019, 9, 16564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Meurice E, Rguiti E, Brutel A, Hornez JC, Leriche A, Descamps M, Bouchart F, J. Mater. Sci. Mater. Med 2012, 23, 2445; [DOI] [PubMed] [Google Scholar]; b) Kaur S, Harjai K, Chhibber S, PLoS One 2016, 11, e0157626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Serra R, Grande R, Butrico L, Rossi A, Settimio UF, Caroleo B, Amato B, Gallelli L, de Franciscis S, Expert Rev. Anti Infect. Ther 2015, 13, 605; [DOI] [PubMed] [Google Scholar]; b) Weiner-Lastinger LM, Abner S, Edwards JR, Kallen AJ, Karlsson M, Magill SS, Pollock D, See I, Soe MM, Walters MS, Dudeck MA, Infect. Control Hosp. Epidemiol 2020, 41, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Weiner-Lastinger LM, Abner S, Benin AL, Edwards JR, Kallen AJ, Karlsson M, Magill SS, Pollock D, See I, Soe MM, Walters MS, Dudeck MA, Infect. Control Hosp. Epidemiol 2020, 41, 19; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Weiner LM, Webb AK, Limbago B, Dudeck MA, Patel J, Kallen AJ, Edwards JR, Sievert DM, Infect. Control Hosp. Epidemiol 2016, 37, 1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Millette G, Langlois JP, Brouillette E, Frost EH, Cantin AM, Malouin F, Front. Microbiol 2019, 10, 2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Cohen TS, Prince A, Nat. Med 2012, 18, 509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cigana C, Bianconi I, Baldan R, De Simone M, Riva C, Sipione B, Rossi G, Cirillo DM, Bragonzi A, J. Infect. Dis 2018, 217, 933. [DOI] [PubMed] [Google Scholar]
- [7].a) Donlan RM, Trends Microbiol. 2009, 17, 66; [DOI] [PubMed] [Google Scholar]; b) Labrie SJ, Samson JE, Moineau S, Nat. Rev. Microbiol 2010, 8, 317. [DOI] [PubMed] [Google Scholar]
- [8].a) Dedrick RM, Guerrero-Bustamante CA, Garlena RA, Russell DA, Ford K, Harris K, Gilmour KC, Soothill J, Jacobs-Sera D, Schooley RT, Hatfull GF, Spencer H, Nat. Med 2019, 25, 730; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schooley RT, Biswas B, Gill JJ, Hernandez-Morales A, Lancaster J, Lessor L, Barr JJ, Reed SL, Rohwer F, Benler S, Segall AM, Taplitz R, Smith DM, Kerr K, Kumaraswamy M, Nizet V, Lin L, McCauley MD, Strathdee SA, Benson CA, Pope RK, Leroux BM, Picel AC, Mateczun AJ, Cilwa KE, Regeimbal JM, Estrella LA, Wolfe DM, Henry MS, Quinones J, Salka S, Bishop-Lilly KA, Young R, Hamilton T, Antimicrob. Agents Chemother 2017, 61; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jennes S, Merabishvili M, Soentjens P, Pang KW, Rose T, Keersebilck E, Soete O, François P-M, Teodorescu S, Verween G, Verbeken G, De Vos D, Pirnay J-P, Critical Care 2017, 21, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Dufour N, Delattre R, Ricard JD, Debarbieux L, Clin. Infect. Dis 2017, 64, 1582; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cho I, Blaser MJ, Nat. Rev. Genet 2012, 13, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kortright KE, Chan BK, Koff JL, Turner PE, Cell Host Microbe 2019, 25, 219. [DOI] [PubMed] [Google Scholar]
- [11].Jault P, Leclerc T, Jennes S, Pirnay JP, Que Y-A, Resch G, Rousseau AF, Ravat F, Carsin H, Le Floch R, Schaal JV, Soler C, Fevre C, Arnaud I, Bretaudeau L, Gabard J, The Lancet Infectious Diseases 2019, 19, 35. [DOI] [PubMed] [Google Scholar]
- [12].Leung SSY, Carrigy NB, Vehring R, Finlay WH, Morales S, Carter EA, Britton WJ, Kutter E, Chan HK, Int. J. Pharm 2019, 554, 322. [DOI] [PubMed] [Google Scholar]
- [13].a) Vinner GK, Richards K, Leppanen M, Sagona AP, Malik DJ, Pharmaceutics 2019, 11; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Vinner GK, Vladisavljević GT, Clokie MRJ, Malik DJ, PLoS One 2017, 12, e0186239; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sarhan WA, Azzazy HM, Nanomedicine (Lond) 2017, 12, 2055. [DOI] [PubMed] [Google Scholar]
- [14].Kalelkar PP, Riddick M, García AJ, Nature Reviews Materials 2021, DOI: 10.1038/s41578-021-00362-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Agarwal R, Johnson CT, Imhoff BR, Donlan RM, McCarty NA, García AJ, Nat. Biomed. Eng 2018, 2, 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Heesterbeek DAC, Martin NI, Velthuizen A, Duijst M, Ruyken M, Wubbolts R, Rooijakkers SHM, Bardoel BW, Sci. Rep 2019, 9, 3074; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Silhavy TJ, Kahne D, Walker S, Cold Spring Harb. Perspect. Biol 2010, 2, a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Chaurasiya B, Zhao Y-Y, Pharmaceutics 2020, 13, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Champion JA, Walker A, Mitragotri S, Pharm. Res 2008, 25, 1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Edwards DA, Hanes J, Caponetti G, Hrkach J, Ben-Jebria A, Eskew ML, Mintzes J, Deaver D, Lotan N, Langer R, Science 1997, 276, 1868. [DOI] [PubMed] [Google Scholar]
- [20].a) Beenken KE, Dunman PM, McAleese F, Macapagal D, Murphy E, Projan SJ, Blevins JS, Smeltzer MS, J. Bacteriol 2004, 186, 4665; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nishitani K, Sutipornpalangkul W, de Mesy Bentley KL, Varrone JJ, Bello-Irizarry SN, Ito H, Matsuda S, Kates SL, Daiss JL, Schwarz EM, J. Orthop. Res 2015, 33, 1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Trizna EY, Yarullina MN, Baidamshina DR, Mironova AV, Akhatova FS, Rozhina EV, Fakhrullin RF, Khabibrakhmanova AM, Kurbangalieva AR, Bogachev MI, Kayumov AR, Sci. Rep 2020, 10, 14849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Palmer GC, Whiteley M, Microbiol Spectr 2015, 3; [DOI] [PubMed] [Google Scholar]; b) Palmer KL, Mashburn LM, Singh PK, Whiteley M, J. Bacteriol 2005, 187, 5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M, Proc. Natl. Acad. Sci 2015, 112, 4110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Pabary R, Singh C, Morales S, Bush A, Alshafi K, Bilton D, Alton EW, Smithyman A, Davies JC, Antimicrob. Agents Chemother 2016, 60, 744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Morello E, Saussereau E, Maura D, Huerre M, Touqui L, Debarbieux L, PLoS One 2011, 6, e16963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang G, Li L, Wang X, Li X, Zhang Y, Yu J, Jiang J, You X, Xiong YQ, Acta Pharm Sin B 2019, 9, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Zabner J, Karp P, Seiler M, Phillips SL, Mitchell CJ, Saavedra M, Welsh M, Klingelhutz AJ, Am. J. Physiol. Lung Cell Mol. Physiol 2003, 284, L844; [DOI] [PubMed] [Google Scholar]; b) Molina SA, Stauffer B, Moriarty HK, Kim AH, McCarty NA, Koval M, Am. J. Physiol. Lung Cell Mol. Physiol 2015, 309, L475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Adriaenssens EM, Lehman SM, Vandersteegen K, Vandenheuvel D, Philippe DL, Cornelissen A, Clokie MR, García AJ, De Proft M, Maes M, Lavigne R, Virology 2012, 434, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rampersad SN, Sensors 2012, 12, 12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.