

Abstract
We report results from the Bipolar Exome (BipEx) collaboration analysis of whole exome sequencing of 13,933 bipolar disorder (BD) cases, matched with 14,422 controls. We find an excess of ultra-rare protein-truncating variants (PTVs) in BD patients among genes under strong evolutionary constraint in both major BD subtypes. We find enrichment of ultra-rare PTVs within genes implicated from a recent schizophrenia (SCZ) exome meta-analysis (SCHEMA; 24,248 cases, 97,322 controls) and among binding targets of CHD8. Genes implicated from GWAS of BD, however, are not significantly enriched for ultra-rare PTVs. Combining gene-level results with SCHEMA, AKAP11 emerges as a definitive risk gene (OR=7.06, P=2.83×10−9). At the protein level, AKAP-11 interacts with GSK3B, the hypothesized target of lithium, a primary treatment for BD. Our results lend support to BDs polygenicity, demonstrating a role for rare coding variation as a significant risk factor in BD etiology.
Introduction
Bipolar disorder (BD) is a heritable neuropsychiatric disorder characterized by episodes of mania and, oftentimes, episodes of depression. BD has a lifetime prevalence of 1–2% in the population, often with onset in early adulthood. BD is a chronic condition that affects individuals across their lifespan and is a significant source of disease burden worldwide1. Meta-analysis of 24 twin studies estimated broad-sense heritability of BD to be ~67%2, while recent molecular genetic analyses estimated the additive heritable component from common SNPs (minor allele frequency (MAF)>1%) to be between 17 and 23%3. This difference between twin-based heritability estimates of BD and additive heritability tagged by common SNPs indicates that a large fraction of genetic risk is still undiscovered. The discrepancy in variance explained likely originates from a variety of sources, including copy number variants (CNVs), heterogeneity in phenotype and diagnosis, and rare, often deleterious, genetic variants of more recent origin4,5. Each of these sources of variation are excluded from common variant based estimates of heritability.
Rare variation, including CNVs and PTVs, have been shown to influence risk for BD, albeit to a lesser degree than other neuropsychiatric illnesses such as schizophrenia and autism spectrum disorders (ASDs)6,7. Negative selection on BD would cause high penetrance alleles to be held at low frequency in the population8,9. In a large Swedish birth cohort, males and females with BD have a lower reproductive rate compared to their unaffected siblings (0.75:1 and 0.85:1, respectively)10. The reduction in offspring observed in BD, however, is markedly more modest than observed for individuals with schizophrenia (0.23 for males, 0.47 for females) or autism (0.25 for males, 0.48 for females), suggesting that the role of rare variation is likely to be smaller in magnitude, as selection is not acting as strongly on BD in aggregate. These conclusions are tempered by uncertainty about the consistency in reduced fecundity over human evolutionary history. Nevertheless, the interrogation of rare variation in BD patients will be pivotal in the discovery of variants with high penetrance for BD risk.
Within BD, two clinical subtypes are recognized: bipolar I disorder (BD1) and bipolar II disorder (BD2) (American Psychological Association (APA) DSM-IV11; World Health Organization (WHO) ICD-1012). BD1 diagnosis necessitates at least one manic episode, although at least one depressive episode is usually present. Psychotic symptoms may occur during the manic and/or depressive episodes. In contrast, a BD2 diagnosis requires at least one depressive episode and one hypomanic (but not manic) episode across the lifetime. In addition, the DSM-5 includes schizoaffective disorder bipolar type as a subtype of schizoaffective disorder. Patients with schizoaffective disorder exhibit psychotic symptoms concurrent with a major mood episode, and depressed mood. To be diagnosed with schizoaffective disorder bipolar type, a manic episode must constitute part of the presentation13–15. Despite distinct diagnostic categories, genetic susceptibility for BD from common SNPs has shown strong overlap with schizophrenia (genetic correlation rg=0.70) and major depressive disorder (MDD) (rg=0.35), with BD1 showing preferential overlap with schizophrenia and BD2 with MDD, reflecting a broad continuum of genetic influence on psychosis and mood disturbance3.
To date, GWAS meta-analysis of common SNPs have identified 64 independent loci that contribute to BD susceptibility, implicating genes encoding ion channels, neurotransmitter transporters, and synaptic and calcium signaling pathways3,5. Evidence of rare variation on BD risk, however, remains inconclusive as sample sizes are substantially smaller than GWAS. Analysis of large rare CNVs (MAF<1%) in 6,353 BD cases found CNV enrichment among schizoaffective disorder bipolar type over both controls and other BD diagnoses, suggesting that increased risk among detectable rare CNVs is restricted to individuals with psychotic symptoms6. Analysis of whole exome and genome sequencing of pedigree and case-control cohorts have shown only nominal enrichment among individual genes and candidate gene-sets16–19.
Here, we report results from the Bipolar Exome (BipEx) collaboration, the largest whole-exome study of BD to date at the time of analysis, comprising 13,933 BD cases and 14,422 controls following aggregation, sequencing, and quality control (QC).
Results
Curated exome sequence data generation
We combined BD case-control whole exome sequencing data from 13 sample collections in 6 countries. The aggregated dataset consists of 33,699 individuals, 16,486 of whom have been diagnosed with BD, and 17,213 who have no known psychiatric diagnosis (Supplementary Table 1). All sample collections have been previously genotyped for common variant analyses3. However, this is the first time that exome-sequencing and joint analysis has been performed on these collections. All exome sequencing data were generated using the same library preparation, sequencing platform, and joint calling pipeline (Methods). Following sequencing and joint calling, we ran a series of quality control steps to filter out low quality variants and samples (Supplementary Table 2–3), and restricted the dataset to unrelated individuals of broad continental European ancestry (Methods; Supplementary Note, Supplementary Figures 1–5). The analysis-ready dataset (Supplementary Table 4) consists of 13,933 bipolar cases (8,238 BD1; 3,446 BD2; 1,288 BD not otherwise specified (BDNOS, which includes disorders with bipolar features that do not meet criteria for any specific bipolar disorder11); 961 BD cases without a finer diagnosis), 277 schizoaffective disorder cases, and 14,422 controls. We exclude schizoaffective disorder cases to obtain more BD specific results and reduce signals more attributable to schizophrenia.
Significant contribution of rare damaging PTVs to BD risk
To test whether BD cases carry an excess of damaging coding variants, we analyzed exome-wide burden relative to controls using a logistic regression model controlling for principal components, sex, and overall coding variant burden (Methods). Drawing from previous exome sequencing studies of psychiatric disease18,20,21, we restricted our analysis to variants with minor allele count (MAC) ≤5 (MAF≈0.01%) across the entirety of the dataset. We annotated variants using the Ensembl Variant Effect Predictor (VEP)22 version 95, and assigned variants to four variant classes: two putatively damaging classes: protein-truncating variants (PTVs) and damaging missense variants, and two likely benign annotations: other missense, and synonymous variants (Methods, Supplementary Table 5). Following this initial restriction, we observed nominally significant enrichment of damaging missense variation in BD cases overall and BD2 cases over controls (OR=1.01, P=0.024 and OR=1.02, P=0.0086 respectively; Figure 1b,c), but not of PTVs. However, stepwise filtering of rare PTVs to those not in the non-neurological portion of the Genome Aggregation Database (gnomAD), hereafter referred to as ‘ultra-rare variants’, and then in constrained genes (defined as pLI≥0.9), showed that case-control PTV enrichment is present once we filter to high pLI genes, a finding in line with that from schizophrenia exomes23 (Figure 1b,c; Supplementary Figure 6). This enrichment is consistent across both BD1 and BD2 subtypes (Figure 1a). A conservative Bonferroni significance threshold (accounting for all analyses in Figure 1) was set at P=0.05/27 ≈ 0.0019. The magnitude of PTV enrichment in BD (OR=1.11, P=5.0×10−5) is considerably lower than in schizophrenia (OR=1.26; Singh et al.23), in line with the decreased selective pressure estimated from higher reproductive rates in BD affected siblings relative to schizophrenia affected siblings10.
Figure 1: Case-control enrichment of ultra-rare variants, split by case status and consequence category.
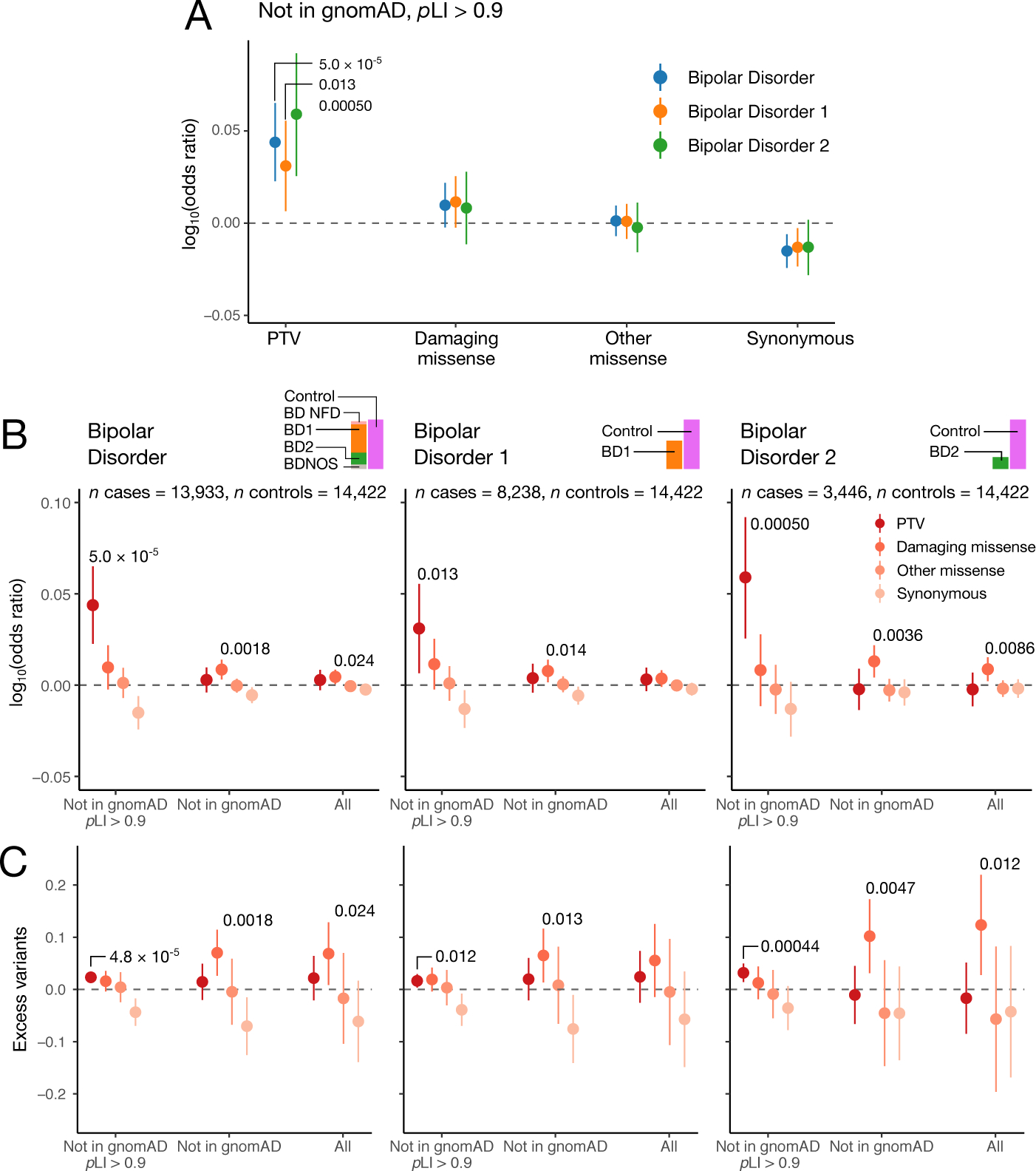
a) Enrichment in cases over controls (n=14,422) in case subsets (BD cases: n=13,933, BD1 cases: n=8,238, BD2 cases: n=3,446), according to the legend. The midpoint displays the logistic regression estimate. Bars show the 95% confidence intervals on the logistic regression estimate of the enrichment of the class of variation labelled on the x-axis. b,c) Case-control enrichment and excess case rare variant burden in increasingly a priori damaging variant subsets using logistic and linear regression, respectively. Consequence categories are stratified by rarity: moving from left to right the putatively damaging nature of the variants reduces from dark red to pink according to the legend, and the rarity reduces from a variant with MAC≤5 in a pLI≥0.9 gene and not in the non-neurological portion of gnomAD (Not in gnomAD pLI≥0.9), to a variant with MAC≤5 (All) according to the x-axis labelling. In (b), midpoints and bars display the logistic regression estimates, and associated 95% confidence intervals of the enrichment of the class of variation labelled on the x-axis. In (c), midpoints and bars show the linear regression estimates on excess variants in cases, and associated 95% confidence intervals for the class of variation labelled on the x-axis. Regressions are run as described in Methods: exome-wide burden, and include sex, 10 principal components and total coding burden with the same rarity as covariates. Nominally significant enrichments or excess variants in cases are labelled with the unadjusted associated two-sided P-value computed using a Wald test.
To attempt to refine the nominally significant damaging missense signal, we sought to further distinguish likely deleterious missense variants from benign missense variants. We annotated variants with a missense deleteriousness predictor which takes into account regional missense constraint: “Missense badness, PolyPhen-2, and regional Constraint score” (MPC)24, and identified a highly deleterious subset of missense variants (MPC≥2), as recommended24. However, upon restriction to this subset of missense variants, we did not observe a significant burden of enrichment at either of the three levels of filtering (MAC≤5, ultra-rare, or ultra-rare in a pLI≥0.9 gene) for either BD1, BD2 or BD (Supplementary Figure 7). This is likely because the MPC≥2 group accounts for a small proportion of missense variants (Methods).
To try to tease apart the signal of excess ultra-rare PTVs in BD cases over controls, we examined whether age of first impairment or presence of psychosis stratified ultra-rare PTV burden (Methods). We found no difference in the distribution of ultra-rare PTV burden or carrier status between earlier onset cases compared to older onset cases (Supplementary Table 6, n=3,134, minimum P-value across 50 Kolmogorov-Smirnov tests was 0.40, minimum P-value across 50 Fisher’s exact tests was 0.067). BD cases with and without psychosis (Supplementary Table 7) displayed significant enrichment of ultra-rare PTV burden in constrained genes (n=4,214, OR=1.12, P=0.0018; n=3,803, OR=1.16, P=6.6×10−5, respectively). There was no significant difference in excess ultra-rare PTV burden between individuals with and without psychosis: a logistic regression of ultra-rare PTV burden in constrained genes on psychosis status was not significant when controlling for BD case status (P=0.42).
Restricting to missense variants, we do not observe a significant signal of enrichment of ultra-rare MPC≥2 variation in BD cases, in contrast to schizophrenia23 (Supplementary Figure 7). However, we did observe nominally significant enrichment of ultra-rare damaging missense variation across both BD subtypes when not filtering to loss of function intolerant genes (pLI≥0.9); (Figure 1b,c; BD: OR=1.02, P=0.0018; BD1: OR=1.02, P=0.014; BD2: OR=1.03, P=0.0036).
Ultra-rare variant burden in tissues and candidate gene-sets
Biologically and empirically informed gene-sets can refine our understanding of how ultra-rare PTVs confer risk for BD and generate potential biological hypotheses for follow-up analyses. Using the Genotype-Tissue Expression (GTEx) portal25, we found weak evidence for enrichment of ultra-rare PTVs in 13,372 genes expressed in brain tissues in BD cases (OR=1.01, P=0.032), but not in genes expressed in non-brain tissues (23,450 genes, OR=1.00, P=0.15). To examine tissue-specific enrichment more broadly, we tested for enrichment of ultra-rare PTVs in 43 GTEx tissues (Finucane et al.26, Supplementary Table 8) in tissue specific expression gene-sets (Figure 2a; Supplementary Figure 8, Methods). Enrichment of damaging ultra-rare variation resides predominantly in brain tissues, with the strongest association in the Amygdala (OR=1.03, P=3.9×10−5), a brain region found to be reduced in size in BD1 cases27.
Figure 2: Biological insights from bipolar case-control whole-exome sequencing data.
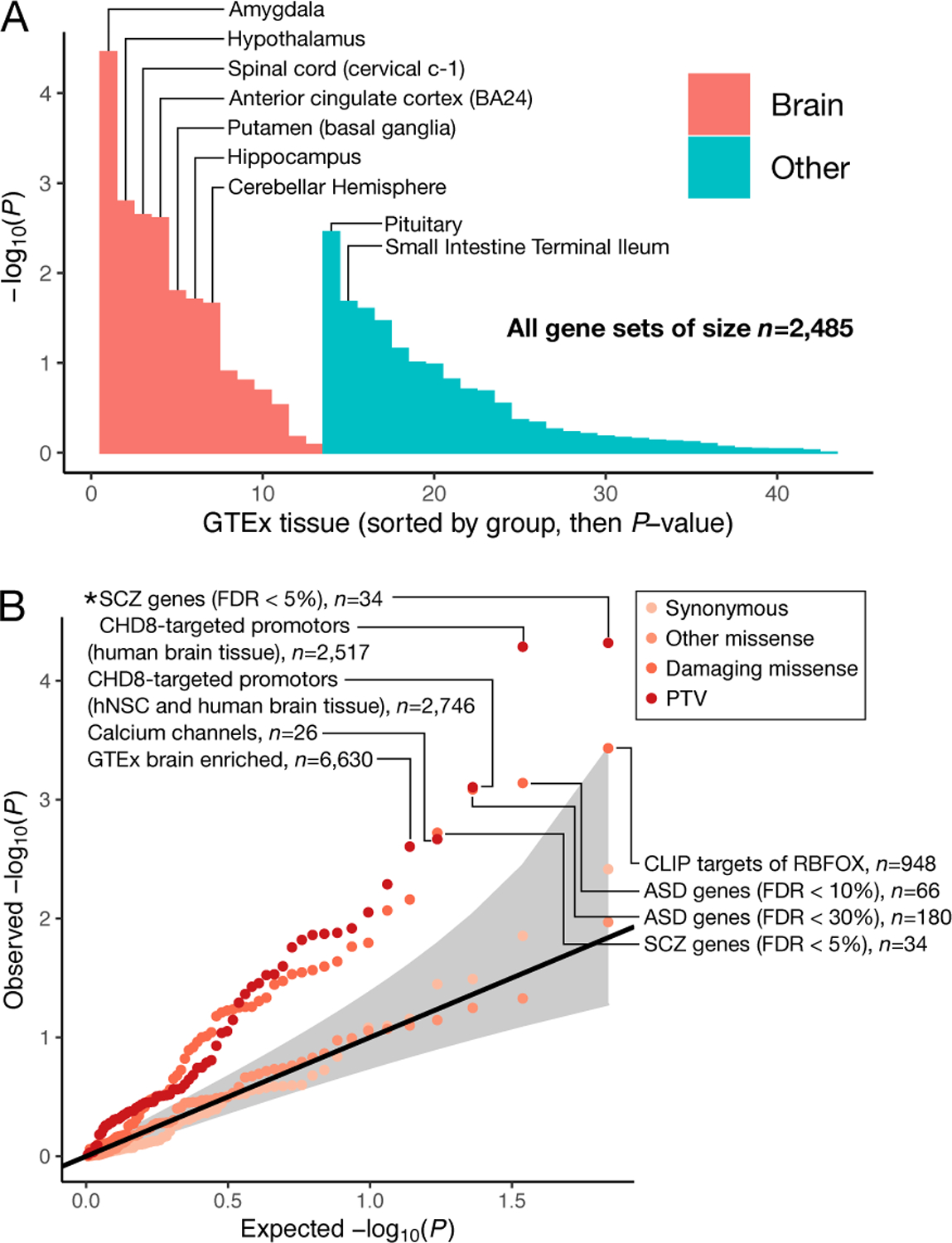
a) Enrichment of ultra-rare PTVs in BD cases over controls in tissue-specific expression gene-sets. We run logistic regressions of case status on ultra-rare PTV burden in tissue-specific expression gene-sets. Logistic regressions were performed as described in the Supplementary Note. Two-sided P-values were obtained via Wald tests. Gene-sets are defined in Finucane et al. 26 in detail. Bars are ordered by P-value, first for brain tissue and then for other tissues. No nominally significant association was enriched in controls over BD cases. b) Enrichment of ultra-rare variants in targeted 68 gene-sets taken from the literature23,43. We run logistic regressions of case status on ultra-rare variant burden in classes of variation labelled in the legend, and display the expected against observed two-sided unadjusted −log(P-values). Logistic regressions were performed as described in Methods. Two-sided P-values were obtained via Wald tests. Top PTV and damaging missense gene-sets are labelled, and annotated with the number of genes in each gene-set. The 95% confidence interval under the null, is shown in grey. Classes of variants tested in each gene-set are colored according to the legend. Gene-sets surpassing Bonferroni test correction are labelled with an asterisk. hNSC, human neuronal stem cells.
We considered 68 candidate gene-sets generated or implicated in previous genetic studies of psychiatric disorders (Figure 2b; Supplementary Figure 9), and a stricter definition of highly brain expressed: average expression over two-fold higher in brain tissues than the average across all GTEx28 tissues. Using this more stringent definition (6,630 genes), we saw stronger ultra-rare PTV enrichment in BD cases (OR=1.04, P=2.49×10−3). Among the 68 candidate gene-sets, we observed significant enrichment (P<0.05/68 ≈ 3.68×10−4) of ultra-rare PTV variation in two gene-sets in BD cases: SCHEMA genes; FDR < 5%23 (34 genes, OR=1.89, P=4.81×10−5), and CHD8 binding targets in human brain29 (2,517 genes, OR=1.09, P=5.18×10−5). For ultra-rare damaging missense variants, the strongest gene-set enrichment was in genes targeted by RBFOX30 (948 genes, OR=1.07, P=3.70×10−4), and ASD FDR<10%31 (66 genes, OR=1.24, P=7.25×10−4), though neither passes multiple testing correction.
Enrichment of ultra-rare PTVs in SCHEMA and damaging missense variants in ASD provides further evidence of convergence of shared signal across psychiatric and neurodevelopmental disorders in the ultra-rare end of the allele frequency spectrum, mirroring the overlapping genetic risk for schizophrenia and BD observed in common variation32, and schizophrenia and ASD in rare variation23.
We did not observe rare-variant enrichment of damaging variation in gene-sets generated from a GWAS of BD of 20,352 cases and 31,358 controls3. However, we do see a nominally significant (OR=1.69, P=0.00215) signal of enrichment of ultra-rare PTVs in calcium channel genes (26 genes), in line with significant common variant signals of enrichment in targets of calcium channel blockers determined from BD GWAS5.
To investigate the overlapping rare-variant signal with schizophrenia further, we considered four distinct gene-sets, each with 50 genes, ordered by P-value in SCHEMA23. We observed ultra-rare PTV enrichment in the top 50 genes, which include the FDR < 5% set (OR=2.05, P=1.25×10−8), but not in the less significant genes in SCHEMA (genes 51–100; OR=1.01, P=0.932, genes 101–150; OR=1.07, P=0.481, genes 151–200; OR=1.06, P=0.703). We also did not observe enrichment of ultra-rare PTVs in the recently fine-mapped schizophrenia genes published by the Psychiatric Genetics Consortium33 (OR=0.867, P=0.192).
To seek to elucidate pathways enriched for damaging variation in BD cases in an agnostic manner we performed an enrichment analysis using gene-sets derived from large pathway databases including Gene Ontology, REACTOME and KEGG (1,697 gene-sets, Supplementary Figure 10). We observed significant (P<0.05/1697 ≈ 2.95×10−5) enrichment of one gene-set: genes involved in the G1/S transition of the mitotic cell cycle (172 genes; OR=1.46, P=1.37×10−5).
AKAP11 implicated by ultra-rare protein truncating variants
To boost power for gene discovery, we again restricted to ultra-rare variants and tested for enrichment of putatively damaging classes of variation: PTVs and damaging missense variants (Methods; Supplementary Figures 11–15). Enrichment in constrained genes remains significant after excluding the top 20 BD-risk associated genes in BipEx with pLI≥0.9 (OR=1.07; P=0.00313, Supplementary Table 9).
In our primary analysis, no gene surpassed exome-wide significance (P<0.05/23,321 ≈ 2.14×10−6, Figure 3). However, we begin to observe deviation from the null in the collection of tests of ultra-rare PTV enrichment in BD cases, particularly in BD1 (Figure 3, Supplementary Figure 16). This deviation was not observed for BD2 (Supplementary Figure 17) despite the genome-wide enrichment of the PTV signal (Figure 1b,c), likely due to the reduced power of Fisher’s exact tests in BD2 case counts (n=3,446). The strongest case-control association we observed was with AKAP11 (P=1.15×10−5, Q=2.02×10−2 in BD, P=5.30×10−6, Q=5.77×10−3 in BD1).
Figure 3: Results of the analysis of ultra-rare PTVs in 13,933 cases and 14,422 controls. Gene-based Manhattan and QQ plot for BD (comprising BD1, BD2 and BDNOS).
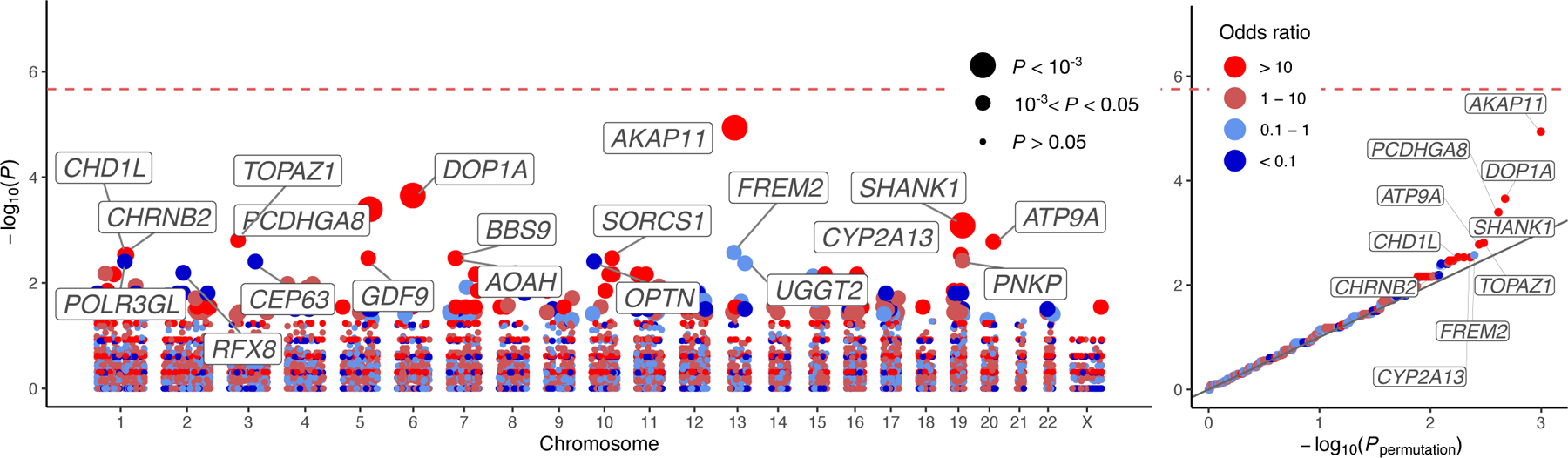
−log10 P-values obtained via two-sided Fisher’s exact tests are plotted against genetic position for each of the analyzed genes. In the QQ plots, observed −log10 P-values are plotted against permutation P-values according to the procedure described in the Methods. Points are colored according to the discrete scale displayed in the legend. In the Manhattan plot and QQ plot, the gene symbols of the top 20 and top ten genes by P-value are labelled, respectively. Points in the Manhattan plot are sized according to P-value as displayed in the legend.
Given the strong overlap in common variant risk between BD and schizophrenia, we looked for a shared signal of enrichment of ultra-rare PTVs in BD and schizophrenia cases. Due to overlap in controls between SCHEMA and BipEx, we meta-analyzed an ultra-rare variant count data which excluded these controls in SCHEMA (Methods). To avoid the schizophrenia ultra-rare PTV case-control enrichment signal overwhelming the BD signal when presenting results, we first sorted on P-value in the primary gene-based BD analysis and displayed the top ten P-values before and after meta-analysis with SCHEMA (Table 1 and Supplementary Table 10). The combined analysis in BD and schizophrenia cases reveals one exome-wide significant gene, AKAP11 (P=2.83×10−9), and one gene which almost attains exome-wide significance, ATP9A (P=5.36×10−6).
Table 1:
BipEx and SCHEMA case-control counts of the top ten most significant genes in the BipEx gene-based analysis. Case and control columns denote the count of ultra-rare PTVs in the gene in the respective dataset. Two-sided P-values are determined using Fisher’s exact and CMH tests for BipEx and SCHEMA (Methods) respectively, and meta-analyzed weighting by effective sample size. Two-sided Q-values for Fisher’s exact test statistics in BipEx were evaluated using the Benjamini and Hochberg adjustment44 applied to all genes with at least 10 ultra-rare PTVs across cases and controls. BipEx: BD case count 13,933, control count 14,422. SCHEMA: schizophrenia case count 24,248, control count 91,960. The SCHEMA Odds ratio (OR) is the estimated OR averaged over strata, whereas the combined OR is the simple OR calculated by combining the BipEx and SCHEMA cases and controls. Note that differential coverage across exome sequencing platforms and whole genome sequencing means that case/control counts differ across genes.
| Gene | BD (BipEx) | SCZ (SCHEMA) | Combined | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Case count n = 13,933 |
Control count n = 14,422 |
P-value | Q-value | OR | Case count n = 24,248 |
Control count n = 91,960 |
P-value | OR | OR | Meta P-value |
|
|
| |||||||||||
| AKAP11 | 16 | 0 | 1.15 × 10−5 | 2.02 × 10−2 | ∞ | 17 | 13 | 2.02 × 10−5 | 5.60 | 7.06 | 2.83 × 10−9 |
|
| |||||||||||
| DOP1A | 15 | 1 | 2.22 × 10−4 | 1.95 × 10−2 | 15.54 | 19 | 43 | 1.47 × 10−1 | 1.59 | 2.11 | 1.44 × 10−4 |
|
| |||||||||||
| PCDHGA8 | 11 | 0 | 4.02 × 10−4 | 2.36 × 10−1 | ∞ | 6 | 44 | 2.19 × 10−1 | 0.54 | 0.99 | 3.38 × 10−3 |
|
| |||||||||||
| SHANK1 | 10 | 0 | 8.19 × 10−4 | 3.60 × 10−1 | ∞ | 4 | 4 | 4.43 × 10−1 | 2.90 | 6.99 | 9.71 × 10−3 |
|
| |||||||||||
| TOPAZ1 | 12 | 1 | 1.56 × 10−3 | 5.48 × 10−1 | 12.43 | 2 | 3 | 6.67 × 10−1 | 0.93 | 3.93 | 2.51 × 10−3 |
|
| |||||||||||
| ATP9A | 9 | 0 | 1.66 × 10−3 | - | ∞ | 15 | 11 | 6.96 × 10−4 | 4.08 | 5.46 | 5.36 × 10−6 |
|
| |||||||||||
| FREM2 | 4 | 19 | 2.67 × 10−3 | 5.77 × 10−1 | 0.22 | 22 | 92 | 5.48 × 10−1 | 0.83 | 0.65 | 3.80 × 10−2 |
|
| |||||||||||
| CHD1L | 11 | 1 | 2.95 × 10−3 | 5.77 × 10−1 | 11.39 | 16 | 73 | 5.99 × 10−1 | 0.82 | 1.01 | 4.57 × 10−2 |
|
| |||||||||||
| CHRNB2 | 11 | 1 | 2.95 × 10−3 | 5.77 × 10−1 | 11.39 | 2 | 17 | 5.54 × 10−1 | 0.52 | 1.88 | 3.04 × 10−2 |
|
| |||||||||||
| CYP2A13 | 11 | 1 | 2.95 × 10−3 | 6.68 × 10−1 | 11.39 | 13 | 28 | 6.30 × 10−1 | 1.29 | 2.27 | 4.61 × 10−2 |
The top gene hit, AKAP11 (the gene encoding A-Kinase Anchoring Protein 11 (AKAP-11, also known as AKAP220)) has only a single isoform, is under evolutionary constraint (LOEUF=0.3, pLI=0.98), and is highly expressed in the brain (cerebellar hemisphere: 38.54 median transcript per million (TPM); frontal cortex (BA9): 31.52 median TPM 25). Additionally, AKAP-11 has been shown to interact with GSK3B, the hypothesized target of lithium therapy34–36. We gathered available lithium response data for carriers of AKAP11 PTVs among BD cases: of the eleven cases for which lithium response information (Methods) was available, seven reported a good response and four did not respond well to lithium. While the percent of good responders in AKAP11 PTV carriers (63.6%) is marginally elevated relative to the background response rate in available BD cases (52%), the sample size is far too small to form any robust conclusions from the data.
To our knowledge and at the time of analysis, there is no signal of enrichment in AKAP11 in other neurodevelopmental disorders at current sample sizes: AKAP11 does not appear to be a prominent risk gene for autism37,38 or epilepsy39 and is not present in a collection of curated ‘developmental disorder genes’40. Furthermore, expression of AKAP11 tends to occur later in development (Supplementary Figure 18).
We examined ultra-rare PTV variant counts in the Bipolar Sequencing Consortium (BSC)18 exome sequencing data (Methods, Supplementary Table 11). Non-zero count data were available for seven of the top ten gene associations (Table 1). One, enriched for ultra-rare PTVs in controls (FREM2) in BipEx, did not display control enrichment in the BSC data. The remaining six displayed case enrichment in BipEx, with four (including AKAP11 and ATP9A), displaying further case enrichment in the BSC data (Supplementary Table 12).
Discussion
In this large BD exome study, ultra-rare PTVs in constrained genes are significantly enriched in BD cases. In fact, enrichment in constrained genes remains significant even after excluding the top 20 BD-risk associated genes (OR=1.07; P=0.00313) with pLI≥0.9 (Supplementary Table 9). This reflects the highly polygenic genetic architecture of BD, a property shared with schizophrenia23, and suggests that the majority of genes involved in BD risk will require larger sample sizes to be discovered. Furthermore, in BD cases, ultra-rare PTVs are significantly enriched in schizophrenia risk genes identified in the SCHEMA consortium, suggesting that rare variation in these genes is not specific to schizophrenia pathophysiology: overlap in risk for schizophrenia and BD is now evident in both rare and common variation. Finally, combining our results with data from SCHEMA reveals strong evidence that haploinsufficiency in AKAP11 confers risk for both BD and schizophrenia, but this does not appear to be the case for early-onset neurodevelopmental disorders.
AKAP11 codes for the AKAP-11 protein (also known as AKAP220), one of a family of scaffolding proteins that bind to the regulatory subunit of the protein kinase A (PKA). These anchoring proteins confine PKA to discrete locations in the cell to target specific substrates for phosphorylation and dephosphorylation. In particular, GSK3B is bound by AKAP-11. GSK3B is hypothesized to be the target of lithium, the primary treatment for BD41. By binding to GSK3B, AKAP-11 mediates PKA-dependent inhibition of GSK3B. PKA inhibits the activity of GSK3B bound to AKAP-11 more strongly than GSK3B in general, and thus modifications to AKAP-11 have the potential to affect downstream pathways. GSK3B is one of two paralogous genes (GSK3A and GSK3B) that encode a serine/threonine protein kinase, glycogen synthase kinase 3. The primary known function of this protein is phosphorylation of over one hundred substrates, affecting myriads of signalling pathways6,41,42.
We see early evidence of enrichment in ultra-rare damaging missense variation, particularly within BD2. This enrichment is evident outside of missense constrained regions (as defined by MPC≥2): perhaps surprising given the signal of association seen for rare (MAF≈2⨉10−5) missense variation in schizophrenia cases is mainly within constrained missense regions (MPC≥2)23. Because BD2 displays a stronger correlation of common variant effects with major depression than BD1, and BD1 is more correlated with schizophrenia than BD2, this missense signal may be capturing something distinct to mood disorders relative to psychotic disorders. However, we should be cautious not to overinterpret differences in ultra-rare damaging missense enrichment across BD subtypes; BD2 sample count (n=3,446) is less than half that of BD1 (n=8,238), and confidence intervals overlap (Figure 1). Furthermore, attempts to refine this exome-wide signal to individual genes or targeted gene-sets did not result in any significant signals of association after correcting for multiple testing (Supplementary Figure 9,17). We expect to see a refinement of the putatively damaging missense signal as sample sizes increase.
Despite sequencing 13,933 BD cases, we did not observe any BD specific risk genes surpassing exome-wide significance. In contrast, the 24,248 schizophrenia cases analyzed in SCHEMA yielded ten significant risk genes. When comparing the observed ultra-rare PTV enrichment among constrained genes in our current sample (OR=1.11) to SCHEMA (OR=1.26), we estimate that roughly double the case sample size of schizophrenia is needed to achieve comparable statistical power to discover individual BD risk genes. We now see convergence of gene overlap for schizophrenia from the common and rare end of the allele frequency spectrum, in large part through increased exome sample sizes23. Genetic overlap between common and rare variation in BD, however, remains uncertain. The BSC examined 3,987 BD case exomes18, and found suggestive enrichment in 165 genes implicated in BD GWAS (OR=1.9, P=6.0×10−4), but we did not replicate this finding (OR=0.9, P=0.40). Prior to SCHEMA, convergence of genes implicated in schizophrenia by common and rare variation was modest20,21,43. As BD sample sizes increase in both common and rare variation analyses, we expect a similar convergence of genes implicated in BD.
In summary, ultra-rare PTVs in constrained genes are significantly enriched in BD patients over controls, a result firmly established in schizophrenia and other early-onset neurodevelopmental disorders. We are beginning to see promising signals among individual genes, despite none surpassing exome-wide significance for BD alone. We observe that shared risk for BD and schizophrenia is present in both common and damaging ultra-rare variation. Our top gene, AKAP11, shows shared evidence of risk for BD and schizophrenia, increasing our confidence that we are discovering true risk factors underlying psychiatric disease. Overall, the current evidence suggests gene discovery in BD is on a similar trajectory to schizophrenia, where increased sample sizes and further collaborative efforts will inevitably lead to biologically meaningful risk genes and pathways underlying BD risk.
Methods
Sequence data production
Exome sequencing was performed at the Broad Institute of Harvard and MIT from July 2017 to September 2018. Processing included sample QC using the picogreen assay to measure for sample volume, concentration and DNA yield. Sample library preparation was carried out using Illumina Nextera, followed by hybrid capture using Illumina rapid capture enrichment of a 37Mb target. Sequencing was performed on HiSeqX instruments to 150bp paired reads. Sample identification checking was carried out to confirm all samples. Sequencing was run until hybrid selection libraries met or exceeded 85% of targets at 20x, comparable to ~55x mean coverage. Data delivery per sample includes a demultiplexed, aggregated into a BAM file and processed through a pipeline based on the Picard 2.19 suite of software tools. The BWA aligner mapped reads onto the human genome build 38 (GRCh38). Single nucleotide polymorphism and insertions/deletions were joint called across all samples using Genome Analysis Toolkit (GATK)45 HaplotypeCaller package version 4.0.10 to produce a version 4.2 variant callset file (VCF). Variant call accuracy was estimated using the GATK Variant Quality Score Recalibration (VQSR) approach46.
Quality control
We perform a series of hard-filters on genotype and variant metrics (Supplementary Table 2), followed by a collection of hard-filters on sample metrics (Supplementary Table 3). We confirm genotype sex with reported sex, remove related individuals, and restrict analysis to samples of continental European ancestry where we have sufficient sample size and balanced case-control counts (Supplementary Table 3), using random forest classifiers. Finally, we filter based on a second collection of sample and variant hard-filters (Supplementary Tables 2–3). Final curated sample counts, split by cohort are provided in Supplementary Table 4. We used Hail 0.2 and PLINK 1.9 to perform all quality control steps, in combination with R (4.0.2) scripts for data filtering and plotting. Data was manipulated in R using data.table (1.13.0) and dplyr (1.0.1), random forest classifiers were trained using the randomForest (4.6) library, and plotting was performed using a ggplot2 (3.3.2) and add-on packages ggsci (2.9) and ggExtra (0.9). Full details are provided in the Supplementary Note.
Variant annotation
We use the Ensembl Variant Effect Predictor (VEP)22 version 95 with the loftee plugin to annotate variants against GRCh38 using hail, including SIFT 47 and Polyphen2 scores 48, according to the GENCODE v19 reference. The configuration file is available in google cloud: gs://hail-us-vep/vep95-GRCh38-loftee-gcloud.json. In addition, we annotate with version 2.1.1 gnomAD site annotations49 and MPC scores24 after lifting the genome coordinates over to GRCh38. MPC is an aggregate score which uses ExAC to identify sub-genic regions that are depleted of missense variation in combination with existing metrics to create a composite predictor. Finally, we annotate with Combined Annotation Dependent Depletion (CADD) version 1.424,50, and annotate constraint using the gnomAD loss of function (LOF) metrics table from release 2.1.149. We then process the VEP annotated consequences, and define variant-specific consequences and gene annotations as the most severe consequence of a canonical transcript on which that variant lies. We then assign variants (where possible) to four distinct consequence classes: PTV, missense, synonymous, and non-coding as defined in Supplementary Table 5. We then subdivide missense variants into ‘damaging missense’ if both the Polyphen2 prediction is ‘probably damaging’ and the SIFT prediction is ‘deleterious’, and ‘other missense’ otherwise. Note that the MPC≥2 group accounts for a small proportion of the total damaging and benign missense variants annotated by PolyPhen-2 and SIFT (e.g. the number of MPC≥2 variants in BipEx following the increasingly stringent filters (MAC ≤ 5, ultra-rare, or ultra-rare and in a pLI ≥ 0.9 gene) are 39,000, 23,000 and 5,000 respectively, compared to 360,000, 159,000 and 31,000 for damaging missense variants).
Exome-wide burden
We ran a series of logistic regressions to test for an association between putatively damaging rare variation and case status, and linear regressions to test for an association between case status and excess burden of damaging variation using base-R and logistf (1.23) in R (4.0.2). Both tests will result in near identical P-values; the motivation here is to ascertain two effect size parameters of rare-variant burden. We then sought to focus on more recent mutations by restricting to rare variation not present in the non-neurological portion of the gnomAD database, and perform the same collection of association tests. Furthermore, we leveraged evolutionary constraint models to enrich for deleterious variation by testing for enrichment of missense variation with MPC≥2 (representing the top ~3.9% pathogenicity of missense variation 24, and restricting our PTV enrichment tests within genes most likely to be loss-of-function intolerant (pLI≥0.9).
Throughout, we test for a signal of enrichment of synonymous, and other-missense as a negative control to confirm that our burden model was well calibrated. For each collection of regressions, we include sex, ten principal components (PCs) and overall burden of MAC≤5 variants in the dataset following the imposed restrictions. All regressions were robust to incorporation of the overall burden covariate: the overall observed patterns did not change if we controlled for overall coding burden or did not control for overall burden.
To ensure that the results in the full dataset were not driven by artefacts introduced by jointly analyzing multiple cohorts or residual population structure, we ran burden tests within each location and meta-analyzed these results. We observed consistent results across the cohorts, and estimated odds ratios and excess burden between the joint analysis and meta-analysis were roughly equivalent.
Multiple studies have shown enrichment of rare damaging coding variation in SCZ cases over controls20,21. As a positive control, we considered the SCZ cases in BipEx and tested for enrichment of ultra-rare PTVs in loss of function intolerant (pLI≥0.9) genes and replicated this result (OR=1.28, P=1.9×10−10).
Age of onset
Three definitions for age of onset were available for subsets of the data: age at first symptoms, age at first diagnosis, and age at first impairment. In each case, two distinct age encodings were used: <18; 18–40; 40+, and <12; 12–24; 24+. Onset definitions were defined differently dependent on cohort and included data from clinical instruments, algorithms using clinician notes, and telephone interviews with patients. Detailed age of onset information split by cohort is provided in the Supplementary Note.
To test for an association between age of onset and burden of rare damaging variation, we first restricted to the class of variation with the strongest signal for excess in cases over controls: PTVs. We considered only ‘age at first impairment’ (Supplementary Table 6) as phenotypic data using this definition was available for the largest number of samples: 3,677. We further split the age of first impairment categories into five discrete age bins across the two age encodings: <12, 12–18, 18–24, 24–40 and >40. We tested all ten possible ‘younger bin’ vs ‘older bin’ pairs across this partition to check for differences in MAC≤5 PTV burden, MAC≤5 not in gnomAD PTV burden, and MAC≤5 not in non-neurological gnomAD PTV in pLI≥0.9 burden, using Kolmogorov-Smirnov tests. We also used Fisher’s exact tests to test for an association between carrier status for the damaging rare PTV categories between the ‘younger’ and ‘older’ bins. All tests were performed in R (4.0.2).
Psychosis definitions
Psychosis was defined by a lifetime history of hallucinations or delusions. Presence of psychosis was evaluated differently across cohorts based on available data:
Boston, USA: validated Natural language processing-based algorithm run on clinical notes51,52.
Cardiff, UK: SCAN interview53 and case records. Definite evidence of lifetime presence of psychotic symptoms and lifetime presence of individual OPCRIT 53,54 psychotic symptoms.
London, UK: OPCRIT53,54 interview: lifetime presence of psychotic symptoms as defined by questions 52, 54, 55, 57–77 of the OPCRIT checklist detailed in the DNA polymorphisms in mental illness (DPIM) bipolar affective disorder (BPAD) questionnaire.
Stockholm, SWE: SWEBIC (Swedish Bipolar Cohort Collection);
SBP: Affective Disorder Evaluation (ADE) question: any psychotic disorder?
BipoläR and HDR: during a structured telephone interview that research nurses conducted, “have you ever lost touch with reality (i.e. have heard or seen things that others have not seen) or experienced things that you later realized were not real?” was asked. Patients were defined as having psychosis if the answer to this question was clear-cut ‘yes’, and not having psychosis if doubtful.
Gene-set burden testing
For each gene-set, we tested for ultra-rare variant enrichment of the following classes of variation: PTV, damaging missense, other missense, and synonymous. For each gene-set, we regressed case status on ultra-rare burden of each variant class in that gene-set using logistic regression, including ultra-rare coding burden in the gene-set (sum of ultra-rare burden of PTVs, damaging missense, other missense, and synonymous variants in the gene-set), sex, and PCs 1–10 as covariates. The resulting logistic regression performed for each (gene-set, variant class) pair is then: case status ∽ burdeng,c + burdeng,coding + sex + PC1+ PC2 + … + PC10, where burdeng,c is the count of ultra-rare variants of variant class c in gene-set g for the sample, and burdeng,coding is the total number of ultra-rare coding variants in the gene-set for the sample.
GTEx tissue specific gene-sets are defined in Finucane et al.26. Briefly, t-statistics for specific expression in each of the focal tissues were determined for each gene; these were then ranked, and the top 10% of t-statistics defined the collection of genes ‘specifically expressed’ in that tissue. For full details see Supplementary Note.
Gene-based analysis
In order to increase power for gene discovery, we filter down to variants not present in the non-neurological portion of the gnomAD dataset49, and we further enriched for pathogenic variants by restricting our analysis to variants with MAC≤5. We then examine case-control enrichment of PTVs or damaging missense variants (missense variants annotated as ‘probably damaging’ in PolyPhen and ‘deleterious’ in SIFT). We further restricted our analysis to the coding exons within the target intervals of the Illumina capture, to reduce potential for artefacts which could potentially be induced due to differential coverage across batches in any padded target interval, using synonymous, and other missense ultra-rare variants in each gene as the negative control (Supplementary Figure 11–12).
Throughout, we use Fisher’s exact tests in each gene. We considered a Cochran–Mantel–Haenszel (CMH) test, using the strata defined by broad geographic location (Supplementary Table 13). We use a permutation approach to determine the null distribution of test statistics throughout our gene-based analysis, and evaluate QQ plots of synonymous and other-missense ultra-rare variants to ensure that tests are well-calibrated (Supplementary Figure 11–12). We used Fisher’s exact tests in our primary analysis, as tests showed the strongest power and also had well calibrated QQ plots across annotation categories (Supplementary Figure 11–15). To determine Q-values we apply the Benjamini and Hochberg adjustment44 to Fisher’s exact test P-values for genes with at least 10 ultra-rare PTVs across cases and controls. We exclude genes with fewer than ten ultra-rare PTVs in the BipEx dataset to guard against incorrect P-value adjustment using the Benjamini and Hochberg procedure. Conservative Q-values occur when applying the Benjamini and Hochberg correction to discrete test statistics with low counts, due to the null distribution of P-values not following a uniform distribution under the null.
We tested for an excess of ultra-rare variation (MAC≤5 and not present in the non-neurological portion of the gnomAD dataset) in each gene using both Fisher’s exact and CMH tests for each phenotype. For each gene, each sample was assessed for carrier status for each of the following consequence classes: synonymous, other missense, damaging missense, and PTV (Supplementary Table 5); individuals harboring at least one copy in the consequence class under analysis were counted as carriers. These counts were then taken through to define 2×2 and 2×2×6 contingency tables for Fisher’s exact and CMH tests respectively, using location as strata (Supplementary Table 13). To ensure that our tests were well calibrated, we randomly permuted case labels (within stratum for CMH) for each gene and reran the test 20 times across all genes and keep track of the summation of the ordered vectors of P-values up to that permutation, before taking an average at the last permutation. This vector of length |n genes| then defines our expected distribution of P-values. Fisher’s exact test P-values and odds-ratio for carrier status are displayed in the gene results tables on the browser: bipex.broadinstitute.org.
To ensure that our tests were robust, we performed a series of checks to see if the Fisher’s exact (Supplementary Figures 11–12) and CMH test results showed an elevated false positive rate. In both tests, we observed the expected null P-value distribution in the collection of gene-based tests when analyzing synonymous and ‘other-missense’ variants with MAC≤5 not in gnomAD non-neurological. To further test calibration of the test statistic, we filtered to genes where we are well powered to detect differences between BD cases and controls. We examined case-control enrichment of synonymous ultra-rare variants in genes with an allele count of >20 and >50 and compared observed P-value to the uniform expectation (Supplementary Figure 15). In each, we did not observe inflation of the test statistic. All tests were performed in R (4.0.2).
Meta-analyzing SCHEMA and BipEx
To examine the extent of shared ultra-rare PTV signal between BD and SCZ we ran separate Fisher and CMH tests for BipEx and SCHEMA separately and meta-analyzed the results using weighted Z-scores, weighing by effective sample sizes. Fisher’s exact and CMH two-sided P-values were halved and converted to signed Z-scores using the OR to define the sign. Weighted Z-score were then evaluated:
where , and is the case proportion in the ith cohort. Associated P-values were then evaluated. As the UK and Ireland controls were present in BipEx and SCHEMA, these controls were excluded from the SCHEMA Z-score in the meta-analysis.
Lithium response
BipoläR and HDR subsets of the SWEBIC (Swedish Bipolar Cohort Collection), SWE: during a structured telephone interview that research nurses conducted, patients prescribed lithium for at least 12 months were asked: “What do you think of the effect (of lithium)? Do not consider side effects.” Patients were partitioned according to the following response options:
-Non-responder ‘None or very doubtful effect’
-Partial-responder ‘Doubtless effect of treatment but additional temporary or continuous treatment needed’
-Good-responder ‘Complete response, recovered’.
Cardiff, UK: patients were partitioned according to the following criteria:
-No evidence of response
-Subjective good response - upon interview, patients reported that lithium helped stabilise their moods
-Objective evidence for beneficial response, i.e., clear reduction in number and/or severity of episodes following introduction of lithium prophylaxis (can only be rated if at least 3 episodes of illness have occurred before lithium prophylaxis and lithium response has been observed for at least 3 years)
-Objective evidence for excellent response to lithium prophylaxis, i.e., frequency of episodes reduced to <10% of frequency after lithium prophylaxis and/or 2 or more episodes of illness occurring within weeks of cessation of lithium (can only be rated if at least 3 episodes of illness have occurred before lithium prophylaxis and lithium response has been observed for at least 5 years).
External validation
To externally check our gene-based PTV results, we obtained PTV counts from the BSC. Specifically, rare variant counts within the top ten genes defined by P-value in the Fisher’s exact tests of enrichment of ultra-rare PTVs in the data were provided by the BSC. To harmonize BSC data with BipEx, we used annotation definitions defined in Supplementary Table 5. We then generate MAC≤5 counts for each gene in the BSC data. The addition of the BSC dataset has some limitations. Frameshift indels were not called for a subset of the cohorts, reducing power to detect an association. Among the BSC cohorts that called indels, only the Rarebliss dataset provided indel calls. Furthermore, library preparation, sequencing platform, and variant calling differed across the BSC cohorts (Supplementary Table 11).
Data availability
We display all of our results, from the variant and gene level in a browser available at https://bipex.broadinstitute.org. Phenotype curation and QC are available at https://astheeggeggs.github.io/BipEx/. Data are available under the following EGS study accession numbers: EGAS00001005838, EGAS00001005841, EGAS00001005842, EGAS00001005843, EGAS00001005844, EGAS00001005845, EGAS00001005851, EGAS00001005852, EGAS00001005853, EGAS00001005854, EGAS00001005855, EGAS00001005856, EGAS00001005857, EGAS00001005858, EGAS00001005859, and EGAS00001005860. Data are also available through contacting the corresponding authors directly. GnomAD database: gnomad.broadinstitute.org. Pathway databases: Gene Ontology: geneontology.org, KEGG: www.genome.jp/kegg, REACTOME: reactome.org/. GTEx tissue specific enrichment gene-sets: data.broadinstitute.org/alkesgroup/LDSCORE/LDSC_SEG_ldscores/.
Code availability
Code to perform QC, analysis, and plot creationis provided at github.com/astheeggeggs/BipEx. Data were manipulated using Hail 0.2 and R (4.0.2) using data.table (1.13.0) and dplyr (1.0.1), and plotted with ggplot2 (3.3.2), ggsci (2.9), ggExtra (0.9), ggrepel (0.8.2), RColorBrewer (1.1–2), and gridExtra (2.3) in R (4.0.2).
Ethics statement
IRB approvals and study consent forms from each of the sample contributing organizations were sent to the Broad Institute before samples were sequenced and analyzed. Contributing organizations include: University of Aberdeen, Trinity College Dublin, University of Edinburgh, University College London, Cardiff University, University of Cambridge, Vrije Universitat Amsterdam, University College of Los Angeles, Universitats Klinikum Frankfurt, Massachusetts General Hospital, Johns Hopkins University, Karolinska Institute, LifeGene Biorepository at Karolinska Institute, and Umea University.
All ethical approvals are on file at the Massachusetts General Brigham (MGB), formerly Partners, IRB office amended to protocol #2014P001342, title: ‘Molecular Profiling of Psychiatric Disease’.
Supplementary Material
Acknowledgements
This study was supported by the Stanley Family Foundation, Kent and Elizabeth Dauten, National Institute of Health (NIH): R01 CA194393 (BMN), R37 MH107649 (BMN), R01 MH085542 (JWS), National Institute of Mental Health (NIMH): R01 MH090553 (RO), R01 MH095034 (EAS), U01 MH105578 (NBF), UK Medical Research Council (MRC): G1000708 (AM), MR/L010305/1 (MJO), MR/P005748/1 (MJO, MCO’D, JW), ongoing grant support from Stanley Medical Research Institute (FD, RY), and The Dalio Foundation (BMN) who have enabled us to rapidly expand our data generation collections with the goal of moving towards better treatments for BD, schizophrenia, and other psychiatric disorders. BSC grant support: NIH grants R01 MH110437 (PZ), R01 MH085543 (CS) and RC2 AG036607 (CS and NR). We thank Willem Ouwehand for contributing control samples for exome sequencing, and Emilie Wigdor for thoughtful comments.
Competing interests
BMN is a member of the scientific advisory board at Deep Genomics and Neumora and consultant for Camp4 Therapeutics, Takeda Pharmaceutical, and Biogen. AMM has received speaker fees from Illumina and Janssen and research grant support from The Sackler Trust. ML has received speakers fees from Lundbeck pharmaceuticals. MJO, MCO’D and JW have received a research grant from Takeda Pharmaceuticals outside the scope of the present study. FSG has received a research grant from Janssen Pharmaceuticals outside the scope of the present study. C-YC is an employee of Biogen. FD is an employee of Sheppard Pratt. AEL and EAS are now employees of Regeneron. XJ is now an employee of Genentech. All other authors declare no competing interests.
References
- 1.Ferrari AJ et al. The prevalence and burden of bipolar disorder: findings from the Global Burden of Disease Study 2013. Bipolar Disord. 18, 440–450 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Polderman TJC et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat. Genet. 47, 702–709 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Stahl EA et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 51, 793–803 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Consortium Brainstorm et al. Analysis of shared heritability in common disorders of the brain. Science 360, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mullins N et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat. Genet. (2021) doi: 10.1038/s41588-021-00857-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charney AW et al. Contribution of Rare Copy Number Variants to Bipolar Disorder Risk Is Limited to Schizoaffective Cases. Biol. Psychiatry 86, 110–119 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ganna A et al. Quantifying the Impact of Rare and Ultra-rare Coding Variation across the Phenotypic Spectrum. Am. J. Hum. Genet. 102, 1204–1211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crow J,A, F. J & M. Kimura An Introduction to Population Genetics Theory. Population (French Edition) vol. 26 977 (1971). [Google Scholar]
- 9.Kryukov GV, Pennacchio LA & Sunyaev SR Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am. J. Hum. Genet. 80, 727–739 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Power RA et al. Fecundity of patients with schizophrenia, autism, bipolar disorder, depression, anorexia nervosa, or substance abuse vs their unaffected siblings. JAMA Psychiatry 70, 22–30 (2013). [DOI] [PubMed] [Google Scholar]
- 11.American Psychiatric Association. Task Force on DSM-IV. DSM-IV Sourcebook. (Amer Psychiatric Pub Incorporated, 1998). [Google Scholar]
- 12.Janca A, Ustün TB, Early TS & Sartorius N The ICD-10 symptom checklist: a companion to the ICD-10 classification of mental and behavioural disorders. Soc. Psychiatry Psychiatr. Epidemiol. 28, 239–242 (1993). [DOI] [PubMed] [Google Scholar]
- 13.Malaspina D et al. Schizoaffective Disorder in the DSM-5. Schizophr. Res. 150, 21–25 (2013). [DOI] [PubMed] [Google Scholar]
- 14.O’Connell KS & Coombes BJ Genetic contributions to bipolar disorder: current status and future directions. Psychol. Med 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5®). (American Psychiatric Pub, 2013). [Google Scholar]
- 16.Husson T et al. Identification of potential genetic risk factors for bipolar disorder by whole-exome sequencing. Transl. Psychiatry 8, 268 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sul JH et al. Contribution of common and rare variants to bipolar disorder susceptibility in extended pedigrees from population isolates. Transl. Psychiatry 10, 74 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jia X et al. Investigating rare pathogenic/likely pathogenic exonic variation in bipolar disorder. Mol. Psychiatry (2021) doi: 10.1038/s41380-020-01006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cruceanu C et al. Rare susceptibility variants for bipolar disorder suggest a role for G protein-coupled receptors. Molecular Psychiatry vol. 23 2050–2056 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Genovese G et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nature Neuroscience vol. 19 1433–1441 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh T et al. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat. Genet. 49, 1167–1173 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McLaren W et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh T et al. Exome sequencing identifies rare coding variants in 10 genes which confer substantial risk for schizophrenia. medRxiv 2020.09.18.20192815 (2020). [Google Scholar]
- 24.Samocha KE, Kosmicki JA & Karczewski KJ Regional missense constraint improves variant deleteriousness prediction. BioRxiv (2017). [Google Scholar]
- 25.Aguet F et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. doi: 10.1101/787903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet. 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hibar DP et al. Subcortical volumetric abnormalities in bipolar disorder. Mol. Psychiatry 21, 1710–1716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ganna A et al. Ultra-rare disruptive and damaging mutations influence educational attainment in the general population. Nat. Neurosci. 19, 1563–1565 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cotney J et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun. 6, 6404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weyn-Vanhentenryck SM et al. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 6, 1139–1152 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanders SJ et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium. Electronic address: douglas.ruderfer@vanderbilt.edu & Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium. Genomic Dissection of Bipolar Disorder and Schizophrenia, Including 28 Subphenotypes. Cell 173, 1705–1715.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ripke S, Walters JTR, O’Donovan MC, the Psychiatric Genomics Consortium, G. SW of & Others. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. MedRxiv (2020). [Google Scholar]
- 34.Freland L & Beaulieu J-M Inhibition of GSK3 by lithium, from single molecules to signaling networks. Front. Mol. Neurosci 5, 14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kishore BK & Ecelbarger CM Lithium: a versatile tool for understanding renal physiology. Am. J. Physiol. Renal Physiol 304, F1139–49 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jope RS Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol. Sci. 24, 441–443 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Satterstrom FK et al. Large-Scale Exome Sequencing Study Implicates Both Developmental and Functional Changes in the Neurobiology of Autism. Cell 180, 568–584.e23 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Satterstrom FK et al. Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat. Neurosci. 22, 1961–1965 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Epi25 Collaborative. Electronic address: s.berkovic@unimelb.edu.au & Epi25 Collaborative. Ultra-Rare Genetic Variation in the Epilepsies: A Whole-Exome Sequencing Study of 17,606 Individuals. Am. J. Hum. Genet. 105, 267–282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Firth HV et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 84, 524–533 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanji C et al. A-kinase anchoring protein AKAP220 binds to glycogen synthase kinase-3beta (GSK-3beta ) and mediates protein kinase A-dependent inhibition of GSK-3beta. J. Biol. Chem. 277, 36955–36961 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Beurel E, Grieco SF & Jope RS Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol. Ther. 148, 114–131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Howrigan DP et al. Exome sequencing in schizophrenia-affected parent-offspring trios reveals risk conferred by protein-coding de novo mutations. Nat. Neurosci. 23, 185–193 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Benjamini Y & Hochberg Y Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 57, 289–300 (1995). [Google Scholar]
Methods references
- 45.Van der Auwera GA et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.DePristo MA et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 43, 491–498 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ng PC & Henikoff S SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adzhubei IA et al. A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rentzsch P, Witten D, Cooper GM, Shendure J & Kircher M CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen C-Y et al. Genetic validation of bipolar disorder identified by automated phenotyping using electronic health records. Transl. Psychiatry 8, 86 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Murphy S et al. Instrumenting the health care enterprise for discovery research in the genomic era. Genome Res. 19, 1675–1681 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wing J SCAN (Schedules for Clinical Assessment in Neuropsychiatry) and the PSE (Present State Examination) Tradition. Mental Health Outcome Measures 123–130 (1996) doi: 10.1007/978-3-642-80202-7_9. [DOI] [Google Scholar]
- 54.McGuffin P, Farmer A & Harvey I A polydiagnostic application of operational criteria in studies of psychotic illness. Development and reliability of the OPCRIT system. Arch. Gen. Psychiatry 48, 764–770 (1991). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
We display all of our results, from the variant and gene level in a browser available at https://bipex.broadinstitute.org. Phenotype curation and QC are available at https://astheeggeggs.github.io/BipEx/. Data are available under the following EGS study accession numbers: EGAS00001005838, EGAS00001005841, EGAS00001005842, EGAS00001005843, EGAS00001005844, EGAS00001005845, EGAS00001005851, EGAS00001005852, EGAS00001005853, EGAS00001005854, EGAS00001005855, EGAS00001005856, EGAS00001005857, EGAS00001005858, EGAS00001005859, and EGAS00001005860. Data are also available through contacting the corresponding authors directly. GnomAD database: gnomad.broadinstitute.org. Pathway databases: Gene Ontology: geneontology.org, KEGG: www.genome.jp/kegg, REACTOME: reactome.org/. GTEx tissue specific enrichment gene-sets: data.broadinstitute.org/alkesgroup/LDSCORE/LDSC_SEG_ldscores/.