

Abstract
Background
Assessing the effect of alcohol consumption on biological age is essential for understanding alcohol use‐related comorbidities and mortality. Previously developed epigenetic clocks are mainly based on DNA methylation in heterogeneous cell types, which provide limited knowledge on the impacts of alcohol consumption at the individual cellular level. Evidence shows that monocytes play an important role in both alcohol‐induced pathophysiology and the aging process. In this study, we developed a novel monocyte‐based DNA methylation clock (MonoDNAmAge) to assess the impact of alcohol consumption on monocyte age.
Methods
A machine learning method was applied to select a set of chronological age‐associated DNA methylation CpG sites from 1202 monocyte methylomes. Pearson correlation was tested between MonoDNAmAge and chronological age in three independent cohorts (N total = 2242). Using the MonoDNAmAge clock and four established clocks (i.e., HorvathDNAmAge, HannumDNAmAge, PhenoDNAmAge, GrimDNAmAge), we then evaluated the effect of alcohol consumption on epigenetic aging in the three cohorts [i.e., Yale Stress Center Community Study (YSCCS), Veteran Aging Cohort Study (VACS), Women's Interagency HIV Study (WIHS)] using linear and quadratic models.
Results
The MonoDNAmAge, comprised of 186 CpG sites, was moderately to strongly correlated with chronological age in the three cohorts (r = 0.90, p = 3.12E−181 in YSCCS; r = 0.54, p = 1.75E−96 in VACS; r = 0.66, p = 1.50E−60 in WIHS). More importantly, we found a nonlinear association between MonoDNAmAge and alcohol consumption (p model = 4.55E−08, = 7.80E−08 in YSCCS; p model = 1.85E−02, = 3.46E−02 in VACS). Heavy alcohol consumption increased EAAMonoDNAmAge up to 1.60 years while light alcohol consumption decreased EAAMonoDNAmAge up to 2.66 years. These results were corroborated by the four established epigenetic clocks (i.e., HorvathDNAmAge, HannumDNAmAge, PhenoDNAmAge, GrimDNAmAge).
Conclusions
The results suggest a nonlinear relationship between alcohol consumption and its effects on epigenetic age. Considering adverse effects of alcohol consumption on health, nonlinear effects of alcohol use should be interpreted with caution. The findings, for the first time, highlight the complex effects of alcohol consumption on biological aging.
Keywords: alcohol consumption, epigenetic age acceleration, monocyte epigenetic clock
Evidence shows that monocyte plays an important role in both alcohol‐induced pathophysiology and aging process. In this study, we developed a novel monocyte‐based DNA methylation clock to assess the impact of alcohol consumption on monocyte age. We found that alcohol use affects epigenetic aging in a nonlinear manner, with heavy consumption increasing and nonheavy use decreasing the epigenetic age. Our study expands previous knowledge and provides new insights into the effect of a spectrum of alcohol use on epigenetic aging.

INTRODUCTION
Alcohol consumption exerts significant adverse effects on health and contributes to increased morbidity and mortality (Collaborators GBDA, 2018). Biological aging has been proposed as an indicator of the adverse effects of alcohol on health and frailty (Shin & Baik, 2016; Strandberg et al., 2012). Recently developed epigenetic “clocks” employ cellular DNA methylation (DNAm) as a measure of the aging process. DNAm‐based epigenetic clocks were shown to be more sensitive and precise measures of cellular age than other genomic measures (e.g., transcriptome and telomere length; Lopez‐Otin et al., 2013). It is well‐established that alcohol consumption alters DNAm in whole blood (Liang et al., 2021; Liu et al., 2018). However, the relationship between alcohol consumption and epigenetic age remains unclear.
To date, more than a dozen DNAm‐based epigenetic clocks have been reported, including four well‐established DNAm‐based age estimators. The Horvath clock (HorvathDNAmAge) is based on 353 CpG sites that capture estimated multi‐tissue biological age (Horvath, 2013). The Hannum clock (HannumDNAmAge) is derived from 71 CpGs in leukocytes (Hannum et al., 2013). The Levine clock (PhenoDNAmAge) employs 513 CpGs to predict lifespan (Levine et al., 2018). Lu's GrimAge clock (GrimDNAmAge) is a linear combination of chronological age, sex, and 1030 CpG sites modeled as surrogate biomarkers for seven plasma proteins and smoking pack‐years, predicting age at death (Lu et al., 2019). These clocks have been applied as biomarkers of the aging process to understand how numerous medical or psychiatric conditions affect biological age. For example, biological age was significantly greater than chronological age by a mean of 5 to 10 years in HIV‐positive (HIV+) compared to HIV‐negative (HIV−) individuals (Boulias et al., 2016; Horvath & Levine, 2015).
Epigenetic age acceleration (EAA) is defined as the residuals of regressing DNAm age on chronological age (Kresovich et al., 2021; Stephenson et al., 2021). The established “clocks” have been recently applied to examine whether alcohol use increases or decreases EAA and reported inconsistent findings. Alcohol consumption was found significantly associated with EAA that was estimated using GrimDNAmAge (Kresovich et al., 2021; Stephenson et al., 2021). An increased EAA has been reported in individuals with heavy alcohol use and in children with fetal alcohol spectrum disorder (Fiorito et al., 2019; Okazaki et al., 2021). Compared to healthy individuals, persons with alcohol use disorder (AUD) show a trend toward higher EAA in liver tissue (Rosen et al., 2018) and a 2.22‐year age increase in blood (Luo et al., 2020). On the other hand, light to moderate alcohol use appears to have no effect on or even slows extrinsic EAA (Quach et al., 2017; Robinson et al., 2020). Interestingly, using HannumDNAmAge, Beach et al. first observed a U‐shaped relationship between alcohol consumption and EAA. Their results demonstrate that light and heavy levels of alcohol consumption increases EAA while intermediate levels of alcohol consumption decrease EAA (Beach et al., 2015). These observations suggest that the effect of alcohol consumption on the biological aging process may differ by the quantity of use and tissue type.
One limitation of prior studies using available epigenetic clocks is their lack of tissue and cell‐type specificity; thus, they may provide limited insight into the mechanisms of tissue‐specific cellular aging affected by alcohol. Alcohol consumption changes immunity and inflammatory functions that may result from alterations in immune cell functions (Sureshchandra et al., 2019). Moderate or heavy alcohol use has been shown to change monocyte function (Badia et al., 2004; Donnadieu‐Rigole et al., 2016; Szabo, 1998). Szabo et al. (Szabo, 1998) reported that alcohol consumption exerts a biphasic effect on interferon inducibility and potentially affects monocyte‐derived inflammatory cytokine production, which changes the course of the aging process. In addition, monocytes show aging‐related gene dysfunction in metabolism, immune function, and inflammation (Saare et al., 2020; Viel et al., 2012), as well as DNAm and transcriptomic alterations (Metcalf et al., 2017; Reynolds et al., 2014). Therefore, we hypothesize that a set of CpG sites selected from monocytes can serve as an indicator of the effect of alcohol consumption on biological age and provide insights into its underlying mechanisms. A monocyte‐based epigenetic clock may serve as a surrogate of peripheral immune function among persons consuming alcohol.
In this study, we aimed to characterize the effect of a range of alcohol consumption levels on monocyte epigenetic age. We developed a novel “clock” derived from the human monocyte DNA methylome (termed “MonoDNAmAge”) using the Elastic Net Regularization (ENR). We evaluated the performance of MonoDNAmAge in estimating HIV‐associated age acceleration as a benchmark. Finally, using data‐driven modeling approach, we assessed the effect of alcohol consumption on MonoDNAmAge, EAA, and apparent methylation age rate (AMAR) in three distinct cohorts: the Yale Stress Center Community Study (YSCCS; Blaine et al., 2019); the Veteran Aging Cohort Study (VACS; Justice et al., 2006); and the Women's Interagency HIV Study (WIHS; Adimora et al., 2018; Bacon et al., 2005). Four well‐established clocks (i.e., HorvathDNAmAge, HannumDNAmAge, PhenoDNAmAge, and GrimDNAmAge) were also used to assess the effect of alcohol consumption on measures of biological age. MonoDNAmAge development and analytic strategy are shown in Figure 1.
FIGURE 1.
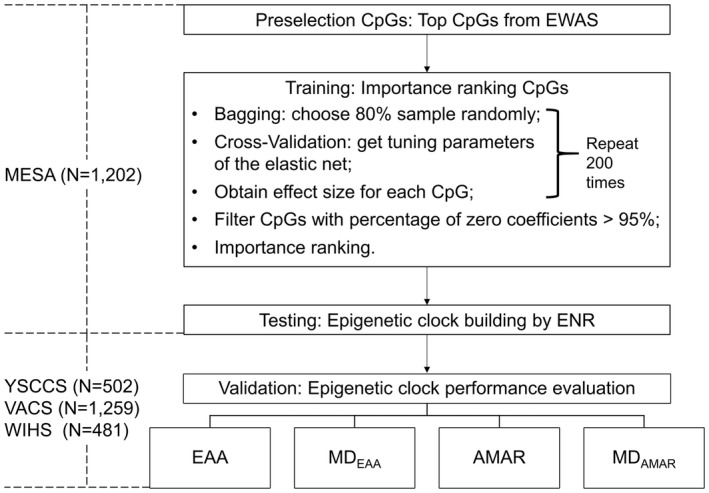
The workflow for establishing a monocyte epigenetic clock using a feature selection method. YSCCS: Yale Stress Center Cohort Study; VACS: Veterans Aging Cohort Study; WIHS: Women's Interagency HIV Study; EWAS: Epigenome‐Wide Association Studies; ENR: Elastic Net Regularization; EAA: Epigenetic Age Acceleration, the residuals of regressing DNA methylation age on chronological age; MDEAA: The mean difference in EAA between different groups for phenotypes of interest; AMAR: Apparent Methylation Age Rate, the ratio of DNA methylated age to chronological age; MDAMAR: The mean difference in AMAR between different groups for phenotypes of interest
MATERIALS AND METHODS
Study cohorts and phenotype assessments
Multi‐ethnic study of atherosclerosis (MESA; N = 1202)
The MESA cohort is a multi‐site, longitudinal study designed to investigate the prevalence, correlates, and progression of subclinical cardiovascular disease (CVD) in a population cohort of 6814 participants (Bild et al., 2002; Liu et al., 2017; Reynolds et al., 2014). The MESA Epigenomics and Transcriptomics Study has been launched to investigate potential gene expression regulatory methylation sites in humans by examining the association between CpG methylation and gene expression in purified human monocytes from the large study population. The DNA samples were obtained from CD14+ monocyte samples collected from 1202 relatively healthy individuals with ages ranging from 44 to 83 years. Among 1202 participants, 51.4% were female. In this cohort, 21.5% were African American, 32.6% were Hispanic, and 45.9% were European descents. Only 5.3% had prevalent CVD. In this study, DNAm data from the MESA Epigenomics and Transcriptomics Study (GSE56046) was used to construct the MonoDNAmAge clock.
The following three cohorts were used to assess the effects of alcohol on DNAm aging. Demographic and clinical characteristics and alcohol assessment for each cohort are described below and presented in Table 1 and Supplementary Information.
TABLE 1.
Demographic and clinical characteristics of the YSCCS, VACS, and WIHS cohorts
| Variable | YSCCS | VACS | WIHS | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HAD | MAD | p | HAD | MAD | p | LAD | NAD | p | |
| Men: AUDIT ≥ 8 Women: AUDIT≥7 (N = 148) | Men: AUDIT < 8 Women: AUDIT<7 (N = 354) | PEth ≥ 20 (N = 299) | PEth < 20 (N = 738) | 0 < NDRNKWK ≤ 7 (N = 196) | NDRNKWK = 0 (N = 255) | ||||
| Age | 26.8 ± 7.13 | 29.8 ± 9.28 | 1.35E−04 | 50.7 ± 7.23 | 51.1 ± 8.14 | 5.03E−01 | 40.3 ± 9.13 | 43.7 ± 9.46 | 1.13E−04 |
| Sex (%male) | 64.9% | 35.3% | 2.18E−09 | 100% | 100%/ | NA | 0% | 0% | NA |
| Race (AA) | 12.2% | 22.4% | 1.29E−02 | 86.6% | 80.2% | 1.90E−02 | 49.0% | 49.8% | 9.69E−01 |
| Smoker | 39.9% | 12.7% | 1.77E−11 | 68.1% | 54.3% | 6.27E−05 | 70.9% | 67.3% | 8.97E−01 |
| HIV‐positive | 0% | 0% | NA | 93.7% | 91.3% | 2.63E−01 | 50.5% | 63.9% | 1.54E−04 |
| log10VL | 0 | 0 | NA | 2.82 ± 1.23 | 2.66 ± 1.24 | 8.44E−02 | 2.16 ± 0.69 | 2.05 ± 0.48 | 1.34E−01 |
| ART adherence | NA | NA | NA | 71.43% | 79.85% | 6.62E−03 | 74.75% | 85.89% | 4.64E−03 |
| Cannabis use | 88.4% | 56.9% | 2.40E−11 | 84.5% | 73.4% | 2.21E−04 | 50.8% | 29.1% | 3.83E−06 |
| Cocaine use | NA | NA | NA | 75.3% | 66.0% | 4.34E−03 | NA | NA | NA |
| Stimulant use | NA | NA | NA | 36.9% | 39.9% | 4.08E−01 | NA | NA | NA |
| Opiate use | NA | NA | NA | 42.3% | 44.2% | 6.27E−01 | NA | NA | NA |
Abbreviations: AA, African American; ART adherence, adherence to antiretroviral therapy; AUDIT, Alcohol Use Disorders Identification Test; AUDIT‐C, first three questions of the Alcohol Use Disorders Identification Test; chi‐square test was used to compare percentages between two groups; HAD, Heavy Alcohol Drinking; LAD, Light Alcohol Drinking; MAD, Moderate Alcohol Drinking; NAD, Non‐Alcohol Drinking; VACS, Veterans Aging Cohort Study; VL, viral load; Welch's two‐sample t‐test was used to compare means between two groups; WIHS, Women's Interagency HIV Study; YSCCS, Yale Stress Center Cohort Study.
Significant phenotypes are shown in bold.
The cannabis, cocaine, stimulant, and opiate use in the VACS cohort were defined as a case (>0) and a control (=0).
Yale stress center cohort study (YSCCS; N = 502)
The cohort served as a community‐based sample to examine the effect of alcohol consumption on DNAm age among healthy participants (Blaine et al., 2019). The 10‐item Alcohol Use Diagnosis Identification Test (AUDIT) was used to assess alcohol use. Heavy alcohol drinking (HAD) was defined as an AUDIT score ≥8 for men and an AUDIT score ≥7 for women (N = 148), moderate alcohol drinking (MAD) was defined as an AUDIT score <8 for men, and an AUDIT score <7 for women (N = 354; Lee et al., 2018). The average AUDIT score was 5.69 among all participants in the cohort. The self‐reported AUDIT‐Consumption (AUDIT‐C, the first three items of AUDIT) score was also used for the analysis.
Veterans aging cohort study (VACS; N = 1259)
The cohort served as a clinic‐based sample to benchmark the effect of HIV infection on MonoDNAmAge and to examine the effect of alcohol consumption on DNAm age (Justice et al., 2006). The participants included both HIV+ (N = 1151) and HIV− (N = 104) individuals. A majority of HIV+ participants were on antiretroviral therapy and were virally suppressed (63.66%). Alcohol consumption was assessed by measuring phosphatidylethanol (PEth) levels, a biomarker for alcohol use (Viel et al., 2012) that is positively correlated with AUDIT scores (Liang et al., 2021; Piano et al., 2015). HAD was defined as PEth levels ≥20 ng/ml (N = 299) and MAD was defined as PEth levels <20 ng/ml (N = 738) according to a previous study (Stewart et al., 2009). The average PEth level was 41.7 ng/ml. The AUDIT‐C score was also collected for each participant.
Women's interagency HIV study (WIHS; N = 481)
The cohort served as a clinic‐ and community‐based sample to examine the effects of HIV infection and alcohol consumption on DNAm age (Adimora et al., 2018; Bacon et al., 2005). This study included HIV+ (N = 272, 90.44% virally suppressed) and HIV− (N = 209) participants. The cohort predominantly reported light alcohol use with an average number of drinks per week (NDRNKWK) of 0.7. Light alcohol drinking (LAD) was defined as 0 < NDRNKWK ≤ 7 (N = 196), and non‐alcohol drinking (NAD) was defined as NDRNKWK = 0 (N = 255; Adams et al., 1996).
DNA methylation in the three cohorts
Epigenome‐wide CpG methylation was profiled using either the Illumina HumanMethylation450 BeadChip (450K) in the MESA (monocyte), YSCCS (blood), and VACS (blood; 57.2% of the sample) cohorts or Illumina HumanMethylation EPIC BeadChip (EPIC) in 42.8% of the VACS samples and the WIHS (peripheral blood mononuclear cells) samples. All samples in the three study cohorts (YSCCS, VACS, and WIHS) were processed at the Yale Center for Genomic Analysis (Zhang et al., 2017). We applied the method described by Houseman et al. (2012; Jaffe & Irizarry, 2014) to estimate the proportions of CD4+ T cells, CD8+ T cells, NK T cells, B cells, monocytes, and granulocytes in each cohort. Quality control of methylation data for each cohort is presented in Supplementary Information.
MonoDNAmAge clock development in the MESA cohort
Preselection features in the entire MESA cohort
We first performed an epigenome‐wide association study (EWAS) on chronological age in the MESA cohort. Linear regression was applied in which DNAm β value was the dependent variable and chronological age was the independent variable. The significance of EWAS was set at nominal p < 1.47E−07 (corresponding to Bonferroni correction p < 0.05).
We then selected the top 1000 significant CpG features from EWAS for feature selection. The number of 1000 was selected because it approached the sample size of 1202 in the MESA cohort. A sensitivity test was performed later to validate the preselection number of CpG sites.
To build a predictive model for estimating the monocyte age, samples in the MESA cohort were divided into a training set (N = 721) and a testing set (N = 481).
Importance ranking of CpGs in the training set
We randomly selected 80% of subjects in the training set without replacement 200 times and constructed a model for each replicate. Only the CpGs present in more than 95% of replicates was considered for inclusion in the final model and ranked based on the coefficients of all replicates. In each replicate, there are two parameters (α and λ) to tune in ENR using the “glmnet” R package (Friedman et al., 2010). We performed a grid search for α values from 0 (ridge regression) to 1 (lasso regression) increasing by 0.05. For each value of α, the 10‐fold cross‐validation procedure was performed to get the tuning parameter λ and the corresponding mean cross‐validated error. We selected the α and λ that have the minimum mean cross‐validated error as the estimations of ENR tuning parameters. We extracted the coefficients for the model with the α and λ values corresponding to the minimum mean cross‐validated error. The CpG importance ranking was based on the sum of the absolute value of the coefficients of all replicates.
MonoDNAmAge construction using ENR in the testing set
Based on the importance ranking, we evaluated the performance of different sets of CpG sites in the testing samples by adding one CpG at a time. The performance was assessed using the Pearson correlation coefficient (Pearson's r) between predicted DNAm age and chronological age. The correlation coefficient was calculated for each set of CpG sites. We selected a set of CpG with the correlation coefficient at the inflection point of the performance curve as the best set of predictive clocks, MonoDNAmAge (Figure 2A).
FIGURE 2.
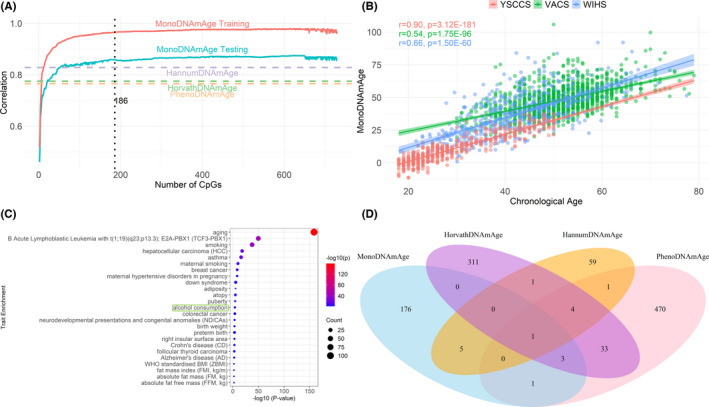
MonoDNAmAge development and performance. (A) Feature selection using Elastic Net Regularization (ENR) for estimating MonoDNAmAge. The figure shows the Pearson correlation coefficients of predicted age and chronological age in the training and testing datasets from the Multi‐Ethnic Study of Atherosclerosis (MESA) cohort. (B) Correlation between MonoDNAmAge and chronological age in three independent cohorts: YSCCS, VACS, and WIHS. (C) Trait enrichment of the selected 186 CpGs in MonoDNAmAge. (D) Overlapping CpG sites among four epigenetic clocks (i.e., MonoDNAmAge, HorvathDNAmAge, HannumDNAmAge, and PhenoDNAmAge)
Further evaluation of MonoDNAmAge performance in the three study cohorts
Performance of the above CpG features selected from the testing set in MESA was evaluated in three independent validation cohorts (YSCCS, VACS, and WIHS) using the four measures: EAA, Mean Difference of EAA (MDEAA), Apparent Methylation Age Rate (AMAR), and Mean Difference of AMAR and (MDAMAR; see the statistical analysis section).
Sensitivity test for preselection CpG features
Different sets of CpGs with p‐value < 1E−30, <6.15E−20, and <1E−10 from EWAS on chronological age were selected for feature selection in the training sample and model evaluation in the testing sample (Figure S1). The performance of each CpG set was evaluated based on the correlation coefficient between MonoDNAmAge predicted age and chronological age. The set of CpG with the best performance was determined in the final model.
Statistical analysis
Epigenetic clocks and assessments
Pearson's r between DNAm age and chronological age was estimated in each cohort. EAA was defined as the residuals of regressing DNAm age on chronological age (Horvath, 2013; Horvath & Raj, 2018). We calculated the mean difference in EAA between heavy users and nonheavy users, between HIV+ and HIV−, and denoted it as MDEAA. The AMAR was defined as the ratio of DNAm age to chronological age (Hannum et al., 2013), an AMAR > 1 represents DNAm age acceleration, and an AMAR < 1 represents DNAm age deceleration. We also calculated the mean difference in AMAR between heavy and nonheavy users, between HIV+ and HIV−, which were denoted as MDAMAR.
Benchmarking the MonoDNAmAge clock using HIV infection
We evaluated whether MonoDNAmAge was able to predict changes in DNAm age for individuals with HIV infection in the two cohorts (VACS and WIHS) using a linear model, with EAA or AMAR as the dependent variable and HIV infection as the independent variable. We adjusted for potential confounding factors, including adherence to antiretroviral therapy (ART adherence), alcohol‐related phenotype, tobacco use, self‐reported ethnicity, and body mass index (BMI) in the model.
Association between alcohol consumption and DNAm‐based biological age
Instead of assuming a linear relationship between alcohol consumption and epigenetic age, we examined both linear and nonlinear relationships using linear and quadratic models, respectively. In the quadratic model, DNAmAge = , where and x represents the continuous variable of each assessment for alcohol consumption. T‐tests were used to test the regression coefficients obtained in both quadratic and linear regression. That is, for the quadratic model, we tested versus using t‐test , where n is the sample size. We also tested the regression model H 0: the regression model was not significant versus H a: the regression model was significant by performing F test, based on ANOVA test, where MSR was the regression mean square and MSE was the error mean square, was the corrected sum of squares for the regression model and was the sum of squares for error.
We also examined EAA and AMAR between different groups of alcohol consumption in each cohort. Dichotomized alcohol assessments were used to assess the different effects of heavy and light alcohol consumption. The model adjusted potential confounding factors including HIV infection, ART adherence, tobacco use, self‐reported ethnicity, and BMI. Because the VACS cohort included only men and the WIHS cohort included only women, sex was adjusted only in the YSCCS cohort that included both men and women.
Correlation between DNAm age and cell types in different alcohol consumption groups
Pearson's r between DNAm‐based estimates of biological age and the proportions of six cell types were estimated in each cohort to address potential confounding effects of the cell types.
RESULTS
A new monocyte‐based epigenetic clock, MonoDNAmAge, validations, and biological interpretations
MonoDNAmAge was derived from the CD14+ monocyte methylome in MESA (N = 1202; GSE56046; Bild et al., 2002; Reynolds et al., 2014). ENR selected a set of 186 age‐associated CpGs as MonoDNAmAge (Table S1). All 186 CpGs were on the 450K array. The majority of the 186 CpGs were also included on the EPIC array except nine CpGs [cg09499629 (KLF14), cg04434593 (LOC100130987; CLCF1), cg12422450 (CHGA), cg26062560 (C3orf21), cg26864395 (RUNX3), cg21922223, cg16567172 (HSPB1), cg14564815 (SLC4A9), cg15428620 (SFXN3)], which were unique to the 450K array. The correlation between DNAm age and chronological age was 0.96 (p < 2.20E−16) in the training set and was 0.86 (p = 1.55E−141) in the testing set (Figure 2A).
MonoDNAmAge was significantly correlated with chronological age in all three cohorts (YSCCS: r = 0.90, p = 3.12E−181; VACS: r = 0.54, p = 1.75E−96; WIHS: r = 0.66, p = 1.50E−60; Figure 2B). We also estimated the correlations of four established clocks with chronological age and correlation between each pair among the five epigenetic clocks in these three cohorts. As expected, all clocks showed significant correlations with chronological age in each cohort (YSCCS: p = 8.42E−215~4.69E−168; VACS: p = 6.82E−205~3.94E−135; WIHS: p = 2.25E−175~1.75E−91) and all clocks were highly correlated with each other (Figure S2).
These 186 CpG sites mapped to 135 genes, including well‐established genes associated with age (e.g., KLF14), transcription factors (e.g., RUNX3), and inflammatory function (e.g., IL17RC). Interestingly, the 186 CpG sites were enriched for 25 complex traits with p < 5.00E−03 in the EWAS Atlas database (Li et al., 2019; Figure 2C). The top significant traits included aging (p = 2.57E−159), smoking (p = 2.39E−38), breast cancer (p = 2.71E−09), and alcohol consumption (p = 8.95E−05). As a comparison, we assessed the CpG enrichments for three established clocks (HorvathDNAmAge, HannumDNAmAge, and PhenoDNAmAge; Figure S3). Identifiers of CpG sites for GrimDNAmAge were not publicly available for comparison. Consistent with the MonoDNAmAge clock, three clocks were enriched for multiple traits including age and smoking. However, only CpGs on HorvathDNAmAge were enriched in alcohol consumption (p = 2.45E−03), while the other two clocks were not significant. Among MonoDNAmAge and three established clocks, one CpG site, SCGN cg06493394, was shared across the four clocks (Figure 2D). The 135 genes harboring the 186 CpG sites were enriched in biological pathways relevant to aging (e.g., regulation of multicellular organismal process) by performing Database for Annotation, Visualization and Integrated Discovery pathway enrichment analysis (Figure S4; Huang et al., 2007).
We evaluated the performance of MonoDNAmAge in estimating HIV‐associated epigenetic age alteration as a benchmark. A detailed description of the approach is included in the Supplementary Information. Briefly, the EAA showed an average of a 10.14‐year increase in HIV+participants compared to HIV− participants in the VACS cohort (p vacs = 1.01E−26) and a 12.17‐year increase in the WIHS cohort (p WIHS = 3.34E−13; Table S2 and Figures S5, S6, S7). The results further confirm the effectiveness of MonoDNAmAge in estimating biological age.
Nonlinear effects of alcohol consumption on DNA methylation age
Instead of just assuming a linear relationship between alcohol consumption and DNAm age, we first compared the linear model (reduced model) with the quadratic model (full model) for modeling the relationship between DNAm age and alcohol consumption [AUDIT score in the YSCCS cohort, the natural logarithm of PEth level (ln(PEth)) in the VACS cohort, and NDRNKWK in the WIHS cohort] using the Akaike information criterion (AIC) and the Bayesian information criterion (BIC) values (Table S3).
Both the AIC and BIC correct the maximum likelihood estimate by introducing a penalty term for the number of parameters in the model to discourage overfitting, and the penalty term is larger in BIC than in AIC (Vrieze, 2012). Given a set of candidate models for the data, the model with the lowest AIC and BIC is preferred. The resulting AIC and BIC showed that the quadratic model fits the data better than the linear model for all five clocks in the YSCCS cohort (ΔAIC = AIClinear − AICquadratic; ΔBIC = BIClinear − BICquadratic; ΔAIC = 20.39~31.99; ΔBIC = 16.17~27.77). In the VACS cohort, compared to the linear model, the quadratic model had smaller AIC for all five clocks (ΔAIC = 2.27~9.21), smaller BIC for HannumDNAmAge (ΔBIC = 4.26), and PhenoDNAmAge (ΔBIC = 4.20). However, both the AIC and BIC of the linear model were smaller than the quadratic model in the WIHS cohort (ΔAIC = −1.39~−0.65; ΔBIC = −5.50~−4.76). Therefore, the values of AIC and BIC showed that the quadratic model fits the data better than the linear model for all five clocks in the YSCCS cohort and most clocks in the VACS cohort but not in the WIHS cohort. Therefore, the quadratic regression was applied to the YSCCS (Figure 3A) and VACS (Figure 3B) cohorts.
FIGURE 3.
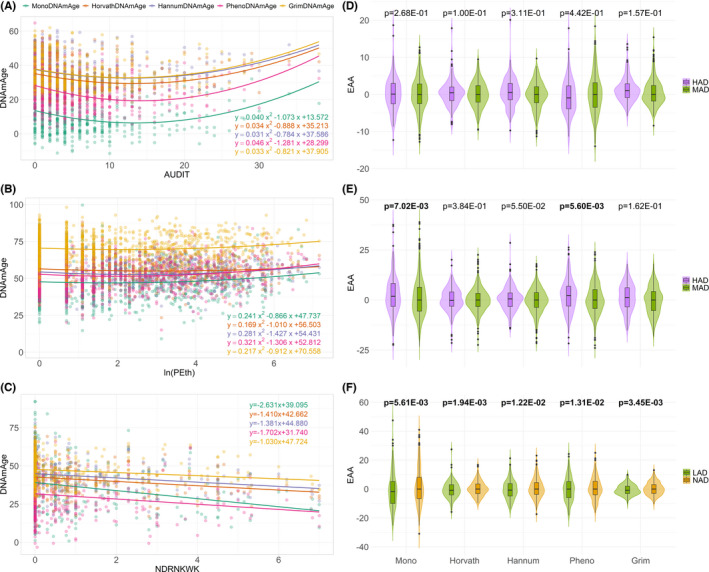
Relationships between DNA methylation age (DNAmAge) and alcohol consumption. The three rows correspond to YSCCS, VACS, and WIHS, respectively. (A) The parabola shows the relationship between DNAmAge and the Alcohol Use Disorders Identification Test (AUDIT) score in the YSCCS cohort. (B) The parabola shows the relationship between DNAmAge and the natural logarithm of phosphatidylethanol levels (ln(PEth)) in the VACS cohort. (C) The linear regression line shows the negative relationship between DNAmAge and the number of drinks per week (NDRNKWK) in the WIHS cohort. (D–F) Violin plots for Epigenetic Age Acceleration (EAA; the residuals of regressing DNA methylation age on chronological age) for different groups of alcohol consumption in the three cohorts. The p‐values were obtained from the linear model to test the association between EAA and continuous measure of alcohol consumption with adjusting the confounding variables. Bold values denote statistical significance at the p < 0.05 level. (D) Comparison of EAA between heavy alcohol drinking (HAD; AUDIT score ≥7 for men and AUDIT score ≥8 for women) and moderate alcohol drinking (MAD; AUDIT score <7 for men and AUDIT score <8 for women) in the YSCCS cohort. (E) Comparison of EAA between HAD (PEth level ≥20) and MAD (PEth level <20) in the VACS cohort. (F) Comparison of EAA between light alcohol drinking (LAD; 0 < NDRNKWK < 7) and non‐alcohol drinking (NAD; NDRNKWK = 0) in the WIHS cohort
In the YSCCS cohort, we observed a nonlinear relationship between MonoDNAmAge and the AUDIT score (p model = 4.55E−08, = 7.80E−08; Figure 3A). The other four DNAm clocks also showed nonlinear associations between DNAm age and the AUDIT score (HorvathDNAmAge: p model = 2.82E−07, = 2.37E−07; HannumDNAmAge: p model = 5.54E−06, = 2.41E−06; PhenoDNAmAge: p model = 4.08E−10, = 6.27E−09; GrimDNAmAge: p model = 1.80E−07, = 6.12E−08).
We then investigated the relationship between MonoDNAmAge and different levels of alcohol consumption by performing two linear regression analyses at the inflection point of the nonlinear distribution at the AUDIT score of 13 in the YSCCS cohort (Figure S8). We found that MonoDNAmAge was negatively correlated with alcohol consumption at an AUDIT score <13 (p = 5.36E−06) and positively correlated with heavy drinking at an AUDIT score >13 (p = 3.70E−03). Each unit change in the AUDIT score <13 was associated with a 1.20‐year decrease in MonoDNAmAge in the nonHAD group, and every unit change in the AUDIT score >13 was associated with a 0.36‐year increase in MonoDNAmAge in the HAD group.
We also observed a significant nonlinear relationship between MonoDNAmAge and the AUDIT‐C score (p model = 2.38E−11, = 3.56E−08) in YSCCS (Figure S9). The other four DNAm clocks also showed nonlinear associations between DNAm age and the AUDIT‐C score (HorvathDNAmAge: p model = 7.11E−09, = 2.53E−06; HannumDNAmAge: p model = 1.65E−08, = 8.45E−07; PhenoDNAmAge: p model = 1.08E−13, = 1.38E−08; GrimDNAmAge: p model = 1.49E−12, = 7.87E−10). The nonlinear associations of each epigenetic clock with alcohol consumption remained significant even with the conservative Bonferroni correction.
In the VACS cohort, the quadratic regression analysis also showed a significant association of MonoDNAmAge with alcohol consumption measured by In(PEth; p model = 1.85E−02, = 3.46E−02; Figure 3B). The results from the three established clocks provided estimates consistent with MonoDNAmAge: HannumDNAmAge (p model = 3.74E−03, = 8.32E−04), PhenoDNAmAge (p model = 4.97E−04, = 8.60E−04), GrimDNAmAge (p model = 3.40E−02, = 2.21E−02). Here, the inflection point for the curve of PEth levels was close to the PEth cut‐off used for defining HAD. We found that the slope below the PEth inflection point (nonHAD: PEth < 20, that is ln(PEth) < 2.996) was not significantly associated with MonoDNAmAge, while the slope above the PEth inflection point (HAD: PEth ≥ 20, that is In(PEth) ≥ 2.996) showed a significant positive association with MonoDNAmAge (p = 4.39E−02; Figure S8). Every unit change in In(PEth) above the inflection point was associated with a 1.31‐year increase in DNAm age. However, DNAm age was not significantly associated with the AUDIT‐C score for all five clocks (p model = 1.79E−01~9.01E−01) in the VACS cohort (Figure S9).
In the WIHS cohort, MonoDNAmAge was linearly associated with light alcohol consumption measured by NDRNKWK (β = −2.63, p model = 2.82E−06, = 2.82E−06; Figure 3C). A negative linear relationship between DNAm age and light alcohol consumption was also observed with the four established clocks (HorvathDNAmAge: β = −1.41, p model = 1.33E−05, px = 1.33E−05; HannumDNAmAge: β = −1.38, p model = 4.99E−05, px = 4.99E−05; PhenoDNAmAge: β = −1.70, p model = 7.74E−05, px = 7.74E−05; GrimDNAmAge: β = −1.03, p model = 1.23E−03, px = 1.23E−03).
Alcohol consumption alters epigenetic age acceleration
In the YSCCS cohort, alcohol consumption was not associated with the EAA estimated using MonoDNAmAge or the four established clocks (Figure 3D; Table S4). However, the AMAR of HorvathDNAmAge, HannumDNAmAge, and GrimDNAmAge showed a greater DNAm age than chronological age and were associated with alcohol consumption (β = 0.003~0.005, p = 3.31E−05~7.29E−03).
In the VACS cohort, the EAA analysis showed that alcohol consumption significantly increased MonoDNAmAge by 1.60 years (β = 0.433, p = 7.02E−03; Figure 3E; Table S4). The EAA estimated using PhenoDNAmAge also showed significant acceleration (β = 0.363, p = 5.60E−03). The AMAR estimated based on MonoDNAmAge was associated with alcohol consumption (β = 0.012, p = 3.55E−04). PhenoDNAmAge showed a similar association (β = 0.011, p = 9.16E−05). Similarly, GrimDNAmAge was also significantly positively correlated with alcohol consumption (β = 0.010, p = 4.89E−04).
In the WIHS cohort, EAA of MonoDNAmAge negatively correlated with alcohol consumption (β = −1.074, p = 5.61E−03, MDEAA = −2.656; Figure 3F; Table S4). All five clocks showed a decrease in DNAm age with an average MDEAA of −1.24 years. AMAR of MonoDNAmAge was negatively correlated with alcohol consumption (MonoDNAmAge: β = −0.046, p = 2.54E−05). Similarly, each AMAR of HorvathDNAmAge and PhenoDNAmAge was significantly negatively correlated with alcohol consumption.
We evaluated whether the impacts of alcohol consumption on epigenetic age were due to heterogeneous cell proportions. We found that DNAm‐based age estimated using MonoDNAmAge and four other clocks were significantly correlated with each of the six cell types in all samples regardless of alcohol consumption status across three cohorts (r = 0.54~0.93, p = 8.42E−215~1.50E−60; Figure S10). We then examined the correlations for each of the five clocks with six estimated cell types in the HAD and MAD groups separately in the YSCCS and VACS cohorts, and LAD and NAD in the WIHS cohort. We found that the correlation patterns were similar between the alcohol use groups (HAD and MAD; LAD and NAD) across the three cohorts, suggesting the effect of cell‐type composition on the performance of the five biological age estimators did not differ significantly between HAD and MAD, or between LAD and NAD groups. Thus, cell type proportion is not considered as a confounding factor for alcohol consumption status on epigenetic age.
DISCUSSION
Our results reveal that a set of DNAm CpG sites in monocytes predict biological age, enabling the detection of the effect of alcohol consumption on DNAm age validated in three distinct cohorts. The MonoDNAmAge clock, benchmarked against HIV infection, showed an approximate 10‐year acceleration in HIV+ participants. More importantly, this novel MonoDNAmAge clock detected a nonlinear relationship between DNAm‐based biological age and alcohol consumption in both a healthy community cohort (YSCCS) and a clinic‐based cohort (VACS) that included heavy alcohol drinkers (~33%). The effect of alcohol consumption on MonoDNAmAge was corroborated using four established epigenetic clocks. Thus, employing a comprehensive approach (i.e., one novel and four established epigenetic clocks, three independent cohorts, and differing but commonly employed measures of self‐reported alcohol exposure, and an objective biological measure), we identified for the first time that alcohol consumption appears to have a complex, nonlinear relationship with DNAm‐based estimates of biological age.
In contrast to previous studies, we examined the effect of alcohol consumption on epigenetic age by applying a novel cell‐type‐specific epigenetic clock related to the biological mechanism of alcohol consumption in the present study. The monocyte methylome plays an important role in epigenetic aging. Recently, differentially methylated regions in CD14+ monocytes between young (24 to 30 years) and older (57 to 70 years) individuals have been reported (Shchukina et al., 2021). These age‐associated CpG sites or DNAm regions are related to transcriptomic changes with aging (Shchukina et al., 2021). Therefore, a CD14+ monocyte epigenetic clock may provide a more precise measure of biological age. Genes that compose the MonoDNAmAge clock have functional implications for the aging process in monocyte‐related phenotypes, such as HIV infection and alcohol consumption. Our results highlight the importance of developing phenotype‐relevant cell/tissue‐specific epigenetic clocks that enable the prediction of organ or tissue functions and disease comorbidities.
Indeed, the 186 MonoDNAmAge CpG sites are enriched in individuals with alcohol consumption and other related traits (e.g., smoking and cancer). Interestingly, although MonoDNAmAge and the established epigenetic clocks are composed of different sets of CpGs with the exception of one site (cg06493394 on SCGN), these clocks showed reasonably consistent but not identical performance in estimating the effect of alcohol on epigenetic age. We speculate that each epigenetic clock estimates different but related facets of biological aging, inferred in part by the overlapping traits that show enrichment for the CpG sites that comprise each DNAm‐based epigenetic clock.
The nonlinear association between alcohol consumption and biological age is intriguing. Both the beneficial and the harmful effects of alcohol consumption have been documented extensively in the literature (Bagnardi et al., 2013; Chiva‐Blanch & Badimon, 2019; Costanzo et al., 2010; Di Castelnuovo et al., 2006; Collaborators GBDA, 2018). While one epidemiological study has shown that any amount of alcohol is harmful to health (Collaborators GBDA, 2018), some studies have shown that alcohol consumption has a U‐shaped relationship with biomarkers, including HDL, LDL, VLDL, and inflammation (Du et al., 2020; Wurtz et al., 2016), underscoring the bidirectional effects on cardiovascular and metabolic disease risks. Consistent with this evidence, we observed a nonlinear association between alcohol consumption and biological age in the two cohorts, YSCCS and VACS, in which the numbers of heavy and nonheavy alcohol consumers were relatively balanced. In the WIHS cohort, the predominance of nondrinkers to light drinkers may explain why a negative association with biological aging was similarly observed; insufficient observations were available to test the relationship between alcohol consumption of heavy drinkers and biological aging. The results of EAA showed that heavy consumption measured by PEth increased the epigenetic age by 1.60 years, consistent with a previous report of a 2‐year acceleration in individuals with AUD (Luo et al., 2020). On the other hand, EAAs were inversely associated with light to moderate alcohol consumption. Our results further support the previously reported nonlinear relationship between epigenetic age and alcohol consumption (Beach et al., 2015) and provide additional insights on alcohol effects on monocyte‐specific age.
It is noted that our results indicate the impacts of alcohol consumption on monocyte function and to some extent on peripheral immune function. We speculate that the nonlinear relationship between alcohol consumption and monocyte age is not contradictory to epidemiological studies. A possible explanation for the observed nonlinear relationship is that individuals with light to moderate alcohol use are more likely to follow a healthier lifestyle compared to heavy alcohol use. For example, in the YSCCS and VACS cohorts, individuals not reporting HAD showed a lower smoking rate and cannabis use than those with HAD, suggesting that a healthy lifestyle may slow or potentially even reverse the epigenetic aging process due to alcohol consumption. Another possibility is underreported alcohol consumption among participants. For example, in the VACS cohort, 17.02% of participants reported an AUDIT‐C score of 0, but the PEth level among those participants was greater than 8 ng/ml (Figure S11), which indicates active alcohol use (Eyawo et al., 2018). Inaccurate self‐reported alcohol consumption may result in biased findings of the slow acceleration of biological age in light‐to‐moderate drinkers. On the other hand, AUDIT and AUDIT‐C assess drinking over the past year whereas PEth is a phospholipid formed in the presence of alcohol that is detectable in blood up to 3 weeks after sustained alcohol intake (Bakhireva & Savage, 2011; Eyawo et al., 2018). These obstacles require further investigation with accurately assessed phenotypes or in longitudinal cohorts. Although the overall harm of alcohol consumption outweighs the benefit, causality has remained elusive (Chiva‐Blanch & Badimon, 2019).
We acknowledge some limitations of this study. A recent study suggests that the accuracy of DNAm‐based epigenetic clock estimation is affected by sample size (Zhang et al., 2019). Validation of the MonoDNAmAge clock in a large independent sample is warranted. DNAm in the three cohorts was measured in DNAm from whole blood. We expect that DNAm derived solely from monocytes may be a more accurate predictor of the effect of alcohol consumption. We are unable to examine DNAm age in past alcohol users, individuals with different patterns of alcohol consumption, or individuals presenting comorbidity with other substance or drug use. It is worth mentioning that DNAm patterns for a particular cell type are inherited through successive cell cycles and extended through a specific lineage (Kim & Costello, 2017). A future study using the within‐family design is warranted to address the heritable effects that might be a confounding factor to assess alcohol consumption on epigenetic age. Furthermore, differences in the effects of alcohol use between women and men are well documented, but the study lacked sufficient power to evaluate sex differences in the effect of alcohol exposure on biological aging.
CONCLUSION
We found that alcohol use affects epigenetic aging in a nonlinear manner, with heavy consumption increasing and nonheavy use decreasing epigenetic age. The use of cell‐type‐specific epigenetic clocks that are known to be directly affected by alcohol exposure may provide more precise information than that provided by more holistic epigenetic clocks that estimate more global effects on biological aging. Considering adverse effects of alcohol consumption on health, findings should be interpreted with caution. Our study expands previous knowledge and provides new insights into the effect of a spectrum of alcohol use on epigenetic aging.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
XL was responsible for bioinformatics data processing, statistical analyses, and manuscript preparation. ACJ provided DNA samples and clinical data and contributed to the interpretation of findings and manuscript preparation. BA contributed to the study design. RS and BA provided DNA methylation and phenotype data and contributed to the interpretation of findings and manuscript preparation. MC contributed to manuscript preparation. KX was responsible for the study design, study protocol, sample preparation, data analysis, interpretation of findings, and manuscript preparation. All authors read and approved the final manuscript.
Supporting information
Supinfo S1
ACKNOWLEDGMENTS
The authors appreciate the support of the Veterans Aging Study Cohort Biomarker Core (VACSBC), Yale Stress Center (YSC), Women’s Interagency HIV Study (WIHS), now the MACS/WIHS Combined Cohort Study (MWCCS), and the Yale Center of Genomic Analysis. Data in this manuscript were collected by YSC, VACSBC, and WIHS/MWCCS. The contents of this publication are solely the responsibility of the authors and do not represent the official views of the National Institutes of Health (NIH).
Liang, X. , Sinha, R. , Justice, A.C. , Cohen, M.H. , Aouizerat, B.E. & Xu, K. (2022) A new monocyte epigenetic clock reveals nonlinear effects of alcohol consumption on biological aging in three independent cohorts (N = 2242). Alcoholism: Clinical and Experimental Research, 46, 736–748. Available from: 10.1111/acer.14803
Funding information
The project was supported by the National Institute on Drug Abuse [R03 DA039745 (Xu), R01 DA038632 (Xu), R01 DA047063 (Xu and Aouizerat), R01 DA047820 (Xu and Aouizerat)], R01 DA013892 (Sinha), PL1 DA024859 (Sinha). COMpAAAS/Veterans Aging Cohort Study, a CHAART Cooperative Agreement, is supported by the National Institutes of Health: National Institute on Alcohol Abuse and Alcoholism (U24‐AA020794, U01‐AA020790, U01‐AA020795, U01‐AA020799; U10‐AA013566‐completed) and in kind by the US Department of Veterans Affairs. In addition to grant support from NIAAA, we gratefully acknowledge the scientific contributions of Dr. Kendall Bryant, our Scientific Collaborator. Additional grant support from the National Institute on Drug Abuse R01‐DA035616 is acknowledged. MWCCS (Principal Investigators): Atlanta CRS (Ighovwerha Ofotokun, Anandi Sheth, and Gina Wingood), U01‐HL146241; Bronx CRS (Kathryn Anastos and Anjali Sharma), U01‐HL146204; Brooklyn CRS (Deborah Gustafson and Tracey Wilson), U01‐HL146202; Data Analysis and Coordination Center (Gypsyamber D’Souza, Stephen Gange and Elizabeth Golub), U01‐HL146193; Chicago‐Cook County CRS (Mardge Cohen and Audrey French), U01‐HL146245; Northern California CRS (Bradley Aouizerat, Jennifer Price, and Phyllis Tien), U01‐HL146242; Metropolitan Washington CRS (Seble Kassaye and Daniel Merenstein), U01‐HL146205; Miami CRS (Maria Alcaide, Margaret Fischl, and Deborah Jones), U01‐HL146203; UAB‐MS CRS (Mirjam‐Colette Kempf, Jodie Dionne‐Odom, and Deborah Konkle‐Parker), U01‐HL146192; UNC CRS (Adaora Adimora), U01‐HL146194. The MWCCS is funded primarily by the National Heart, Lung, and Blood Institute (NHLBI), with additional co‐funding from the Eunice Kennedy Shriver National Institute Of Child Health & Human Development (NICHD), National Institute On Aging (NIA), National Institute Of Dental & Craniofacial Research (NIDCR), National Institute Of Allergy And Infectious Diseases (NIAID), National Institute Of Neurological Disorders And Stroke (NINDS), National Institute Of Mental Health (NIMH), National Institute On Drug Abuse (NIDA), National Institute Of Nursing Research (NINR), National Cancer Institute (NCI), National Institute on Alcohol Abuse and Alcoholism (NIAAA), National Institute on Deafness and Other Communication Disorders (NIDCD), National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute on Minority Health and Health Disparities (NIMHD), and in coordination and alignment with the research priorities of the National Institutes of Health, Office of AIDS Research (OAR). MWCCS data collection is also supported by UL1‐TR000004 (UCSF CTSA), P30‐AI‐050409 (Atlanta CFAR), P30‐AI‐050410 (UNC CFAR), and P30‐AI‐027767 (UAB CFAR).
REFERENCES
- Adams, W.L. , Barry, K.L. & Fleming, M.F. (1996) Screening for problem drinking in older primary care patients. JAMA, 276, 1964–1967. [PubMed] [Google Scholar]
- Adimora, A.A. , Ramirez, C. , Benning, L. , Greenblatt, R.M. , Kempf, M.C. , Tien, P.C. et al. (2018) Cohort Profile: The Women's Interagency HIV Study (WIHS). International Journal of Epidemiology, 47, 393–394i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon, M.C. , von Wyl, V. , Alden, C. , Sharp, G. , Robison, E. , Hessol, N. et al. (2005) The Women's Interagency HIV Study: an observational cohort brings clinical sciences to the bench. Clinical and Diagnostic Laboratory Immunology, 12, 1013–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badia, E. , Sacanella, E. , Fernandez‐Sola, J. , Nicolas, J.M. , Antunez, E. , Rotilio, D. et al. (2004) Decreased tumor necrosis factor‐induced adhesion of human monocytes to endothelial cells after moderate alcohol consumption. American Journal of Clinical Nutrition, 80, 225–230. [DOI] [PubMed] [Google Scholar]
- Bagnardi, V. , Rota, M. , Botteri, E. , Tramacere, I. , Islami, F. , Fedirko, V. et al. (2013) Light alcohol drinking and cancer: a meta‐analysis. Annals of Oncology, 24, 301–308. [DOI] [PubMed] [Google Scholar]
- Bakhireva, L.N. & Savage, D.D. (2011) Focus on: biomarkers of fetal alcohol exposure and fetal alcohol effects. Alcohol Research & Health, 34, 56–63. [PMC free article] [PubMed] [Google Scholar]
- Beach, S.R. , Dogan, M.V. , Lei, M.K. , Cutrona, C.E. , Gerrard, M. , Gibbons, F.X. et al. (2015) Methylomic aging as a window onto the influence of lifestyle: tobacco and alcohol use alter the rate of biological aging. Journal of the American Geriatrics Society, 63, 2519–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bild, D.E. , Bluemke, D.A. , Burke, G.L. , Detrano, R. , Diez Roux, A.V. , Folsom, A.R. et al. (2002) Multi‐ethnic study of atherosclerosis: objectives and design. American Journal of Epidemiology, 156, 871–881. [DOI] [PubMed] [Google Scholar]
- Blaine, S.K. , Nautiyal, N. , Hart, R. , Guarnaccia, J.B. & Sinha, R. (2019) Craving, cortisol and behavioral alcohol motivation responses to stress and alcohol cue contexts and discrete cues in binge and non‐binge drinkers. Addiction Biology, 24, 1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulias, K. , Lieberman, J. & Greer, E.L. (2016) An epigenetic clock measures accelerated aging in treated HIV infection. Molecular Cell, 62, 153–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiva‐Blanch, G. & Badimon, L. (2019) Benefits and risks of moderate alcohol consumption on cardiovascular disease: current findings and controversies. Nutrients, 12, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo, S. , Di Castelnuovo, A. , Donati, M.B. , Iacoviello, L. & de Gaetano, G. (2010) Alcohol consumption and mortality in patients with cardiovascular disease: a meta‐analysis. Journal of the American College of Cardiology, 55, 1339–1347. [DOI] [PubMed] [Google Scholar]
- Di Castelnuovo, A. , Costanzo, S. , Bagnardi, V. , Donati, M.B. , Iacoviello, L. & de Gaetano, G. (2006) Alcohol dosing and total mortality in men and women: an updated meta‐analysis of 34 prospective studies. Archives of Internal Medicine, 166, 2437–2445. [DOI] [PubMed] [Google Scholar]
- Donnadieu‐Rigole, H. , Mura, T. , Portales, P. , Duroux‐Richard, I. , Bouthier, M. , Eliaou, J.F. et al. (2016) Effects of alcohol withdrawal on monocyte subset defects in chronic alcohol users. Journal of Leukocyte Biology, 100, 1191–1199. [DOI] [PubMed] [Google Scholar]
- Du, D. , Bruno, R. , Blizzard, L. , Venn, A. , Dwyer, T. , Smith, K.J. et al. (2020) The metabolomic signatures of alcohol consumption in young adults. European Journal of Preventive Cardiology, 27, 840–849. [DOI] [PubMed] [Google Scholar]
- Eyawo, O. , McGinnis, K.A. , Justice, A.C. , Fiellin, D.A. , Hahn, J.A. , Williams, E.C. et al. (2018) Alcohol and mortality: combining self‐reported (AUDIT‐C) and biomarker detected (PEth) alcohol measures among HIV infected and uninfected. Journal of Acquired Immune Deficiency Syndromes, 77, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorito, G. , McCrory, C. , Robinson, O. , Carmeli, C. , Rosales, C.O. , Zhang, Y. et al. (2019) Socioeconomic position, lifestyle habits and biomarkers of epigenetic aging: a multi‐cohort analysis. Aging, 11, 2045–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, J. , Hastie, T. & Tibshirani, R. (2010) Regularization paths for generalized linear models via coordinate descent. Journal of Statistical Software, 33, 1–22. [PMC free article] [PubMed] [Google Scholar]
- Collaborators GBDA . (2018) Alcohol use and burden for 195 countries and territories, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. The Lancet, 392, 1015–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum, G. , Guinney, J. , Zhao, L. , Zhang, L. , Hughes, G. , Sadda, S. et al. (2013) Genome‐wide methylation profiles reveal quantitative views of human aging rates. Molecular Cell, 49, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath, S. (2013) DNA methylation age of human tissues and cell types. Genome Biology, 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath, S. & Levine, A.J. (2015) HIV‐1 infection accelerates age according to the epigenetic clock. Journal of Infectious Diseases, 212, 1563–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath, S. & Raj, K. (2018) DNA methylation‐based biomarkers and the epigenetic clock theory of ageing. Nature Reviews Genetics, 19, 371–384. [DOI] [PubMed] [Google Scholar]
- Houseman, E.A. , Accomando, W.P. , Koestler, D.C. , Christensen, B.C. , Marsit, C.J. , Nelson, H.H. et al. (2012) DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics, 13, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, D.W. , Sherman, B.T. , Tan, Q. , Kir, J. , Liu, D. , Bryant, D. et al. (2007) DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Research, 35, W169–W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe, A.E. & Irizarry, R.A. (2014) Accounting for cellular heterogeneity is critical in epigenome‐wide association studies. Genome Biology, 15, R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice, A.C. , Dombrowski, E. , Conigliaro, J. , Fultz, S.L. , Gibson, D. , Madenwald, T. et al. (2006) Veterans Aging Cohort Study (VACS): overview and description. Medical Care, 44, S13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, M. & Costello, J. (2017) DNA methylation: an epigenetic mark of cellular memory. Experimental & Molecular Medicine, 49, e322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kresovich, J.K. , Martinez Lopez, A.M. , Garval, E.L. , Xu, Z. , White, A.J. , Sandler, D.P. et al. (2021) Alcohol consumption and methylation‐based measures of biological age. Journals of Gerontology. Series A, Biological Sciences and Medical Sciences, 76, 2107–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J.H. , Kong, K.A. , Lee, D.H. , Choi, Y.H. & Jung, K.Y. (2018) Validation and proposal for cut‐off values of an abbreviated version of the alcohol use disorder identification test using the Korean National Health and Nutrition Examination Survey. Clinical and Experimental Emergency Medicine, 5, 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, M.E. , Lu, A.T. , Quach, A. , Chen, B.H. , Assimes, T.L. , Bandinelli, S. et al. (2018) An epigenetic biomarker of aging for lifespan and healthspan. Aging, 10, 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Zou, D. , Li, Z. , Gao, R. , Sang, J. , Zhang, Y. et al. (2019) EWAS Atlas: a curated knowledgebase of epigenome‐wide association studies. Nucleic Acids Research, 47, D983–D988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, X. , Justice, A.C. , So‐Armah, K. , Krystal, J.H. , Sinha, R. & Xu, K. (2021) DNA methylation signature on phosphatidylethanol, not on self‐reported alcohol consumption, predicts hazardous alcohol consumption in two distinct populations. Molecular Psychiatry, 26(6), 2238–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. , Marioni, R.E. , Hedman, Å.K. , Pfeiffer, L. , Tsai, P.‐C. , Reynolds, L.M. et al. (2018) A DNA methylation biomarker of alcohol consumption. Molecular Psychiatry, 23, 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Reynolds, L.M. , Ding, J. , Hou, L. , Lohman, K. , Young, T. et al. (2017) Blood monocyte transcriptome and epigenome analyses reveal loci associated with human atherosclerosis. Nature Communications, 8, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Otin, C. , Blasco, M.A. , Partridge, L. , Serrano, M. & Kroemer, G. (2013) The hallmarks of aging. Cell, 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, A.T. , Quach, A. , Wilson, J.G. , Reiner, A.P. , Aviv, A. , Raj, K. et al. (2019) DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging, 11, 303–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, A. , Jung, J. , Longley, M. , Rosoff, D.B. , Charlet, K. , Muench, C. et al. (2020) Epigenetic aging is accelerated in alcohol use disorder and regulated by genetic variation in APOL2. Neuropsychopharmacology, 45, 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf, T.U. , Wilkinson, P.A. , Cameron, M.J. , Ghneim, K. , Chiang, C. , Wertheimer, A.M. et al. (2017) Human monocyte subsets are transcriptionally and functionally altered in aging in response to pattern recognition receptor agonists. The Journal of Immunology, 199, 1405–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki, S. , Otsuka, I. , Shinko, Y. , Horai, T. , Hirata, T. , Yamaki, N. et al. (2021) Epigenetic clock analysis in children with fetal alcohol spectrum disorder. Alcoholism, Clinical and Experimental Research, 45, 329–337. [DOI] [PubMed] [Google Scholar]
- Piano, M.R. , Tiwari, S. , Nevoral, L. & Phillips, S.A. (2015) Phosphatidylethanol levels are elevated and correlate strongly with AUDIT scores in young adult binge drinkers. Alcohol and Alcoholism, 50, 519–525. [DOI] [PubMed] [Google Scholar]
- Quach, A. , Levine, M.E. , Tanaka, T. , Lu, A.T. , Chen, B.H. , Ferrucci, L. et al. (2017) Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging, 9, 419–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds, L.M. , Taylor, J.R. , Ding, J. , Lohman, K. , Johnson, C. , Siscovick, D. et al. (2014) Age‐related variations in the methylome associated with gene expression in human monocytes and T cells. Nature Communications, 5, 5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, O. , Chadeau Hyam, M. , Karaman, I. , Climaco Pinto, R. , Ala‐Korpela, M. , Handakas, E. et al. (2020) Determinants of accelerated metabolomic and epigenetic aging in a UK cohort. Aging Cell, 19, e13149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen, A.D. , Robertson, K.D. , Hlady, R.A. , Muench, C. , Lee, J. , Philibert, R. et al. (2018) DNA methylation age is accelerated in alcohol dependence. Transl Psychiatry, 8, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saare, M. , Tserel, L. , Haljasmagi, L. , Taalberg, E. , Peet, N. , Eimre, M. et al. (2020) Monocytes present age‐related changes in phospholipid concentration and decreased energy metabolism. Aging Cell, 19, e13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchukina, I. , Bagaitkar, J. , Shpynov, O. , Loginicheva, E. , Porter, S. , Mogilenko, D.A. et al. (2021) Enhanced epigenetic profiling of classical human monocytes reveals a specific signature of healthy aging in the DNA methylome. Nature Aging, 1, 124–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, C. & Baik, I. (2016) Associations between alcohol consumption and leukocyte telomere length modified by a common polymorphism of ALDH2. Alcoholism, Clinical and Experimental Research, 40, 765–771. [DOI] [PubMed] [Google Scholar]
- Stephenson, M. , Bollepalli, S. , Cazaly, E. , Salvatore, J.E. , Barr, P. , Rose, R.J. et al. (2021) Associations of alcohol consumption with epigenome‐wide DNA methylation and epigenetic age acceleration: individual‐level and co‐twin comparison analyses. Alcoholism, Clinical and Experimental Research, 45, 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, S.H. , Reuben, A. , Brzezinski, W.A. , Koch, D.G. , Basile, J. , Randall, P.K. et al. (2009) Preliminary evaluation of phosphatidylethanol and alcohol consumption in patients with liver disease and hypertension. Alcohol and Alcoholism, 44, 464–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strandberg, T.E. , Strandberg, A.Y. , Saijonmaa, O. , Tilvis, R.S. , Pitkala, K.H. & Fyhrquist, F. (2012) Association between alcohol consumption in healthy midlife and telomere length in older men. The Helsinki Businessmen Study. European Journal of Epidemiology, 27, 815–822. [DOI] [PubMed] [Google Scholar]
- Sureshchandra, S. , Raus, A. , Jankeel, A. , Ligh, B.J.K. , Walter, N.A.R. , Newman, N. et al. (2019) Dose‐dependent effects of chronic alcohol drinking on peripheral immune responses. Scientific Reports, 9, 7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo, G. (1998) Monocytes, alcohol use, and altered immunity. Alcoholism, Clinical and Experimental Research, 22, 216S–219S. [DOI] [PubMed] [Google Scholar]
- Viel, G. , Boscolo‐Berto, R. , Cecchetto, G. , Fais, P. , Nalesso, A. & Ferrara, S.D. (2012) Phosphatidylethanol in blood as a marker of chronic alcohol use: a systematic review and meta‐analysis. International Journal of Molecular Sciences, 13, 14788–14812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze, S.I. (2012) Model selection and psychological theory: a discussion of the differences between the Akaike information criterion (AIC) and the Bayesian information criterion (BIC). Psychological Methods, 17, 228–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtz, P. , Cook, S. , Wang, Q. , Tiainen, M. , Tynkkynen, T. , Kangas, A.J. et al. (2016) Metabolic profiling of alcohol consumption in 9778 young adults. International Journal of Epidemiology, 45, 1493–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Vallerga, C.L. , Walker, R.M. , Lin, T. , Henders, A.K. , Montgomery, G.W. et al. (2019) Improved precision of epigenetic clock estimates across tissues and its implication for biological ageing. Genome Medicine, 11, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Hu, Y. , Justice, A.C. , Li, B. , Wang, Z. , Zhao, H. et al. (2017) DNA methylation signatures of illicit drug injection and hepatitis C are associated with HIV frailty. Nature Communications, 8, 2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supinfo S1