

Abstract
Background
High doses of doxorubicin (DOX) put cancer patients at risk for developing dilated cardiomyopathy (DC). Previously, we showed that DOX treatment decreases SIRT3, the main mitochondrial deacetylase and increases protein acetylation in rat cardiomyocytes. Here we hypothesize that SIRT3 expression can attenuate DOX induced DC in vivo by preventing the acetylation of mitochondrial proteins.
Methods
Non-transgenic, M3-SIRT3 (short isoform) and M1-SIRT3 (mitochondrial localized) transgenic mice were treated with DOX for four weeks (8mg/kg of body weight per week). Echocardiography was performed to assess cardiac structure and function and validated by immunohistochemistry and immunofluorescence (n=4–10). Mass spectrometry was performed on cardiac mitochondrial peptides in saline (n=6) and DOX (n=5) treated hearts. Validation was performed in DOX treated primary rat and human induced stem cell derived cardiomyocytes transduced with adenoviruses for M3-SIRT3 and M1-SIRT3 and deacetylase deficient mutants. (n=4–10).
Results
Echocardiography revealed that M3-SIRT3 transgenic mice were partially resistant to DOX induced changes to cardiac structure and function whereas M1-SIRT3 expression prevented cardiac remodelling and dysfunction. In DOX hearts, 37 unique acetylation sites on mitochondrial proteins were altered. Pathway analysis revealed these proteins are involved in energy production, fatty acid metabolism, and oxidative stress resistance. Increased M1-SIRT3 expression in primary rat and human cardiomyocytes attenuated DOX-induced superoxide formation, whereas deacetylase deficient mutants were unable to prevent oxidative stress.
Conclusion
DOX reduced SIRT3 expression and markedly affected the cardiac mitochondrial acetylome. Increased M1-SIRT3 expression in vivo prevented DOX-induced cardiac dysfunction, suggesting that SIRT3 could be a potential therapeutic target for mitigating DOX-induced DC.
Keywords: Dilated Cardiomyopathy, Doxorubicin, Sirtuins, Acetylation, Mass Spectrometry, Reactive Oxygen Species, Superoxide Dismutase
1. Introduction
Doxorubicin (DOX) is an anthracycline chemotherapeutic used in the treatment of cancer1,2. Clinical use of DOX is limited by the dose-dependent risk for progressive dilated cardiomyopathy (DC)2,3. DC is characterized by enlargement of the left ventricular (LV) chamber and ventricular wall thinning as a result of cardiomyocyte death, leading to inefficient systolic and diastolic function4,5. Therapies that prevent chemotherapy induced DC are limited and molecular mechanisms responsible for the pathogenesis of DOX-induced cardiotoxicity have not been fully elucidated, though mitochondrial dysfunction and oxidative stress are thought to be involved6,7.
Sirtuins (SIRT) are nicotinamide adenine dinucleotide-regulated lysine deacetylases, which remove acetyl-groups from lysine residues of proteins8. In the heart, ~60% of mitochondrial proteins undergo-reversible lysine acetylation9. SIRT3 regulates the reversible acetylation of a range of mitochondrial enzymes in the heart8. The murine SIRT3 gene codes for three transcripts that produce three SIRT3 isoforms (M1, M2, M3)10. M1-/M2-SIRT3 encode the full-length isoform which is localized to the mitochondria and act as the main mitochondrial deacetylase. The shorter M3-SIRT3 form has deacetylase activity, but lacks the N-terminal mitochondrial localization signal and is poorly localized to mitochondria10. Previously, we showed that increased expression of M1-SIRT3 attenuates DOX-induced mitochondrial ROS production and improves mitochondrial respiration in H9c2 rat cardiomyocytes6.
In this study we investigated how increased SIRT3 expression affects the murine heart following DOX treatment. Using mass spectrometry, we are the first to identify how DOX affects the cardiac mitochondrial acetylome in vivo. Moreover, we show that increased M1-SIRT3 expression can prevent DOX-induced cardiac dysfunction and oxidative stress in human and primary rodent cardiomyocytes and hearts.
2. Methods
2.1. Data Availability
The source data that support the findings of this study are available from the corresponding author upon reasonable request.
2.2. Generation of M3-SIRT3 and M1-SIRT3 Transgenic Mice
Transgenic mice were generated by injecting transgene fragments (αMHC -M3-SIRT3 and MCK-M1-SIRT3) into fertilized C57BL/6 mouse oocytes by the Genetically Engineered Mouse Core at Baylor College of Medicine. Details about mouse generation are in the data supplement11–13.
2.3. Animal Housing and DOX Treatment
Ten-week-old mice were treated intraperitoneally (i.p.) with 8.0mg/kg body weight of DOX for 4 weeks and compared to non-transgenic saline (0.9%) treated littermates. Mice were euthanized by a single i.p. dose of sodium pentobarbital (90mg/kg). All animal procedures were conducted in accordance with the University of Manitoba Animal Welfare Committee and the Canadian Council on Animal Care. Additional details in data supplement (Fig S1).
2.4. Mitochondrial Peptide and Mass Spectrometry Preparation
Mitochondrial protein was isolated from freshly harvested heart tissue using the Sigma mitochondrial isolation kit (MITOISO1). Mitochondrial protein concentration was determined by Pierce Detergent Compatible Bradford Assay Reagent (Thermo, 1863028). Protein clean-up was performed using Sera-Mag Carboxylate-Modified Magnetic SpeedBeads (GE Life Sciences, 45152105050350, 65152105050250). Proteins were trypsinzed to peptides using mass spec grade Trypsin Lys-C Mix (Promega, V5071).
Immunoprecipitation of acetylated peptides was performed using acetyl-lysine antibody prebound to agarose beads (ImmuneChem, ICP0388). 200 fmol of acetyl-lysine peptide standard (LVSSVSDLPacKR) was added to the resuspended peptides. Peptides were immunoprecipitated overnight at 4°C and eluted in 0.15% trifluoroacetic acid. Detailed sample preparation appears in the data supplement.
2.5. Mass Spectrometry of Acetylated Cardiac Mitochondrial Peptides
Analysis of peptide digests was performed on an Orbitrap Q Exactive HF-X instrument (Thermo Fisher Scientific, Bremen, Germany) at the Manitoba Center for Proteomics and Systems Biology. Data acquisition on the Orbitrap Q Exactive HF-X instrument was configured for data-dependent method using the full MS/DD−MS/MS setup in a positive mode. Detailed mass spectrometry methods are in the data supplement.
2.6. Mass Spectrometry Bioinformatic Analysis
Raw spectra for each of the 1D LC-MS runs in experiment (n=6 Non-TG-CON, n=5 Non-Tg-DOX) were extracted using the Proteome Discoverer bundled tool and searched against the Uniprot database of mouse protein sequences (June 2016). Label-free quantitation following the peptide identification was done using in-house tools. Intensity was assigned as the sum of all MS2 fragment intensities in all sequence-matching spectra, converted into a log2 scale. The raw mass spectrometry data (MGF format) and peptide expression matrix have been deposited to the University of California-San Diego MassIVE data repository (massive.ucsd.edu) under accession MSV000086892. Detailed bioinformatic methods in data supplement.
2.7. Transthoracic Echocardiography
Echocardiography (Vevo 2100, Fujifilm-Visual Sonics, ON Canada) to assess cardiac morphology and function was performed on 10–14 week-old female mice that were imaged under mild anesthesia (induced with 3% isoflurane and 1.0L/min oxygen and maintained at 1 – 1.5% isoflurane and 1.0 L/min oxygen) during echocardiography as described previously14. Additional details are in the data supplement.
2.8. Trichrome Staining for Cardiac Fibrosis
Hearts were excised and fixed in TheraLin tissue fixative for 24 hours. Fixed tissue was parafilm embedded and sectioned. Cardiac fibrosis was assessed using Trichrome Stain Kit (Sigma, HT15–1KT). A minimum of four fields of view were imaged from each mouse using a Zeiss brightfield microscope and values were averaged.
2.9. Cleaved Caspase-3 Immunohistochemistry and 4-Hydroxynonenal Staining
Cardiac tissue sections were immunostained with Cleaved caspase-3 (9661S, Cell Signalling Tech.), or 4-hydroxy-2,3-transnonenal (4-HNE; ab46545, Abcam). Imaging was performed on Zeiss Brightfield microscope. A minimum of four cardiac regions were imaged from each mouse and values were averaged.
2.10. Cell Culture
Primary rat neonatal cardiomyocytes (PRNCs) were isolated by enzymatic dissociation from one-to-two day old Sprague-Dawley rats as previously described15. After plating PRNC were transduced with adenoviruses for 24h and then treated with 10μM DOX. Human induced pluripotent stem cell (hiPSC) derived cardiomyocyte iCells (Fujifilm Cellular Dynamics, CMC-100-010-000.5) were thawed and plated according to manufactures guidelines. Seven days post-plating cells were transduced. Full-length SIRT3 (Ad.M1-SIRT3) and truncated (Ad.M3-SIRT3) as well as deacetylase-deficient SIRT3 mutant adenoviruses (Ad.M1-SIRT3-N164A and Ad.M3-SIRT3-N87A) were prepared as described6. Details of cell culture methods are in the data supplement.
2.11. Mitochondrial H2O2 Efflux and Superoxide Formation
H2O2 efflux by isolated mitochondria was measured using an extramitochondrial fluorescence detection system based on16–18, but using 10 μM Amplex Ultrared. Superoxide production by isolated mitochondria was measured directly with MitoSOX™ using a fluorescence-based-HPLC method based on19–23 with modifications. Detailed methods can be found in the data supplement. Staining for mitochondrial ROS in live cells was performed using 5μM MitoSOX™ Red mitochondrial superoxide indicator (M36008) in HBSS/Ca/Mg (Gibco, 14025–092). Cells were stain was applied for 15min in at 37°C and 5% CO2 and washed with warm HBSS, then fixed with 4% PFA for 15mins at 4°C. Images were taken under Cy3 (red) and DAPI (blue) channels on the Zeiss (Axio Observer Z1) Epifluorescence microscope.
2.12. RNA extraction and quantitative RT-PCR
RNA was isolated from tissues using a Qiashredder column and further purified using the RNeasy kit (Qiagen, Valencia CA). cDNA was synthesized using the Protoscript kit (New England Biolabs). The QuantiTect SYBR Green PCR kit (Qiagen) was used to monitor amplification of cDNA on a CFX96 real-time PCR detection machine (Bio-Rad). A geomean of CypA, Gapdh, and Eif2a was used to normalize gene expression. Primer sequences are listed in Table S1 in data supplement.
2.13. Western Immunoblotting
Western blotting was performed as previously described6. Briefly, 15–20μg of total cardiac protein was used for SDS-PAGE, transferred to nitrocellulose and probed using antibodies specified. For chemiluminescence, secondary antibodies (Santa Cruz Biotechnology) were applied, and SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) was used for detection. Detailed immunoblotting methods and reagents can be found in data supplement.
2.14. Statistical Analysis
Figures were made using Prism 9 (Graphpad, San Diego, CA, USA), NetworkAnalyst 3.024, and R programming visualised in RStudio (Version 1.3.1093). Graphical figures created with BioRender.com. Two-tailed Student’s t test, or two-way analysis of variance followed by Tukey’s post-hoc analysis was used to determine statistical difference between groups. Values are mean ± SD. Bars show multiple comparisons between groups. A p-value of < 0.05 was considered statistically significant.
3. Results
3.1. DOX Alters the Acetylation of Cardiac Mitochondrial Peptides in Mice
DOX inhibits SIRT3 expression in mouse hearts (Fig. S2A–C in data supplement)6, therefore we investigated how DOX affected cardiac protein acetylation. Immunoblotting of cardiac protein using an acetylated lysine antibody revealed a ~1.5-fold (p=0.001) increase in the hearts of Non-Tg DOX treated mice compared to saline controls (Fig 1A, B). Furthermore, M3-SIRT3 and M1-SIRT3 mice were resistant (p<0.001 and p<0.001 vs Non-Tg-DOX) to increases in lysine acetylation (Fig. 1A, B).
Figure 1. The Cardiac Mitochondrial Acetylome is Involved in Metabolic Processes.
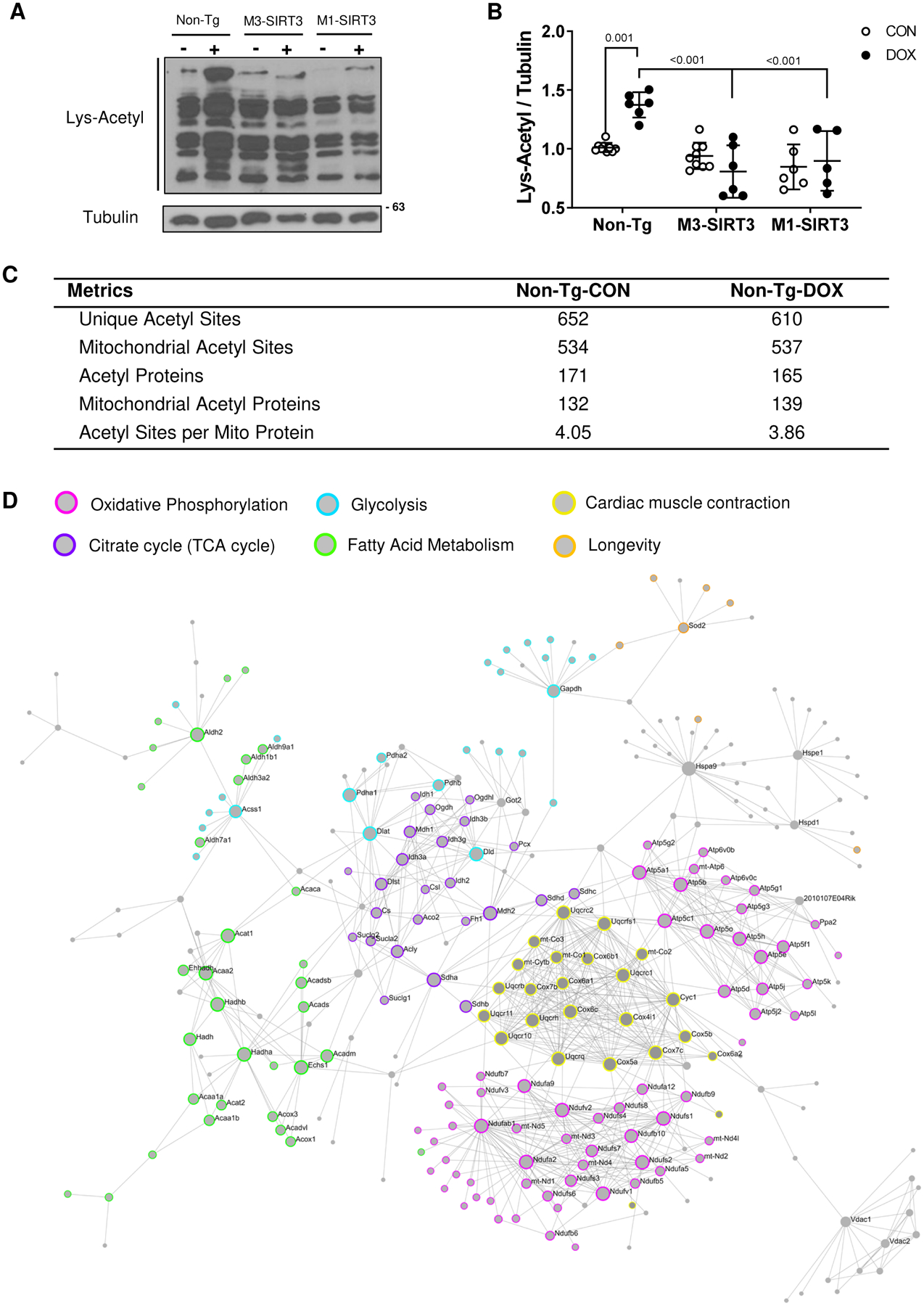
(A) Representative image of acetylated lysine western blot from total cardiac lysates. (B) Optical density quantification of Lys-Acetyl blot relative to tubulin (n=6–10, females) (C) Acetyl-proteomic metrics for saline and DOX treated Non-Tg mice (n=6 CON males, n=5 DOX males) (D) First order protein interaction network of mitochondrial cardiac acetylproteome. Identified proteins from both saline and DOX groups were used as seeds to generate the network using Network Analyst 3.0. The STRING Interactome function was used with a 95% confidence score and requiring experimental evidence. Colours used to highlight KEGG pathways of identified proteins in the network. Pink (oxidative phosphorylation), Purple (TCA cycle), Blue (glycolysis), Green (fatty acid metabolism), Yellow (cardiac muscle contraction), Orange (longevity). Sample size refers to biological replicates. Values are mean ± SD. Statistics are two-way ANOVA with Tukey’s post-hoc test.
Previous mass spectrometry studies evaluated the role of SIRT3 on protein acetylation; however, the effect of DOX on the cardiac mitochondrial acetylome was not examined. Using non-label quantitative mass spectrometry, we examined how DOX affects the acetylation of cardiac mitochondrial proteins (Fig. S3 in data supplement). We identified 652 unique acetylation sites in controls samples and 610 in DOX samples, respectively (Fig 1C). Among these, 534 mitochondrial acetylation sites were detected in the controls and 537 sites were observed in the DOX heart (Fig. 1C). We identified 171 and 165 acetylated proteins in Non-Tg-CON and Non-Tg-DOX samples respectively (Fig 1C). Of those, 132 and 139 were mitochondrial proteins. Interestingly, we did not see an increase in the average acetylation sites per mitochondrial protein in the DOX treated group (3.86) when compared to the controls (4.05). Using all identified acetylated mitochondrial proteins, a first order protein interaction network was generated to represent the cardiac mitochondrial acetylome and their potential protein interactions (Fig. 1D). A KEGG pathway analysis of the network identified that the acetylated seed proteins and their protein interactions were involved in mitochondrial processes such as oxidative phosphorylation, carbon metabolism, TCA cycle, glycolysis, and fatty acid metabolism (Table S2 in data supplement). Pathways of relevance to mitochondria and cardiac energy production were highlighted within the network (Fig 1D). These data suggest that acetylation modifications likely have key roles in the dysregulation of mitochondrial processes.
3.2. Increased SIRT3 Protein Prevents DOX Induced Cardiac Remodelling and Dysfunction
Four weekly i.p. injections of DOX (8.0mg/kg of body weight) were administered to M3-SIRT3, M1-SIRT3 and non-transgenic (Non-Tg) mice. Consistent with previous findings6, DOX significantly (p=0.005) reduced cardiac SIRT3 protein levels in Non-Tg mice, whereas hearts from both M3-SIRT3 and M1-SIRT3 transgenic animals were resistant to decreased SIRT3 protein expression (Fig. S2B, S2C in data supplement).
All mice regardless of genotype exhibited a progressive decrease in body weight over the four-week treatment period (Table 1, Fig S2D in data supplement). The weights of hearts isolated from Non-Tg and M3-SIRT3 mice decreased significantly (greater than 20%, p=0.007) with DOX when compared to Non-Tg saline controls (Table 1). Hearts from M1-SIRT3 treated with DOX also exhibited marginal decreases (~15%) that were not statistically significant (n.s., Table 1). However, when comparing the ratio of heart weight to body weight as a percentage there was no change among the groups, suggesting that DOX treated hearts decrease proportionally with body size (Table 1).
Table 1.
Body Characteristics and Echocardiography Measurements for Non-Tg, M3-SIRT3 and M1-SIRT3 Mice Treated with 8.0mg/kg of DOX for 4 Weeks.
| Genotype-Treatment | Non-Tg-CON | Non-Tg-DOX | M3-SIRT3-CON | M3-SIRT3-DOX | M1-SIRT3-CON | M1-SIRT3-DOX |
|---|---|---|---|---|---|---|
| Number of animals | (n=8) | (n=8) | (n=8) | (n=9) | (n=9) | (n=10) |
| Body Characteristics | ||||||
| Body weight (g) | 24.8±2.9 | 17.0±2.4* | 24.2±2.3† | 19.2±2.5*,‡ | 24.1±3.5† | 19.8±2.2*,‡ |
| Heart weight (mg) | 123±17 | 94±12* | 125±25† | 97±14*,‡ | 134±13† | 114±10 |
| HW/BW (%) | 0.50±0.08 | 0.56±0.09 | 0.52±0.10 | 0.51±0.09 | 0.57±0.11 | 0.57±0.06 |
| Cardiac Structure | ||||||
| LVPWd (mm) | 0.71±0.08 | 0.59±0.07* | 0.69±0.08 | 0.64±0.08 | 0.70±0.07† | 0.67±0.12 |
| LVAWs (mm) | 1.06±0.09 | 0.88±0.15* | 1.05±0.14 | 0.97±0.19 | 1.13±0.13† | 1.10±0.15† |
| LVAWd (mm) | 0.74±0.03 | 0.61±0.05 | 0.74±0.09 | 0.66±0.12 | 0.81±0.15† | 0.73±0.11 |
| LVIDs (mm) | 2.79±0.30 | 2.80±0.24 | 2.65±0.34 | 2.67±0.33 | 2.98±0.19 | 2.84±0.14 |
| LVIDd (mm) | 3.85±0.28 | 3.74±0.33 | 3.81±0.28 | 3.75±0.24 | 3.95±0.18 | 3.92±0.23 |
| IVSs (mm) | 0.95±0.09 | 0.80±0.10 | 0.98±0.05 | 0.96±0.11 | 0.99±0.20† | 1.02±0.14† |
| IVSd (mm) | 0.66±0.07 | 0.58±0.01 | 0.66±0.05 | 0.64±0.12 | 0.73±0.17 | 0.72±0.11 |
| LV Vols (μL) | 29.8±8.1 | 29.8±6.0 | 26.4±8.1 | 26.8±7.7 | 34.7±5.4 | 30.8±3.8 |
| LV Vold (μL) | 64.3±11.7 | 60.3±12.1 | 62.8±10.4 | 60.3±8.9 | 68.2±7.4 | 67.1±9.5 |
| Cardiac Function | ||||||
| CO (mL/min) | 18.6±2.4 | 12.8±3.6* | 18.4±2.7† | 16.8±2.7 | 17.9±2.6† | 19.7±4.2† |
| FS (%) | 29.1±2.5 | 23.9±2.0* | 30.3±4.0† | 28.9±4.1† | 26.2±3.0 | 28.1±3.3 |
| SV (μL) | 36.9±4.8 | 28.5±5.8 | 36.6±4.8 | 34.3±4.8 | 36.4±5.6 | 38.2±8.6† |
| E’/A’ Ratio | 1.4±0.3 | 1.1±0.2* | 1.5±0.3† | 1.4±0.3 | 1.4±0.3 | 1.4±0.2 |
| LV MPI | 0.45±0.08 | 0.65±0.10* | 0.43±0.12† | 0.41±0.06† | 0.41±0.10† | 0.42±0.08† |
| BPM | 505±20 | 445±52* | 501±32 | 489±39 | 492±28 | 518±49† |
HW, Heart Weight; BW, Body Weight; LVAW, left ventricular anterior wall; LVPW, left ventricular posterior wall; IVS, intraventricular septum; LV Vol, left ventricular volume; s, systole; d, diastole; CO, cardiac output; FS, fraction shortening; SV, stroke volume; LVMPI, left ventricular myocardial performance index; BPM, beats per minute. Female mice.
p<0.05 vs Non-Tg-CON,
p<0.05 vs Non-Tg-DOX,
p<0.05 vs related CON group, values are mean +/− SD. Comparisons by Two-way ANOVA with Tukey’s post-hoc test.
Transthoracic echocardiography measurements were performed on mice from all six experimental groups and revealed severe cardiac remodelling in DOX treated Non-Tg mice as indicated by decreased LV posterior and anterior wall thickness (17% and 18% respectively, p=0.034 and p=0.023, Fig. 2A, Table 1). Expression of the M3-SIRT3 transgene partially attenuated (LVPWd 10% decrease vs Non-Tg-CON, n.s.), whereas M1-SIRT3 expression rescued (LVPWd n.s. vs Non-Tg-CON) DOX-induced cardiac remodelling (Fig.2A, Table1). Similarly, DOX treatment markedly decreased LV mass in Non-Tg mice (p=0.025). Interestingly, M1-SIRT3 expression prevented these effects of DOX on LV mass (p=0.004 vs Non-Tg-DOX), whereas M3-SIRT3 expression only partially prevented (n.s. vs Non-Tg-DOX) the effects of DOX (Fig 2B).
Figure 2. M1-SIR3 Transgenic Mice are Resistant to DOX Induced Cardiac Remodelling and Dysfunction.
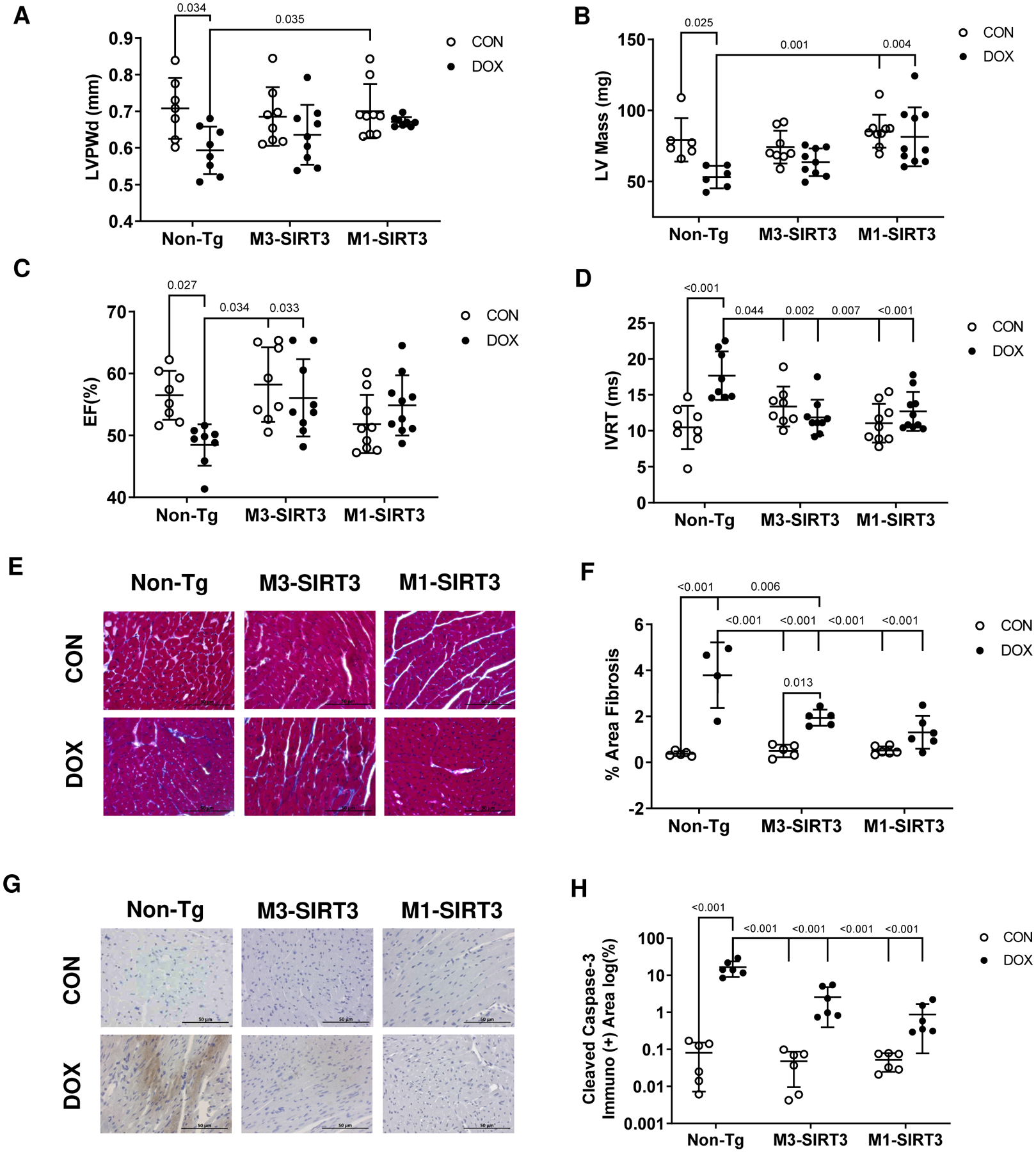
M3-SIRT3, M1-SIRT3 and Non-Tg controls treated with 8.0mg/kg of DOX or saline once a week for four weeks. (A) Left ventricular posterior wall thickness during diastole. (B) Left ventricular mass. (C) Ejection fraction. (D) Isovolumetric relaxation time (n=8–10) (E) Representative images of trichrome staining. (F) Area fibrosis quantification of trichrome staining (n=4–6) (G) Representative images of cleaved caspase-3 staining. (H) Area quantification of immuno-positive cleaved caspase-3 immunohistochemistry (n=6). Scale bars = 50μm. Sample size refers to biological replicates. Female mice. Values are mean ± SD. Statistics are two-way ANOVA with Tukey’s post-hoc test.
In Non-Tg mice, DOX treatment reduced ejection fraction below 50% compared to saline controls (p=0.027), whereas both M1-and M3-SIRT3 mice were resistant (n.s. vs Non-Tg-CON) to the DOX-induced impairment of systolic function (Fig 2C). Additionally, cardiac output (p=0.007) and LV fractional shortening (p=0.031) were decreased in Non-Tg mice treated with DOX, whereas M3- and M1-SIRT3 transgenic mice were resistant to these perturbations (Table 1). An increase in isovolumetric relaxation time, indicative of diastolic dysfunction, was observed in the hearts of Non-Tg DOX treated animals (p<0.001, Fig 2D). Expression of M3- and M1-SIRT3 transgenes in mice prevented DOX-induced increases in isovolumetric relaxation time (n.s. vs Non-Tg-CON, Fig 2D). The myocardial performance index indicated overall cardiac dysfunction in DOX treated Non-Tg mice, whereas the myocardial performance index was preserved in both M3- and M1-SIRT3 mice (n.s. vs Non-Tg-CON). To further examine how DOX impacts cardiac function we performed radial and longitudinal strain, and strain rate measurements at baseline (Fig S4A–D in data supplement) and after four weeks of treatment (Fig S4E–H in data supplement). Post treatment DOX significantly decreased radial strain (p=0.013), increased longitudinal strain (p=0.007). Similarly, there was a decrease in radial (p=0.012) strain rate and an increase in longitudinal strain rate (p=0.026, Video 1 in data supplement). The M1-SIRT3 mice were resistant to all these changes (n.s. vs Non-Tg-CON, Video 2 in data supplement). Therefore, DOX treatment induced cardiac remodeling as well as cardiac dysfunction in mice that was characteristic of DC. These findings show that increased cardiac expression of mitochondrial M1-SIRT3 preserved LV mass and cardiac function in the presence of DOX.
To investigate whether cardiac dysfunction was a result of DOX-induced fibrosis, we performed trichrome staining of cardiac tissue in the mice (Fig. 2E). Although, DOX increased the area of fibrosis in all groups, M1-SIRT3 hearts exhibited significantly less fibrotic staining (1.3% area fibrosis) when compared to hearts from DOX-treated Non-Tg (3.79% area fibrosis) and M3-SIRT3 mice (1.9% area fibrosis, Fig. 2E, F). In addition to fibrosis, cardiomyocyte death is a hallmark of DC. Using cleaved-caspase-3 as a marker of cardiac apoptosis, we performed immunohistochemistry (Fig. 2G). DOX treatment increased immuno-positive staining of cleaved-caspase-3 compared to saline treatment in all groups (p<0.001 vs Non-Tg-CON); however, the area of staining was reduced in M1-SIRT3 hearts (p<0.001 vs Non-Tg-DOX, Fig. 2G, H). Additionally, gene expression for proteins involved in apoptosis Casp4 (~2-fold, p=0.017) and Dapl1 (~8-fold, p<0.001) were increased in the hearts of DOX treated Non-Tg mice and attenuated in M3-SIRT3 and M1-SIRT3 mice (Fig S5A, S5B in data supplement). These data suggest SIRT3 prevents DOX-induced apoptosis.
3.3. DOX Alters Acetylation of Cardiac Mitochondrial Enzymes
To further investigate how SIRT3 attenuates cardiac dysfunction, we assessed mitochondrial protein acetylation. Using mass spectrometry, we identified 36 peptides which exhibited altered acetylation following DOX treatment in Non-Tg hearts (Fig 3A). These peptides corresponded to 25 unique mitochondrial proteins (Fig 3A, Table 2). Among the identified peptides, 12 exhibited increased acetylation, while 24 demonstrated reduced acetylation (Fig 3A, Table 2). These peptides corresponded to a total of 37 unique acetylation sites as two lysine-acetyl sites were detected on the same peptide (Table 2; K82, K90 on GOT2). The identified peptides belong to proteins involved in the beta-oxidation of fatty acids (HADHA, HADHB), tricarboxylic acid cycle (IDH2, CS, MDH2), and oxidative stress resistance (superoxide dismutase 2, SOD2) which are reported in Table 2. Interestingly, we observed that DOX significantly altered the acetylation of multiple separate lysines on the same protein (e.g. IDH2, HADA and GOT2, Table 2).
Figure 3. DOX Treatment Alters Acetylation of Cardiac Mitochondrial Peptides Involved in Oxidative Stress in vivo.
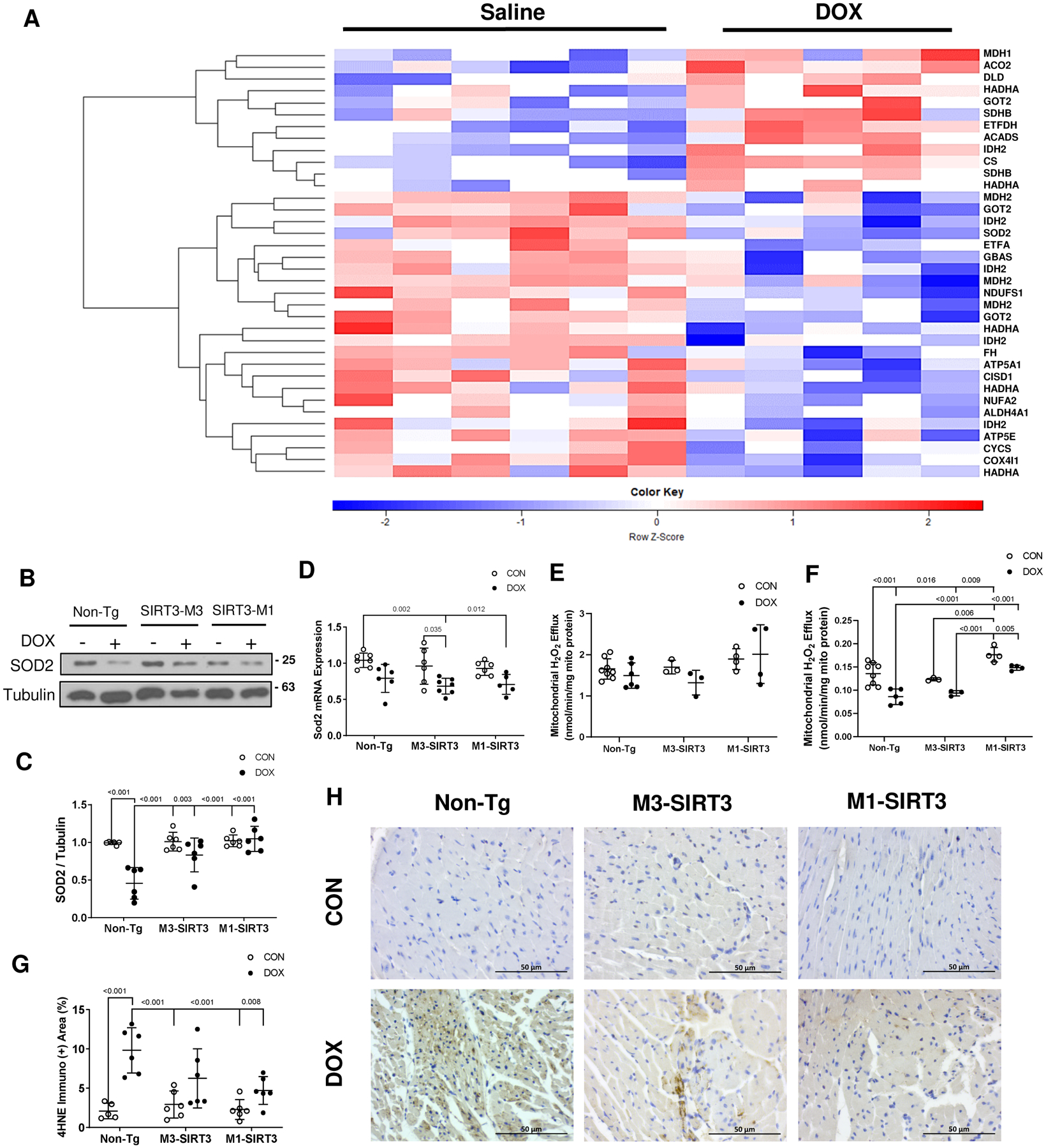
(A) Heatmap of acetylated mitochondrial proteins in non-transgenic DOX treated mouse hearts identified by mass spectrometry. Blank cut-off set at 40%. Data represented as scaled log2 value of normalized peak intensity where values have been z-scored. Red indicates hyperacetylated peptide. Blue represents hypoacetylated peptides. Statistics are unpaired student t-test. P<0.05, Non-Tg-CON n=6 (males), Non-Tg-DOX n=5 (males) (B) Representative image of superoxide dismutase 2 (SOD2) western blot from total cardiac lysates. (C) Optical density quantification of SOD2 blot relative to tubulin n=6 (females). (D) SOD2 mRNA expression as measured by qPCR (n=6 females). (E) Mitochondrial hydrogen peroxide (H2O2) efflux after treatment with 5 μM succinate (nmol/min/mg mito protein, n=3–8 females). (F) Mitochondrial H2O2 efflux after treatment with 5 μM succinate and 4 μM rotenone (nmol/min/mg mito protein, n=3–8 females). (G) Area quantification of immuno-positive 4HNE staining (n=4–6 females). Scale bars = 50μm. (H) Representative images of 4-hydroxy-2,3-transnonenal (4HNE) staining. Sample size refers to biological replicates. Values are mean ± SD. Statistics are two-way ANOVA with Tukey’s post-hoc test.
Table 2.
Differentially acetylated peptides identified with LC-MS/MS after DOX treatment in Non-Tg Mice
| Description | Gene ID | Protein ID* | Peptide Sequence† | Lys-Acetyl Location‡ | Fold Change§ |
|---|---|---|---|---|---|
| Hyperacetylated | |||||
| Isocitrate dehydrogenase [NADP] | Idh2 | P54071 | TDFDkNK | K280 | 6.26 |
| Succinate dehydrogenase [ubiquinone] iron-sulfur subunit | Sdhb | Q9CQA3 | kFAIYR | K43 | 2.62 |
| Citrate synthase | Cs | Q9CZU6 | TFkQQHGK | K53 | 2.22 |
| Dihydrolipoyl dehydrogenase | Dld | O08749 | MMEQkHSAVK | K127 | 2.00 |
| Trifunctional enzyme subunit alpha, | Hadha | Q8BMS1 | VLkEVESVTPEHCIFASNTSALPINQIAAVSK | K460 | 1.64 |
| Electron transfer flavoprotein-ubiquinone oxidoreductase | Etfdh | Q921G7 | FAILTEkHR | K152 | 1.56 |
| Aspartate aminotransferase | Got2 | P05202 | kAEAQIAAkNLDK | K82, K90 | 1.49 |
| Aconitate hydratase | Aco2 | Q99KI0 | AIITkSFAR | K689 | 1.33 |
| Short-chain specific acyl-CoA dehydrogenase | Acadsd | Q07417 | AAMLkDNK | K335 | 0.95 |
| Malate dehydrogenase, cytoplasmic | Mdh1 | P14152 | SQGTALEkYAK | K118 | 0.69 |
| Succinate dehydrogenase [ubiquinone] iron-sulfur subunit | Sdhb | Q9CQA3 | WDPDkTGDKPR | K53 | 0.58 |
| Trifunctional enzyme subunit alpha | Hadha | Q8BMS1 | TSkDTTASAVAVGLR | K519 | 0.48 |
| Hypoacetylated | |||||
| Trifunctional enzyme subunit alpha | Hadha | Q8BMS1 | ILQEGVDPkK | K569 | −0.28 |
| ATP synthase subunit epsilon | Atp5e | P56382 | FSQICAkAVR | K21 | −0.68 |
| Isocitrate dehydrogenase [NADP] | Idh2 | P54071 | GkLDGNQDLIR | K384 | −0.77 |
| Cytochrome c, somatic | Cycs | P62897 | MIFAGIkK | K87 | −0.83 |
| Cytochrome c oxidase subunit 4 isoform 1 | Cox4i1 | P19783 | EKEkADWSSLSR | K67 | −0.87 |
| Superoxide dismutase [Mn] | Sod2 | P09671 | GELLEAIkR | K122 | −0.88 |
| Trifunctional enzyme subunit alpha | Hadha | Q8BMS1 | KYESAYGTQFTPCQLLLDHANNSSKk | K760 | −0.91 |
| Electron transfer flavoprotein subunit alpha | Etfa | Q99LC5 | LGGEVSCLVAGTkCDK | K59 | −1.12 |
| NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 2 | Ndufa2 | Q9CQ75 | ECSEVQPkLWAR | K64 | −1.15 |
| Trifunctional enzyme subunit alpha | Hadha | Q8BMS1 | FVDLYGAQkVVDR | K728 | −1.23 |
| Fumarate hydratase | Fh | P97807 | KPVHPNDHVNkSQSSNDTFPTAMHIAAAVEVHK | K180 | −1.24 |
| NADH-ubiquinone oxidoreductase 75 kDa subunit | Ndufs1 | Q91VD9 | MCLVEIEkAPK | K84 | −1.29 |
| Protein NipSnap homolog 2 | Gbas | O55126 | LQFHNVKPECLDAYNkICQEVLPK | K86 | −1.42 |
| Aspartate aminotransferase | Got2 | P05202 | ASAELALGENNEVLkSGR | K122 | −1.45 |
| CDGSH iron-sulfur domain-containing protein 1 | Cisd1 | Q91WS0 | VVHAFDMEDLGDkAVYCR | K68 | −1.47 |
| Isocitrate dehydrogenase [NADP | Idh2 | P54071 | NTILkAYDGR | K256 | −1.50 |
| ATP synthase subunit alpha | Atp5a1 | Q03265 | DNGkHALIIYDDLSK | K315 | −1.63 |
| Aspartate aminotransferase | Got2 | P05202 | LTkEFSVYMTK | K396 | −1.74 |
| Delta-1-pyrroline-5-carboxylate dehydrogenase | Aldh4a1 | Q8CHT0 | ETHkPLGDWR | K560 | −1.77 |
| Malate dehydrogenase | Mdh2 | P08249 | VNVPVIGGHAGkTIIPLISQCTPK | K213 | −1.78 |
| Isocitrate dehydrogenase [NADP] | Idh2 | P54071 | VEkPVVEMDGDEMTR | K48 | −1.97 |
| Isocitrate dehydrogenase [NADP] | Idh2 | P54071 | FAQTLEkVCVQTVESGAMTK | K400 | −2.05 |
| Malate dehydrogenase | Mdh2 | P08249 | ANVkGYLGPEQLPDCLK | K78 | −2.08 |
| Malate dehydrogenase | Mdh2 | P08249 | ANTFVAELkGLDPAR | K185 | −2.43 |
Protein ID refers to UniProt ID.
Peptide sequence: Bolded lowercase k represents position of acetylation in peptide sequence. Location refers to amino acid position within total protein peptide sequence.
Lys-Acetyl: Lysine Acetylation. Location identified using UniProt.
Fold change calculated DOX-CON using normalized and scaled down log2 values. Statistics are unpaired student t-test. P<0.05, Non-Tg-CON n=6 (males), Non-Tg-DOX n=5 (males).
3.4. SIRT3 Modulates Oxidative Stress Resistance
We identified six peptides corresponding to six acetylation sites (K53, K68, K89, K114, K122, K130) on the oxidative stress resistance protein SOD2 (Table S3 in data supplement). Specifically, K122 showed decreased acetylation of −0.88 log2 fold change while K130 showed increased log2 1.31-fold in acetylation status in DOX hearts (Table 2). Isocitrate dehydrogenase 2 (IDH2) is a protein involved in cardiac energy production through the citric acid cycle and whose acetylation has also been linked to oxidative stress25. In total we identified 19 peptides that corresponded to 18 unique acetylation sites on the protein IDH2 (Table S3 in data supplement). We also identified 2 acetylation sites on the alpha and gamma subunits of IDH3A and IDH3G (Table S3 in data supplement). Five of the sites on IDH2 exhibited significantly altered acetylation following DOX treatment (K48, K256, K280, K384, K400). K280 had the largest increase in acetylation with a log2 fold change of 4.11, while K400 showed the largest decrease in acetylation with a log2 fold change of −2.05. SOD2 and IDH2 are important regulators of oxidative stress in the heart26,27, which suggests that inhibition of SIRT3 by DOX and the deacetylation of SOD2 and IDH2 could affect ROS levels. SOD2 protein levels were decreased in Non-Tg-DOX (p<0.001), while M3-SIRT3 and M1-SIRT3 hearts were resistant to these decreases (Fig 3B, C). DOX also decreased SOD2 gene expression (Fig 3D) in all groups. This suggests that SIRT3 could prevent DOX-induced oxidative stress through the regulation of K122 and K130 acetylation on SOD2 as well as by preventing reduced SOD2 protein levels. To investigate this further we measured H2O2 levels in cardiac mitochondria. Using succinate as the substrate, DOX did not affect mitochondrial H2O2 efflux (Fig 3E). Measuring H2O2 levels in the presence of succinate and rotenone to assess superoxide production downstream of ubiquinone reduction, we found that DOX inhibited H2O2 efflux (Fig 3F, p<0.001), which is consistent with the DOX-induced alterations in SOD2 acetylation and expression in Non-Tg mouse hearts. Importantly, mitochondria from M1-SIRT3 hearts, but not M3-SIRT3, were resistant to the effects of DOX on H2O2 efflux (p<0.001 vs Non-Tg-DOX).
We utilized HPLC based fluorescence methods to investigate MitoSOX products in isolated mitochondria but did not examine differences in hydroxyethidium and ethidium production (Fig S6A–F in data supplement). Short superoxide half-life, high reactivity and insufficient H2O2 production could lead to lipid peroxidation and ultimately cardiomyocyte death. Thus, we performed immunohistochemistry for the product of lipid peroxidation, 4-HNE as a marker of cardiac tissue oxidative stress. Consistent with our previous findings, DOX increased cardiac 4-HNE staining in the hearts of Non-Tg mice (p<0.001), while M1-SIRT3 hearts exhibited reduced levels in 4-HNE staining in DOX treated hearts compared to Non-Tg-DOX treated animals. (Fig 3G, H). These results suggest increased cardiac SIRT3 expression is protective against DOX-induced oxidative stress in vivo.
3.5. M1-SIRT3 Prevents Mitochondrial ROS Production in PRNCs and hiPSC Derived Cardiomyocytes
To further investigate how oxidative stress might contribute to DOX-induced cardiotoxicity we measured mitochondrial ROS production in primary cardiomyocytes by MitoSOX staining. DOX treatment in PRNCs increased mitochondrial ROS production (~1.5 fold, p<0.001, Fig. 4A, D) and expression of mitochondrial localized M1-SIRT3 prevented DOX induced mitochondrial ROS production (p=0.002 vs Ad.GFP DOX) whereas truncated M3-SIRT3 resulted in a partial (n.s. vs ad.GFP) decrease in mitochondrial ROS staining (Fig. 4B, D).
Figure 4. M1-SIRT3 Overexpression Protects Against DOX-Induced Mitochondrial Oxidative Stress in Neonatal Rat Cardiomyocytes.
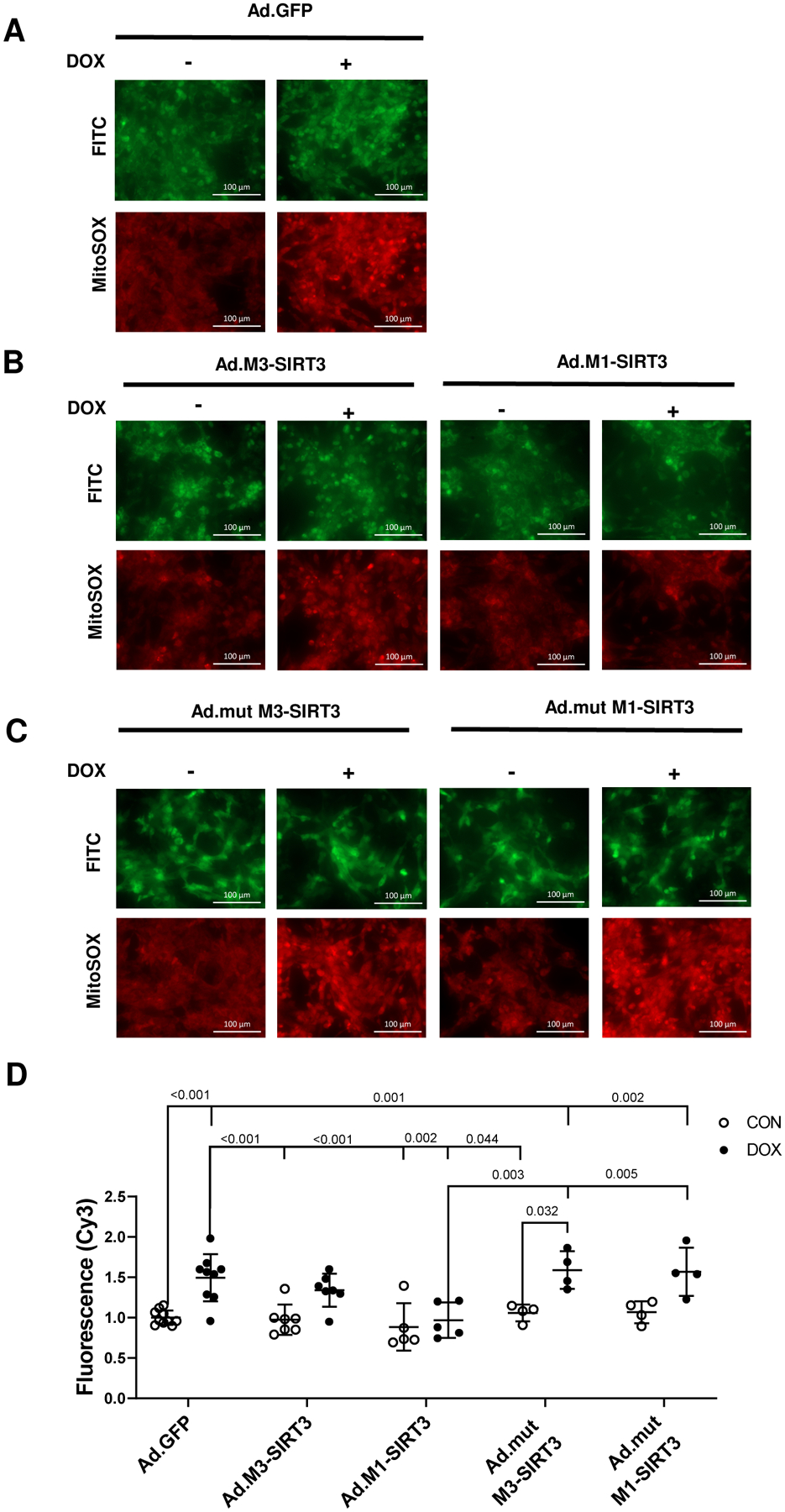
Primary Rat Neonatal Cardiomyocytes (PRNCs) transduced with adenoviruses containing green fluorescent protein (Ad.GFP), M1-SIRT3 (Ad.M1-SIRT3), M3-SIRT3 (Ad.M3-SIRT3), mutant M1-SIRT3 (Ad.mutM1-SIRT3), or mutant M3-SIRT3 (Ad.mutM3-SIRT3) for 24 h; treated with vehicle, DMSO (CON), or DOX (10 μM) for 10–24 h; and stained with MitoSOX (5 μM). Representative images of cardiomyocytes transduced with (A) Ad.GFP (B) Ad.M1-SIRT3 or Ad.M3-SIRT3 (C) Ad.mutM1-SIRT3, or Ad.mutM3-SIRT3 and treated with vehicle or DOX. (D) Quantification of mitochondrial reactive oxygen species levels determined by epifluorescence imaging. Scale bars = 100μm. n = 4–10 biological replicates. Values are mean ± SD. Statistics are two-way ANOVA with Tukey’s post-hoc test.
To examine whether the decrease in mitochondrial ROS is a direct effect of SIRT3 deacetylase activity we transduced PRNCs with mutant SIRT3 (mut-M3-SIRT3, mut-M1-SIRT3) adenoviruses. Consistent with our hypothesis, expression of mut-M3-SIRT3 (p=0.001 vs ad.GFP CON) and mut-M1-SIRT3 (p=0.002 vs ad.GFP CON) did not attenuate DOX induced mitochondrial ROS production (Fig 4C, D).
To validate our findings in a human cell-based model, we utilized hiPSC derived cardiomyocytes. DOX increased MitoSOX staining of hiPSC cardiomyocytes indicating increased mitochondrial ROS production (~2.5 fold, p=0.001 vs ad. GFP CON, Fig. 5A, D). Expression of M3-SIRT3 and M1-SIRT3 isoforms in hiPSC cardiomyocytes attenuated DOX-induced MitoSOX staining compared to Ad.GFP transduced cells (Fig 5B, D) whereas, expression of mutant SIRT3 forms (specifically M1-SIRT3) did not prevent mitochondrial ROS production (p<0.001 vs Ad.GFP, Fig. 5C, D). These findings provide further evidence that mitochondrial M1-SIRT3 could preserve cardiac function by regulating cardiomyocyte oxidative stress resistance, possibly by deacetylating SOD2.
Figure 5. M1-SIRT3 Overexpression Prevents Mitochondrial ROS Production in Human iPSC-Derived Cardiomyocytes.
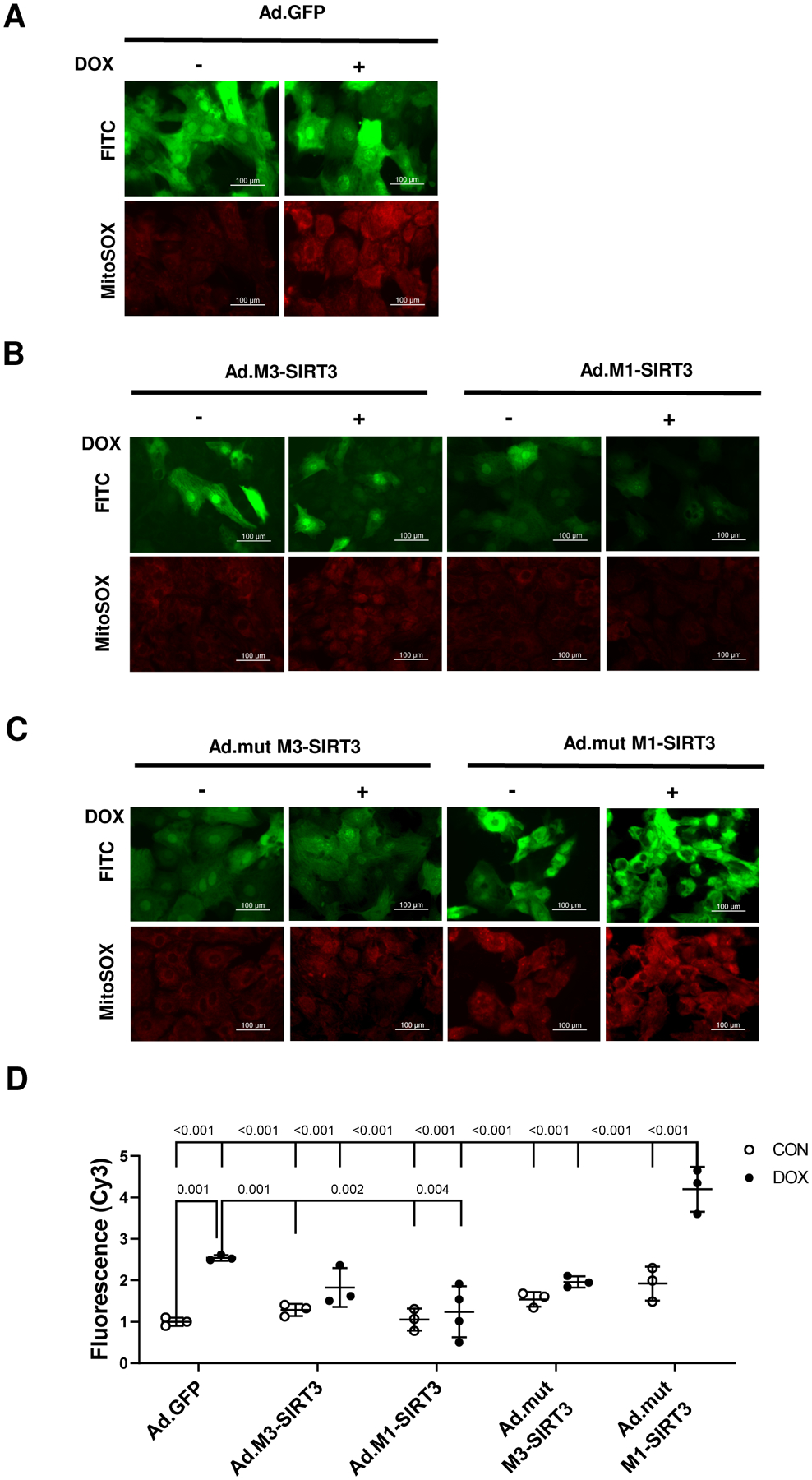
Human induced Pluripotent Stem Cell (hi-PSC) -derived cardiomyocytes transduced with adenoviruses containing green fluorescent protein (Ad.GFP), M1-SIRT3 (Ad.M1-SIRT3), M3-SIRT3 (Ad.M3-SIRT3), mutant M1-SIRT3 (Ad.mutM1-SIRT3), or mutant M3-SIRT3 (Ad.mutM3-SIRT3) for 24 h; treated with vehicle, DMSO (CON), or DOX (10 μM) for 24h and stained with MitoSOX (5 μM). Representative images of cardiomyocytes transduced with (A) Ad.GFP (B) Ad.M1-SIRT3 or Ad.M3-SIRT3 (C) Ad.mutM1-SIRT3 or Ad.mutM3-SIRT3 and treated with vehicle or DOX. (D) Quantification of mitochondrial reactive oxygen species levels determined by epifluorescence imaging. n = 4 biological replicates. Scale bars = 100 μm, Values are mean ± SD. Statistics are two-way ANOVA with Tukey’s post-hoc test.
4. Discussion
The DC caused by anthracyclines has been attributed to mitochondrial dysfunction and oxidative stress6,28. SIRT3 is the main mitochondrial lysine deacetylase and regulates mitochondrial function and oxidative stress resistance8. Previously, we reported that DOX inhibits SIRT3 and increased SIRT3 expression prevents DOX-induced mitochondrial dysfunction and ROS production6. Subsequently, Pillai et al.28 reported that SIRT3 protects against DOX induced cell death and ROS production in primary rat cardiomyocytes. Using SIRT3 transgenic and knockout (KO) models, they showed that SIRT3 deficiency exacerbated mitochondrial DNA damage in the mouse heart and SIRT3 expression attenuated DOX induced cardiac damage28.
Here, we unveil the first study of DOX on the cardiac mitochondrial acetylproteome and how full-length mitochondrial SIRT3 expression can prevent DOX-induced ROS production and cardiac dysfunction. We also show that M1-SIRT3 could prevent DOX-induced cardiac remodelling and dysfunction in mice. Our results show that M3-SIRT3 also conferred partial protection against DOX-induced cardiac dysfunction. Increases in SIRT3 prevented DOX-induced fibrosis whereas SIRT3-KO intensifies fibrotic scarring28–30 and similarly we report that M1-SIRT3 expression decreased DOX-induced cardiac fibrosis. Using PRNC and hiPSC-derived cardiomyocytes we show that SIRT3 expression precluded oxidative stress whereas the deacetylase deficient mutant had no protective effect.
Mass spectrometry revealed that at least one acetylation site is present on ~60% of cardiac mitochondrial proteins9,31. Comprehensive quantification of acetylation in SIRT3-KO tissues, including cardiac tissue, reported increased acetylation of proteins involved in key metabolic processes, including IDH232. We addressed the paucity of research about how DOX affects the mitochondrial acetylome and the role of SIRT3 in DOX-induced DC. Our data suggests a dynamic response in acetylation as both hyperacetylation and hypoacetylation of mitochondrial peptides were observed. Similar to the acetyl-proteomic studies performed in SIRT3-KO mice31–35, DOX reduced cardiac SIRT3 expression and altered acetylation of proteins involved in cardiac energy metabolism (e.g. SDHB, HADHA, ACADSSD) and oxidative stress resistance (e.g. SOD2, IDH2, Table 2), that could be a consequence of reduced SIRT3-mediated deacetylation. However, DOX also impairs energy metabolism, limiting the amount of acetate available for acetylation of proteins. Consequently, a second group of peptides showed reduced acetylation. Among the 18 acetylated lysines on IDH2, only four exhibited increased acetylation. Similarly, SOD2 exhibited both decreased K122 and increased K130 acetylation. The divergent acetylation patterns of these lysine sites could have implications for protein function and ultimately affect mitochondrial superoxide formation. Previous studies identified multiple SOD2 lysine acetylation modifications36–39. Similarly, our study identified acetylation sites on K53, K63, K89, K114, K122, and K130 on SOD2. SIRT3 mediated deacetylation of K53, K58, K68, K122, has been shown to regulate SOD2 activity in various tissue systems36–39. Cardiomyocyte restricted SOD2-knockout mice exhibit increased oxidative damage and a lethal dilated cardiomyopathy26. Relatively little is known about the regulation of SOD2 through acetylation modifications in cardiac tissue, but SIRT3 has been associated with altered K68 and K122 SOD2 acetylation39. A separate set of studies determined the effect of SIRT3 and SOD2 acetylation in aortic and kidney tissues40,41. Although mass spectrometry was only performed in kidney mitochondria, increases in K69, K75 and decreases in K122 on SOD2 were reported. Interestingly we show that cardiac mitochondria from DOX treated mice also exhibited decreases in K122 on SOD2. In human HepG2 cells K122 was determined to be ubiquitinated and this was inversely correlated with acetylation modifications; therefore it is possible that K122 acetylation could protect against ubiquitination and protein degradation42. Together this suggests that alterations of SOD2 acetylation at K122 could have implications for both SOD2 protein levels and its activity.
IDH2 is also involved in oxidative stress resistance and contains a plethora of acetylation modifications43. IDH2 knockout mice develop accelerated heart failure in association with high levels of ROS27. We identified 18 acetylation sites on IDH2. In our model K280, which has been reported by mass spectrometry44 but not characterized in heart disease, showed increased acetylation following DOX treatment. DOX also increased acetylation at K48, K256, K384 and K400, suggesting altered IDH2 could play a key role in DOX-induced oxidative stress within the heart.
This is the first study to examine the effect of SIRT3 downregulation by DOX on the mitochondrial acetylome in an in vivo model using mass spectrometry. Limitations of our study include a relatively small sample size in our mass spectrometry analysis that could explain the lack of statistical significance for K130 acetylation on SOD2, though it exhibited a log2 1.31-fold increase in acetylation when compared to the control group. In addition, mass spectrometry represents a snapshot of the mitochondrial acetylome at the end of the four week DOX treatment. The reversible nature of acetylation modifications and compensatory mechanisms within the cell could explain the dynamic changes exhibited in our acetylomics dataset. Our present study focused on SIRT3, however additional sirtuins could be at play.
5. Conclusion
Collectively, our data shows that increased expression of M1-SIRT3 can prevent DOX-induced cardiac remodelling and dysfunction that is characteristic of DC through the inhibition of mitochondrial oxidative stress. Our findings suggest mitochondrial M1-SIRT3 or its deacetylation substrates could be potential therapeutic targets for the prevention of DOX-induced DC.
Supplementary Material
Clinical Perspective:
What’s New?
Doxorubicin (DOX) is a widely used anthracycline chemotherapeutic that increases the risk for the development of progressive dilated cardiomyopathy. In this study we show that cardiac restricted expression of full-length M1-SIRT3 prevents DOX-induced cardiac remodelling and dysfunction.
This is the first acetyl-proteomics study which examines how DOX affects mitochondrial protein acetylation within the heart. We identified that DOX alters metabolic and oxidative stress resistance enzymes.
We provide evidence that SIRT3 deacetylase activity affects SOD2 acetylation and preventing DOX induced oxidative stress within the heart.
What are the Clinical Implications?
Therapies for DOX induced dilated cardiomyopathy are limited therefore, by targeting cardiac oxidative stress and energy production pathways, SIRT3 activation could be a unique strategy to circumvent dilated cardiomyopathy in patients undergoing anthracycline treatment.
Acknowledgements
We thank Dr. John Wilkins and the Manitoba Center for Proteomics and System Biology for developing the mass spectrometry protocols and Ying (Tenny) Lao for the technical assistance. We thank Dr. Wai Hei Tse from the CHRIM microscopy platform, Mario Fonseca, Gabriel Brawerman and Jack Lee Ward III for technical advice and assistance. This work was conducted at the Children’s Hospital Research Institute of Manitoba.
Sources of Funding
This work was supported by funding to V.W.D. from the Heart and Stroke Foundation of Canada (HSFC) Grant GIA16-00013990 and the Canadian Foundation for Innovation and funding to J.T. from the Canada Research Chairs Program (CRC-2015-147). Q. T. was supported by U.S. Department of Agriculture (3092-5-001-059) and NIH (DK075978). M.M.T. and S.M.K were supported by Research Manitoba Studentships. V.W.D. is the Allen Rouse-Manitoba Medical Services Foundation Basic Scientist.
Abbreviations:
- DOX
Doxorubicin
- DC
Dilated cardiomyopathy
- LV
Left ventricle
- SIRT3
Sirtuin 3
- i.p.
Intraperitoneal
- n.s.
non-significant
- Non-Tg
Non transgenic
- M3-SIRT3
Truncated SIRT3
- M1-SIRT3
Full length SIRT3 (mitochondrial localized)
- SOD2
Superoxide dismutase 2
- IDH2
Isocitrate dehydrogenase 2
- ROS
Reactive oxygen species
- Lys-Acetyl
Lysine-acetylation
- Ad.
Adenovirus
- Ad.mut
Adenovirus mutant
- PRNC
Primary rat neonatal cardiomyocytes
- hiPSC
human induced pluripotent stem cell
- KO
knock-out
Footnotes
References
- 1.Kremer LCM, Van Dalen EC, Offringa M, Voûte PA. Frequency and risk factors of anthracycline-induced clinical heart failure in children: a systematic review. Ann Oncol 2002;13:503–512. doi: 10.1093/annonc/mdf118. [DOI] [PubMed] [Google Scholar]
- 2.Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003;97:2869–2879. doi: 10.1002/cncr.11407. [DOI] [PubMed] [Google Scholar]
- 3.Gradman AH, Alfayoumi F. From Left Ventricular Hypertrophy to Congestive Heart Failure: Management of Hypertensive Heart Disease. Prog Cardiovasc Dis 2006;48:326–341. doi: 10.1016/j.pcad.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 4.Dadson K, Hauck L, Billia F. Molecular mechanisms in cardiomyopathy. Clin Sci 2017;131:1375–1392. doi: 10.1042/CS20160170. [DOI] [PubMed] [Google Scholar]
- 5.Lenneman AJ, Wang L, Wigger M, Frangoul H, Harrell FE, Silverstein C, Sawyer DB, Lenneman CG. Heart transplant survival outcomes for adriamycin-dilated cardiomyopathy. Am J Cardiol 2013;111:609–612. doi: 10.1016/j.amjcard.2012.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheung KG, Cole LK, Xiang B, Chen K, Ma X, Myal Y, Hatch GM, Tong Q, Dolinsky VW. Sirtuin-3 (SIRT3) protein attenuates doxorubicin-induced oxidative stress and improves mitochondrial respiration in H9c2 cardiomyocytes. J Biol Chem 2015;290:10981–10993. doi: 10.1074/jbc.M114.607960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wallace KB, Sardão VA, Oliveira PJ. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ Res 2020;126:926–941. doi: 10.1161/CIRCRESAHA.119.314681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dolinsky VW. The role of sirtuins in mitochondrial function and doxorubicin-induced cardiac dysfunction. Biol Chem 2017;398:955–974. doi: 10.1515/hsz-2016-0316. [DOI] [PubMed] [Google Scholar]
- 9.Foster DB, Liu T, Rucker J, O’Meally RN, Devine LR, Cole RN, O’Rourke B. The Cardiac Acetyl-Lysine Proteome. PLoS One 2013;8:e67513. doi: 10.1371/journal.pone.0067513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang Y, Hubbard BP, Sinclair DA, Tong Q. Characterization of Murine SIRT3 Transcript Variants and Corresponding Protein Products. J Cell Biochem 2010;111:1051–1058. doi: 10.1002/jcb.22795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi T, Wang F, Stieren E, Tong Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J Biol Chem 2005;280:13560–13567. doi: 10.1074/jbc.M414670200. [DOI] [PubMed] [Google Scholar]
- 12.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the α-myosin heavy chain gene promoter in transgenic mice. J Biol Chem 1991;266:24613–24620. doi: 10.1016/s0021-9258(18)54273-3. [DOI] [PubMed] [Google Scholar]
- 13.Cox GA, Cole NM, Matsumura K, Phelps SF, Hauschka SD, Campbell KP, Faulkner JA, Chamberlain JS. Overexpression of dystrophin in transgenic mdx mice eliminates dystrophic symptoms without toxicity. Nature 1993;364:725–729. doi: 10.1038/364725a0. [DOI] [PubMed] [Google Scholar]
- 14.Cole LK, Mejia EM, Sparagna GC, Vandel M, Xiang B, Han X, Dedousis N, Kaufman BA, Dolinsky VW, Hatch GM. Cardiolipin deficiency elevates susceptibility to a lipotoxic hypertrophic cardiomyopathy. J Mol Cell Cardiol 2020;144:24–34. doi: 10.1016/j.yjmcc.2020.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Kovacic S, Soltys CLM, Barr AJ, Shiojima I, Walsh K, Dyck JRB. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem 2003;278:39422–39427. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 16.Munro D, Banh S, Sotiri E, Tamanna N, Treberg J. The thioredoxin and glutathione-dependent H2O2 consumption pathways in muscle mitochondria: Involvement in H2O2 metabolism and consequence to H2O2 efflux assays. Free Radic Biol Med 2016;96:334–346. doi: 10.1016/J.FREERADBIOMED.2016.04.014. [DOI] [PubMed] [Google Scholar]
- 17.Munro D, Baldy C, Pamenter ME, Treberg JR. The exceptional longevity of the naked mole-rat may be explained by mitochondrial antioxidant defenses. Aging Cell 2019;18:e12916. doi: 10.1111/ACEL.12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 2016;100:14–31. doi: 10.1016/J.FREERADBIOMED.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Kalinovic S, Oelze M, Kröller-Schön S, Steven S, Vujacic-Mirski K, Kvandová M, Schmal I, Zuabi A Al, Münzel T, Daiber A. Comparison of Mitochondrial Superoxide Detection Ex Vivo/In Vivo by mitoSOX HPLC Method with Classical Assays in Three Different Animal Models of Oxidative Stress. Antioxidants 2019;8:514. doi: 10.3390/ANTIOX8110514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dikalov S, Griendling KK, Harrison DG. Measurement of Reactive Oxygen Species in Cardiovascular Studies. Hypertension 2007;49:717–727. doi: 10.1161/01.HYP.0000258594.87211.6B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zielonka J, Srinivasan S, Hardy M, Ouari O, Lopez M, Vasquez-Vivar J, Avadhani NG, Kalyanaraman B. Cytochrome c-mediated oxidation of hydroethidine and mito-hydroethidine in mitochondria: Identification of homo- and heterodimers. Free Radic Biol Med 2008;44:835–846. doi: 10.1016/J.FREERADBIOMED.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci 2006;103:15038–15043. doi: 10.1073/PNAS.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Treberg JR, Braun K, Zacharias P, Kroeker K. Multidimensional mitochondrial energetics: Application to the study of electron leak and hydrogen peroxide metabolism. Comp Biochem Physiol Part B Biochem Mol Biol 2018;224:121–128. doi: 10.1016/J.CBPB.2017.12.013. [DOI] [PubMed] [Google Scholar]
- 24.Zhou G, Soufan O, Ewald J, Hancock REW, Basu N, Xia J. NetworkAnalyst 3.0: A visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res 2019;47:W234–W241. doi: 10.1093/nar/gkz240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogura Y, Kitada M, Monno I, Kanasaki K, Watanabe A, Koya D. Renal mitochondrial oxidative stress is enhanced by the reduction of Sirt3 activity, in Zucker diabetic fatty rats. Redox Rep 2018;23:153–159. doi: 10.1080/13510002.2018.1487174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma S, Bhattarai S, Ara H, Sun G, St Clair DK, Bhuiyan MS, Kevil C, Watts MN, Dominic P, Shimizu T, et al. SOD2 deficiency in cardiomyocytes defines defective mitochondrial bioenergetics as a cause of lethal dilated cardiomyopathy. Redox Biol 2020;37:101740. doi: 10.1016/j.redox.2020.101740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ku H, Ahn Y, Lee J, Park K, Park JW. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free Radic Biol Med 2015;80:84–92. doi: 10.1016/j.freeradbiomed.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 28.Pillai VB, Bindu S, Sharp W, Fang YH, Kim G, Gupta M, Samant S, Gupta MP. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. Am J Physiol Circ Physiol 2016;310:H962–H972. doi: 10.1152/ajpheart.00832.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pillai VB, Kanwal A, Fang YH, Sharp WW, Samant S, Arbiser J, Gupta MP. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget 2017;8:34082–34098. doi: 10.18632/oncotarget.16133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pillai VB, Samant S, Sundaresan NR, Raghuraman H, Kim G, Bonner MY, Arbiser JL, Walker DI, Jones DP, Gius D, et al. Honokiol blocks and reverses cardiac hypertrophy in mice by activating mitochondrial Sirt3. Nat Commun 2015;6:6656. doi: 10.1038/ncomms7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, et al. Calorie Restriction and SIRT3 Trigger Global Reprogramming of the Mitochondrial Protein Acetylome. Mol Cell 2013;49:186–199. doi: 10.1016/j.molcel.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dittenhafer-Reed KE, Richards AL, Coon JJ, Denu JM. SIRT3 Mediates Multi-Tissue Coupling for Metabolic Fuel Switching. Cell Metab 2015;21:637–646. doi: 10.1016/j.cmet.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang W, Nagasawa K, Münch C, Xu Y, Satterstrom K, Jeong S, Hayes SD, Jedrychowski MP, Vyas FS, Zaganjor E, et al. Mitochondrial Sirtuin Network Reveals Dynamic SIRT3-Dependent Deacetylation in Response to Membrane Depolarization. Cell 2016;167:985–1000.e21. doi: 10.1016/j.cell.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, Schilling B, Mooney SD, Kahn CR, Verdin E, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proc Natl Acad Sci U S A 2013;110:6601–6606. doi: 10.1073/pnas.1302961110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davidson MT, Grimsrud PA, Lai L, Draper JA, Fisher-Wellman KH, Narowski TM, Abraham DM, Koves TR, Kelly DP, Muoio DM. Extreme Acetylation of the Cardiac Mitochondrial Proteome Does Not Promote Heart Failure. Circ Res 2020;127:1094–1108. doi: 10.1161/CIRCRESAHA.120.317293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiu X, Brown K, Hirschey MD, Verdin E, Chen D. Cell Metabolism Calorie Restriction Reduces Oxidative Stress by SIRT3-Mediated SOD2 Activation. Cell Metab 2010;12:662–667. doi: 10.1016/j.cmet.2010.11.015. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Zhang J, Lin Y, Lei Q, Guan KL, Zhao S, Xiong Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep 2011;12:534–541. doi: 10.1038/embor.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, McDonald WH, et al. Sirt3-Mediated Deacetylation of Evolutionarily Conserved Lysine 122 Regulates MnSOD Activity in Response to Stress. Mol Cell 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang L, Chen CL, Kang PT, Jin Z, Chen YR. Differential protein acetylation assists import of excess SOD2 into mitochondria and mediates SOD2 aggregation associated with cardiac hypertrophy in the murine SOD2-tg heart. Free Radic Biol Med 2017;108:595–609. doi: 10.1016/j.freeradbiomed.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dikalova AE, Itani HA, Nazarewicz RR, McMaster WG, Flynn CR, Uzhachenko R, Fessel JP, Gamboa JL, Harrison DG, Dikalov SI. Sirt3 impairment and SOD2 hyperacetylation in vascular oxidative stress and hypertension. Circ Res 2017;121:564–574. doi: 10.1161/CIRCRESAHA.117.310933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dikalova AE, Pandey A, Xiao L, Arslanbaeva L, Sidorova T, Lopez MG, Billings FT, Verdin E, Auwerx J, Harrison DG, et al. Mitochondrial Deacetylase Sirt3 Reduces Vascular Dysfunction and Hypertension While Sirt3 Depletion in Essential Hypertension Is Linked to Vascular Inflammation and Oxidative Stress. Circ Res 2020;126:439–452. doi: 10.1161/CIRCRESAHA.119.315767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akimov V, Barrio-Hernandez I, Hansen SVF, Hallenborg P, Pedersen AK, Bekker-Jensen DB, Puglia M, Christensen SDK, Vanselow JT, Nielsen MM, et al. Ubisite approach for comprehensive mapping of lysine and n-terminal ubiquitination sites. Nat Struct Mol Biol 2018;25:631–640. doi: 10.1038/s41594-018-0084-y. [DOI] [PubMed] [Google Scholar]
- 43.Yu W, Dittenhafer-Reed KE, Denu JM. SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem 2012;287:14078–14086. doi: 10.1074/jbc.M112.355206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, Burgess MW, Gillette MA, Jaffe JD, Carr SA. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods 2013;10:634–637. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source data that support the findings of this study are available from the corresponding author upon reasonable request.