

Abstract
Background:
Secreted mucin 5AC (MUC5AC) promotes pancreatic cancer (PC) progression and chemoresistance, suggesting its clinical association with poor prognosis. RNA sequencing analysis from the autochthonous pancreatic tumors showed a significant stromal alteration upon genetic ablation of Muc5ac. Previously, depletion or targeting the stromal fibroblasts showed an ambiguous effect on PC pathogenesis. Hence, identifying the molecular players and mechanisms driving fibroblast heterogeneity is critical for improved clinical outcomes.
Methods:
Autochthonous murine models of PC [KrasG12D, Pdx1-Cre (KC); KrasG12D, Pdx1-Cre, Muc5ac−/− (KCM)] and co-implanted allografts of murine PC cell lines (Muc5ac-WT and CRISPR/Cas-KO) with adipose-derived mesenchymal stem cells (AD-MSCs) were used to assess the role of Muc5ac in stromal heterogeneity. Proliferation, migration, surface expression of cell-adhesion markers on AD-MSCs were measured using live-cell imaging and flow cytometry. MUC5AC-interactome was investigated using mass-spectrometry and ELISA.
Results:
The KCM tumors showed a significant decrease in the expression of α-smooth muscle actin, and fibronectin, as compared to histology-matched KC tumors. Our study showed that MUC5AC, carrying tumor secretome, gets enriched in the adipose tissues of tumor-bearing mice and patients, where it promotes CD44/CD29 (integrin-β1) clustering leading to Rac1 activation and migration of AD-MSCs. Further, treatment with KC-derived serum enhanced proliferation and migration of AD-MSCs, which was abolished upon Muc5ac-depletion or pharmacological inhibition of CXCR2 and Rac1, respectively. The AD-MSCs significantly contribute towards α-SMA-positive CAFs population in Muc5ac-dependent manner, as suggested by autochthonous tumors, co-implantation xenografts, and patient tumors.
Conclusion:
MUC5AC, secreted during PC progression, enriches in adipose and enhances the mobilization of AD-MSCs. Upon recruitment to pancreatic tumors, AD-MSCs proliferate and contribute towards stromal heterogeneity.
Keywords: Pancreatic Cancer, MUC5AC, Stroma, Mesenchymal stem cells, cancer-associated fibroblast, chemokines
Introduction
Pancreatic cancer (PC) continues to rank as one of the most lethal malignancies, with a dismal 5-year survival rate of 10%1. It is thought to progress through a series of histologically defined precursor lesions known as pancreatic intraepithelial neoplasias (PanINs)2. As PanINs progress to carcinoma, an intense desmoplastic stroma develops around the malignant cells, which eventually encompasses 70–90% of the bulk tumor volume3. Given the significant contribution of desmoplasia in driving the aggressiveness and therapy-refractory nature of the disease4, several efforts have been made to target the stroma per se in combination with conventional chemotherapy5, 6. However, preclinical studies attempting the stromal ablation led to unfavorable outcomes with increased tumor aggressiveness.7–9. Since then, several groups have investigated the heterogeneity and plasticity of cancer-associated fibroblasts (CAFs), the primary architects of the stroma, using virtual microdissection and single-cell RNA sequencing approaches. These studies have defined functionally diverse CAFs based on their cellular origin, phenotypic nature, associated transcriptome, and intra-tumoral spatial organization10–12. Parallel findings from murine models have demonstrated that from neoplastic initiation, the tumor mesenchyme is primed by the CAF-precursor cells, known as the mesenchymal stem cells (MSCs), from distant sites3. Reprogramming of the recruited MSCs and maturation of the tissue-resident pancreatic stellate cells (PSCs) eventually give rise to the CAFs in the primary tumor. Hence, identifying the molecular mechanisms that drive the diverse origin of CAFs and define their contribution towards stromal heterogeneity is essential for a better understanding and improved therapeutic outcomes in PC.
The Kras oncogene-mediated reprogramming leads to the aberrant overexpression of high molecular weight glycoproteins called mucins in the epithelial cells, a characteristic feature of PC13, 14. Among the mucin family members, secreted mucin MUC5AC is expressed de novo in the early neoplastic lesions15. Driven by its strong clinical association with patient survival, we generated KCM (KrasG12D, Pdx-1cre, Muc5ac−/−) mice by genetically ablating Muc5ac from the well-characterized autochthonous murine model (KC: KrasG12D, Pdx-1cre) to investigate its mechanistic contribution in PC pathogenesis. While Muc5ac was involved in the enrichment of cancer stemness in the epithelial compartment during PanIN progression16, we were intrigued by the transcriptomic analysis that revealed “extracellular matrix (ECM) organization” as the most downregulated pathway in KCM compared to KC tumors. Alongside the extent of stroma, KC tumors had significantly higher α-smooth muscle actin (α-SMA)-positive CAFs than the KCM, while other stromal markers like fibroblast activating protein (FAP) remained unaltered, suggesting a phenotypic heterogeneity in the stromal compartment. Conceptually, we envisioned that MUC5AC, as a secreted factor, may influence the non-malignant stromal compartment locally in pancreatic tumors as well as systemically. Interestingly, we observed that tumor-borne Muc5ac present in the systemic circulation gets enriched in the adipose tissue and acts as a carrier of the tumor secretome by entrapping heat-labile chemokines in its polymeric gel. Accumulating evidence from various solid malignancies like pancreatic, prostate, and breast cancer suggests that MSCs from the bone marrow and adipose tissue get recruited to the primary tumor and express diverse CAF markers upon maturation. Several factors behind the recruitment of circulating MSCs to the primary tumor have been suggested before17; however, the precise systemic crosstalk between tumor and distant organs leading to MSC mobilization and their maturation to different CAF subtypes is inadequately characterized in PC.
MUC5AC’s gel-forming ability, high abundance in the serum, and proximity to the MSCs in adipose tissue made us question whether MUC5AC contributes to PC stromal heterogeneity via adipose-derived MSC (AD-MSC) mobilization. Observations from our autochthonous murine model, co-implantation allografts, and primary murine AD-MSCs, suggested that Muc5ac-mediated clustering of CD44 and CD29 with Rac1 activation in AD-MSCs led to their mobilization. Upon recruitment to the primary tumor, these MSCs expand and contribute towards the α-SMA-positive myofibroblast population of CAFs. The current study is the first to report the multifaceted roles of bulky glycosylated mucin, MUC5AC in orchestrating the stromal heterogeneity by mobilizing fibroblast precursor cells from adipose tissue, which will significantly contribute towards our current understanding and pave the way for better patient prognostication and therapeutic strategies.
Materials and Methods
Animal studies
Animal studies were approved by the UNMC’s Institutional Animal Care and Use Committee. PC autochthonous murine models, KC (KrasG12D, Pdx-1cre) and KCM (KrasG12D,Pdx-1cre, Muc5ac−/−) mice were maintained in our lab, as mentioned previously16.
Pancreatic tumor sections and homogenates were generated from randomized KC and KCM mice (50 weeks, male and female). RNA was isolated from KC and KCM tumors at early (20 weeks) and late (50 weeks) stages of disease progression. RNA seq. performed on the KC and KCM tumors (GSE160029)16, was used to analyze the top differential pathways. Blood MSCs were analyzed using flow cytometry18, after magnetic bead-based CD45-depletion (ThermoFisher, Cat# 8804-6864-74) from mice at 20, 30, and 50 weeks. For serum studies, whole blood from KC and KCM mice (50 weeks, male and female) was collected in Citrate-Plus blood collection tubes (BD Vacutainer, Cat# 363080) by terminal cardiac puncture, spun at 5000 rpm for 15 minutes at 4°C, and stored in −80°C for future analyses.
For the co-implantation allograft experiment, the right and left flanks of Muc5ac−/− mice were implanted (1:1:1 ratio) with 0.25 × 106 syngeneic murine PC cell line (either WT or Muc5ac−/− KCT 3266), 0.25 × 106 RFP-labelled pancreatic stellate cells (generated in the lab using a Lentiviral construct, rTA LVP400-RP) and 0.25 × 106 AD-MSCs (isolated from the gonadal and peritoneal fat pads of whole body GFP-expressing mice) suspended together in 50ul PBS (per flank/ mouse). Age and sex-matched 6 Muc5ac−/− mice were utilized in each group (WT and KO grafts). The constitutively expressing GFP mice [GFP expression under ubiquitin promoter, C57BL/6-TG (UBC-GFP) 30 Scha/J] were obtained from the Jackson Laboratory (Stock # 004353). The tumors were allowed to grow for 4 weeks. At the endpoint, the mice were then euthanized, subcutaneous tumors and adipose tissues (gonadal and peritoneal) were resected from these mice and fixed in 10% Neutral-Buffered Formalin (FisherScientific, Cat# SF 100-4) for blinded immunohistochemical analyses.
Patients’ samples
Snap-frozen omentum adipose tissues from de-identified PC patients (n=32) were procured from the Rapid Autopsy Program at UNMC. Unclassified PC patients’ sera (n=29) were procured from the Nebraska Biobank.
Mass Spectrometry-based interactome analysis
Serum from 3 randomized KC mice (50 weeks) were independently used to remove albumin using a bead-based approach, followed by immunoprecipitating Muc5ac (in 4–5 rounds) from the KC mice serum using 10 μg 45M1 (anti-Muc5ac antibody) immobilized with magnetic beads. The eluents from all the immunoprecipitation rounds from each sample were combined together, desiccated, mixed with loading dye, and ran in SDS-PAGE. Protein bands from high (above 50 kDa), medium (30–55kDa), and low (below 30 kDa) molecular weights were excised from the gel, destained, alkylated using iodoacetamide, and digested overnight with sequencing-grade trypsin (Pierce). After elution using C18 spin columns (Pierce), eluted peptides were concentrated and analyzed using a high-resolution mass spectrometry nano-LC-MS/MS, Orbitrap Fusion, coupled with UltiMate 3000 HPLC system (Thermo Scientific). For identification of specific peptides enriched in the Muc5ac interactome, MS/MS peaks were matched with the SwissProt+ TrEMBL database for Mus musculus. The identified peptides from the low molecular-weight proteins were fed in the GeneOntology PANTHER classification system to identify the enriched pathways.
Human proteomic dataset analysis
We downloaded the average normalized protein expression data for the CPTAC PDA Discovery Study – Proteome dataset (https://proteomic.datacommons.cancer.gov/pdc/study/PDC000270). The 137 tumor samples (after discarding disqualified samples) were partitioned into a high MUC5AC – high CXCL5 set and a low MUC5AC – low CXCL5. The high MUC5AC – high CXCL5 set was computed as the intersection of samples with normalized protein expression value equal to or higher than the median for both MUC5AC and CXCL5, whereas the low MUC5AC – low CXCL5 set was computed as the intersection of samples with normalized protein expression value lower than the median for both MUC5AC and CXCL5. Boxplots comparing the normalized protein expression value for ACTA2 in the high MUC5AC – high CXCL5 set and low MUC5AC – low CXCL5 set were produced in R Bioconductor, and p-values were computed using the Wilcoxon signed ranked test.
Quantification and Statistical Analyses
Image J analysis was used to quantify the intensity of periductal α-SMA expression in the human PC patient tissue samples. Details of sample size and statistical tests used in each experiment are explained in the respective method details and figure legends. A p-value of <0.05 was considered significant. P-values for comparisons of two groups or samples were determined by a two-tailed Student’s t-test. All technical and biological replicates were included in the analyses.
Results
MUC5AC is associated with stromal modulation in PC
Transcriptomic analysis (GSE160029) performed in KC and KCM pancreatic tumors demonstrated that extracellular matrix (ECM) organization was the top downregulated pathway after Muc5ac depletion (Supplementary Figure 1A). Histologic assessment of the pancreatic tumor sections revealed a reduction in the periductal stromal compartment, with a 2–3 folds decrease in the expression of classical stromal markers, α-SMA, fibronectin (FN1), hyaluronic acid (HA) in the KCM group, as compared to KC, while expression of fibroblast activating protein (FAP) increased concomitantly, albeit not significantly in the KCM tumors (Figure 1A, B). The stromal alterations in tumors were further corroborated at the transcript level (Figure 1C). Our previous study on KC and KCM mice revealed a significant delay in PanIN progression upon Muc5ac ablation16. Hence, to confirm that the stromal alterations were not simply due to delayed progression to PC, we compared the expression of stromal markers between early-stage (20-week) KC mice and that of late-stage (50-week) KCM mice. The variations in stromal markers observed in age-matched KC and KCM were sustained in histology-matched tumor tissues (Supplementary Figure 1B). Interestingly, primary CAFs isolated from KC and KCM tumors demonstrated a similar pattern of stromal marker expression under in vitro conditions, indicating that the CAFs from Muc5ac-expressing and depleted tumors were inherently different (Figure 1D).
Figure 1. Muc5ac is associated with stromal modulation in PC.
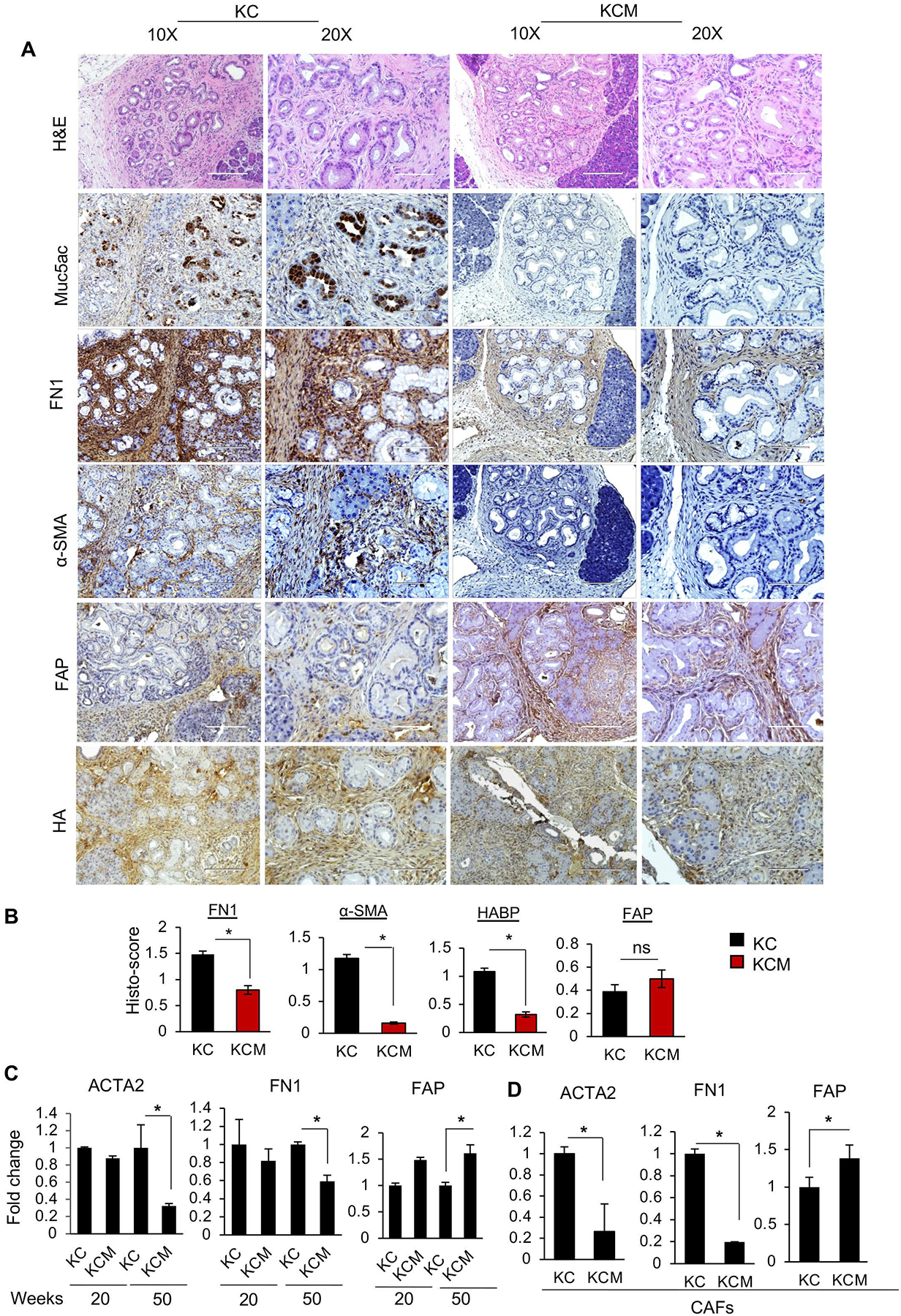
(A) Representative images and (B) quantitative analyses of immunohistochemistry (IHC) for stromal markers on tumor sections from KC and KCM mice at 50 weeks of age (n=4/ group). Expression of α-SMA, FN1, and HA was significantly reduced, accompanied by a nonsignificant increase in expression of FAP in KCM tumors, as compared to KC tumors. (C) Quantitative PCR analysis on RNA isolated from KC/ KCM tumors (n=4/ group) and (D) CAFs (isolated from 50 weeks KC and KCM tumors, n=4/ group) demonstrate heterogeneity in stromal markers. t-test *p<0.05.
Secreted Muc5ac accumulates in the adipose tissue and modulates the mesenchymal stem cells (MSCs) behavior
We first questioned whether MUC5AC could modulate CAF proliferation. A normal murine pancreatic stellate cell (PSC) line, ImPaSCs, and KC and KCM CAFs showed significantly higher proliferation upon treatment with Muc5ac-enriched conditioned media (CM) from a murine PC cell line (KCT 3266) (Supplementary Figure 1C). However, PSCs cultured with Muc5ac-enriched CM did not recapitulate the expression of stromal markers and ECM proteins as observed in KC tumors (Supplementary Figure 1D). Recent studies have indicated that the CAFs in pancreatic tumors originate either from the tissue-resident stellate cells or the MSCs from bone marrow and adipose tissue recruited to the primary tumor via systemic circulation19–23. Muc5ac’s ability to increase CAF proliferation and failure to mirror the KC fibrosis from the stellate cells suggested that Muc5ac may participate in PC stromagenesis by acting on precursor cells. As MUC5AC is abundantly present in the serum of PC patients and murine models from an early neoplastic stage24, we first evaluated the systemic presence of Muc5ac. Strikingly, Muc5ac gets enriched in the adipose tissue (AT) of KC mice, compared to other organs, including spleen, kidney, liver, or bone marrow (Figure 2A). Immunoblot and immunohistochemistry further showed Muc5ac’s presence in the pancreatic tumor, serum, and AT of KC mice while remaining absent in the AT of wild-type mice (Figure 2B, C). Driven by Muc5ac’s presence in close proximity to the MSCs in AT, we asked if Muc5ac enriched in adipose could regulate the biology of adipose-derived MSCs (AD-MSCs). The MSCs isolated from KC and KCM ATs revealed that KCM AD-MSCs had a significantly (2-folds) lower CD44 and CD29/ITGB1 expression compared to KC AD-MSCs (Figure 2D, E). To investigate the impact of tumor-borne Muc5ac on AD-MSCs, we specifically depleted Muc5ac from the KC mouse serum (Supplementary Figure 1E, F, G). While the treatment with 5% KC serum enhanced CD44 and CD29 expression on AD-MSCs by 2–3 folds, treatment with Muc5ac-depleted KC serum failed to increase the same (Figure 2F, G). We next genetically ablated Muc5ac from a murine PC cell line (KCT 3266) using CRISPR-Cas9 technology (Supplementary Figure 2A, B). The CM from parental and Muc5ac-KO KCT 3266 cells had a similar influence on CD44 and CD29 expression like the Muc5ac-proficient and depleted KC serum, respectively (Supplementary Figure 2C, D), indicating the plausible role of Muc5ac in modulating MSC biology in AT.
Figure 2. Secreted Muc5ac enriches in AT and modulates AD-MSCs of tumor-bearing mice.

(A) ELISA analysis of the tissue homogenates from KC mice (n=4) demonstrates the abundant expression of Muc5ac in the pancreatic tumor and adipose tissue. The stomach serves as a positive control. (B) Immunoblot from WT (n=2) and KC mice (n=4) serum and tissue homogenates. Muc5ac was absent in WT mouse adipose and pancreas; Liver and stomach homogenates served as negative and positive controls, respectively. Arrows indicate MUC5AC protein bands. (C) Representative images of IHC for Muc5ac demonstrating its spatial localization proximal to the AD-MSCs in the KC AT, with no trace of Muc5ac in KCM and WT adipose. (D) Representative histograms and quantitative analyses from flow cytometry demonstrate significant reductions in the expression of CD44 and CD29 on the AD-MSCs isolated from KCM adipose, as compared to KC. (I) Representative histograms and (F) Quantitative analyses from flow cytometry demonstrate a significant increase in the expression of CD44 and CD29 on the AD-MSCs after treatment with KC serum with significant reductions in the Muc5ac-depleted KC serum-treated AD-MSCs. t-test *p<0.05
Muc5ac promotes the migratory propensity of AD-MSCs via the Rac 1 axis
Apart from the Muc5ac-mediated increase in expression, CD44 and CD29 were co-localized as clusters on the MSCs in the KC adipose, while such colocalization was absent in KCM and WT AT (Figure 3A). This interaction was further validated when CD44 and CD29 co-immunoprecipitated directly and reciprocally from primary AD-MSCs upon treatment with Muc5ac-containing KC serum (Figure 3B, Supplementary Figure 2E, F). A recent study using quantum-dot / near-field scanning optical microscopy (QD/NSOM) suggests that CD44 and CD29 clustering on AD-MSCs leads to cytoskeletal rearrangement and migration of the MSCs25. Hence, we speculated that secreted Muc5ac, accumulated in the adipose tissue of tumor-bearing mice, leads to an increase in the expression and clustering of CD44 and CD29 on AD-MSCs, which may activate downstream focal adhesions and migratory cues via phosphorylation of myosin light chain proteins (pMLC2) and focal adhesion kinases (pFAK) (Figure 3C). Indeed, KC adipose tissue exhibited a significant upregulation in pMLC2 expression as compared to KCM adipose tissue (Figure 3D, E). Similar increases in pFAK and pMLC2 expressions were also observed on isolated AD-MSCs treated with KC serum (compare lanes 1 and 5, Figure 3F), which were significantly reduced upon Rac1 inhibition in a dose-dependent manner. Phenotypically, the migration propensity (percentage wound closure) of AD-MSCs, decreased significantly upon treatment with Muc5ac-depleted KC serum and Rac1 inhibitor, as compared to KC serum-treated group (Figure 3G, H, Supplementary Figure 2G, H). As direct mechanistic evidence, treatment with CD44-neutralizing antibody significantly reduced the migration of KC serum-treated AD-MSCs, in a dose-dependent manner. Concurrently, there was a drastic decline in the phosphorylation of FAK and MLC2, upon treatment with CD44-neutralizing antibody (Supplementary Figure 2I), suggesting that Muc5ac-mediated AD-MSC migration is significantly dependent on CD44 and CD29/ITGB1 clustering.
Figure 3. Muc5ac promotes AD-MSCs’ migration via CD44/CD29 clustering and Rac1 activation.
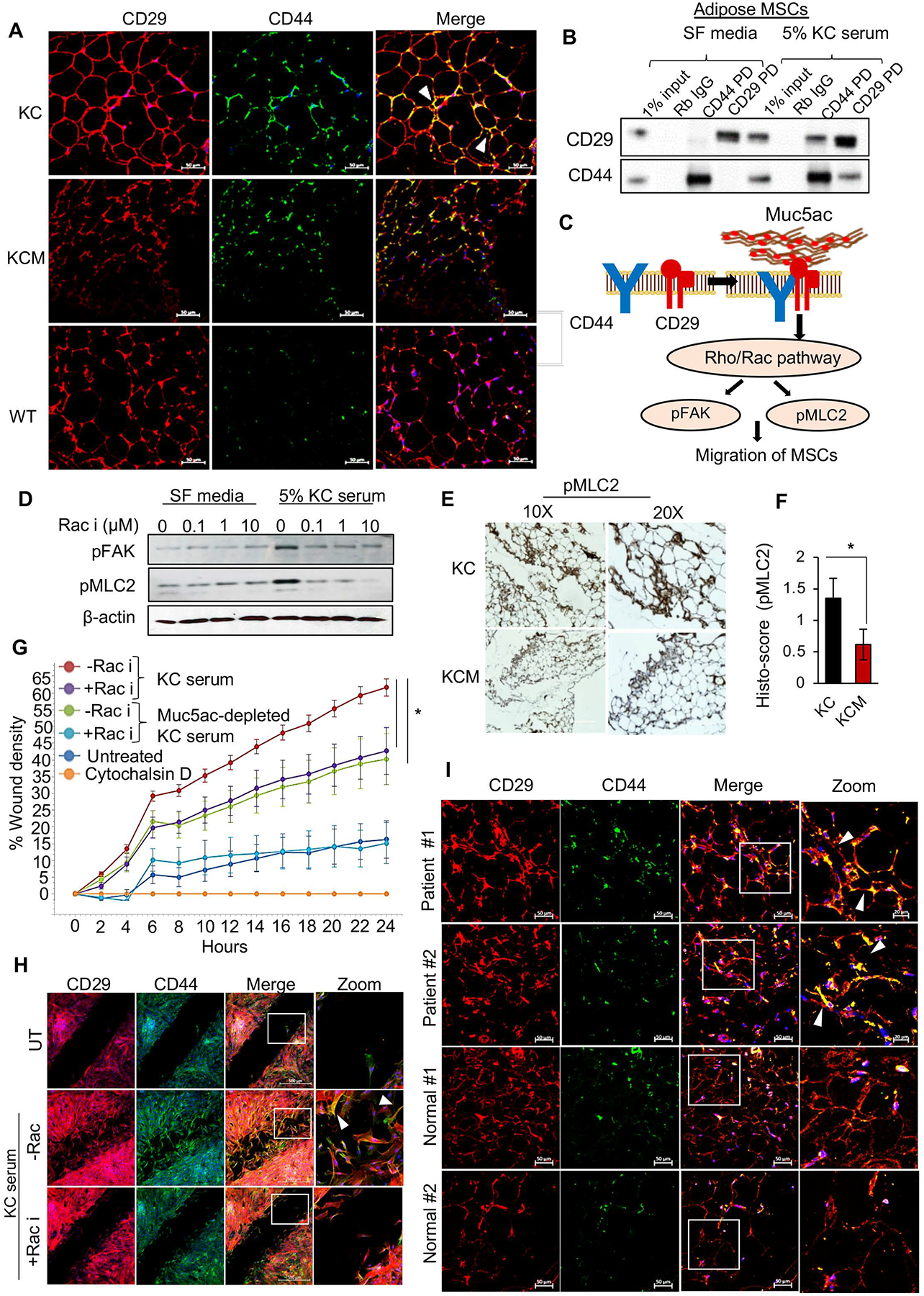
(A) Immunofluorescence images demonstrate the colocalization of CD44 and CD29 (focal clusters marked by white arrowhead) in KC, absent in KCM and WT ATs. (B) Direct and reciprocal co-immunoprecipitation demonstrate the interaction of CD44 and CD29 on AD-MSCs treated with KC serum. Rabbit IgG serves as a negative control, and 1% input demonstrates endogenous expression of the target molecules. (C) Schematic working model of Muc5ac-mediated clustering of CD44 and CD29 on AD-MSCs leading to the activation of Rac 1 pathway, manifested by the enhanced expression of pMLC2 and pFAK. (D) Representative images and (E) quantitative analysis from immunohistochemistry show a significant decline in the expression of pMLC2 in KCM adipose compared to KC. (F) Immunoblot demonstrate upregulation of pMLC2 and pFAK in AD-MSCs treated with 5% KC serum and subsequent decline in these markers upon Rac1 inhibition in a dose-dependent manner. (G) Live-cell imaging-based kinetic wound-healing assay demonstrates a significant decline in migratory propensity (manifested by a lower percentage of wound density) of AD-MSCs treated with Muc5ac-depleted KC serum and Rac1 inhibitor, as compared to those treated with 5% KC serum. Treatment with Cytochalasin D, an F-actin blocker, was used as a negative control. (H) Immunofluorescence images from the migration assay at the endpoint (24 hours) demonstrate co-localization of CD44 and CD29 (white arrowheads) on the migratory AD-MSCs treated with KC serum. t-test *p<0.05.
Secreted MUC5AC sequesters chemokines in the systemic circulation and mobilizes MSCs from adipose tissue
Muc5ac is a sponge-like glycan-rich molecule and therefore, we wondered if Muc5ac gets enriched in the adipose tissue alone or if it sequesters other factors while in the systemic circulation of tumor-bearing mice. To address this, we performed mass spectrometry analysis of immunoprecipitated Muc5ac from KC mouse serum, which revealed significant enrichment of soluble factors involved in the “response to stimulus” pathway. Further, the analysis of enriched pathways and soluble factors revealed Chemokine-Subfamily B Cys-X-Cys protein (Gene name: Ppbp or Cxcl7), a prominent ligand for CXCR2, as one of the top hits in the Muc5ac interactome (Figure 4A). In a sandwich ELISA-based approach, we captured Muc5ac from KC and KCM serum, adipose, and pancreatic homogenates and measured Cxcl1, Cxcl5, and Cxcl7 (detection antibody), the most abundantly expressed CXCR2 ligands in PC. Cxcl7 was significantly enriched with captured Muc5ac from KC mouse serum (Figure 4B). Muc5ac was further observed to enrich Cxcl7 in the tumor homogenate of KC mice (Figure 4B), suggesting that Muc5ac shed from the tumor may act as a gel-forming scaffold to carry tumor-derived chemical messengers into the systemic circulation. These results were confirmed by reciprocal ELISAs (Supplementary Figure 3A). Since Muc5ac was enriched in the AT of tumor-bearing mice, we investigated whether it sustains its interactions with the CXCR2 ligands in the AT. Surprisingly, the enrichment of Cxcl5 was higher compared to Cxcl7 or Cxcl1 in the AT of KC mice (Figure 4B). This switch in chemokine enrichment with Muc5ac may result from the elevated secretion of Cxcl5 by the inflamed adipose tissue26. Furthermore, in the high MUC5AC-expressing PC patient samples, the abundance of Muc5ac-bound forms of Cxcl7 in the serum and Cxcl5 in the adipose homogenates were significantly higher as compared to their respective free forms, while no such enrichment was observed in the low MUC5AC-expressing samples, corroborating the entrapment of CXCR2 ligands in the MUC5AC gel (Figure 4C, D). Interestingly, PNGase treatment significantly abrogated the ability of MUC5AC to sequester Cxcl7 (Figure 4E, F), without any alterations in the abundance of captured MUC5AC (Supplementary Figure 3B), suggesting that Muc5ac-chemokine interactions are mostly glycan-based.
Figure 4. Muc5ac scaffolds chemokines and promotes AD-MSCs’ mobilization in the systemic circulation.
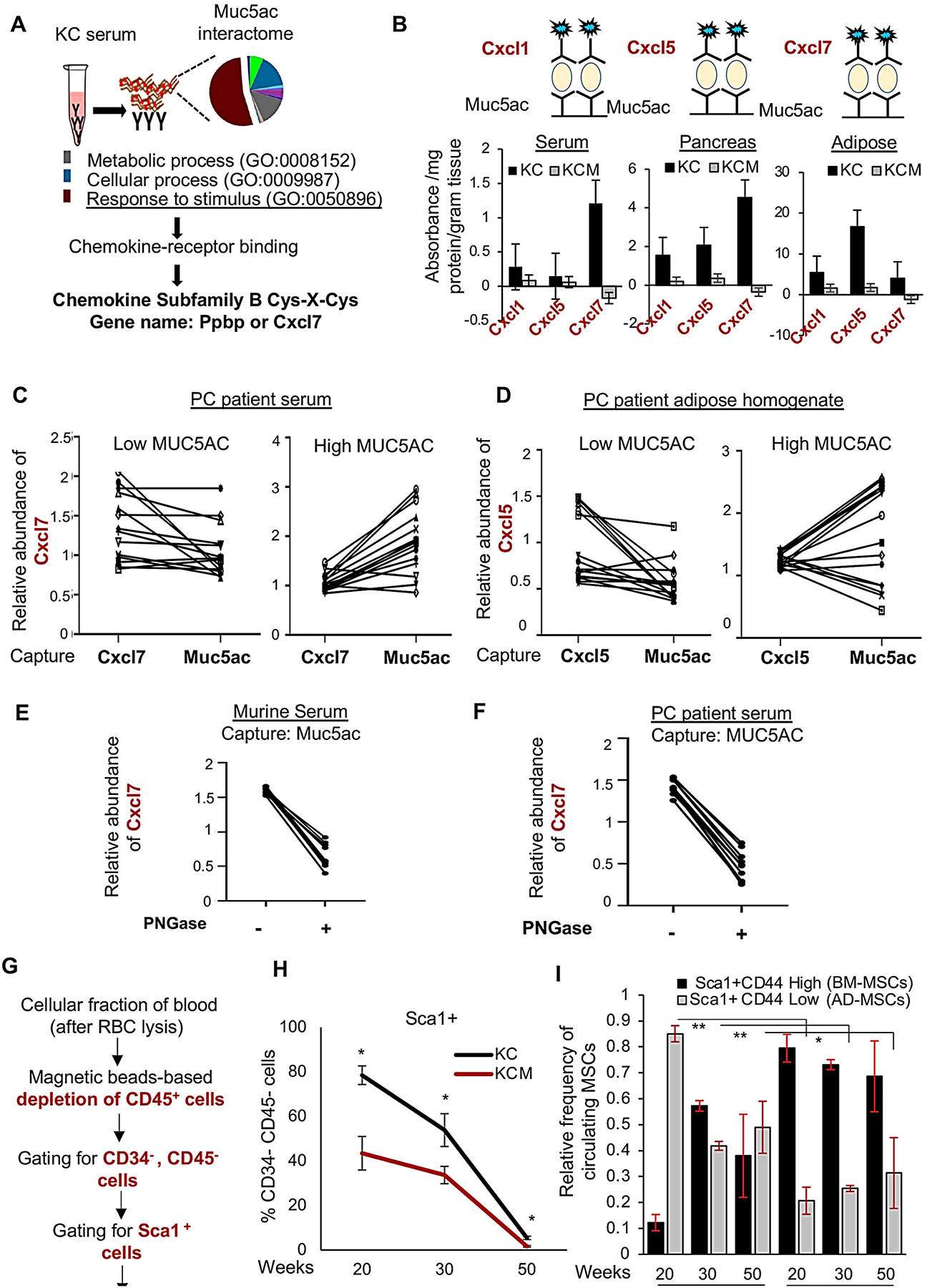
(A) The peptide hits from mass spectrometry-analysis on Muc5ac immunoprecipitate from the KC serum were subjected to pathway analysis using Panther webtool. “Response to stimulus” was the top-most enriched pathway that led to “chemokine-receptor binding” upon functional classification. Subsequent candidate-based analysis led to the identification of Cxcl7 in the secreted Muc5ac interactome. (B) Sandwich-ELISA analysis demonstrates a significant abundance of CXCR2 ligands bound with Muc5ac captured from KC pancreas, serum, and adipose tissues. (C, D) Correlation plots demonstrate enrichments in the Muc5ac-bound form of Cxcl7 and Cxcl5, compared to their total (free and bound) forms in the serum and adipose of human PC patients with high MUC5AC expression; without any enrichments in the low MUC5AC-expressing group. (E, F) Correlation plot demonstrating a significant decline in the abundance of Muc5ac-bound Cxcl7 in the murine and human PC serum upon incubation with PNGase. (G) Workflow of isolation and flow cytometry-based detection of circulating MSCs. (H) Line graph demonstrates significantly higher Sca1+ MSCs in the blood of KC mice at 20 and 30 weeks, compared to age-matched KCM group (n=3/time-point/group). Subsequent analysis shows a significant decline in the CD44+CD29+ MSCs in KC blood, compared to KCM, at 30 and 50 weeks. t-test *p<0.05.
The ability of MUC5AC to cluster CD44/CD29 on the AD-MSCs and increase their migratory potential suggests that Muc5ac can mobilize AD-MSCs into the systemic circulation. Indeed, the Sca1+ (CD45−/ CD34−) MSC population was 8-folds and 3-folds higher in KC circulation at 20 and 30 weeks, respectively, as compared to KCM (Figure 4G, H). However, at 30 weeks, CD44+CD29+ MSCs declined significantly in KC blood as compared to KCM, which may suggest faster recruitment of this specific subset to the KC tumors that may contribute to stromal heterogeneity in the KC tumors.
Secreted MUC5AC expands AD-MSCs and enhances α-SMA subtype of CAFs in PC stroma
Studies in solid tumors suggest that AD-MSCs are recruited from systemic circulation and mature into CAFs in the primary tumor3. To investigate the involvement of Muc5ac in differentiating the tissue-resident stellate cells and AD-MSCs towards α-SMA+ CAFs, Muc5ac-WT and -KO murine PC cell lines (KCT-3266) were each co-implanted subcutaneously with AD-MSCs isolated from whole-body GFP-expressing mice (GFP-MSCs) and exogenously RFP-labeled stellate cells (RFP-PSCs) in a 1:0.5:0.5 ratio, respectively into Muc5ac KO animals (Figure 5A). The notion that tumor-borne Muc5ac reaches the AT via systemic circulation in the PC autochthonous murine model was supported by our observation of Muc5ac presence in the gonadal and peritoneal AT of Muc5ac−/− mice only when implanted with 3266-WT cells, but not with 3266-KO cells (Figure 5B). Interestingly, the MSCs in the AT of Muc5ac−/− mice implanted with 3266-WT and not the 3266-KO cells demonstrated clusters of co-localized CD44 and CD29 with elevated expression of pMLC2 (Figure 5B, Supplementary 3D). IHC analysis further demonstrated that 3266-WT tumors, compared to 3266-KO tumors, had a 5-folds higher area covered by GFP+ (AD-MSC) cells compared to 2-folds area for RFP+ stellate cells (Figure 5C, D), suggesting that Muc5ac may preferentially promote the expansion of MSCs over PSCs. Furthermore, significant reductions in the percentage of the tumor area and staining intensity of α-SMA+ cells were observed in the Muc5ac-KO allografts (Figure 5C, D). Notably, the 3266-WT and KO tumors exhibited differential spatial distribution of GFP-MSCs, identical to the distribution of α-SMA+ cells, with no marked difference in the distribution of RFP-PSCs (Supplementary Figure 3C). While the GFP+ and α-SMA+ cells were integrated inside the WT tumors, showing patterns of infiltration near the malignant cells, both these markers were more displaced towards the periphery of the 3266-KO tumors. The specific contribution of AD-MSCs to α-SMA+ CAF generation in the PC stroma was further demonstrated by significantly higher colocalization of GFP and α-SMA, as compared to RFP and α-SMA (Figure 5E).
Figure 5. Muc5ac promotes AD-MSC expansion and maturation towards α-SMA-expressing CAFs.
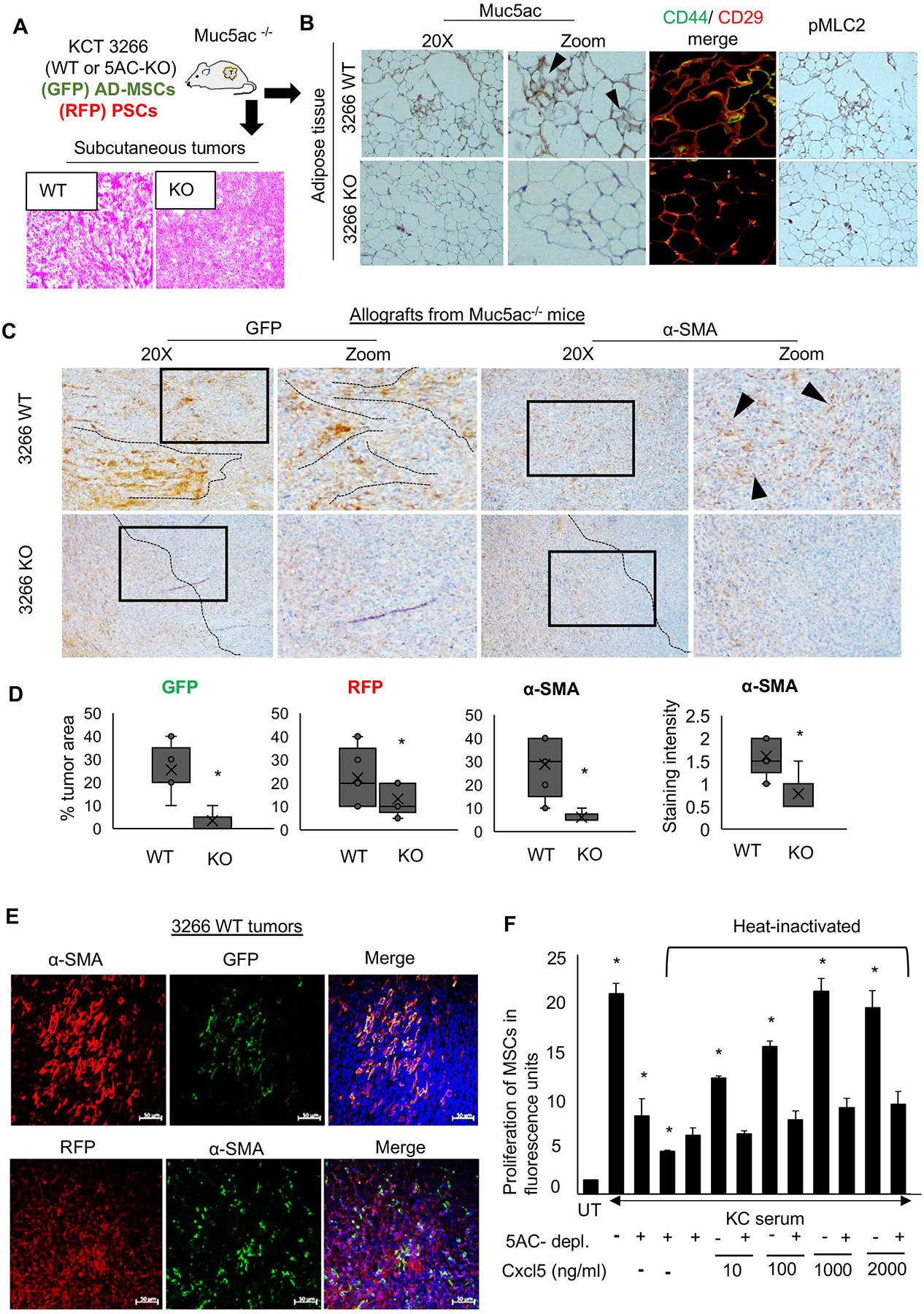
(A) Schematic experimental plan for the co-implantation allograft model in Muc5ac−/− mice, using murine PC cell line KCT 3266 (WT and MUC5AC-KO) admixed with AD-MSCs isolated from GFP mice and RFP-labelled pancreatic stellate cells. Representative H&E images demonstrating the histology of WT and KO tumors (n=10/group). (B) Immunohistochemistry and immunofluorescence images from adipose tissue isolated from Muc5ac−/− animals demonstrate the presence of Muc5ac, with higher CD44/CD29 colocalization and pMLC2 expression in the mice grafted with Muc5ac-expressing cells, as compared to those receiving Muc5ac-KO cells. (C) Representative images and (D) quantitative analysis (n=10/group) from immunohistochemistry demonstrate the expansion of AD-MSCs (GFP), pancreatic stellate cells (RFP), and α-SMA+ CAFs with increased α-SMA staining in 3266 WT tumors, as compared to the KO group. Black dotted lines and arrowheads demonstrate the pattern of GFP-MSCs and α-SMA-CAFs, localized near the malignant cells in the WT tumors but dispersed towards the periphery of the KO tumors. (E) Immunofluorescence images show co-expression of GFP and α-SMA in the WT tumors, with no significant co-expression of RFP and α-SMA, suggesting AD-MSCs contribute more than the resident stellate cells towards the α-SMA+ stromal population in the Muc5ac-expressing pancreatic tumors. (F) The bar graph demonstrates that the proliferation of AD-MSCs significantly increased when treated with KC serum, which subsequently declined upon Muc5ac depletion and heat inactivation of KC serum. Ectopic addition of Cxcl5 at increasing concentrations of 10, 100, 1000, and 2000 ng/ml in heat-inactivated KC serum rescued the proliferation of AD-MSCs in a dose-dependent manner. t-test *p<0.05
Guided by the observations from our implantation study, we asked how MUC5AC promoted the expansion of MSCs. While Muc5ac-enriched CM from a murine PC cell line significantly increased the proliferation of KC and KCM MSCs, heat-inactivation of the CM prior to treatment significantly reduced the proliferation of the MSCs (Supplementary Figure 3E). Similarly, Muc5ac from the KC serum enhanced the proliferation of the AD-MSCs by 2.5–5-folds. However, Muc5ac depletion significantly dampened MSC proliferation to an extent similar to heat inactivation (Supplementary Figure 3F), suggesting that Muc5ac’s ability to induce MSC expansion involves heat-labile factors like Cxcr2-ligands entrapped in its glycosylated polymeric gel. To test this, AD-MSCs were treated with KC serum, with and without heat inactivation and Muc5ac depletion. As noted previously, Muc5ac depletion and heat activation decreased the ability of KC serum to induce MSC proliferation (Figure 5F). Interestingly, ectopic addition of Cxcl5 (Supplementary Figure 3G) was able to rescue the MSC proliferation in a dose-dependent manner only in the presence of Muc5ac-proficient KC serum; however, the addition of Cxcl5 in the Muc5ac-depleted serum could marginally rescue MSC proliferation (Figure 5F). Together, these data suggest that Muc5ac-entrapped Cxcr2 ligands contribute to AD-MSC expansion. Pre-treatment of AD-MSCs with a pharmacologic inhibitor of CXCR2 prior to treatment with KC serum reduced MSC proliferation, similar to Muc5ac depletion and heat inactivation groups (Supplementary Figure 4A). Thus, these data collectively suggest that Muc5ac-trapped ligands induce AD-MSC proliferation primarily via the Cxcr2 signaling axis.
While revisiting the autochthonous tumors, we observed significant co-localization of CD29 and α-SMA in the KC tumor, indicating CD29+ cells contribute significantly to the α-SMA-expressing CAF population in the PC tumor. On the other hand, while CD29 expression was retained in the epithelial compartment, its expression was almost absent in the stromal compartment of the KCM tumors (Figure 6A). This finding supports our working model that Muc5ac shed from the primary tumor promotes CD44/CD29 clustering on AD-MSCs, leading to their mobilization from AT. The specific recruitment of CD44+CD29+ MSCs from systemic circulation may give rise to the α-SMA+ CAFs in the MUC5AC-expressing pancreatic tumors.
Figure 6. MUC5AC expression significantly correlates with peri-ductal α-SMA+-expressing CAFs in murine and human pancreatic tumors.
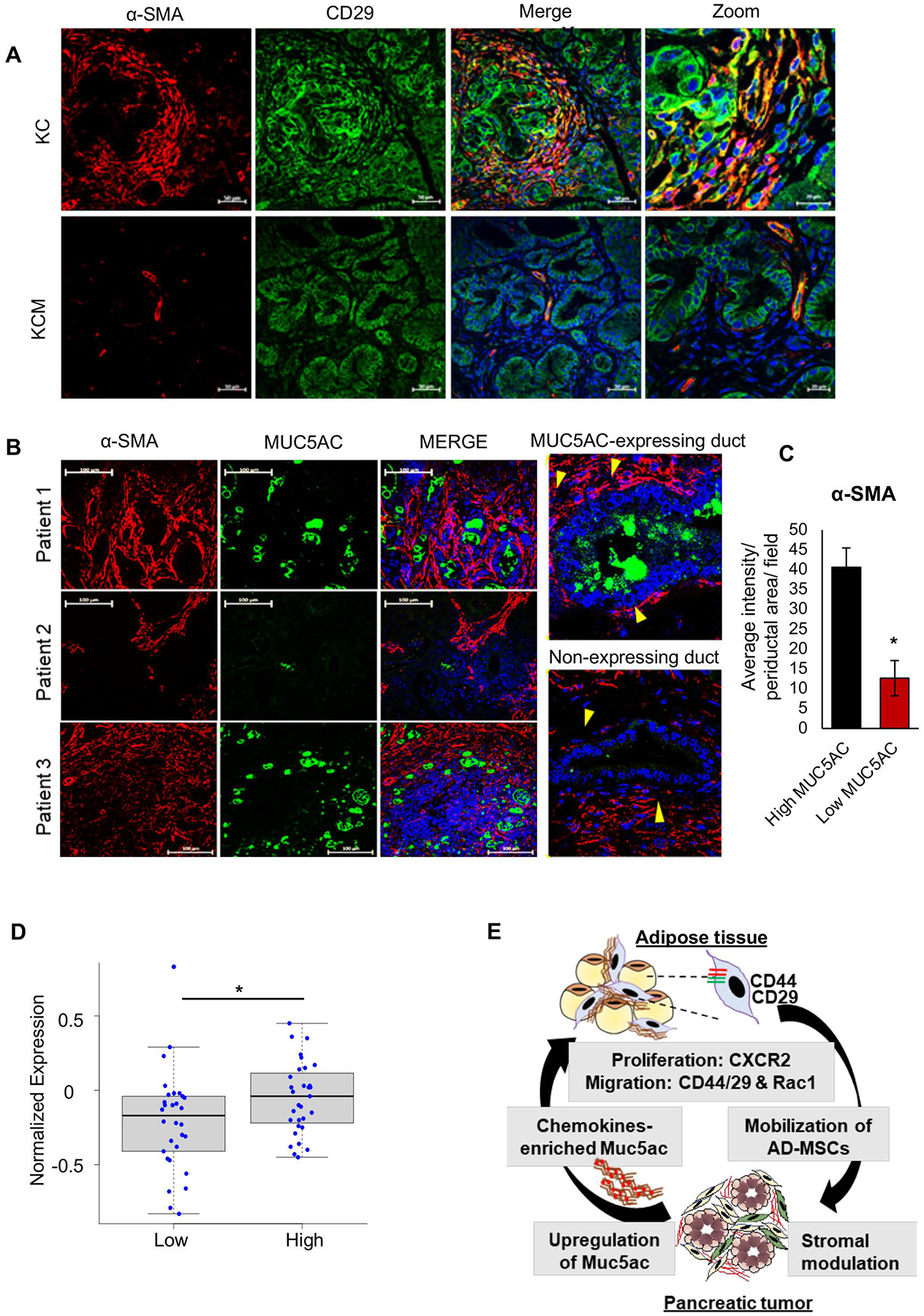
(A) Immunofluorescence images show co-expression of CD29 and periductal α-SMA expressing population in the KC tumor stroma, but not in KCM tumors. (B) Representative immunofluorescence images from PC patient tumor sections (n=6) demonstrate the correlation of MUC5AC expression in the malignant ducts and α-SMA expression in the periductal CAFs. Yellow arrowheads demonstrate the α-SMA+ CAF clusters lying in the juxtacrine position of the malignant cells in MUC5AC-expressing ducts, unlike in non-MUC5AC- expressing ducts. (C) ImageJ-based quantitation demonstrates an increased intensity of α-SMA+ expression in the periductal area/field (n=10 areas/ field, n=3 fields, n=4 patients) in high MUC5AC-expressing ducts, as compared to low-expressing ducts. t-test *p<0.05. (D) Boxplot from PC patients’ proteomic data demonstrate a significant association of MUC5AChighCXCL5high expression with α-SMA+ expression. p=0.03 (E) Schematic workflow of MUC5AC-mediated crosstalk between adipose tissue and pancreatic tumors. Abundantly expressed MUC5AC, with entrapped chemokines, is secreted in the systemic circulation from the pancreatic tumors. The Muc5ac-entrapped tumor secretome reaches the adipose tissue, where it causes the expansion and migration of AD-MSCs via the CXCR2 and Rac1 axis. Muc5ac-mediated CD44/CD29 clustering mobilizes the AD-MSCs into circulation, from where they home to the pancreatic tumors and phenotypically mature into α-SMA+ CAF subtypes.
Interestingly, an analogous association of MUC5AC and α-SMA-expressing CAFs (also known as myofibroblasts27) was observed in the human pancreatic tumor sections. While dense clusters of myofibroblasts surrounded the MUC5AC-expressing ducts, there was sparse α-SMA expression around the non-MUC5AC-expressing ducts (Figure 6B, C, Supplementary Figure 4B). Furthermore, the proteomic analysis from the PC patients (CPTAC, PDC000270) suggested a strong significant correlation between MUC5AChighCxcl5high and α-SMA expression (Figure 6D). Cumulatively, using autochthonous murine models and patient samples, we report a novel systemic association of MUC5AC in PC stromagenesis via modulation of adipose biology (Figure 6E).
Discussion
Transgenic, reporter mice models, and lineage-tracing studies in several solid malignancies have elucidated that the tissue-resident fibroblasts and the MSCs recruited via systemic circulation contribute towards the CAFs28. Our study has demonstrated a unique mechanistic involvement of tumor cell-derived secreted mucin, MUC5AC, in driving stromal heterogeneity by mediating systemic crosstalk between the primary tumor and distant sites. The tumor transcriptomic data and histological analysis from the Muc5ac-KO murine model led us to speculate that the role of MUC5AC in the PC is not restricted to the epithelial compartment; rather, it significantly contributes to stromagenesis. Although recent studies have recognized the contribution of adipose-derived MSCs’ in stromal heterogeneity in several solid malignancies, including PC23, 29, the intricate molecular players and mechanisms remained elusive.
The abundance of MUC5AC in the serum and specific enrichment in adipose tissues, especially in proximity to the MSCs, enhances the expression and clustering of cell-adhesion molecules (CAMs) like CD44 and CD29 (integrinβ1) on AD-MSCs and prompts their cytoskeletal reorganization via the Rac1 axis leading to their mobilization into the systemic circulation. This is supported by a significantly higher number of MSCs in the blood of KC mice as compared to the KCM. A recent finding from an experimental obesity model suggests that secreted protein acidic and rich in cysteine (SPARC) simultaneously binds to the ECM of adipose tissue and integrinβ1 on AD-MSCs, leading to their deadhesion and migration30. Biophysical studies have also suggested that CD44 and CD29 clustering on the AD-MSCs are required to induce their migratory propensity25.
In addition to the protein backbone, another unique attribute of MUC5AC is the negatively charged glycans, which can dock positively charged chemokines and other soluble factors from pancreatic tumors and carry them through systemic circulation to distant sites like adipose tissues. Indeed, increased proliferation of AD-MSCs by Muc5ac- enriched serum and CM was drastically reduced upon heat-inactivation and co-administration with CXCR2 inhibitor. Hence, tumor-borne MUC5AC increases the localized concentration of chemokines in the adipose that promotes local inflammation and drives AD-MSC expansion and mobilization, which modulates stromal heterogeneity at the primary tumor from an early-stage of PC initiation. While previous findings demonstrated that inflamed adipose-derived factors like IL-1β, IL-6, TNF-α, promote the proliferation of PC cells26, a recent study31 proposed a mechanistic link between obesity, inflammation, and PC fibrosis, which further supports our working model.
Owing to the natural propensity of MSCs to home to the sites of inflammation, the perpetual enrichment of chemokines in the MUC5AC-expressing tumors may enhance the recruitment of AD-MSCs from circulation. Another parallel mechanism behind the faster recruitment of AD-MSCs in KC compared to KCM tumors may be the pre-conditioning of the endothelial boundaries both at distant and primary sites by MUC5AC-bound chemokines. Further, our co-implantation allograft studies demonstrated that the AD-MSCs infiltrated the Muc5ac-expressing tumors, abundantly expressed α-SMA, and localized juxtacrine to the malignant cells. This was supported by the significant abundance of CD29+ cells and their co-localization with the α-SMA+ population in the KC tumor stroma. Although AD-MSCs were recently demonstrated to colonize pancreatic tumors and serve as a major source of α-SMA+ cells19, 20, our study provides the mechanistic link driving their mobilization from the local organ and maturation to CAFs in the tumor bed. Guided by the decrease in the periductal clusters of α-SMA+ CAFs around the low-MUC5AC expressing ducts in PC patients’ tumors, we envision that the polymeric gel of MUC5AC can promote the maturation of recruited AD-MSCs to an α-SMAhigh CAF phenotype via juxtacrine signaling22.
Further research should be directed towards understanding the contribution of AD-MSCs in the development of fibrotic scar in benign pathologies like a cutaneous wound, smoking-associated lung fibrosis, and chronic pancreatitis, which mark the proficient expression of secreted mucins. The ability of MUC5AC to entrap inflammatory mediators and act as a systemic messenger of the tumor secretome to distant sites deserves further attention in the field of pancreatic pathologies and beyond. Other possible avenues of investigation are the role of mechanical pressure exhibited by the abundant accumulation of MUC5AC in the pancreatic tumor that can prime PSCs and recruited MSCs for reprogramming into specific CAF subtypes.
Supplementary Material
Acknowledgments:
We are thankful to Dr. Sunil Hingorani at Fred Hutchinson Cancer Research Center for discussion and critical reading of the manuscript. We are grateful to Dr. Christopher M. Evans at the University of Colorado School of Medicine for providing the Muc5ac knockout mouse model. We are also thankful to the Flow Cytometry, Mass Spectrometry, and Proteomic core facilities at UNMC for providing the technical assistance. We also thank Dr. Michel Ouellette and Dr. Rakesh Singh at UNMC for providing Rac 1 and CXCR 2 inhibitors, respectively. We thank Jessica Mercer for her editorial contributions.
Funding:
The authors/work on this manuscript were supported, in parts, by grants from the NIH (R01 CA247471, RO1 CA210637, RO1CA206444, RO1 CA183459, UO1 CA200466, PO1 CA217798, R44 CA235991).
Footnotes
Conflicts of Interest: Authors have no conflicts of interest to declare.
References
- 1.Neoptolemos JP, Kleeff J, Michl P, et al. Therapeutic developments in pancreatic cancer: current and future perspectives. Nature reviews Gastroenterology & hepatology 2018;15:333–348. [DOI] [PubMed] [Google Scholar]
- 2.Patra KC, Bardeesy N, Mizukami Y. Diversity of precursor lesions for pancreatic cancer: the genetics and biology of intraductal papillary mucinous neoplasm. Clinical and translational gastroenterology 2017;8:e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sahai E, Astsaturov I, Cukierman E, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nature Reviews Cancer 2020;20:174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gore J, Korc M. Pancreatic cancer stroma: friend or foe? Cancer cell 2014;25:711–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neesse A, Bauer CA, Öhlund D, et al. Stromal biology and therapy in pancreatic cancer: ready for clinical translation? Gut 2019;68:159–171. [DOI] [PubMed] [Google Scholar]
- 6.Rucki AA, Zheng L. Pancreatic cancer stroma: understanding biology leads to new therapeutic strategies. World journal of gastroenterology: WJG 2014;20:2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Özdemir BC, Pentcheva-Hoang T, Carstens JL, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer cell 2014;25:719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu J, Saiyin H, Fu D, et al. Stroma—A double-edged sword in pancreatic cancer: A lesson from targeting stroma in pancreatic cancer with Hedgehog signaling inhibitors. Pancreas 2018;47:382–389. [DOI] [PubMed] [Google Scholar]
- 9.Cannon A, Thompson C, Hall BR, et al. Desmoplasia in pancreatic ductal adenocarcinoma: insight into pathological function and therapeutic potential. Genes & cancer 2018;9:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sunami Y, Häußler J, Kleeff J. Cellular Heterogeneity of Pancreatic Stellate Cells, Mesenchymal Stem Cells, and Cancer-Associated Fibroblasts in Pancreatic Cancer. Cancers 2020;12:3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helms E, Onate MK, Sherman MH. Fibroblast heterogeneity in the pancreatic tumor microenvironment. Cancer discovery 2020;10:648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elyada E, Bolisetty M, Laise P, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer discovery 2019;9:1102–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ganguly K, Rauth S, Marimuthu S, et al. Unraveling mucin domains in cancer and metastasis: when protectors become predators. Cancer and Metastasis Reviews 2020:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaur S, Kumar S, Momi N, et al. Mucins in pancreatic cancer and its microenvironment. Nature reviews Gastroenterology & hepatology 2013;10:607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krishn SR, Ganguly K, Kaur S, et al. Ramifications of secreted mucin MUC5AC in malignant journey: a holistic view. Carcinogenesis 2018;39:633–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ganguly K, Krishn SR, Rachagani S, et al. Secretory mucin 5AC promotes neoplastic progression by augmenting KLF4-mediated pancreatic cancer cell stemness. Cancer Research 2021;81:91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuiffo BG, Karnoub AE. Mesenchymal stem cells in tumor development: emerging roles and concepts. Cell adhesion & migration 2012;6:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nardi NB, da Silva Meirelles L. Mesenchymal stem cells: isolation, in vitro expansion and characterization. Stem cells 2008:249–282. [PubMed] [Google Scholar]
- 19.Okumura T, Ohuchida K, Kibe S, et al. Adipose tissue‐derived stromal cells are sources of cancer‐associated fibroblasts and enhance tumor progression by dense collagen matrix. International journal of cancer 2019;144:1401–1413. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Daquinag A, Traktuev DO, et al. White adipose tissue cells are recruited by experimental tumors and promote cancer progression in mouse models. Cancer research 2009;69:5259–5266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arina A, Idel C, Hyjek EM, et al. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proceedings of the National Academy of Sciences 2016;113:7551–7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miyazaki Y, Oda T, Mori N, et al. Adipose‐derived mesenchymal stem cells differentiate into pancreatic cancer‐associated fibroblasts in vitro. FEBS Open bio 2020;10:2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kidd S, Spaeth E, Watson K, et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow–derived stroma. PloS one 2012;7:e30563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaur S, Smith LM, Patel A, et al. A combination of MUC5AC and CA19–9 improves the diagnosis of pancreatic cancer: a multicenter study. The American journal of gastroenterology 2017;112:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ke C, Chen J, Guo Y, et al. Migration mechanism of mesenchymal stem cells studied by QD/NSOM. Biochimica et Biophysica Acta (BBA)-Biomembranes 2015;1848:859–868. [DOI] [PubMed] [Google Scholar]
- 26.Brocco D, Florio R, De Lellis L, et al. The role of dysfunctional adipose tissue in pancreatic cancer: A molecular perspective. Cancers 2020;12:1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Öhlund D, Handly-Santana A, Biffi G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. Journal of Experimental Medicine 2017;214:579–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pereira BA, Vennin C, Papanicolaou M, et al. CAF subpopulations: a new reservoir of stromal targets in pancreatic cancer. Trends in cancer 2019;5:724–741. [DOI] [PubMed] [Google Scholar]
- 29.Nieman KM, Kenny HA, Penicka CV, et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nature medicine 2011;17:1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tseng C, Kolonin MG. Proteolytic Isoforms of SPARC induce adipose stromal cell mobilization in obesity. Stem cells 2016;34:174–190. [DOI] [PubMed] [Google Scholar]
- 31.Incio J, Liu H, Suboj P, et al. Obesity-induced inflammation and desmoplasia promote pancreatic cancer progression and resistance to chemotherapy. Cancer discovery 2016;6:852–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.