

Abstract
Background & Aims:
Hepatic energy metabolism is a dynamic process modulated by multiple stimuli. In non-alcoholic fatty liver disease (NAFLD), human studies typically focus on the static fasting state. We hypothesized that unique postprandial alterations in hepatic lipid metabolism are present in NAFLD.
Methods:
In a prospective clinical study, 37 NAFLD and 10 healthy control subjects ingested a standardized liquid meal with pre- and postprandial blood sampling. Postprandial plasma lipid kinetics were characterized at the molecular lipid species level by untargeted lipidomics, cluster analysis and lipid particle isolation then confirmed in a mouse model.
Results:
There was a specific increase of multiple plasma diacylglycerols (DAG) species at 4 hours postprandially in NAFLD subjects but not in controls. This was replicated in a NASH mouse model, where postprandial DAGs increased in plasma and concomitantly decreased in the liver. The increase in plasma DAGs appears early in the disease course, is dissociated from NAFLD severity and obesity, and correlates with postprandial insulin levels. Immunocapture isolation of VLDL in human samples and stable-isotope tracer studies in mice revealed that elevated postprandial plasma DAGs reflect hepatic secretion of endogenous, rather than meal-derived lipids.
Conclusions:
We identified a selective insulin-related increase in hepatic secretion of endogenously-derived DAGs after a mixed meal as a unique feature of NAFLD. DAGs are known to be lipotoxic and associated with atherosclerosis. While it is still unknown whether the increased exposure to hepatic DAGs contributes to extrahepatic manifestations and cardiovascular risk in NAFLD, our study highlights the importance of extending NAFLD research beyond the fasting state.
Keywords: Diacylglycerols, postprandial lipids, NAFLD, lipidomics, mixed meal
Graphical Abstract
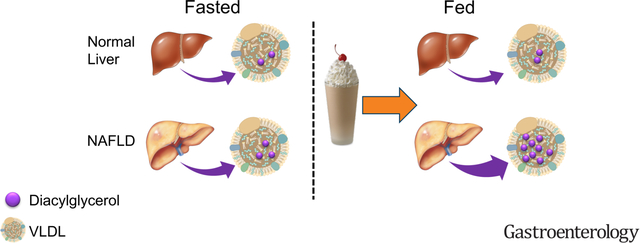
LAY SUMMARY
In humans with NAFLD and in mouse models of the disease, mixed-meal leads to selective secretion of diacylglycerols (DAG) from the liver to the plasma through VLDL.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most common liver disease in western societies, the fastest rising cause for hepatocellular carcinoma and a leading indication for liver transplantation making it a major public health problem1. Despite its high prevalence and impact, significant gaps still exist in our understanding of this disease and its pathophysiology.
Energy imbalance with resultant obesity and insulin resistance are important initiating events in the development of NAFLD. It is unclear whether the progression from steatosis to nonalcoholic steatohepatitis (NASH), which only occurs in a subset of NAFLD patients, results from the same drivers that promote hepatic fat accumulation or whether a “second hit” in the form of lipotoxic metabolites is needed to trigger inflammation, intracellular stress responses and oxidative stress2. The effects of total energy intake and specific nutritional components on development and progression of NAFLD have been studied before3; however, studies typically focused on average or long-term intake, whereas the dynamic effects of a single standardized oral load have rarely been studied. Similarly, in the continuing search for biomarkers that identify NAFLD or distinguish NASH from steatosis, studies predominantly rely on a static one-time assessment, typically in the fasting state4. This is easy to standardize and yields important information; however, significant additional information can be obtained with dynamic tests that employ a perturbation from the steady state. Indeed, postprandial dyslipidemia may not be predicted by fasting triacylglycerols (TAGs) in obese subjects5.
The study of postprandial lipid metabolism has mainly focused on lipoprotein particles, TAGs, and free fatty acids (FFA) as a class; however, several other lipid species are generated during absorption of dietary fat. These include intermediary lipids from intestinal lipolysis and TAG re-synthesis as well as partitioning into cholesterol esters (CEs) and phospholipids6. Beyond lipid absorption, peripheral lipases and hepatic TAG recycling contribute to the intricate process of lipid metabolism but postprandial kinetics of other lipid species remain poorly understood.
We hypothesized that postprandial lipid metabolism will be altered in subjects with NAFLD, independent of the fasted state. To test this hypothesis, we applied an untargeted lipidomic assessment to study the dynamic response of subjects to a standardized mixed-meal challenge, aiming to shed light on the pathogenesis of fat accumulation in NAFLD and to look for possible biomarkers of disease presence or severity. In addition, we systemically evaluated the postprandial interplay between endogenous and meal-derived lipids in clinical plasma samples and in a mouse model of NASH. We show that NAFLD is associated with a sustained increase in postprandial diacylglycerols (DAGs) secreted by the liver and found in VLDL particles. Moreover, the postprandial increase in plasma DAGs appears to manifest early in disease progression and correlate with insulin levels. Finally, using stable isotope tracers, we demonstrate that elevated postprandial plasma DAGs are derived from endogenous lipids and are not a product of lipid metabolism from the current or previous meal.
Material and Methods
Clinical trial design
Adult subjects with NAFLD or healthy controls were enrolled in a prospective single-center mechanistic study (clinicaltrials.gov NCT02520609). After an overnight fast, subjects received a single dose of a liquid meal (Ensure Plus, Abbott) containing 30% of their total estimated daily energy needs. Blood samples were collected prior to the meal and at 0.5, 1, 2 and 4 hours after it and stored at −80C for processing.
Animal Studies
Eight-week-old C57Bl/6N male mice were randomized to the Gubra Amylin NASH (GAN) diet (D09100310) or a matching control diet (D17112301R, Research Diets, Inc.) for 26 weeks. At the endpoint, mice were fasted from 12:00–18:00, gavaged with a single dose of Ensure Plus (25 μL/g lean body mass) at 18:00, and euthanized 0.5, 1, 2, or 4 hours later. Mice euthanized at 0-hours were given 25 μL/g water by gavage immediately prior to euthanasia. Blood was collected and centrifuged to isolate plasma. Liver tissue was divided and flash-frozen, embedded in OCT-medium or formalin-fixed in 10% phosphate-buffered formalin for 24 hours.
Sample processing and lipidomic analysis
Plasma lipids were extracted using the Folch method. Liver lipids were extracted using a modified Bligh & Dyer method. 5 μL or 1 μL of extracted plasma or liver lipids, respectively, were subjected to ultra-performance liquid chromatography coupled to quadrupole time of flight mass spectrometry (UPLC-QTOFMS) for untargeted lipidomics or tandem mass spectrometry (UPLC-MS/MS) for targeted measurement of DAGs. For untargeted lipidomics analysis, peak picking, deconvolution, adduct annotation and retention time alignment was performed using Progenesis QI software.
In targeted analyses, abundance of DAG species is reported as absolute concentration if a standard was available. In the absence of standards, abundance of DAG species is reported relative to the average abundance at baseline in controls.
Stable isotope enrichment analysis of plasma and liver lipids
Neutral and stable isotope-labelled TAGs, DAG(36:2)(18:1–18:1) and oleic acid were measured in plasma and liver samples by UPLC-QTOFMS. Natural isotope abundance was corrected using IsoCorrectoR7.
Statistics
Clinical data were summarized using means and standard deviation (SD) for continuous variables and percentages for discrete variables. Continuous variables were assessed using parametric and non-parametric tests and discrete variables were analyzed using Fisher’s exact test. Significance was set at α=0.05.
R (version 3.6.2) and SIMCA 15 were used to analyze data from untargeted lipidomics. Principal component analysis (PCA) and multilevel partial least squares discriminant analysis (mPLS-DA) were performed using SIMCA and mixOmics (6.10.8), respectively. Individual lipid features were statistically analyzed using a mixed-model repeated measures ANOVA (nlme 3.1) with Benjamini-Hochberg false discovery correction (multtest 2.42). Cluster analysis was performed to identify postprandial lipid patterns on features that were significantly different by group, time, and interaction using the degPatterns function, which correlates repeated measure feature abundance and performs divisive hierarchical clustering (DEGreport 1.22). Z-scores were calculated by centering the mean and dividing by the standard deviation for each lipid feature then calculating the average per group within each cluster.
Linear and multiple regressions for association of clinical factors with postprandial change lipids and receiver operating characteristic (ROC) curve evaluating performance of postprandial change in lipids for discrimination of NAFLD were obtained using GraphPad Prism versions 8.4.1 and 9.0.0 (GraphPad Software).
Study Approval
The clinical study was approved by the NIDDK/NIAMS Institutional Review Board and all subjects provided written informed consent (clinicaltrials.gov NCT02520609). All animal studies and procedures were carried out in accordance with Institute of Laboratory Animal Resources guidelines under protocols approved by the National Cancer Institute Animal Care and Use Committee.
Additional details are provided in the supplementary methods.
Results
Between November 2015 and October 2018, 52 individuals (40 NAFLD and 12 controls) were screened and 37 NAFLD patients and 10 controls completed the study. Baseline characteristics are described in Table 1. With the exception of sex, a statistically significant difference between both groups was noted among all clinically relevant baseline characteristics. Hispanics were overrepresented in the NAFLD group, as expected.
Table 1.
Baseline Demographic and Clinical Data
| Controls (n=10) | NAFLD (n=37) | p-value | |
|---|---|---|---|
| Age (years) | 33 (11.7) | 51 (12.0) | <0.0001 |
| Sex - (female %) | 4 (40%) | 20 (54%) | 0.49 |
| Ethnicity [n (%)] | |||
| Caucasian | 5 (50%) | 17 (46%) | 0.009 |
| Hispanic | 0 (0%) | 14 (38%) | |
| African-American | 3 (30%) | 1 (3%) | |
| Other | 2 (20%) | 5 (13%) | |
| BMI (kg/m 2 ) | 21.8 (2.0) | 33.3 (5.5) | <0.0001 |
| ALT (U/L) | 16 (7.7) | 62 (54) | <0.0001 |
| HbA1C (%) | 5.3 (0.3) | 5.9 (0.9) | 0.03 |
| Fasting glucose (mg/dL) | 89 (5.2) | 108 (21.6) | <0.0001 |
| Fasting Insulin (mcU/mL) | 7.9 (5.3) | 23.9 (9.6) | <0.0001 |
| HOMA-IR | 1.8 (1.2) | 6.5 (3.2) | <0.0001 |
| Triglycerides (mg/dL) | 61 (18) | 171 (70) | <0.0001 |
| Waist circumference (cm) | 77.4 (5.6) | 108.4 (13.4) | <0.0001 |
| Percent Fat (%) | 24 (8.7) | 40 (7.4) | <0.0001 |
| Fat mass (kg) | 14.6 (4.8) | 37.0 (12.9) | <0.0001 |
| Lean mass (kg) | 44.9 (10.4) | 51.3 (10.1) | 0.09 |
| Fat Free Mass (kg) | 47.5 (10.9) | 53.9 (10.5) | 0.10 |
| Visceral Adipose Tissue volume (cm 3 ) | 256 (127) | 2257 (1114) | <0.0001 |
| Baseline Energy Expenditure (kcal/min) a | 1.57 (0.47) | 1.88 (0.33) | 0.03 |
| Baseline Respiratory Quotient A | 0.87 (0.02) | 0.89 (0.04) | 0.12 |
| Postprandial Change in Respiratory Quotient | 0.08 (0.26) | −0.01 (0.03) | 0.31 |
| Mixed Meal Dose (kCal) | 613 (101) | 683 (127) | 0.11 |
Demographic and clinical characteristics are presented as mean (SD) unless otherwise stated.
n=9 Controls and n=36 NAFLD
Within the NAFLD group, 15 subjects had a liver biopsy available for review with 13 of 15 demonstrating NASH (Table S1). Transient elastography was obtained in 32 patients, with a mean score of 8.0 Kilopascals (SD 6.3) and average controlled attenuation parameter (CAP) of 323 dB/m (SD 53.1), reflecting significant steatosis. The NAFLD fibrosis score (NFS) was <−1.455 (minimal fibrosis) in 46% (17 patients), indeterminate in 51% (19 patients) and >0.675 (advanced fibrosis) in 3% (1 patient).
Untargeted lipidomics identifies clusters of altered postprandial plasma lipids in human NAFLD
At the baseline fasting state, plasma lipidomics demonstrated clear separation between control and NAFLD subjects. The lipidome variation was primarily (76%) driven by elevated triacylglycerol and phosphatidylcholine species in the NAFLD samples (Fig. 1A, B). Postprandial lipid kinetics, assessed by mPLS-DA, revealed a progressive temporal shift in lipid profile between baseline and 4 hours with a return to baseline at 24 hours for both control and NAFLD subjects (Fig. 1C).
Figure 1. Untargeted lipidomic analysis reveals postprandial plasma diacylglycerols are significantly altered in NAFLD subjects.
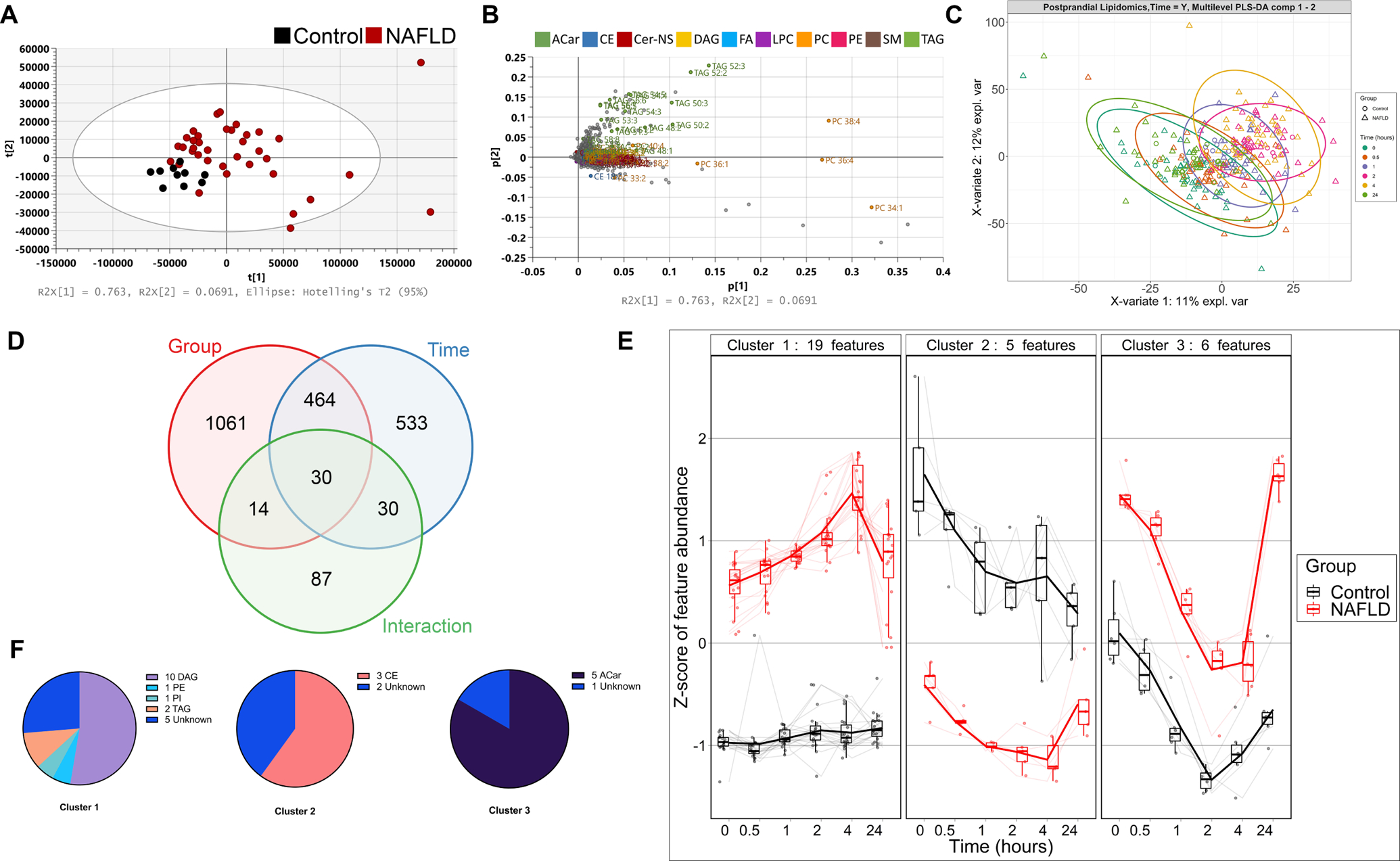
(A) Principal component analysis shows separation between fasting control and NAFLD plasma, driven by increased TAG and PC lipid species (B). (C) Multilevel partial least squares discriminant analysis (mPLS-DA) demonstrates time-dependent changes in postprandial plasma lipids from 0 to 4 hours with a return to baseline at 24 hours. (D) Repeated measures mixed model ANOVA identified 30 features that are significantly different by group, time and interaction. (E) Cluster analysis of the 30 significantly different lipid features demonstrates three distinct postprandial kinetic patterns, grouped mainly by class (F). Control n=10, NAFLD n=37. Lipid classes are colored and abbreviated as follows: acylcarnitine (ACar), cholesterol ester (CE), diacylglycerol (DAG), fatty acids (FA), phosphatidylcholine (PC), phosphatidylethanolamine (PE), triacylglycerol (TAG). Z-scores were calculated by centering the mean and dividing by the standard deviation for each lipid feature then calculating the average per group. Box and swarm – median, interquartile range (IQR) and 1.5xIQR whiskers. Thin lines - individual feature trajectories; thick lines - mean z-score for all features in the cluster.
We applied a repeated measure mixed-model ANOVA to serial postprandial samples on all lipid features generated by untargeted analysis (n=3469) to identify features that demonstrate postprandial lipid kinetic differences between control and NAFLD subjects. A total of 30 features were found to be significantly different by group (control vs. NAFLD) (q<0.05), time (q<0.05) and by interaction (P<0.05) (Fig. 1D).
Since lipid species that follow similar postprandial kinetics may be related, we performed a cluster analysis on significantly altered lipid features and identified three groups of features with distinct postprandial kinetic patterns consisting of 19, 5 and 6 lipid features, respectively (Fig. 1E). Cluster 1 features were mainly comprised of DAGs, which were identified to the isomer level (Fig. 1F, Table S2). Cluster 1 features were elevated at baseline in NAFLD compared to controls. Independently of the elevated baseline, cluster 1 features increased markedly to 4 hours in NAFLD subjects before returning to baseline at 24 hours whereas in control subjects, only a slight increase was seen, peaking at 2 hours. In cluster 2, three CE species were lower in NAFLD subjects compared to controls at baseline and decreased over the postprandial time course for both NAFLD and control subjects. Cluster 3 contained five acylcarnitine features from three acylcarnitine species (Table S2). These species were initially elevated in NAFLD plasma but similarly decreased over the time course in both experimental groups prior to a return to baseline at 24 hours (Fig. 1E).
Human and mouse NAFLD associated with sustained postprandial increase in plasma diacylglycerols
The difference in postprandial plasma kinetics between control and NAFLD subjects of individual DAGs was confirmed by targeted analysis of DAG isomers. As expected from the selection criteria, requiring that features will be significantly different between groups, plasma DAGs from cluster 1 were elevated in NAFLD plasma at baseline compared to controls (Fig. S1). In addition, postprandial plasma DAG levels demonstrated a continued rise from 2 to 4 hours in NAFLD while levels in control subjects decreased or were unaffected over time (shown as fold-change from baseline in Fig. 2 and abundance in Fig. S1). The targeted analysis allowed us to identify additional DAG species, even those that were not initially identified within cluster 1, with a similar pattern in most species measured (Fig. S2). Importantly, the postprandial increase in DAGs was not secondary to delayed absorption or overall lipid kinetics, as most TAG species, the predominant glycerolipid species in plasma, followed a similar temporal pattern in both NAFLD and controls or remained unchanged during the time course (Fig. S3). Thus, we demonstrate a specific dysregulation of DAG kinetics in NAFLD.
Figure 2. Human NAFLD causes a sustained postprandial increase in plasma diacylglycerols.
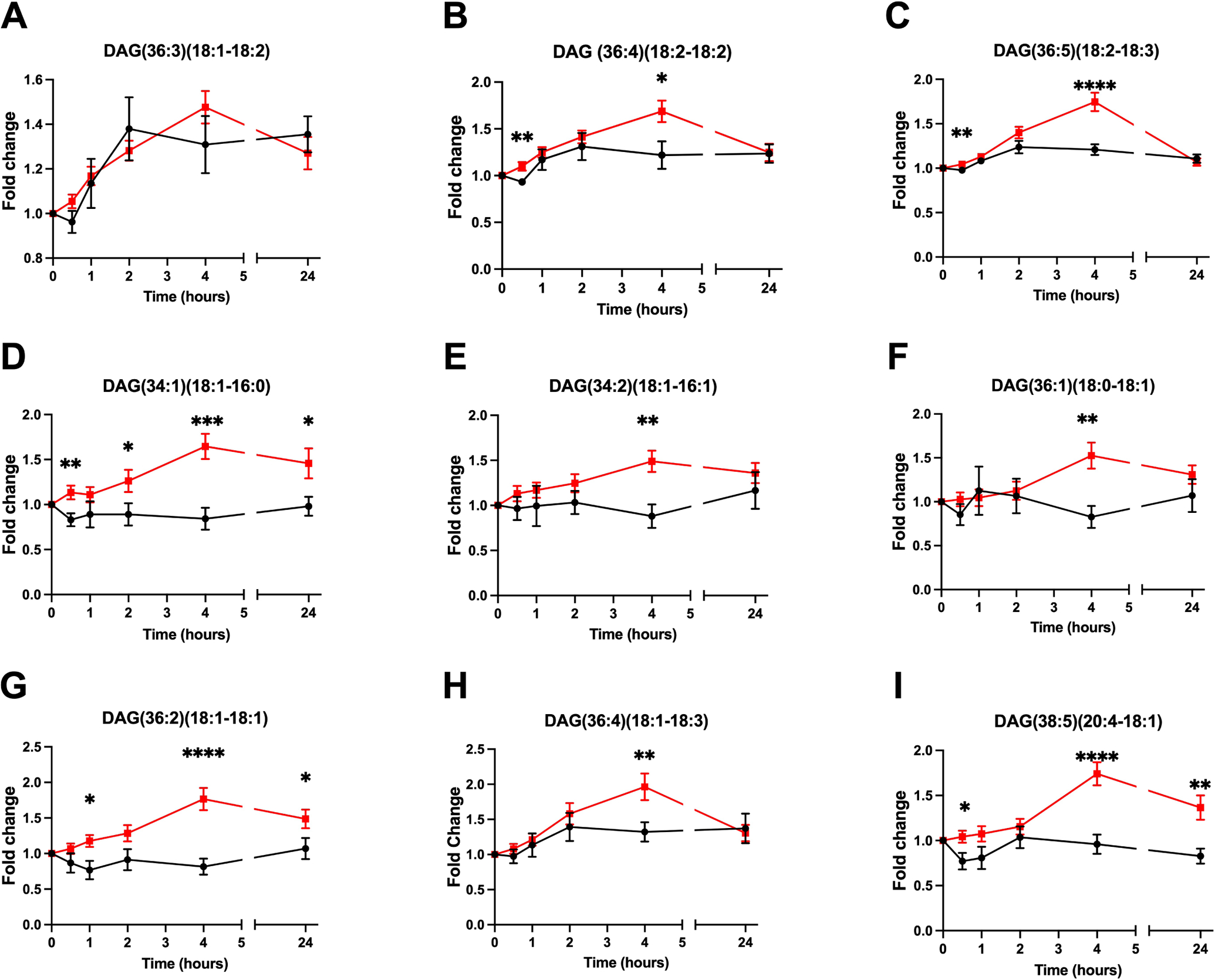
Fold-change from baseline in postprandial plasma diacylglycerols levels obtained using targeted measurements. Data are presented as mean±SEM. Statistical analysis was performed using repeated-measures two-way ANOVA, * P<.05, ** P<.01, *** P<.001, **** P<.0001 compared to control. Control (black) n=10, NAFLD (red) n=37. Abundance of the same DAG species is presented in Figure S1.
To further characterize postprandial DAG kinetics in NAFLD, we established a similar model in mice. Mice fed GAN-diet for 26 weeks developed NASH with hepatic steatosis, fibrosis and inflammation as well as a considerable increase in body weight, liver weight, fat mass, plasma AST and ALT compared to controls (Fig. S4). Mice ingest their largest meal soon after the end of the light cycle (18:00) and were therefore fasted from 12:00–18:00 and then administered the same liquid meal provided to human subjects. Several plasma DAGs were significantly increased in NASH mice 1 hour postprandially compared to controls (Fig. 3A–G). The increase in plasma DAGs coincided with a marked and significant decrease in hepatic DAGs (Fig. 3H–K). Of note, some DAG isomers had differing postprandial kinetics in human and animal studies, such as DAG(36:5)(18:2–18:3), which was significantly elevated in human NAFLD subjects but decreased in NASH mice compared to controls (Figs. 2C and 3C, respectively).
Figure 3. Murine NASH causes a concurrent postprandial diacylglycerol plasma increase and hepatic decrease.
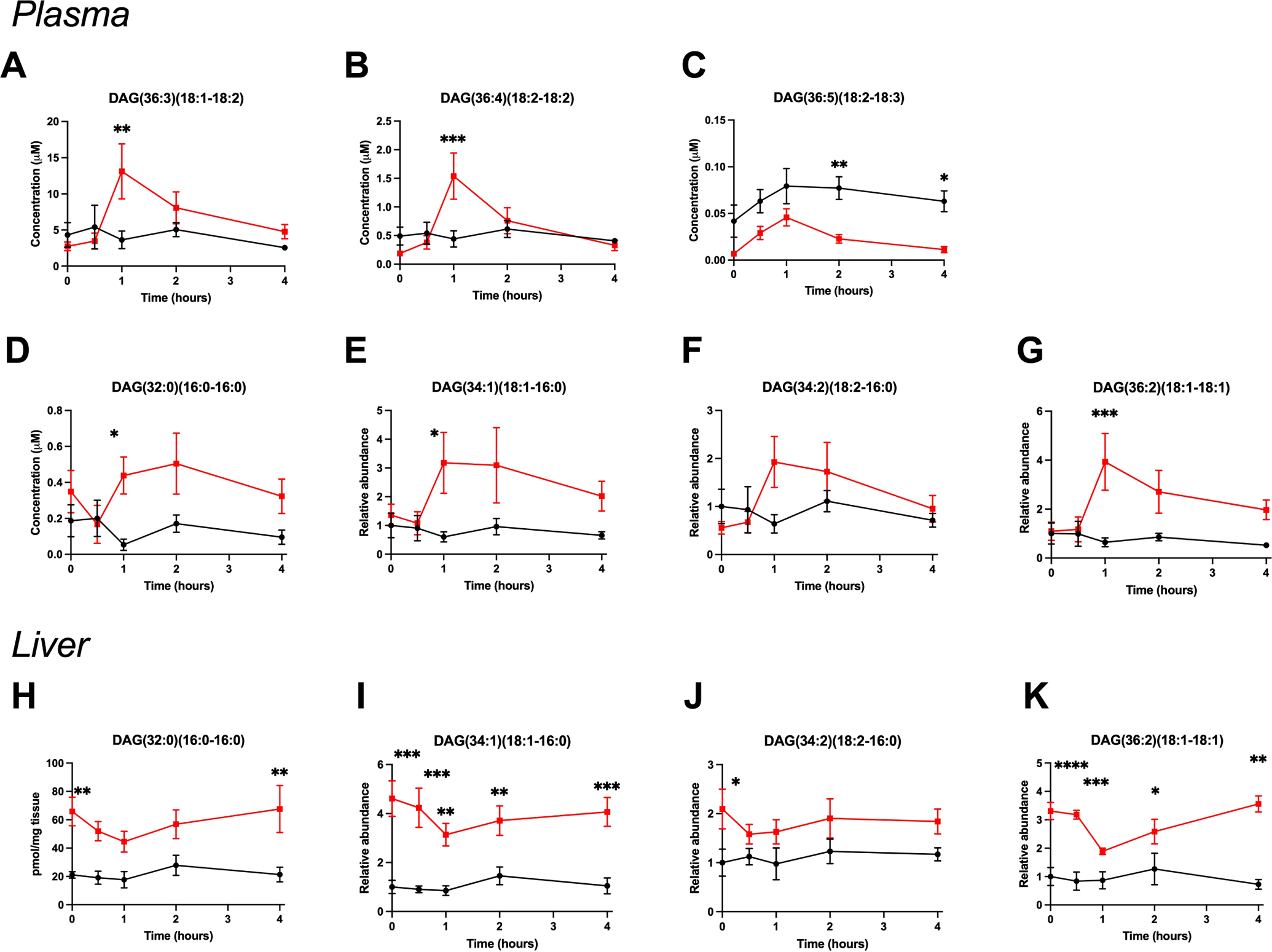
(A-G) Postprandial plasma diacylglycerol kinetics in control (black) and NASH (red) mice fasted for 6 hours and given a bolus of Ensure Plus (25μL/g lean body mass) by gavage. (H-K) Postprandial liver diacylglycerols comprised of the most abundant fatty acids in the GAN-diet. Data are presented as mean±SEM. Statistical analysis was performed by two-way ANOVA, * P<.05, ** P<.01, *** P<.001, **** P<.0001 compared to control, n=4–5.
The postprandial diacylglycerol increase in NAFLD occurs in particles derived from the liver and not from the meal
Plasma DAGs are expected to be present predominantly in VLDL or chylomicrons (CM) particles, both components of the triacylglycerol-rich fraction (TRF). To determine if the postprandial increase in DAGs in NAFLD is a liver-derived or meal-derived process, we aimed to study it separately in VLDL or CM, respectively. The ability of size and density alone to separate VLDL from CM is limited; we therefore established an immune capture assay to purify VLDL (Fig. S5A–C). TRF and VLDL were isolated from postprandial human control and NAFLD plasma samples (n=3) by differential centrifugation and immunocapture, respectively (Fig. S5D). Four hours postprandially, DAGs significantly increased from baseline in purified VLDL isolates from NAFLD subjects but not in controls (Fig. 4A, B). Thus, a postprandial increase in DAGs after a meal occurs in the VLDL plasma compartment in humans.
Figure 4. The postprandial increase in NAFLD plasma diacylglycerols is comprised of endogenous lipids secreted in VLDL.
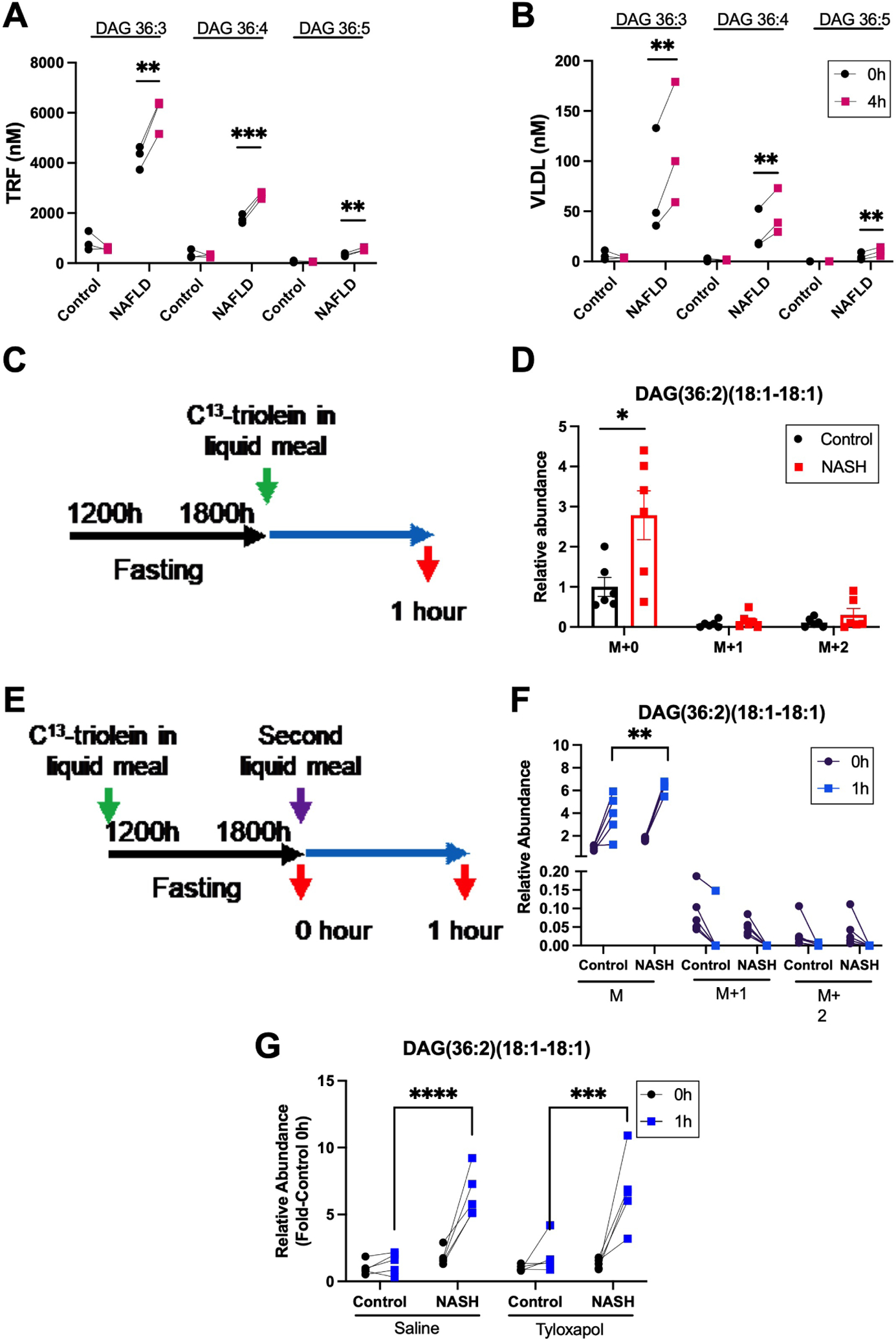
(A) Plasma diacylglycerols in the triacylglycerol rich fraction (TRF) and (B) VLDL fraction at 0 and 4 hours following food bolus ingestion by control and NAFLD human subjects. (C) Schematic of 13C-triolein given in the current or (E) previous liquid meal to control and NASH mice. (D) Stable isotope labelling of postprandial plasma DAG(36:2)(18:1–18:1) in control and NASH mice 1 hour after a liquid meal bolus containing 13C-triolein. (F) Labelling of plasma DAG(36:2)(18:1–18:1) 1 hour after a second liquid meal when the prior liquid meal contained 13C-triolein. (G) Postprandial plasma DAG(36:2)(18:1–18:1) in control and NASH mice treated with saline or tyloxapol 30 minutes prior to liquid meal. M denotes unlabeled DAG; M+1, DAG with one labeled oleic acid; and M+2, DAG with both oleic acids labeled. Data are presented mean±SEM or individual values, statistical analysis was performed using a paired two-tailed Student’s t-test, n=3 (A, B), n=6 (D), n=5 (F, G) ** P<.01, *** P<.001, **** P<.0001.
To determine whether meal-derived lipids also contribute to postprandial plasma DAGs we fed NASH or control mice a liquid meal containing 12C or 13C-triolein and measured DAG(36:2)(18:1–18:1) one hour postprandially (Fig. 4C). Despite an increase in total DAG(36:2)(18:1–18:1) as expected, no increase was seen in 13C-labeled DAG, and only the unlabeled plasma DAG(36:2)(18:1–18:1) was significantly increased in NASH compared to controls (Fig. 4D). Furthermore, only 15% of plasma DAG(36:2)(18:1–18:1) were labelled by the tracer in control and NASH mice, suggesting that meal-derived lipids did not contribute to the postprandial increase in plasma DAGs (Fig. S6B).
A “second meal effect” was described, where postprandial plasma lipids are derived from a previous and not the most recent meal. To determine if increased postprandial DAGs in NAFLD derive from lipids consumed in the previous meal, mice were administered a liquid meal containing 13C-triolein, followed 6 hours later by a second liquid meal containing unlabeled lipids (Fig. 4E), and plasma DAG(36:2)(18:1–18:1) was measured pre- and post-prandially. Only the unlabeled DAG(36:2)(18:1–18:1) fraction was significantly increased in mice with NASH compared to controls (Fig. 4E) while no increase was seen in the labeled fraction. Furthermore, DAG(36:2)(18:1–18:1) preprandial isotope enrichment could be found in plasma from control and NASH mice but was mostly diminished 1 hour postprandial (Fig. S6F). In addition, the percentage of pre-prandial DAG(36:2)(18:1–18:1) isotope enrichment was significantly increased in controls as compared to mice with NASH. Thus, the human and mouse data taken together suggest that elevated postprandial plasma DAGs in NAFLD are derived from the liver and not from the current or previous meal.
Peripheral lipases do not contribute to increased DAGs in NAFLD in mice
The increase in VLDL-DAGs postprandially could reflect increased secretion of DAGs from the liver or incomplete lipolysis of liver-secreted TAGs by peripheral lipases. When peripheral lipases were inhibited by tyloxapol, mice with diet-induced NASH continued to demonstrate a significant postprandial increase in plasma DAGs compared to controls (Fig. 4G). Thus, DAGs in postprandial plasma are mostly derived from liver secretion.
Postprandial DAG kinetics are not associated with severity of human or mouse NAFLD
Since baseline and postprandial DAG kinetics differed markedly between controls and NAFLD, we tested their association with parameters of disease severity within the NAFLD cohort. We generated a compound average score of the 19 lipid features in cluster 1 (Table S2) for each subject. Baseline cluster 1 score was not associated with any of the clinical features of obesity, NAFLD or insulin resistance.
No significant correlation was found between the postprandial change in cluster 1 score from baseline to 4 hours and markers of hepatic inflammation (ALT, r=0.19, p=0.25) (Fig. S7); non-invasive markers of fibrosis such as liver stiffness by transient elastography (r=−0.005, p=0.98) or NAFLD fibrosis score (r=−0.054, p=0.75); steatosis (CAP, r=0.21, p=0.33) (Fig. S7), or hepatic insulin resistance (HOMA-IR r=−0.12, p=0.48). In the subset of subjects who had liver biopsy data, no correlation was seen with histological scores. The postprandial change in cluster 1 score was also not associated with other clinical features such as BMI (r=0.19, p=0.26), serum TAG (r=0.00005 p=1.0) (Fig. S7), body fat mass (r=0.21, p=0.21), energy expenditure (r=0.05, p=0.79) or change in RQ post meal (r=−0.11, p=0.52).
We similarly evaluated the association of clinical characteristics with total DAG levels and a single DAG, using the baseline-to-4 hour change in DAG(36:4)(18:2–18:2) (Fig. 2B) as a representative feature. Baseline total DAG levels were negatively correlated with BMI (R=−0.37, P=0.02) but no other clinical parameter. The 4-hour change in total DAG or DAG(36:4)(18:2–18:2) was not correlated with any of the tested clinical characteristics (data not shown). We thus conclude that the increase in postprandial plasma DAGs seen in subjects with NAFLD is not associated with the severity of their liver disease. This was further supported by our mouse model, where an increase in 1-hour postprandial plasma DAGs compared to controls was already observed in mice after 8 or 16 weeks of GAN diet, at an earlier stage of disease and prior to development of fibrosis (Fig. S8).
The 4-hour change in the cluster 1 score significantly discriminated between NAFLD and controls using a receiver-operator characteristics (ROC) analysis with an area under the curve of 0.88 (95% CI 0.76–1.0) (Fig. S7E).
Genomic DNA was not collected as part of this study but was available for 21 NAFLD subjects from prior studies. For the TM6SF2 SNP rs58542926, 3 subjects carried the minor allele T while 21 were homozygous for the wild type C allele. 16 subjects were carriers of the PNPLA3 rs738409-G allele and 5 were rs738409-CC homozygotes. PNPLA3 or TM6SF2 genotype were not associated with postprandial changes in cluster 1 score or individual DAG levels (data not shown).
Human postprandial DAGs are correlated with insulin response to a meal
Patients with NAFLD are typically insulin resistant, manifested as higher HOMA-IR (Table 1) and insulin levels (fasting and postprandial, Fig S9A–B). To determine whether postprandial insulin affects hepatic DAG secretion, we evaluated the association between serum insulin and the change in the DAG-predominant cluster 1 score from baseline to 4 hours postprandially. The change in cluster 1 score was significantly associated with fasting (R2=0.16, p=0.005) and 2-hour insulin levels (R2=0.15, p=0.007), but not with the 4-hour insulin (Fig. 5A–C). Similarly, the 4-hour changes from baseline in the individual concentrations of DAG(36:4)(18:2–18:2) and DAG(36:5)(18:2–18:3) were also highly correlated with 2-hour insulin levels (Fig. 5D–E)). On a multivariate linear regression, cluster 1 increase was independently associated with fasting insulin (p=0.0001), 2-hour insulin (p=0.0005) and their interaction term (p=0.0004) and the model explained a significant amount of the variability (R2=0.40, P<.0001).
Figure 5. Postprandial insulin correlates with DAG secretion.
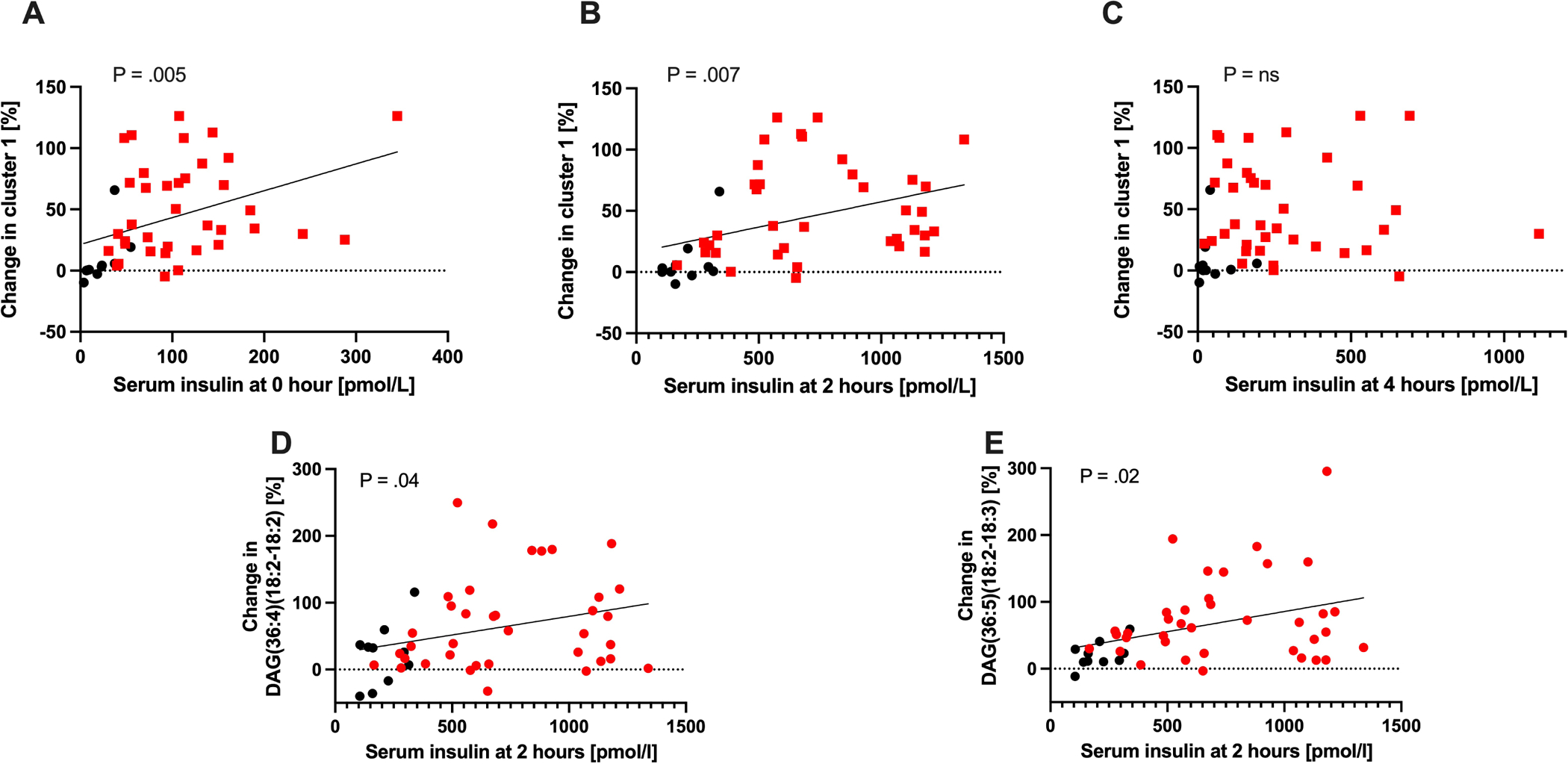
Serum levels of insulin at (A) fasting, (B) 2-hours and (C) 4-hours after a mixed meal and their association with the 4-hour increase in the DAG-predominant Cluster 1 score. (D) Serum levels of insulin at 2-hours after a mixed meal and association with the 4-hour increases of DAG(36:4)(18:2–18:2) and (E) DAG(36:5)(18:2–18:3). Red – NAFLD, black – healthy controls. P-value from Pearson correlation.
Postprandial DAG kinetics are a feature of human NAFLD and not of obesity
NAFLD is commonly associated with obesity and the metabolic syndrome but postprandial DAGs were not associated with BMI (Fig. S7A) or body fat mass. To confirm that the aberrant postprandial increase in plasma DAGs is a unique feature of NAFLD and not of obesity, we performed a similar analysis in subjects with obesity but no evidence of excess liver fat. The obese non-NAFLD cohort was not significantly different from the NAFLD patients in BMI (P=0.20) and age (P=.43) but had significantly different HOMA-IR (P<.0001) and TAGs (P=.02) (Table S3, Figure S9) and liver fat percentage of 1.8% (range 0.5–3.8%). Samples for these subjects were available before the meal challenge and 3-hours postprandially; we therefore compared them to the change from baseline to the average of the 2- and 4-hour time points in NAFLD and healthy control subjects.
In contrast to subjects with NAFLD, plasma levels of DAG(36:3)(18:2–18:1), DAG(36:4)(18:2–18:2) and DAG(36:5)(18:2–18:3) did not increase significantly in obese non-NAFLD subjects after a liquid mixed-meal (Fig. 6). Similarly, 2-hour insulin levels in non-NAFLD obese did not correlate with the 3-hour increase in individual DAGs (data not shown). Thus, the increase in postprandial plasma DAGs appears to require the presence of NAFLD.
Figure 6. Obesity does not cause a postprandial increase in plasma diacylglycerols.
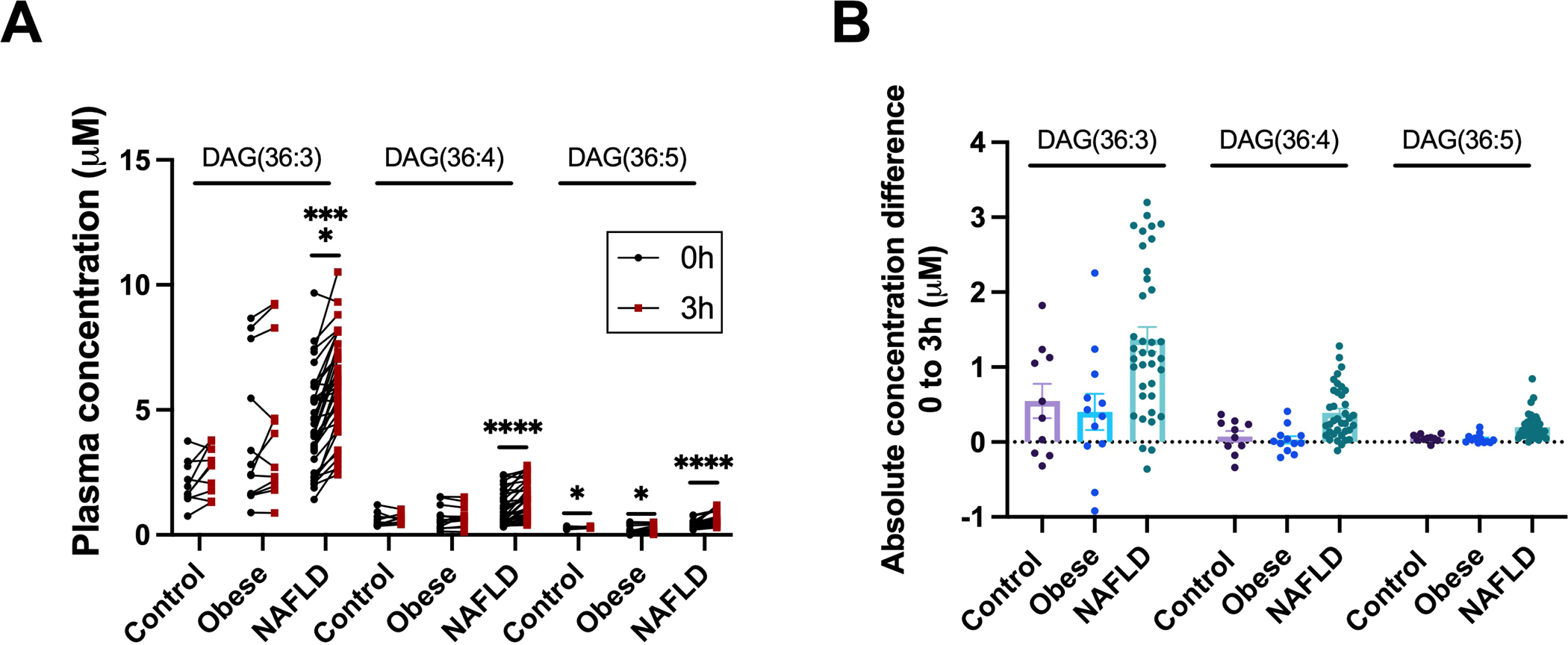
(A) Quantified concentrations and (B) absolute concentration differences from 0 to 3 hours of pre- and postprandial plasma diacylglycerols in control, obese non-NAFLD, and NAFLD study subjects. The 3-hour time point for control and NAFLD subjects was calculated as an average of 2 and 4-hours. Data are presented as mean±SEM. Statistical analysis was performed using a paired two-tailed Student’s t-test, * P<.05, **** P<.0001. Control, n=10; obese non-NAFLD, n=12; and NAFLD, n=37.
Discussion
Human NAFLD is typically assessed in the fasting state, whereas not much is known about events that occur postprandially8, and studies that do evaluate postprandial hepatic lipid metabolism typically focus on lipoprotein or lipid class levels. In this study, we assessed postprandial plasma lipidomics after a mixed-meal in subjects with NAFLD and compared them to healthy controls, applying a rigorous analysis to identify and measure lipids at the molecular species level9.
The postprandial lipidome was clearly distinct between the two groups. Unsurprisingly, the levels of many lipid species differ at baseline between NAFLD and controls, and similarly, it was expected that the levels of many lipids will change temporally after a meal. Our focus was on species with a temporal pattern that differs between NAFLD and controls. Using this untargeted approach we identified a cluster of lipids, comprised predominantly of DAGs, that meets this criterion and confirmed that this is a feature common to most DAGs in a more targeted approach. DAG levels were higher at all time points in NAFLD compared to controls, increased for 2 hours after a meal and, in NAFLD patients, continued to increase at least to the 4-hour mark. We were further able to replicate the postprandial plasma DAG increase in a NASH mouse model, where it also corresponded with a transient DAG decrease in liver tissue. Previous studies have demonstrated elevated fasting DAGs in NAFLD and NASH10, 11, which is further supported by our study. Elevated baseline levels indicate constant exposure to elevated DAGs, but the postprandial increase suggests that each meal may have an additional impact. To our knowledge, this is the first study to demonstrate a prolonged increase in postprandial plasma DAGs.
Postprandial plasma lipids are contained predominantly in liver-derived VLDL and in intestine-derived chylomicrons. We provide several lines of evidence to suggest that the postprandial increase in DAGs occurs in VLDL and not in CM, supporting a liver-related event. First, when testing immune-purified VLDL from human subjects we were able to detect the rise in DAGs, which exceeded VLDL-TAG levels. Second, 13C-labeled lipids in the mixed-meal did not contribute to the postprandial increase in plasma DAG in the mouse model. A “second meal effect” has been described, where intestinal-released CM contain lipids from a previous meal6, 12, but was ruled out in our mouse model using an earlier 13C-labeled meal. Third, in the mouse model, we saw a decrease in hepatic DAGs that paralleled the increase in their peripheral blood levels; whether a similar postprandial change in liver DAGs is present in humans and whether its kinetics are as rapid is yet unknown. Finally, most of the DAG species elevated postprandially in NASH mice contained 16:0 acyl groups. Palm oil is the major lipid source in the GAN-diet, which is mostly comprised of 16:0 lipids. Conversely, Ensure Plus, used for the mixed-meal in both mice and humans in this study is mainly composed of 18:1 and 18:2 lipid species with low levels of 16:0. Therefore, the increase in postprandial DAGs in these mice is more consistent with an endogenous lipid store (driven by the GAN diet), and less likely to be from the acute Ensure Plus load. Overall, our data shows that the postprandial increase in DAG species in NAFLD is not from absorption of meal-derived lipids and is occurring in liver-derived VLDL.
VLDL particles in the circulation undergo continuous remodeling by peripheral lipases. Theoretically, competition for peripheral lipases by postprandial CM6 could lead to incomplete hydrolysis of VLDL-TAG and generation of DAG species as intermediary products. However, when peripheral lipases were inhibited, mice with NASH continued to demonstrate a postprandial increase in plasma DAG compared to control mice, confirming that the postprandial plasma DAGs are secreted as such from the liver.
Studies of postprandial hepatic lipid metabolism typically focus on lipoprotein or lipid class levels. Multiple studies assessed the regulation of hepatic VLDL secretion. In healthy individuals, postprandial release of VLDL is inhibited by insulin through modulation of apoB availability13, 14. In NAFLD, VLDL-secretion is increased in the basal state15. Insulin suppression of VLDL secretion is lost in NAFLD16, likely due to underlying insulin resistance17. Consistent with that, Musso18 demonstrated a marked postprandial increase in plasma VLDL in non-obese NAFLD compared to matched controls; importantly, although post-prandial levels of VLDL cholesterol were higher in NAFLD, the temporal pattern and time of peak were similar in both groups. In contrast, we demonstrate a protracted increase in multiple individual DAG species that appears to be decoupled from VLDL kinetics. In contrast to studies on regulation of hepatic VLDL secretion, no studies addressed the mechanisms regulating which lipid species are packaged into it prior to secretion. DAGs have previously been shown to be present in VLDL and other lipoproteins19, 20 but mechanistic studies of VLDL lipidation focused on packaging of TAG21; whether the same mechanisms apply to DAG packaging is unknown. We show that there is a selective increase in DAG species within VLDL after a meal, and that in mice, this corresponds to a decrease in DAG species in the liver. The lipid species contained in the secreted VLDL particle are therefore not merely a passive reflection of hepatic lipid composition but likely reflect an actively regulated process. Our study is thus the first evidence of a meal-induced change in the lipid content of VLDL.
Genetic heterogeneity affects hepatic lipid metabolism. The TM6SF2-E167K missense variant leads to impaired VLDL lipidation, increased intrahepatic TAGs and decreased plasma total TAG and cholesterol in the fasting22 and postprandial23 states. The PNPLA3-I148M mutation is associated with incidence and severity of NAFLD24, lower fasting plasma VLDL and total TAGs25 and decreased VLDL secretion26 but does not affect postprandial VLDL secretion27. We did not identify an effect of PNPLA3 or TM6SF2 genotype on postprandial DAG secretion but our study was neither designed nor powered to detect such an effect.
Diacylglycerols are an intermediate lipid species in the generation and lipolysis of TAGs and have been studied in skeletal muscle and the liver as potential mediators of lipid-induced insulin resistance28. Liver fat and fasting HOMA-IR have been shown to correlate with total hepatic DAGs, and DAG-mediated activation of PKCε was suggested as a driver of hepatic insulin resistance29, 30. Importantly, hepatic DAG species that have previously been shown to correlate with HOMA-IR such as DAG(34:1)(18:1–16:0), DAG(36:2)(18:1–18:1) and DAG(36:3)(18:1–18:2) were similarly elevated in NAFLD postprandial plasma in our study30.
In healthy subjects, adipose-derived fatty acids are the main source of VLDL-TAG before and after a meal, in lieu of de-novo lipogenesis (DNL) and meal-derived lipids31. In NAFLD, DNL is elevated but adipose-derived lipids continue to be the major source of postprandial VLDL-TAGs32. In our study, postprandial DAGs containing both essential (such as C18:2 and C18:3) and non-essential fatty acid acyl groups were similarly elevated in NAFLD subjects compared to controls. Moreover, in mice given 13C-triolein, the tracer was undetected in liver oleic acid-containing lipid species, indicating that meal-derived fatty acids did not undergo liver recycling to increase postprandial plasma DAGs. Therefore, our data suggests, albeit indirectly, that adipose-derived fatty acids are the major source of postprandial DAGs. The contribution of DNL to postprandial DAGs was not assessed directly in our study and requires further study. It was recently suggested that compartmentalization of hepatic DAGs to lipid droplets protects from their effect on insulin resistance33. If postprandial hepatic DAGs increase acutely in response to unsuppressed adipose tissue lipolysis or intrahepatic repartitioning in NAFLD, it is plausible that this will be reflected in a selective increase in their release with VLDL.
Insulin has a complex multifactorial effect on VLDL secretion, as evident from the marked difference in plasma TAGs between subjects with insulin receptor mutations and those with lipodystrophy, two disorders of extreme insulin resistance caused by different molecular mechanisms34. We identified an association between insulin levels and the rise in postprandial DAGs. Importantly, insulin levels 2 hours after the mixed-meal predicted a later rise in cluster 1 score and in individual DAGs at 4 hours, suggesting a true association and possible causality. The association with postprandial insulin could reflect a direct effect of insulin on DAG secretion, or an indirect effect where hyperinsulinemia reflects insulin resistance. Whether the difference in postprandial DAG secretion between NAFLD and controls is solely due to the presence of insulin resistance is unclear. In non-NAFLD obese subjects with milder insulin resistance, fasting insulin was associated with fasting DAG levels, but no association was seen between the 2-hour insulin and postprandial rise in DAGs. This could suggest that the presence of NAFLD is required for the impact of insulin resistance on postprandial DAG secretion. The molecular mechanism by which postprandial insulin resistance leads to enhanced hepatic DAG secretion remains to be established.
It is interesting that the magnitude of postprandial increase in DAGs was not associated with parameters of severity of the liver disease itself. This is consistent with previously published data, where fasting-state DAGs were shown to be elevated in the liver10, 17 and plasma11 of patients with NAFLD and NASH, but did not differ between the two groups, although recently it was suggested that long-chain highly-desaturated plasma DAGs are decreased in NASH compared to NAFLD35. Importantly, none of these studies assessed subjects in the fed state. Furthermore, in our mouse model, elevated postprandial plasma DAGs were already seen in mice fed the GAN-diet for 8- and 16-weeks, at earlier stages of the development of NASH. Finally, we have shown that a postprandial rise in DAGs is unique to patients with NAFLD and is not seen in comparable obese subjects. Thus, our data suggests this effect is an early feature in the development of NAFLD and not related to further progression of the disease.
Although the postprandial DAGs were not associated with severity of the liver disease, and are likely a reflection of hepatic insulin resistance, their increased plasma levels could confer risk in extra-hepatic tissues. For example, fasting blood levels of several DAGs are associated with incident cardiovascular disease36, 37 and within atherosclerotic plaques, DAGs are associated with plaque thrombus38. Cardiovascular disease is the major cause of death in patients with NAFLD and NAFLD is considered an independent risk factor for its development39. It is plausible that the postprandial increase in circulating DAGs in these patients contributes to the acceleration of their atherosclerosis.
There are several limitations to our work. First, we do not have histological evaluation of the liver disease in many of the subjects to reliably separate NASH from NAFL, nor do we have a quantitative measure of their liver fat content at the time of the mixed-meal study. Second, we did not perform tracer-labeling studies in our patients and therefore our conclusions regarding liver source for the DAG increase are derived from rigorous VLDL isolation in humans and from tracer studies in mice. Results obtained solely in mice should therefore be interpreted with caution until confirmed by clinical studies in humans. Third, the obese non-NAFLD cohort was obtained from a separate trial, with a different methodology and a lower dose of the mixed-meal which limits our ability to directly compare the findings in them to those of NAFLD patients and controls. Fourth, it is unclear whether the data obtained from a liquid meal challenge is applicable to solid meals40. Finally, postprandial DAGs were elevated in human NAFLD and mice at various stages of NASH development compared to controls; however, baseline DAGs were unaltered in mouse studies, limiting the relevance of this model in assessing fasting DAG levels in NAFLD.
Our study also has several important strengths. First, the clinical trial was performed in a highly controlled environment including standardized meals and food intake to limit confounding variables, integrated concomitant energy expenditure and utilization, and insulin levels were measured in the same time frame. Second, the controls were carefully selected to ensure they have no evidence of NAFLD, as we have previously shown that NAFLD is common in clinical trial “healthy volunteers”41.Third, the repeated-measures design increased our statistical power, allowing us to assess the large number of lipid features generated by an untargeted lipidomics approach. Fourth, we were able to replicate our findings in a mouse NASH model using the same mixed-meal bolus, allowing us to further characterize the source of postprandial DAGs. Finally, while most studies measure tracer-labels in total fatty acids after saponification under the assumption that they are mostly derived from TAGs, we directly measured tracer labels in esterified lipids to characterize TAG, DAG and FFA isotope enrichment.
In conclusion, using an unbiased lipidomic approach we discovered a selective increase in hepatic secretion of DAGs to the plasma after a mixed-meal in patients with NAFLD, likely due to hepatic insulin resistance.
Supplementary Material
WHAT YOU NEED TO KNOW.
BACKGROUND AND CONTEXT
Excess energy intake is a major driver of non-alcoholic fatty liver disease (NAFLD), but human NAFLD is typically studied in the fasting state.
NEW FINDINGS
After a meal there is a selective increase in hepatic secretion of diacylglycerols in very-low density lipoproteins (VLDL) in human and mouse NAFLD.
LIMITATIONS
The mechanism and regulation of diacylglycerol packaging into VLDL is unclear. Mouse findings may not be fully translatable to human physiology.
IMPACT
Studying subjects with NAFLD in the fed state yields additional insights into disease pathophysiology.
Acknowledgements
The graphic abstract was created with Biorender.com. We thank Drs. Robert Shamburek and Lita Freeman for assistance in separating VLDL, Dr. Oksana Gavrilova for advice and John Buckley for technical assistance. We are indebted to the study participants and NIH Clinical Center staff for their support.
Funding and conflict of interests
The study was funded by the intramural research programs of NIDDK and CCR, NCI. TJV was supported by the Canadian Institutes of Health Research (CIHR) Postdoctoral Fellowship. The authors have declared that no conflict of interest exists.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability
All data and code are available from the corresponding author upon request
References
- 1.Younossi Z, Stepanova M, Ong JP, et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin Gastroenterol Hepatol 2019;17:748–755 e3. [DOI] [PubMed] [Google Scholar]
- 2.Berlanga A, Guiu-Jurado E, Porras JA, et al. Molecular pathways in non-alcoholic fatty liver disease. Clin Exp Gastroenterol 2014;7:221–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fan JG, Cao HX. Role of diet and nutritional management in non-alcoholic fatty liver disease. J Gastroenterol Hepatol 2013;28 Suppl 4:81–7. [DOI] [PubMed] [Google Scholar]
- 4.Dumas ME, Kinross J, Nicholson JK. Metabolic phenotyping and systems biology approaches to understanding metabolic syndrome and fatty liver disease. Gastroenterology 2014;146:46–62. [DOI] [PubMed] [Google Scholar]
- 5.Guerci B, Verges B, Durlach V, et al. Relationship between altered postprandial lipemia and insulin resistance in normolipidemic and normoglucose tolerant obese patients. Int J Obes Relat Metab Disord 2000;24:468–78. [DOI] [PubMed] [Google Scholar]
- 6.Lambert JE, Parks EJ. Postprandial metabolism of meal triglyceride in humans. Biochim Biophys Acta 2012;1821:721–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinrich P, Kohler C, Ellmann L, et al. Correcting for natural isotope abundance and tracer impurity in MS-, MS/MS- and high-resolution-multiple-tracer-data from stable isotope labeling experiments with IsoCorrectoR. Sci Rep 2018;8:17910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Naguib G, Morris N, Yang S, et al. Dietary fatty acid oxidation is decreased in non-alcoholic fatty liver disease: A palmitate breath test study. Liver Int 2020;40:590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liebisch G, Fahy E, Aoki J, et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J Lipid Res 2020;61:1539–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puri P, Baillie RA, Wiest MM, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology 2007;46:1081–90. [DOI] [PubMed] [Google Scholar]
- 11.Puri P, Wiest MM, Cheung O, et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009;50:1827–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heath RB, Karpe F, Milne RW, et al. Dietary fatty acids make a rapid and substantial contribution to VLDL-triacylglycerol in the fed state. Am J Physiol Endocrinol Metab 2007;292:E732–9. [DOI] [PubMed] [Google Scholar]
- 13.Sparks JD, Sparks CE, Adeli K. Selective hepatic insulin resistance, VLDL overproduction, and hypertriglyceridemia. Arterioscler Thromb Vasc Biol 2012;32:2104–12. [DOI] [PubMed] [Google Scholar]
- 14.Choi SH, Ginsberg HN. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol Metab 2011;22:353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fabbrini E, Mohammed BS, Magkos F, et al. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008;134:424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adiels M, Westerbacka J, Soro-Paavonen A, et al. Acute suppression of VLDL1 secretion rate by insulin is associated with hepatic fat content and insulin resistance. Diabetologia 2007;50:2356–65. [DOI] [PubMed] [Google Scholar]
- 17.Hodson L Hepatic fatty acid synthesis and partitioning: the effect of metabolic and nutritional state. Proc Nutr Soc 2019;78:126–134. [DOI] [PubMed] [Google Scholar]
- 18.Smagris E, Gilyard S, BasuRay S, et al. Inactivation of Tm6sf2, a Gene Defective in Fatty Liver Disease, Impairs Lipidation but Not Secretion of Very Low Density Lipoproteins. J Biol Chem 2016;291:10659–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lalanne F, Pruneta V, Bernard S, et al. Distribution of diacylglycerols among plasma lipoproteins in control subjects and in patients with non-insulin-dependent diabetes. Eur J Clin Invest 1999;29:139–44. [DOI] [PubMed] [Google Scholar]
- 20.Mucinski JM, Manrique-Acevedo C, Kasumov T, et al. Relationships between Very Low-Density Lipoproteins-Ceramides, -Diacylglycerols, and - Triacylglycerols in Insulin-Resistant Men. Lipids 2020;55:387–393. [DOI] [PubMed] [Google Scholar]
- 21.Irshad Z, Chmel N, Adya R, et al. Hepatic VLDL secretion: DGAT1 determines particle size but not particle number, which can be supported entirely by DGAT2. J Lipid Res 2019;60:111–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozlitina J, Smagris E, Stender S, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Musso G, Cipolla U, Cassader M, et al. TM6SF2 rs58542926 variant affects postprandial lipoprotein metabolism and glucose homeostasis in NAFLD. J Lipid Res 2017;58:1221–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rotman Y, Koh C, Zmuda JM, et al. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010;52:894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luukkonen PK, Qadri S, Lehtimaki TE, et al. The PNPLA3-I148M Variant Confers an Antiatherogenic Lipid Profile in Insulin-resistant Patients. J Clin Endocrinol Metab 2021;106:e300–e315. [DOI] [PubMed] [Google Scholar]
- 26.Pirazzi C, Adiels M, Burza MA, et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol 2012;57:1276–82. [DOI] [PubMed] [Google Scholar]
- 27.Luukkonen PK, Nick A, Holtta-Vuori M, et al. Human PNPLA3-I148M variant increases hepatic retention of polyunsaturated fatty acids. JCI Insight 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petersen MC, Shulman GI. Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol Sci 2017;38:649–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Magkos F, Su X, Bradley D, et al. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012;142:1444–6 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumashiro N, Erion DM, Zhang D, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2011;108:16381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barrows BR, Parks EJ. Contributions of different fatty acid sources to very low-density lipoprotein-triacylglycerol in the fasted and fed states. J Clin Endocrinol Metab 2006;91:1446–52. [DOI] [PubMed] [Google Scholar]
- 32.Lambert JE, Ramos-Roman MA, Browning JD, et al. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146:726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abulizi A, Vatner DF, Ye Z, et al. Membrane-bound sn-1,2-diacylglycerols explain the dissociation of hepatic insulin resistance from hepatic steatosis in MTTP knockout mice. J Lipid Res 2020;61:1565–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sekizkardes H, Chung ST, Chacko S, et al. Free fatty acid processing diverges in human pathologic insulin resistance conditions. J Clin Invest 2020;130:3592–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung Y, Lee MK, Puri P, et al. Circulating lipidomic alterations in obese and non-obese subjects with non-alcoholic fatty liver disease. Aliment Pharmacol Ther 2020;52:1603–1614. [DOI] [PubMed] [Google Scholar]
- 36.Paynter NP, Balasubramanian R, Giulianini F, et al. Metabolic Predictors of Incident Coronary Heart Disease in Women. Circulation 2018;137:841–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toledo E, Wang DD, Ruiz-Canela M, et al. Plasma lipidomic profiles and cardiovascular events in a randomized intervention trial with the Mediterranean diet. Am J Clin Nutr 2017;106:973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moerman AM, Visscher M, Slijkhuis N, et al. Lipid signature of advanced human carotid atherosclerosis assessed by mass spectrometry imaging. J Lipid Res 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai J, Zhang XJ, Ji YX, et al. Nonalcoholic Fatty Liver Disease Pandemic Fuels the Upsurge in Cardiovascular Diseases. Circ Res 2020;126:679–704. [DOI] [PubMed] [Google Scholar]
- 40.LaBarre JL, Singer K, Burant CF. Advantages of Studying the Metabolome in Response to Mixed-Macronutrient Challenges and Suggestions for Future Research Designs. J Nutr 2021;151:2868–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takyar V, Nath A, Beri A, et al. How healthy are the “Healthy volunteers”? Penetrance of NAFLD in the biomedical research volunteer pool. Hepatology 2017;66:825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and code are available from the corresponding author upon request