

Abstract
Background:
Rare sequence variation in genes underlying cardiac repolarization and common polygenic variation influence QT interval duration. However, current clinical genetic testing of individuals with unexplained QT prolongation is restricted to examination of monogenic rare variants. The recent emergence of large-scale biorepositories with sequence data enables examination of the joint contribution of rare and common variation to the QT interval in the population.
Methods:
We performed a genome wide association study (GWAS) of the QTc in 84,630 United Kingdom Biobank (UKB) participants and created a polygenic risk score (PRS). Among 26,976 participants with whole genome sequencing and electrocardiogram data in the Trans-Omics for Precision Medicine (TOPMed) program, we identified 160 carriers of putative pathogenic rare variants in 10 genes known to be associated with the QT interval. We examined QTc associations with the PRS and with rare variants in TOPMed.
Results:
Fifty-four independent loci were identified by GWAS in the UKB. Twenty-one loci were novel, of which 12 were replicated in TOPMed. The PRS comprising 1,110,494 common variants was significantly associated with the QTc in TOPMed (ΔQTc/decile of PRS =1.4 ms, 95% CI 1.3 −1.5; p-value=1.1×10−196). Carriers of putative pathogenic rare variants had longer QTc than non-carriers (ΔQTc=10.9 ms [7.4–14.4]). 23.7% of individuals with QTc>480 ms carried either a monogenic rare variant or had a PRS in the top decile (3.4% monogenic, 21% top decile of PRS).
Conclusions:
QTc duration in the population is influenced by both rare variants in genes underlying cardiac repolarization and polygenic risk, with a sizeable contribution from polygenic risk. Comprehensive assessment of the genetic determinants of QTc prolongation includes incorporation of both polygenic and monogenic risk.
Keywords: QT interval, Polygenic, Monogenic, Long QT Syndrome, Sudden Cardiac Death
Introduction
Perturbation of pathways involved in cardiac repolarization can lead to significant prolongation of the electrocardiographic QT interval with associated risks of cardiac arrhythmias, sudden cardiac death and major adverse cardiovascular events.1–3 While multiple extrinsic factors such as medications, hormones and serum electrolytes influence cardiac ion channels, rare variants within genes encoding subunits that comprise these channels (e.g., KCNQ1, KCNH2, and SCN5A) and other proteins are implicated in QT prolongation and the long QT syndrome (LQTS). Recently, common genetic variants have been implicated in QT interval duration as well via genome wide association studies (GWAS) in the general population.4 However, the joint contribution of rare monogenic and common polygenic variation to the QT interval in the general population remains unknown.
Current clinical genetic testing of individuals with otherwise unexplained prolonged QT interval in the population is restricted to examination of monogenic rare variants.5 Recently, biorepositories with large-scale sequence data have emerged which enable an unprecedented examination of the relations between both rare and common variation, and QT duration, in the general population. Here, we leverage paired electrocardiogram (ECG) and genomic data from over 100,000 individuals in the United Kingdom Biobank (UKB) and Trans-Omics for Precision Medicine (TOPMed) program to comprehensively examine the joint contributions of monogenic and polygenic variation to QT interval duration.
Methods
Data Availability
UKB data are made available to researchers from research institutions with genuine research inquiries, following institutional review board (IRB) and UKB approval. All other data are contained within the article and its supplementary material, or are available upon reasonable request to the corresponding author.
Study Design and Patient Population
United Kingdom Biobank Cohort
The UKB is a prospective cohort of ~500,000 individuals from the UK with deep phenotyping and multiple genomic data types.6 ECGs with automated QT interval measurements were available on 94,486 study participants. Two types of ECGs were performed on partially overlapping subsets of the UKB: supine resting 12-lead ECGs (N=42,635) and 3-lead upright resting ECGs prior to a bicycle exercise protocol (N=60,799). Among individuals who underwent both 12-lead and 3-lead ECGs (N=8,948), their QT interval was extracted from the 12-lead ECGs for this study. Heart rate corrected QT intervals were calculated using the Bazett formula, defined as QTc = . Additional details pertaining to ECG methods and automated QT interval measurement are summarized in the Supplemental Methods.
We included participants from the UKB with both imputed genomic data and ECG data. Details of genotype arrays, initial data quality control and imputation in the UKB have been published previously6 and are briefly summarized in the Supplemental Methods. We excluded individuals with poor quality genetic data (Supplemental Methods) and with characteristics that would affect their QT interval such as Wolff-Parkinson-White Syndrome, a paced rhythm, QRS interval >120 ms, digoxin use or class I or III antiarrhythmic drug use. Individuals with 2nd or 3rd degree AV block and those with extreme heart rates, <40 or >120 beats per minute, were also excluded (Figure S1A). The final UKB cohort consisted of 84,630 individuals. Use of UKB data was performed under application number 17488 and was approved by the local Massachusetts General Hospital IRB.
Trans-Omics for Precision Medicine Cohort
The TOPMed cohort included participants from the National Heart Lung and Blood Institute’s (NHLBI) TOPMed program with both whole-genome sequencing (WGS) and QTc data (N=29,511). The study sample consisted of participants from 9 NHLBI cardiovascular cohorts (Supplemental Methods). In brief, we excluded participants with poor quality genetic data, and with characteristics that would affect their QT interval as detailed above. The final TOPMed study sample included 26,976 individuals (Figure S1B). All participants provided written informed consent, and all participating studies obtained study approval from their local IRB.
Rare Variant Annotation in TOPMed
WGS quality control was performed on TOPMed data included in Freeze 6. Details of sequencing methods, variant calling and initial sequencing data quality control have been previously described by the TOPMed Informatics Research Center.7 Rare variant annotation methods are summarized in detail in the Supplemental Methods. To enrich for disease-causing rare variants, we defined putative pathogenic rare variants as those classified as pathogenic or likely pathogenic in ClinVar,8 or that were classified as high confidence loss-of-function (LOF) rare variants (minor allele frequency (MAF) < 0.01) in canonical transcripts using Loss-Of-Function Transcript Effect Estimator.9 LOF variants within SCN5A were excluded as they are mechanistically not expected to be associated with prolongation of the QT interval. After subsetting to pathogenic/likely pathogenic variants and high confidence LOF variants in canonical transcripts, we identified 160 carriers of rare putative pathogenic variants in 10 (KCNQ1, KCNH2, SCN5A, KCNJ2, KCNE2, CACNA1C, CAV3, TRDN, CALM3, ANK2) out of 13 total genes included in a commercial (Invitae) LQTS panel in the TOPMed cohort (no putative pathogenic rare variant carriers in CALM1, CALM2 or KCNE1 were identified within TOPMed). In a sensitivity analysis further restricting to putative pathogenic rare variants with MAF <0.001, we identified the same variants and number of carriers.
Statistical Analysis
In the UKB, we first examined the unexplained variance in QTc for the 3-lead and 12-lead ECG subgroups, separately, following adjustment for covariates included in our genetic association model (age, sex, beta blocker use, calcium channel blocker use, history of myocardial infarction, history of heart failure and the first twelve principal components of genetic ancestry). We found that the residual unexplained variance in the QTc was similar for the two subgroups suggesting that there is no significant bias between the two subgroups and supporting a joint analysis of the QTc from these two ECG modalities (Figure S2). We also calculated the genetic correlation between the QTc in the 3-lead and 12-lead ECG subgroups using linkage disequilibrium (LD) score regression, which was very strong (rg=0.99, p=4.3×10−47). Next, we combined the 3-lead and 12-lead subgroups and performed a joint common variant (MAF ≥ 0.01) GWAS for the QTc using SAIGE10 adjusting for age, genetically determined sex, beta blocker use, calcium channel blocker use, history of myocardial infarction, history of heart failure, genotyping array, ECG type (12-lead vs. 3-lead) and the first twelve principal components of genetic ancestry. LD score regression analysis was performed using ldsc version 1.0.0.11 With ldsc, the genomic control factor (lambda GC) was partitioned into components reflecting polygenicity and inflation, using the software’s defaults. We calculated SNP-heritability of the QTc using BOLT-REML v2.3.4.12 We performed replication analysis of the novel loci in TOPMed using GENESIS.13
Using the results of the GWAS and after subsetting to overlapping variants in the UKB and TOPMed (7,457,978 out of 8,595,785 variants), we generated a genome-wide polygenic risk score (PRS) for the QTc using a Bayesian regression framework with continuous shrinkage priors on SNP effect sizes implemented in PRS-CS.14 We used PRS-CS to generate a genome-wide PRS since unlike other approaches such as pruning and thresholding, PRS-CS does not require defining a derivation and validation sub-cohort. Our approach allowed us to maximize our GWAS sample size and power for genetic discovery. In PRS-CS, we used the European ancestry LD reference panel from 1000 Genomes Project Phase 3. Using the --score function in PLINK v1.9 we calculated the PRS for all study participants in TOPMed. Derivation of a new PRS for the QTc in the UKB was necessary to avoid overfitting that would be induced by using prior available genetic risk scores15 and GWAS results4 which were derived from studies that overlapped with our TOPMed cohort.
We examined the joint contribution of monogenic and polygenic variation to the QTc in TOPMed (Figure 1). Correlation between the derived QTc PRS and the QTc was assessed using Pearson’s correlation coefficient. We tested the association between the QTc PRS and putative pathogenic rare variants with the QTc using multiple linear regression with adjustment for age, sex, beta blocker use, calcium channel blocker use, history of heart failure, history of myocardial infarction and the first twelve principal components of genetic ancestry. We collapsed putative pathogenic rare variants by gene and defined monogenic rare variant carriers as carriers of putative pathogenic rare variants in any gene within the Invitae commercial LQTS panel (N=160).
Figure 1. Study design and flowchart.
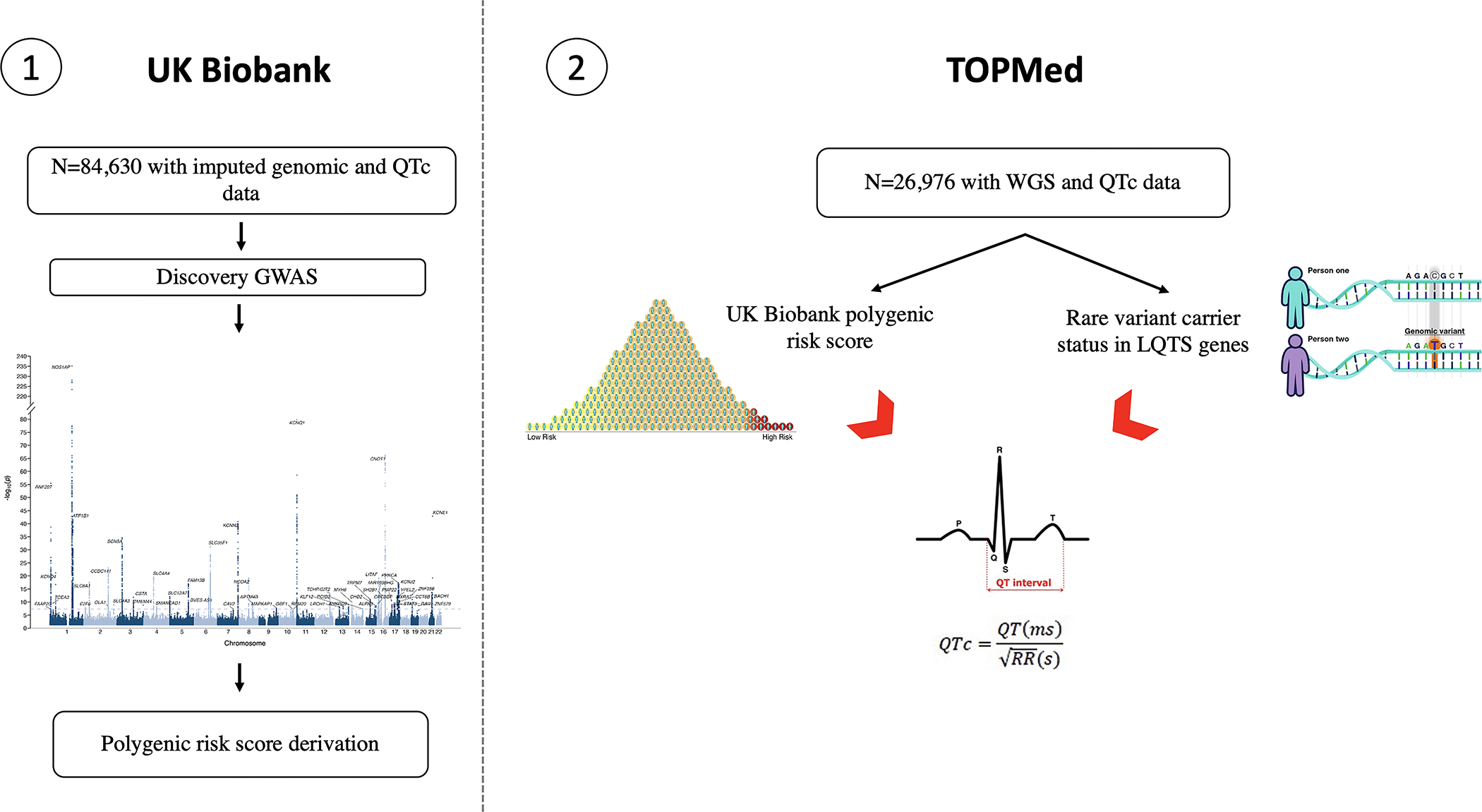
GWAS: genome-wide association study; PRS: polygenic risk score; TOPMed: Trans-Omics for Precision Medicine; UKB: United Kingdom Biobank.
All statistical analyses were performed using R version 4.0.2 (R, Core Team 2020) unless otherwise specified. A two-sided p-value < 5×10−8 was used to identify genome-wide significant common variants and <0.05 was used as the p-value threshold for replication of novel loci in TOPMed. For the remaining statistical analyses two-sided p-values <0.05 were considered statistically significant.
Results
UKB Cohort
In the UKB, mean age of participants was 57.9 ± 8.8 and 48.6% were male. The majority of participants (93.5%) were of European ancestry. Prevalence of cardiovascular comorbidities was relatively low with 4.4%, 2.6% and 1.6% having a diagnosis of atrial fibrillation, myocardial infarction or heart failure, respectively, at the time of ECG. The study sample mean QT duration was 398.1 ± 34.3 ms and mean QTc duration was 415.8 ± 23.3 ms (Table 1).
Table 1.
Characteristics of the UK Biobank and TOPMed cohorts
| UKB Cohort (N=84,630) | TOPMed Cohort (N=26,976) | |
|---|---|---|
| Age (Years) | 57.9 ± 8.8 | 59.8 ± 12.5 |
| Height (cm) | 169.2 ± 9.2 | 166.0 ± 9.5 |
| Weight (kg) | 78.0 ± 15.1 | 79.5 ± 18.9 |
| Male (n, (%)) | 41,108 (48.6) | 17,644 (65.4) |
| Ancestry (n, (%)) | ||
| European | 79,167 (93.5) | 16,074 (59.6) |
| African | 1,614 (1.9) | 4,887 (18.1) |
| Admixed American | 78 (0.1) | 596 (2.2) |
| Asian | 1500 (1.8) | 768 (2.9) |
| Amish | -- | 998 (3.7) |
| Undetermined | 2,271 (2.7) | 3,653 (13.5) |
| Heart failure (n, (%)) | 1,327 (1.6) | 1,788 (6.6) |
| Myocardial Infarction (n, (%)) | 2,182 (2.6) | 2,361 (8.8) |
| Atrial Fibrillation (n, (%)) | 3,730 (4.4) | 1,450 (5.4) |
| Beta Blocker (n, (%)) | 3,986 (4.7) | 3,415 (12.7) |
| Calcium Channel Blocker (n, (%)) | 5,514 (6.5) | 3,043 (11.3) |
| QRS duration (ms) | 87.4 ± 11.0 | 89.6 ± 10.2 |
| Heart rate (beats/minute) | 66.8 ± 12.0 | 65.9 ± 11.1 |
| QT interval (ms) | 398.1 ± 34.3 | 408.4 ± 31.6 |
| QTc interval (ms) | 415.8 ± 23.3 | 423.8 ± 22.9 |
Continuous variables are presented as mean ± standard deviation.
In the QTc GWAS, we identified 54 independent loci that exceeded genome-wide significance (Figure 2). We replicated 33 loci previously reported to be associated with the QTc and identified 21 novel loci (Table S1). The genomic control factor λGC was1.16 with an LD-score regression intercept of 1.07, suggesting polygenicity of the QTc as opposed to inflation (Figure S3). In TOPMed we replicated 12 out of the 21 novel loci (Table S2). The SNP-heritability of the QTc was 0.24 (standard error 0.007).
Figure 2. QTc genome-wide association results across 22 autosomes in the UKB.
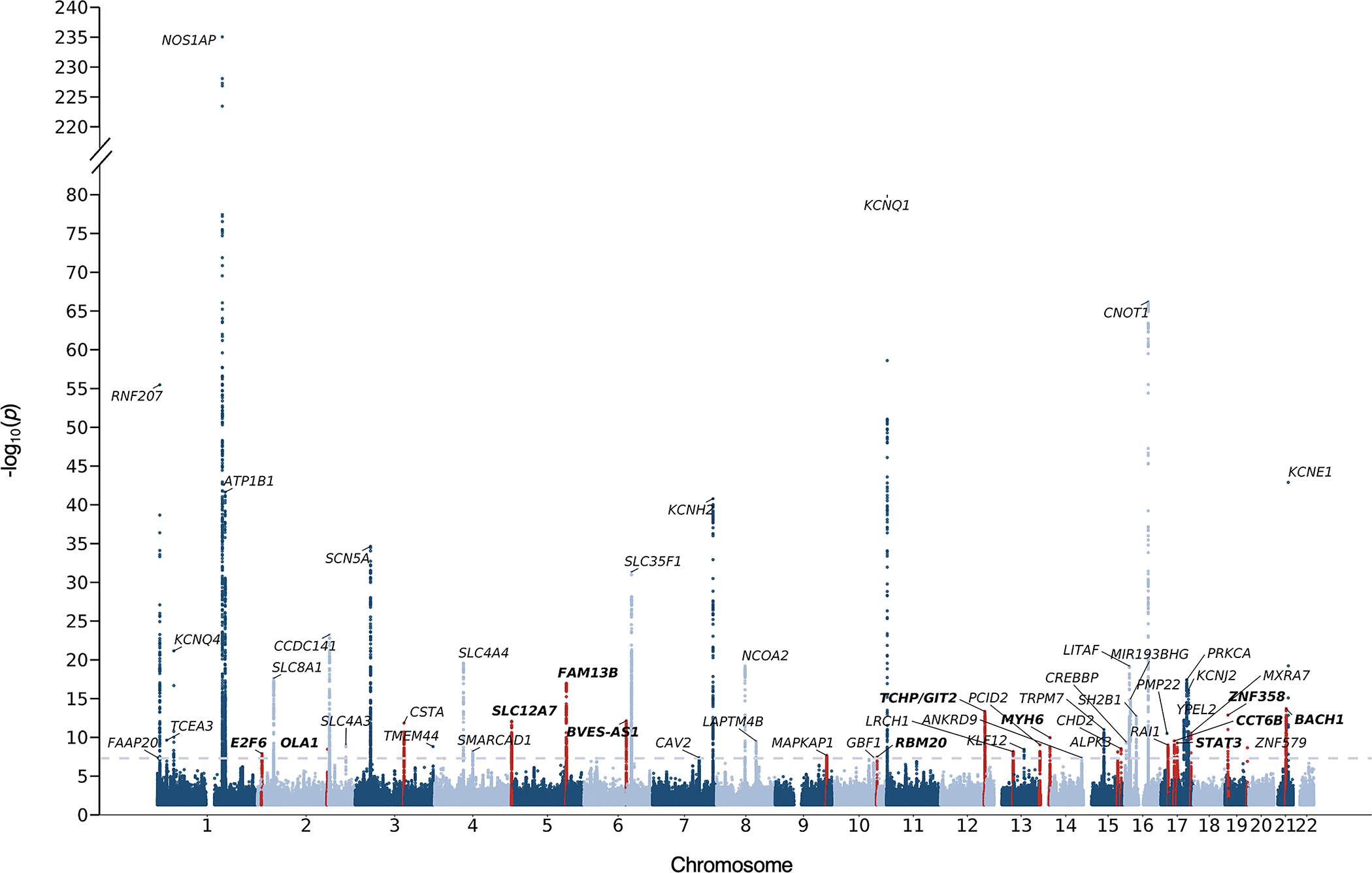
Nearest genes are used for annotation. The dashed grey line represents the threshold for genome-wide significance (P<5×10−8). Red peaks correspond to novel loci. Bolded labels represent loci that were replicated in TOPMed.
Utilizing the GWAS results and after subsetting to overlapping variants between UKB and TOPMed, we derived a genome-wide PRS comprising 1,110,494 common variants.
TOPMed Cohort
Ancestral diversity was more prominent in TOPMed as compared to the UKB. Individuals of European (N= 16,074, 59.6%) and African (N=4,887, 18.1%) ancestries constituted the largest ancestry groups in the TOPMed cohort. There was a substantial degree of admixture, with genetically-inferred ancestry unable to be determined for 3,653 (13.5%) individuals. The study sample mean QT duration was 408.4 ± 31.6 ms and mean QTc duration was 423.8 ± 22.9 ms (Table 1).
The QTc PRS, derived in the UKB and comprising 1,110,494 common variants, was normally distributed across all ancestries in TOPMed and was significantly associated with the QTc (Pearson correlation coefficient 0.18, 95% CI 0.17 – 0.19; p-value=2×10−196) (Figures S4, S5). The QTc PRS remained significantly associated with the QTc (ΔQTc/decile of PRS =1.4 ms, 95% CI 1.3 −1.5; p-value=1.1×10−196) following adjustment for age, sex, beta blocker use, calcium channel blocker use, history of heart failure, history of myocardial infarction and the first twelve principal components of genetic ancestry with a model R2 of 0.087. Across the different genetic ancestries in TOPMed, increasing tertiles of the PRS stratified study participants in to higher QTc strata (Figure 3). The performance of the PRS was comparable across different ancestries albeit a relatively lower predictive power in individuals with African ancestry (Table S3).
Figure 3. Distribution of QTc duration across tertiles of the polygenic risk score within genetic ancestries in TOPMed.
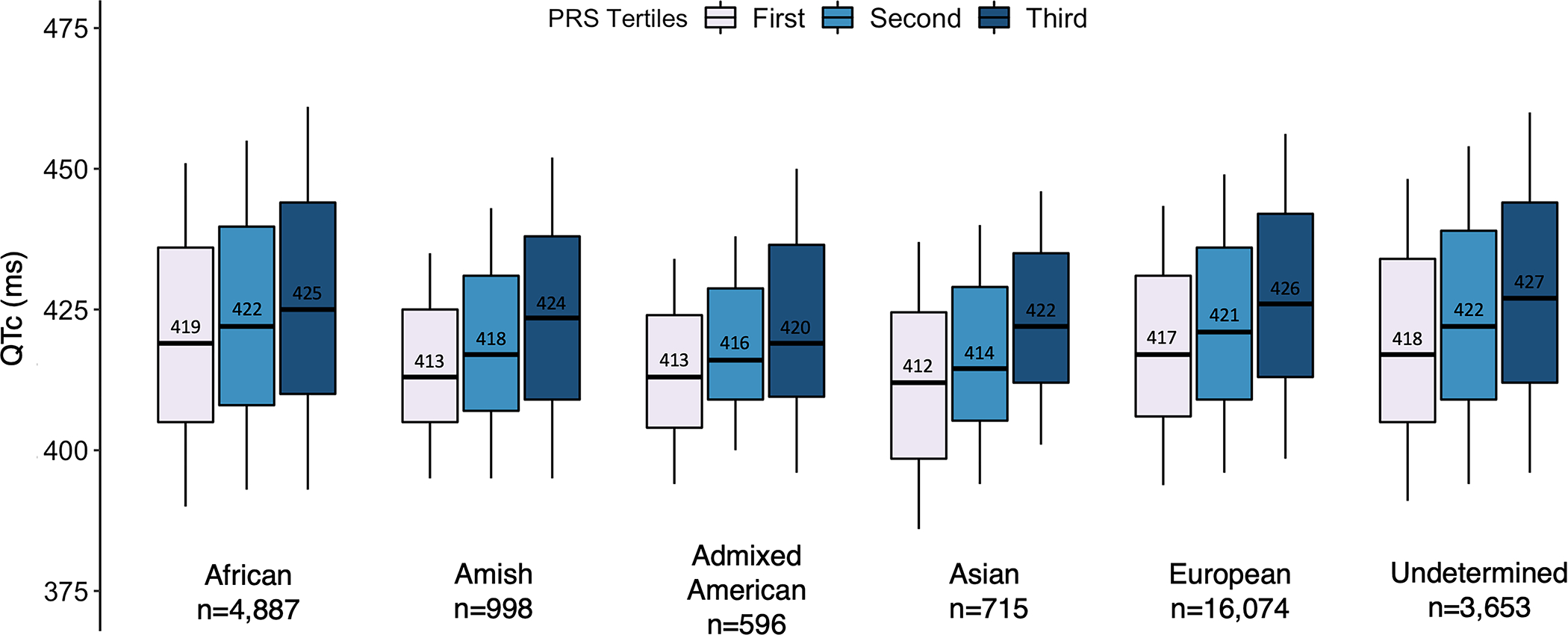
PRS: polygenic risk score.
We collapsed putative pathogenic rare variants by gene and examined the association of carriers of putative pathogenic rare variants by gene with the QTc (Table S4). In multivariable analysis, carriers of putative pathogenic rare variants in any of the 10 LQTS genes had a 10.9 ms (95% CI, 7.4 – 14.4, p-value=1.1×10−9) increase in their QTc as compared to non-carriers (Table S4).
To approximate monogenic and polygenic risk, we categorized the PRS into two categories (highest decile vs. lower 90th percentile). The multivariable adjusted-mean difference in QTc interval between the top PRS decile and lower 90th PRS percentile (8.7 ms 95% CI, 7.8 – 9.5, p-value=3.6×10−80) approximates the delta QTc observed among carriers of putative pathogenic rare variants compared to non-carriers. In multivariable analysis, we found an additive effect of monogenic and polygenic risk. Carriers of a monogenic rare variant who fell in the top decile of polygenic risk (n=14) had a 21.7 ms (95% CI, 10.1 – 33.3, p-value=2.4×10−4) higher QTc compared to non-carriers with polygenic risk in the lower 90th percentile (Figure S6).
We then examined the distribution of polygenic and monogenic risk across the spectrum of QTc duration in the study sample to assess the contribution of genetic determinants to the QTc. Across increasing strata of the QTc in the study sample, there was a consistent increase in the proportion of individuals with a PRS in the top decile (p-trend=9.6×10−59) and the proportion of individuals who were carriers of monogenic putative pathogenic rare variants (p-trend=4.1×10−14) (Figure 4). We found that only 3.4% of individuals with pronounced QTc prolongation (>480 ms) carried a monogenic rare variant, 21% had a PRS in the top decile and 23.7% had either a monogenic rare variant or a PRS in the top decile (Figure 4). Thus, approximately 75% of individuals with pronounced QTc prolongation (>480 ms) do not have an identified rare genetic variant or burden of polygenic risk equivalent to a monogenic variant to explain the QTc prolongation.
Figure 4. Top polygenic risk score decile and putative pathogenic rare variant carrier distribution across QTc strata.
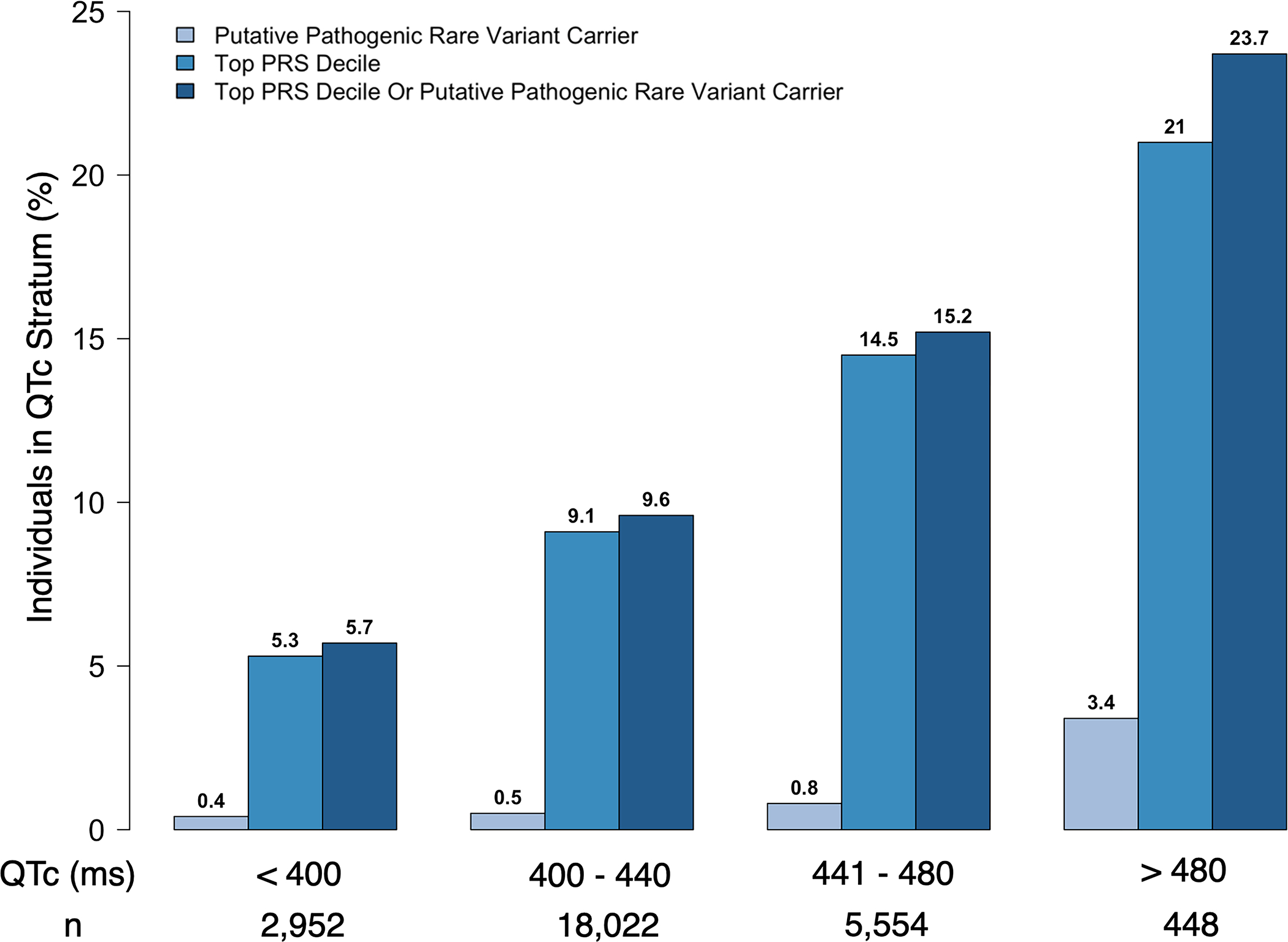
P-trend across QTc strata for putative pathogenic rare variant carrier, top PRS decile and putative pathogenic rare variant carrier Or top PRS decile: 4.1×10−14, 9.6 × 10−59, 8.2 × 10−65, respectively. PRS: polygenic risk score.
Discussion
In this study, we performed a large genome-wide association analysis of the QTc in 84,630 UKB participants and identified 21 novel loci, of which 12 replicated in an independent multi-ancestry TOPMed cohort. We demonstrate that a genome-wide PRS derived from the UKB GWAS and applied in an independent multi-ancestry TOPMed cohort had comparable performance across ancestries. Leveraging whole genome sequencing and ECG data in TOPMed, we characterized the joint contribution of common and rare variants to QTc prolongation in an adult population (mean age 59.8 years). Approximately 3% of individuals with marked QTc prolongation beyond 480 msec carried a putative pathogenic rare variant, whereas nearly 21% of such individuals carried a large-effect polygenic risk equivalent. Notably, approximately three in four individuals with pronounced QTc prolongation >480 ms did not have a yet identified genetic risk factor.
Genetic Determinants of the QT Interval
Prior studies examined the association, individually, of either common or rare variants to the QTc,4,16,17 whereas we uniquely leveraged whole genome sequencing data in TOPMed to examine both rare and common variation jointly in relation to QTc duration in large multi-ancestry population-based studies. We found that a substantial proportion of individuals with pronounced QTc prolongation (> 480 ms) in the population have a high polygenic risk equivalent. Indeed, the proportion of individuals with high polygenic risk equivalent exceeded the proportion of monogenic rare variant carriers.
Our findings highlight a need to understand both the clinical implications of polygenic predisposition to QT prolongation and mechanisms by which common variation affects QT interval duration. With enhanced access to sequencing in clinical settings and genomic sequence data emerging from large-scale biobanks, long-term correlation between genetic predisposition to QT prolongation and clinical outcomes will be feasible. A recent LQTS case-control genetic association study revealed strong genetic correlation between the QTc and LQTS. The study also demonstrated that high polygenic predisposition to QT prolongation contributed to susceptibility to LQTS, particularly among LQTS cases without a monogenic disease-causing variant.18 In our current study, we additionally identified an additive effect of QTc polygenic risk on the QTc among carriers of putative pathogenic rare variants in genes associated with the QTc highlighting the contribution of QTc polygenic risk to the QTc even among monogenic rare variant carriers. In light of the shared polygenic architecture of the QTc and LQTS, development of strong polygenic risk instruments of the QTc, a more readily available endophenotype, may aid in better characterizing polygenic susceptibility to LQTS. Additionally, QTc polygenic risk may have important implications for determining susceptibility to QT prolonging drugs. A study demonstrated the utility of a QTc PRS, comprising 61 variants, in predicting the extent of QTc prolongation associated with different anti-arrhythmic drugs and the associated risk of Torsade des Pointes.15 Prospective assessment of management approaches for individuals carrying high polygenic risk are needed.
Moreover, our findings may have implications for the approach to clinical genetic testing when evaluating individuals with an otherwise unexplained prolonged QTc. Current genetic testing approaches for individuals with an unexplained prolonged QTc focus on examining monogenic rare variants in genes associated with the LQTS.5 Extrapolating from our findings, we anticipate that incorporation of polygenic risk into genetic testing for individuals with pronounced QTc prolongation (>480 ms) could substantially increase the yield of identifying a genetic determinant of QTc prolongation by ~7-fold, from 1 in 30 to 1 in 4. While many factors contribute to QTc prolongation, our findings emphasize the potential importance of assessing polygenic risk for comprehensive evaluation of a genetic basis of QTc prolongation. However, we note that our study was not focused on individuals in whom genetic testing for suspected LQTS is considered clinically indicated. Further studies are needed to establish the role of polygenic risk assessment for the evaluation of individuals with suspected LQTS.
Polygenic and Monogenic Contribution to Other Genomic Disorders
Recently, the contribution of monogenic and polygenic risk to a number of genomic disorders has been described. A recent trans-ancestry GWAS of LQTS described the polygenic contribution to LQTS and found that approximately 15% of variation in LQTS susceptibility is attributable to common genetic variation.18 The study findings highlight the polygenic architecture of LQTS in addition to the established monogenic architecture of LQTS. A contemporary study examining a spectrum of diseases including familial hypercholesterolemia, hereditary breast and ovarian cancer and Lynch syndrome found that polygenic risk had an additive effect on monogenic risk and aided in better risk estimation among monogenic rare variant carriers.19 Our group has previously reported higher prevalence of atrial fibrillation among TTN LOF carriers within higher strata of polygenic risk of atrial fibrillation.20 The current study extends this body of work and is of particular relevance given the implications of living with the potential risk of sudden cardiac death among carriers of monogenic rare variants associated with QT prolongation.
Non-Genetic Determinants of the QT Interval
Over 75% of individuals with pronounced QTc prolongation (>480 ms) did not carry a polygenic risk equivalent or monogenic putative pathogenic rare variant in this study. Our findings support the possibility that additional genetic contributions to QTc duration remain undiscovered and highlight the important contribution of non-genetic factors to the QTc. These findings should be interpreted in the context of the average age of the study sample which included middle-aged and older individuals, where comorbid cardiovascular disease and polypharmacy are prevalent and among other non-genetic factors are important determinants of the QTc. Future increasingly large-scale common and rare variant population-based association studies of the QTc may identify additional novel genetic determinants of the QTc.
Novel Genome-Wide Association Loci Implicate Genes Associated with Cardiovascular Disease and ECG Traits
We identified 21 novel loci associated with the QT interval in the UKB QTc GWAS of which 12 replicated in TOPMed. The replicated loci implicated genes that have been previously associated with a number of cardiovascular diseases and ECG traits, further validating them as true signals associated with the QTc. The lead novel SNP (p=1.1 × 10−17), rs17171711, is an intronic variant in FAM13B which has been previously associated with atrial fibrillation21 and implicated as an eQTL locus in a previous QTc GWAS.4 rs422068, another intronic variant in MYH6, has also been associated with atrial fibrillation22 and resting heart rate23 in genome-wide association studies. The lead SNP in the ZNF358 locus, rs113394178, has been shown to be associated with the PR interval24 and more recently associated with trabecular morphology and myocardial fractal dimension.25 The SLC12A7 locus which encodes a solute carrier that mediates electroneutral potassium-chloride transport was among the novel replicated loci and has been previously associated with the PR24 and JT intervals.26 Additionally, loci previously associated with atrial fibrillation21,22 (RBM20, MYH6 and FAM13B) and myocardial infarction27,28 (BACH1, STAT3, TCHP/GIT2) were among the novel replicated loci associated with the QTc.
Limitations
Our results should be interpreted in the context of the study design. The study findings apply to individuals who enrolled in the included population-based cohorts, and do not necessarily apply to patients seeking clinical evaluations for evident or possible LQTS. The TOPMed cohort did not ascertain LQTS diagnosis among study participants which prohibited performing an analysis examining the contribution of polygenic and monogenic risk to LQTS in the general population and our findings strictly apply to the QTc. Accordingly, we noted a higher prevalence of individuals with monogenic rare variants in genes associated with the QTc in TOPMed when compared to a prior study examining the prevalence of LQTS in the population by Schwartz et al,29 findings likely due to incomplete penetrance of LQTS, broader panel of genes examined in our study, study design and the increased ancestral diversity of participants in TOPMed. Further population-based studies with LQTS phenotyping are needed to examine the monogenic and polygenic contribution to LQTS in the population. To enable large-scale curation of QT intervals we relied on automated QT measurements rather than manual measurement of the QT interval which is the gold standard in clinical practice and is particularly important in the setting of irregular rhythms such as atrial fibrillation. However, only a very small fraction of our study sample had a history of atrial fibrillation. Additionally, despite studying a large multi-ancestry cohort from the UKB to derive our PRS, the population was predominantly of European ancestry which may not accurately characterize polygenic risk in the TOPMed cohort. However, we demonstrate that our derived PRS was normally distributed and had overall comparable performance across all TOPMed ancestries. Future large-scale genetic association studies in non-European ancestries are needed to enhance the performance of polygenic risk scores across ancestries and allow for better characterization of common variation in non-European ancestries. Our analyses of monogenic variation focused on coding variants within genes associated with the QT interval. Additional studies are needed to evaluate the contribution of non-coding rare variation, copy-number variation and structural variation to the QTc in the general population. We relied on the ClinVar database’s assertions of variant pathogenicity to classify pathogenic/likely pathogenic rare variants, which may be subject to bias and heterogeneity associated with different reporting laboratories/groups. This study focused on exploring genetic determinants of the QTc, however, multiple non-genetic factors contribute to the QTc, particularly among older individuals with multiple cardiovascular comorbidities and who are exposed to polypharmacy, which we were unable to fully account for in this analysis. Additionally, the findings from this study should be interpreted in the context of the average age of the study sample as the genetic contribution to cardiovascular traits tends to vary with age. The samples included in this study predominantly included middle-aged to older adults which limited our ability to perform a meaningful age-stratified analysis of monogenic and polygenic contribution to the QT interval, particularly in adolescents and young adults. Lastly, further studies are needed to examine the impact of QTc polygenic risk on clinical outcomes in the general population.
Conclusions
QTc duration is influenced by both polygenic risk and rare variation in genes underlying cardiac repolarization. A substantial proportion of individuals with pronounced QTc prolongation (> 480 ms) in the population have a high polygenic risk equivalent. Incorporation of polygenic risk assessment may aid in increasing the yield of genetic testing for QTc prolongation. Further studies are needed to uncover novel genetic determinants of the QTc and establish the role of polygenic risk assessment in clinical evaluation of individuals with prolonged QTc in the general population.
Supplementary Material
What is new?
12 novel loci associated with the QTc were identified in a genome-wide association analysis in the UK Biobank and replicated in an independent multi-ancestry cohort, TOPMed.
A genome-wide polygenic risk score of the QTc derived in the UK Biobank and applied in TOPMed captured polygenic variation associated with the QTc across genetic ancestries.
Among individuals with a QTc >480 ms, 3.4% carried a monogenic rare variant, 21% had a polygenic risk equivalent (top PRS decile), and 23.7% had either a monogenic rare variant or a polygenic risk equivalent.
What are the clinical implications?
Individuals in the population with QTc prolongation are more likely to have a common polygenic risk equivalent than a rare monogenic variant in an established QTc prolonging gene.
Comprehensive assessment of the genetic determinants of QTc prolongation may require assessment of both monogenic rare variants and common polygenic risk variants, which we estimate may increase the yield of identifying a genetic determinant of QTc prolongation by ~7-fold, from 1 in 30 to 1 in 4.
Acknowledgements
We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed and the study participants of the UKB.
Sources of Funding
This work was supported by National Institutes of Heatlh (NIH) grant 1R01HL139731 and American Heart Association (AHA) grant 18SFRN34250007 to Dr. Lubitz. Dr. Ellinor is funded by the Fondation Leducq (14CVD01), the NIH (1RO1HL092577,R01HL128914,K24HL105780), and by a grant from the AHA Strategically Focused Research Networks (18SFRN34110082). Dr. Sotoodehnia is funded by the NIH (R01HL141989). Dr. Nauffal is funded by a training grant from the NIH (T32HL007604). Dr. Weng is supported by National Institutes of Heatlh (NIH) grant 1R01HL139731 and American Heart Association (AHA) postdoctoral fellowship 18SFRN34110082. Dr. Loos is funded by the NIH (R01DK110113; R01DK075787; R01DK107786; R01HL142302; R01HG010297; R01DK124097; R01HL151152). Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626–02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I).
Disclaimer
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services.
Non-standard Abbreviations and Acronyms
- ECG
Electrocardiogram
- GWAS
Genome-Wide Association Study
- GC
Genomic Control
- IRB
Institutional Review Board
- LOF
Loss-Of-Function
- LQTS
Long QT Syndrome
- MAF
Minor Allele Frequency
- NHLBI
National Heart Lung and Blood Institute
- PRS
Polygenic Risk Score
- TOPMed
Trans-Omics for Precision Medicine
- UKB
United Kingdom Biobank
- WGS
Whole-Genome Sequencing
Appendix
List of TOPMed investigators to be indexed in PubMed.gov as collaborators
Stephen S Rich PhD
Jerome I Rotter MD
Henry J Lin MD
Footnotes
Disclosures
Dr. Psaty serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson. Drs. Haggerty and Fornwalt receive sponsored research support from Tempus Labs. Dr. Lubitz receives sponsored research support from Bristol Myers Squibb / Pfizer, Bayer AG, Boehringer Ingelheim, Fitbit, and IBM, and has consulted for Bristol Myers Squibb / Pfizer, Bayer AG, and Blackstone Life Sciences. Dr. Ellinor receives sponsored research support from Bayer AG, Novartis, Myokardia and Quest. Remaining authors have nothing to disclose. Dr. Weng receives sponsored research support from IBM to the Broad Institute.
References
- 1.Roden DM, Yang T. Protecting the Heart Against Arrhythmias: Potassium Current Physiology and Repolarization Reserve. Circulation. 2005;112:1376–1378. [DOI] [PubMed] [Google Scholar]
- 2.Beinart R, Zhang Y, Lima JAC, Bluemke DA, Soliman EZ, Heckbert SR, Post WS, Guallar E, Nazarian S. The QT interval is associated with incident cardiovascular events: the MESA study. J Am Coll Cardiol. 2014;64:2111–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Giudicessi JR, Noseworthy PA, Ackerman MJ. The QT Interval: An Emerging Vital Sign for the Precision Medicine Era? Circulation. 2019;139:2711–2713. [DOI] [PubMed] [Google Scholar]
- 4.CARe Consortium, COGENT Consortium, DCCT/EDIC, eMERGE Consortium, HRGEN Consortium, Arking DE, Pulit SL, Crotti L, van der Harst P, Munroe PB, et al. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet. 2014;46:826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang C-E, Huikuri H, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–1963. [DOI] [PubMed] [Google Scholar]
- 6.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.ToPMed Consortium. TOPMed Whole Genome Sequencing Methods: Freeze 6 [Internet]. 2021. [cited 2022 Apr 5];Available from:https://www.nhlbiwgs.org/topmed-whole-genome-sequencing-methods-freeze-6 [Google Scholar]
- 8.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. Genome Aggregation Database Consortium, Neale BM, Daly MJ, MacArthur DG. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN, LeFaive J, VandeHaar P, Gagliano SA, Gifford A, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet. 2018;50:1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schizophrenia Working Group of the Psychiatric Genomics Consortium, Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Patterson N, Daly MJ, Price AL, Neale BM. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loh P-R, Bhatia G, Gusev A, Finucane HK, Bulik-Sullivan BK, Pollack SJ, Schizophrenia Working Group of Psychiatric Genomics Consortium, de Candia TR, Lee SH, Wray NR, et al. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nat Genet. 2015;47:1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gogarten SM, Sofer T, Chen H, Yu C, Brody JA, Thornton TA, Rice KM, Conomos MP. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics. 2019;35:5346–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ge T, Chen C-Y, Ni Y, Feng Y-CA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat Commun. 2019;10:1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Strauss DG, Vicente J, Johannesen L, Blinova K, Mason JW, Weeke P, Behr ER, M. Roden D, Woosley R, Kosova G, et al. Common Genetic Variant Risk Score Is Associated With Drug-Induced QT Prolongation and Torsade de Pointes Risk: A Pilot Study. Circulation. 2017;135:1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi SH, Jurgens SJ, Haggerty CM, Hall AW, Halford JL, Morrill VN, Weng L-C, Lagerman B, Mirshahi T, Pettinger M, et al. Rare Coding Variants Associated With Electrocardiographic Intervals Identify Monogenic Arrhythmia Susceptibility Genes: A Multi-Ancestry Analysis. Circ Genom Precis Med 2021;14:e003300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenberg MA, Lubitz SA, Lin H, Kosova G, Castro VM, Huang P, Ellinor PT, Perlis RH, Newton-Cheh C. Validation of Polygenic Scores for QT Interval in Clinical Populations. Circ Cardiovasc Genet 2017;10:e001724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lahrouchi N, Tadros R, Crotti L, Mizusawa Y, Postema PG, Beekman L, Walsh R, Hasegawa K, Barc J, Ernsting M, et al. Transethnic Genome-Wide Association Study Provides Insights in the Genetic Architecture and Heritability of Long QT Syndrome. Circulation. 2020;142:324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fahed AC, Wang M, Homburger JR, Patel AP, Bick AG, Neben CL, Lai C, Brockman D, Philippakis A, Ellinor PT, et al. Polygenic background modifies penetrance of monogenic variants for tier 1 genomic conditions. Nat Commun. 2020;11:3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi SH, Jurgens SJ, Weng L-C, Pirruccello JP, Roselli C, Chaffin M, Lee CJ-Y, Hall AW, Khera AV, Lunetta KL, et al. Monogenic and Polygenic Contributions to Atrial Fibrillation Risk: Results From a National Biobank. Circ Res. 2020;126:200–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu J, Gore-Panter S, Tchou G, Castel L, Lovano B, Moravec CS, Pettersson GB, Roselli EE, Gillinov AM, McCurry KR, et al. Genetic Control of Left Atrial Gene Expression Yields Insights into the Genetic Susceptibility for Atrial Fibrillation. Circ Genom Precis Med. 2018;11:e002107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nielsen JB, Thorolfsdottir RB, Fritsche LG, Zhou W, Skov MW, Graham SE, Herron TJ, McCarthy S, Schmidt EM, Sveinbjornsson G, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50:1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eppinga RN, Hagemeijer Y, Burgess S, Hinds DA, Stefansson K, Gudbjartsson DF, van Veldhuisen DJ, Munroe PB, Verweij N, van der Harst P. Identification of genomic loci associated with resting heart rate and shared genetic predictors with all-cause mortality. Nat Genet. 2016;48:1557–1563. [DOI] [PubMed] [Google Scholar]
- 24.Ntalla I, Weng L-C, Cartwright JH, Hall AW, Sveinbjornsson G, Tucker NR, Choi SH, Chaffin MD, Roselli C, Barnes MR, et al. Multi-ancestry GWAS of the electrocardiographic PR interval identifies 202 loci underlying cardiac conduction. Nat Commun. 2020;11:2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer HV, Dawes TJW, Serrani M, Bai W, Tokarczuk P, Cai J, de Marvao A, Henry A, Lumbers RT, Gierten J, et al. Genetic and functional insights into the fractal structure of the heart. Nature. 2020;584:589–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bihlmeyer NA, Brody JA, Smith AV, Warren HR, Lin H, Isaacs A, Liu C-T, Marten J, Radmanesh F, Hall LM, et al. ExomeChip-Wide Analysis of 95 626 Individuals Identifies 10 Novel Loci Associated With QT and JT Intervals. Circ Genom Precis Med 2018;11:e001758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hartiala JA, Han Y, Jia Q, Hilser JR, Huang P, Gukasyan J, Schwartzman WS, Cai Z, Biswas S, Trégouët D-A, et al. Genome-wide analysis identifies novel susceptibility loci for myocardial infarction. Eur Heart J. 2021;42:919–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamada Y, Kato K, Oguri M, Horibe H, Fujimaki T, Yasukochi Y, Takeuchi I, Sakuma J. Identification of 13 novel susceptibility loci for early-onset myocardial infarction, hypertension, or chronic kidney disease. Int J Mol Med. 2018;42:2415–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, et al. Prevalence of the Congenital Long-QT Syndrome. Circulation. 2009;120:1761–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
UKB data are made available to researchers from research institutions with genuine research inquiries, following institutional review board (IRB) and UKB approval. All other data are contained within the article and its supplementary material, or are available upon reasonable request to the corresponding author.