

Abstract
Chronic lymphoproliferative disorder of natural killer cells (CLPD-NK) is characterized by clonal expansion of NK cells where the underlying genetic mechanisms are incompletely understood. Here, we report somatic mutations in the chemokine gene CCL22 as the hallmark of a distinct subset of CLPD-NK. CCL22 mutations were enriched at highly conserved residues, were mutually exclusive of STAT3 mutations, and were associated with gene expression programs that resembled normal CD16dim/CD56bright NK cells. Mechanistically, the mutations resulted in ligand-biased chemokine receptor signaling, with decreased internalization of the G-protein-coupled receptor for CCL22, CCR4, via impaired β-arrestin recruitment. This resulted in increased cell chemotaxis in vitro, bidirectional crosstalk with the hematopoietic microenvironment, and enhanced NK cell proliferation in vivo in transgenic human IL-15 mice. Somatic CCL22 mutations illustrate a unique mechanism of tumor formation in which gain-of-function chemokine mutations promote tumorigenesis by biased G-protein-coupled receptor signaling and dysregulation of microenvironmental crosstalk.
Chronic lymphoproliferative disorder of natural killer (NK)-cells (CLPD-NK) is a subset of large granular lymphocyte (LGL) leukemia included as a provisional entity in the 2016 World Health Organization (WHO) classification of lymphoid neoplasms1. CLPD-NK is characterized by the presence of a persistent clonal expansion of CD3 negative (CD3−) NK cells. In contrast to aggressive NK large granular lymphocyte leukemia (ANKL), it is mostly an indolent disease of the elderly, absent in East Asian countries, and not associated with Epstein-Barr virus infection2–4. CLPD-NK is associated with autoimmune phenomena including immune-mediated cytopenia and can coexist with other neoplasms2. Patients with symptomatic cytopenia or autoimmune diseases are often treated with immunosuppressive drugs2,3,5. Classification of CLPD-NK is mainly based on cell surface expression of CD16, CD56 and CD57, markers that define subsets of normal NK cells: CD16dim/CD56bright; CD16+/CD56dim/CD57+; CD16+/CD56dim/CD57− (refs 6–8). The CD56bright subset of normal NK cells is associated with production of cytokines and chemokines including interferon gamma, tumor necrosis factor alpha, CSF2, XCL1, and XCL2 that facilitate communication with other immune cells9,10. In contrast, the CD56dim/low subset is a potent cytolytic effector that can further differentiate into a terminally differentiated CD57+ adaptive subset11.
Several mutations have been identified in CLPD-NK including STAT3 (refs. 12,13) and TET2 (refs. 14–16), however, these mutations are each observed in only one third of cases, leaving the genetic basis of the remaining cases unknown. Here we performed a comprehensive genomic and transcriptomic analysis of CLPD-NK to elucidate the genomic landscape, investigate mechanism of pathogenesis, and improve diagnostic classification and treatment strategies. We identified recurrent somatic mutations in the chemokine gene CCL22 in 21.5% (discovery cohort, 16/59 [27%]; validation cohort, 10/62 [16%]) of cases which define a distinct subgroup of CD56bright CLPD-NK. Functionally, we show that CCL22 mutations were associated with dysregulated immune signaling with the hematopoietic microenvironment resulting from altered G protein-coupled receptor (GPCR) signaling that facilitates NK cell expansion.
Results
Recurrent CCL22 mutations in CLPD-NK
Analysis of genomic alterations of 59 CLPD-NK cases identified previously reported mutations in STAT3 (n=18), STAT5B (n=3), TET2 (n=12), ATM (n=4), ASXL1 (n=3), DNMT3A (n=3), and PPM1D (n=3), whereas no recurrent fusions or copy number changes were found (Figure 1a, Supplementary figure 1, and Supplementary table 1). In addition, we identified sequence mutations of the C-C chemokine-encoding gene CCL22 in 16 (27%) cases, which were mutually exclusive of STAT3 and STAT5B mutations but not TET2 mutations (Figure 1a). By sequencing of fluorescence-activated cell sorted CD56+ NK and CD3+ T cells from two cases, the mutations were shown to be present only in NK cells and thus presumed to be somatic (Supplementary figure 2). Notably, CCL22 mutations were not found in 2837 cases of other hematological malignancies including T-LGL (n=120)17 and other NK leukemia (n=61), such as myeloid/NK cell acute leukemia (n=22), extranodal NK/T cell lymphoma, nasal type (n=25) and ANKL (n=14)18–20. The CCL22 mutations were located at the conserved Leu45, Pro46, and Pro79 residues (Figure 1b,c), and were clonal (Figure 1d). To confirm the occurrence of disease-restricted CCL22 mutations in CLPD-NK, and their exclusivity with STAT3 mutations, we examined a second cohort of 62 patients, which identified CCL22 mutations in 10 patients (16%), also at the Leu45 and Pro46 residues (Figure 1b and Supplementary table 1). In contrast to STAT3-mutated cases (CD16bright/CD56dim), CCL22-mutated CLPD-NK patients were most commonly of CD16dim/CD56bright phenotype, and not associated with anemia (Table 1 and Extended data figure 1). The different hotspot CCL22 mutations did not show unique clinical characteristics (Supplementary table 2). There was a trend to an association with cutaneous complications (psoriasis) and/or neurological symptoms (polyneuropathy, tremor) (p = 0.04) in CCL22-mutated CLPD-NK patients (Table 1), which supports the role of CCL22-CCR4 signaling in skin or neurological inflammation21,22. There were no morphological differences of leukemic cells between cases harboring different mutations.
Figure 1. Mutational landscape of CLPD-NK and CCL22 mutations.
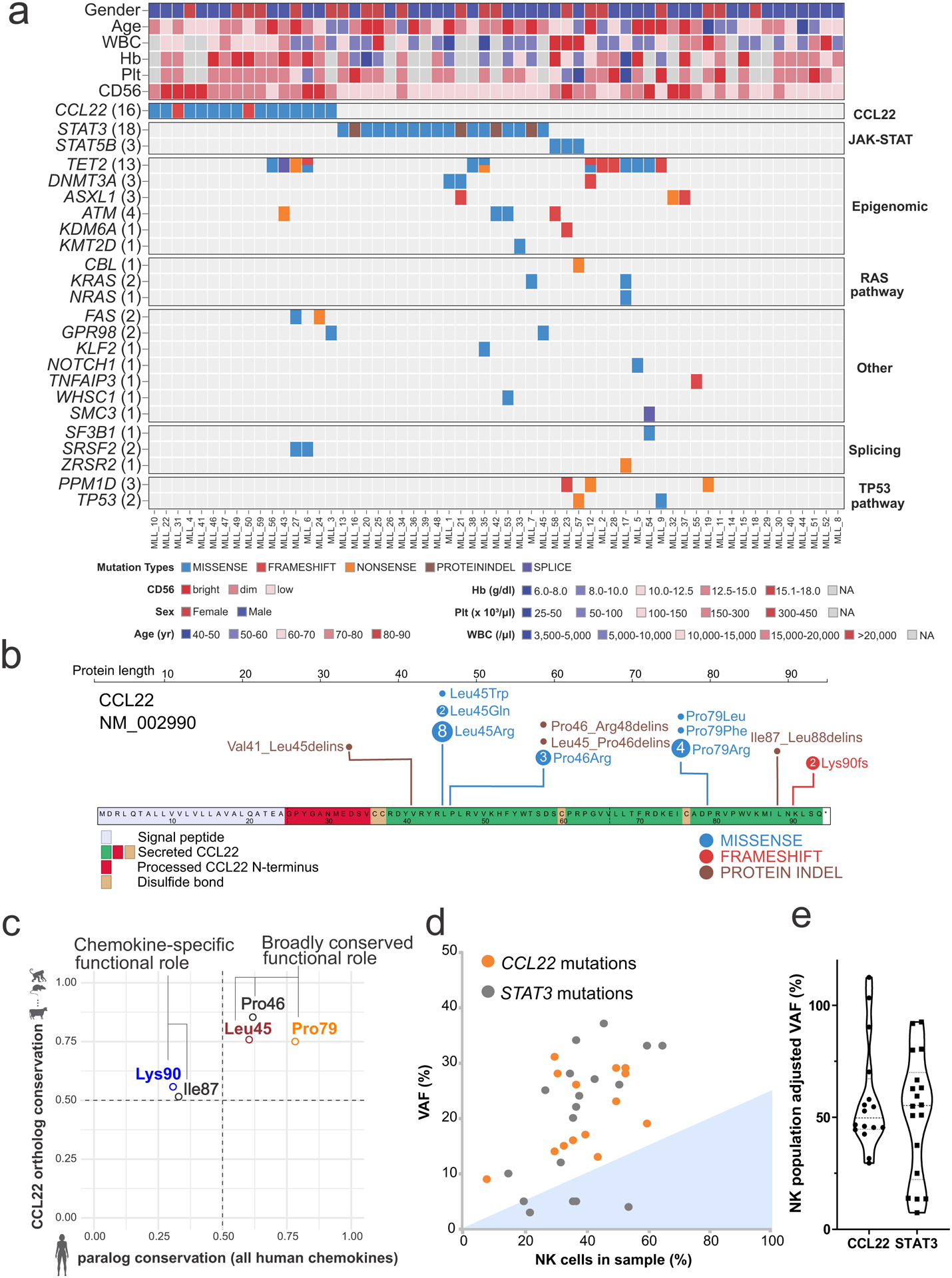
a, Heat map showing the mutational landscape and clinical parameters of the 59 cases with CLPD-NK in the discovery cohort. CCL22 mutations were detected in 16 out of 59 (27%) of CLPD-NK and were mutually exclusive of STAT3 mutations. Higher CD56 expression by immunophenotype was observed in CCL22 mutant cases. Hb, hemoglobin; Plt, platelet; WBC, white blood cell; yr, year. b, Location of detected CCL22 mutations in both discovery (n = 59) and validation (n = 62) cohort. Most CCL22 mutations were located at the Leu45, Pro46 and Pro79 residues. c, Comparison of residue conservation among CCL22 orthologs and human chemokine paralogs. The Leu45, Pro46, and Pro79 residues (all missense mutations) were well conserved among both CCL22 orthologs (i.e. different species) and paralogs among all human chemokines, suggesting a broadly conserved functional role. The Ile87 and Lys90 residues (indels and frameshift mutation, respectively) were conserved among CCL22 orthologs but not among human paralogs, suggesting a chemokine-specific functional role. d,e, Comparison of variant allele frequency (VAF) and NK cell populations in each sample with CCL22 mutations or STAT3 mutations. d, Dotted line indicates VAF of 0.5 ratio. Filling color represents subclonal VAF, defined as average minus standard deviation. e, In each sample with CCL22 mutations (n=16) or STAT3 mutations (n=18), VAF normalized by NK cell population is shown. All the CCL22 mutations are clonal while some STAT3 mutations are subclonal.
Table 1.
Clinical characteristics of CLPD-NK cases with CCL22 mutations.
| Discovery (n = 59) | Validation (n = 62) | |||||||
|---|---|---|---|---|---|---|---|---|
| CCL22 mutation | STAT3 mutation | Others | ANOVA (Tukey)/Fisher (Holm) | CCL22 mutation | STAT3 mutation | Others | ANOVA (Tukey)/Fisher (Holm) | |
| n=16 | n=18 | n=25 | n=10 | n=20 | n=32 | |||
| Age (median) | 72 (55–81) | 75 (54–89) | 68 (41–89) | n.s. | 59.5 (36–69) | 68 (21–91) | 64 (7–80) | n.s. |
| Sex | ||||||||
| Male | 10 | 11 | 18 | n.s. | 6 | 17 | 16 | n.s. |
| Female | 6 | 7 | 7 | 4 | 3 | 16 | ||
| Immunophenotype | ||||||||
| CD56 bright | 6 | 0 | 4 | p = 0.02# | 6 | 2 | 3 | p = 0.02# |
| CD56 dim/neg | 10 | 18 | 21 | 3 | 15 | 21 | p = 0.01## | |
| CD16 bright | 2 | 13 | 12 | p = 0.001# | 2 | 13 | 7 | p = 0.03# |
| CD16 dim/neg | 10 | 2 | 5 | p = 0.02## | 7 | 4 | 16 | p = 0.03### |
| CD57 bright | 0 | 2 | 2 | n.s. | 0 | 1 | 3 | n.s. |
| CD57 dim/neg | 16 | 14 | 16 | 4 | 6 | 12 | ||
| Complete blood count | ||||||||
| WBC (/μl) | 12,200 (7,200–16,700) | 7,450 (3,960–21,880) | 12,170 (6,600–78,400) | n.s. | 9,280 (2,200–21,400) | 6,985 (2,000–24,850) | 5,490 (1,030–34,100) | n.s. |
| ANC (/μl) | 2,213 (1,224–8,000) | 1,457 (141–3,720) | 1,548 (580–10,192) | n.s. | 3,601 (558–12,056) | 1,618 (688–4,738) | 1,756 (280–16,368) | n.s. |
| Hb (g/dl) | 15 (12.8–17.5) | 9.0 (7.9–14.9) | 14 (6.7–16.2) | p < 0.001# p = 0.001### |
13.9 (8–15.9) | 11.6 (7.1–14.6) | 12.1 (7.2–16.5) | p = 0.049# |
| Plt (×103/μl) | 218.5 (85–277) | 180 (125–329) | 194 (26–353) | n.s. | 221 (26–321) | 215 (9–681) | 178 (47–550) | n.s. |
| Treatment§ | ||||||||
| Yes | 2 | 7 | 3 | n.s. | 3 | 10 | 17 | n.s. |
| No | 7 | 6 | 8 | 7 | 10 | 15 | ||
| Complications | No data | CCL22 vs non-CCL22 | ||||||
| Skin disease | 3 | 3 | 2 | n.s. (p = 0.11) | ||||
| Neurological symptoms | 3 | 0 | 4 | n.s. (p = 0.08) | ||||
| Skin or Neurological disease | 5 | 3 | 6 | p = 0.04 | ||||
| Autoimmune disease | 1 | 2 | 8 | n.s. | ||||
| Cytopenia | 0 | 9 | 7 | n.s. (p = 0.051) | ||||
| Other malignancies | 1 | 1 | 7 | n.s. | ||||
CCL22 versus STAT3.
CCL22 versus Others.
STAT3 versus Others.
WBC, white blood cell count; ANC, absolute neutrophil count; Hb, hemoglobin; Plt, platelet.
Treatment includes methotrexate or cyclophosphamide or Cyclosporine A or prednisone (5mg/day higher) or another agent for related autoimmune disease. Categorical variables were examined with the use of Fisher’s exact test with correction of P values for multiple testing (Holm). Associations between continuous variables were compared with the use of the one-way ANOVA with Tukey’s honestly significant difference post hoc test.
Distinct expression profiles in cytokine signaling networks
Analysis of transcriptome sequencing data of 59 CLPD-NK cases, including 16 CCL22 mutated cases and 43 cases without CCL22 mutations (Supplementary table 3), identified 110 upregulated and 197 downregulated differentially expressed genes (adjusted P value <0.01 and log2 fold change >1.5; Figure 2a and Supplementary table 4). The gene expression profile and immunophenotype were similar to CD56bright normal NK cells, with higher expression of NCAM1 (CD56) and low FCGR3A (CD16) in CCL22 mutant cases (Figure 2a,b, Extended data figure 2, Table 1, and Supplementary tables 4,5). Additional overexpressed genes and enriched pathways included cytokines and chemokines important for cell-to-cell communication (CCR4, CCR5, CXCR3, XCL1, XCL2, XCR1), immune cell maturation and activation (XCL1, XCL2, CSF2, XCR1), transcription factors (TCF7, HOXA9), and GZMK (granzyme K) similar to CD56bright normal NK cells (Figure 2b–d, and Supplementary tables 4,6). Deregulation of selected genes was validated in NK-92 cells engineered to express the CCL22-Pro79Arg mutation (Extended data figure 3). In contrast, STAT3-mutated samples were more similar to CD56dim normal NK cells in expression of cell surface markers, the cytotoxic genes FASLG encoding Fas ligand, and PRF1 encoding perforin (Figure 2b). Accordingly, reduced FASLG expression and NK-mediated cytotoxicity was observed in CCL22-Pro79Arg compared to CCL22 wild type NK-92 cells (Figure 2e and Extended data figure 3a,b). In addition, we observed enriched expression of mediators of cytokine signaling, NF-κB, and dendritic cell (DC) maturation in CCL22-mutated samples including CSF2, CCL2, CXCL3, XCL1 and XCL2 (Figure 2f and Supplementary table 7). DCs are considered to be one of the pivotal providers of IL-15 by cell-to-cell contact with NK cells using IL-15 receptor α chain (IL-15RA)23; conversely, several cytokines from NK cells activate DCs. This DC/NK cell crosstalk involving IL-15, PGDF, XCL1, and interferons plays an important role in mutual activation and maturation of both cell types23–25. Furthermore, DCs efficiently stimulate NK cell proliferation, and cell-to-cell DC/NK cell contact has been demonstrated by bone marrow biopsies in CLPD-NK patients but not in healthy individuals26. Thus, integrated genomic analysis demonstrated that CCL22 mutations define a distinct subtype of CLPD-NK, recapitulating the phenotype of CD16dim/CD56bright NK cells, with the observation of similar overexpressed genes and enriched expression profiles in cytokine/chemokine signaling, suggesting a potential interaction with other immune cell populations that may facilitate NK cell proliferation.
Figure 2. Similar expression profiles of CCL22 mutant CLPD-NK to normal CD56bright NK cells.
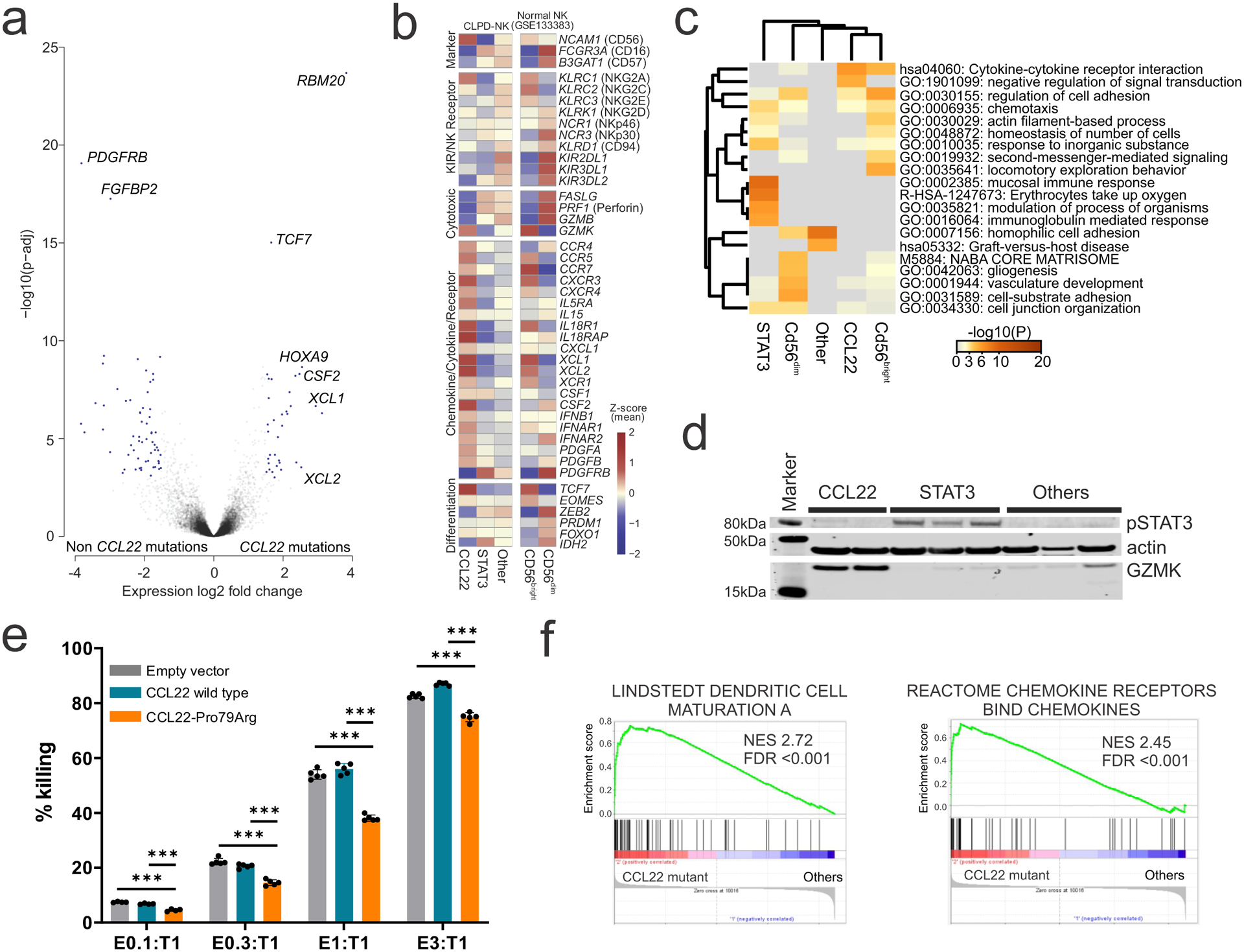
a, Differentially expressed genes (blue dots; adjusted P value (p-adj) <0.01 and log2 fold change >1.5 analyzed by DESeq2 R package61) between CLPD-NK cases with CCL22 mutations and those with non-CCL22 mutations are shown in the volcano plot. Cytokine and chemokine genes (XCL1, XCL2, CSF2) and transcription factors (TCF7, HOXA9) were differentially expressed in CCL22 mutated samples. b, Heatmap showing the expression of representative core NK cell signature genes. Mean z-scores are represented in each subgroup; CLPD-NK with CCL22 mutations (CCL22), STAT3 mutations (STAT3), non-CCL22/STAT3 mutations (Other), and normal CD56bright and CD56dim NK cells. CCL22 mutated cases showed similar expression pattern to CD56bright normal NK cells in high expression of NCAM1 (CD56), chemokine/cytokine related genes with low expression of FCGR3A (CD16) and most cytotoxic genes. In contrast, STAT3 mutated cases showed similar expression pattern to CD56dim normal NK cells in high expression of cytotoxic genes. c, Heatmap comparing enriched GO terms of upregulated genes in each subgroup. There was a similarity of enriched GO terms in CCL22 mutated cases and CD56bright normal NK cells compared to non-CCL22 mutated cases. d, High GZMK expression in CCL22 mutant primary CLPD-NK samples and increased pSTAT3 in STAT3 mutant primary CLPD-NK samples were validated by immunoblotting. e, Reduced cytotoxicity against CellTrace Far Red treated K562 cells (T; target) in CCL22-Pro79Arg transduced NK-92 cell line (E; effector) compared to empty vector or CCL22-wild type transduced NK-92 cells (E) were observed in all the tested E to T ratio. The mean (± SD) killing rate (%) from negative control (T only; 0%) and positive control (T only without CellTrace Far Red treatment; 100%) is shown (n=5). P value was calculated by t-test. ***0.0001 < p < 0.001. f, Gene Set Enrichment Analysis of enriched gene sets in CLPD-NK cases with CCL22 mutations compared to non-CCL22 mutant cases. Dendritic cell maturation pathway and chemokine signaling pathway were enriched in CCL22 mutated cases.
CCL22 mutations at CCR4 enhance cellular chemotaxis
To assess the functional consequences of CCL22 mutations, we first examined their effect on cell line proliferation in vitro. Wild type and mutant CCL22 transcripts were virally expressed with its sole G protein-coupled receptor CCR4 in the IL-3 dependent Ba/F3 mouse hematopoietic cell line, with no discernible effect on growth factor dependence (Extended data figure 4a). A similar lack of effect on cellular proliferation was observed in mutant CCL22 transduced NK-92 cells (Extended data figure 4b–e), indicating that a solely autocrine effect of mutant CCL22 on proliferation of CCR4 expressing cells was unlikely. CCL22 is a member of the family of C-C chemokines with two adjacent cysteine residues originally known as macrophage-derived chemokine (MDC)27. We mapped the identified CCL22 mutations onto a representative chemokine-GPCR complex28 (PDB 5UIW) to infer possible mutational effects on CCL22-CCR4 interactions. Mutations were clustered into three secondary structural elements, including the N-loop (Leu45, Pro46), the 50s-loop (Pro79), and the C-terminal alpha helix (Ile87, Lys90) (Figure 3a). The three commonly mutated residues Leu45, Pro46 and Pro79 were broadly conserved among all chemokines (Figure 1c and Extended data figure 5a–c). CCL22 mutations were enriched at basic residues (Extended data figure 5a), which may enhance CCL22 interactions with negatively charged glycosaminoglycans in the formation of chemokine gradients29. In particular, the CCL22-Leu45Arg and Pro46Arg mutations were predicted to introduce basic residues in close proximity to a putative CCR4 sulfotyrosine motif comprised of Glu23 and Tyr22, which would likely promote electrostatic interactions between mutant CCL22 and CCR4 compared to wild type (Figure 3b and Extended data figure 5d,e). The CCL22-Pro79 mutations were expected to enhance interaction with CCR4-extracellular loop 2 (Extended data figure 5f–h), which has profound effects on biased signaling through G protein and β-arrestin effectors30,31.
Figure 3. Structural mapping and in vitro functional analysis of CCL22 mutants.
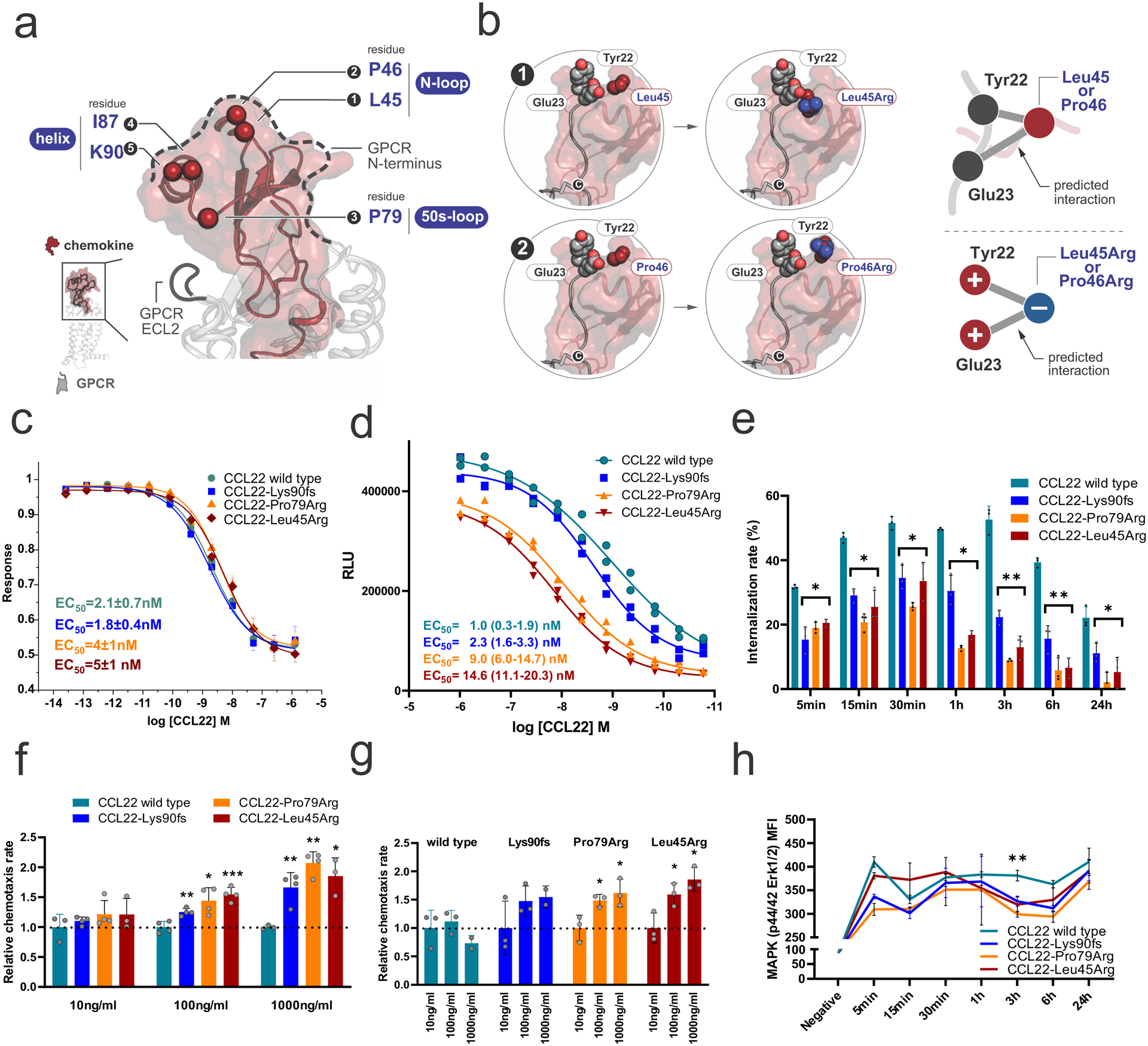
a, A representative chemokine-GPCR complex28 (PDB 5UIW) depicting the positions of the mutated residues on the chemokine structure as spheres. Residues are labeled by number as well as with their corresponding Secondary Structural Element (SSE). b, Mapping of Leu45Arg and Pro46Arg mutations onto a model of a representative chemokine-GPCR complex28 shows the relative positions of the putative CCR4 sulfotyrosine motif Tyr22/Glu23 to the CCL22 mutants. Leu45/Pro46 mutations place these basic residues in close proximity to negatively charged Glu23 and the adjacent putative Tyr22, likely enhancing CCR4 N-terminal interactions with mutated CCL22 through electrostatic interactions. c, GloSensor assay with exogenous recombinant CCL22 representing GPCR activity. All the recombinant CCL22 proteins induced Gi-coupled receptor activation in a dose dependent manner. The mean (± SD) decrease in normalized luminescence signal is plotted (n=3). d, β-arrestin assay with exogenous recombinant CCL22 representing β-arrestin recruitment based on enzyme fragment complementation technology (PathHunter®). β-arrestin recruitment was impaired in mutant CCL22. Mean RLU (relative light units) value (± SD) is shown (n=2). e, Internalization assay of CCR4 over time with exogenous recombinant CCL22 in Ba/F3-CCR4 cells. Recombinant mutant CCL22 proteins showed significant prolonged attenuation of CCR4 internalization. The mean (± SD) difference from positive control (Ba/F3-CCR4 without treatment; 0%) and negative control (isotype; 100%) is shown (n=3). f,g, Cellular chemotaxis of Ba/F3-CCR4 cells in response to exogenous recombinant mutant CCL22 compared to wild type CCL22 in a transwell cell migration assay. Recombinant mutant CCL22 proteins showed enhanced cellular chemotaxis in six-hour incubation. f, The mean (± SD) relative chemotaxis rate of recombinant mutant CCL22 to wild type CCL22 in each concentration is shown (n=4). g, The mean (± SD) relative chemotaxis rate in each recombinant CCL22 protein (wild type and three mutants) is shown (n=4). h, Ba/F3-CCR4 cells were stimulated over time with exogenous recombinant CCL22 and MAPK (p44/42 Erk1/2) was assessed by phosphoflow cytometric analysis. Phosphorylation of p44/42 Erk1/2 was suppressed in recombinant mutant CCL22 protein in three-hour continuous stimulation. The mean (± SD) fluorescence intensity (MFI) is shown (n=3). *0.01 < p < 0.05; **0.001 < p < 0.01; ***0.0001 < p < 0.001.
CCL22 acts as a chemotactic protein to recruit cells expressing CCR4 cells (e.g., T helper type 2 cells (Th2) and regulatory T cells (Treg))32,33. In addition to cellular chemotaxis induced by G protein signaling, engagement of CCR4 also induces interaction of CCR4 with β-arrestin leading to receptor endocytosis to desensitize G protein signaling34. Importantly, the CCL22-CCR4 axis has been implicated in tumor-induced immunomodulation by promoting direct and/or indirect communication with several types of immune cells such as DCs, regulatory T cells, cytotoxic T cells, and NK cells35–37. Furthermore, somatic CCR4 alterations resulting in truncation of the C-terminal cytoplasmic domain are frequently found in adult T cell leukemia (ATL), which not only prevent receptor internalization upon ligand stimulation but also play an important role in leukemogenesis38. We next examined the effect of exogenous recombinant wild type or mutant CCL22 on CCR4 internalization in Ba/F3-CCR4 cells. All recombinant CCL22 proteins (wild type, Leu45Arg, Pro79Arg, and Lys90fs) were able to bind CCR4 and induce dose-dependent Gi-coupled receptor activation with similar potency and efficacy as measured by the GloSensor cAMP assay (Figure 3c). In contrast, β-arrestin recruitment was significantly impaired by recombinant mutant compared to wild type CCL22 protein, especially the missense variants Leu45Arg and Pro79Arg (Figure 3d). Accordingly, although both recombinant wild type and mutant CCL22 showed dose-dependent ligand-induced internalization of CCR4, all three mutants resulted in significant and prolonged attenuation of CCR4 internalization compared to wild type CCL22 (Figure 3e and Extended data figure 6). This effect was most prominent between three and six hours in a 24-hour window. We observed significantly increased chemotaxis of Ba/F3-CCR4 cells treated with recombinant mutant compared to wild type CCL22 in a transwell cell migration assay (Figure 3f). However, maximal (1,000 ng/ml) stimulation by recombinant mutant CCL22 did not further enhance chemotaxis, and attenuated this effect for wild type CCL22 (Figure 3g). This is consistent with a dichotomy between the effects of wild type and mutant chemokine, with reduced internalization effects of mutant CCL22, and concentration-dependent functional effects of CCL22. Transduction of wild type or mutant CCL22 into CCR4-expressing cells resulted in decreased CCR4 internalization of mutant CCL22 in immunofluorescence and serum swapping assay, indicating a paracrine mechanism of action of the CCL22 mutations (Extended data figure 7).
We then investigated activation of intracellular signaling pathways in Ba/F3-CCR4 cells after incubation with recombinant wild type or mutant CCL22. Incubation with CCL22 augmented AKT and mitogen-activated protein kinase (MAPK) (p44/42 extracellular signal-regulated kinase (Erk) 1/2) phosphorylation (Extended data figure 8a,b). Consistent with the result of the GloSensor assay (Figure 3c), we did not observe a difference in augmented phosphorylation level of AKT, which is dependent upon G protein signaling39 (Extended data figure 8a,b). In contrast, as suggested by the β-arrestin assay (Figure 3d), phosphorylation of ERK was reduced in all the tested mutants compared to recombinant wild type CCL22 at 3 hours incubation by flow cytometry and single cell phosphoproteomics (Figure 3h, Extended data figure 8c,d, and Supplementary table 8). This may partly explain attenuation of CCR4 internalization, as β-arrestin governs both receptor internalization and GPCR-mediated ERK phosphorylation40,41. Thus, the lack of difference between wild type and mutant CCL22 in G protein signaling is in contrast to the effects of the mutations on internalization, chemotaxis, and reduced ERK phosphorylation, which supports the notion of CCL22 mutation-specific biased CCR4/GPCR signaling through impaired β-arrestin recruitment and signaling.
CCL22 mutations drive NK cell proliferation in vivo
We next transplanted NK-92 cells expressing wild type or mutant CCL22, or only GFP-expressing lentiviral vector into NSG mice, and NSG-Tg(Hu-IL15) mice that constitutively express human IL-15, a growth factor for human NK cells. Limited viable primary CCL22-mutated CLPD-NK cells precluded establishment of patient-derived xenografts. Hu-IL-15 expressing mice but not non-hIL-15-transgenic NSG supported engraftment of NK-92 cells in bone marrow (BM) and spleen though few circulating cells were observed, and those transplanted with CCL22-Pro79Arg expressing cells became moribund with robust engraftment and splenomegaly 2 months after transplantation (Figure 4a–c and Extended data figure 9). Expression of CCL22-Pro79Arg, but not wild type CCL22 or empty vector also augmented cell surface CD56 expression of engrafted NK-92 cells, which is a characteristic of CLPD-NK patients with CCL22 mutations (Figure 4d). Engrafted CCL22-Pro79Arg NK-92 cells exhibited distinct expression profiles compared to engrafted empty vector- and wild type CCL22 NK-92 transduced cells with higher expression of XCR1 and GZMK as well as deregulation of type I interferon response pathways indicating reactive cytokine production (Figure 4e–g, Extended data figure 3c and Supplementary tables 9,10). Furthermore, engrafted CCL22-Pro79Arg NK-92 cells recapitulated downregulated genes in CLPD-NK patients with CCL22 mutations (Figure 4h, and Supplementary table 11).
Figure 4. CCL22 mutations drive proliferation and CD56+ phenotype in vivo.
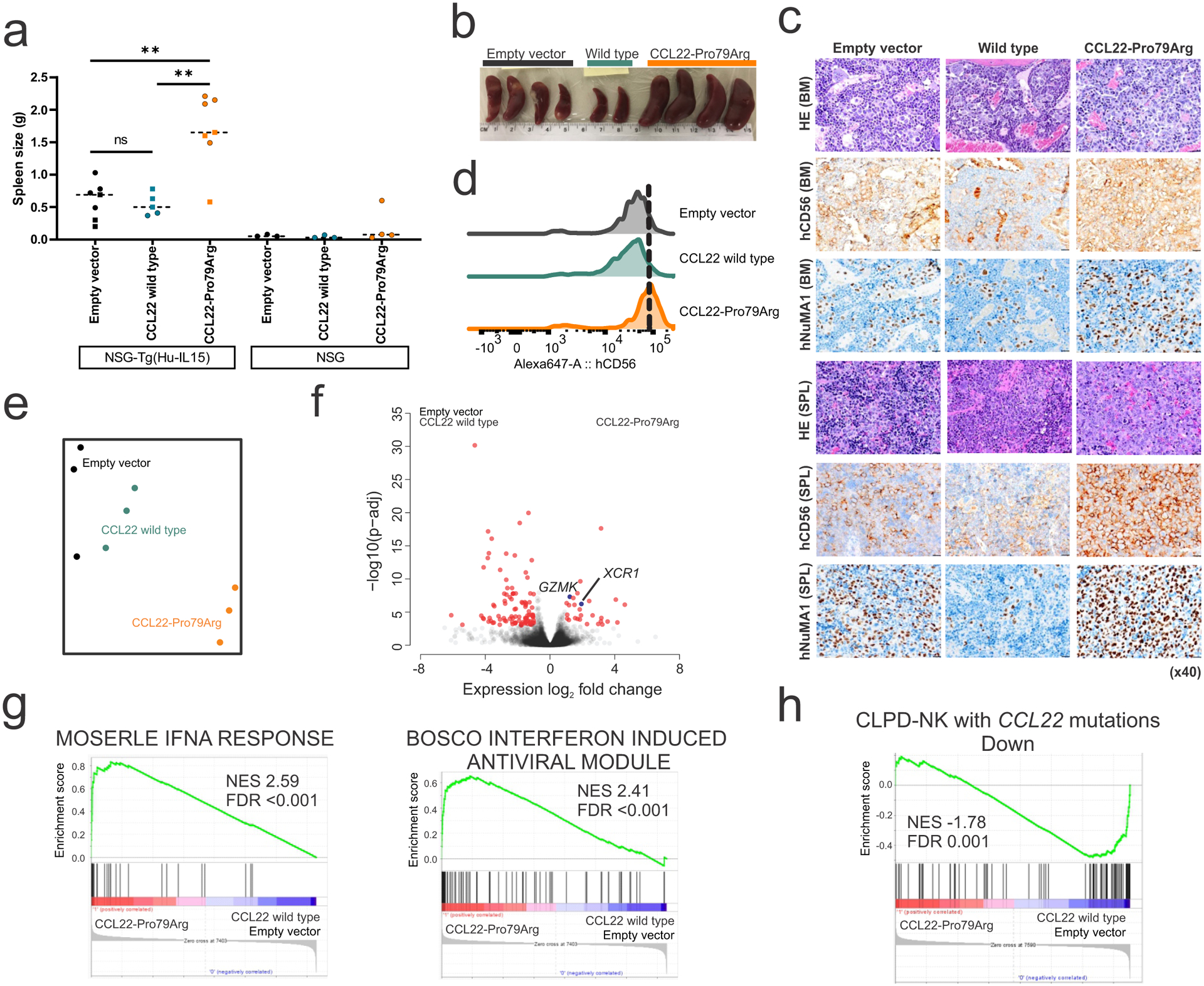
a,b, Spleen weight at the end of study (batch 1; day 57, batch 2; day 44) in NSG mice (3 mice for each group) and NSG-Tg(Hu-IL15) mice that constitutively express human IL-15 transplanted with NK-92 cells expressing wild type (n=3 (batch 1, square), 2 (batch 2, round), biologically independent animals) or mutant CCL22 (Pro79Arg; n=3, 4), or GFP-expressing lentiviral vector (empty vector; n=3, 4). NK-92 cells expressing mutant, but not wild type CCL22 resulted in marked splenomegaly when engrafted in NSG-Tg(Hu-IL15) mice, but not in NSG mice not expressing Hu-IL15. The mean value is shown in dotted line. P value was calculated by t-test. **0.001 < p < 0.01. c, Immunohistochemistry images (x40, scale bar 20 μm) for bone marrow (BM) and spleen (SPL) of engrafted NSG-Tg(Hu-IL15) mice transplanted with NK-92 cells expressing wild type or mutant CCL22 (Pro79Arg), or GFP-expressing lentiviral vector (empty vector). Robust engrafted human CD56 positive NK cells were observed in CCL22-Pro79Arg. d, CD56 levels of engrafted NK-92 cells in spleen of NSG-Tg(Hu-IL15) mice were assessed by flow cytometry. Higher CD56 expression was observed in mutant CCL22 than wild type CCL22 (wtCCL22) and empty vector. e, Gene expression profiling of engrafted NK-92 cells is represented in a two-dimensional tSNE plot, showing the distinct gene expression profile of mutant CCL22 expressing NK-92 cells from wild type CCL22 or empty vector bearing cells. f, Differentially expressed genes (red dots; adjusted P value (p-adj) <0.01 and log2 fold change >1 analyzed by DESeq2 R package61) between engrafted NK-92 cells expressing CCL22-Pro79Arg and those expressing wild type CCL22/empty vector. High expression of XCR1 and GZMK in mutant CCL22 recapitulated expression profile of primary samples with CCL22 mutations. g, Gene Set Enrichment Analysis (GSEA) in engrafted NK-92 cells expressing CCL22-Pro79Arg compared to wild type CCL22 and empty vector. Type I interferon related pathways were enriched in mutant CCL22, indicating reactive cytokine production. h, GSEA of custom gene sets down-regulated in CLPD-NK cases with CCL22 mutations compared to non-CCL22 mutant cases in engrafted NK-92 cells expressing CCL22-Pro79Arg. Engrafted mutant CCL22 cells recapitulated dysregulated genes found in primary CLPD-NK cases with CCL22 mutations.
Crosstalk with NK cells leads to microenvironmental deregulation
In contrast to the lack of an in vitro autocrine effect on cell proliferation in cells expressing CCL22 mutations (Extended data figure 4), the results from the in vivo transplant assay showed robust engraftment and splenomegaly in CCL22-Pro79Arg expressing NK-92 cells. This suggested enhanced chemokine/cytokine gradients between NK and microenvironment cells in BM and lymphoid tissues, including the spleen (Figure 4 and Extended data figure 9). In support of this hypothesis, higher expression of several chemokine receptors including CXCR4 and CCR5, which are also overexpressed in primary CCL22-mutant CLPD-NK samples (Figure 2b), was observed in the NK cell line NKL with CCL22 mutation (Arg44_Leu45insSer) but not in isogenic CCL22 wild type NKL cells generated by genome editing (Extended data figure 10). These chemokine receptors are known to favor migration of hematopoietic cells into BM and lymphoid tissues42,43. Profiling of secreted chemokines showed higher secretion of several cytokines that are known activators of NK cells and DC in CCL22 mutant compared to wild type NKL cells (Extended data figure 10b and Supplementary table 12). This is consistent with the upregulation and enrichment of chemokine signaling and DC maturation pathway in primary CCL22 mutated samples (Figure 2f and Supplementary table 7).
As many cytokines/chemokines including IL-15 share amino acid sequence homology between human and mouse (IL-15 homology 73%, ref.44) we examined effects of mouse and human IL-15 on CCL22 wild type and mutant NK-92 cells in vitro, showing that mouse IL-15 supported human NK-92 cell proliferation, albeit less efficaciously than human IL-15 (Extended data figure 4e). To further explore a potential cell-crosstalk in engineered human NK-92 and mouse cells, we analyzed the phenotype of mouse cells harvested from spleen of engrafted mice by flow cytometry. We observed a higher percentage of IL-15-providing cells, including macrophages, and a trend to a higher percentage of plasmacytoid DC (pDC) in mice transplanted with CCL22-Pro79Arg expressing cells (Figure 5a and Supplementary figure 3). In contrast, the frequency of neutrophils was decreased in CCL22-Pro79Arg, possibly reflecting neutrophil apoptosis induced by NK cells45,46 (Figure 5a). We also observed a trend of higher frequency of endothelial cells, follicular dendritic cells (FDC), marginal zone reticular cells (MRC), and perivascular reticular cells (PRC) in CCL22-Pro79Arg, which are known to produce IL-1547, to regulate migration of DCs48, and to support macrophages49 (Figure 5b). Increased CD163 expressing cells (including macrophages) by immunohistochemistry in both BM and spleen cells from mice transplanted with CCL22-Pro79Arg-expressing cells also supports the notion of CCL22 mutation induced bidirectional NK-microenvironment crosstalk (Figure 5c). Finally, we directly examined the effects of wild type or mutant (Pro79Arg) CCL22 and/or co-expressed cytokines/chemokines (CSF1, CSF2, XCL1, XCL2; Figure 2b) on cell migration of bone marrow-derived mononuclear cells. Consistent with data from mouse spleen cells, we observed a trend of enhanced migration of conventional DC (cDC), pDC and CD4+ T cells, which facilitate NK cell proliferation (Figure 5d). Thus, collectively these in vitro and in vivo data indicate that exogenous IL-15 supports proliferation of CCL22-mutant NK cells, and that CCL22-mutated cells liberate a cytokine/chemokine milieu that stimulates microenvironmental cell subsets that further supports NK cell proliferation.
Figure 5. Enhanced cell crosstalk for CCL22 mutant NK cells.
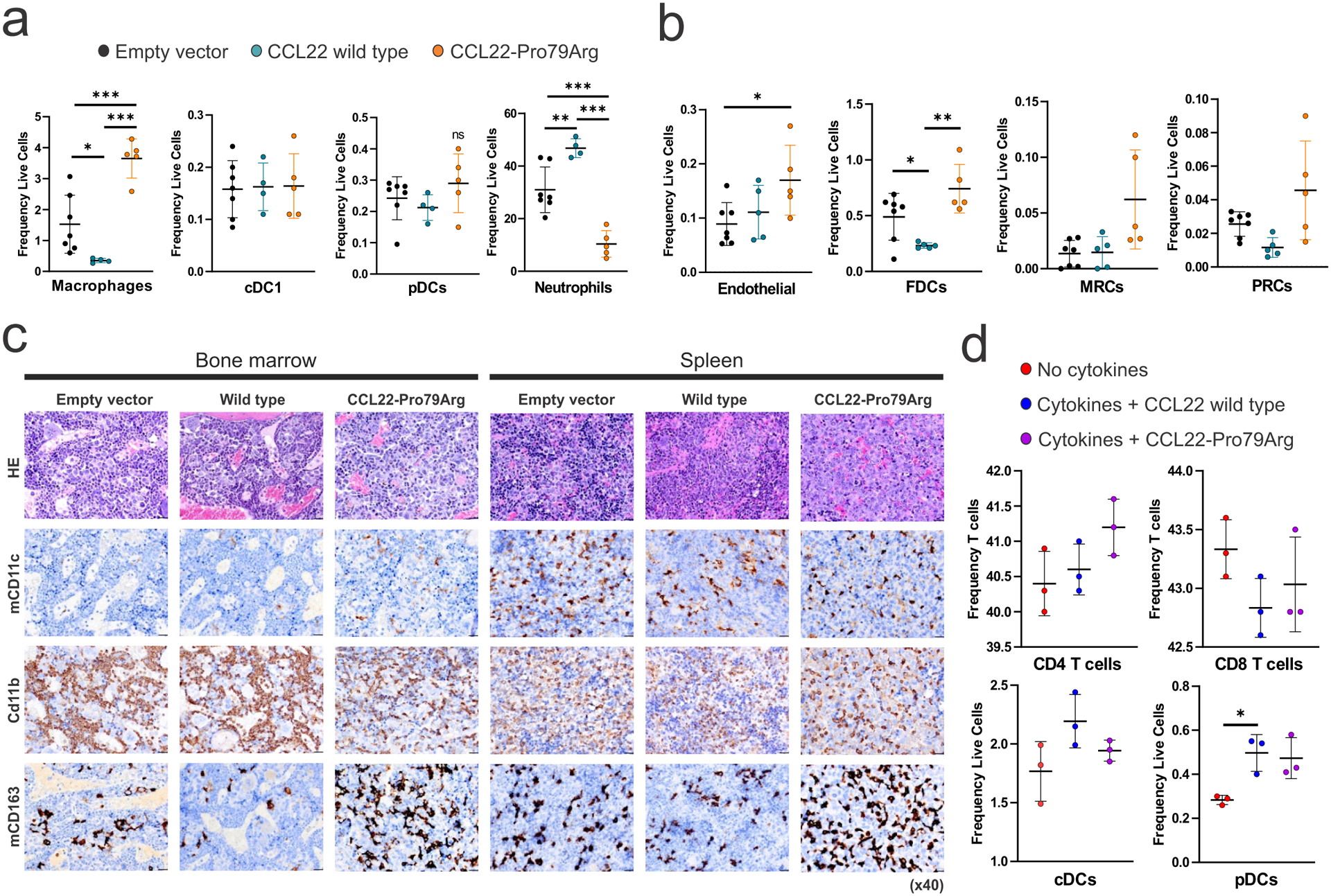
Infiltrated (a) immune cell and (b) stromal cell populations (%) in spleens of engrafted NSG-Tg(Hu-IL15) mice transplanted with NK-92 cells expressing wild type (n=4, biologically independent animal samples in a single experiment) or mutant CCL22 (Pro79Arg, n=5), or GFP-expressing lentiviral vector (empty vector, n=7). Significant increases of the number of macrophages, endothelial cells, and follicular dendritic cells (FDC) were detected in NK-92 cells expressing mutant, but not wild type CCL22 or empty vector. A trend of higher plasmacytoid DC (pDC), marginal zone reticular cells (MRC), and perivascular reticular cells (PRC) were observed. In contrast, neutrophils were decreased in CCL22-Pro79Arg NK cell engrafted mice. The mean (± SD) values are shown in black lines. Representative single experiment results in two independent experiments are shown. P values were calculated by two-sided t-test. *0.01 < p < 0.05; **0.001 < p < 0.01; ***0.0001 < p < 0.001. c, Immunohistochemistry images (x40, scale bar 20 μm) stained for mouse cells in bone marrow and spleen of engrafted NSG-Tg(Hu-IL15) mice. Increased CD163 expressing cells (including macrophages) were observed in CCL22-Pro79Arg. d, Frequency (%) of chemoattracted immune cells among human bone marrow mononuclear cells after overnight incubation with or without mixture of highly expressed cytokines/chemokines (M-CSF, GM-CSF, XCL1, XCL2) in CCL22 mutant primary samples and recombinant CCL22 protein (wild type or CCL22-Pro79Arg) in Transwell® 6-well plate with 8.0 μm pore, performed in triplicate in a single experiment. Each group A trend of higher migration of conventional DC (cDC), pDC and CD4+ T cells were observed. The mean (± SD) values are shown in black lines. Representative single experiment results in two independent experiments are shown. P values were calculated by two-sided t-test. *0.01 < p < 0.05; **0.001 < p < 0.01; ***0.0001 < p < 0.001.
Discussion
Genomic sequencing has revealed the genomic landscapes of many malignant and nonmalignant diseases50 with the identification of driving alterations promoting enhanced survival and proliferation of mutated cells. Due to its relative rarity and commonly indolent clinical course2,3,5, detailed genomic analyses of CLPD-NK have been lacking. Similar to T-cell large granular lymphocyte leukemia, STAT3 mutations were most frequently observed in CLPD-NK with relatively severe clinical course compared to non-STAT3 mutant cases commonly of CD16bright/CD56dim immunophenotype7,12,13. In addition, TET2 mutations have also been reported in CLPD-NK14,15. Here, using comprehensive genomic analysis of more than 100 cases including both CD56bright and CD56dim phenotypes, we identified CCL22 mutations that define a subset of CLPD-NK of CD16dim/CD56bright phenotype.
In contrast to other leukemias in which mutations commonly directly perturb gene expression and cellular pathways in leukemic cells harboring the mutations, including CCR4-mutated adult T cell leukemia/lymphoma (ATL)51, CCL22 mutations do not exert leukemogenic effects through direct cell intrinsic or autocrine effects, which is further supported by the lack of CCR4 mutations in CLPD-NK. In contrast, our data support a model in which sequential induction of mutually tropic NK and hematopoietic microenvironment cells is required to induce CCL22-mutant CLPD-NK. This model is supported by multiple lines of evidence from this study and prior data. First, NK cells with CCL22 mutations secrete CCL22 mutant protein which binds to CCR4. Several immune effectors including DC, Th2, Treg, and skin-homing T cells express CCR421,34,52 and can bind mutant CCL22 (schematically represented in Figure 6).
Figure 6. Schema of CCL22-mutant CLPD-NK leukemogenesis.
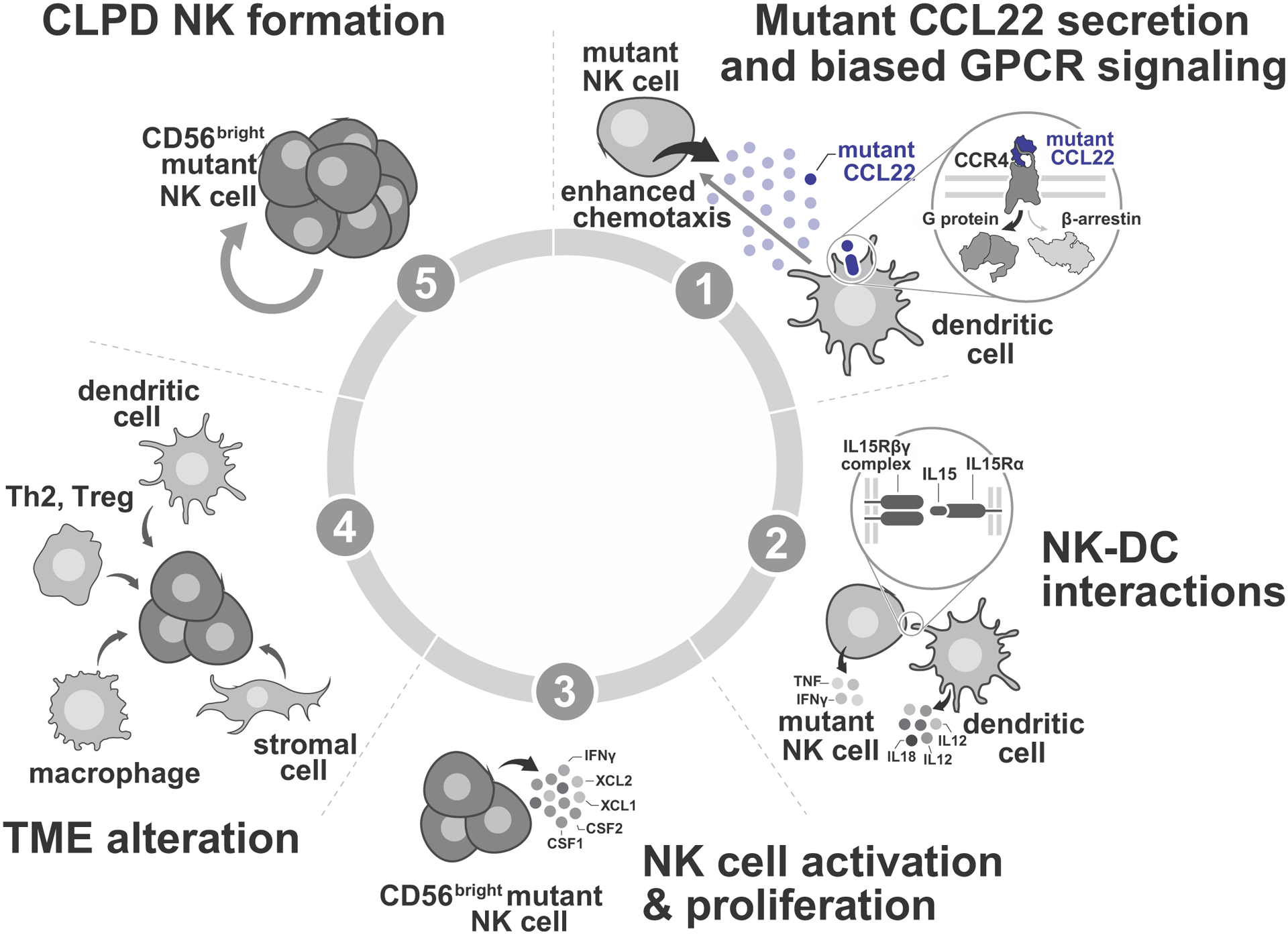
(1) NK cells with CCL22 mutations secret mutant CCL22 protein. The sole receptor for CCL22, CCR4, is expressed by immune effectors including dendritic cells (DC), T helper 2 cells (Th2), regulatory T cells (Treg), and skin-homing T cells21,34,52. No evidence for autocrine effects of CCL22 in CCL22-mutated NK cells was observed in proliferation assays. Instead, mutant CCL22 impairs beta arrestin recruitment and subsequent CCR4 internalization, leading to enhanced chemotaxis effect of CCR4-expressing cells including DC34,62, and ligand biased GPCR signaling. (2) Chemoattraction of DC mediated by the CCL22-CCR4 axis results in direct provision of IL-15 to CCL22-mutated NK cells by cell-to-cell contact23,63. IL-15 is the most important cytokine for NK cell activation, differentiation, and proliferation55,56,64, and bone marrow biopsies of CLPD-NK have shown NK-DC direct interaction26. Additional cytokines (IL-12, IL-18, type 1 interferon (IFN)) are further stimulate NK cells24,56,58. Conversely, stimulated NK cells release TNF and IFN-γ to induce activation of DC, consistent with bidirectional interaction of NK-DC24,58. (3) IL-15 signaling promotes proliferation of NK cells55,56,64. NK cells stimulated by IL-15 acquire CD56bright phenotype24, and release additional cytokines/chemokines including CSF1, CSF2, XCL1, XCL2, and INF-γ11,65,66. (4) Several immune effectors including DC, macrophages, Th2, Treg are attracted by cytokines/chemokines released by activated NK cells to enhance microenvironmental crosstalk11,58,67–69. Stromal cells including endothelial cells, follicular dendritic cells (FDC), marginal zone reticular cells (MRC), and perivascular reticular cells (PRC) also participate in microenvironmental crosstalk47–49. Dysregulated microenvironmental crosstalk induced by cytokines/chemokines released by activated NK cells results in induction of a NK-tropic milieu of other immune cells (DC, macrophages, stromal cells) that provide cytokines (IL-15, IL-2, IL-6, IL-12, IL-18, PDGF) to CCL22 mutated CD56bright NK cells59,70. (5) CCL22 mutated CD56bright NK cells proliferate in response to this NK-tropic milieu of other immune cells (DC, macrophages, stromal cells), leading to CLPD-NK2,3,57,71.
Secondly, mutant CCL22 impairs β-arrestin recruitment and subsequent CCR4 internalization, leading to enhanced chemotaxis of CCR4 expressing cells including DC (Figure 6). Notably, the functional effects of CCL22 mutations were most prominent for the Leu45 and Pro79 mutations which are at residues more broadly conserved among all human chemokines than the Lys90 mutation. This supports a role for biased CCR4/GPCR signaling in the pathogenesis of CLPD-NK via gain-of-function mutations in ligands – an exceedingly rare phenomenon53. The concept of ligand-biased agonism describes the ability of ligands acting at the same G protein-coupled receptor to elicit distinct cellular signaling profiles by stabilizing different active conformational states of the receptor, thereby altering G protein and β-arrestin signaling profiles54. Accordingly, structural modeling of mutant CCL22 predicted enhanced interaction with CCR4, which in turn likely drives ligand biased CCL22-CCR4 signaling.
Third, and consistent with the lack of a direct autocrine effect, chemoattraction of DC by CCL22-CCR4 axis results in direct provision of IL-15 to CCL22-mutated NK cells by cell-to-cell contact. IL-15 is an essential cytokine for development, proliferation, and activation of NK cells and robustly stimulates several pathways including phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) and Janus kinase (JAK)/signal transducer and activator of transcription (STAT), especially in the CD56bright subset of NK cells9,55,56. Excessive stimulation of IL-15 can in turn drive aberrant NK cell proliferation resulting in large granular lymphocyte leukemia57. IL-15 is provided from several cells but primarily from DCs by cell-to-cell contact with NK cells23. In addition to IL-15, several other cytokines (IL-12, IL-18, type 1 interferon (IFN)) are indirectly provided to further stimulate NK cells24,56,58. Conversely, stimulated NK cells release tumor necrosis factor (TNF) and IFN-γ to induce activation of DCs (Figure 6). Cell-to-cell DC/NK cell contact in CLPD-NK patients has been also demonstrated by bone marrow biopsies26. Fourth, NK cells stimulated by IL-15 acquire CD56bright phenotype24, and release additional cytokines/chemokines including CSF1, CSF2, XCL1, XCL2, and INF-γ, resulting in chemoattraction of other immune cells (DC, macrophages, Th2) (Figure 6). This is supported by CD56bright phenotype and higher XCL1/2 and CSF1/2 expression in CLPD-NK samples with CCL22 mutations and transwell migration assay of human normal bone marrow cells by using these cytokines/chemokines. Consistent with this model, our xenograft model successfully recapitulated the phenotype of primary CLPD-NK with CCL22 mutations in dysregulated gene expression and pathways that resemble CD56bright cytokine productive NK cells.
Finally, dysregulated microenvironmental crosstalk induced by cytokines/chemokines released by activated NK cells results in induction of a NK-tropic milieu of other immune cells (DC, macrophages, stromal cells) that further drive proliferation and migration of NK-tropic hematopoietic cells that provide IL-15 to CCL22 mutated CD56bright NK cells, favoring the chronic expansion of NK cells characteristic of CLPD-NK (Figure 6). The immune cell crosstalk induced by CCL22 is supported by an independent study in the context of antibody affinity maturation in germinal centers (GCs). Upon stimulation of CD40, GC B cells upregulate the chemokine CCL22 that binds to CCR4 on follicular helper T (TFH) cells and attract multiple helper cells from a distance to increase productive help. Blockade of TFH cells helps in turn to decrease CCL22 expression by GC B cells59.
Collectively, our genomic and mechanistic observations support a model of leukemogenesis in which a mutated chemokine, CCL22, does not directly perturb gene expression and cellular pathways in cell-intrinsic manner, but promotes a bidirectional NK-immune cell crosstalk which ultimately promotes NK cell expansion. The role of microenvironment in supporting leukemia growth and influencing drug response has been reported in multiple studies60, however this is the unique example of a mutated chemokine promoting tumor expansion.
Our study has generated experimental models to dissect the mechanism of CCL22-mutant CLPD-NK, and specifically, direct evidence of NK-microenvironment interaction mediated by mutant CCL22. However, a limitation was the lack of primary patient samples, including bone marrow and skin biopsies, and patient-derived xenografts, to perform additional studies to further support the roles of these cellular mediators of microenvironmental crosstalk implicated by our studies using normal bone marrow. Further studies in this vein will provide additional mechanistic insight and enable testing of strategies disrupting this interaction as a therapeutic approach.
In conclusion, we showed that somatic mutations in the chemokine CCL22 define a distinct subgroup of CLPD-NK resembling the immunophenotype and gene expression of CD56bright normal NK cells, and driven by biased G protein-coupled receptor signaling and pathogenic dysregulation of crosstalk between CCL22-mutated NK cells and the NK-tropic hematopoietic microenvironment.
Online Methods
Patients and primary material preparation
Fifty-nine (discovery cohort) and 62 (validation cohort16) patients with CLPD-NK (Supplementary Table 1) were studied. All patients provided written informed consent in accordance with the Declaration of Helsinki. Compensation was not provided to patients. The study was approved by the Institutional Review Board of St. Jude Children’s Research Hospital (XPD13-068) and the Internal Review Board and by the Bavarian Ethics Commitee, the Bavarian State Medical Association (Bayerische Landesärztekammer) with the number 05117. All human subjects in validation cohort were consented and samples were studied under IRB-approved protocols for the LGL Leukemia Registry at the University of Virginia (IRBHSR#17000 “Large Granular Lymphocyte Leukemia Registry” and IRB #17070 “Pathogenesis of Large Granular Lymphocyte Leukemia”).
For the discovery cohort, cases were selected retrospectively from sample repository of Munich Leukemia Laboratory (MLL) based on (i) suspected lymphoid neoplasm by referring physician due to persistent lymphocytosis and (ii) increased NK cell percentage confirmed by flow cytometric analysis at MLL. For immunophenotyping, peripheral blood or bone marrow samples were each separated into three tubes for staining with different sets of antibodies. All samples were subjected to erythrocyte lysis. For intracellular staining, samples were fixed and permeabilized. The samples were then stained with various antibodies at room temperature in the dark for 15–20 min, washed 1x in PBS and measured on Navios flow cytometers (Beckman Coulter, Miami, FL). Analysis was conducted using Kaluza analysis software (Beckman Coulter, Miami, FL). Besides forward and side scatter signals the following antigens were analyzed according to the manufacturer’s instructions: CD1a, CD2, sCD3, cyCD3, CD4, CD5, CD7, CD8, CD16, CD19, CD22, CD30, CD34, CD45, CD56, CD57, CD79a, CD79b, TCRab, TCRgd, MPO, TdT and IgM. Detailed antibody list with dilution is shown in Supplementary table 13. For the validation cohort, a diagnosis of CLPD-NK was made according to one or more of the following criteria; greater than 2 × 109/L atypical lymphocytes in peripheral blood, evidence of NK cell invasion of the marrow, and aberrant NK populations detected by flow cytometry.
DNA and RNA were isolated from peripheral blood or bone marrow samples according to standard protocols (MagNA Pure Systeme, Roche, Mannheim/Penzberg, Germany) or PBMC DNA was extracted by magnetic bead isolation (Anaprep, BioChain Institute). For sorting, CD45/SSC was used to gate on lymphoid cells. CD56 positive CD3 negative cells (NK cells) and CD56 negative CD3 positive cells (T-cells) were separately sorted for further molecular genetic analysis in two cases with CCL22 mutations.
Data analysis of whole genome and targeted sequencing
Whole genome sequencing (WGS) data analysis was conducted on Illumina’s BaseSpace Sequence Hub (alignment: human genome build GRCh37/hg19 with Isaac3).72 A pool of sex-matched genomic DNA (Promega, Madison, WI) was used for unmatched normal variant calling with Strelka2.73 For the initial screening we considered coding variant with a VAF >15%, and a global population frequency ≤0.00005 (genome aggregation database; gnomAD).74,75 Non-protein truncating single nucleotide variant were included, if the in silico prediction by HePPy was ≥0.5.76 To test the presence of CCL22 mutations in other hematological malignancies, we used our MLL 5K cohort, which was sequenced and analyzed under the same conditions77. For analysis of somatic mutations besides STAT3 and CCL22, we analyzed known hematologic gens from panel sequencing or WGS and included manual variant curation based on the following databases: COSMIC DB (v91), UMD TP53 DB (2017_R2), ClinVar (2020-03) gnomAD (non-cancer, v2.1.1). The schema of data analysis was shown in Supplementary figure 1. Detected mutations were visualized using GenomePaint78.
Analysis of whole transcriptome sequencing
Paired-end reads from whole transcriptome sequencing of 59 CLPD-NK cases and 12 normal NK samples79 (GSE133383, 6 CD56bright, 6 CD56dim) were mapped and aligned to human reference genome (GRCh37) by using STAR80. Gene-level read counts were generated using HTseq-count81 and normalized using DESeq2 Bioconductor R package61. A regularized log-transformed (rlog) value was calculated by variance-stabilizing transformation (VST) of DESeq2. The ComBat function in the sva R package82 was used to correct the batch effect due to different library preparation methods. Differentially expressed genes (adjusted P value <0.01 and log2 fold change >1.5) were extracted by DESeq2 with default parameters and subsequently imported to the gene signature analysis and pathway analysis with Metascape83. Gene-set enrichment and pathway analysis was carried out by using GSEA84. To perform GSEA, we used normalized gene counts and log2_Ratio_of_Classes for the metric for ranking genes with mSigDB C2 all sets or custom gene sets generated by differentially expressed genes between CCL22 mutant and non-CCL22/STAT3 mutant (Supplementary table 11).
Cell culture and proliferation assays
The Ba/F3, 293T, K562 and NK-92 cells were purchased from the American Type Culture Collection (#HB-283, #CRL-3216, #CCL-243, ATCC, Manassas, VA) or Leibniz Institute DSMZ (#ACC 488). The cell lines were authenticated by Short Tandem Repeat (STR) analysis. Ba/F3 cells were cultured in RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS, Cytiva, #SH30396.03HI), 100 U/mL penicillin-streptomycin, 2 mM L-glutamine (Gibco, #10378016), and 10 ng/mL recombinant murine IL-3 (Peprotech, #213-13). HEK 293T cells were cultured in Dulbecco’s modified Eagle’s Medium (BioWhittaker, #BW12-614F) with 10% FBS and L-glutamine, 100 U/mL penicillin-streptomycin. The K562 cells were cultured in Iscove’s Modified Dulbecco’s Medium (Gibco, #12440-053) supplemented with 10% heat-inactivated FBS, and 100 U/mL penicillin-streptomycin. The NK-92 and NKL cell lines were cultured in Minimum Essential Medium Eagle - alpha modification (STEMCELL Technologies, #36463) supplemented with 12.5% heat-inactivated FBS, 12.5% heat-inactivated horse serum (Gibco, #16050122), and 100 U/mL penicillin-streptomycin in the presence of 100 (NK-92) and 500 (NKL) U/mL recombinant human IL-2 (Protech, #200-2) at 37°C and 5% carbon dioxide. To evaluate cytokine-independent proliferation and growth advantage in presence of cytokine, cells were washed two times with phosphate-buffered saline (PBS) and seeded in triplicate at the concentrations of 0.2 × 106 cells/well in 24-wells plate with or without a cytokine. mIL-3 (10ng/mL), hIL-2 (100 U/mL), mIL-15 (Peprotech, #210-15, 10 ng/mL), hIL-15 (Peprotech, #200-15, 10 ng/mL), and hIL-18 (50 ng/mL) were added accordingly. Cell proliferation and viability were examined every two or three days using trypan blue and a TC10 cell counter (Bio Rad).
Structural analysis of CCL22 mutations
Structural locations of mutated CCL22 residues were mapped on to the structures of CCL5[5P7]-CCR5 complex (PDB 5UIW) or models of the WT-CCL5-CCR5 complex from the same study28. Structurally equivalent positions between CCL22 and WT-CCL5 or CCL5[5P7] were identified from a sequence alignment of human chemokine paralogs that was generated via a hybrid sequence- and structure-based sequence alignment approach employing both MUSTANG and Clustal Omega85,86. Human paralog chemokine sequences were obtained from Uniprot. CCL22 ortholog sequences were obtained from the Orthologous MAtrix (OMA) database (www.omabrowser.org). Conservation scoring among (1) human chemokine paralogs and (2) CCL22 orthologs was performed using MstatX using default settings and the trident scoring algorithm (https://github.com/gcollet/MstatX)87. Endogenous and mutated CCL22 and CCR4 residues were mapped on the CCL5[5P7]-CCR5 complex or WT-CCL5-CCR5 model using the “Mutagenesis” tool in PyMol. Sequence “logos” were generated using the human chemokine paralog sequence alignment using the ggseqlogo package in R88.
Internalization assay
Ba/F3 cells expressing wild-type CCR4 (Ba/F3-CCR4) were seeded in triplicate at a concentration of 1 × 106 cells/mL and incubated with increasing dose (ranging from 10 to 1,000 ng/mL) of wild type or mutant CCL22 (Lys90Argfs, Pro79Arg, Leu45Arg) for 30 minutes (min) at 37°C. In a time-course assay, cells were incubated with 50 ng/mL of wild type or mutant CCL22 from 5 min to 24 hours. For serum swapping assay, serum of CCL22 wild type or mutant (Pro79Arg,Leu45Arg) expressing Ba/F3-CCR4 cells was collected after 24 hour incubation at a concentration of 1 × 106 cells/mL. Cells were washed twice with PBS and incubated at 37C for 60 min with indicated serum (wild type to mutant, mutant to wild type) at a concentration of 1 × 106 cells/mL. After these treatments, cells were washed and incubated with PE-conjugated anti-human CD194 (CCR4) antibody (BD, #561110) at 4°C for 30 min according to the manufacturer’s instructions. Surface CCR4 expression levels were immediately evaluated by flow cytometry on LSR II flow cytometer (BD Biosciences) using DIVA software (BD Biosciences), and analyzed with FlowJo v.10.0 (Tree Star). CCR4 internalization rate was calculated by the difference from positive control (Ba/F3-CCR4 without treatment; 0%) and negative control (Ba/F3 and Ba/F3-CCR4 with isotype; 100%).
Chemotaxis assay
Chemotaxis of Ba/F3-CCR4 cells towards wild type or mutant CCL22 (Lys90Argfs, Pro79Arg, Leu45Arg) was measured by using 5 μm transwell plates of ChemoTx Disposable Chemotaxis System (Neuro Probe, #106-5) and Transwell® 24-well plate with 5.0 μm pore (Coster, #3421) according to the manufacturer’s instructions. Cell culture medium was loaded into the lower wells with 10 ng/mL, 100 ng/mL, and 1000 ng/mL of wild type, mutant CCL22. Ba/F3-CCR4 cells at a concentration of 1 × 106 cells/mL were placed above the filter. Wells incubated without chemokine or input cells were set as background control. Chemotaxis assay was performed at least in triplicate. After 6 hours of incubation at 37°C, migrated cells in each condition were evaluated by Resazurin Cell Viability assay (abcam, #ab129732) according to the manufacturer’s instructions.
Bone marrow mononuclear cells were tested for chemotaxis toward mixture of M-CSF (Peprotech, #300-25, 200 ng/mL), GM-CSF (Peprotech, #300-03, 2 ng/mL), XCL1 (Peprotech, #300-20, 100 ng/mL), XCL2 (Novus Biologicals, #8418-X2-025, 50 ng/mL) by using Transwell® 6-well plate with 8.0 μm pore (Thermo Scientific, #140668) according to the manufacturer’s instructions. After overnight incubation at 37°C, migrated cells were examined by flow cytometry. Besides forward and side scatter signals the following antigens were analyzed according to the manufacturer’s instructions: CD45, CD3, CD19, CD33, CD56, CD16, CCR4, CCR6, CCR7, CD103, CD10, CD117, CD11b, CD11c, CD123, CD124, CD13, CD14, CD141, CD157, CD15, CD16, CD16/32 (Fc block), CD1c, CD20, CD27, CD317, CD31, CD34, CD35, CD4, CD8a, CD69, CD7, CD95, CXCR4, CXCR5, F4/80, HLA-DR, Ia/Ie, LDA, NKp46, Ly6c, Ly6g, MADCAM-1, NKG2A, NKG2D, PD-1, PDGFRa, PD-L1, PDPN, Sca-1, CD172, TCRgd, TER119, and XCR1. A detailed antibody list with dilutions is shown in Supplementary table 13.
Immunoblotting
Ba/F3-CCR4 cells at a concentration of 2 × 106 cells/mL were starved in medium containing RPMI 1640 supplemented with 2% BSA, 50 μM 2-Mercaptoethanol, 2 mM L-glutamine, and 100U/mL penicillin-streptomycin (starvation medium) for 24 hours before treatment with 100 ng/mL of wild type or mutant CCL22 (Lys90Argfs, Pro79Arg, Leu45Arg) for 5 min. Treated Ba/F3-CCR4 cells or engineered NK-92 cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors (Thermo Scientific, #1861281). For Western blotting, 20 μg protein of cell lysate was electrophoresed through 4–12% NuPage Bis-Tris gels (Life Technologies) at 110 V for 100 min. Blots were probed with anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (Cell Signaling, #9101), anti-Phospho-Akt (Ser473) (Cell Signaling, #4060), anti-Granzyme K (Thermo Fisher, #PA5-50980), anti- Phospho-Stat3 (Tyr705) (D3A7) (Cell Signaling, # 9145S), and anti-beta actin (C4) (Santa Cruz Biotechnology, #sc-47778) antibodies. For imaging and signal quantification, Odyssey DLx (LI-COR) and Image Studio (LI-COR) were used. To evaluate expression of each condition, relative expression was calculated by comparing the signal of beta actin for each condition.
Phosphoflow cytometry
Intracellular signaling after treatment with wild type or mutant CCL22 (Lys90Argfs, Pro79Arg, Leu45Arg) was assessed in Ba/F3-CCR4 cells. Cells were seeded at a concentration of 2 × 106 cells/mL and starved with starvation medium for 24 h. After the treatment with 100 ng/mL of wild type or mutant CCL22 for 5 min to 24h, cells were immediately fixed and permeabilized with BD™ Phosflow Fix Buffer I (BD). Fixed cells were stained with Alexa Fluor 647-conjugated anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (BD, #561992) or Alexa Fluor 647-conjugated anti-Phospho-Akt (Ser473) (BD, #560343) antibodies according to the manufacturer’s instructions. Cellular fluorescence data were collected on an LSR II flow cytometer (BD Biosciences) using DIVA software (BD Biosciences), and analyzed with FlowJo v.10.0 (Tree Star).
Phosphoproteomic Detection via IsoLight
Ba/F3-CCR4 cells at a concentration of 2 × 106 cells/mL were starved in medium containing RPMI 1640 supplemented with 2% BSA, 50 μM 2-Mercaptoethanol, 2 mM L-glutamine, and 100U/mL penicillin-streptomycin (starvation medium) for 24 hours before treatment with 100 ng/mL of wild type or mutant CCL22 (Lys90Argfs, Pro79Arg, Leu45Arg) for 3 hours. Briefly, 200,000 cells were labeled with CellTrace Violet 1:500 (Thermo Fisher Scientific, Waltham, MA, USA). Cells were removed and immediately loaded onto an IsoLight single-cell proteome chip (IsoPlexis, Branford, #PANEL-4L01-4), according to the manufacture’s instruction. The IsoLight single-cell proteome chip detects 16 distinct phosphoproteins89, 13 of which are cross-reactive to mouse proteins. Results were analyzed using IsoSpeak v2.8.0.0.
NK cytotoxicity assay
K562 cells as the target (T) cells were incubated with CellTrace™ Far Red Cell Proliferation Kit (Thermo Fisher Scientific, #C34564) and 0.5 × 105 K562 cells were seeded on 96 well plate, according to the manufacture’s instruction. NK-92 cells as effector (E) cells expressing wild type CCL22-IRES-GFP, mutant CCL22-IRES-GFP (Pro79Arg), and empty vector (GFP) were added to have appropriate E to T ratio (3:1, 1:1, 0.3:1, and 0.1:1) and incubated in a medium containing Minimum Essential Medium Eagle - alpha modification supplemented with 12.5% heat-inactivated FBS, 12.5% heat-inactivated horse serum, and 100 U/mL penicillin-streptomycin in the presence of 500 U/ml recombinant human IL-2 at 37°C for overnight. Far Red positive cells (live cells) were analyzed by flowcytometry and the extent (%) of killing was calculated by using the ratio of live cells in test wells (T and E) to control wells (T only, no E). CountBright™ Absolute Counting Beads (Thermo Fisher Scientific, #C36950) were used to normalize data.
Mouse modeling of CCL22-mutated NK cells
Mice were housed in an American Association of Laboratory Animal Care-accredited facility and were treated on Institutional Animal Care and Use Committee-approved protocols in accordance with NIH guidelines. The study was approved by institutional review board of St Jude Children’s Research Hospital. All mice were housed in sterilized conditions at 20–23 °C and 40–60% humidity, and applied a 12-h light–12-h dark cycle. Human NK NK-92 cells (0.5 × 106 cells) expressing wild type CCL22-IRES-GFP, mutant CCL22-IRES-GFP (Pro79Arg), and empty vector (GFP) were transplanted by tail vein injection into 8–10 week-old female immunodeficient NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ(NOD-SCID gamma-null, or NSG) mice90 or NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(IL15)1Sz/SzJ mice (The Jackson Laboratory, #030890) which continuously express human IL-15. To monitor the development of NK cell proliferation in transplanted mice, flow cytometric analysis of peripheral blood cells stained for mCD45-APC-Cy7 (BD, #557659), hCD45-PerCP (BD, #347464), hCD19-BV605 (BD, #740394), hCD3-PE (BD, #347347), hCD33-PE-Cy7 (BD, #333946), hCD56-Alexa Fluor 647 (BD, #557711), and hCD16-Alexa Fluor 700 (BD, #557920) were performed every two or four weeks. Animals that became moribund or reached the defined endpoint (batch 1, n=9, day 57; batch 2, n=18, day 44) were killed, and blood, bone marrow and spleen samples were analyzed for evidence of NK cell proliferation by using flow cytometry and histopathologic analyses. Post-mortem flow analysis for a panel of multilineage markers was performed as described above, and the GFP-positive population was used to calculate the rate of engrafted cells in each sample. Engrafted transduced human NK-92 cells were isolated with EasySep Mouse/Human Chimera Isolation Kit (STEMCELL Technologies, #19849A). RNA was extracted from isolated human engrafted cells by AllPrep DNA/RNA Mini kit (Qiagen, #80204) according to the manufacturer’s instructions and subsequently processed for RNA-seq. Sequenced RNA-seq data were mapped to the GRCh37 reference human genome assembly by using STAR80. Gene-level read counts were generated using After generating gene-level read counts by HTseq-count81, normalization and calculation of a rlog value by VST were performed by DESeq2 Bioconductor R package61. The top 400 most variable genes (on the basis of median absolute deviation) were mapped into a two-dimensional tSNE plot (the R package Rtsne with perplexity parameter 2). Differentially expressed genes (adjusted P value <0.01 and log2 fold change >1) were extracted by DESeq2. Gene-set enrichment and pathway analysis was carried out by using GSEA84 with mSigDB C2 gene sets or originally generated gene sets (Supplementary table 9) from the differentially expressed gene analysis from primary CLPD-NK samples between CCL22 mutant and others.
Statistics and reproducibility
Statistical analyses were performed using R v3.6.3 software. Categorical variables were analyzed using Fisher’s exact test with correction of P values for multiple testing (Holm91). Associations between continuous variables were compared by the one-way ANOVA with Tukey’s honestly significant difference post hoc test. Two-sided Student’s t-test was used to compare mean values of different groups. P values, adjusted P value, and/or q value of differentially expressed gene analysis and pathway analysis were analyzed by using DESeq2 Bioconductor R package61, Metascape83, and GSEA84. The results were considered statistically significant at P < 0.05. Unless indicated otherwise, each experiment was independently repeated twice and representative results are shown. No statistical method was used to predetermine the sample size but we included all samples applicable and our sample sizes are similar to those reported in previous publications7,15,16. No data were excluded from the analyses. Data distribution was assumed to be normal but this was not formally tested. Randomization was not used for in vivo modeling experiments. The investigators were not blinded to allocation during experiments and outcome assessment.
Extended Data
Extended Data Fig. 1. The mutational landscape and clinical parameters CLPD-NK patients.
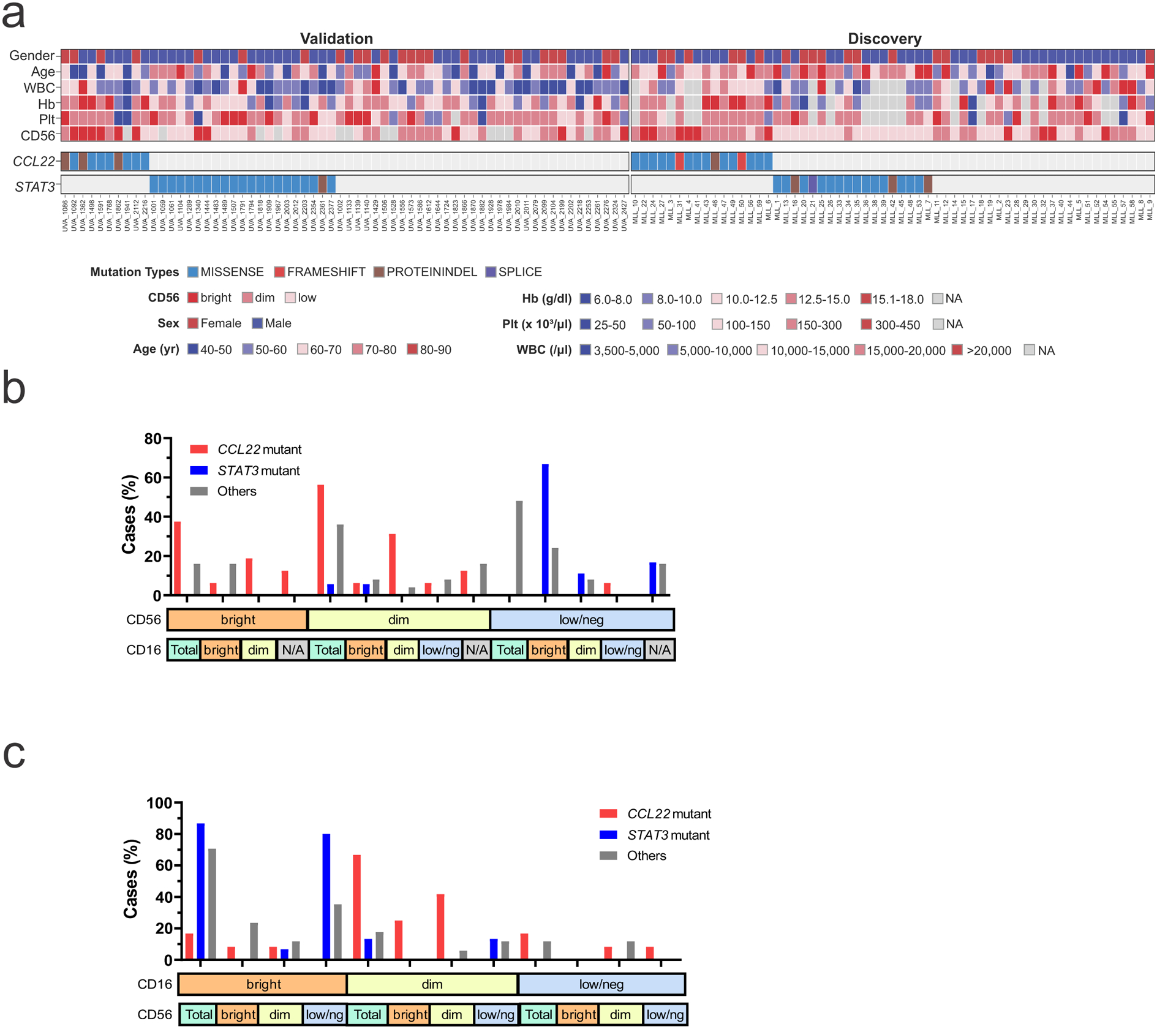
a, Heatmap of the 59 cases with CLPD-NK in the discovery cohort and 62 CLPD-NK cases in the validation cohort. CCL22 mutations were detected in 16 out of 59 (27%) and 10 out of 62 (16%) cases and were mutually exclusive of STAT3 mutations. Higher CD56 expression was observed in CCL22 mutant cases. b,c, Distribution of CD56 and CD16 expression in the discovery cohort. Hb, hemoglobin; Plt, platelet; WBC, white blood cell; yr, year.
Extended Data Fig. 2. Gene expression profiling of 59 CLPD-NK cases and 12 normal NK samples (6 CD56bright, 6 CD56dim).
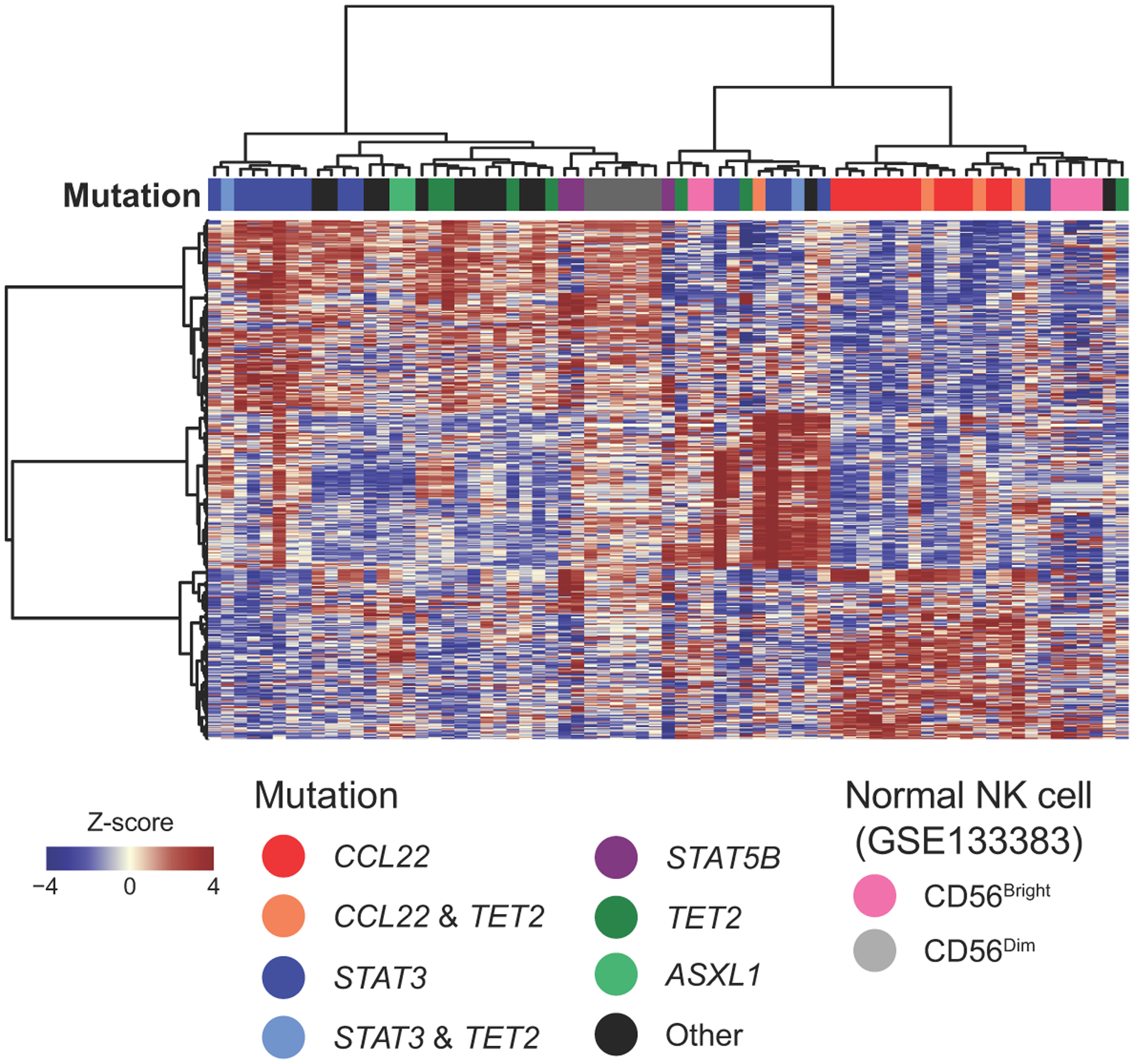
A total of 495 differentially expressed genes between CCL22 mutant vs other cases, STAT3 mutant vs other cases, and non-CCL22/STAT3 mutant vs other cases were used for the hierarchical clustering. CCL22 mutant cases were clustered with CD56bright normal NK cells.
Extended Data Fig. 3. CCL-22-Pro79Arg transduced NK-92 recapitulated primary CLPD-NK samples with CCL22 mutations in the expression of several important genes.
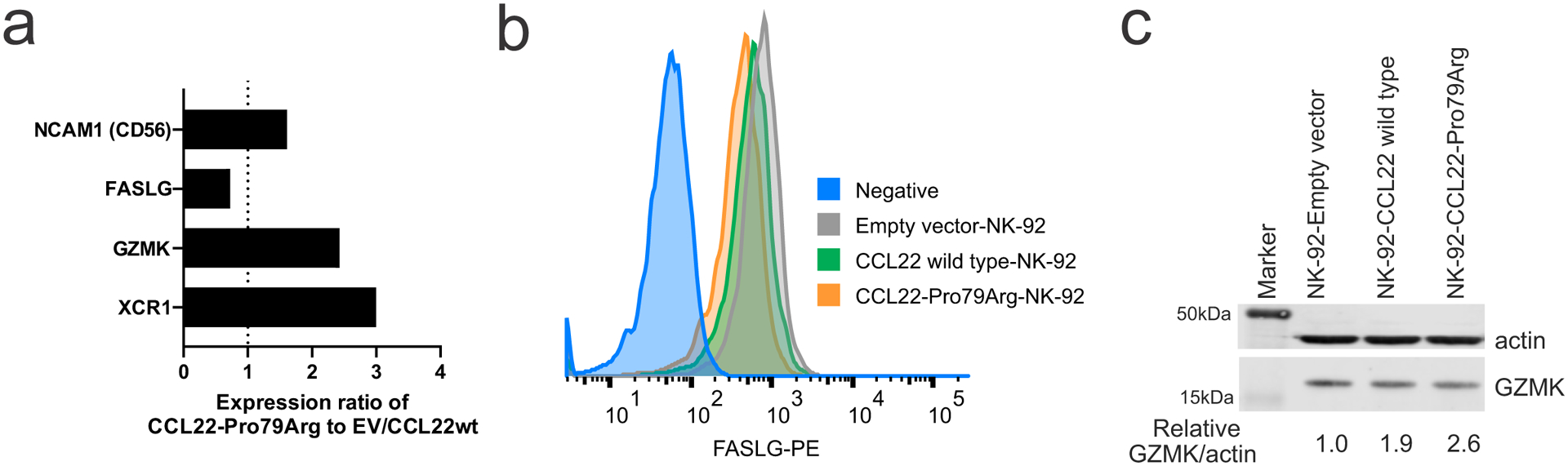
a, Gene expression ratio of CCL22-Pro79Arg NK-92 cells compared to CCL22-wild type (wt) and empty vector (EV)- transduced NK-92 cells. b, Reduced FASLG expression of CCL22-Pro79Arg NK-92 cells compared to CCL22wt and EV NK-92 cells was confirmed by flow cytometry. CCL22-Pro79Arg NK-92 cells without staining is shown as Negative. c, Immunoblotting confirmed higher GZMK expression in engrafted CCL22-Pro79Arg NK-92 cells. The samples derive from the same experiment and were processed in parallel.
Extended Data Fig. 4. Comparison of growth advantage in CCL22-Pro79arg, wild type, and empty vector transduced cells.
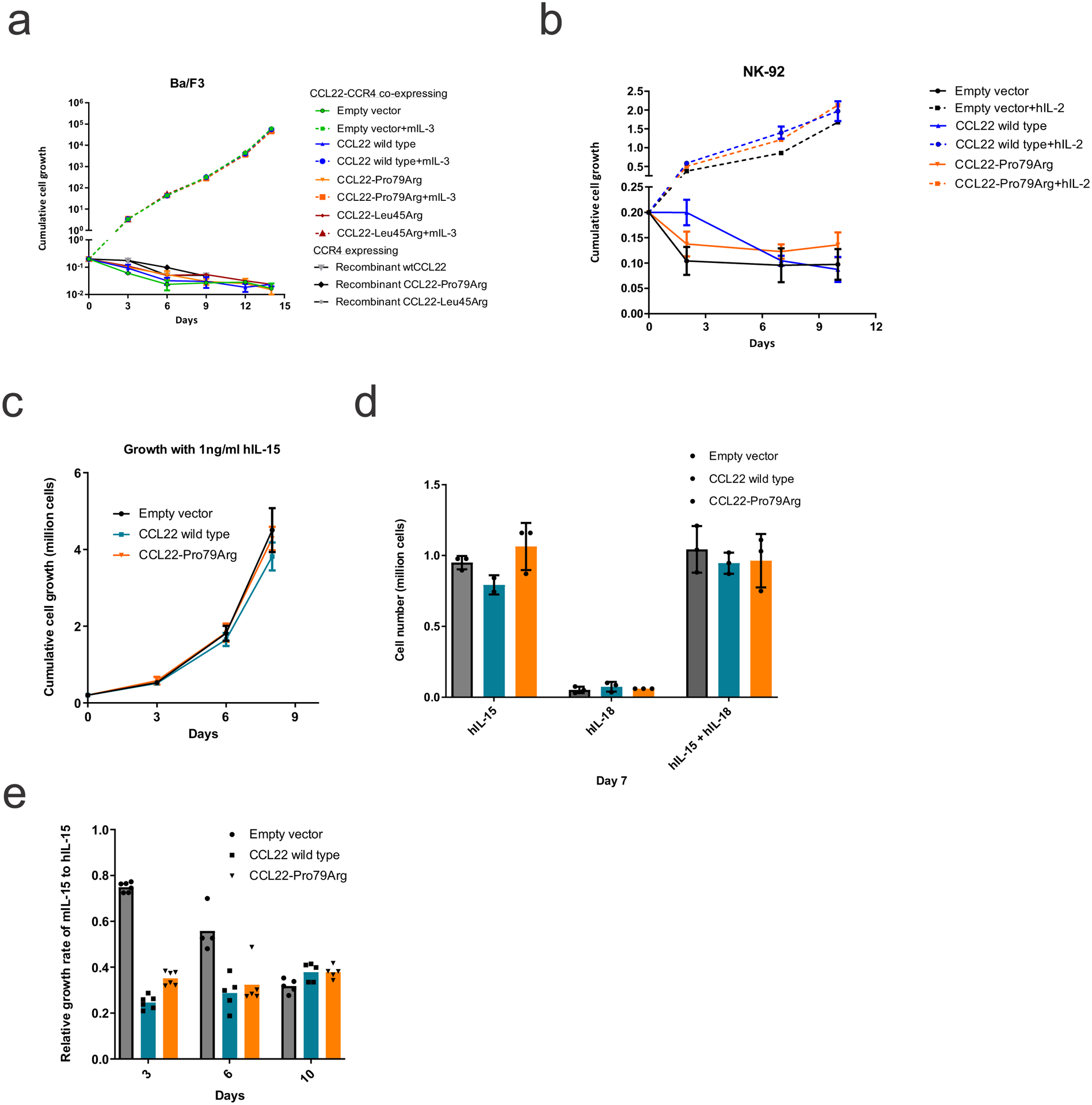
Cytokine independent assay in CCR4 expressing (a) Ba/F3 cells with murine Il-3 or recombinant CCL22 proteins and (b) NK-92 cells with human IL-2, co-expressing wild type or mutant CCL22 (Pro79Arg, Leu45Arg), or GFP-expressing lentiviral vector (Empty vector). No cytokine independent growth was observed. The mean (± SD) cell count (x 1,000,000) is shown (n=3). c,d, No growth advantage was observed by adding hIL-15 and/or hIL-18 to engineered NK-92 cells. The mean (± SD) cell count (x 1,000,000) is shown (n=3). e, Recombinant murine IL-15 showed cross-reactivity to human engineered NK-92 cells with lower efficiency than human IL-15. The mean (± SD) relative proliferation rate of murine IL-15 compared to human IL-15 is shown (n=6, except for empty vector at day6 n=4 due to technical issue).
Extended Data Fig. 5. Inferring likely functional impact of the mutations on CCL22-CCR4 interactions.
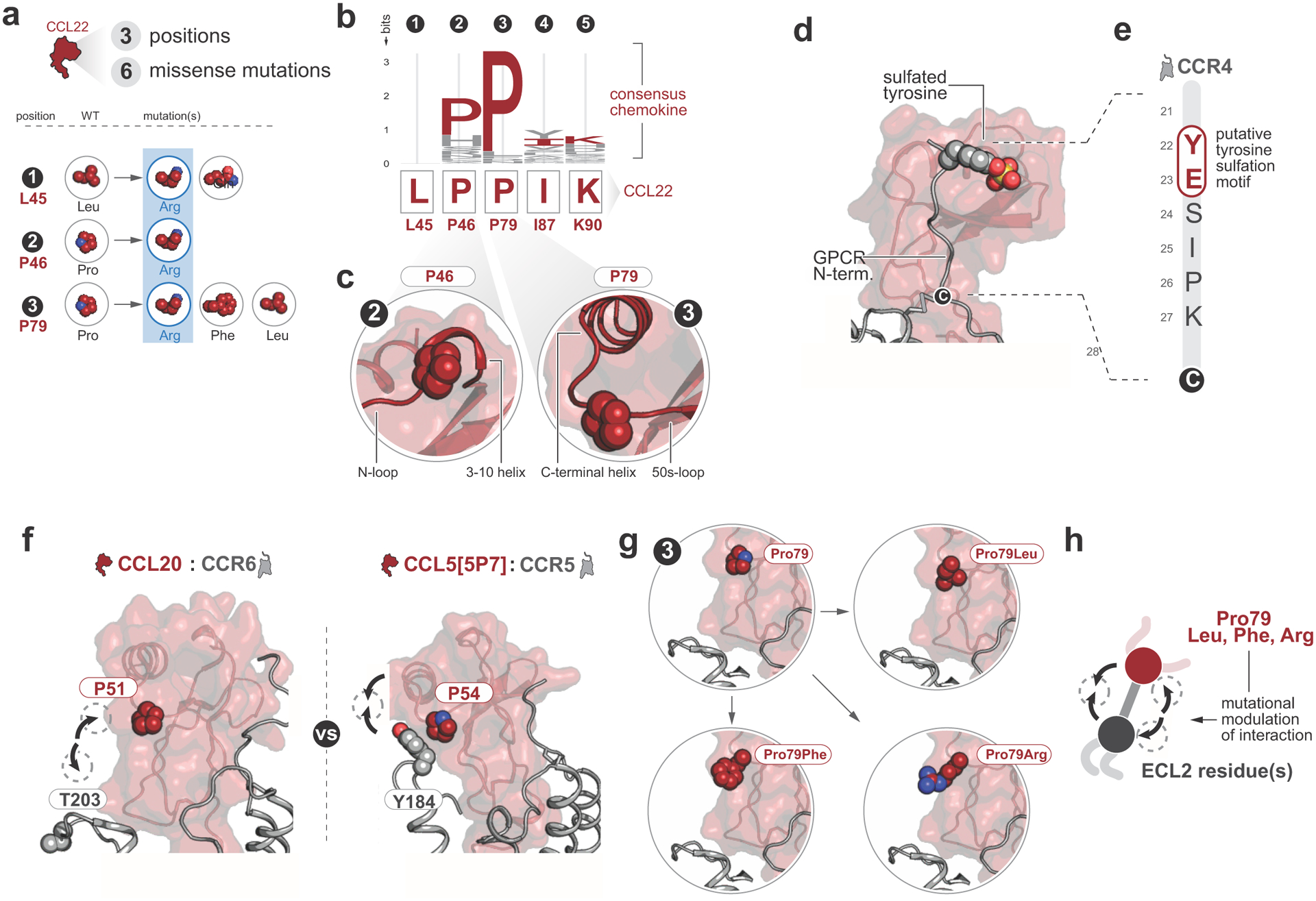
a, 6 missense mutations were identified in CCL22 at 3 positions. The positions at which each mutation occurs are indicated with numbers 1–3, and residue identities for both WT and mutated residues are indicated. Basic residues are highlighted with blue. Mutations of Leu45Arg, Pro46Arg, and Pro79Arg were substitutions to basic residues, suggesting that additional basic residues might modulate chemokine-glycosaminoglycans interactions. b,c, Sequence logo depicting residue conservation among human chemokine paralogs at the five mutated positions. Pro46 and Pro79 were well conserved among all human chemokine paralogs. Structural context of these two conserved Proline residues relative to the chemokine secondary structure: both Prolines occur in loops adjacent to alpha helices and likely facilitate loop turns that orient the adjacent alpha helices. d, Chemokine-GPCR model of the CCL5-CCR5 complex depicting a sulfated Tyrosine in the chemokine receptor N-terminus. Tyrosine sulfation is a common modification of chemokine receptor N-termini with significant effects on chemokine-GPCR interactions and functional properties. e, CCR4 N-terminus with putative sulfotyrosine motif labeled. Chemokine receptors are known to undergo sulfation at tyrosine residues that are adjacent to acidic residues in chemokine receptor N-termini, and sulfated tyrosine residues enhance chemokine-GPCR interactions. f, Comparison of CCL20-CCR6 and CCL5[5P7]-CCR5 chemokine-GPCR complexes reveals that the residue corresponding to CCL22 P79 interacts with the receptor ECL2 in the CCL5[5P7]-CCR5 complex but not the CCL20-CCR6 complex due to different orientations of the chemokine and receptor relative to one another (arrows). g,h, Mutation of the CCL22 residue at P79 may alter the ability of CCL22 to interact with CCR4 at extracellular loop 2. The CCR4 extracellular loop 2 is rich in hydroxyl-containing residues, such that Pro79Arg mutation might be expected to enhance interactions with CCR4 extracellular loop 2 whereas Pro79Leu or Pro79Phe mutations might have minimal or inhibitory effects on CCL22-CCR4 interactions relative to the endogenous Pro79.
Extended Data Fig. 6. Internalization assay with exogenous recombinant wild type or mutant CCL22 in Ba/F3-CCR4 cells.
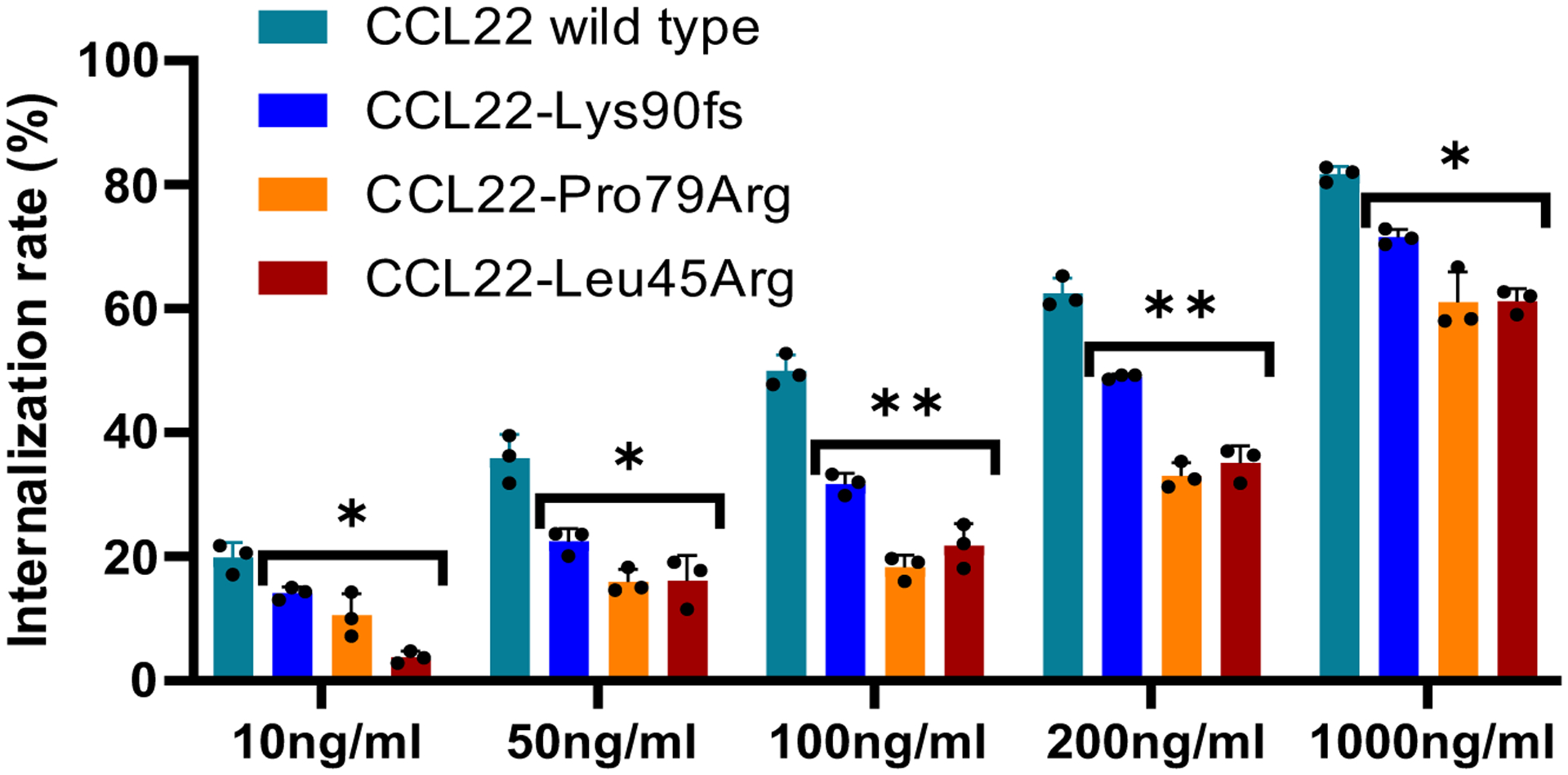
Both exogenous recombinant wild type and mutant CCL22 internalize CCR4 dose dependently. Mutant CCL22 showed weaker effect than wild type CCL22 in all the tested condition. The mean (± SD) difference from positive control (Ba/F3-CCR4 without treatment; 0%) and negative control (Ba/F3 and Ba/F3-CCR4 with isotype; 100%) is shown (n=3). P value was calculated by t-test. *0.01 < p < 0.05; **0.001 < p < 0.01.
Extended Data Fig. 7. Paracrine mechanism with endogenous CCL22 induced internalization of CCR4.
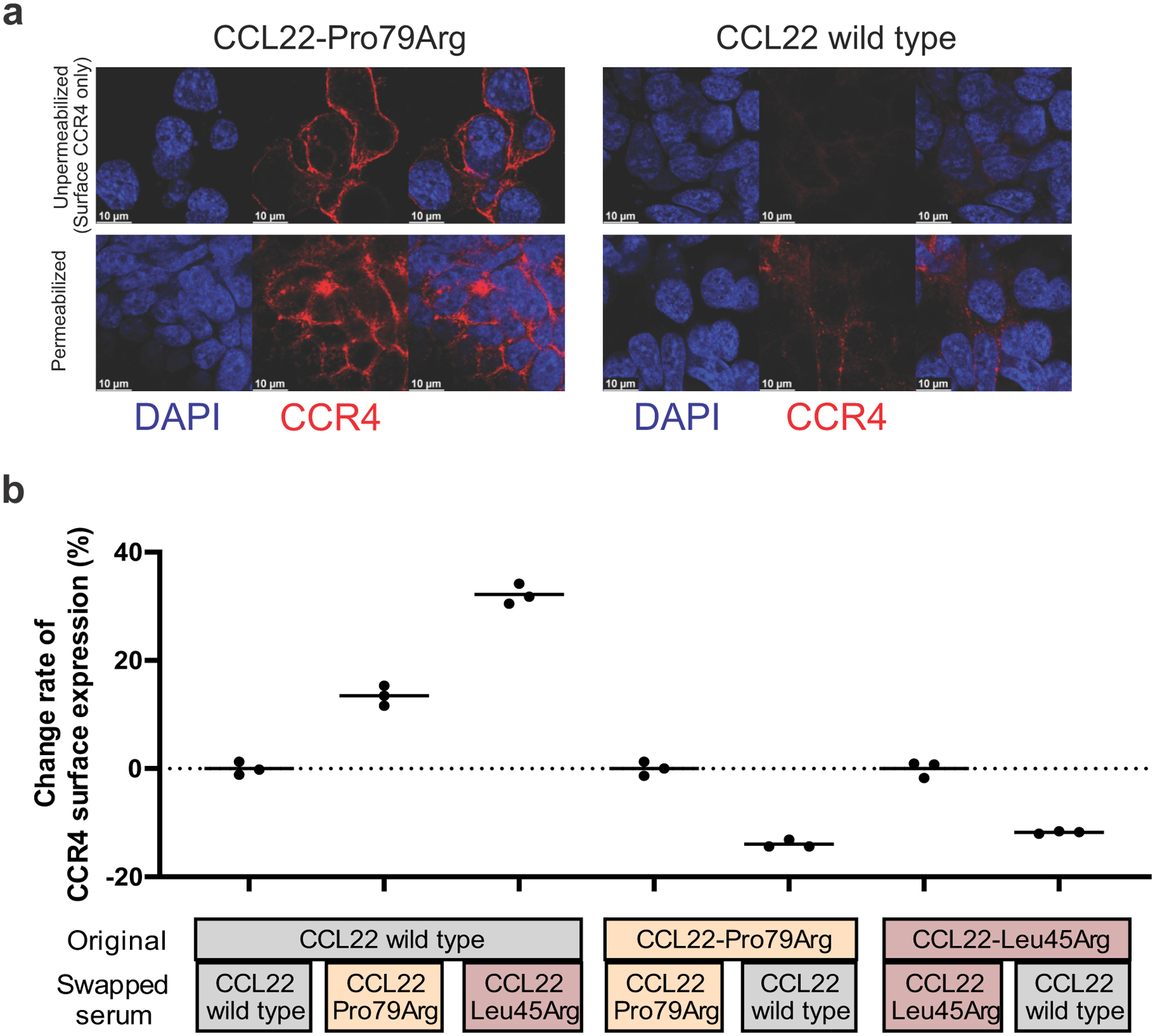
a, Immunofluorescence showing (scale bar 10 μm) the difference of CCR4 internalization between CCL22-Pro79Arg and wild type in unpermeabilized and permeabilized condition. CCR4-expressing 293T cells were transiently transfected with the vector containing CCL22 wild type or Pro79Arg. Unpermeabilized condition only detect surface CCR4, while permeabilized condition can detect both surface and internalized CCR4. Surface CCR4 expression was decreased in wild type CCL22 but not in Pro79Arg-CCL22 mutation after 48 hours incubation. b, Serum swapping assay showing change of CCR4 expression by replacing serum of CCL22 wild type and mutant (Pro79Arg, Leu45Arg) expressing Ba/F3-CCR4 cells. Decreased CCR4 internalization (increased CCR4 expression) was observed by replacing serum of mutant CCL22 from that of CCL22 wild type, and vice versa. The mean (± SD) change rate from serum non-swapped data is shown (n=3).
Extended Data Fig. 8. CCL22-CCR4 downstream signaling.
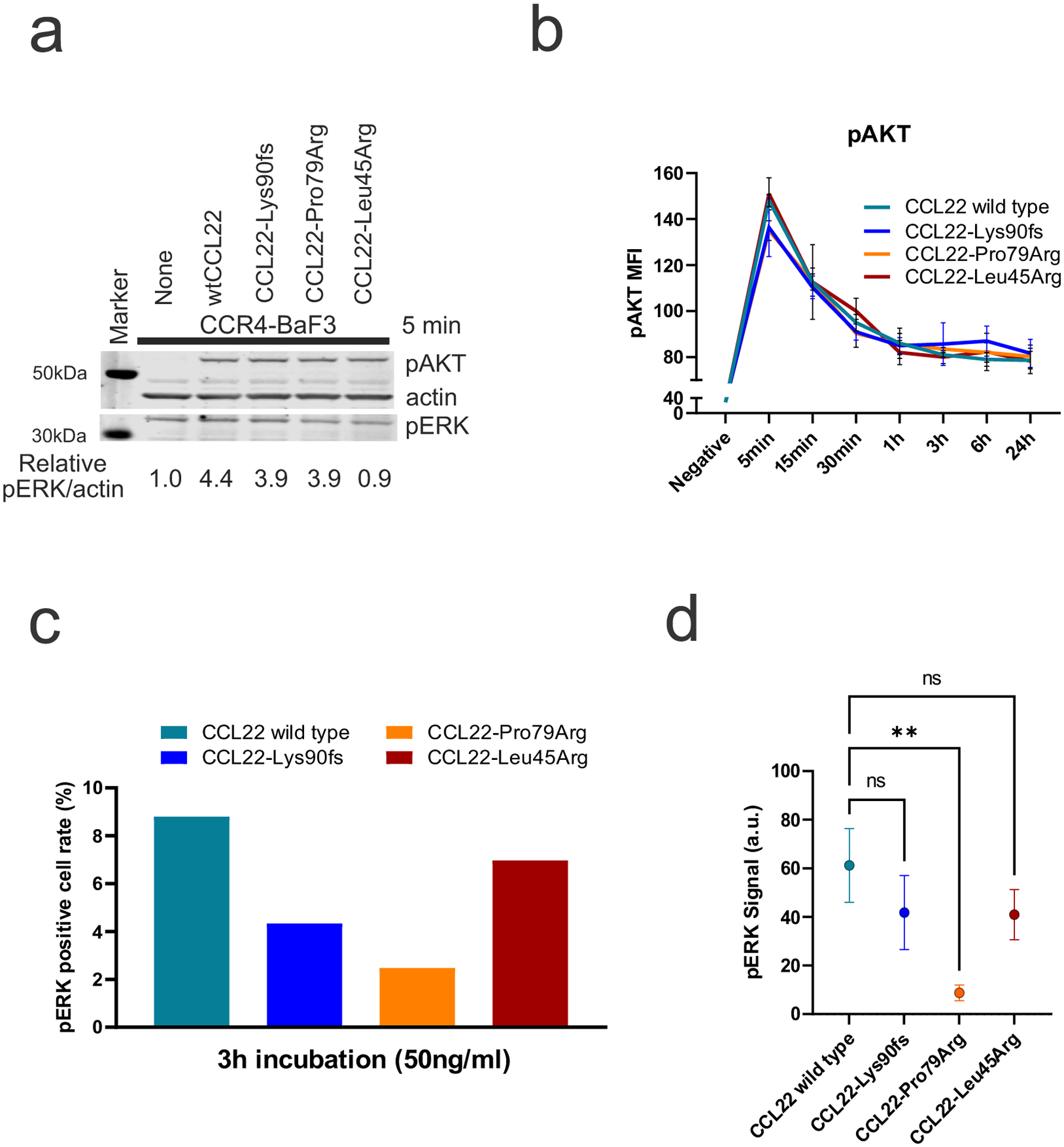
a, Phosphorylation of AKT, and mitogen-activated protein kinase (MAPK) (p44/42 extracellular signal-regulated kinase (Erk) 1/2) by immunoblotting. Ba/F3-CCR4 cells were stimulated with exogenous recombinant wild type or mutant CCL22 (50 ng/ml) for 5 minutes (min). Both exogenous recombinant wild type and mutant CCL22 activated pAKT in a similar way, but p42/44 ERK was slightly reduced by mutant CCL22. The samples derive from the same experiment and were processed in parallel. b, Ba/F3-CCR4 cells were stimulated over time with exogenous recombinant wild type or mutant CCL22 (100 ng/ml) and pAKT was assessed by phosphoflow cytometric analysis. Activation of pAKT was not different between exogenous recombinant wild type and mutant CCL22. The mean (± SD) fluorescence intensity (MFI) is shown (n=3). c,d, Single cell phosphoproteomic detection via IsoLight validated reduced phosphorylation of ERK in mutant CCL22 (50 ng/ml) after three-hour (h) incubation. c, Positive cell rate of pERK and (d) the mean of detected pERK signal are shown. P value was calculated by t-test. **0.001 < p < 0.01.
Extended Data Fig. 9. CCL22 mutations drive proliferation in vivo.
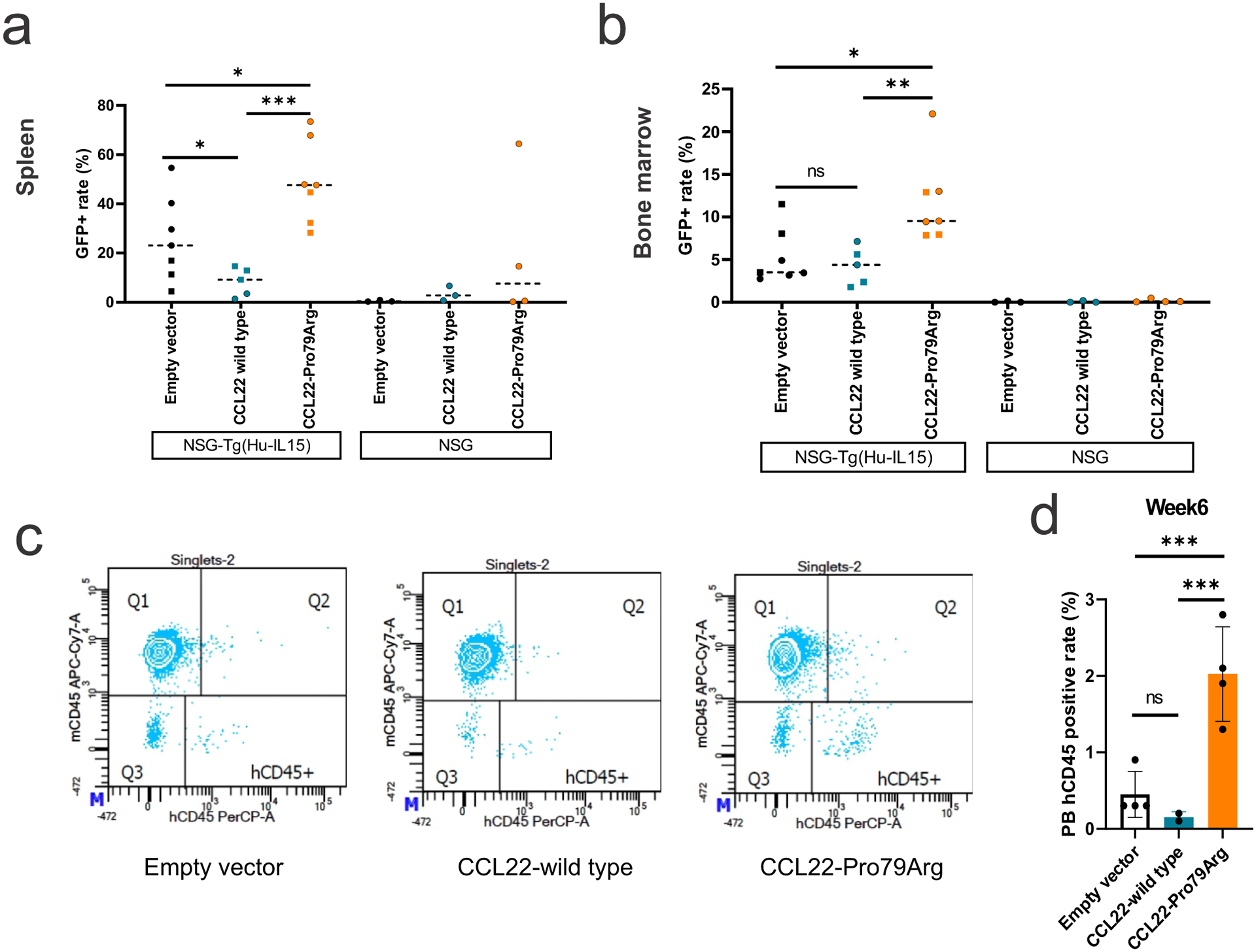
a,b, GFP positive cell rate in (a) spleen and (b) bone marrow cells at the end of study (batch 1; day 57, batch 2; day 44) in NSG mice (3 mice for each group) and NSG-Tg(Hu-IL15) mice that constitutively express human IL-15. Mice were transplanted with NK-92 cells expressing wild type (n=3 (batch 1, square), 2 (batch 2, round), biologically independent animals) or mutant CCL22 (Pro79Arg; n=3, 4), or GFP-expressing lentiviral vector (empty vector; n=3, 4). NK-92 cells expressing mutant CCL22 showed higher GFP positive cell rate than wild type CCL22 and empty vector expressing NK-92 cells in both spleen and bone marrow. GFP positive cells were observed in NSG-Tg(Hu-IL15) mice but not or few in regular NSG mice. The mean GFP positive rate is shown. c,d, Peripheral blood analysis from retroorbital bleeds detected only few circulating human cells at week 6 (batch 2 day 44; the same day of the end of study, empty vector n=4, wild type CCL22 n=2, mutant CCL22 n=4, biologically independent animal samples). Higher circulating human cells were observed in mutant CCL22 although at lower percentage than those observed in spleen and bone marrow. The mean (± SD) GFP positive rate is shown. P value was calculated by t-test. *0.01 < p < 0.05; **0.001 < p < 0.01; ***0.0001 < p < 0.001.
Extended Data Fig. 10. Correction by genome editing of mutated CCL22 reverts the higher expression of chemokine/cytokine and their receptors in primary CLPD-NK samples.
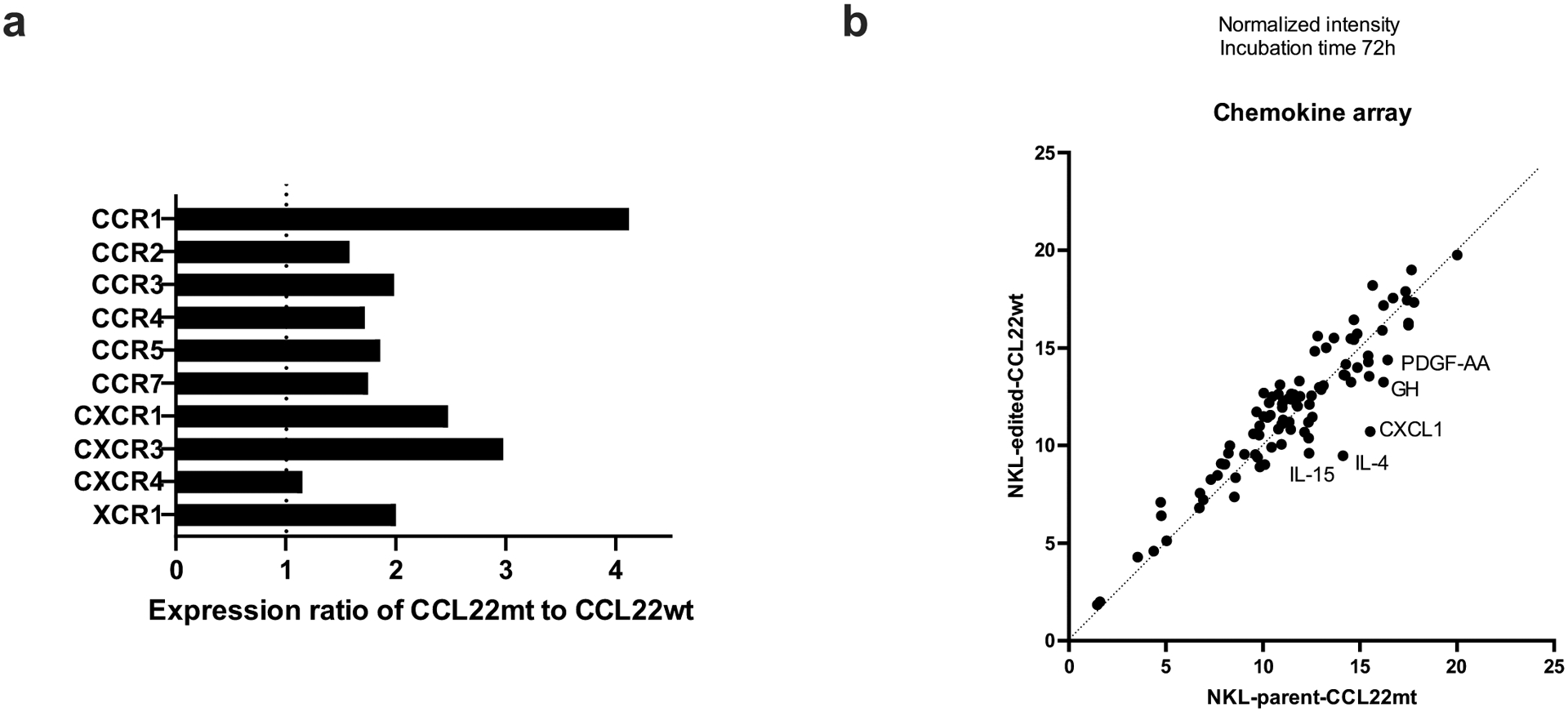
NK cell line NKL which has the hotspot CCL22 mutation (Arg44_Leu45insSer) was edited (CRISPR/Cas9) to revert the mutation (mt) into the wild type (wt) codon. a, Relative gene expression of chemokine receptors in CCL22 mutant compared to wild type was shown. CCL22 mutant NKL represented higher expression of several chemokine receptors. b, Comparison of chemokine and cytokine secretion between CCL22 mutant (NKL-parent-CCL22mt) and wild type NKL (NKL-edited-CCL22wt) by using chemokine array. Cell culture media after 72-hour incubation was analyzed. Several cytokines showed enhanced secretion in CCL22 mutant that related to dendritic cell maturation.
Supplementary Material
Supplementary Table 1. Patients’ data in discovery and validation cohort.
Supplementary Table 2. Summary of clinical features of CLPD-NK cases with hotspot CCL22 mutations (at residues Leu45, Pro79).
Supplementary Table 3. Z-score of 59 cases with CLPD-NK and 12 normal NK cells from peripheral blood and bone marrow (GSE133383)68. This tables describes normalized gene expression of each sample.
Supplementary Table 4. Differentially expressed genes analyzed by DESeq2 between CCL22 mutations vs others, STAT3 mutations vs others, and non-CCL22/STAT3 mutations (Others) vs others.
Supplementary Table 5. Differentially expressed genes analyzed by DESeq2 between sorted BM/PB CD56bright and CD56dim normal NK cells from the public database (GSE133383)68.
Supplementary Table 6. Pathway analysis using differentially expressed genes in CLPD-NK cases with CCL22 mutations, STAT3 mutations, and Other (non-CCL22/STAT3 mutations), compared with normal NK cell.
Supplementary Table 7. Gene set enrichment analysis of 59 cases of primary CLPD-NK patients with CCL22 mutations v.s. others (c2all)..
Supplementary Table 8. The results of intracellular phosphorylation of pERK in BaF3-CCR4 cells after treatment of CCL22 wild type/mutant proteins detected by isoplexis.
Supplementary Table 9. Differentially expressed genes of engrafted NK-92 cells in NSG-Tg(Hu-IL15) mice analyzed by DESeq2 between CCL22 mutant v.s. wild type/empty vector transduced NK-92 cells.
Supplementary Table 10. Gene set enrichment analysis of engrafted NK-92 cells in NSG-Tg(Hu-IL15) mice between mutant v.s. wild type/empty vector transduced NK-92 cells..
Supplementary Table 11. Gene sets generated by differentially expressed genes for GSEA.
Supplementary Table 12. CCL22-dependent chemokine secretion. This tables describes the results from chemokine array performed to compare chemokine secretion between gene edited CCL22 wild type and mutant NKL cell line.
Supplementary Table 13. Antibody list used in the manuscript.
Supplementary Table 14. Primer sequences for cloning and gene editing.
Acknowledgements
The authors were supported by the American and Lebanese Syrian Associated Charities of St Jude Children’s Research Hospital and the National Cancer Institute of the National Institutes of Health under award numbers: P30 CA021765, R35 CA197695 (C.G.M.), R01 CA178393 and P30 CA044579 (T.P.L.), UG1CA189859, U10CA180820 and UG1CA232760 (J.F., E.P.), and F30CA196040 (A.B.K.); NIGMS grant T32 GM080202 (A.B.K.); and NLM grant T32 LM012416 (H.C); the Henry Schueler 41&9 Foundation (C.G.M.); a St. Baldrick’s Foundation Robert J. Arceci Innovation Award (C.G.M.); a Garwood Postdoctoral Fellowship of the Hematological Malignancies Program of the St Jude Children’s Research Hospital Comprehensive Cancer Center (S.K.); the Bess Family Charitable Fund, the LGL Leukemia Foundation and a generous anonymous donor (T.P.L.). We thank ECOG-ACRIN Cancer Research Group for acute NK leukemia samples. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Sherri Surman (Department of Infectious Diseases, St. Jude) for flow cytometry, and Shaina N. Porter (Center for Advanced Genome Editing, St. Jude) for genome editing; Bryna Shemo for LGL Leukemia Registry support, Matt Schmachtenberg for technical support, and Dr. Suna Onengut-Gumuscu and Emily Farber for targeted sequencing of the validation cohort at the University of Virginia.
Competing Interests Statement
C.B., W.W., M.M., M.L.M.: Employment by MLL Munich Leukemia Laboratory; C.H., W.K., T.H.: Equity ownership of MLL Munich Leukemia Laboratory. I.I. received honoraria from Amgen and Mission Bio. C.G.M. received research funding from Loxo Oncology, Pfizer, AbbVie; honoraria from Amgen and Illumina, and holds stock in Amgen. T.P.L. is on the Scientific Advisory Board and has stock options for Keystone Nano, Bioniz Therapeutics and Dren Bio. T.P.L. and D.J.F. received honoraria from Kymera Therapeutics. D.J.F. has research funding from AstraZeneca. There are no conflicts of interest with the work presented in this manuscript. The remaining authors declare no competing interests.
Footnotes
Code availability
All details of all analysis code used in the manuscript has been provided in the methods section.
Data availability
Primary sample whole genome and transcriptome data have been deposited in the European Genome-phenome Archive (EGA), accession EGAS00001006009, and are also housed in a protected Cloud environment in accordance with European General Data Protection regulations. Transcriptome sequencing data (genome build GRCh37) of engrafted NK-92 cells have been deposited in the Gene Expression Omnibus, accession GSE163864. Normal NK cell data were downloaded from GSE133383 (ref.79). These data were used for Figures 1, 2, 4, Extended data figures 1, 2, 3, 10. Source data are provided with this paper.
References
- 1.Swerdlow SH et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127, 2375–90 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barila G et al. T cell large granular lymphocyte leukemia and chronic NK lymphocytosis. Best Pract Res Clin Haematol 32, 207–216 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Lamy T, Moignet A & Loughran TP Jr. LGL leukemia: from pathogenesis to treatment. Blood 129, 1082–1094 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Giussani E et al. Lack of Viral Load Within Chronic Lymphoproliferative Disorder of Natural Killer Cells: What Is Outside the Leukemic Clone? Front Oncol 10, 613570 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamy T & Loughran TP Jr. How I treat LGL leukemia. Blood 117, 2764–74 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morice WG et al. Chronic lymphoproliferative disorder of natural killer cells: a distinct entity with subtypes correlating with normal natural killer cell subsets. Leukemia 24, 881–4 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Barila G et al. Dominant cytotoxic NK cell subset within CLPD-NK patients identifies a more aggressive NK cell proliferation. Blood Cancer J 8, 51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith SL et al. Diversity of peripheral blood human NK cells identified by single-cell RNA sequencing. Blood Adv 4, 1388–1406 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner JA et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J Clin Invest 127, 4042–4058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michel T et al. Human CD56bright NK Cells: An Update. J Immunol 196, 2923–31 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Dogra P et al. Tissue Determinants of Human NK Cell Development, Function, and Residence. Cell 180, 749–763 e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koskela HL et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med 366, 1905–13 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jerez A et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T-cell large granular lymphocyte leukemia. Blood 120, 3048–57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gasparini VR et al. A high definition picture of somatic mutations in chronic lymphoproliferative disorder of natural killer cells. Blood Cancer J 10, 42 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pastoret C et al. Linking the KIR phenotype with STAT3 and TET2 mutations to identify chronic lymphoproliferative disorders of NK cells. Blood (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olson TL et al. Frequent Somatic TET2 Mutations in Chronic NK-LGL Leukemia with Distinct Patterns of Cytopenias. Blood 138, 662–673 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheon H et al. Analysis of Genomic Landscape of Large Granular Lymphocyte Leukemia Reveals Etiologic Insights. Blood 136, 27–28 (2020). [Google Scholar]
- 18.Stengel A, Meggendorfer M, Kern W, Haferlach T & Haferlach C Correlation of Mutation Patterns with Patient Age in 2656 Cases with 11 Different Hematological Malignancies. Blood 136, 16–17 (2020). [Google Scholar]
- 19.Jiang L et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet 47, 1061–6 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Dufva O et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat Commun 9, 1567 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scheu S, Ali S, Ruland C, Arolt V & Alferink J The C-C Chemokines CCL17 and CCL22 and Their Receptor CCR4 in CNS Autoimmunity. Int J Mol Sci 18(2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Homey B, Steinhoff M, Ruzicka T & Leung DY Cytokines and chemokines orchestrate atopic skin inflammation. J Allergy Clin Immunol 118, 178–89 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Lucas M, Schachterle W, Oberle K, Aichele P & Diefenbach A Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 26, 503–17 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anguille S et al. Interleukin-15 Dendritic Cells Harness NK Cell Cytotoxic Effector Function in a Contact- and IL-15-Dependent Manner. PLoS One 10, e0123340 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andoniou CE et al. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol 6, 1011–9 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Zambello R et al. Phenotypic and functional analyses of dendritic cells in patients with lymphoproliferative disease of granular lymphocytes (LDGL). Blood 106, 3926–31 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Godiska R et al. Human Macrophage–derived Chemokine (MDC), a Novel Chemoattractant for Monocytes, Monocyte-derived Dendritic Cells, and Natural Killer Cells. Journal of Experimental Medicine 185, 1595–1604 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zheng Y et al. Structure of CC Chemokine Receptor 5 with a Potent Chemokine Antagonist Reveals Mechanisms of Chemokine Recognition and Molecular Mimicry by HIV. Immunity 46, 1005–1017 e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salanga CL & Handel TM Chemokine oligomerization and interactions with receptors and glycosaminoglycans: the role of structural dynamics in function. Exp Cell Res 317, 590–601 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wacker D et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 168, 377–389 e12 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCorvy JD et al. Structural determinants of 5-HT2B receptor activation and biased agonism. Nat Struct Mol Biol 25, 787–796 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamashita U & Kuroda E Regulation of macrophage-derived chemokine (MDC, CCL22) production. Crit Rev Immunol 22, 105–14 (2002). [PubMed] [Google Scholar]
- 33.Yoshie O & Matsushima K CCR4 and its ligands: from bench to bedside. International Immunology 27, 11–20 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Mariani M, Lang R, Binda E, Panina-Bordignon P & D’Ambrosio D Dominance of CCL22 over CCL17 in induction of chemokine receptor CCR4 desensitization and internalization on human Th2 cells. European Journal of Immunology 34, 231–240 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Rapp M et al. CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. Journal of Experimental Medicine 216, 1170–1181 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Curiel TJ et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature Medicine 10, 942–949 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Mailloux AW & Young MRI NK-Dependent Increases in CCL22 Secretion Selectively Recruits Regulatory T Cells to the Tumor Microenvironment. The Journal of Immunology 182, 2753–2765 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakagawa M et al. Gain-of-function CCR4 mutations in adult T cell leukemia/lymphoma. Journal of Experimental Medicine 211, 2497–2505 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith JS, Lefkowitz RJ & Rajagopal S Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov 17, 243–260 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lefkowitz RJ & Shenoy SK Transduction of receptor signals by beta-arrestins. Science 308, 512–7 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Drury LJ et al. Monomeric and dimeric CXCL12 inhibit metastasis through distinct CXCR4 interactions and signaling pathways. Proc Natl Acad Sci U S A 108, 17655–60 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernardini G, Sciume G & Santoni A Differential chemotactic receptor requirements for NK cell subset trafficking into bone marrow. Front Immunol 4, 12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wright DE, Bowman EP, Wagers AJ, Butcher EC & Weissman IL Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med 195, 1145–54 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eisenman J et al. Interleukin-15 interactions with interleukin-15 receptor complexes: characterization and species specificity. Cytokine 20, 121–9 (2002). [DOI] [PubMed] [Google Scholar]
- 45.Costantini C et al. Neutrophil activation and survival are modulated by interaction with NK cells. Int Immunol 22, 827–38 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Thoren FB et al. Human NK Cells induce neutrophil apoptosis via an NKp46- and Fas-dependent mechanism. J Immunol 188, 1668–74 (2012). [DOI] [PubMed] [Google Scholar]
- 47.Park CS, Yoon SO, Armitage RJ & Choi YS Follicular dendritic cells produce IL-15 that enhances germinal center B cell proliferation in membrane-bound form. J Immunol 173, 6676–83 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Katakai T Marginal reticular cells: a stromal subset directly descended from the lymphoid tissue organizer. Front Immunol 3, 200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L, Wu J, Abdi R, Jewell CM & Bromberg JS Lymph node fibroblastic reticular cells steer immune responses. Trends Immunol 42, 723–734 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pan-cancer analysis of whole genomes. Nature 578, 82–93 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nakagawa M et al. Gain-of-function CCR4 mutations in adult T cell leukemia/lymphoma. J Exp Med 211, 2497–505 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yoshie O & Matsushima K CCR4 and its ligands: from bench to bedside. Int Immunol 27, 11–20 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Wang ZQ et al. Gain-of-function mutation of KIT ligand on melanin synthesis causes familial progressive hyperpigmentation. Am J Hum Genet 84, 672–7 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wootten D, Christopoulos A, Marti-Solano M, Babu MM & Sexton PM Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat Rev Mol Cell Biol 19, 638–653 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Caligiuri MA Human natural killer cells. Blood 112, 461–9 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ali AK, Nandagopal N & Lee SH IL-15-PI3K-AKT-mTOR: A Critical Pathway in the Life Journey of Natural Killer Cells. Front Immunol 6, 355 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mishra A et al. Aberrant overexpression of IL-15 initiates large granular lymphocyte leukemia through chromosomal instability and DNA hypermethylation. Cancer Cell 22, 645–55 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ferlazzo G & Morandi B Cross-Talks between Natural Killer Cells and Distinct Subsets of Dendritic Cells. Front Immunol 5, 159 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu B et al. Affinity-coupled CCL22 promotes positive selection in germinal centres. Nature 592, 133–137 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Bakker E, Qattan M, Mutti L, Demonacos C & Krstic-Demonacos M The role of microenvironment and immunity in drug response in leukemia. Biochim Biophys Acta 1863, 414–426 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ajram L et al. Internalization of the chemokine receptor CCR4 can be evoked by orthosteric and allosteric receptor antagonists. European Journal of Pharmacology 729, 75–85 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mortier E, Woo T, Advincula R, Gozalo S & Ma A IL-15Ralpha chaperones IL-15 to stable dendritic cell membrane complexes that activate NK cells via trans presentation. J Exp Med 205, 1213–25 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nandagopal N, Ali AK, Komal AK & Lee SH The Critical Role of IL-15-PI3K-mTOR Pathway in Natural Killer Cell Effector Functions. Front Immunol 5, 187 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crinier A, Narni-Mancinelli E, Ugolini S & Vivier E SnapShot: Natural Killer Cells. Cell 180, 1280–1280 e1 (2020). [DOI] [PubMed] [Google Scholar]
- 66.Freud AG, Mundy-Bosse BL, Yu J & Caligiuri MA The Broad Spectrum of Human Natural Killer Cell Diversity. Immunity 47, 820–833 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Geiger TL & Sun JC Development and maturation of natural killer cells. Curr Opin Immunol 39, 82–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gaggero S, Witt K, Carlsten M & Mitra S Cytokines Orchestrating the Natural Killer-Myeloid Cell Crosstalk in the Tumor Microenvironment: Implications for Natural Killer Cell-Based Cancer Immunotherapy. Front Immunol 11, 621225 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Michel T, Hentges F & Zimmer J Consequences of the crosstalk between monocytes/macrophages and natural killer cells. Front Immunol 3, 403 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rohrle N, Knott MML & Anz D CCL22 Signaling in the Tumor Environment. Adv Exp Med Biol 1231, 79–96 (2020). [DOI] [PubMed] [Google Scholar]
- 71.Fehniger TA et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med 193, 219–31 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raczy C et al. Isaac: ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics 29, 2041–3 (2013). [DOI] [PubMed] [Google Scholar]
- 73.Kim S et al. Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15, 591–594 (2018). [DOI] [PubMed] [Google Scholar]
- 74.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Haferlach T, Hutter S & Meggendorfer M Genome Sequencing in Myeloid Cancers. N Engl J Med 384, e106 (2021). [DOI] [PubMed] [Google Scholar]
- 76.Hutter S et al. A Novel Machine Learning Based in silico Pathogenicity Predictor for Missense Variants in a Hematological Setting Blood 134(2019). [Google Scholar]
- 77.Parida L et al. Dark-matter matters: Discriminating subtle blood cancers using the darkest DNA. PLoS Comput Biol 15, e1007332 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhou X et al. Exploration of Coding and Non-coding Variants in Cancer Using GenomePaint. Cancer Cell 39, 83–95 e4 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dogra P et al. Tissue Determinants of Human NK Cell Development, Function, and Residence. Cell 180, 749–763.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leek JT, Johnson WE, Parker HS, Jaffe AE & Storey JD The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou Y et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–50 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Konagurthu AS, Whisstock JC, Stuckey PJ & Lesk AM MUSTANG: a multiple structural alignment algorithm. Proteins 64, 559–74 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Sievers F et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Valdar WS Scoring residue conservation. Proteins 48, 227–41 (2002). [DOI] [PubMed] [Google Scholar]
- 88.Wagih O ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 33, 3645–3647 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Liu D, Paczkowski P, Mackay S, Ng C & Zhou J Single-Cell Multiplexed Proteomics on the IsoLight Resolves Cellular Functional Heterogeneity to Reveal Clinical Responses of Cancer Patients to Immunotherapies. Methods Mol Biol 2055, 413–431 (2020). [DOI] [PubMed] [Google Scholar]
- 90.Shultz LD et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174, 6477–89 (2005). [DOI] [PubMed] [Google Scholar]
- 91.Aickin M & Gensler H Adjusting for multiple testing when reporting research results: the Bonferroni vs Holm methods. Am J Public Health 86, 726–8 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Patients’ data in discovery and validation cohort.
Supplementary Table 2. Summary of clinical features of CLPD-NK cases with hotspot CCL22 mutations (at residues Leu45, Pro79).
Supplementary Table 3. Z-score of 59 cases with CLPD-NK and 12 normal NK cells from peripheral blood and bone marrow (GSE133383)68. This tables describes normalized gene expression of each sample.
Supplementary Table 4. Differentially expressed genes analyzed by DESeq2 between CCL22 mutations vs others, STAT3 mutations vs others, and non-CCL22/STAT3 mutations (Others) vs others.
Supplementary Table 5. Differentially expressed genes analyzed by DESeq2 between sorted BM/PB CD56bright and CD56dim normal NK cells from the public database (GSE133383)68.
Supplementary Table 6. Pathway analysis using differentially expressed genes in CLPD-NK cases with CCL22 mutations, STAT3 mutations, and Other (non-CCL22/STAT3 mutations), compared with normal NK cell.
Supplementary Table 7. Gene set enrichment analysis of 59 cases of primary CLPD-NK patients with CCL22 mutations v.s. others (c2all)..
Supplementary Table 8. The results of intracellular phosphorylation of pERK in BaF3-CCR4 cells after treatment of CCL22 wild type/mutant proteins detected by isoplexis.
Supplementary Table 9. Differentially expressed genes of engrafted NK-92 cells in NSG-Tg(Hu-IL15) mice analyzed by DESeq2 between CCL22 mutant v.s. wild type/empty vector transduced NK-92 cells.
Supplementary Table 10. Gene set enrichment analysis of engrafted NK-92 cells in NSG-Tg(Hu-IL15) mice between mutant v.s. wild type/empty vector transduced NK-92 cells..
Supplementary Table 11. Gene sets generated by differentially expressed genes for GSEA.
Supplementary Table 12. CCL22-dependent chemokine secretion. This tables describes the results from chemokine array performed to compare chemokine secretion between gene edited CCL22 wild type and mutant NKL cell line.
Supplementary Table 13. Antibody list used in the manuscript.
Supplementary Table 14. Primer sequences for cloning and gene editing.
Data Availability Statement
Primary sample whole genome and transcriptome data have been deposited in the European Genome-phenome Archive (EGA), accession EGAS00001006009, and are also housed in a protected Cloud environment in accordance with European General Data Protection regulations. Transcriptome sequencing data (genome build GRCh37) of engrafted NK-92 cells have been deposited in the Gene Expression Omnibus, accession GSE163864. Normal NK cell data were downloaded from GSE133383 (ref.79). These data were used for Figures 1, 2, 4, Extended data figures 1, 2, 3, 10. Source data are provided with this paper.