

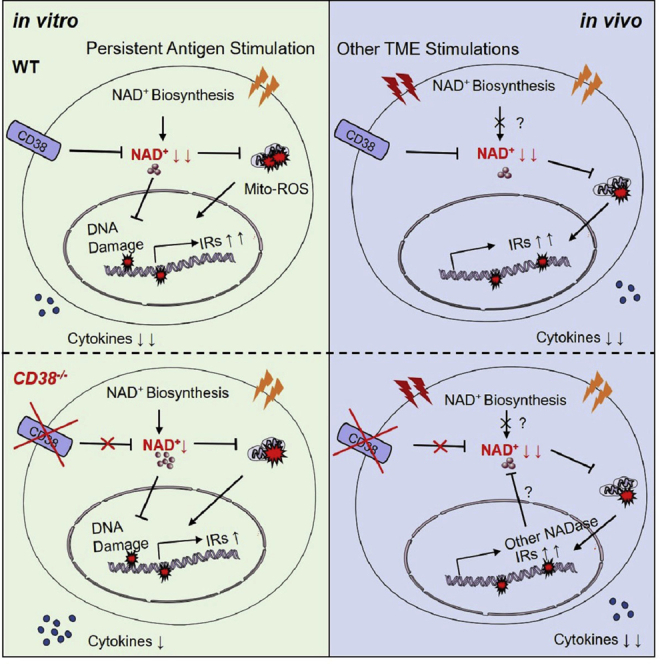
Summary
Exhausted CD8+ T cells with limited effector functions and high expression of multiple co-inhibitory receptors are one of the main barriers hindering antitumor immunity. The NADase CD38 has received considerable attention as a biomarker of CD8+ T cell exhaustion, but it remains unclear whether the increased CD38 directly promotes T cell dysfunctionality. Here, we surprisingly found that although Cd38 deficiency partially reverses NAD+ degradation and T cell dysfunction in vitro, the terminal exhausted differentiation of adoptively transferred CD8+ T cells in tumor is not impacted by either deficiency or overexpression of CD38. Monitoring the dynamic NAD+ levels shows that NAD+ levels are comparable between tumor infiltrated WT and Cd38−/− OT-1 cells. Therefore, our results suggest that decreased NAD+ are correlated with T cell dysfunction, but deficiency of CD38 is not enough for rescuing NAD+ in tumor infiltrated CD8+ T cells and fails to increase the efficacy of antitumor T cell therapy.
Subject areas: Immunology, Cell biology, Cancer
Graphical abstract
Highlights
-
•
CD38 is upregulated on CD8+ T cells by persistent antigen stimulation
-
•
Deletion of CD38 partially reverses NAD+ degradation and T cell dysfunction in vitro
-
•
CD38 deficiency fails to prevent or delay CD8+ T cell exhaustion within tumor
-
•
NAD+ levels in tumor infiltrated T cells are regulated by CD38 and other NADases
Immunology; Cell biology; Cancer
Introduction
Activated CD8+ T cells rapidly produce large amounts of effector cytokines (e.g., IL-2, TNFα, and IFNγ) and Granzyme B to effectively kill transformed cells (Golstein and Griffiths, 2018). However, multiple factors within the tumor microenvironment (TME) such as persistent antigen stimulation, hypoxia, and nutrient limitations (low glucose etc.) result in the functional exhaustion of tumor antigen-specific CD8+ T cells, which is the major barrier for mounting an effective antitumor immune response (Cheng et al., 2021; Duan et al., 2020; Moller et al., 2022). Generally, exhausted CD8+ T cells in the TME are classified into two distinct subsets: Ly108+TCF-1+ memory-like progenitors (TPRO) and Ly108−TIM-3+ or PD-1+TIM-3+ terminally exhausted cells (TEXH) (Chen et al., 2019; McLane et al., 2019). In contrast to TPRO, which retains certain proliferative potential and can be partially reinvigorated by immune checkpoint blockade (ICB), TEXH are largely resistant to respond to ICB immunotherapies (Beltra et al., 2020; Siddiqui et al., 2019; Verma et al., 2019). With mouse spontaneous HCC model, human melanoma and non-small cell lung cancer (NSCLC), Philip et al. identified CD38 as an important TEXH surface maker and demonstrated that CD38 was associated with the fixed dysfunctional state of T cells (Philip et al., 2017). Moreover, depleting the PD-1+CD38hiCD8+ cells that were induced by PD-1 blockade before vaccination proved to significantly promote subsequent immunotherapeutic outcomes in mice, demonstrating that CD38 may reflect the dysfunctionality of CD8+ T cells (Verma et al., 2019).
As a classical nicotinamide adenine dinucleotide (NAD+) consuming enzyme, CD38 converts NAD+ to ADP-ribose (ADPR) and cADPR, which participates in the regulation of intracellular Ca2+, extracellular metabolites, and signal transduction such as the KEAP1-NRF2 pathway (Gao et al., 2021; Malavasi et al., 2008). Above all, the degradation of NAD+ not only destroys oxidative phosphorylation and redox balance but also restricts the activity of Sirtuins (SIRTs) family members, DNA repair, and DNA demethylation in various mammalian cells, and thus plays an important role in aging, diseases, and cancers (Chiarugi et al., 2012; Verdin, 2015). Specifically, it was reported that high levels of CD38 in CD8+ T cells resulted in the decrease of NAD+ concentration, which further reduced CD8+ T cell-mediated cytotoxicity by NAD+-SIRT1-EZH2 pathway and increased propensity to infections in patients with systemic lupus erythematosus (SLE) (Katsuyama et al., 2020). In addition, the upregulation of intracellular NAD+ levels via targeting CD38 in hybrid Th1/17 cells would enhance the antitumor efficacy of adoptively transferred CD4+ T cells through NAD+-SIRT1-FOXO1 pathway (Chatterjee et al., 2018). Beyond the regulation of NAD+, high level expression of CD38 in tumor cells was also found to inhibit CD8+ T cell mediated antitumor response via modulation of adenosine receptor signaling (Chen et al., 2018). However, it remains unclear whether CD38 is functionally relevant to CD8+ T cell exhaustion within the TME or it solely acts as a phenotypic marker to better define exhausted T cells together with PD-1 and TIM-3.
Here, by utilizing Cd38−/− OT-1 mice and retroviral expression of CD38 specifically in CD8+ T cells, we characterized the specific role of CD38 in regulating CD8+ T cell exhaustion and functionality. We demonstrated that deficiency of CD38 led to increased resistance to exhaustion induction by chronic antigen stimulation in vitro as suggested by reduced expression of co-inhibitory molecules (IRs) and partially rescued cytokine production. Of note, the exhaustion phenotypes of tumor infiltrated CD8+ T cells were not impacted with either deficiency or overexpression of CD38 in vivo. We further demonstrated that the intracellular NAD+ levels of CD38 knockout T cells were slightly but significantly increased as compared with WT counterparts in vitro but not in vivo. Therefore, we proposed that the NADase CD38, which was defined as a TEXH marker, was upregulated by persistent TCR stimulation in the TME but was less functionally relevant with T cell exhaustion, because deficiency of CD38 failed to restore the antitumor ability of T cells.
Results
CD38 is barely expressed in naive CD8+ T cells but is upregulated in the TME
To interrogate the precise role of CD38 in CD8+ tumor infiltrated lymphocytes (TILs), we first analyzed the expression and dynamic changes of CD38 in CD8+ T cells. First, by using gene expression profiling interactive analysis 2 (GEPIA2) tool (Tang et al., 2019), we observed striking correlation between CD38 expression and exhausted T cell signature genes (Figure S1A) in multiple types of cancer, such as human colon adenocarcinoma (COAD), breast invasive carcinoma (BRCA), or skin cutaneous melanoma (SKCM). Next, we demonstrated that CD38 was barely expressed in naive CD8+ T cells as compared with B lymphocytes (Figure 1A), which express the high level of CD38. We further confirmed the efficient deletion of CD38 with our Cd38−/− mouse model, as CD38 expression was completely lost on Cd38−/− B cells (Figure S1B). Moreover, the mRNA and protein expression levels of CD38 were not changed in CD8+ T cells upon αCD3/CD28 stimulation for 24 h, which was distinct from PD-1 expression (Figures 1B and 1C). Therefore, we measured the CD38 expression with ovalbumin (OVA) peptide-stimulated TCR-transgenic OT-1 cells, and Cd38−/− OT-1 cells were utilized as control to standardize its expression (Figure 1D). We confirmed that CD38 expression was slightly increased after T cell activation but the frequency of CD38+CD8+ T cells was still limited, suggesting that CD38 expression is not modulated significantly during T cell activation (Figures 1D and S1C).
Figure 1.

The dynamic expression of CD38 in CD8+ T cells
(A) Representative FACS plots showing CD38 expression in WT mice indicated lymphocytes. B cells with high CD38 as a positive control.
(B and C) Purified WT CD8+ T cells were primed by αCD3/CD28 for 24 h, then the protein expression and mRNA transcription levels of CD38 were analyzed via flow cytometry or qPCR, respectively.
(D) Left: the representative histogram of CD38 expression in indicated cells. Right: quantified the dynamic change of CD38 expression of OT-1 cells during OVA peptide activation. The ΔMFI (mean fluorescence intensity) of CD38 was calculated from the difference values between WT and CD38 knockout cells. Data are shown as Mean ± SD (n = 3), Student’s t test, ∗∗∗∗: p < 0.0001; ∗: p < 0.05; ns: not significant.
(E) Left: activated WT or Cd38−/− OT-1 cells were stimulated with OVA peptide continuously, hypoxia or 2 mM glucose for 4 days. The representative FACS plots of CD38 expression were shown. Right: The ΔMFI was analyzed by flow cytometry and summarized. Data are shown as Mean ± SD (n = 3), Student’s t test, ∗∗∗∗: p < 0.0001; ∗∗∗: p < 0.001; ns: not significant.
(F) Left: representative histogram of CD38 expression in the spleen or B16-F10 tumor-infiltrated CD8+ T cells. Right: Statistical analysis of CD38 MFI in indicated cells. Data are shown as Mean ± SD (n = 5), Student’s t test, ∗∗∗: p < 0.001.
(G) Left: CD8+ TILs were divided into PD-1−TIM-3+ TPRO and PD-1+TIM-3+ TEXH in contour plots. Middle: representative FACS plots of CD38 expression in TPRO or TEXH cells. Right: the levels of CD38 in these two subsets were compared. Data are shown as Mean ± SD (n = 5), Student’s t test, ∗∗: p < 0.01.
Next, we sought to examine CD38 expression with an in vitro exhaustion induction model by chronic antigen stimulation, hypoxia, and low glucose, which were the critical factors in the TME for T cell exhaustion. To mimic persistent antigen stimulation in vitro, we treated OT-1 cells with continuous OVA peptide stimulation and confirmed their exhausted phenotype by flow cytometry as suggested by markedly increased expression of inhibitory receptors and decreased cytokine production ability (Figures S1D and S1E). In comparison to hypoxia and low glucose, persistent antigen stimulation significantly induced the expression of CD38 (Figure 1E), we again confirmed the efficient deletion of CD38 with our Cd38−/− mice as Cd38−/− OT-1 cells fail to upregulate CD38 in response to chronic antigen stimulation, glucose restriction, or hypoxia. Consistent with a previous report (Philip et al., 2017), we also confirmed the high levels of CD38 in tumor infiltrated CD8+ T cells, especially in PD-1+TIM-3+ terminal exhausted T cells (Figures 1F and 1G). Taken together, these data confirmed that the expression of CD38 during CD8+ T cell activation was mild but persistent antigen stimulation could specifically upregulate its expression.
CD38 knockout does not impact the development and activation of CD8+ T cells
Given the role of NAD+ in T cell differentiation (Wang et al., 2021), we asked whether NADase CD38 deficiency impacted the CD8+ T cell development/homeostasis and function. Analyses of T cell development in the thymus have shown that CD38 knockout did not disturb the development of T cells including the maturation of CD4+ and CD8+ single-positive population (Figures 2A and 2B). In addition, the proportions of B cells, CD4+, and CD8+ T cells were similar between WT and CD38 knockout mice both in spleen and lymph nodes (Figures 2C, 2D and S2A). CD62L and CD44 have been employed to define functionally distinct CD8+ T cell populations involving naive T (CD62LhiCD44lo), activated T (CD62LloCD44hi), and central memory T (CD62LhiCD44hi) (Sckisel et al., 2017). Likewise, we observed comparable frequencies of naive, activated, and memory CD8+ T cell populations that were distinguished by CD62L and CD44 between WT and CD38 knockout mice (Figures 2E and S2B). Moreover, we monitored the dynamic changes of surface proteins such as CD25, CD44, CD69, as well as CD62L, and observed that the activation process of CD38 knockout OT-1 cells was almost indistinguishable with WT OT-1 cells (Figures 2F and S2C). However, the ability of proliferation was decreased in CD38 deficient OT-1 cells upon activation (Figure 2G). Next, we assessed the cytokine production ability of WT and Cd38−/− OT-1 cells in vitro and did not find significant differences between them (Figures 2H and 2I). Hence, these observations revealed that depletion of CD38 does not significantly disturb the T cell development, homeostasis, and CD8+ T cell activation.
Figure 2.

CD8+ T cells development and activation in the absence of CD38
(A) Flow cytometry analysis of thymocytes isolated from 8-week-old WT and Cd38−/− mice (up). Percentages of cells in various thymic populations were shown (down). Data are shown as Mean ± SD (n = 4), Student’s t test, ns: not significant.
(B) Then gating on CD4/CD8+ double-negative thymocytes, representative contour plots and statistical ratios of DN1-4 as shown. Data are shown as Mean ± SD (n = 4), Student’s t test, ns: not significant.
(C and D) Percentages of CD4, CD8+ or B cells in splenocytes or lymph node cells from 8-week-old mice of the indicated genotypes, respectively. Data are shown as Mean ± SD (n = 4), Student’s t test, ns: not significant.
(E) CD8+ T cells from the spleen of WT or Cd38−/− mice were stained with CD62L and CD44, the indicated populations were analyzed. Data are shown as Mean ± SD (n = 4), Student’s t test, ns: not significant.
(F) Statistical analysis of surface activation marker CD25 expression in WT or Cd38−/− OT-1 cells during αCD3/CD28 activation. Data are shown as Mean ± SD (n = 3), Student’s t test, ns: not significant.
(G) Naive WT or Cd38−/− OT-1 cells were labeled by cell proliferation dye (CPD 450). After activation by αCD3/CD28 3 days, flow cytometry analyzed the proliferation index of the indicated genotypes.
(H and I) Left: representative contour plots of WT or Cd38−/− OT-1 cells producing cytokines TNFα, IFNγ, or IL-2 on the sixth day after activation. Right: summary of the relative levels of OT-1 cells producing cytokines. Data are shown as Mean ± SD (n = 3), Student’s t test, ns: not significant.
CD38 deficiency ameliorates the exhaustion state of CD8+ T cells in vitro
Our previous data have shown that chronic antigen stimulation caused exhausted T cell phenotypes in vitro, which were accompanied by the upregulation of CD38. We thus tested the role of CD38 in T cell differentiation under persistent antigen stimulation. Of note, we observed that deficiency of CD38 could significantly reduce the proportion of multi-IRs expressing subset or the terminally exhausted Ly108−TIM-3+ population (Figures 3A, 3B, S3A, and S3B). Beyond IRs, exhausted T cells display unique transcriptional and epigenetic modifications, among which T cell factor 1 (TCF-1) and thymocyte-selection-associated high mobility group-box protein (TOX) are two key transcription factors. TCF-1 deficiency in CD8+ TILs resulted in loss of self-renewal capacity of intratumoral T cells (Siddiqui et al., 2019), and increased TOX was demonstrated to drive TEXH generation in chronic infections and tumors (Khan et al., 2019). Here, we found that the expression TOX but not TCF-1 had a different expression in CD38 knockout OT-1 cells under treatment, indicating that Cd38−/− cells possibly displayed a distinguished transcription program (Figure 3C). Although the cytokine production capacity was decreased both in WT and Cd38−/− OT-1 cells under persistent OVA peptide stimulation, the decline of TNFα and IFNγ was partially reversed in absence of CD38 (Figures 3D, 3E, and S3C). Given the upregulation of CD38 is often coordinated with the degradation of NAD+, which plays a critical role in both mitochondria oxidation and DNA damage that are associated with T cell exhaustion or cellular senescence (Hernandez-Segura et al., 2018; Scharping et al., 2021; Verdin, 2015). Therefore, we measured the levels of mitoROS production and DNA damage by MitoSOX and γ-H2AX, respectively. As expected, the increased level of mitoROS and DNA damage in OT-1 cells were induced by persistent antigen stimulation, which was partially inhibited in CD38 deficient cells (Figures 3F and 3G). Altogether, we found that CD38 deficiency could partially protect CD8+ T cells from TCR chronic stimulation mediated exhaustion program in vitro.
Figure 3.

The genetic absence of CD38 inhibited the antigen persistent stimulation-induced T dysfunction
In this figure, all representative contour plots of indicated protein expression were placed to the left, and statistical results were on the right. All error bars indicate Mean ± SD (n = 3), Student’s t test, ∗∗∗∗: p < 0.0001; ∗∗∗: p < 0.001; ∗∗: p < 0.01; ∗: p < 0.05; ns: not significant.
(A and B) Activated WT or CD38 knockout OT-1 cells were treated with antigen chronic stimulation as shown in Figure S1D, and then following flow cytometry analysis of PD-1, TIM-3, and Ly108 expression. Percentages of PD-1+TIM-3+ and Ly108−TIM-3+ were compared.
(C) The expression and MFI of transcript factors TCF-1 and TOX in WT or CD38 knockout OT-1 cells under continuous OVA peptides treatment or not were analyzed by flow cytometry.
(D and E) The ability of cytokine production of antigen persistent stimulated-WT or Cd38−/− OT-1 cells were analyzed by comparing the percentages of TNFα+IFNγ+ or IL-2+ frequencies in the indicated cells.
(F) The mitochondria redox levels in OT-1 cells with indicated treatment or not were labeled by Mito-SOX, and then percentages of Mito-SOX+ in these four groups were compared.
(G) γ-H2AX was utilized as a marker to reflect the DNA damage levels of indicated cells under antigen continuous stimulation or not. Values indicate the MFI of γ-H2AX in OT-1 cells.
CD38 deficient CD8+ T cells display comparable exhaustion differentiation and antitumor immunity
Based on previous findings in vitro, we performed an adoptive cell transfer (ACT) experiment to verify the antitumor ability of Cd38−/− T cells (Figure 4A). To our surprise, in the B16-OVA tumor model, although CD38 expression was also significantly increased in WT OT-1 cells as compared to CD38 deficient cells, Cd38−/− OT-1 cells did not show any considerable potential of better tumor control (Figures 4B and S3E). The absolute numbers of transferred Cd38−/− cells were even less than WT in the spleen (Figure 4C), in which they show clearly the central memory phenotype that was defined by CD62L+CD44+ (Figure 4D). In addition, adoptively transferred cells in draining lymph nodes (DLN) had the comparable ability of cytokine production (Figures 4E and S3D). Given that CD38 was specifically expressed in CD8+ TILs and involved in TEXH differentiation induced by persistent antigen stimulation, we thus further investigated the effects of CD38 deficiency on tumor-infiltrating CD8+ T cells. First, we counted the number of transferred cells in the tumor and noticed that there was no statistical significance between the two genotypes (Figure 4F). In fact, the frequency of the terminal exhausted population was similar between WT and Cd38−/− T cells as indicated by PD-1+TIM-3+ or Ly108−TIM-3+ (Figures 4G and 4H). Next, we analyzed the cytokine production of the transferred population and did not find any difference between WT and Cd38−/− cells (Figures 4I and 4J). These results strongly support the fact that CD38 deficiency did not regulate CD8+ T cell exhaustion differentiation within the TME, at least in our settings.
Figure 4.

CD38 knockout fails to promote the tumor control ability of adoptively transferred OT-1 cells
All error bars indicate Mean ± SD (n = 5), Student’s t test, ∗: p < 0.05; ns: not significant.
(A) Diagram of the experimental strategy used in ACT.
(B) Tumor growth curves in C57BL/6 mice B16-OVA tumor models before and after adoptively transferring WT or CD38 knockout OT-1 cells.
(C and D) The total numbers and frequencies of CD62L+CD44+ of adoptive transferred OT-1 cells in the spleen of recipient mice at day 14 post i.v. as shown.
(E) Values indicate the frequencies of WT or Cd38−/− OT-1 cells in the DLN producing IFNγ and TNFα or IL-2.
(F) Absolute numbers of indicated genotypes OT-1 cells per mg or mm3 in the tumor at day 14 post-adoptive transfer.
(G) Representative contour plots of PD-1 and TIM-3 expression in indicated OT-1 TILs (left), and percentage of OT-1 TILs in the PD-1+TIM-3+ TEXH subset (right).
(H) The Ly108+TIM-3- TPRO and Ly108−TIM-3+ TEXH frequencies of WT or Cd38−/− OT-1 TILs were analyzed by flow cytometry for individual (left) and multiple mice (right).
(I and J) Intracellular cytokines of indicated TILs staining for TNFα, IFNγ and IL-2 after stimulation with PMA and Ionomycin. Values indicate the frequency of WT or CD38 knockout OT-1 cells coproducing IFNγ and TNFα, or producing IL-2 for individual mice (left) and multiple mice (right).
CD38 deficiency impacts on survival rather than exhaustion differentiation of the transferred T cells
To avoid any effects caused by different host microenvironments, we utilized a cotransfer strategy, in which a 1:1 ratio of WT and Cd38−/− OT-1 cells were transferred into the same B16-OVA bearing mice (Figures 5A and S4A). After two weeks, the ratio of CD38 knockout OT-1 cells was significantly decreased both in the spleen, DLN, and the tumor tissues (Figure 5B). Moreover, both WT and CD38 knockout cells were differentiated into memory phenotype and highly capable of producing cytokines in the spleen (Figure S4B). We further analyzed the phenotype of mixed transferred cells in the tumor and observed that although CD38-deficient OT-1 cells displayed a significant reduction in PD-1 expression, the percentage of TEXH and TPRO was similar to WT OT-1 cells (Figures 5C and 5D). Moreover, consistent with the separate adoptive transfer model, all tumor-infiltrated OT-1 cells exhibited comparable cytokine production of TNFα and IFNγ, indicating that CD38 deficiency did not prevent the differentiation of TEXH in the TME (Figure 5E).
Figure 5.

Reduced competitive advantage but similar exhaustion differentiation of Cd38−/− OT-1 cells during cotransfer model
(A) Schematic representation of co-adoptive transfer T cells in the tumor model.
(B) Upper: representative FACS plots of WT and CD38 knockout OT-1 cells in total transferred OT-1 cells of recipient mice spleen, DLN, and tumor. Down: kinetics of the cotransferred OT-1 cells in recipient mice spleen, DLN, and tumor at 14 days post-i.v., respectively. Data are shown as Mean ± SD (n = 5), Student’s t test, ∗∗∗∗: p < 0.0001.
(C) Representative FACS plots of PD-1 and TIM-3 expression in indicated OT-1 TILs (left). Values indicate the frequency of PD-1+TIM-3+ or relative MFI levels of PD-1 in OT-1 TILs (right). Data are shown as Mean ± SD (n = 5), Student’s t test, ∗∗∗∗: p < 0.0001; ns: not significant.
(D) Expression of Ly108 and TIM-3 in WT or Cd38−/− OT-1 TILs for the individual as shown on the left. Summary of the Ly108+TIM-3- TPRO and Ly108−TIM-3+ TEXH frequencies of indicated OT-1 TILs for multiple mice as shown on right. Data are shown as Mean ± SD (n = 5), Student’s t test, ns: not significant.
(E) Left: representative contour plots of indicated OT-1 TILs producing cytokines TNFα, IFNγ after stimulation with PMA and Ionomycin. Right: summary of the relative levels of OT-1 TILs producing cytokines. Data are shown as Mean ± SD (n = 5), Student’s t test, ns: not significant.
(F) Schematic representation of co-adoptive transfer model.
(G) Kinetics of the cotransferred OT-1 cells in the blood post-primary activated by OVA peptide. Mean ± SD (n = 5), Student’s t test, ∗∗∗∗: p < 0.0001; ∗∗∗: p < 0.001. (H) Naive WT or Cd38−/− OT-1 cells stained with CFSE and then co-adoptive transferred in recipient mice with OVA peptide stimulation. FACS analysis of CFSE signal in indicated OT-1 cells from recipient mice spleen at day 3 and 6 post OVA peptide injection.
Of note, the percentage of CD38-deficient cells was lower in this co-transfer model, we thus explored whether this decreased level of CD38-deficient cells was because of impaired migration, proliferation, or apoptosis of WT and Cd38−/− OT-1 cells after antigen stimulation in vivo (Figure 5F). First, the migration of OT-1 cells was not affected in the absence of CD38 (Figure S4C) as measured 24 h after being transferred. We noticed that the ratios of WT and Cd38−/− OT-1 cells were changed in the early stage and then kept constant in the spleen (Figure 5G). Next, we compared the proliferation of transferred cells at different time points and found that WT cells displayed slightly increased proliferation ability than Cd38−/− OT-1 cells at the early stage (3th day) after antigen stimulation (Figures 5H and S4D). In addition, the expression of antiapoptosis protein BCL-2 and the frequency of Annexin V+ apoptotic cells were comparable in transferred cells on sixth day (Figures S4E and S4F),suggesting that CD38 deficiency mainly affected the CD8+ T cell expansion at early stage after antigen stimulation, thus resulting in decreased accumulation of Cd38−/− OT-1 in the cotransfer model.
Ectopic expression of CD38 does not impact T cell function and differentiation
Because Cd38−/− mice are generated by CRISPR-Cas9 method with a potential risk of uncertain off-target effect, and CD8+ T cells are derived from the environment in which all cells lack the expression of CD38, we thus took advantage of retroviral transduction to overexpress CD38 in OT-1 cells to further confirm the function of CD38 in regulating T cell exhaustion within the TME. Notably, overexpression of CD38 in Cd38−/− OT-1 cells neither altered the frequency of the CD62L+CD44+ population nor increased the DNA damage in vitro (Figures 6A, 6B, and 6C). In addition, CD38 overexpressing OT-1 cells were capable of producing cytokines including TNFα, IFNγ, and IL-2 in vitro (Figures 6D and 6E). The ACT experiment further supported the notion that enforced expression of CD38 did not impact T cell exhaustion in the TME (Figure S5A). All adoptively transferred OT-1 cells with vector or CD38 overexpression displayed similar phenotype and cytokine production ability in the spleen of recipients (Figures 6F, 6G, and S5B). In the tumor tissue, we confirmed much higher CD38 levels with ectopic expression in Cd38−/− OT-1 cells than the physiological levels of CD38 in WT OT-1 cells (Figure 6H). However, the frequency of PD-1+TIM-3+ population was comparable between all groups (Figure 6I). Likewise, the proportions of transferred cells with dual TNFα and IFNγ production were similar in tumors (Figure 6J). In this regard, enforced upregulation of CD38 in CD8+ T cells did not critically regulate T cell functionality or exhaustion differentiation.
Figure 6.

Enforced CD38 expression did not impact the exhaustion differentiation of adoptively transferred OT-1 cells
Thy1.1-Vector or Thy1.1-CD38 were ectopically expressed in Cd38−/− OT-1 cells by retrovirus infection.
(A) Histogram of CD38 expression in Thy1.1+ subsets of indicated two groups.
(B) Representative FACS plots of CD62L and CD44 expression in Vector and CD38 expressed Cd38−/− OT-1 cells (left). Percentage of CD62L+CD44+ Thy1.1+ cells in two groups (right). Mean ± SD (n = 3), Student’s t test, ns: not significant.
(C) Flow cytometry analysis of γ-H2AX expression in indicated two groups.
(D and E) Intracellular cytokines staining for TNFα, IFNγ, and IL-2 in Vector or CD38-overexpressed OT-1 cells at day 3 postinfection (left). Values indicate the TNFα+IFNγ+ or IL-2+ frequencies of two groups (right). Mean ± SD (n = 3), Student’s t test, ns: not significant.
(F and G) The three groups of WT OT-1 cells with Thy1.1-Vector expression, Cd38−/− OT-1 cells with Thy1.1-Vector or Thy1.1-CD38 expression were adoptive transferred in B16-OVA tumor-bearing mice, respectively. Summary of CD62L+CD44+ Thy1.1+, or TNFα+IFNγ+ Thy1.1+ frequencies of indicated transferred OT-1 cells in recipient mice spleen. Data are shown as Mean ± SD (n = 5), Student’s t test, ns: not significant.
(H) Flow cytometry analysis of CD38 expression in indicated Thy1.1+ OT-1 TILs.
(I) Representative FACS plots of PD-1 and TIM-3 expression in indicated OT-1 TILs (left). Values indicate the frequency of PD-1+TIM-3+ in OT-1 TILs (right). Data are shown as Mean ± SD (n = 5), Student’s t test, ns: not significant.
(J) Left: representative contour plots of indicated OT-1 TILs producing cytokines TNFα, IFNγ. Right: summary of the frequencies of indicated Thy1.1+ OT-1 TILs producing cytokines. Data are shown as Mean ± SD (n = 3), Student’s t test, ns: not significant.
Genetic deficiency of CD38 fails to rescue the degradation of NAD+ in the TME
Despite the futility of CD38 overexpression in CD8+ T cells, the role of CD38 in antigen chronic stimulation experiment in vitro was inconsistent with ACT experiment in vivo. Therefore, we used the NAD/NADH assay kit and FiNad sensor in CD8+ T cells to measure NAD+, which was a key mediator for energy metabolism and signal transduction but generally downregulated by CD38 (Canto et al., 2015). To begin with, we confirmed that the NAD+ levels in Cd38−/− liver and spleen were significantly upregulated (Figure 7A). In addition, we validated that FiNad sensor—a genetically encoded fluorescent indicator—can rapidly and specifically denote the intracellular NAD+/AXP dynamics in various organisms by assessing the green/red fluorescence ratio (Zou et al., 2020). We treated WT OT-1 cells with NAD+ precursor nicotinamide mononucleotide (NMN) and found NMN treatment could indeed increase the normalized green/red ratio without affecting the AXP (ATP + ADP) levels (Figures S6A, S6B, and S6C), suggesting that FiNad sensor could indicate the NAD+ changes in CD8+ T cells together with AXP level measurements. Next, we further measured the NAD+ levels in OT-1 cells overexpressing CD38 via FiNad sensor and NAD/NADH assay kit and demonstrated that CD38 overexpression would significantly downregulate the NAD+ levels (Figures 7B, 7C, and 7D). Moreover, consistent with low expression of CD38 in normal culture conditions, the NAD+ levels in WT OT-1 cells had no significant differences from CD38-deficient cells in vitro, but the all-trans retinoic acid (ATRA) treated-WT cells could induce the decrease of NAD+ levels with the CD38 upregulation (Figure S6D). Notably, CD38 overexpression caused the decrease of NAD+ levels but did not alter T cell phenotype and DNA damage in Figure 6, indicating that NAD+ degradation alone is likely not enough to induce T cell dysfunction independent of antigen chronic stimulation.
Figure 7.

The dynamic NAD+ levels of Cd38−/− OT-1 cells in different models
(A) Left: livers from 5-month-old male WT or Cd38−/− mice were lysed by cold lysis buffer and the NAD+ levels were then measured and quantified according to the standard curve. Right: spleens from indicated mice were harvested and red cells were removed by RBC lysis, then an equal number of indicated cells were collected for NAD+ measurements. Data are shown as Mean ± SD (n = 3), Student’s t test, ∗∗: p < 0.01; ∗: p < 0.05.
(B) Representative FACS plots of mCherry-YFP and mCherry-FiNad fluorescence intensity in Vector or CD38 expressed Cd38−/− OT-1 cells.
(C) Left: comparing fold changes of the normalized mCherry-FiNad (green to red) ratios in indicated mCherry+ OT-1 cells. Data are shown as Mean ± SD (n = 5), Student’s t test, ∗∗: p < 0.01. Right: Vector or CD38 expressed Cd38−/− OT-1 cells were collected and detected ATP levels and ADP/ATP ratio. Calculating and summarizing the relative AXP ([ATP]+[ATP × ADP/ATP ratio]) levels of two groups. Data are shown as Mean ± SD (n = 4), Student’s t test, ns: not significant.
(D) Measurement of NAD+ levels with indicated Cd38−/− OT-1 cells. Data are shown as Mean ± SD (n = 3), Student’s t test, ∗: p < 0.05.
(E) Quantification of the relative NAD+ levels from the chronically stimulated cells, according to the values of AXP and normalized mCherry-FiNad (green to red) ratios as shown in Figure S6F. Data are shown as Mean ± SD (n = 3), Student’s t test, ∗∗∗∗: p < 0.0001; ∗∗: p < 0.01; ns: not significant.
(F) Representative FACS plots of mCherry-FiNad fluorescence intensity in mCherry+ WT or Cd38−/− OT-1 cells from recipient mice spleen or tumor.
(G) Summary of the relative green to red ratios of mCherry-FiNad in indicated mCherry+ OT-1 cells. Data are shown as Mean ± SD (n = 5), Student’s t test, ns: not significant.
(H) The B16-F10 tumors were implanted in WT or Cd38−/− C57BL/6 female mice, tumor infiltrated CD8+ T cells were purified by PE-anti-CD8a antibody and anti-PE beads at day 15 post engraftment. The NAD+ levels were measured as indicated in methods. Data are shown as Mean ± SD (n = 3), Student’s t test, ns: not significant.
(I and J) qPCR experiment showed the relative mRNA levels of Bst1, Sarm1, Slc12a8 and Nampt in WT or Cd38−/− OT-1 TILs. Mean ± SD (n = 3), Student’s t test, ∗∗: p < 0.01; ns: not significant.
With chronic antigen stimulation model, we observed that AXP levels were decreased both in WT and Cd38−/− OT-1 cells (Figure S6F), and the normalized green/red ratio of FiNad sensor was also evidently decreased in OT-1 cells under chronic antigen stimulation, but deficiency of CD38 partially inhibited this trend (Figures S6E and S6F). Thus, calculating the relative NAD+ levels from FiNad sensor and AXP results confirmed that CD38 knockout partially relieved NAD+ degradation under chronic antigen stimulation in vitro (Figure 7E). However, the FiNad sensor revealed comparable NAD+/AXP in mixed transferred cells regardless of CD38 deficiency or not (Figures 7F, 7G, and S6G). Moreover, we demonstrated that NAD+ levels are comparable between B16-F10 tumor-infiltrated WT and Cd38−/− CD8+ T cells with NAD+ assay kit (Figures 7H and S6H), suggesting that the alternations of NAD+ levels in CD8+ TILs were not solely dependent on CD38 activity in vivo.
Further investigations indeed showed that the transcription levels of the other two NADase, CD38 homologue CD157 (encode by Bst1) and Sarm1, were significantly higher in tumor infiltrated Cd38−/− OT-1 cells than WT cells (Figure 7I). However, the expression of NMN transporter Slc12a8 (Grozio et al., 2019) and nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme of the NAD+ salvage pathway (Burgos, 2011), was similar in two genotypes (Figure 7J). Altogether, those findings supported the fact that depletion CD38 in OT-1 cells could partially rescue NAD+ levels in vitro treatment but not in the TME, which might induce other NADase compensating effects.
Discussion
NAD+ supplementation has been proved to promote CD8+ TILs-mediated cytotoxicity by improving mitochondria metabolism and maintaining appropriate redox balance (Vardhana et al., 2020; Yu et al., 2020). Recently, Wang et al. reported that the disturbance of NAD+ metabolism impacted the activation and tumor-killing function of CD8+ T cells in vivo (Wang et al., 2021). As one of the well-known NAD+ consuming enzymes, CD38 was positively correlated with the dysfunctionality of CD8+ T cells in the tumor (Philip et al., 2017; Verma et al., 2019). However, it remains unclear whether targeting CD38 on CD8+ TILs could increase the efficacy of antitumor adoptive T cell therapy. In this study, we found that CD38 deficiency partially rescued the NAD+ consumption and T cell dysfunctional phenotype caused by TCR persistent stimulation in vitro. However, the efficacy of adoptive transferred Cd38−/− OT-1 cells in the B16-OVA tumor model was not increased. Moreover, CD38 overexpression in CD8+ T cells did not impact the exhaustion state of T cells in the TME. In brief, our data supported that CD38 was a biomarker of TEXH, but it was not an effective target for preventing the terminal exhaustion of CD8+ TILs in the tumor.
The upregulation of CD38 was induced by multiple factors in mammalian cells. In tumor cells, ATRA and IFN-β in the TME could increase the CD38 expression, which possibly mediated the resistance to ICB treatment (Chen et al., 2018). We also observed that ATRA greatly upregulated CD38 expression in CD8+ T cells (Figure S6D). Moreover, it was well shown that the CD38 expression in pro-inflammatory M1-like or tissue-resident macrophages was upregulated by senescent cells-secreted inflammation cytokines, leading to declined NAD+ concentration in metabolic tissues (Chini et al., 2020; Covarrubias et al., 2020). In addition, PD-1+CD38hiCD8+ T cells that result in resistance to anti-PD-1 antibody and therapeutic failure was induced by PD-1 blockade under unprimed or suboptimally primed CD8+ T cell conditions (Verma et al., 2019). In-depth profiling of the chromatin states of tumor-specific T cells via ATAC-seq demonstrated that the high expression of CD38 in fixed exhausted CD8+ T cells was associated with discrete dysfunctional chromatin states (Philip et al., 2017). Moreover, accumulated evidence has indicated that the prolonged antigen exposure contributed to epigenetic and transcriptomic reprogramming of exhausted CD8+ T cells (Ghoneim et al., 2017; Vardhana et al., 2020). Therefore, we compared the persistent antigenic stimulation induced effects of CD38 in CD8+ T cells with other two major factors in the TME—hypoxia and glucose restriction—and verified that only chronic antigen stimulation could significantly increase the CD38 levels.
Yet, our data (Figure S1E) and previous studies have shown that persistent antigenic stimulation could induce the CD8+ T cell dysfunction in vitro, which mimics the key exhaustion features of T cells in the TME (Vardhana et al., 2020). However, we obtained conflicting conclusions from antigen chronic stimulation treatment in vitro and adoptive transfer of Cd38−/− OT-1 cells in the B16-OVA model. First, deficiency of CD38 fails to reverse CD8+ T dysfunction in vivo was not caused by the development difference between Cd38−/− and WT OT-1 cells as shown in Figure 2. Second, the CD38-deficient cells kept similar disadvantages of proliferation in vitro and in vivo at the early stage after antigen stimulation (Figures 2G and 5H). Third, ectopic expression of CD38 in ACT experiments did not regulate adoptively transferred OT-1 cell differentiation and effector function both in spleen or tumor (Figures 6F–6J), further confirming that CD38 did not play an instructive role in driving CD8+ TIL exhaustion.
In addition, the NAD+ metabolism is not only regulated by CD38 but also modulated by synthesis pathways and other consuming enzymes (Chiarugi et al., 2012). Therefore, we investigated the expression pattern of the other two NADases and key factors in NAD+ salvage pathway. As a result, we observed that the transcript levels of NADase, Bst1 and Sarm1, were specifically higher in tumor infiltrated Cd38−/− OT-1cells than WT cells but similar in the spleen of adoptively transferred cells (Figure S6I). In addition, the transcription levels of Bst1 and Sarm1 were even decreased in CD38-deficient cells when treated with chronic antigen stimulation in vitro (Figure S6J), suggesting that antigen persistent stimulation could not totally mimic the exhaustion of T cells within the TME.
In conclusion, our present study aimed to illustrate the cell-autonomous role of CD38 in T cell exhaustion, and we further explored the possibility of targeting CD38 on CD8+ T cells in adoptive T cell therapy. Surprisingly, we found that preventing NAD+ degradation in CD8+ T cells by deletion of CD38 could partially relieve the T cell dysfunction that was induced by antigen chronic stimulation in vitro, but NAD+ levels and the exhausted phenotypes of adoptively transferred T cells were not changed with CD38 deficiency or overexpression. In this regard, the genetic absence of PD-1 had a similar conclusion, which did not inhibit but rather promote the accumulation of terminally differentiated exhausted CD8+ T cells (Odorizzi et al., 2015).
Limitations of the study
In this study, we utilized FiNad sensor to better understand how changes of NAD+ levels under different T cell differentiation settings (Zou et al., 2020). Yet, the FiNad sensor is reflecting the NAD+/AXP ratio, which is related to the changes of AXP levels. Thus, the measurements with FiNad sensor in the tumor cotransferred OT-1 cells are likely indicative of NAD+/AXP ratio (Figures 7F and 7G). In addition, it is worth noting that the decrease of NAD+ levels in CD8+ TILs is not solely regulated by CD38, which deficiency may compensate for upregulating the expression of other NADase. Notably, intracellular NAD+ levels are highly coordinated by the consumption processes and biosynthesis, such as de novo synthesis and salvage pathways (Morandi et al., 2021). Recently, Wang et al. reported that the downregulation of NAMPT—the rate-limiting enzyme of NAD+ salvage pathway—could decrease NAD+ levels in tumor-infiltrated T cells (Wang et al., 2021). Thus, it remains to be addressed whether simultaneous targeting of several NADase is able to relieve CD8+ T cell exhaustion state. Moreover, it is also possible that the expression of NADase in other types of cells within the TME may further decrease NAD+ levels, which subsequently induces or stabilizes the exhaustion state of tumor infiltrated CD8+ T cells.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Brilliant Violet 711-anti-CD8a | Biolegend | Cat# 100748; RRID: AB_2562100 |
| FITC-anti-CD4 | Biolegend | Cat# 100406; RRID: AB_312691 |
| APC-anti-CD45R | Biolegend | Cat# 103212; RRID: AB_312997 |
| PE-Cy7-anti-CD19 | Biolegend | Cat# 115520; RRID: AB_313655 |
| PB-anti-CD38 | Biolegend | Cat# 102720; RRID: AB_10613468 |
| FITC-anti-CD69 | Biolegend | Cat# 104506; RRID: AB_313109 |
| APC-anti-CD62L | Biolegend | Cat# 104412; RRID: AB_313099 |
| Percp-eflour 710-anti-PD-1 | Invitrogen | Cat# 46-9981-82; RRID: AB_11151142 |
| PE-Cy7-anti-TIM-3 | Invitrogen | Cat# 25-5870-82; RRID: AB_2573483 |
| APC-anti-Ly108 | Biolegend | Cat# 134610; RRID: AB_2728155 |
| PE-anti-CD45.1 | Biolegend | Cat# 110708; RRID: AB_313497 |
| PerCP-Cy5.5-anti-CD45.1 | Biolegend | Cat# 110728; RRID: AB_893346 |
| FITC-anti-CD45.2 | Invitrogen | Cat# 11-0454-85; RRID: AB_465062 |
| PB-anti-CD45.2 | Biolegend | Cat# 109820; RRID: AB_492872 |
| APC-Cy7-anti-CD44 | Biolegend | Cat# 103028; RRID: AB_830785 |
| AF700-anti-Thy1.1(CD90.1) | Biolegend | Cat# 202528; RRID: AB_1626241 |
| FITC-anti-TNAα | Biolegend | Cat# 506304; RRID: AB_315425 |
| APC-anti-IFNγ | Invitrogen | Cat# 17-7311-82; RRID: AB_469504 |
| PE-IL-2 | Biolegend | Cat# 503808; RRID: AB_315302 |
| Alexa Fluor647-anti-TCF-1 | Cell Signaling Technology | Cat# 6709S; RRID: AB_2797631 |
| PE-TOX | Invitrogen | Cat# 12-6502-82; RRID: AB_10855034 |
| PerCP-Cy5.5-anti-Ki67 | Biolegend | Cat# 652424; RRID: AB_2629531 |
| anti-γH2AX | Cell Signaling Technology | Cat# 2577S; RRID: AB_2118010 |
| Chemicals, peptides, and recombinant proteins | ||
| PBS | HyClone | Cat# SH30013.04 |
| DMEM Media (High Glucose) | Gibco | Cat# C11995500CP |
| RPMI Medium (1640) | Gibco | Cat# 11875-093 |
| Fetal Bovine Serum (FBS) | Royacel | Cat# RY-F22-05 |
| Penicillin-Streptomycin Solution | Hyclone | Cat# SV30010 |
| Trypsin-EDTA | Gibco | Cat# 25200-072 |
| Hepes Buffer Solution | Bioind | Cat# 03-025-1B |
| Sodium Pyruvate | Gibco | Cat# 11360-070 |
| L-Glutamine | Gibco | Cat# 25030081 |
| MEM Non-Essential Amino Acids Solution | Gibco | Cat# 11140-050 |
| β-mercaptoethanol | Sigma | Cat# B6891 |
| Live FDV506 | Invitrogen | Cat# 65-0866-18 |
| CFSE | Selleck | Cat# S8269 |
| Cell Proliferation Dye 450 | Invitrogen | Cat# 65-0842-85 |
| Mito-Sox | Invitrogen | Cat# M36008 |
| PMA | Sigama | Cat# P8139-5MG |
| Ionomycin | Fcmacs | Cat# FMS-FZ208 |
| eBioscience Brefeldin A | Biolegend | Cat# 420601 |
| Fixation Buffer | Biolegend | Cat# 420801 |
| Permeabilization Buffer | Biolegend | Cat# 421002 |
| Foxp3/Transcription Factor Staining Buffer | Invitrogen | Cat# 00-5223-56; Cat# 00-5123-43 |
| Foxp3/Transcription Factor Permeabilization Buffer | Invitrogen | Cat# 00-8333-56 |
| TransIT-293 | Mirusbio | Cat# MIR2700 |
| Polybrene | Santa Cruz | Cat# SC-134220 |
| PerfectStart Green qPCR SuperMix | TransGen Bitoech | Cat# AQ601-04 |
| NMN | Yeasen Biotechnology | Cat# 60303ES80 |
| NAD+ | MCE | Cat# HY-B0445 |
| Ficoll-Paque | GE | Cat# 17-1440-03 |
| Percoll | Cytiva | Cat# 17089109 |
| OVA peptides | Abcepta Biotech | N/A |
| Human IL-2 | Pepro Tech | Cat# 200-02-500 |
| Human IL-7 | Novoprotein | Cat# CX47 |
| RetroNectin Recombinant Human Fibronectin Fragment | TaKaRa | Cat# T100A |
| Critical commercial assays | ||
| Negative CD8 Sorting kit | Biolegend | Cat# 480044 |
| PI/Annexin V staining kit | Life technologies | Cat# V13245 |
| Anti-PE | Biolegend | Cat# 480080 |
| PrimeScript RT Master Mix | TaKaRa | Cat# RR036B |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| NAD+/NADH Assay kit | Beyotime | Cat# S0175 |
| ATP Assay kit | Beyotime | Cat# S0026 |
| ADP/ATP -Lite Assay kit | Vigorous Biotechnology | Cat# T008 |
| Experimental models: Cell lines | ||
| 293FT | ATCC | CRL-3249 |
| B16-F10 | ATCC | CRL-6475 |
| B16-OVA | Bo Huang Lab | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6 | Vital River | N/A |
| Oligonucleotides | ||
| Cas9 for Cd38 see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pMSGV | Addgene | Cat# 64269 |
| pMSGV-Thy1.1 | This paper | N/A |
| pMSGV-Thy1.1-CD38 | This paper | N/A |
| pLVX-mCherry-YFP | Yuzheng Zhao Lab | N/A |
| pLVX-mCherry-FiNad | Yuzheng Zhao Lab | N/A |
| pMSGV-mCherry-YFP | This paper | Recombinated form pLVX-mCherry-YFP |
| pMSGV-mCherry FiNad | This paper | Recombinated form pLVX-mCherry-FiNad |
| pEco | Clontech | Cat# HG-VNC0832 |
| Software and algorithms | ||
| Prism8 | Graphpad-prism | Prism - GraphPad |
| Flowjo | BD | FlowJo | FlowJo, LLC |
| GEPIA 2 online tool | Zemin Zhang Lab | http://gepia2.cancer-pku.cn/#correlation |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Lianjun Zhang (zlj@ism.cams.cn).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
Animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Suzhou Institute of System Medicine (ISM-IACUC-0018 and ISM-IACUC-0055). Female C57BL/6J CD38 knockout mice were kindly provided by Prof. Xiaofeng Qin's laboratory. In brief, the mouse line was generated by Nanjing Biomedical Research Institute of Nanjing University, China, with the CRISPR-Cas9 based genetic engineering strategy. The four sgRNA (sequence shown in Table S1) direct Cas9 endonuclease cleavage in intron1-2 and intron 3-4 of Cd38 gene and create a DSB (double strand break). Such breaks will be repaired by non-homologous end joining (NHEJ), which will lead to the disruption of Cd38. The genotype identification of Cd38-/- mice by sequencing the edited genomic regions confirmed Cd38-/- mice with the complete deletion of CD38 exons 2 and 3. The CD45.1+ or CD45.2+ OT-1 TCR transgenic male and female mice on a C57BL/6N background were housed. CD45.1+ Cd38-/- mice crossing with CD45.1+ or CD45.2+ OT-1 TCR transgenic mice to generate CD45.1+ or CD45.1/2+ Cd38-/- OT-1 mice. CD45.2+ female C57BL/6N mice (6-8 weeks old, WT) were purchased from Vital River Co, Ltd (Beijing, China) as recipients. All of the mice were bred and maintained under specific pathogen-free conditions in the animal facility of Suzhou Institute of Systems Medicine (Suzhou, China).
Cell lines
293FT cells were grown in DMEM supplemented with 10% FBS and Pen/Strep at 37°C with 5% CO2. B16-F10 or B16-OVA cells were cultured in RPMI medium supplemented with 10% FBS and Penicillin-Streptomycin and grown in the same conditions as noted above.
CD8+ T cell activation and culture
WT and Cd38-/- CD8+ T cells were sorted from the spleens of male or female WT or Cd38-/- mice (6-8 weeks old) by commercial kit. 1 × 106 CD8+ T cells were plated into each well of 48-well plate in 1 mL RPMI medium with IL-2 (10 ng/mL), which was precoated with αCD3 plus αCD28 antibodies. For activating OT-1 splenocytes, 2 × 106 splenocytes of indicated OT-1 mice were plated into each well of 24-well plate in 2 mL medium with IL-2 and OVA peptide (1 μg/mL). On 3rd day, dead cells were removed by Ficoll-Paque and the remaining live CD8+ T cells were further cultured in the medium containing IL-2 and IL-7 (10 ng/mL). The culture RPMI medium contains 10% FBS, Penicillin-Streptomycin, Hepes (10 mM), Sodium Pyruvate (1 mM), MEM Non-Essential Amino Acids (1 ×), L-Glutamine (2 mM), and β-mercaptoethanol (50 μM).
Antigen chronic stimulation in vitro
1 × 106 activated OT-1 cells were plated into each well of 24-well plate in 2 mL medium with IL-2, and IL-7. Meanwhile, cells were treated with OVA peptide (10 ng/mL) every other day for 4 days. The chronic phenotype was confirmed by upregulation of IRs and downregulation of cytokine production.
Tumor implantation and adoptive T cell therapy
Tumors were implanted in C57BL/6 female mice by injecting 0.1 × 106 B16-OVA tumor cells per mouse s.c. into the right flank at day 0. When tumors measured approximately 4–5 mm in diameter, equal numbers of indicated cells (2 × 106 per mice) from female WT or Cd38-/- OT-1 mice (6-8 weeks old) were transferred (i.v.) into recipient mice at the day after irradiation (5 Gy). Tumors were measured every 3 days using a digital Vernier caliper; tumor volume was calculated using the formula V = (L × W2)/2, where V is tumor volume, L is the length of the tumor (longer diameter) and W is the width of the tumor (shorter diameter).
Co-transfer strategy
Naïve CD8+ T cells were isolated from indicated mice spleen. After the irradiation, WT and CD38 knockout OT-1 cells were mixed at a 1:1 ratio, a total of 4 × 106 cells and transferred into naive recipient mice with s.c. OVA peptide (10 μg per mice) and CPG (30 μg per mice), or B16-OVA tumor bearing mice as indicated.
Method details
DNA cloning
For overexpressing CD38 in OT-1 cells, mouse CD38 was cloned into Xho I and HindIII sites of the pMSGV-Thy1.1 vector. For construction of retrovirus vectors of FiNad sensor, the mCherry-YFP and mCherry-FiNad were cloned into EcoRI and HindIII sites of the pMSGV vector. The primers were listed in Table S1.
Flow cytometry analysis
Single-cell suspension from spleens, lymphocyte nodes, thymus or tumor tissues were prepared. All samples were stained with Live/Dead dye for 20 min on ice. For cell surface staining, all antibodies were diluted 1:200 with FACS buffer (PBS containing 2% FBS) for 25 min on ice. For intracellular cytokine staining, cells were incubated with PMA (10 ng/mL), ionomycin (50 μg/mL), Brefeldin A and Monensin or 3.5 h at 37°C. Then, these cells were fixed with Fixation Buffer for 20 min on ice, permeabilized with Permeabilization Buffer and stained with indicated antibodies. For intracellular transcription factor staining, cells were fixed, permeabilized by using Foxp3/Transcription Factor Staining Buffer and Permeabilization Buffer,and then stained with antibody. All samples were resuspended in FACS buffer and loaded in an LSR Fortessa flow cytometer (Becton-Dickinson, San Jose, CA) and analyzed by FlowJo software.
Retroviral transduction
The helper plasmid pCL-Eco and indicated pMSGV retroviral plasmids were co-transferred into 293FT cells using Trans-IT 293 transfection reagent. The supernatants containing viruses were mixed with anti-CD3/CD28-activated WT or CD38 knockout OT-1 cells (MOI = 5:1) with polybrene (1 μg/mL), and then mixtures were added to a 24-well plate, which precoated with RetroNectin (0.1 mg/mL). After centrifugation at 2000 g for 90 min, cells were cultured at 37°C under 5% CO2 for 24 h. Infected cells were detected by flow cytometry.
Analysis NAD+ levels via FiNad sensor
The mCherry-YFP or mCherry-FiNad were ectopic expressed in indicated OT-1 cells for different treatments. Cells were thus collected for flow cytometry analysis. The ratio of green MFI to red MFI in each group was calculated, and then normalized based on the mCherry-FiNad ratio values to mCherry-YFP ratio values in the same treatment.
Measurement of NAD+ levels
The indicated cells were collected and intracellular NAD+ levels were measured by using NAD+/NADH assay kit (Beyotime, S0175) with WST-8, an upgraded alternative to MTT, according to the manufacturer’s protocol. In brief, the cellular NAD+ were converted to NADH under alcohol dehydrogenase and reaction buffer, then the WST-8 was reverted to formazan under the NADH and 1-mPMS (1-Methoxy-5-methylphenazinium Methyl Sulfate) function. To analyze the total NAD+/NADH contents, the absorbance values were measured at 450 nm and analyzed on a plate reader (SpectraMax I3; Molecular Devices, Shanghai, China). NADH contents were obtained by incubating the cell lysates at 60 °C for 30 min that to remove the NAD+. The NAD+ levels were derived by subtracting NADH from total NAD+/NADH.
Detection of cellular ATP and ADP levels
The equal number of indicated cells were collected and intracellular ATP levels were measured by a commercial ATP assay kit (Beyotime, S0026). Briefly, luciferase catalyzes the conversion of ATP and D-luciferin to light, which is measured by a chemiluminescence type microplate meter (SpectraMax L; Molecular Devices, Shanghai, China). The ADP/ATP ratio in cells was measured by the ADP/ATP-Lite assay kit (Vigorous Biotechnology, Cat# T008). In brief, firefly luciferase catalyzes the conversion of ATP and luciferin to light, and then the ADP was converted to ATP by converting enzyme. The relative light units (RUL) were measured by a single channel microplate meter (SpectraMax L; Molecular Devices, Shanghai, China). ADP levels were derived by ATP levels multiplying by ADP/ATP ratio.
Real-time quantitative-PCR
WT or Cd38-/- OT-1 cells from spleen or tumor tissues were purified by PE-anti-CD45.1 antibody and anti-PE beads. For analyzing in vitro antigen stimulated-OT-1 cells, dead OT-1 cells were removed by Ficoll-Paque. Total RNA of live indicated cells was first isolated using RNeasy Mini Kit, and then subjected to reverse transcription by PrimeScript RT Master Mix Kit. Reactions were run on a real-time PCR system (Roche LC480) using PerfectStart Green qPCR SuperMix. Sequences of primers were listed in Table S1.
Quantification and statistical analysis
Data were analyzed from at least three independent experiments and are shown as the mean ± SD. The comparison of two groups were performed using Student’s two-tailed t-test, with p values < 0.05 (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001) being considered significant in the figures. All analyses were performed with GraphPad Prism software.
Acknowledgments
The authors would like to thank Prof. Yuzheng Zhao and Prof. Yi Yang at East China University of Science and Technology for kindly providing pLVX-mCherry-YFP and pLVX-mCherry-FiNad plasmids. We are also grateful to Prof. Bo Huang from CAMS for kindly providing B16-OVA cells. K.M. was supported by the Special Research Fund for Central Universities, Peking Union Medical College (3332021074) and Natural Science Foundation of China (32100724). L.Z. was in part supported by CAMS Innovation Fund for Medical Sciences (CIFMS 2021-I2M-1-061), the Special Research Fund for Central Universities, Peking Union Medical College (2021-PT180-001), and the National Natural Science Foundation of China grants (81971466). F.G. was supported by Suzhou Municipal Science and Technology Bureau (Grant Number SLJ202011 and SLT201959) and Jiangsu Commission of Health (Grant Number M2020043).
Author contributions
L.Z. and K.M. conceived and designed the experiments. K.M., L.S., and M.S. performed most of the experiments and data analysis with assistance from X.Z., Z.X., and J.W. L.X. helped to maintain the mouse line. K.J., F.Q., and F.G. helped for data analysis and interpretation. L.Z., F.G., and B.Z. supervised the study. K.M., B.Z., and L.Z. wrote the manuscript. All authors provided intellectual input to the manuscript.
Declaration of interests
The authors declare no competing interests.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104347.
Contributor Information
Feng Guo, Email: guofeng27@gmail.com.
Baojun Zhang, Email: bj.zhang@mail.xjtu.edu.cn.
Lianjun Zhang, Email: zlj@ism.cams.cn.
Supplemental information
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. The analysis of the correlation between the CD38 and exhausted T-cell signature genes in human was performed using the GEPIA 2 online tool (http://gepia2.cancer-pku.cn/#correlation). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Beltra J.C., Manne S., Abdel-Hakeem M.S., Kurachi M., Giles J.R., Chen Z., Casella V., Ngiow S.F., Khan O., Huang Y.J., et al. Developmental relationships of four exhausted CD8(+) T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity. 2020;52:825–841.e8. doi: 10.1016/j.immuni.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos E.S. NAMPT in regulated NAD biosynthesis and its pivotal role in human metabolism. Curr. Med. Chem. 2011;18:1947–1961. doi: 10.2174/092986711795590101. [DOI] [PubMed] [Google Scholar]
- Canto C., Menzies K., Auwerx J. NAD(+) metabolism and the control of energy homeostasis: a balancing act between mitochondria and the nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S., Daenthanasanmak A., Chakraborty P., Wyatt M.W., Dhar P., Selvam S.P., Fu J., Zhang J., Nguyen H., Kang I., et al. CD38-NAD(+)Axis regulates immunotherapeutic anti-tumor T cell response. Cell Metab. 2018;27:85–100.e8. doi: 10.1016/j.cmet.2017.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L., Diao L., Yang Y., Yi X., Rodriguez B.L., Li Y., Villalobos P.A., Cascone T., Liu X., Tan L., et al. CD38-Mediated immunosuppression as a mechanism of tumor cell escape from PD-1/PD-L1 blockade. Cancer Discov. 2018;8:1156–1175. doi: 10.1158/2159-8290.CD-17-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Ji Z., Ngiow S.F., Manne S., Cai Z., Huang A.C., Johnson J., Staupe R.P., Bengsch B., Xu C., et al. TCF-1-Centered transcriptional network drives an effector versus exhausted CD8 T cell-fate decision. Immunity. 2019;51:840–855.e5. doi: 10.1016/j.immuni.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H., Ma K., Zhang L., Li G. The tumor microenvironment shapes the molecular characteristics of exhausted CD8(+) T cells. Cancer Lett. 2021;506:55–66. doi: 10.1016/j.canlet.2021.02.013. [DOI] [PubMed] [Google Scholar]
- Chiarugi A., Dolle C., Felici R., Ziegler M. The NAD metabolome--a key determinant of cancer cell biology. Nat. Rev. Cancer. 2012;12:741–752. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- Chini C.C.S., Peclat T.R., Warner G.M., Kashyap S., Espindola-Netto J.M., de Oliveira G.C., Gomez L.S., Hogan K.A., Tarrago M.G., Puranik A.S., et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab. 2020;2:1284–1304. doi: 10.1038/s42255-020-00298-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covarrubias A.J., Kale A., Perrone R., Lopez-Dominguez J.A., Pisco A.O., Kasler H.G., Schmidt M.S., Heckenbach I., Kwok R., Wiley C.D., et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat. Metab. 2020;2:1265–1283. doi: 10.1038/s42255-020-00305-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Q., Zhang H., Zheng J., Zhang L. Turning cold into hot: firing up the tumor microenvironment. Trends Cancer. 2020;6:605–618. doi: 10.1016/j.trecan.2020.02.022. [DOI] [PubMed] [Google Scholar]
- Gao L., Liu Y., Du X., Ma S., Ge M., Tang H., Han C., Zhao X., Liu Y., Shao Y., et al. The intrinsic role and mechanism of tumor expressed-CD38 on lung adenocarcinoma progression. Cell Death Dis. 2021;12:680. doi: 10.1038/s41419-021-03968-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoneim H.E., Fan Y., Moustaki A., Abdelsamed H.A., Dash P., Dogra P., Carter R., Awad W., Neale G., Thomas P.G., Youngblood B. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. 2017;170:142–157.e19. doi: 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golstein P., Griffiths G.M. An early history of T cell-mediated cytotoxicity. Nat. Rev. Immunol. 2018;18:527–535. doi: 10.1038/s41577-018-0009-3. [DOI] [PubMed] [Google Scholar]
- Grozio A., Mills K.F., Yoshino J., Bruzzone S., Sociali G., Tokizane K., Lei H.C., Cunningham R., Sasaki Y., Migaud M.E., Imai S.I. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019;1:47–57. doi: 10.1038/s42255-018-0009-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Segura A., Nehme J., Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–453. doi: 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- Katsuyama E., Suarez-Fueyo A., Bradley S.J., Mizui M., Marin A.V., Mulki L., Krishfield S., Malavasi F., Yoon J., Sui S.J.H., et al. The CD38/NAD/SIRTUIN1/EZH2 Axis mitigates cytotoxic CD8 T cell function and identifies patients with SLE prone to infections. Cell Rep. 2020;30:112–123.e4. doi: 10.1016/j.celrep.2019.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan O., Giles J.R., McDonald S., Manne S., Ngiow S.F., Patel K.P., Werner M.T., Huang A.C., Alexander K.A., Wu J.E., et al. TOX transcriptionally and epigenetically programs CD8(+) T cell exhaustion. Nature. 2019;571:211–218. doi: 10.1038/s41586-019-1325-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malavasi F., Deaglio S., Funaro A., Ferrero E., Horenstein A.L., Ortolan E., Vaisitti T., Aydin S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008;88:841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- McLane L.M., Abdel-Hakeem M.S., Wherry E.J. CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 2019;37:457–495. doi: 10.1146/annurev-immunol-041015-055318. [DOI] [PubMed] [Google Scholar]
- Moller S.H., Hsueh P.C., Yu Y.R., Zhang L., Ho P.C. Metabolic programs tailor T cell immunity in viral infection, cancer, and aging. Cell Metab. 2022;34:378–395. doi: 10.1016/j.cmet.2022.02.003. [DOI] [PubMed] [Google Scholar]
- Morandi F., Horenstein A.L., Malavasi F. The key role of NAD(+) in anti-tumor immune response: an update. Front Immunol. 2021;12:658263. doi: 10.3389/fimmu.2021.658263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odorizzi P.M., Pauken K.E., Paley M.A., Sharpe A., Wherry E.J. Genetic absence of PD-1 promotes accumulation of terminally differentiated exhausted CD8+ T cells. J Exp Med. 2015;212:1125–1137. doi: 10.1084/jem.20142237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M., Fairchild L., Sun L., Horste E.L., Camara S., Shakiba M., Scott A.C., Viale A., Lauer P., Merghoub T., et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017;545:452–456. doi: 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping N.E., Rivadeneira D.B., Menk A.V., Vignali P.D.A., Ford B.R., Rittenhouse N.L., Peralta R., Wang Y., Wang Y., DePeaux K., et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021;22:205–215. doi: 10.1038/s41590-020-00834-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sckisel G.D., Mirsoian A., Minnar C.M., Crittenden M., Curti B., Chen J.Q., Blazar B.R., Borowsky A.D., Monjazeb A.M., Murphy W.J. Differential phenotypes of memory CD4 and CD8 T cells in the spleen and peripheral tissues following immunostimulatory therapy. J. Immunother. Cancer. 2017;5:33. doi: 10.1186/s40425-017-0235-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui I., Schaeuble K., Chennupati V., Fuertes Marraco S.A., Calderon-Copete S., Pais Ferreira D., Carmona S.J., Scarpellino L., Gfeller D., Pradervand S., et al. Intratumoral Tcf1(+)PD-1(+)CD8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50:195–211.e10. doi: 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- Tang Z., Kang B., Li C., Chen T., Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47:W556–W560. doi: 10.1093/nar/gkz430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardhana S.A., Hwee M.A., Berisa M., Wells D.K., Yost K.E., King B., Smith M., Herrera P.S., Chang H.Y., Satpathy A.T., et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 2020;21:1022–1033. doi: 10.1038/s41590-020-0725-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- Verma V., Shrimali R.K., Ahmad S., Dai W., Wang H., Lu S., Nandre R., Gaur P., Lopez J., Sade-Feldman M., et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat. Immunol. 2019;20:1231–1243. doi: 10.1038/s41590-019-0441-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Wang F., Wang L., Qiu S., Yao Y., Yan C., Xiong X., Chen X., Ji Q., Cao J., et al. NAD(+) supplement potentiates tumor-killing function by rescuing defective TUB-mediated NAMPT transcription in tumor-infiltrated T cells. Cell Rep. 2021;36:109516. doi: 10.1016/j.celrep.2021.109516. [DOI] [PubMed] [Google Scholar]
- Yu Y.R., Imrichova H., Wang H., Chao T., Xiao Z., Gao M., Rincon-Restrepo M., Franco F., Genolet R., Cheng W.C., et al. Disturbed mitochondrial dynamics in CD8(+) TILs reinforce T cell exhaustion. Nat. Immunol. 2020;21:1540–1551. doi: 10.1038/s41590-020-0793-3. [DOI] [PubMed] [Google Scholar]
- Zou Y., Wang A., Huang L., Zhu X., Hu Q., Zhang Y., Chen X., Li F., Wang Q., Wang H., et al. Illuminating NAD(+) metabolism in live cells and in vivo using a genetically encoded fluorescent sensor. Dev. Cell. 2020;53:240–252.e7. doi: 10.1016/j.devcel.2020.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. The analysis of the correlation between the CD38 and exhausted T-cell signature genes in human was performed using the GEPIA 2 online tool (http://gepia2.cancer-pku.cn/#correlation). This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.