

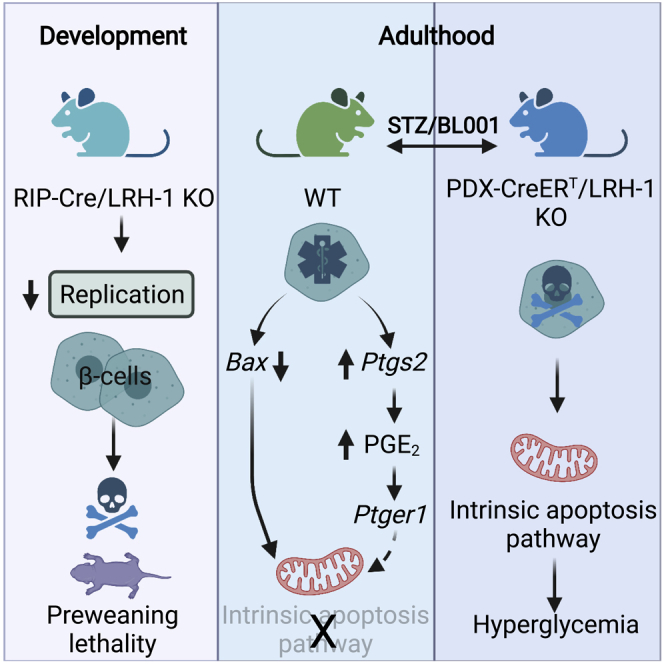
Summary
LRH-1/NR5A2 is implicated in islet morphogenesis postnatally, and its activation using the agonist BL001 protects islets against apoptosis, reverting hyperglycemia in mouse models of Type 1 Diabetes Mellitus. Islet transcriptome profiling revealed that the expression of PTGS2/COX2 is increased by BL001. Herein, we sought to define the role of LRH-1 in postnatal islet morphogenesis and chart the BL001 mode of action conferring beta cell protection. LRH-1 ablation within developing beta cells impeded beta cell proliferation, correlating with mouse growth retardation, weight loss, and hypoglycemia leading to lethality. LRH-1 deletion in adult beta cells abolished the BL001 antidiabetic action, correlating with beta cell destruction and blunted Ptgs2 induction. Islet PTGS2 inactivation led to reduced PGE2 levels and loss of BL001 protection against cytokines as evidenced by increased cytochrome c release and cleaved-PARP. The PTGER1 antagonist—ONO-8130—negated BL001-mediated islet survival. Our results define the LRH-1/PTGS2/PGE2/PTGER1 signaling axis as a key pathway mediating BL001 survival properties.
Subject areas: Physiology, Molecular biology
Graphical abstract
Highlights
-
•
LRH-1 ablation during development impedes neonatal beta cell replication
-
•
LRH-1 knockout in adult beta cells negates BL001-mediated antidiabetic properties
-
•
Islets lacking PTGS2 are refractory to BL001-mediated protection against cytokines
-
•
PTGER1 relays the BL001/LRH-1/PTGS2/PGE2 signaling axis to islet survival
Physiology; Molecular biology
Introduction
Nuclear receptors (NRs) are key factors of a vast number of physiological and pathophysiological processes. At the cellular level, they control energy homeostasis, survival, and plasticity via genetic and epigenetic regulation. At the whole organism level, NRs participate in development, reproduction, metabolism, immune response, and tissue regeneration (Huang et al., 2010). Ligand dependent regulation of NRs activity has forged them as druggable factors with promising therapeutic value for cancerous, metabolic, and immune diseases. Of particular interest is the liver receptor homolog 1 (LRH-1, also known as NR5A2) that belongs to the NR5A family of nuclear receptors (Fayard et al., 2004). LRH-1/NR5A2 is highly expressed in the liver, intestine, and pancreas, whereas lower expression levels are detected in the brain and immune cell types such as macrophages and T cells (Michalek and Brunner, 2021). LRH-1/NR5A2 is essential for embryonic development, whereas in adults it regulates key metabolic pathways. In the liver, LRH-1/NR5A2 modulates the expression of target genes involved in glucose, cholesterol, and bile acid metabolism, attenuates the hepatic acute phase response triggered by pro-inflammatory cytokines, and protects against ER stress (Sun et al., 2021). In the intestine, LRH-1/NR5A2 modulates the enterocyte renewal and regulates the local immune system via production of glucocorticoids (Fernandez-Marcos et al., 2011). In the pancreas, LRH-1/NR5A2 regulates the expression of genes involved in digestive functions and protects the endocrine islets against cytokine- and streptozotocin-induced apoptosis (Baquie et al., 2011; Mellado-Gil et al., 2012). Haploinsufficiency of NR5A2 sensitizes mice to pancreatic cancer highlighting the role of NR5A2/LRH1 as a pancreatic tumor suppressor rather than an oncogenic factor (Cobo et al., 2018, 2021).
Given the role of LRH-1/NR5A2 in beta cell survival, glucose homeostasis, and in attenuating inflammatory processes, attempts to regulate its activity are of important therapeutic value for the treatment of diabetes. Accordingly, administration of dilauroyl phosphatidylcholine (DLPC, the natural ligand of LRH-1/NR5A2) was shown to decrease hepatic steatosis and improve glucose homeostasis in two mouse models of insulin resistance and T2DM (Lee et al., 2011). These results prompted the development of hybrid phospholipid mimics as well as small nonpolar bicyclic compounds that act as potent LRH-1/NR5A2 ligands (Cornelison et al., 2020; Flynn et al., 2018; Whitby et al., 2006). Based on one of these published structures that are able to bind to the ligand binding pocket of NR5A2/LRH1, we synthesized a small chemical agonist of LRH-1/NR5A2, denoted as BL001, and assessed its antidiabetic therapeutic value for T1DM. We found that BL001 treatment either prophylactically or therapeutically reduced the incidence of diabetes in three independent preclinical mouse models of T1DM and protected human islets against stress-induced apoptosis (Cobo-Vuilleumier et al., 2018a, 2018b). These antidiabetic benefits of BL001 were conveyed via immune cell tolerization and regression of insulitis combined with enhanced islet beta cell expansion and survival, a process that we have coined as ‘immune-coupled trans-regeneration’ (Cobo-Vuilleumier and Gauthier, 2020). Such coupling was further substantiated by the finding that most of differentially regulated genes in BL001-treated mouse islets were involved in immunomodulation, indicating a dialogue between islet cells and immune cells (Cobo-Vuilleumier et al., 2018b). Among these target genes stimulated by BL001 was the inducible prostaglandin endoperoxide synthase-2 (PTGS2 a.k.a. COX-2) that has been previously shown to partially protect mouse islets from streptozotocin (STZ)-mediated diabetogenic toxicity as well as to enhance human islet survival in the presence of cytokines through interaction with one or multiple prostaglandin E2 receptors (PTGERs) (Carboneau et al., 2017a; Vennemann et al., 2012). Notwithstanding, other studies contend that inhibition or deletion of PTGS2 shields the human islet against cytokine-mediated beta cell dismay (Ma et al., 2017). Interestingly, in breast cancer cells, the downstream metabolic pathway product of PTGS2/COX2—prostaglandin E2 (PGE2)—induces LRH-1 mRNA expression by recruitment of multiple transcriptional activators to the LRH-1 promoter (Michalek and Brunner, 2021). Taken together, whether the axis LRH1/PTGS2 is of importance in pancreatic beta cells remains to be clarified.
LRH-1/NR5A2 ablation in beta cells using the constitutive RIP-Cre/LRH-1lox/lox mouse model resulted in early lethality of the mice (before weaning) (Cobo-Vuilleumier et al., 2018b), hampering the attempt to demonstrate both the specificity of LRH-1/NR5A2 to convey BL001-mediated islet cell survival and more importantly to delineate its mode of action. Analysis of the pancreatic islets of these mice at P1 (postnatal day 1) revealed a reduced number of pancreatic beta cells, indicating that LRH-1/NR5A2 deletion at the onset of beta cell identity commitment likely perturbs beta cell function or mass (Cobo-Vuilleumier et al., 2018b). Herein, we sought to: 1) further delineate the role of LRH-1/NR5A2 in islet morphology and architecture postnatally, 2) determine whether the antiapoptotic and antidiabetic effects of BL001 are specifically conveyed via LRH-1/NR5A2 activation in beta cells, and 3) delineate the BL001/LRH-1/NR5A2 antiapoptotic mode of action in beta cells with an emphasis on the potential implication of the PTGS2 signaling cascade (Cobo-Vuilleumier and Gauthier, 2020). We report that deletion of LRH-1/NR5A2 blunts neonatal beta cell proliferation consistent with growth retardation, weight loss, and hypoglycemia. Noteworthy, Cre expression and thus LRH-1/NR5A2 deletion was also detected in the brain, suggesting a potential contribution of this organ to the severe phenotype. We further report that LRH-1/NR5A2 deletion in adult beta cells abolishes the antidiabetic properties of BL001, an effect that involves the pro-survival PTGS2/PGE2/PTGER1 signaling axis.
Results
LRH-1/NR5A2 ablation disrupts neonatal beta cell replication
Constitutive deletion of the Lrh1/Nr5a2 gene subsequent to beta cell commitment during development using the RIP-Cre:Lrh1lox/lox mouse model results in offspring lethality between P1 and P21 (Cobo-Vuilleumier et al., 2018b), which was further evidenced by a decrease in homozygous knockout genotype frequency observed between P1 and P7 (Figure 1A). Prior morphometric analysis performed at P1 indicated that premature death may ensue from an inadequate postnatal islet beta cell pool (Cobo-Vuilleumier et al., 2018b), a premise we now extended to P14 and P21 in the RIP-Cre:Lrh1lox/lox:ROSA-YFP triple transgenic mouse model (ConβLRH-1KO mice) (Figures 1B–1E). Although, in the ConβLRH-1KO mice, the number of total number of cells per islet as well as the number of alpha cells were decreased at P14 but normalized at P21 as compared to WT mice (litter match control mice with normal LRH-1/NR5A2 expression, including the RIP-Cre:ROSA-YFP and Lrh1/Nr5a2lox/lox:ROSA-YFP mice), the beta cell mass was significantly reduced at both time points in homozygous ConβLRH-1−/−KO-R26Y mice (Figures 1B–1E). Interestingly, the percentage of YFP+/INS+ cells in homozygous ConβLRH-1−/−KO mice was significantly decreased as compared to heterozygous ConβLRH-1−/+ mice at both at P7 and P14, attaining approximately 60% of the overall beta cell population (Figure 1F). Although not significant, the percentage of YFP+/INS+ cells in heterozygous animals gradually decreased from 70% to 60% between P7 and P21 (Figure 1F). Active beta cell proliferation is a hallmark of neonatal islets that establishes the beta mass during adulthood both in humans and rodents (Georgia and Bhushan, 2004; Meier et al., 2008; Svenstrup et al., 2002). As such, we reasoned that deletion of LRH1/NR5A2 may impact neonatal beta cell expansion. This premise was confirmed as evidenced by a drastic decrease in the percentage of Ki67+/INS+ cells in islets of homozygous ConβLRH-1−/−KO mice as compared to either heterozygous ConβLRH-1−/+ or WT mice (Figure 1G). Consistent with reduced beta cell numbers, ConβLRH-1−/−KO mice displayed growth retardation, correlating with a hypoglycemic state and weight loss as compared to either WT or heterozygous ConβLRH-1−/+ animals (Figures 1H–1J). Noteworthy, the Cre transgene driven by the RIP was shown to be expressed also in the CNS (Schwartz et al., 2010; Song et al., 2010). Consistent with these reports, we observed expression of YFP in several regions of the brain including the hypothalamus and cortex (Figure S1A). Because LRH-1/NR5A2 expression was also detected in the CNS, albeit at lower levels than in islets and liver (Figure S1B), we cannot exclude the involvement of neuronal LRH-1/NR5A2 ablation in the observed phenotype of ConβLRH-1−/−KO mice. Taken together, these results demonstrate that LRH-1/NR5A2 contributes to neonatal beta cell expansion via cell replication.
Figure 1.

LRH-1 contributes to islet formation
(A) Representation of the genotypes frequency distribution: WT, LRH1w/w; HET, LRH1wt/ko; HOMO, and LRH1ko/ko). n = 23 mice for P1 and n = 347 mice for P7.
(B) Pancreases were extracted at P14 and P21 and immunostaining for insulin (INS, red), glucagon (GLUC, white) and YFP (green) was performed on fixed sections. Scale bar: 25 μmM.
(C-F) Morphometric analysis was performed to assess (C) islet cell composition, (D) b-cell mass, (E) α-cell mass, and (F) percentage of INS+/YFP+ cells.
(G) Co-staining of Ki67 and Insulin was performed on P7 pancreas and percentage of double-positives was assessed over total insulin positive cells.
(H) Representative images of wild type mice (WT) and ConbLRH-1 transgenic pups with either 1 (−/+) or 2 (−/−) Nr5a2 allele disruption 21 days postnatal (P21).
(I) Blood glucose levels (P21).
(J) weight of P14 and P21 WT, heterozygous and homozygous pups.
Statistics: Data are represented as the mean ± SEM ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 ∗∗∗∗p < 0.0001, one-way ANOVA, Tukey multiple comparisons test (C,D,E, H, and I) and unpaired Student’s t test, HET versus HOMO (F).
See also Figure S1.
TAM-induced LRH1/NR5A2 deletion does not compromise the metabolic status of healthy IndβLRH-1KO mice
The goal of generating the ConβLRH-1KO transgenic mouse model was to demonstrate the specificity of BL001-mediated LRH-1 activation in conveying islet survival. However, as we uncovered that LRH-1/NR5A2 is important for postnatal islet beta cell mass expansion and its deletion compromised survival (Figure 1), we generated a second transgenic mouse model, the IndβLRH-1KO mice, in which LRH-1/NR5A2 is spatially and temporally deleted in adult beta cells in a TAM-dependent inducible manner using the PDX1-CreERT mouse model. This deletion of LRH-1/NR5A2 in beta cells from adult animals avoids the developmental consequences of the lack of this nuclear receptor and minimizes off-target deletion. A 5-day TAM treatment did not alter body weight, glycemia, or glucose tolerance of IndβLRH-1KO mice either during or after treatment (Figures 2A–2C), independent of gender (data not shown). In addition, liver toxicity, measured by ALT and AST levels, was not observed at either one or 8 weeks post TAM-treatment (Figures 2D and 2E). Therefore, all subsequent experiments were performed at 4 weeks after TAM treatment to ensure the elimination of any traces of the drug. TAM treatment of IndβLRH-1KO mice induced YFP expression in 70–80% of beta cells with a concomitant 80% reduction in NR5A2/LRH1 transcript levels in islet but not in the liver and more importantly not in the brain (Figures 2F–2I). Altogether, these results indicate that TAM administration and beta cell specific ablation LRH-1/NR5A2 in adult beta cells do not compromise the metabolic health status of IndβLRH-1KO mice under normal physiological/environmental conditions.
Figure 2.

TAM treatment does not compromise the metabolic health status of 8 weeks old IndβLRH-1 mice
(A) The body weight of IndβLRH-1 treated or not with TAM was monitored for several weeks before (negative weeks) and after (positive weeks) treatment. TAM started at week 0 for five consecutive days.
(B) Blood glucose levels were also monitored starting at week one post TAM treatment.
(C) An OGTT was conducted 4 weeks post TAM treatment.
(D and E) Circulating levels of (D) ALT and (E) AST were assessed 1 week and 8 weeks post TAM treatment. As positive control, liver damage was induced using diquat dibromide (125 mg/kg body weight). Results are expressed as means ± SEM (F) Representative images of pancreas sections from IndβLRH-1 treated or not with TAM co-stained for YFP (green), insulin (INS, red), and glucagon (GLUC, white). Nuclei were stained with DAPI. Scale bar: 25 μM. (G-I) LRH-1 transcript levels were assessed in (G) islets, (H) liver, and (I whole brain extracted from IndβLRH-1 mice treated or not with TAM. Expression levels were normalized to the housekeeping gene Cyclophilin (CYCLO) or β-Actin. Statistics: Results are expressed as means ± SEM ∗p < 0.05, unpaired Student’s t test, vehicle versus TAM.
Conditional beta cell specific ablation of LRH-1/NR5A2 negates BL001-mediated cell survival and antidiabetic properties
We next assessed the contribution of LRH-1/NR5A2 in mediating the beneficial anti-diabetic and cell survival effects of BL001 in the IndβLRH-1 mice. As previously reported for WT mice after BL001 and STZ administration, only 20–30% of BL001 and non-TAM-treated IndβLRH-1 developed hyperglycemia (Figure 3A). In contrast, after TAM treatment, and thus depletion LRH-1/NR5A2, 80% of mice developed hyperglycemia subsequent to BL001/STZ treatment (Figure 3B). Therefore, the depletion of LRH-1/NR5A2 in beta cells blunts the antidiabetic effect of BL001 treatment. Immunohistochemistry combined with morphometric analysis of the pancreas of these mice revealed that hyperglycemic TAM/BL001/STZ-treated IndβLRH-1KO mice exhibited a drastic decrease in beta cell number, comparable to that of STZ-treated mice (Figures 3C and 3D). In contrast, normoglycemic non-TAM/BL001/STZ-treated IndβLRH1 mice harbored normal islet beta cell numbers similar to that of control non-STZ mice (Figures 3C and 3D). Interestingly, the number of alpha cells was increased in TAM BL001/STZ-treated mice as compared to non-TAM BL001/STZ-treated mice resulting in a decreased ratio of INS+/GCG+ cells (Figures 3E and 3F). This increase in alpha cell mass was not because of increased proliferation (Figure S2). These results establish that the antiapoptotic and pro-survival properties of BL001 are specifically conveyed through its interaction with LRH-1/NR5A2 in beta cells.
Figure 3.

β-cell-specific LRH-1 ablation in adult mice negates the antidiabetic properties of BL001
(A and B) Diabetes incidence of IndβLRH-1 mice either (A) not treated or (B) treated with TAM and then subjected to an STZ and/or BL001 regimen for 5 weeks. BL001 (10 mg/kg body weight) treatment started 4 weeks after finishing TAM treatment. Diabetes was induced at week one post-BL001 treatment with a single high dose of STZ (175 mg/kg body weight).
(C) Representative immunofluorescence images of pancreas sections from the various experimental groups co-stained for insulin (INS, red) and glucagon (GLUC, green). Nuclei were stained with DAPI (blue). Scale bar: 50 μM.
(D–F) Quantification of (D) β-cells, (E) α-cells and (F) ratio thereof in the different groups. n = individual islets counted from three to six independent mice. Statistics: Data are represented as the mean ± SEM ∗p < 0.05 and ∗∗∗∗p < 0.0001, unpaired Student’s t test between (−) and (+) TAM provided in the various treatments.
See also Figure S2.
PTGS2 is a downstream target of both cytokines and the BL001/LRH-1/NR5A2 signaling pathway
Comparative global transcriptome profiling of BL001-treated and untreated mouse islets revealed that most of the differentially expressed genes are involved in immunomodulation pathways (Cobo-Vuilleumier et al., 2018b). Among these is Ptgs2 that generates PGE2 and that depending upon the levels of expression of the four different PGE2 receptors will convey either a pro-inflammatory or anti-inflammatory stimulus (Carboneau et al., 2017b; Markovic et al., 2017). To further dissect the molecular mechanism implicated in the BL001/LRH-1/NR5A2 signaling pathway conferring beta cell survival, we focused on PTGS2 as a downstream effector. Consistent with previous results, BL001 treatment increased Ptgs2 expression, an effect that was abrogated in islets isolated from TAM-treated IndβLRH-1KO mice that lack LRH1/NR5A2, validating Ptgs2 as a downstream target of LRH1/NR5A2 (Figure 4A). Islet Ptgs2 expression was also increased by cytokines attaining a 3-fold stimulation at 24 h after treatment (Figure 4B). The latter time point was selected for subsequent experiments, as most in vitro BL001 studies have been performed between 24 and 72 h (Cobo-Vuilleumier et al., 2018b). Interestingly, the combined BL001/cytokine treatment did not further stimulate Ptgs2 expression above levels detected with cytokine treatment alone (Figure 4C). Attempts were made, albeit unsuccessfully, to detect the PTGS2 protein in islets (Figures S3A and S3B). In contrast, PTGS2 was discerned in LPS-treated RAW264.7 cells correlating with a robust increase in its transcript levels, suggesting that PTGS2 levels in islets are below the detection threshold (Figures S2C–S2E). To assess the contribution of PTGS2 in the BL001/LRH1 signaling pathway, the transcript was silenced using siRNA in isolated WT islets that were then challenged, or not, with a cytokine cocktail and further treated with BL001. Silencing of Ptgs2 resulted in the dampened induction of the transcript by both cytokines and BL001 (Figure 4D). Remarkably, although Ptgs2 transcript levels were similarly induced by cytokines alone or in combination with BL001, the secretion of the PTGS2 product PGE2 was 5-fold higher in the combined treatment (BL001 and cytokines) as compared to levels in cytokine-treated islets (Figure 4E). This BL001-mediated and specific increase in PGE2 was abolished in Ptgs2 silenced islets (Figure 4E).
Figure 4.

PTGS2 is a downstream target of the BL001/LRH1 signaling pathway
(A–C) Ptgs2 expression levels were assessed in (A) islets isolated from IndbLRH-1 mice treated or not with TAM, (B) mouse islets treated with a cytokine cocktail (CTK) for 2, 6, and 24 h, and (C) islets treated with or without cytokines and BL001 for 24 h.
(D) siSC or siPTGS2 transfected mouse islets treated or not with cytokines and BL001. Ptgs2 expression levels were normalized to either the housekeeping gene Gapdh or Cyclophilin.
(E) Secreted PGE2 levels were assessed by ELISA in the culture media of siSC and siPTGS2 transfected islets treated or not with cytokines and BL001. n = number of mouse islet preparation.
Statistics: Results are expressed as means ± SEM ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.0001 Student’s t test.
See also Figure S3.
Islets lacking PTGS2 are refractory to BL001-mediated protection against cytokines
We next assessed the impact of silencing PTGS2 on BL001-mediated islet cell survival after cytokines challenge. Hallmarks of cytokine-mediated cell death via the intrinsic apoptotic pathway include the release of cytochrome c from mitochondria resulting in cleavage of PARP (Kroemer et al., 2007). Consistent with this, cytochrome c release and PARP cleavage were induced/increased in cytokine exposed siSC-treated islets, whereas BL001 rescinded these apoptotic events (Figures 5A–5D). In contrast, PTGS2 silenced islets were completely refractory to the protective effect of BL001 under cytokine attack as assessed by the presence of cytochrome c release and increased PARP cleavage (Figures 5A–5D). In the intrinsic apoptotic pathway, cellular stress leads to the stimulation and translocation of BAX to the mitochondria, an event that permeabilizes the outer membrane, resulting in the release of cytochrome c. Accordingly, BL001 inhibited Bax expression levels in siSC-treated islets;however, in PTGS2 silenced islets, BL001 treatment failed to repress Bax transcript levels, consistent with increased apoptosis (Figure 5E).
Figure 5.

PTGS2 silenced islets are refractory to the protective effect of BL001 under cytokine attack
(A and B) Representative immunofluorescence images of embedded islets transfected with either a control siRNA (siSC) or siPTGS2 and treated or not with cytokines (CTK) or/and with BL001. Islet slices were stained for either (A) Cytochrome c (CytoC, green) or (B) cleaved PARP (Cl.PARP, green) along with insulin (INS, red). Nuclei were stained with DAPI (blue). Scale bar: 25 μM.
(C and D) Protein levels of cleaved PARP (Cl.PARP) (C) and (D) quantification thereof were assessed in whole islet cell extracts that were transfected with either a control siRNA (siSC) or siPTGS2 and treated or not with CTK or/and with BL001. Relative protein levels were normalized to ACT. Each line represents an independent experiment. Results are expressed as means ± SEM ∗∗p < 0.01 unpaired two-tailed t-test siSC/CTK versus siSC/CTK/BL001.
(E) Transcript levels of Bax were also determined in the same experimental groups. Expression levels were normalized to the housekeeping gene Cyclophilin. Results are expressed as means ± SEM ∗∗∗∗p < 0.0001, unpaired two-tailed t-test siCT/CTK versus siSC/CTK/BL001.
PTGER1 partially relays the BL001/LRH-1/NR5A2/PTGS2/PGE2 signaling axis to islet survival
Several studies indicate that the pro-survival effects of PTGS2 and its product PGE2 on islets are conveyed mainly by PTGER4 (a.k.a. EP4), whereas the proapoptotic events are transmitted by PTGER3 (a.k.a. EP3) for which blockade improves cell survival (Bosma et al., 2021; Carboneau et al., 2017a; Vennemann et al., 2012). Therefore, we next evaluated whether the islet pro-survival effects of BL001/LRH-1/NR5A2/PTGS2/PGE2 were mediated by PTGER4 downstream pro-survival signaling cascade, which include phosphorylation of PKA, CREB, and AKT (Konya et al., 2013). Remarkably, only pAKT levels were significantly increased in siSC-treated islets exposed to cytokines and BL001, whereas pPKA and pCREB were unaffected (Figures 6A–6D). In contrast, phosphorylation of CREB was increased in cytokines/BL001-treated islets in which PTGS2 was silenced, whereas pAKT levels were completely abolished in these islets independent of treatment (Figures 6A, 6C, and 6D). Phosphorylation of PKA was unaffected by silencing and/or treatment (Figures 6A and 6B). As BL001 failed to stimulate both PKA and CREB phosphorylation, we reasoned that PTGR4 may not relay the pro-survival benefits of the LRH-1/NR5A2 agonist. This premise was confirmed as evidenced by the inability of the PTGER4 specific antagonist, L-161,982, to block BL001-mediated islet protection against cytokines (Figure 6E).
Figure 6.

The BL001/LRH-1/NR5A2/PTGS2 anti-apoptotic benefits are not conveyed via PTGER4 signaling
(A–D) Phosphorylated protein levels of PKA (pPKA), CREB (pCREB), and AKT (pAKT) as well as total AKT and quantification thereof (B) pPKA, (C) pCREB, and (D) pAKT/AKT were assessed in whole islet cell extracts that were transfected with either a control siRNA (siSC) or siPTGS2 and treated or not with CTK or/and with BL001. Relative protein levels were normalized to ACTIN (ACT) for both siCT and siPTGS2. Only one representative ACT blot is shown, whereas quantification was performed with matched ACT for each blot. Each lane represents an independent sample. Results are expressed as means ± SEM ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 unpaired two-tailed t-test siPTGS2 versus siPTGS2/CTK/BL001.
(E) Representative immunofluorescence images of embedded islets treated or not with a cytokine cocktail (CTK), BL001 and/or the PTGER4 antagonist L-161,982. Islet slices were stained for cleaved PARP (Cl.PARP, green) along with DAPI (blue) for nuclei staining. Scale bar: 25 μM.
These findings led us to consider whether BL001 could alter the overall expression profile of the four PTGER members inducing a shift in PGE2 selectivity toward either PTGER1 or 2, also implicated in cell survival. Although not significant, BL001 treated islets displayed an increase in PTGER1, whereas PTGER2, PTGER3, and PTGER3 were decreased as compared to control islets (Figure 7A). In view of these results, we focused on the potential role of PTGER1 in BL001-mediated cell survival in response to cytokines using the PTGER1-specific inhibitor, ONO-8130. Consistent with our premise, the PTGER1 inhibitor, ONO-8130, blocked the BL001-mediated cell protection against cytokines as gauged by increased cytoplasmic histone-associated DNA fragments and cleaved PARP (Figures 7B and 7C). In contrast, L-161,982 was ineffective in reverting BL001-mediated survival (Figure 7B). We are currently assessing the impact of ONO-8130 on human induced pluripotent stem cell (iPSC)-derived organoid islets in which we demonstrate reduced cytokine-mediated apoptosis in the presence of BL001 (Figure S4). Taken together, these results indicate that PTGR1 is involved in relaying the BL001/LRH-1/NR5A2/PTGS2/PGE2 signaling to islet survival.
Figure 7.

The PGE2/PTGER1 signaling pathway mediates the cell protective effect of BL001
(A) Expression levels of PTGERs were assessed in islets treated or not with 1uM BL001. Expression levels were normalized to the housekeeping gene Gapdh. Results are expressed as means ± SEM.
(B) Isolated mouse islets were treated or not with a cocktail of cytokines (CTK), BL001 and antagonists for either PTGER1 (ONO-8130) or PTGER4 (L-161,982). Cell death was assessed by ELISA quantification of mono-nucleosomes and oligo-nucleosomes released by apoptotic cells. Results are expressed as means ± SEM ∗∗p < 0.01, unpaired two-tailed t-test CTK versus BL001 and L-161,982 groups.
(C) Representative immunofluorescence images of embedded islets treated or not with a cytokine cocktail, BL001 and/or the PTGER1 antagonist ONO-8130. Islet slices were stained for cleaved PARP (Cl.PARP, green) along with DAPI (blue) for nuclei staining. Scale bar: 25 μM. See also Figure S4.
Discussion
Here, we disclose that LRH-1/NR5A2 is essential for proper postnatal islet beta cell expansion and that its specific interaction with BL001 regulates the PTGS2-PGE2-PTGER1 signaling axis, contributing to islet survival and antdiabetic actions of the agonist. These conclusions are based on the results that: 1) the proliferative capacity of postnatal beta cells is severely hampered in the absence of LRH-1/NR5A2, 2) deletion of LRH-1/NR5A2 in adult islets abolishes the pro-survival and antidiabetic effects of BL001 in STZ-treated mice, 3) silencing of PTGS2 sensitizes BL001-treated islets to cytokine-induce apoptosis, and 4) pharmacological blockade of PTGER1 but not PTGR4 partially blocks the antiapoptotic effects of BL001 in cytokine-treated islets.
A hallmark of neonatal beta cells is their high replicative capacity associated with islet remodeling, organization, and maturation which is crucial for subsequent functionality of adult islets in both rodents and humans (Mezza and Kulkarni, 2014). Although our previous studies did not reveal a role of LRH-1/NR5A2 in beta cell replication in adult islets (Baquie et al., 2011; Cobo-Vuilleumier et al., 2018b), we now demonstrate that the nuclear receptor contributes to this process in neonatal islets, and its deletion in beta cells resulted in reduced beta cell number, hypoglycemia, and premature death. Of particular interest is the development of hypoglycemia in pups, which is counterintuitive with the observed decrease in the beta cell mass. Such conundrum could be rationalized from deregulated insulin secretion that ensues from LRH-1/NR5A2 ablation early on during beta cell development that may interfere with secretory pathways implicated in this process. In line with this premise, we previously showed that overexpression of LRH-1/NR5A2 in adult mouse islets impeded both basal and glucose-induced insulin secretion (Baquie et al., 2011). Therefore, we can envisage that deletion of LRH-1/NR5A2 may have the opposite effect resulting in higher basal insulin secretion. The latter highlights the importance of fine-tuning LRH-1/NR5A2 expression levels to prevent beta cell dysfunction. However, as we detected RIP-Cre activity in the brain, we cannot exclude that the severe growth retardation and hypoglycemia may be, at least in part, consequential to the neuronal deletion of LRH-1/NR5A2, because this nuclear receptor was shown to be implicated in neurogenesis (Stergiopoulos and Politis, 2016). Interestingly, we previously demonstrated that LRH-1/NR5A2 overexpression in islets or its activation using BL001 increases the expression of key genes implicated in glucocorticoid (GC) biosynthesis as well as secretion and is likely involved in beta cell survival (Baquie et al., 2011; Cobo-Vuilleumier et al., 2018a). Systemic GC secretion, under the control of the hypothalamo-pituitary-adrenal (HPA) axis, is critical for the endocrine regulation of glucose homeostasis (de Guia et al., 2014). As such, central nervous deletion of LRH-1/NR5A2 during development may have an impact on global GC production and glucose homeostasis via various target tissues. In line with this premise, mice lacking the GC receptor (GR) in hepatocytes (mimicking the lack of GC), exhibit hypoglycemia after prolonged fasting (Opherk et al., 2004), whereas overexpression of GR in beta cells within the insulin temporal expression domain decreased insulin secretion (Blondeau et al., 2012). Therefore, the hypoglycemic phenotype of RIP-Cre/LRH-1−/− may partly ensue from deregulated central GC secretion. Further studies will be required to elucidate the impact of LRH-1/NR5A2 deletion in the brain on energy homeostasis. Notwithstanding, Dabernat and colleagues reported a comparable phenotype, including hypoglycemia and premature death, in new born mice in which the floxed beta catenin gene (Catnblox/lox) was ablated in developing beta cells using the same RIP-Cre transgenic mouse model used here. They conclude that deletion of beta catenin within the developmental insulin expression domain impedes expansion and subsequent maturation of beta cells, an effect that they argue to be independent of the off-target activity of the RIP promoter detected in the CNS (Dabernat et al., 2009). Substantiating this premise, repression of TCF7L2 (a.k.a TCF4, a nuclear coactivator of beta catenin) in rat newborn pups using antisense morpholinos blunted beta cell replication resulting in an almost 30% reduction in beta cell mass (Figeac et al., 2010). Contextually, LRH-1/NR5A2 cooperates with the beta catenin/TCF7L2 complex to promote cell proliferation through activation of c-myc, Ccne1, and Ccnd1 (Botrugno et al., 2004). High expression levels of c-myc were shown to be correlated with the high replicative capacity of juvenile islets, whereas low levels associated with poor replication in adult islets (Puri et al., 2018). Therefore, the failure of beta-catenin/TCF7L2 to complex with LRH-1/NR5A2 and downstream activation c-myc, as well as Ccne1 and Ccnd1 may explain the reduced neonatal beta cell proliferation in ConβLRH-1−/−KO mice. Interestingly, in adult islets, we have shown that overexpression of LRH-1/NR5A2 was unable to enhance Ccne1 and Ccnd1 expression and beta cell replication, likely because of low levels of beta catenin (Baquie et al., 2011). Our findings reinforce the concept that LRH-1/NR5A2 regulatory networks are temporally and cell type restricted through interaction partners that are differentially expressed during development, adulthood, and aging. The latter is further evidenced by the fact that deletion of LRH-1/NR5A2 in adult mice beta cells results in a different phenotype to that of its deletion during development.
Although our previous in vivo studies established that BL001 treatment blocked/reverted progression of hyperglycemia in three independent mouse models of diabetes, the specificity of the agonist for LRH-1/NR5A2 and its MoA remained elusive. We now authenticate the exclusivity of LRH-1/NR5A2 to convey the pro-survival and antidiabetic therapeutic benefits of BL001 in islets rebutting any potential off-target effects of BL001 via binding to other receptors such as SF-1 (a.k.a. NR5A1), the second NR5A family member that shares similarities with LRH-1/NR5A2 in both the protein ligand binding domain and DNA binding site (Meinsohn et al., 2019). Interestingly, TAM/BL001/STZ-treated ConβLRH-1KO in which LRH-1/NR5A2 is specifically ablated in beta cells displayed a higher number of alpha cells as compared to BL001/STZ-treated mice, a process independent of cell proliferation. Although unclear, the loss of beta cells may stimulate alpha cell mass expansion through ductal neogenesis as we previously reported in the RIP-B7.1 mouse model of experimental autoimmune diabetes (Bru-Tari et al., 2019).
Of particular importance was the finding that PTGS2 expression was blunted in BL001/TAM-treated IndβLRH-1−/−KO mice. As a consequence, cytochrome c release and PARP cleavage were increased upon cytokine insult. These results confirm that PTGS2 is a bona fide downstream target of the BL001/LRH-1/NR5A2 antiapoptotic signaling pathway in islets. Remarkably, in silico analysis failed to identify LRH-1/NR5A2 consensus DNA binding sequence (T/C-CAAGG-T/C-CA) within the proximal promoter region of the Ptgs2 gene, indicating that the nuclear receptor does not directly regulate this gene. Notwithstanding, expression levels of several transcription factors including NF-κB1 (Fold change 1.23, p = 0.015), TCF7L2/TCF4 (Fold change 1.36, p = 0.032), C/EBP (Fold change 1.37, p = 0.002), ATF4 (Fold change 1.3, p = 0.029), and ATF5 (Fold change 1.38, p = 0.005) that bind to and regulate Ptgs2 transcription were significantly increased in a transcriptomic analysis of BL001-treated islets (Cobo-Vuilleumier et al., 2018b; Tsatsanis et al., 2006). Noteworthy, TCF7L2/TCF4, ATF4, and ATF5 were all implicated in rodent beta cell survival under stress conditions, independent of their regulatory action on the Ptgs2 gene (Juliana et al., 2017; Sachdeva et al., 2009; Zhou et al., 2012). Therefore, we argue that pro-survival signals in response to stress converge onto Ptgs2 expression in an attempt to reestablish cellular homeostasis and preventing apoptosis, a process that is facilitated by BL001-mediated activation of LRH-1/NR5A2. Such pro-survival function of Ptgs2 was highlighted in a study in which its deletion sensitized transgenic mice to STZ-induced hyperglycemia (Vennemann et al., 2012). In the same vein, PGE2 supplementation reduced caspase 3 activity in mouse islets (Papadimitriou et al., 2007). In contrast, another study reported that sustained Ptgs2 overexpression in islet beta cells induced hyperglycemia in hemizygous transgenic mice. Notwithstanding, these mice did not exhibit increased apoptosis (Oshima et al., 2006). These data argue against a role of PTGS2 in beta cell apoptosis and emphasize the importance of fine-tuning its levels and activity to prevent beta cell dysfunction. The latter is also likely contingent on the expression levels and activity of the four PTGERs that dictate the overall impact of PTGS2, as revealed by either blockade of the proapoptotic PTGER3 or activation of the antiapoptotic PTGER4 that increase beta cell survival in the presence of cytokines (Carboneau et al., 2017a).
Our data reveal that BL001 did not augment Ptgs2 expression above levels induced by cytokines alone, whereas its product, PGE2, was robustly increased by the LRH-1/NR5A2 agonist. The latter indicates a posttranslational regulation of PTGS2 as reported for the case of inducible nitric oxide synthase that binds s-nitrosylates, and enhances PTGS2 activity (Kim et al., 2005). Similarly, the nonreceptor Src-family kinase FYN was recently shown to phosphorylate and increase PTGS2 activity resulting in elevated production of PGE2 without altering protein steady state levels in prostate cancer cells (Alexanian et al., 2014). As a consequence of increased PGE2, prostate cancer cells were refractory to sanguinarine-induced apoptosis similar to our findings with BL001/cytokine-treated islets (Huh et al., 2006). In parallel, another member of the Src-family of kinases—LYN—was also shown to phosphorylate PTGS2, albeit at a different site than FYN; however, its impact on PTGS2 activity has not yet been studied (Alexanian et al., 2014). LYN is regarded as a master suppressor of immune cell activation and its global knockout in mice leads to an autoimmune disorder with similarities to human systemic lupus erythematosus (Brian and Freedman, 2021). In addition to immune cells, LYN is also expressed in pancreatic endocrine cells (https://www.proteinatlas.org/ENSG00000254087-LYN/celltype/pancreas), and more importantly, its levels are mildly but significantly increased by BL001 treatment (Fold change 1.17, p = 0.008) (Cobo-Vuilleumier et al., 2018b). Taken together, it is tempting to speculate that BL001-mediated activation of LRH-1/NR5A2 enhances LYN expression in islets, which under stress conditions such as exposure to cytokines will phosphorylate PTGS2 increasing its activity and PGE2 production, leading to immune modulation and increased cell survival. This hypothesis is currently under investigation.
Downstream signaling of PGE2 is conveyed via binding to one or several of the four PTGERs with the net cellular physiological outcome, i.e., survival versus death, contingent on the expression levels of these receptors as well as their individual affinity for the prostaglandin in a define environment. For example, expression of the PTGER3 is increased in T2DM patient islets as well as in T2DM mouse models and is associated with beta cell dysfunction and apoptosis (Carboneau et al., 2017a). Blockade of PTGER3 in db/db mice prevents beta cell death (Bosma et al., 2021; Carboneau et al., 2017b). In contrast, activation of PTGER4 enhanced beta cell survival in response to cytokines (Carboneau et al., 2017a); therefore, we selected this receptor as the ideal candidate to mediate the effect of BL001. This assumption was refuted as BL001 did not increase phosphorylation of PKA and CREB, two downstream target kinases of PTGER4 signaling, and the pharmacological inhibition of PTGER4 failed to block BL001-mediated cell survival in response to cytokines. In contrast, AKT phosphorylation—another downstream target of PTGER4—was increased by cytokines alone and in combination with BL001, whereas silencing of PTGS2 completely abolished phosphorylation independent of treatment, indicating that PGE2 is a key regulator of AKT phosphorylation levels in beta cells. Nevertheless, AKT phosphorylation can also be conveyed by PKCε via the PI3 kinase-dependent pathway and has been associated with both beta cell survival and proliferation (Fatrai et al., 2006; Moriya et al., 1996; Tuttle et al., 2001). As PKC is a downstream effector of PTGER1, we argue that AKT phosphorylation could be routed through this receptor, reconciling our finding that BL001-mediated islet survival is blocked by an antagonist of PTGER1. Surprisingly, BL001 in combination with cytokines significantly increased CREB phosphorylation as compared to cytokines alone in PTGS2 silenced islets. The latter likely arose as a consequence of alleviating PGE2-induced PTGER3 activity, releasing the break on adenylate cyclase activity and enhancing CREB phosphorylation (Carboneau et al., 2017b). These results emphasize the complex cross talk and promiscuity among the various receptors in dictating the overall outcome of PGE2 on cell physiology pending environmental inputs. This complex networking is further underlined by findings that PGE2 stimulates LRH-1/NR5A2 expression levels, indicating a feedforward mechanism between the nuclear receptor and PTGS2 (Michalek and Brunner, 2021).
As opposed to PTGER4, pharmacological inhibition of PTGER1 negated BL001-dependent beta cell survival under cytokine attack. In contrast to PTGER2, PTGER3, and PTGER4 that modulate cAMP levels, PTGER1 downstream signaling pathway include increasing intracellular calcium as well as diacylglycerol (DAG) levels via the Gαq coupling protein, leading to the activation of PKC as well as NF-κB (Sugimoto and Narumiya, 2007). Consistent with the PTGER1 signaling cascade being activated by BL001, NF-κB1 was significantly increased in a transcriptomic analysis of BL001 treated islets (Cobo-Vuilleumier et al., 2018b). Moreover, PKCε activators improve islet survival during isolation as well as function after transplantation in mice (Hamilton et al., 2014; Kvezereli et al., 2008). In addition to substantiating the implication of PTGER1 in relaying the BL001/LRH-1/NR5A2/PTGS2/PGE2 survival signal, GLP-1-mediated activation of PKC was shown to inhibit the intrinsic apoptotic pathway via modulation of BAX, whereas an activated variant of AKT prevented PARP cleavage, two events we report herein (Kennedy et al., 1997; Zhang et al., 2015). Oddly, PTGER1 exhibits the lowest affinity for PGE2. We reason that high concentrations of PGE2 generated in response to BL001 combined with the mild increase in PTGER1 expression with the concomitant decrease in PTGER2, PTGER3, and PTGER4 favors a shift in receptor selectivity toward PTGER1 as observed for high concentrations of prostacyclin that favor interaction with the IP receptor coupled to the Gαq protein (Narumiya and FitzGerald, 2001).
In summary, our data refine the cellular mechanism by which LRH-1/NR5A2 is implicated in neonatal beta cell mass establishment through cell replication and define the molecular MoA by which the small chemical agonist BL001 specifically activates LRH-1/NR5A2 and the downstream PTGS2/PGE2/PTGER1 signaling axis to foster beta cell survival in response to cytokines. Historically, the role of PTGS2 has been associated with pro-inflammatory response and as such selective inhibitors were developed to treat inflammatory diseases such as osteoarthritis and rheumatoid arthritis (Gilroy and Colville-Nash, 2000). Notwithstanding, other studies have shown that PTGS2 expression is essential for the resolution phase of the pro-inflammatory response in wound healing, raising concerns on the use of such PTGS2 inhibitors for the treatment of inflammatory diseases (Gilman and Limesand, 2021). The data herein support this opinion and substantiate our concept recently brought forth that the BL001/LRH-1/NR5A2 axis on one hand protects islet cells against cytokines and on the other hand incites the exit of the perpetual unresolved wound healing of T1DM, i.e., continuous pro-inflammatory attack, toward an anti-inflammatory and pro-regenerative environment, likely conveyed in part by PGE2 secretion and signaling between immune and islet cells (Cobo-Vuilleumier and Gauthier, 2020).
Limitations of the study
Herein, we delineate the LRH-1/NR5A2/PTGS2/PGE2/PTGER1 signaling cascade as a key axis conveying BL001 protection to mouse islet against stress-induced apoptosis. Whether these results can be translated to human islet remains to be assessed. Finally, such studies are impeded by the current challenging world background limiting availability of high quality human islets for research (Dafoe et al., 2022). To circumvent such caveat, we have launched human studies using induced pluripotent stem cell (iPSC)-derived pancreatic endocrine cells that were recently shown to be a robust model recapitulating the detrimental effects of cytokines on primary human beta cells (Demine et al., 2020). We recently demonstrated that BL001 protects iPSC-derived islet cells against cytokine-induced apoptosis validating this model to pursue BL001 mode of action in human islet-like cells (Figure S4).
Although our results highlight the importance of PTGER1, but not of PTGER4, in conveying BL001-mediated increased islet survival against cytokines, we have not yet assessed the functional role of PTGER2 and three in the LRH-1/NR5A2/PTGS2/PGE2 axis. Consequently, their contribution in this overall survival signaling axis remains to be elucidated.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit mAb PTGS2 Cox2 (D5H5) XP® | Cell Signalling | Cat#12282 (WB 1:1000; IF 1:150) |
| Rabbit mAb PARP | Cell Signalling | Cat#9532 (WB 1:1000) |
| Rabbit mAb Phospho-PKA C (Thr197) (D45D3) | Cell Signalling | Cat#5661 (WB 1:1000) |
| Rabbit mAb Phospho-CREB (Ser133) (87G3) | Cell Signalling | Cat#9198 (WB 1:1000) |
| Rabbit mAb GAPDH (14C10) | Cell Signalling | Cat#2118 (WB 1:10000) |
| Mouse mAb b-Actin | Sigma-Aldrich | Cat#A5441 (WB 1:20000) |
| Mouse mAb Glucagon | Sigma-Aldrich | Cat#G2654 (IF 1:200) |
| Rabbit anti-Glucagon | Cell Signalling | Cat#G2760 (IF 1:150) |
| Mouse mAb Insulin | Sigma-Aldrich | Cat#I2018 (IF 1:300) |
| Mouse mAb Cytochrome C | MBL | Cat#BV-3026-3 (IF 1:100) |
| Rabbit mAb Ki-67 (SP6) | ThermoFisher Scientific | Cat# MA5-14520 (IF: 1:150) |
| Anti-Mouse IgG (whole molecule)–Peroxidase antibody produced in rabbit | Sigma-Aldrich | Cat#A9044 (WB 1:5000) |
| Anti-Rabbit IgG (whole molecule)–Peroxidase antibody | Sigma-Aldrich | Cat#A0545 (WB 1:5000) |
| IRDye® 680RD Goat anti-Mouse IgG (H + L) | LICOR | Cat#926-68070 (WB 1:15000) |
| IRDye® 800CW Goat anti-Rabbit IgG (H + L) | LICOR | Cat#926-32211 (WB 1:15000) |
| Goat anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | ThermoFisher Scientific | Cat#A32731 (IF 1:800) |
| Goat anti-Mouse IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | ThermoFisher Scientific | Cat#A11004 (IF 1:800) |
| Donkey anti-Mouse IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 555 | ThermoFisher Scientific | Cat#A32773 (IF 1:800) |
| Donkey anti-Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | ThermoFisher Scientific | Cat#A31573 (IF 1:800) |
| Donkey anti-Goat IgG (H + L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | ThermoFisher Scientific | Cat#A11055 (IF 1:800) |
| Chemicals, peptides, and recombinant proteins | ||
| ONO 8139 (PTGER1 antagonist) | Tocris | Cat#5406 |
| TNFα | ProSpec-Tany TechnoGene | Cat#CYT-252 |
| BL001 | In house | N/A |
| L-161,982 (PTGER4 antagonist) | Caymen Chemical | Cat#10011565 |
| IFNϒ | ProSpec-Tany TechnoGene | Cat#CYT-358 |
| IL1β | ProSpec-Tany TechnoGene | Cat#CYT-273 |
| Critical commercial assays | ||
| Prostaglandin E2 (PGE2) ELISA | R&D Systems | Cat#KGE004B |
| Cell Death Detection ELISA | Merck | Cat#11544675001 |
| Deposited data | ||
| Microarray data | Gene Expression Omnibus repository | GSE94505; GSE94505; GSE104322 |
| Experimental models: cell lines | ||
| 1023A iPSCs | Prof. D. Eiziric, ULB Center for Diabetes Research, Medical Faculty, Universite Libre de Bruxelles (ULB), Belgium | N/A |
| RAW264.7 | ATCC | https://www.atcc.org/products/tib-71 |
| Experimental models: organisms/strains | ||
| RIPcre | In house | N/A |
| LRH-1lox/lox | Procured from Dr. Schoonjans, EPFL, CH, in house | N/A |
| pdx1PBCreERTM | Procured from Dr. Gannon, Vanderbilt University, USA, in house | N/A |
| B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J | The Jackson Laboratory | Strain #:006148 RRID:IMSR_JAX:006148 |
| C57BL/6J | Charles Rivers/Janvier Labs | strain code 027/SC-C57J-F |
| Oligonucleotides | ||
| See Table S1 for primer sequences | Metabion International AG, DE | N/A |
| Software and algorithms | ||
| Fiji | ImageJ | https://imagej.net/software/fiji/downloads |
| Prism | GraphPad | https://www.graphpad.com/ |
| Adobe Photoshop | Adobe | https://www.adobe.com/es/ |
| ImageJ | Imagej | https://imagej.nih.gov/ij/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Benoit R. Gauthier (Benoit.gauthier@cabimer.es).
Materials availability
All unique/stable reagents generated herein are available from the Lead contact upon request. Mouse lines can also be procured from the Lead contact pending approval from the original laboratory from which they were generated.
Experimental model and subject details
Ethical approval
All experimental mouse procedures were approved by the Institutional Animal Care Committee of the Andalusian Center of Molecular Biology and Regenerative Medicine (CABIMER) and performed according to the Spanish law on animal use RD 53/2013. Animal studies were performed in compliance with the ARRIVE guidelines (Kilkenny et al., 2010).
Experimental animals
C57BL/6J mice were purchased either from Charles Rivers (L'Arbresle Cedex, France) or Janvier Labs (Saint-Berthevin Cedex, France). Two triple transgenic mouse models were generated: 1) a constitutive and beta cell specific LRH-1/NR5A2 ablation mouse model, in which the LRH-1lox/lox mouse line was bred to the hemizygous RIP-Cre mouse line that constitutively express the Cre recombinase under the control of the rat insulin promoter (RIP) and 2) an inducible cell specific LRH-1/NR5A2 ablation mouse model, in which the LRH-1lox/lox mouse line was bred to the pdx1PBCreERTM mouse line that harbours a tamoxifen (TAM)-inducible Cre recombinase/estrogen receptor fusion protein under the transcriptional control of the PDX1 promoter (Zhang et al., 2005). These two double transgenic mouse lines were then bred to the Rosa26-YFP reporter mouse line to generate the ConβLRH-1KO and IndβLRH-1KO mouse lines respectively.
Animal husbandry
Mice were housed in ventilated plastic cages under a 12-h light/dark cycle and given food and water ad libitum.
Regimen and blood biochemistry
For experiments conducted with ConβLRH-1 mice, P1, P7, P14 and P21 pups generated from wild type (LRH-1+/+), heterozygous (LRH-1−/+) and null (LRH-1−/−) were euthanized and pancreas extracted to perform immunofluorescence (IF) and morphometric analyses. Of note, as aging RIP-Cre mice may display beta cell impairment, control WT pups included RIP-Cre as well as Lrh1/Nr5a2lox/lox pups (Lee et al., 2006). For experiments using IndβLRH-1KO mice, TAM (Sigma-Aldrich, Madrid, Spain, 20 mg/mL dissolved in corn oil) was administered via gavage for 5 consecutive days to 8-week old homozygous IndβLRH-1−/− male or female mice (0.2 mg TAM/g body weight). This age was selected in order to have beta cell specific deletion of LRH-1/NR5A2 in adult mice. Animals were allowed a 4 weeks recovery period in order to eliminate any trace of TAM that may impact metabolism (Ceasrine et al., 2019; Cole et al., 2010). Mice were then intraperitoneally (i.p.) injected with 10 mg/kg body weight BL001/Vehicle daily for 5 weeks (Cobo-Vuilleumier et al., 2018b). BL001 treatment was initiated 1 week prior to an i.p. injection of a single high dose of STZ (175 mg/kg body weight). Circulating glucose levels were measured from venepuncture-extracted blood samples using a Precision Xceed glucometer (Abbott Scientifica SA, Barcelona, Spain). Mice with blood glucose above 250 mg/dL for two consecutive measurements were considered hyperglycaemic. For Oral Glucose Tolerance Tests (OGTTs), mice were fasted for 5 hours and then received a glucose bolus (3 g/kg body weight) by gavage. Blood was taken by venepuncture and circulating glucose was determined at different time points. At termination of experiments, mice were euthanized and organs (pancreas, liver and brain) extracted for further analysis (QRT-PCR and immunofluorescence). For transaminases analysis, blood was collected from the tail vein in a sterile environment. Liver damage was induced using diquat dibromide (i.p. 125 mg/kg body weight). The analysis was performed using a Cobas Mira autoanalyzer and reagents from Spinreact (Spinreact S.A.U., Girona, Spain) and Biosystems (Biosystems S.A., Barcelona, Spain).
Method details
Islet isolation and cell culture
Islets were isolated from either C57BL/6J mice or TAM and vehicle-treated IndβLRH-1KO mice and cultured as followed: The isolation was performed by infusing the pancreatic duct with collagenase followed by pancreas digestion, and islet purification. For the infusion, the ligation of the common bile duct in close proximity to the pancreas was executed with one suture. The duodenum wall, near to the edge of the ampulla, was then clamped both sides. An incision into the ampulla was performed with a 30-G needle. Two to three mL of collagenase V solution (Sigma, 1 mg/mL) was infused trough the incision with a blunt 30-G needle coupled to a 3 mL syringe. Pancreas was then removed and digested at 37°C for 5 + 5 minutes shaking 20 times in between the two times. Islets were then handpicked and maintained in 11.1 mM glucose/RPMI-1640 (ThermoFisher Scientific, Madrid, Spain) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, Madrid, Spain), 100 U/mL penicillin (Sigma-Aldrich) and 100 mg/mL streptomycin (Sigma-Aldrich). The RAW264.7 mouse macrophage cell line (ATCC, USA) was cultured in high glucose DMEM supplemented with 10% FBS and 2 mM L-glutamine, 100 U/mL penicillin and100 mg/mL streptomycin.
Culture of the induced pluripotent stem cell (iPSCs) line and differentiation
The iPSCs line 1023A was derived from bone marrow. The iPSCs had normal karyotype (XY), classical stem cell colony morphology and expressed pluripotency markers, as characterized by Lytrivi et al (Lytrivi et al., 2021). iPSCs were maintained in essential E8 medium (ThermoFisher Scientific, ES) on matrigel-coated plates. iPSCs were differentiated into pancreatic beta-like cells using a 7-step protocol (De Franco et al., 2020; Demine et al., 2020; Igoillo-Esteve et al., 2020; Lytrivi et al., 2021). After differentiation, stage 7 aggregates were dispersed. Briefly, aggregates were incubated in a phosphate-buffered saline solution containing 0.5 mM ethylenediamine tetraacetic acid (EDTA) at room temperature for 6 minutes (min), and then exposed to Accumax (Sigma-Aldrich) for 8 min followed by gentle pipetting to detach cells. Knockout serum (ThermoFisher Scientific, ES) was added to quench the dissociation process, and cell were seeded at 5 × 104 cells per 6.4 mm well in stage 7 medium. Results shown for iPSCs-derived beta-like cells are derived from independent biological samples, i.e., independent differentiations.
RNA interference
Islets were transfected with either 50 μMol of a pool of PTGS2 small interfering (si)RNAs ON-Target plus Mouse Ptgs2 (19225) siRNA-SMARTpool (Dharmacon/Horizon Discovery, Cultek, Madrid, ES) or a scramble siRNA (Sigma-Aldrich). Transfections were performed using the Viromer Blue siRNA/miRNA transfection kit following the manufacturers’ instructions (Origene, Rockville, USA). Forty eight hours post siRNA treatment, islets were treated or not with cytokines and/or BL001. Twenty four hours later islets were processed for RNA isolation, protein extraction or IF analysis. Media was also collected to measure PGE2.
In vitro cell treatments
Islets or iPCS-derived islet cells were cultured in the presence of: 1) either 1 or 10 μM BL001 [optimal doses previously used (Cobo-Vuilleumier et al., 2018b)] and/or 2) a cocktail of cytokines (2 ng/mL IL1β, 28 ng/mL TNFα and 833 ng/mL IFNγ, ProSpec-Tany TechnoGene Ltd, Ness-Ziona, IL) and/or 3) 100 nM L-161,982 (Cayman Chemical Coorp, Ann Arbor, USA) and/or 4) 100 nM ONO-830 (Tocris Bioscience, Bristol, UK). In the latter two conditions, islets were treated for 12 hours with the respective antagonists (L-161,982 and ONO-8130) followed by a second treatment with the antagonists along with cytokines. BL001/vehicle was added 30 minutes later and cells were further incubated for for 24 hours. RAW264.7 cells were treated, or not, with 1 μg/mL LPS for 2, 6 and 24 hours. Cells were then processed for ELISA, RNA isolation, protein extraction or immunofluorescence analysis. In some instances, the media was also collected for ELISA analysis.
Cell death
Cell death (apoptosis) was measured using the Cell Death Detection ELISA kit (Merck/Roche Diagnostics, Madrid, Spain) (Lorenzo et al., 2021).
RNA extraction and quantitative real-time PCR
Total RNA from islets was extracted using the RNeasy Micro Kit (Qiagen) while RNA from liver and brain was extracted using TRIzol (Sigma-Aldrich). Complementary DNA using 0.1 to 1 μg RNA was synthesized using the Superscript III Reverse Transcriptase (Invitrogen-Thermo Fisher Scientific, Madrid, Spain). The QRT-PCR was performed on individual cDNAs using SYBR green (Roche) (Cobo-Vuilleumier et al., 2018b). Gene-specific primers were designed using Primer3Web (https://primer3.ut.ee/) and the sequences are listed in Table S1. Expression levels were normalized to various reference genes including Cyclophilin, Rsp9, Gapdh and Actin. The relative gene expression was calculated using the standard curve-based method (Mellado-Gil et al., 2018).
Immunofluorescence analyses
For immunostaining, pancreas or brain were fixed overnight in 4% paraformaldehyde at 4°C. Pancreas were dehydrated, paraffin embedded, and sectioned at 5 μm thickness. Brains were coronally sectioned into six series of 50 μm slices using a vibratome (Leica). Isolated islets were processed and paraffin embedded as previously described (Lorenzo et al., 2015). RAW264.7 cells and iPCS-derived islet cells were washed with PBS and fixed 15 minutes in 4% paraformaldehyde at room temperature. They were washed again with PBS and permeabilized with PBS-0.1% triton 15 minutes on ice. Tissue sections/cells/free floating brain sections were blocked in PBS containing 5% donkey/goat serum and 0.2% Triton ×-100 for 1h at R/T. Immunostaining was then performed overnight at 4°C using a combination of primary antibodies (Key resources table). Subsequently, secondary antibodies were incubated for 1 hour at room temperature in PBS 0.2% TritonX100 (Key resources table). Nuclei were stained with 0.0001% of 4′,6-diamidino-2-phenylindole (DAPI, Sigma-Aldrich) and cover slips were mounted using fluorescent mounting medium (DAKO). Epifluorescence microscopy images were acquired with a Leica DM6000B microscope and z-stack images were acquired using a Confocal Leica TCS SP5 (AOBS) microscope.
Morphometric analyses
For the assessment of the whole pancreatic area, images of pancreatic sections were automatically acquired using the NIS-Elements imaging software. Morphometric quantification was performed using Photoshop, ImageJ and FIJI softwares. For ConsβLRH-1 mouse line, sections from 3 independent mice with an average of about 50 islets and 5000 cells per mice were used for quantifications. For IndβLRH-1-R26Y mouse line, sections from 3 to 6 independent animals with an average of about 50 islets and 4000 cells per group were used for quantifications.
Western blot
Islets and RAW264.7 cells were disrupted in a lysis buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate) containing protease and phosphatase inhibitors P0044, P5725 and P8340. Western blots were performed according to standard methods (Lopez-Noriega et al., 2019). Antibodies and dilutions employed are provided in the Key resources table.
Prostaglandin E2 (PGE2) ELISA
Media was collected from cultured islets after treatments, sieved through a 0.22 μm filter and diluted 3-fold in assay buffer. Secreted PGE2 levels were measured by ELISA, following the manufacturer’s instructions (R&D systems, Minneapolis, USA). Absorbance in each well was measured by using a Varioskan Flash Spectral Scanning Multimode (ThermoFisher Scientific) and serum PGE2 concentrations were calculated from the standard curve.
Quantification and statistical analysis
Data are presented as means ± SEM. Student’s t-test or One-Way ANOVA were used as described in figure legends. p values less than or equal to 0.05 were considered statistically significant. Statistical analyses were performed using the GraphPad Prism software version 8 (GraphPad Software, La Jolla, USA).
Acknowledgments
We thank Dr. Maria José Quintero and Dr. Paloma Dominguez from the Cytometry and Microscopy Core Facility of CABIMER as well as Irene Diaz Contreras for their excellent technical and analysis support. Special thanks to Dr. Jantje M. Gerdes, Dr. Francisco Real, Dr. Jorge Ferrer and Ms. Irene Millan for their expert advice. We acknowledge the support of the pancreatic islet study group of the Spanish Association of Diabetes. The authors are supported by grants from the Consejería de Salud, Fundación Pública Andaluza Progreso y Salud, Junta de Andalucía (PI-0727-2010 to B.R.G., PI-0085-2013 to P.I.L., PI-0247-2016 to F.J.B.S.), the Consejería de Economía, Innovación y Ciencia (P10.CTS.6359 to B.R.G.), the Ministerio de Ciencia e Innovación co-funded by Fondos FEDER (PI10/00871, PI13/00593 and BFU2017-83588-P to B.R.G and PI17/01004 to F.J.B.S.), Vencer el Cancer (B.R.G), DiabetesCero (B.R.G.) and the Juvenile Diabetes Research Foundation Ltd (17-2013-372 and 2-SRA-2019-837-S-B to B.R.G.). E.M.V. is recipient of a Fellowship from the Ministerio de Ciencia e Innovación co-funded by Fondos FEDER (PRE2018-084907). F.J.B.S. is a recipient of a "Nicolás Monardes" research contracts from Consejería de Salud Junta de Andalucía, (C-0070-2012). A.M.M. is supported by CPII19/00023 and PI18/01590 from the Instituto de Salud Carlos III co-funded by Fondos FEDER. V.C. is supported by a AECC investigator award. CIBERDEM is an initiative of the Instituto de Salud Carlos III.
Author contributions
N.C.V., P.I.L., E.M.V., D.E., S.M.C., and B.R.G. were involved in research design. N.C.V., P.I.L., E.M.V., E.M., L.L.B., M.G., A.M.M., E.N., V.C., A.C., A.L.G., S.Y.R.Z., A.D., and F.J.B.S. contributed to conducting experiments. N.C.V., P.I.L., E.M.V., M.G., F.J.B.S., D.E., S.M.C., and B.R.G. contributed to data analysis. A.R., M.G., and F.M. supplied mouse strains and reagents. E.M.V., N.C.V., and B.R.G. wrote the manuscript. All authors commented on the manuscript. B.R.G. is the guarantor of this work, and as such, has full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Declaration of interests
Two patents (WO, 2011 144725 A2 and WO, 2016 156531 A1) related to BL001 have been published of which B.R.G. and N.C.V. are inventors. These patents have been licensed to ARIDDAD Therapeutics, a Biotech spinoff cofounded by B.R.G. and N.C.V. along with European business partners. The other authors declare no competing interests.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104345.
Supplemental information
Data and code availability
-
•
All data reported in this paper will be shared by the Lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.
References
- Alexanian A., Miller B., Chesnik M., Mirza S., Sorokin A. Post-translational regulation of COX2 activity by FYN in prostate cancer cells. Oncotarget. 2014;5:4232–4243. doi: 10.18632/oncotarget.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baquie M., St-Onge L., Kerr-Conte J., Cobo-Vuilleumier N., Lorenzo P.I., Jimenez Moreno C.M., Cederroth C.R., Nef S., Borot S., Bosco D., et al. The liver receptor homolog-1 (LRH-1) is expressed in human islets and protects β-cells against stress-induced apoptosis. Hum. Mol. Genet. 2011;20:2823–2833. doi: 10.1093/hmg/ddr193. ddr193 [pii]10.1093/hmg/ddr193. [DOI] [PubMed] [Google Scholar]
- Blondeau B., Sahly I., Massourides E., Singh-Estivalet A., Valtat B., Dorchene D., Jaisser F., Breant B., Tronche F. Novel transgenic mice for inducible gene overexpression in pancreatic cells define glucocorticoid receptor-mediated regulations of beta cells. PLoS One. 2012;7:e30210. doi: 10.1371/journal.pone.0030210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosma K.J., Andrei S.R., Katz L.S., Smith A.A., Dunn J.C., Ricciardi V.F., Ramirez M.A., Baumel-Alterzon S., Pace W.A., Carroll D.T., Overway E.M., Wolf E.M., Kimple M.E., Sheng Q., Scott D.K., Breyer R.M., Gannon M. Pharmacological blockade of the EP3 prostaglandin E2 receptor in the setting of Type 2 diabetes enhances beta-cell proliferation and identity, and relieves oxidative damage. Mol. Metab. 2021;54:101347. doi: 10.1016/j.molmet.2021.101347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botrugno O.A., Fayard E., Annicotte J.S., Haby C., Brennan T., Wendling O., Tanaka T., Kodama T., Thomas W., Auwerx J., Schoonjans K. Synergy between LRH-1 and beta-catenin induces G1 cyclin-mediated cell proliferation. Mol. Cell. 2004;15:499–509. doi: 10.1016/j.molcel.2004.07.009. [pii] [DOI] [PubMed] [Google Scholar]
- Brian B.F., Freedman T.S. The src-family kinase lyn in immunoreceptor signaling. Endocrinology. 2021;162:bqab152. doi: 10.1210/endocr/bqab152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bru-Tari E., Cobo-Vuilleumier N., Alonso-Magdalena P., Dos Santos R.S., Marroqui L., Nadal A., Gauthier B.R., Quesada I. Pancreatic alpha-cell mass in the early-onset and advanced stage of a mouse model of experimental autoimmune diabetes. Sci. Rep. 2019;9:9515. doi: 10.1038/s41598-019-45853-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboneau B.A., Allan J.A., Townsend S.E., Kimple M.E., Breyer R.M., Gannon M. Opposing effects of prostaglandin E2 receptors EP3 and EP4 on mouse and human beta-cell survival and proliferation. Mol. Metab. 2017;6:548–559. doi: 10.1016/j.molmet.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboneau B.A., Breyer R.M., Gannon M. Regulation of pancreatic beta-cell function and mass dynamics by prostaglandin signaling. J. Cell Commun Signal. 2017;11:105–116. doi: 10.1007/s12079-017-0377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceasrine A.M., Ruiz-Otero N., Lin E.E., Lumelsky D.N., Boehm E.D., Kuruvilla R. Tamoxifen improves glucose tolerance in a delivery-sex-and strain-dependent manner in mice. Endocrinology. 2019;160:782–790. doi: 10.1210/en.2018-00985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobo I., Iglesias M., Flandez M., Verbeke C., Del Pozo N., Llorente M., Lawlor R., Luchini C., Rusev B., Scarpa A., Real F.X. Epithelial Nr5a2 heterozygosity cooperates with mutant Kras in the development of pancreatic cystic lesions. J. Pathol. 2021;253:174–185. doi: 10.1002/path.5570. [DOI] [PubMed] [Google Scholar]
- Cobo I., Martinelli P., Flandez M., Bakiri L., Zhang M., Carrillo-de-Santa-Pau E., Jia J., Sanchez-Arevalo Lobo V.J., Megias D., Felipe I., et al. Transcriptional regulation by NR5A2 links differentiation and inflammation in the pancreas. Nature. 2018;554:533–537. doi: 10.1038/nature25751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobo-Vuilleumier N., Gauthier B.R. Time for a paradigm shift in treating type 1 diabetes mellitus: coupling inflammation to islet regeneration. Metabolism. 2020;104:154137. doi: 10.1016/j.metabol.2020.154137. [DOI] [PubMed] [Google Scholar]
- Cobo-Vuilleumier N., Lorenzo P.I., Gauthier B.R. Targeting LRH-1/NR5A2 to treat type 1 diabetes mellitus. Cell Stress. 2018;2:141–143. doi: 10.15698/cst2018.06.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobo-Vuilleumier N., Lorenzo P.I., Rodriguez N.G., Herrera Gómez I.d.G., Fuente-Martin E., Lopez-Noriega L., Mellado-Gil J.M., Romero-Zerbo S.Y., Baquie M., Lachaud C.C., et al. LRH-1 agonism favours an immune-islet dialogue which protects against diabetes mellitus. Nat. Commun. 2018;9:1488. doi: 10.1038/s41467-018-03943-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole L.K., Jacobs R.L., Vance D.E. Tamoxifen induces triacylglycerol accumulation in the mouse liver by activation of fatty acid synthesis. Hepatology. 2010;52:1258–1265. doi: 10.1002/hep.23813. [DOI] [PubMed] [Google Scholar]
- Cornelison J.L., Cato M.L., Johnson A.M., D'Agostino E.H., Melchers D., Patel A.B., Mays S.G., Houtman R., Ortlund E.A., Jui N.T. Development of a new class of liver receptor homolog-1 (LRH-1) agonists by photoredox conjugate addition. Bioorg. Med. Chem. Lett. 2020;30:127293. doi: 10.1016/j.bmcl.2020.127293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabernat S., Secrest P., Peuchant E., Moreau-Gaudry F., Dubus P., Sarvetnick N. Lack of beta-catenin in early life induces abnormal glucose homeostasis in mice. Diabetologia. 2009;52:1608–1617. doi: 10.1007/s00125-009-1411-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafoe T.J., Dos Santos T., Spigelman A.F., Lyon J., Smith N., Bautista A., MacDonald P.E., Manning Fox J.E. Impacts of the COVID-19 pandemic on a human research islet program. Islets. 2022;14:101–113. doi: 10.1080/19382014.2022.2047571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Franco E., Lytrivi M., Ibrahim H., Montaser H., Wakeling M.N., Fantuzzi F., Patel K., Demarez C., Cai Y., Igoillo-Esteve M., et al. YIPF5 mutations cause neonatal diabetes and microcephaly through endoplasmic reticulum stress. J. Clin. Invest. 2020;130:6338–6353. doi: 10.1172/JCI141455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Guia R.M., Rose A.J., Herzig S. Glucocorticoid hormones and energy homeostasis. Horm. Mol. Biol. Clin. Investig. 2014;19:117–128. doi: 10.1515/hmbci-2014-0021. [DOI] [PubMed] [Google Scholar]
- Demine S., Schiavo A.A., Marin-Canas S., Marchetti P., Cnop M., Eizirik D.L. Pro-inflammatory cytokines induce cell death, inflammatory responses, and endoplasmic reticulum stress in human iPSC-derived beta cells. Stem Cell Res. Ther. 2020;11:7. doi: 10.1186/s13287-019-1523-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatrai S., Elghazi L., Balcazar N., Cras-Meneur C., Krits I., Kiyokawa H., Bernal-Mizrachi E. Akt induces beta-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes. 2006;55:318–325. doi: 10.2337/diabetes.55.02.06.db05-0757. [DOI] [PubMed] [Google Scholar]
- Fayard E., Auwerx J., Schoonjans K. LRH-1: an orphan nuclear receptor involved in development, metabolism and steroidogenesis. Trends Cell Biol. 2004;14:250–260. doi: 10.1016/j.tcb.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Fernandez-Marcos P.J., Auwerx J., Schoonjans K. Emerging actions of the nuclear receptor LRH-1 in the gut. Biochim. Biophys. Acta. 2011;1812:947–955. doi: 10.1016/j.bbadis.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figeac F., Uzan B., Faro M., Chelali N., Portha B., Movassat J. Neonatal growth and regeneration of β-cells are regulated by the Wnt/β-catenin signaling in normal and diabetic rats. Am. J. Physiol. Endocrinol. Metab. 2010;298:E245–E256. doi: 10.1152/ajpendo.00538.2009. [DOI] [PubMed] [Google Scholar]
- Flynn A.R., Mays S.G., Ortlund E.A., Jui N.T. Development of hybrid phospholipid mimics as effective agonists for liver receptor homologue-1. ACS Med. Chem. Lett. 2018;9:1051–1056. doi: 10.1021/acsmedchemlett.8b00361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgia S., Bhushan A. β cell replication is the primary mechanism for maintaining postnatal β cell mass. J. Clin. Invest. 2004;114:963–968. doi: 10.1172/jci22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman K.E., Limesand K.H. The complex role of prostaglandin E2-EP receptor signaling in wound healing. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2021;320:R287–R296. doi: 10.1152/ajpregu.00185.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilroy D.W., Colville-Nash P.R. New insights into the role of COX 2 in inflammation. J. Mol. Med. (Berl) 2000;78:121–129. doi: 10.1007/s001090000094. [DOI] [PubMed] [Google Scholar]
- Hamilton D., Rugg C., Davis N., Kvezereli M., Tafti B.A., Busque S., Fontaine M. A preconditioning regimen with a PKCε activator improves islet graft function in a mouse transplant model. Cell Transpl. 2014;23:913–919. doi: 10.3727/096368913X665567. [DOI] [PubMed] [Google Scholar]
- Huang P., Chandra V., Rastinejad F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu. Rev. Physiol. 2010;72:247–272. doi: 10.1146/annurev-physiol-021909-135917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh J., Liepins A., Zielonka J., Andrekopoulos C., Kalyanaraman B., Sorokin A. Cyclooxygenase 2 rescues LNCaP prostate cancer cells from sanguinarine-induced apoptosis by a mechanism involving inhibition of nitric oxide synthase activity. Cancer Res. 2006;66:3726–3736. doi: 10.1158/0008-5472.CAN-05-4033. [DOI] [PubMed] [Google Scholar]
- Igoillo-Esteve M., Oliveira A.F., Cosentino C., Fantuzzi F., Demarez C., Toivonen S., Hu A., Chintawar S., Lopes M., Pachera N., et al. Exenatide induces frataxin expression and improves mitochondrial function in Friedreich ataxia. JCI Insight. 2020;5:e134221. doi: 10.1172/jci.insight.134221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliana C.A., Yang J., Rozo A.V., Good A., Groff D.N., Wang S.Z., Green M.R., Stoffers D.A. ATF5 regulates beta-cell survival during stress. Proc. Natl. Acad. Sci. U S A. 2017;114:1341–1346. doi: 10.1073/pnas.1620705114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy S.G., Wagner A.J., Conzen S.D., Jordan J., Bellacosa A., Tsichlis P.N., Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- Kilkenny C., Browne W., Cuthill I.C., Emerson M., Altman D.G., NC3Rs Reporting Guidelines Working Group Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br. J. Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.F., Huri D.A., Snyder S.H. Inducible nitric oxide synthase binds, S-nitrosylates, and activates cyclooxygenase-2. Science. 2005;310:1966–1970. doi: 10.1126/science.1119407. [DOI] [PubMed] [Google Scholar]
- Konya V., Marsche G., Schuligoi R., Heinemann A. E-type prostanoid receptor 4 (EP4) in disease and therapy. Pharmacol. Ther. 2013;138:485–502. doi: 10.1016/j.pharmthera.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G., Galluzzi L., Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- Kvezereli M., Vallentin A., Mochly-Rosen D., Busque S., Fontaine M.J. Islet cell survival during isolation improved through protein kinase C epsilon activation. Transpl. Proc. 2008;40:375–378. doi: 10.1016/j.transproceed.2008.01.014. [DOI] [PubMed] [Google Scholar]
- Lee J.M., Lee Y.K., Mamrosh J.L., Busby S.A., Griffin P.R., Pathak M.C., Ortlund E.A., Moore D.D. A nuclear-receptor-dependent phosphatidylcholine pathway with antidiabetic effects. Nature. 2011;474:506–510. doi: 10.1038/nature10111. nature10111 [pii]10.1038/nature10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.Y., Ristow M., Lin X., White M.F., Magnuson M.A., Hennighausen L. RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J. Biol. Chem. 2006;281:2649–2653. doi: 10.1074/jbc.M512373200. [DOI] [PubMed] [Google Scholar]
- Lopez-Noriega L., Capilla-Gonzalez V., Cobo-Vuilleumier N., Martin-Vazquez E., Lorenzo P.I., Martinez-Force E., Soriano-Navarro M., Garcia-Fernandez M., Romero-Zerbo S.Y., Bermudez-Silva F.J., et al. Inadequate control of thyroid hormones sensitizes to hepatocarcinogenesis and unhealthy aging. Aging (Albany NY) 2019;11:7746–7779. doi: 10.18632/aging.102285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo P.I., Fuente-Martin E., Brun T., Cobo-Vuilleumier N., Jimenez-Moreno C.M., G Herrera Gomez I., Lopez Noriega L., Mellado-Gil J.M., Martin-Montalvo A., Soria B., Gauthier B.R. PAX4 defines an expandable beta-cell subpopulation in the adult pancreatic islet. Sci. Rep. 2015;5:15672. doi: 10.1038/srep15672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo P.I., Martin Vazquez E., Lopez Noriega L., Fuente Martin E., Mellado-Gil J.M., Franco J.M., Cobo Vuilleumier N., Guerrero Martinez J.A., Romero-Zerbo S.Y., Perez Cabello J.A., et al. The metabesity factor HMG20A potentiates astrocyte survival and reactive astrogliosis preserving neuronal integrity. Theranostics. 2021;11:6983–7004. doi: 10.7150/thno.57237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytrivi M., Senee V., Salpea P., Fantuzzi F., Philippi A., Abdulkarim B., Sawatani T., Marin-Canas S., Pachera N., Degavre A., et al. DNAJC3 deficiency induces beta-cell mitochondrial apoptosis and causes syndromic young-onset diabetes. Eur. J. Endocrinol. 2021;184:455–468. doi: 10.1530/EJE-20-0636. [DOI] [PubMed] [Google Scholar]
- Ma K., Xiao A., Park S.H., Glenn L., Jackson L., Barot T., Weaver J.R., Taylor-Fishwick D.A., Luci D.K., Maloney D.J., et al. 12-Lipoxygenase inhibitor improves functions of cytokine-treated human islets and type 2 diabetic islets. J. Clin. Endocrinol. Metab. 2017;102:2789–2797. doi: 10.1210/jc.2017-00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic T., Jakopin Z., Dolenc M.S., Mlinaric-Rascan I. Structural features of subtype-selective EP receptor modulators. Drug Discov. Today. 2017;22:57–71. doi: 10.1016/j.drudis.2016.08.003. [DOI] [PubMed] [Google Scholar]
- Meier J.J., Butler A.E., Saisho Y., Monchamp T., Galasso R., Bhushan A., Rizza R.A., Butler P.C. β-Cell replication is the primary mechanism subserving the postnatal expansion of β-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. db07-1369 [pii]10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinsohn M.C., Smith O.E., Bertolin K., Murphy B.D. The orphan nuclear receptors steroidogenic factor-1 and liver receptor homolog-1: structure, regulation, and essential roles in mammalian reproduction. Physiol. Rev. 2019;99:1249–1279. doi: 10.1152/physrev.00019.2018. [DOI] [PubMed] [Google Scholar]
- Mellado-Gil J., Cobo-Vuilleumier N., Gauthier B.R. Islet β-cell mass preservation and regeneration in diabetes mellitus: four factors with potential therapeutic interest. J. Transplant. 2012;2012:230870. doi: 10.1155/2012/230870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellado-Gil J.M., Fuente-Martin E., Lorenzo P.I., Cobo-Vuilleumier N., Lopez-Noriega L., Martin-Montalvo A., Gómez I.d.G., Ceballos-Chavez M., Gomez-Jaramillo L., Campos-Caro A., et al. The type 2 diabetes-associated HMG20A gene is mandatory for islet beta cell functional maturity. Cell Death Dis. 2018;9:279. doi: 10.1038/s41419-018-0272-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezza T., Kulkarni R.N. The regulation of pre- and post-maturational plasticity of mammalian islet cell mass. Diabetologia. 2014;57:1291–1303. doi: 10.1007/s00125-014-3251-7. [DOI] [PubMed] [Google Scholar]
- Michalek S., Brunner T. Nuclear-mitochondrial crosstalk: on the role of the nuclear receptor liver receptor homolog-1 (NR5A2) in the regulation of mitochondrial metabolism, cell survival, and cancer. IUBMB Life. 2021;73:592–610. doi: 10.1002/iub.2386. [DOI] [PubMed] [Google Scholar]
- Moriya S., Kazlauskas A., Akimoto K., Hirai S., Mizuno K., Takenawa T., Fukui Y., Watanabe Y., Ozaki S., Ohno S. Platelet-derived growth factor activates protein kinase C epsilon through redundant and independent signaling pathways involving phospholipase C gamma or phosphatidylinositol 3-kinase. Proc. Natl. Acad. Sci. U S A. 1996;93:151–155. doi: 10.1073/pnas.93.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narumiya S., FitzGerald G.A. Genetic and pharmacological analysis of prostanoid receptor function. J. Clin. Invest. 2001;108:25–30. doi: 10.1172/jci200113455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opherk C., Tronche F., Kellendonk C., Kohlmuller D., Schulze A., Schmid W., Schutz G. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol. Endocrinol. 2004;18:1346–1353. doi: 10.1210/me.2003-0283. [DOI] [PubMed] [Google Scholar]
- Oshima H., Taketo M.M., Oshima M. Destruction of pancreatic beta-cells by transgenic induction of prostaglandin E2 in the islets. J. Biol. Chem. 2006;281:29330–29336. doi: 10.1074/jbc.M602424200. [DOI] [PubMed] [Google Scholar]
- Papadimitriou A., King A.J., Jones P.M., Persaud S.J. Anti-apoptotic effects of arachidonic acid and prostaglandin E2 in pancreatic beta-cells. Cell Physiol. Biochem. 2007;20:607–616. doi: 10.1159/000107544. 000107544 [pii] [DOI] [PubMed] [Google Scholar]
- Puri S., Roy N., Russ H.A., Leonhardt L., French E.K., Roy R., Bengtsson H., Scott D.K., Stewart A.F., Hebrok M. Replication confers beta cell immaturity. Nat. Commun. 2018;9:485. doi: 10.1038/s41467-018-02939-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdeva M.M., Claiborn K.C., Khoo C., Yang J., Groff D.N., Mirmira R.G., Stoffers D.A. Pdx1 (MODY4) regulates pancreatic beta cell susceptibility to ER stress. Proc. Natl. Acad. Sci. U S A. 2009;106:19090–19095. doi: 10.1073/pnas.0904849106. 0904849106 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M.W., Guyenet S.J., Cirulli V. The hypothalamus and β-cell connection in the gene-targeting era. Diabetes. 2010;59:2991–2993. doi: 10.2337/db10-1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J., Xu Y., Hu X., Choi B., Tong Q. Brain expression of Cre recombinase driven by pancreas-specific promoters. Genesis. 2010;48:628–634. doi: 10.1002/dvg.20672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergiopoulos A., Politis P.K. Nuclear receptor NR5A2 controls neural stem cell fate decisions during development. Nat. Commun. 2016;7:12230. doi: 10.1038/ncomms12230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y., Narumiya S. Prostaglandin E receptors. J. Biol. Chem. 2007;282:11613–11617. doi: 10.1074/jbc.R600038200. [DOI] [PubMed] [Google Scholar]
- Sun Y., Demagny H., Schoonjans K. Emerging functions of the nuclear receptor LRH-1 in liver physiology and pathology. Biochim. Biophys. Acta Mol. Basis Dis. 2021;1867:166145. doi: 10.1016/j.bbadis.2021.166145. [DOI] [PubMed] [Google Scholar]
- Svenstrup K., Skau M., Pakkenberg B., Buschard K., Bock T. Postnatal development of beta-cells in rats. Proposed explanatory model. APMIS. 2002;110:372–378. doi: 10.1034/j.1600-0463.2002.100502.x. [DOI] [PubMed] [Google Scholar]
- Tsatsanis C., Androulidaki A., Venihaki M., Margioris A.N. Signalling networks regulating cyclooxygenase-2. Int. J. Biochem. Cell Biol. 2006;38:1654–1661. doi: 10.1016/j.biocel.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Tuttle R.L., Gill N.S., Pugh W., Lee J.P., Koeberlein B., Furth E.E., Polonsky K.S., Naji A., Birnbaum M.J. Regulation of pancreatic β-cell growth and survival by the serine/threonine protein kinase Akt1/PKBα. Nat. Med. 2001;7:1133–1137. doi: 10.1038/nm1001-1133. [DOI] [PubMed] [Google Scholar]
- Vennemann A., Gerstner A., Kern N., Ferreiros Bouzas N., Narumiya S., Maruyama T., Nusing R.M. PTGS-2-PTGER2/4 signaling pathway partially protects from diabetogenic toxicity of streptozotocin in mice. Diabetes. 2012;61:1879–1887. doi: 10.2337/db11-1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitby R.J., Dixon S., Maloney P.R., Delerive P., Goodwin B.J., Parks D.J., Willson T.M. Identification of small molecule agonists of the orphan nuclear receptors liver receptor homolog-1 and steroidogenic factor-1. J. Med. Chem. 2006;49:6652–6655. doi: 10.1021/jm060990k. [DOI] [PubMed] [Google Scholar]
- Zhang H., Fujitani Y., Wright C.V., Gannon M. Efficient recombination in pancreatic islets by a tamoxifen-inducible Cre-recombinase. Genesis. 2005;42:210–217. doi: 10.1002/gene.20137. [DOI] [PubMed] [Google Scholar]
- Zhang L., Wang Y., Wang J., Liu Y., Yin Y. Protein kinase C pathway mediates the protective effects of glucagon-like peptide-1 on the apoptosis of islet beta-cells. Mol. Med. Rep. 2015;12:7589–7594. doi: 10.3892/mmr.2015.4355. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Zhang E., Berggreen C., Jing X., Osmark P., Lang S., Cilio C.M., Goransson O., Groop L., Renstrom E., Hansson O. Survival of pancreatic beta cells is partly controlled by a TCF7L2-p53-p53INP1-dependent pathway. Hum. Mol. Genet. 2012;21:196–207. doi: 10.1093/hmg/ddr454. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
All data reported in this paper will be shared by the Lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.