

This cohort study examines data from the UK Biobank and Mass General Brigham Biobank to assess the contribution of rare and common genetic variants to risk of hypertrophic cardiomyopathy in the general population.
Key Points
Question
What are the contributions of rare and common genetic variation to risk of hypertrophic cardiomyopathy (HCM)?
Findings
In this cohort study, rare variants in 14 genes prioritized by the American College of Medical Genetics and Genomics conferred the greatest risk of HCM, while a common variant (polygenic) score accounted for the greatest proportion of HCM susceptibility. Together, rare variants and the polygenic score enhanced the prediction of incident HCM when added to clinical factors.
Meaning
Efforts to improve HCM prevention may benefit from the development of prediction algorithms that combine rare and common genetic variants with clinical risk factors.
Abstract
Importance
Hypertrophic cardiomyopathy (HCM) is a leading cause of sudden cardiac death in young people. Although rare genetic variants are well-established contributors to HCM risk, common genetic variants have recently been implicated in disease pathogenesis.
Objective
To assess the contributions of rare and common genetic variation to risk of HCM in the general population.
Design, Setting, and Participants
This cohort study of the UK Biobank (data from 2006-2010) and the Mass General Brigham Biobank (2010-2019) assessed the relative and joint contributions of rare genetic variants and a common variant (polygenic) score to risk of HCM. Both rare and common variant predictors were then evaluated in the context of relevant clinical risk factors. Data analysis was conducted from May 2021 to February 2022.
Exposures
Pathogenic rare variants, common-variant (polygenic) score, and clinical risk factors.
Main Outcomes and Measures
Risk of HCM.
Results
The primary study population comprised 184 511 individuals from the UK Biobank. Mean (SD) age was 56 (8) years, 83 690 (45%) of participants were men, and 204 (0.1%) participants had HCM. Of 51 genes included in clinical genetic testing panels for HCM, pathogenic or likely pathogenic variants in 14 core genes (designated by the American College of Medical Genetics and Genomics [ACMG]) were associated with 55-fold higher odds (95% CI, 35-83) of HCM, while those in the remaining 37 non-ACMG genes were not significantly associated with HCM (OR, 1.8; 95% CI, 0.6-4.0). ClinVar pathogenic or likely pathogenic mutations in MYBPC3 (OR, 72; 95% CI, 39-124) and MYH7 (OR, 61; 95% CI, 26-121) were strongly associated with HCM, as were loss-of-function variants in ALPK3 (OR, 13; 95% CI, 4.4-28). A polygenic score was strongly associated with HCM (OR per SD increase in score, 1.6; 95% CI, 1.4-1.8), with concordant results in the Mass General Brigham Biobank. Genetic factors enhanced clinical risk prediction for HCM: addition of rare variant carrier status and the polygenic score to clinical risk factors (obesity, hypertension, atrial fibrillation, and coronary artery disease) improved the area under the receiver operator characteristic curve from 0.71 (95% CI, 0.65-0.77) to 0.82 (95% CI, 0.77-0.87).
Conclusions and Relevance
Both rare and common genetic variants contribute substantially to HCM susceptibility in the general population and improve HCM risk prediction beyond that achieved with clinical factors.
Introduction
Hypertrophic cardiomyopathy (HCM) is a leading cause of sudden cardiac death, affecting up to 1 in 500 individuals.1 Rare genetic mutations are thought to account for up to 40% of HCM cases.2,3,4,5,6 Currently, rare variants in 8 sarcomeric genes are classified as having strong evidence for a causal, pathogenic role in HCM.7 However, many variants in established genes remain classified as variants of unknown significance, and clinical screening panels may include up to 39 additional genes with low to moderate levels of evidence for HCM, contributing to variable interpretations of results across different testing laboratories.7,8,9,10,11,12
More recently, genome-wide association studies (GWAS) have demonstrated that common genetic variants also contribute to HCM risk.1,13 The identification of common variants associated with HCM now enables their aggregation into a single genetic predictor (a polygenic score). However, it remains unclear whether a polygenic score can enhance stratification of risk for HCM in unselected populations alongside rare genetic variants and clinical factors.7
The 2020 American Heart Association/American College of Cardiology guidelines on the diagnosis and management of HCM endorse integrated risk assessments, combining large-scale clinical and genetic data, to improve HCM prognostication and prevention.7 Toward that goal, here we first reassessed the contributions of rare pathogenic variants to HCM risk in the UK Biobank, a large, unselected, cohort population. Next, we evaluated a polygenic risk score for HCM in both population-based and hospital-based settings. Then, we assessed the relative and combined contributions of genetic and clinical factors to risk of HCM.
Methods
Study Population
Analyses were conducted in the UK Biobank and the Mass General Brigham (MGB) Biobank. The UK Biobank is a large, population-based prospective cohort study from the United Kingdom with approximately 500 000 participants.14 We restricted analyses of UK Biobank to 184 511 unrelated participants for whom both high-quality genotyping array and exome sequencing data were available (eMethods in the Supplement).15,16 MGB Biobank is a hospital-based biobank that currently consists of approximately 120 000 participants.17 Here, we included 30 716 participants from MGB Biobank with high-quality genotyping-array and clinical data (eMethods in the Supplement). Informed consent was obtained by the UK Biobank for all study participants, and analysis was approved by the Mass General Brigham institutional review board (protocol 2013P001840; UK Biobank application 7089). This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.
Phenotype Definitions
In the UK Biobank, HCM cases were identified by the presence of International Statistical Classification of Diseases and Related Health Problems, Tenth Revision (ICD-10), billing code I42.1 (hypertrophic obstructive cardiomyopathy) or I42.2 (other hypertrophic cardiomyopathy). In MGB Biobank, HCM was defined as having an ICD-10 code of I42.1 or I42.2, in addition to a mention of “hypertrophic cardiomyopathy,” “hypertrophic obstructive cardiomyopathy,” “HCM,” or “HOCM” in the medical record. Complete disease definitions are available in eTable 1 in the Supplement.
Rare Variant Curation
We considered 51 nonmitochondrial genes present on at least 1 of 3 clinical genetic testing panels for HCM (GeneDx, Invitae, and Blueprint) (eTable 1 in the Supplement).8,9,10 Three primary rare variant masks were analyzed: pathogenic or likely pathogenic variants by ClinVar (ClinVarPLP), predicted loss-of-function (LOF) variants, and predicted-deleterious missense variants (eMethods in the Supplement).16
We then curated a subset of high-impact rare variants (cumulatively termed HCM-ACMG) denoting the presence of a putatively pathogenic rare variant within 14 core genes for HCM as designated by the American College of Medical Genetics and Genomics (ACMG) (eMethods and eTable 2 in the Supplement).11 Rare variant carrier status was defined as presence of (1) a ClinVarPLP variant in any of these genes or (2) LOF variant in 3 genes with prior literature implicating a truncating mutation in HCM pathogenesis (MYBPC3, TNNT2, or PLN; eTable 3 in the Supplement).4,5,18 Sensitivity analyses were conducted using different definitions for rare variant carriers, such as (1) a broader set including ClinVarPLP variants for all of the 51 tested genes (HCM-Panel, eTable 2 in the Supplement), (2) only MYBPC3 rare variant carriers (ClinVarPLP and LOF), and (3) ClinVarPLP rare variants in HCM-Panel genes that were not included in the HCM-ACMG definition (ie, rare variants in noncore genes, termed HCM–non-ACMG). Sensitivity analyses to account for potential limitations of our rare variant classifications are described in the eMethods in the Supplement.
Polygenic Risk Score Estimation
A polygenic risk score (PRS) was constructed using data from a recent GWAS of HCM.1 Conditionally independent common genetic variants associated with HCM below a 5% false discovery rate threshold (27 common variants, in total) were included in the PRS. Weighted allele scores were calculated as the sum of, for each common variant, an individual’s number of risk alleles multiplied by the reported β coefficient (log odds ratio) for that variant (eMethods in the Supplement). The PRS were residualized for the first 5 principal components (PCs) of genetic ancestry and normalized so that the SD was equal to 1.
Statistical Analysis
Per variant mask, rare variants were tested for association with HCM in UK Biobank using gene-based collapsing tests (eMethods in the Supplement). To account for both case-control imbalance and carrier-noncarrier imbalance, we used a Firth bias-reduced logistic regression, adjusting for age, sex, and the first 5 PCs of genetic ancestry.19 In gene-based testing, there were 51 genes × 3 variant classes = 153 total masks. We only considered variant masks “testable” if they had 10 or more rare variant carriers, which means we only performed the test if there were 10 or more carriers for a mask. Eighty masks fulfilled this requirement, and therefore the significance threshold for the gene-based analysis was determined at α = 0.05 ÷ 80 testable masks = 6.25 × 10−4.
We used the same Firth regression model and the same fixed-effects covariates to assess the associations with HCM for the HCM-ACMG, HCM-Panel, and HCM–non-ACMG masks. The HCM PRS was validated in UK Biobank and MGB Biobank. We evaluated the odds of HCM per 1-SD increase in the PRS, and the area under the receiver operator characteristic curve (AUC) based on logistic regression models adjusted for age, sex, genotyping array, and PCs 1 through 5.
Next, the odds of HCM were compared across strata of the PRS and by HCM-ACMG rare variant carrier status. Individuals at low polygenic risk (bottom quintile of the PRS), intermediate risk (middle 3 quintiles), or high risk (top quintile) were further subdivided into rare variant carriers and noncarriers, resulting in 6 distinct categories of genetic risk. The relative odds of HCM were assessed by logistic regression adjusting for age, sex, genotyping array, and PCs 1 through 5 using noncarriers with intermediate polygenic risk as reference.
Longitudinal risk trajectories were modeled across the 6 strata of genetic risk as lifetime risk at age 80 years and by cumulative incidence. Both cumulative incidence and lifetime absolute risks were calculated using Cox regression models, adjusted for sex, genotyping array, and PCs 1 through 5, and using age as the time scale.20
Rare and common variant predictors were then compared with clinical risk factors for HCM. To choose clinical risk factors most relevant for HCM, we first tested several baseline risk factors (obesity, hypertension, atrial fibrillation, chronic kidney disease, type 2 diabetes, coronary artery disease, physical activity, alcohol consumption, and smoking) for association with incident HCM using Cox regression models adjusted for age and sex and selected those reaching nominal significance (P < .05) for analysis alongside genetic factors. We assessed genetic and clinical factors based on (1) hazard ratios for incident disease from Cox regression models adjusted for age, sex, genotyping array, and PCs 1 through 5; (2) AUCs from logistic regression models on top of age, sex, genotyping array, and PCs 1 through 5; and (3) the population-wide variance in HCM susceptibility explained.21 Net reclassification improvement was calculated to compare the predictive improvement of adding genetic factors (rare variants and a PRS) with a 5-year clinical risk model (eMethods in the Supplement). For primary analyses, polygenic risk was dichotomized (high/low PRS) using the top quintile of the PRS as cutoff. Statistical analyses were conducted using R version 3.5; data analysis was conducted from May 2021 to February 2022.
Results
Baseline Characteristics
Study characteristics for UK Biobank are shown in Table 1. Participants in UK Biobank had a mean (SD) age of 56.51 (8.05) years, 83 680 (45.35%) were men, and 204 (0.11%) had HCM. Participants in MGB Biobank had a mean (SD) age of 57.23 (17.26) years, 13 935 (45.37%) were male, and 292 (0.95%) had HCM. Study characteristics for MGB Biobank are shown in eTable 3 in the Supplement.
Table 1. Baseline Characteristics of Individuals in the Biobank Study Populations.
| Characteristic | No. (%)a | |
|---|---|---|
| UK Biobank (n = 184 511) | MGB Biobank (n = 30 716) | |
| Age, mean (SD), y | 56.51 (8.05) | 57.23 (17.26) |
| Men | 83 680 (45.35) | 13 935 (45.37) |
| Women | 100 831 (54.65) | 16 779 (54.63) |
| Self-reported White ethnicity | 161 401 (87.48) | 25 797 (84.04) |
| Smoking initiation | 81 909 (44.44) | NA |
| High alcohol consumption | 69 059 (37.43) | NA |
| Obesity | 44 119 (24.01) | 4134 (28.64) |
| Heart failure | 3157 (1.71) | 5165 (16.82) |
| Hypertension | 59 542 (32.27) | 13 820 (45.03) |
| Atrial defibrillation | 6393 (3.46) | 5662 (18.45) |
| Type 2 diabetes | 8981 (4.87) | 2996 (9.76) |
| Coronary artery disease | 11 692 (6.34) | 3300 (10.74) |
| Hypertrophic cardiomyopathy | 204 (0.11) | 292 (0.95) |
Abbreviations: MGB, Mass General Brigham; NA, not available.
Disease counts are of all cases (prevalent and incident).
Association of Rare Genetic Variants With HCM
We first performed gene-based, rare variant testing for 51 genes typically included on HCM sequencing panels, using 3 different variant masks. ClinVarPLP variants in MYBPC3 (OR, 71.99; 95% CI, 38.66-123.64; P = 1.90 × 10−43) and MYH7 (OR, 60.67; 95% CI, 26.07-121.09; P = 1.20 × 10−11) were associated with HCM at a Bonferroni-corrected level of significance (Table 2 and eTable 4 in the Supplement); variants in DES, PTPN11, TNNI3, and TTR were associated at nominal significance (P < .05). Similarly, predicted LOF variants in MYBPC3 (OR, 89.39; 95% CI, 37.64-187.2; P = 2.78 × 10−14) and ALPK3 (OR, 13.33; 95% CI, 4.37-28.38; P = 2.89 × 10−4) also demonstrated strong associations with HCM, while predicted LOF variants in PLN, ACTN2, and MYH6 reached nominal significance. Predicted-deleterious missense mutations in FLNC, MYH7, and PTPN11 also reached nominal significance.
Table 2. Genes Associated With Hypertrophic Cardiomyopathy in UK Biobanka.
| Gene | Variant mask | No. (%) | OR (95% CI) | P value | |
|---|---|---|---|---|---|
| Carriers | Cases among carriers | ||||
| MYBPC3 | ClinVarPLP | 205 (0.11) | 13 (6.34) | 71.99 (38.66-123.64) | 1.90 × 10−43 |
| MYH7 | ClinVarPLP | 130 (0.07) | 7 (5.38) | 60.67 (26.07-121.09) | 1.20 × 10−11 |
| DES | ClinVarPLP | 23 (0.01) | 1 (4.35) | 63.53 (6.99-253.62) | 2.00 × 10−3 |
| PTPN11 | ClinVarPLP | 25 (0.01) | 1 (4) | 50.16 (5.4-205.86) | 3.00 × 10−3 |
| TNNI3 | ClinVarPLP | 31 (0.02) | 1 (3.23) | 45.13 (5-175.63) | .004 |
| TTR | ClinVarPLP | 27 (0.01) | 1 (3.7) | 71.89 (7.92-285.44) | .002 |
| MYBPC3 | pLOF | 77 (0.04) | 8 (10.39) | 89.39 (37.64-187.2) | 2.78 × 10−14 |
| ALPK3 | pLOF | 213 (0.12) | 3 (1.41) | 13.33 (4.37-28.38) | 2.89 × 10−4 |
| PLN | pLOF | 17 (0.01) | 1 (5.88) | 105.16 (11.36-440.71) | 1.00 × 10−3 |
| ACTN2 | pLOF | 22 (0.01) | 1 (4.55) | 59.85 (6.58-238.81) | .002 |
| MYH6 | pLOF | 162 (0.09) | 1 (0.62) | 7.89 (0.96-26.14) | 5.30 × 10−2 |
| FLNC | Missense | 75 (0.04) | 1 (1.33) | 12.46 (1.39-47.4) | 3.00 × 10−2 |
| MYH7 | Missense | 54 (0.03) | 1 (1.85) | 31.93 (3.59-119.6) | 6.00 × 10−3 |
| PTPN11 | Missense | 16 (0.01) | 1 (6.25) | 147.07 (15.97-609.42) | 5.89 × 10−4 |
Abbreviations: ClinVarPLP, pathogenic or likely pathogenic variants by ClinVar; OR, odds ratio; pLOF, predicted loss of function.
Results of gene-based rare variant associations with hypertrophic cardiomyopathy are presented for genes with P < .05 and with ≥10 rare variant carriers.
In aggregate, the presence of any HCM-Panel variant was associated with 10.18-fold increased odds of HCM (95% CI, 6.60-15.09; P = 1.76 × 10−26). Specifically, presence of an HCM-ACMG variant was associated with 55.04-fold increased odds of HCM (95% CI, 35.09-82.92; P = 1.13 × 10−67). A more stringent definition restricting to ClinVarPLP variants reported specifically for HCM and excluding TNNT2 LOFs yielded a slightly higher effect estimate for the HCM-ACMG class (OR, 68; 95% CI, 43.09-103.92).
In contrast, harboring a ClinVarPLP rare variant among the HCM–non-ACMG genes (ie, genes on the HCM-Panel list but not the HCM-ACMG list) was not significantly associated with HCM (OR, 1.77; 95% CI, 0.59-4.01; P = .27). When restricting to ClinVarPLP variants with HCM-specific assertions, we identified 1 HCM–non-ACMG carrier with HCM. This individual carried a known pathogenic variant in PTPN11 underlying Noonan syndrome, a congenital syndrome with a high risk of ventricular hypertrophy.22
Association of Polygenic Score With HCM
We evaluated a PRS derived from a recent GWAS of HCM. A 1-SD increase in the PRS was associated with 1.56-fold increased odds of HCM (95% CI, 1.36-1.78; P = 8.22 × 10−11) in UK Biobank (eTable 5 in the Supplement) and 1.35-fold increased odds of HCM (95% CI, 1.21-1.51; P = 1.36 × 10−7) in MGB Biobank. Addition of the PRS resulted in an AUC of 0.706 (95% CI, 0.669-0.742) as compared with a reference model including age, sex, and the top 5 PCs (AUC, 0.670; 95% CI, 0.635-0.705) (eFigure 1 in the Supplement). Results were consistent after excluding individuals from the first tranche of whole-exome sequencing data in UK Biobank (n = 50 000) (eTable 5 in the Supplement).
Assessments of Aggregate Genetic Risk for HCM
The PRS was strongly associated with HCM among noncarriers of HCM-ACMG mutations (OR, 1.59; P = 2.39 × 10−10). The lifetime risks of HCM for noncarriers with a low, intermediate, and high PRS were 0.09% (95% CI, 0.03%-0.15%), 0.14% (95% CI, 0.09%-0.18%), and 0.43% (95% CI, 0.29%-0.56%), respectively (Figure 1 and eFigure 2 in the Supplement). Although the association of the PRS with HCM did not reach statistical significance in HCM-ACMG mutation carriers (OR, 1.35; 95% CI, 0.91-1.99; P = .14), there was a trend toward increased lifetime risk of HCM for carriers with a low PRS (2.94%; 95% CI, 0.00%-8.49%), intermediate PRS (8.22%; 95% CI, 2.42%-13.67%), and high PRS (16.64%; 95% CI, 3.31%-28.15%) (Figure 1). This trend toward increased odds of HCM across PRS strata was observed among carriers of mutations in HCM-Panel genes, HCM-ACMG genes, and MYBPC3 alone (eFigure 2 in the Supplement).
Figure 1. Lifetime Risk of Hypertrophic Cardiomyopathy (HCM) by Rare Variant Carrier Status and Strata of Polygenic Risk.
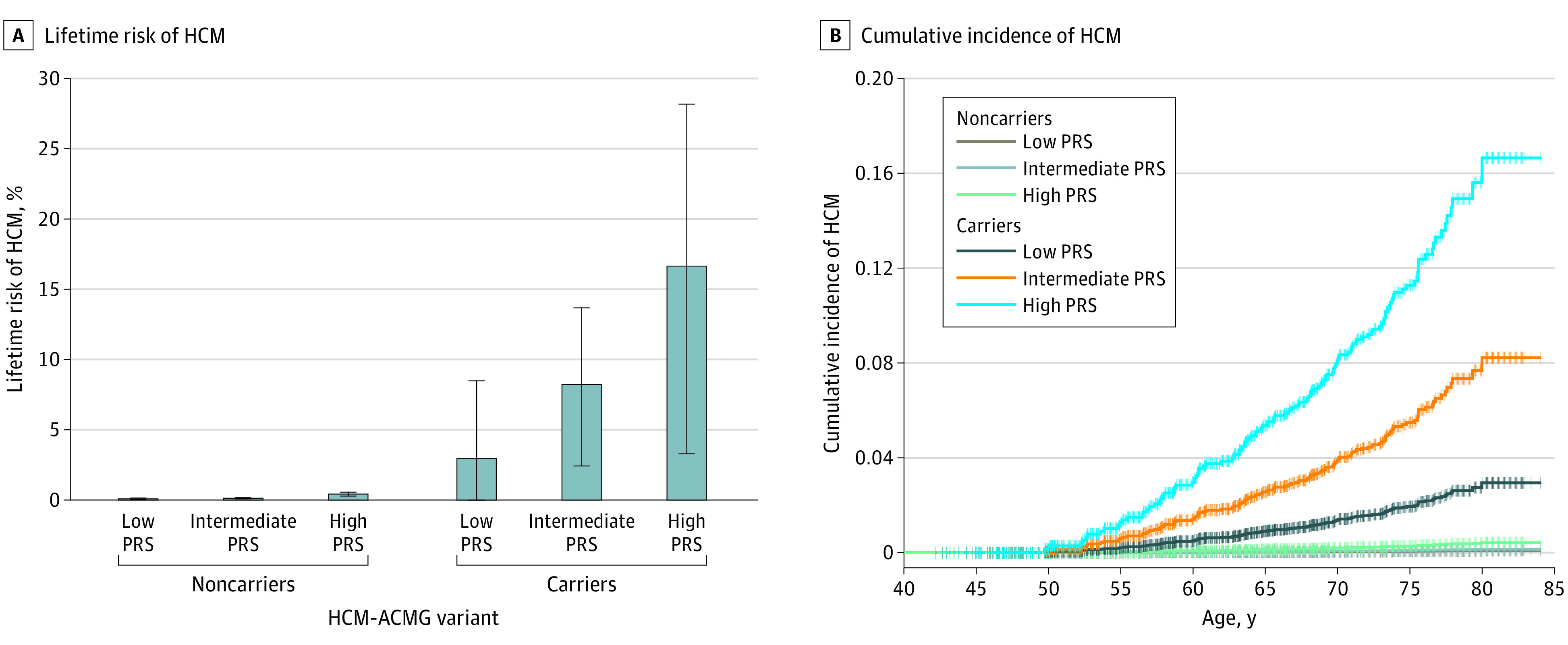
A, Lifetime risk of HCM, defined as the percent chance of a diagnosis of hypertrophic cardiomyopathy by age 80 years (n = 129 incident cases), in strata of rare variant carrier status and polygenic risk. B, Cumulative incidence plots for HCM in strata of rare variant carrier status and polygenic risk. Rare variant carrier status was defined as the presence of an HCM–American College of Medical Genetics and Genomics (ACMG) variant. Low polygenic risk was defined as the lowest quintile, intermediate risk as quintiles 2 through 4, and high risk as the top quintile of the polygenic risk score (PRS) distribution. Error bars denote 95% CI. Data analysis was conducted in the UK Biobank.
Combining Genetic and Nongenetic Risk Factors for HCM
In epidemiological analyses, baseline obesity, hypertension, atrial fibrillation, and coronary artery disease were associated with incident HCM (eTable 6 in the Supplement). We then compared the relative contributions of rare variant carrier status, PRS, and the abovementioned clinical risk factors to risk for HCM (Figure 2). The single strongest risk factor for HCM was harboring an HCM-ACMG rare variant, which conferred a 49.9-fold increase in risk of incident HCM (95% CI, 29.08-85.61; P = 9.64 × 10−46); this was followed by baseline atrial fibrillation (HR, 3.84; 95% CI, 1.84-7.99; P = 3.26 × 10−4) and a high PRS (top quintile of score distribution) (HR, 3.32; 95% CI, 2.35-4.70; P = 1.21 × 10−11). However, a high PRS (r2 = 5.38%) individually explained the greatest proportion of HCM susceptibility in UK Biobank; this was followed by baseline hypertension (r2 = 3.43%) and HCM-ACMG rare variant carrier status (r2 = 1.62%) (Figure 2). The greatest change in AUC over a reference model (comprising age, sex, and PCs 1-5, AUC, 0.657; 95% CI, 0.609-0.704) was seen for a high PRS (AUC, 0.725; 95% CI, 0.678-0.771), followed by HCM-ACMG rare variant carrier status (AUC, 0.712; 95% CI, 0.663-0.760) (eFigure 3 in the Supplement).
Figure 2. Comparisons of Individual Genetic and Nongenetic Risk Factors for Hypertrophic Cardiomyopathy (HCM).
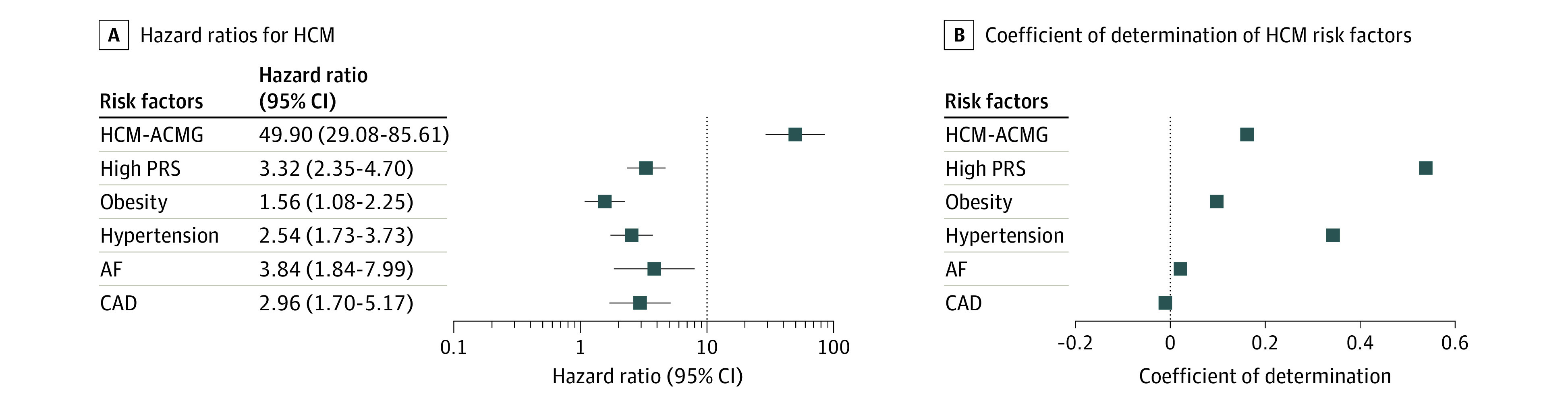
A, Hazard ratios for HCM (n = 129 incident cases), calculated using Cox proportional-hazards models adjusting for age, sex, genotyping array, and principal components 1 through 5. B, Proportion of population-wide HCM susceptibility explained by genetic and nongenetic risk factors. High polygenic risk is defined as a score in the top quintile of polygenic risk score (PRS) distribution. Error bars denote 95% CI. Data analysis was conducted in the UK Biobank. ACMG indicates American College of Medical Genetics and Genomics; AF, atrial fibrillation; CAD, coronary artery disease.
We then assessed the combined utility of clinical and genetic factors for prognostication of HCM. Compared with the reference model (AUC = 0.657, as above), a model incorporating clinical risk factors yielded an AUC of 0.709 (95% CI, 0.649-0.768). Progressive addition of HCM-ACMG rare variant carrier status (AUC, 0.756; 95% CI, 0.696-0.816) and the PRS (AUC, 0.821; 95% CI, 0.772-0.871) resulted in improved discrimination on top of clinical risk factors (Figure 3). The 4 clinical risk factors jointly explained 2.28% of the population-wide variance in HCM susceptibility. Addition of HCM-ACMG rare variants to the model increased this proportion to 3.99%, and further addition of the PRS improved the variance explained to 10.46% (Figure 3). Results were concordant when using HCM-Panel or MYBPC3 rare variants (instead of HCM-ACMG rare variants), when using continuous risk factors where relevant (ie, body mass index instead of obesity, continuous PRS instead of dichotomized or high PRS), and after excluding the first tranche (n = 50 000) of UK Biobank whole-exome sequencing data (eFigures 4-8 in the Supplement). Using the median of a 5-year clinical risk score as reference, the addition of genetic factors to the clinical prediction model resulted in improved net reclassification (improvement, 0.233; 95% CI, 0.136-0.401) (eTable 7 in the Supplement), albeit with a modest number of cases reclassified.
Figure 3. Cumulative Performance of Genetic and Nongenetic Risk Factors for Hypertrophic Cardiomyopathy (HCM).
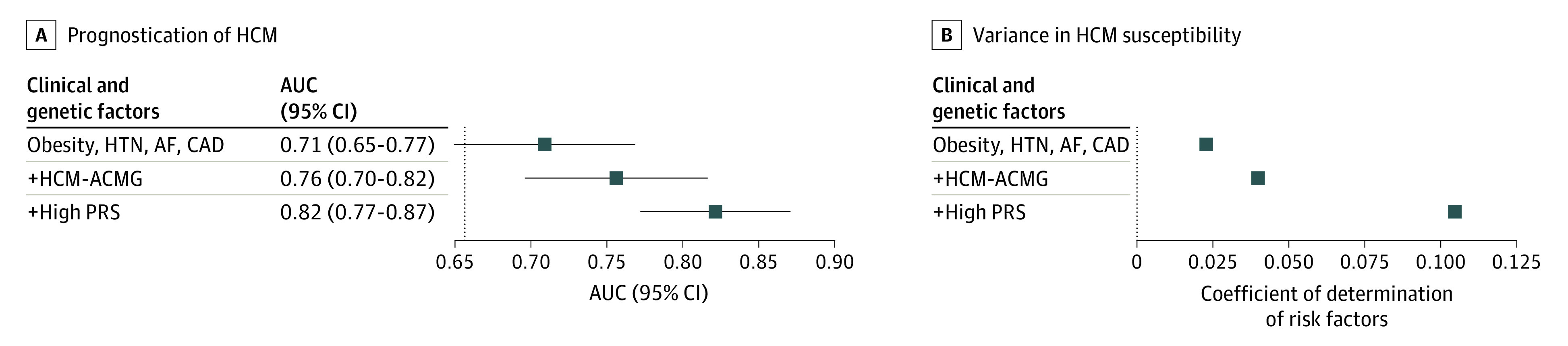
A, Cumulative area under the receiver operator characteristic curve (AUC) for HCM (n = 129 incident cases). The vertical line represents the AUC for the baseline logistic regression model inclusive of age, sex, and principal components (PCs) 1 through 5. Clinical and genetic factors were then added sequentially as predictors on top of the baseline model. B, Cumulative variance in disease susceptibility explained. Plotted values are calculated as improvement in r2 on the liability scale on adding risk factors to a logistic regression model consisting of age, sex, and PCs 1-5 in a stepwise fashion. High polygenic risk is defined as an individual with a score in the top quintile of polygenic risk score (PRS) distribution. Data analysis was conducted in the UK Biobank. Error bars denote 95% CI. ACMG indicates American College of Medical Genetics and Genomics; AF, atrial fibrillation; CAD, coronary artery disease; HTN, hypertension.
Discussion
We assessed the relative and joint contributions of rare and common genetic variation to HCM risk. Our findings reveal the marked effect of harboring an established rare variant and/or a high burden of common variants on population-level risk of HCM. Furthermore, we demonstrate the prognostic importance of both forms of genetic risk in the context of relevant clinical factors.
These results permit several conclusions. First, regular population-based assessments of rare genetic variants are important for prioritizing those mutations of greatest clinical relevance to HCM. Parallel advances with sequencing technologies and the assembly of disease-specific cohorts have led to rapid increases in the numbers of genes implicated in HCM, albeit often with scant supportive evidence.12 A recent analysis illustrated the value of large-scale, population-based sequencing for reassessing putative pathogenic genes for HCM.6 Here, we extend the findings of this recent study by evaluating the influence of different rare variant classes and also of individual genes on HCM risk. For example, our analysis demonstrated that rare variants limited to the 14 HCM genes designated as high confidence by the ACMG were strongly associated with HCM (cumulatively, a 55-fold increased odds of disease), while variants in the remaining 39 non-ACMG genes were not significantly associated with disease. Regarding individual genes, we confirmed that rare variants in MYBPC3 (ClinVarPLP and predicted LOF variants) and MYH7 (ClinVarPLP and missense variants) were the most prevalent among HCM cases. In addition, rare variants in several other genes, including DES, PTPN11, TNNI3, TTR, PLN, ACTN2, MYH6, and FLNC, were nominally associated with HCM, although many of these genes had only 1 case among rare variant carriers. Notably, a strong and statistically significant association with HCM was observed for LOF variants in ALPK3, a gene not frequently cited in prior HCM case-control analyses.3,4,5,11 Coupled with recent studies linking ALPK3 truncating variants to both pediatric and adult-onset HCM, this observation may support reprioritization of ALPK3 for genetic screening of HCM.23,24
Second, common genetic variants contribute substantially to risk of HCM in the general population. A recent study demonstrated that the vast majority of individuals in the general population with unexplained left ventricular hypertrophy lack a disease-relevant, rare sarcomeric mutation, implicating other genetic and nongenetic mechanisms.6 In addition, 2 recent case-control analyses1,13 have uncovered common genetic variants associated with HCM, with one study (Harper et al1) positing the role of a common-variant (polygenic) predictor for stratifying disease risk. Here, we validate this polygenic predictor by showing its association with HCM in population-based (UK Biobank) and health system–based (MGB Biobank) settings. We also newly demonstrate the relative contributions of rare and common genetic variants to population-level risk of HCM and provide estimates of lifetime disease risk based on a cumulative genetic susceptibility. We found that high polygenic risk (defined as the top quintile of the PRS distribution), which is not presently measured in the clinical setting, accounted for more than 5% of the population-wide variance in HCM, greater than the combined variance explained of the most important genetic and clinical predictors in use today (rare variant carrier status and hypertension, respectively). In particular, we observed a strong association of the PRS with HCM among noncarriers of pathogenic, rare variants, suggesting a new avenue for genetic risk stratification of HCM and one with expanded reach.25 Although rare variant carriers with high polygenic risk trended toward having the greatest risk of HCM, these associations did not reach statistical significance in this study. Additional analyses leveraging larger sample sizes and advanced techniques for generating polygenic scores are needed to further evaluate the utility of polygenic risk prediction of HCM among carriers of rare, pathogenic variants.
Third, rare and common genetic variants provide information that is complementary to clinical factors for the prognostication of HCM. To date, risk prediction for HCM has tended to focus on the prevention of secondary events, ie, identifying patients with HCM at risk for sudden cardiac death who might benefit from placement of an implantable cardioverter-defibrillator.7 However, more available genetic testing coupled with both existing and novel HCM therapies may afford new opportunities to preempt this condition at the population level.26,27 With the establishment of an HCM polygenic predictor in a recent case-control study, we now evaluate polygenic risk for HCM in the general population and in the context of rare genetic variants and clinical factors.1 Our analyses demonstrate the significant individual and additive contributions of prominent clinical factors (in particular, hypertension), rare variant carrier status, and a high polygenic burden to HCM risk. While key clinical factors alone demonstrated an AUC of 0.71 (on top of age, sex, and PCs), the addition of genetic factors improved the AUC by 10 points and resulted in a 4-fold increase in the percent variance explained. These findings therefore suggest potential prognostic benefit in combining genetic and clinical data to identify individuals requiring more aggressive and regular screening for detection of preclinical HCM. Additional studies are required to determine whether such high-risk patients stand to benefit from early introduction of HCM therapies.28
The difference in HCM prevalence between the UK Biobank (0.11%) and MGB Biobank (1.0%) is not unexpected given the population-based recruitment of relatively healthy, middle-aged individuals in UK Biobank, which likely biased against the recruitment of young-onset and severe HCM cases. Despite these known limitations, risk factor associations in the UK Biobank are thought to be generalizable.29 In contrast, the high HCM prevalence in MGB Biobank likely reflects an enrichment for patients with HCM in a health system–based cohort. We further note that ICD-code–based phenotyping may underdiagnose asymptomatic HCM. Indeed, a higher prevalence of HCM (0.19%) was recently estimated in UK Biobank when incorporating magnetic resonance imaging ascertainment of left ventricular hypertrophy.6 Further assessments of expressivity are warranted as the number of participants with available sequencing and imaging data continues to grow.
We also highlight several clinical factors that are associated with incident HCM, including well-established risk factors like obesity and hypertension, but also less-established predictors such as atrial fibrillation and coronary artery disease. Atrial fibrillation is a common arrhythmia affecting patients with HCM.30 Shared biological processes between the 2 conditions (ie, atrial and ventricular fibrosis) may yield an observational association between HCM and both antecedent and subsequent atrial fibrillation. However, the observed link between coronary artery disease and incident HCM is not well established and requires further study.
Limitations
Our study has other potential limitations. First, our study cohorts comprised largely individuals of European genetic ancestry. Additional studies are required to generalize findings to other ancestral groups. Second, the designation of high-impact HCM genes and putatively pathogenic rare variants may not be exhaustive, as additional causative HCM genes may exist. Similarly, our definition of high-impact variants was based on bioinformatic predictions and ClinVar reports, and therefore, potentially relevant rare variants may have been excluded. Accordingly, rare variant grouping in the present study should be taken as illustrative. Third, the PRS was based on 27 common variants associated with HCM past a 1% false discovery rate threshold. We anticipate that larger GWAS discovery sets and improved polygenic scoring methods will yield better polygenic predictors for HCM, which may enhance risk prediction in the clinical setting. Fourth, combined analyses of monogenic and polygenic risk were limited in power; additional studies are needed to assess whether a PRS can stratify risk in carriers of rare, disease-causing variants.
Conclusions
Both rare and common genetic variants contribute significantly to risk of HCM in the general population. Future efforts should seek to integrate clinical and genetic factors to improve the prognostication of HCM and facilitate disease prevention.
eMethods
eTable 1. Disease definitions in UK Biobank
eTable 2. Gene categorizations for hypertrophic cardiomyopathy
eTable 3. Prioritized rare variants for hypertrophic cardiomyopathy
eTable 4. Gene-based associations of rare variants with hypertrophic cardiomyopathy in UK Biobank
eTable 5. Associations of the polygenic score with hypertrophic cardiomyopathy in carriers and noncarriers of an HCM-ACMG rare variant
eTable 6. Survey of clinical risk factors for HCM
eTable 7. Net reclassification improvement of clinical and genetic factors over clinical factors
eFigure 1. Receiver-operator curve for HCM PRS
eFigure 2. Combined odds of polygenic risk score and carrier status
eFigure 3. Discriminative benefit of individual risk factors for HCM in the UK Biobank
eFigure 4. Comparison of genetic and nongenetic risk factors for hypertrophic cardiomyopathy
eFigure 5. Cumulative predictive capabilities of genetic and nongenetic risk factors for hypertrophic cardiomyopathy
eFigure 6. Comparisons of genetic and clinical risk factors, using continuous values for risk factors where applicable
eFigure 7. Comparisons of genetic and clinical risk factors, excluding the first UK Biobank 50,000 exome-sequencing tranche
eFigure 8. Receiver-operator curve for the clinical vs. genetic and clinical factors for HCM
eReferences
References
- 1.Harper AR, Goel A, Grace C, et al. ; HCMR Investigators . Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet. 2021;53(2):135-142. doi: 10.1038/s41588-020-00764-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neubauer S, Kolm P, Ho CY, et al. ; HCMR Investigators . Distinct subgroups in hypertrophic cardiomyopathy in the NHLBI HCM Registry. J Am Coll Cardiol. 2019;74(19):2333-2345. doi: 10.1016/j.jacc.2019.08.1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomson KL, Ormondroyd E, Harper AR, et al. ; NIHR BioResource – Rare Diseases Consortium . Analysis of 51 proposed hypertrophic cardiomyopathy genes from genome sequencing data in sarcomere negative cases has negligible diagnostic yield. Genet Med. 2019;21(7):1576-1584. doi: 10.1038/s41436-018-0375-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alfares AA, Kelly MA, McDermott G, et al. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17(11):880-888. doi: 10.1038/gim.2014.205 [DOI] [PubMed] [Google Scholar]
- 5.Walsh R, Thomson KL, Ware JS, et al. ; Exome Aggregation Consortium . Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med. 2017;19(2):192-203. doi: 10.1038/gim.2016.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Marvao A, McGurk KA, Zheng SL, et al. Phenotypic expression and outcomes in individuals with rare genetic variants of hypertrophic cardiomyopathy. J Am Coll Cardiol. 2021;78(11):1097-1110. doi: 10.1016/j.jacc.2021.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ommen SR, Mital S, Burke MA, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2020;142(25):e558-e631. doi: 10.1161/CIR.0000000000000937 [DOI] [PubMed] [Google Scholar]
- 8.Invitae . Invitae hypertrophic cardiomyopathy panel. Accessed April 7, 2021. https://www.invitae.com/en/physician/tests/02261/
- 9.Blueprint Genetics . Genetic testing for hypertrophic cardiomyopathy. Accessed April 7, 2021. https://blueprintgenetics.com/tests/panels/cardiology/hypertrophic-cardiomyopathy-hcm-panel/
- 10.GeneDx . Hypertrophic cardiomyopathy (HCM) panel. Accessed April 7, 2021. https://www.genedx.com/tests/detail/hypertrophic-cardiomyopathy-hcm-panel-863
- 11.Hershberger RE, Givertz MM, Ho CY, et al. ; ACMG Professional Practice and Guidelines Committee . Genetic evaluation of cardiomyopathy: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018;20(9):899-909. doi: 10.1038/s41436-018-0039-z [DOI] [PubMed] [Google Scholar]
- 12.Mazzarotto F, Olivotto I, Boschi B, et al. Contemporary insights into the genetics of hypertrophic cardiomyopathy: toward a new era in clinical testing? J Am Heart Assoc. 2020;9(8):e015473. doi: 10.1161/JAHA.119.015473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tadros R, Francis C, Xu X, et al. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet. 2021;53(2):128-134. doi: 10.1038/s41588-020-00762-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203-209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szustakowski JD, Balasubramanian S, Kvikstad E, et al. ; UKB-ESC Research Team . Advancing human genetics research and drug discovery through exome sequencing of the UK Biobank. Nat Genet. 2021;53(7):942-948. doi: 10.1038/s41588-021-00885-0 [DOI] [PubMed] [Google Scholar]
- 16.Jurgens SJ, Choi SH, Morrill VN, et al. Analysis of rare genetic variation underlying cardiometabolic diseases and traits among 200,000 individuals in the UK Biobank. Nat Genet. 2022;54(3):240-250. doi: 10.1038/s41588-021-01011-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karlson EW, Boutin NT, Hoffnagle AG, Allen NL. Building the Partners HealthCare Biobank at Partners Personalized Medicine: informed consent, return of research results, recruitment lessons and operational considerations. J Pers Med. 2016;6(1):E2. doi: 10.3390/jpm6010002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morita H, Rehm HL, Menesses A, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med. 2008;358(18):1899-1908. doi: 10.1056/NEJMoa075463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinze G. A comparative investigation of methods for logistic regression with separated or nearly separated data. Stat Med. 2006;25(24):4216-4226. doi: 10.1002/sim.2687 [DOI] [PubMed] [Google Scholar]
- 20.Hindy G, Aragam KG, Ng K, et al. ; Regeneron Genetics Center . Genome-wide polygenic score, clinical risk factors, and long-term trajectories of coronary artery disease. Arterioscler Thromb Vasc Biol. 2020;40(11):2738-2746. doi: 10.1161/ATVBAHA.120.314856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Choi SH, Jurgens SJ, Weng LC, et al. Monogenic and polygenic contributions to atrial fibrillation risk: results from a national biobank. Circ Res. 2020;126(2):200-209. doi: 10.1161/CIRCRESAHA.119.315686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nishikawa T, Ishiyama S, Shimojo T, Takeda K, Kasajima T, Momma K. Hypertrophic cardiomyopathy in Noonan syndrome. Acta Paediatr Jpn. 1996;38(1):91-98. doi: 10.1111/j.1442-200X.1996.tb03445.x [DOI] [PubMed] [Google Scholar]
- 23.Lopes LR, Garcia-Hernández S, Lorenzini M, et al. Alpha-protein kinase 3 (ALPK3)-truncating variants are a cause of autosomal dominant hypertrophic cardiomyopathy. Eur Heart J. 2021;42(32):3063-3073. doi: 10.1093/eurheartj/ehab424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheawsamoot C, Phokaew C, Chetruengchai W, et al. A pathogenic variant in ALPK3 is associated with an autosomal dominant adult-onset hypertrophic cardiomyopathy. Circ Genom Precis Med. 2020;13(6):e003127. doi: 10.1161/CIRCGEN.120.003127 [DOI] [PubMed] [Google Scholar]
- 25.Watkins H. Time to think differently about sarcomere-negative hypertrophic cardiomyopathy. Circulation. 2021;143(25):2415-2417. doi: 10.1161/CIRCULATIONAHA.121.053527 [DOI] [PubMed] [Google Scholar]
- 26.Olivotto I, Oreziak A, Barriales-Villa R, et al. ; EXPLORER-HCM study investigators . Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2020;396(10253):759-769. doi: 10.1016/S0140-6736(20)31792-X [DOI] [PubMed] [Google Scholar]
- 27.Ho CY, Day SM, Axelsson A, et al. ; VANISH Investigators . Valsartan in early-stage hypertrophic cardiomyopathy: a randomized phase 2 trial. Nat Med. 2021;27(10):1818-1824. doi: 10.1038/s41591-021-01505-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magnusson P, Palm A, Branden E, Mörner S. Misclassification of hypertrophic cardiomyopathy: validation of diagnostic codes. Clin Epidemiol. 2017;9:403-410. doi: 10.2147/CLEP.S139300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Batty GD, Gale CR, Kivimäki M, Deary IJ, Bell S. Comparison of risk factor associations in UK Biobank against representative, general population based studies with conventional response rates: prospective cohort study and individual participant meta-analysis. BMJ. 2020;368:m131. doi: 10.1136/bmj.m131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rowin EJ, Hausvater A, Link MS, et al. Clinical profile and consequences of atrial fibrillation in hypertrophic cardiomyopathy. Circulation. 2017;136(25):2420-2436. doi: 10.1161/CIRCULATIONAHA.117.029267 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods
eTable 1. Disease definitions in UK Biobank
eTable 2. Gene categorizations for hypertrophic cardiomyopathy
eTable 3. Prioritized rare variants for hypertrophic cardiomyopathy
eTable 4. Gene-based associations of rare variants with hypertrophic cardiomyopathy in UK Biobank
eTable 5. Associations of the polygenic score with hypertrophic cardiomyopathy in carriers and noncarriers of an HCM-ACMG rare variant
eTable 6. Survey of clinical risk factors for HCM
eTable 7. Net reclassification improvement of clinical and genetic factors over clinical factors
eFigure 1. Receiver-operator curve for HCM PRS
eFigure 2. Combined odds of polygenic risk score and carrier status
eFigure 3. Discriminative benefit of individual risk factors for HCM in the UK Biobank
eFigure 4. Comparison of genetic and nongenetic risk factors for hypertrophic cardiomyopathy
eFigure 5. Cumulative predictive capabilities of genetic and nongenetic risk factors for hypertrophic cardiomyopathy
eFigure 6. Comparisons of genetic and clinical risk factors, using continuous values for risk factors where applicable
eFigure 7. Comparisons of genetic and clinical risk factors, excluding the first UK Biobank 50,000 exome-sequencing tranche
eFigure 8. Receiver-operator curve for the clinical vs. genetic and clinical factors for HCM
eReferences