

Summary
While polygenic risk scores (PRSs) enable early identification of genetic risk for chronic obstructive pulmonary disease (COPD), predictive performance is limited when the discovery and target populations are not well matched. Hypothesizing that the biological mechanisms of disease are shared across ancestry groups, we introduce a PrediXcan-derived polygenic transcriptome risk score (PTRS) to improve cross-ethnic portability of risk prediction. We constructed the PTRS using summary statistics from application of PrediXcan on large-scale GWASs of lung function (forced expiratory volume in 1 s [FEV1] and its ratio to forced vital capacity [FEV1/FVC]) in the UK Biobank. We examined prediction performance and cross-ethnic portability of PTRS through smoking-stratified analyses both on 29,381 multi-ethnic participants from TOPMed population/family-based cohorts and on 11,771 multi-ethnic participants from TOPMed COPD-enriched studies. Analyses were carried out for two dichotomous COPD traits (moderate-to-severe and severe COPD) and two quantitative lung function traits (FEV1 and FEV1/FVC). While the proposed PTRS showed weaker associations with disease than PRS for European ancestry, the PTRS showed stronger association with COPD than PRS for African Americans (e.g., odds ratio [OR] = 1.24 [95% confidence interval [CI]: 1.08–1.43] for PTRS versus 1.10 [0.96–1.26] for PRS among heavy smokers with ≥ 40 pack-years of smoking) for moderate-to-severe COPD. Cross-ethnic portability of the PTRS was significantly higher than the PRS (paired t test p < 2.2 × 10−16 with portability gains ranging from 5% to 28%) for both dichotomous COPD traits and across all smoking strata. Our study demonstrates the value of PTRS for improved cross-ethnic portability compared to PRS in predicting COPD risk.
Keywords: polygenic transcriptome risk score, risk prediction, cross-ethnic portability, integrative analysis, gene expression, pulmonary disease
Graphical abstract
Introduction
Chronic obstructive pulmonary disease (COPD), characterized by irreversible airflow obstruction, is currently a leading cause of death in the United States1,2 and worldwide.3 COPD is diagnosed using two spirometric measures of lung function, namely forced expiratory volume in one second (FEV1) and its ratio to forced vital capacity (FEV1/FVC). While the main risk factor for COPD is cigarette smoking, non-smokers can also develop COPD,4,5 which suggests genetic variation in susceptibility to the disease. Furthermore, COPD is a highly heterogeneous disease with heritability estimates ranging from 35% to 60% even after accounting for smoking behavior.6, 7, 8 Currently, there is no convincing therapy that prevents the development and progression of COPD, which reflects limited understanding of its biological mechanisms. Thus, early diagnosis can provide a crucial path toward prevention of more severe disease.
Large-scale genome-wide association studies (GWASs) of COPD and lung function have identified numerous genetic variants associated with COPD risk.9, 10, 11, 12, 13 However, the individual contribution of the identified disease-associated variants to complex disease is generally very small.14,15 The polygenic risk score (PRS) framework, aggregating the cumulative effects of genetic variants, tends to capture a reasonable proportion of variation in COPD risk and exhibits generally stronger association with disease when more genetic variants are included in the risk score.9,11,16, 17, 18
Additionally, PRS has the benefit that it can translate the results of GWASs into clinical application for the early identification of genetic risk of complex diseases. With recent increases in the scale of GWASs, the PRS approach has become more powerful. Many studies have demonstrated the predictive power of PRS on a wide range of complex diseases or traits.19, 20, 21, 22, 23 However, populations with varying genetic ancestry may possess different allelic frequencies and linkage structures, and as a result, the predictive power of PRS is limited when the discovery and target populations are from different genetic ancestry groups, which is referred to as limited cross-ethnic portability. For example, a study of seventeen anthropometric and blood-panel quantitative traits in the UK Biobank has shown that prediction accuracy was far lower for non-European-ancestry populations when the PRS was derived from summary statistics for studies of individuals with European ancestry.24
For COPD, Shrine and colleagues derived a 279-variant weighted PRS from large-scale GWASs of lung function carried out in individuals with European ancestry from the UK Biobank.11 Their results show that the derived PRS performed significantly better for individuals with European ancestry than for individuals with African ancestry in the external validation cohorts. In addition, Moll and colleagues derived an expanded PRS for COPD using Shrine’s GWAS11 results and showed that the gap in odds ratio of COPD between individuals with European and non-European ancestry increased with the decile of PRS.18 As the majority of GWASs have been performed in individuals with European ancestry, disparity in prediction accuracy across non-European-ancestry individuals from GWAS-derived PRS is a major concern in consideration of potential clinical applications.24,25,26
While heterogeneity in genetic architectures limits cross-ethnic portability of PRS,27, 28, 29, 30 the results from several cross-ethnic GWAS replication studies provide evidence for some causal variants shared across populations.31, 32, 33, 34 Given that a substantial proportion of GWAS variants demonstrate gene regulation effects,35 constructing risk scores built on expression quantitative-trait locus (eQTL) variants presents a promising path toward incorporating biological information in genetic prediction. Motivated by the hypothesis that the underlying biological mechanisms of trait or disease are shared across ancestry groups, Liang and colleagues proposed to improve cross-ethnic risk prediction using a polygenic transcriptome risk score (PTRS) constructed using multi-SNP predictors of gene expression.36 The proposed PTRS builds on the widely used PrediXcan approach, an integrative method that leverages gene expression information to identify trait-associated genes from GWAS.37 Compared with more conventional PRS approaches, the PTRS uses the cumulative effect of genes to construct genetic predictors of traits. Their results, which focused on seventeen anthropomorphic and blood phenotypes, showed substantial benefits in cross-ethnic portability from PTRS prediction compared with standard PRS approaches.36. Another benefit of the PTRS as a gene-based risk score is that it provides an additional layer of biological interpretability, as the PrediXcan-based predictors can be tied directly to gene expression traits corresponding to specific genes.
In this paper, we explored the benefits of PTRS for predicting COPD risk across self-reported race/ethnic groups by adapting the PTRS approach proposed by Liang et al.,36 with the main difference being that the current work leverages summary statistics from published GWASs rather than individual-level data. We constructed PTRSs for prediction of two quantitative lung function traits (FEV1 and FEV1/FVC ratio) and two definitions of COPD (moderate-to-severe COPD and severe COPD) using summary statistics from the recent large-scale GWAS of pulmonary function traits (FEV1 and FEV1/FVC ratio) conducted in individuals with European ancestry from the UK Biobank.11. We focused on these specific quantitative traits and definitions of COPD (1) for consistency with prior genetic studies11, 12, 13 and risk scores for COPD11,18 and (2) for their clinical relevance to diagnosis of COPD. We further proposed multiple approaches for the construction of PTRS, leveraging gene expression prediction functions from GTEx38 and the Multi-Ethnic Study of Atherosclerosis (MESA).39 To assess the prediction performance and the cross-ethnic portability of our proposed PTRS candidates, we leveraged multi-ethnic participants in the NHLBI Trans-Omics for Precision Medicine (TOPMed) program to select the best-performing candidates from population/family-based cohorts and then tested their performance on COPD-enriched studies.
Material and methods
Overview of approach
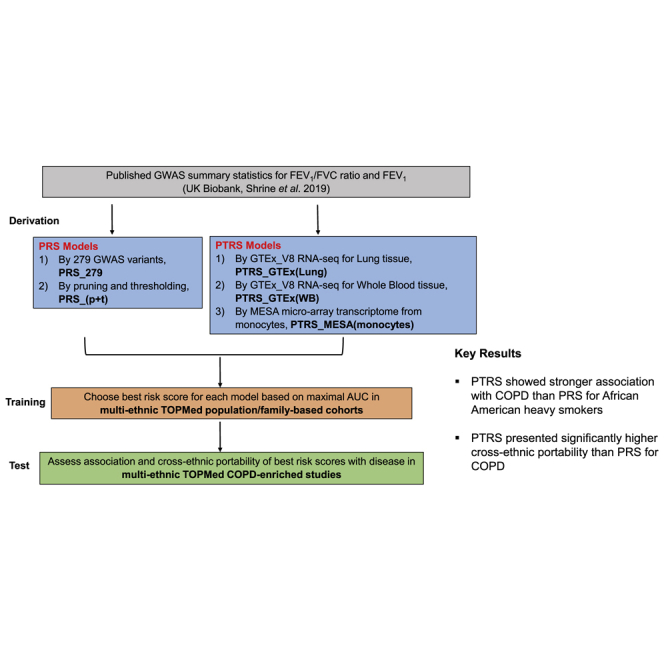
An overview of the study design is shown in Figure 1. For the derivation of risk scores (both PRS and PTRS), we leveraged summary statistics from the recent large-scale GWASs of pulmonary function traits (FEV1 and FEV1/FVC ratio) conducted in individuals with European ancestry from the UK Biobank.11 For the two COPD definitions (moderate-to-severe COPD and severe COPD), the risk-score candidates were constructed using GWAS results for FEV1/FVC ratio. We assessed these candidate scores on the TOPMed population/family-based cohorts (training data) to select the best-performing candidate by maximal area under the curve (AUC) from the set of risk scores derived for each model and trait. Both association and cross-ethnic portability (prediction accuracy for non-European ancestry relative to European ancestry) of the best risk scores with disease were then tested on TOPMed COPD-enriched studies.
Figure 1.

Study design
(A) Study workflow.
(B) Four methods of selecting genes included in each PTRS model.
PRS, polygenic risk score; PTRS, polygenic transcriptome risk score; 279 variants are the SNPs used to derive weighted genetic risk score for COPD in Shrine’s work; TWAS, transcriptome-wide association study; S-PrediXcan, Summary-PrediXcan.
PTRS derivation
Gene expression prediction models for integrative analysis
We selected two existing types of gene expression prediction models for investigation in construction of the PTRS:
-
•
PTRS_GTEx models: based on European ancestry-dominant GTEx_V8 RNA-seq data. We used MASHR-based40 prediction models that are recommended by the PrediXcan team for GTEx_V8 RNA-seq data.38 In the analysis, both lung (n = 444, PTRS_GTEx[Lung]) and whole blood (n = 573, PTRS_GTEx[WB]) tissues of MASHR-based GTEx models were applied.
-
•
PTRS_MESA model: based on multi-ethnic microarray transcriptome data collected from monocytes (total, n = 1,163; non-Hispanic Whites [NHW], n = 578; African Americans [AA], n = 233; and Hispanics/Latinos [HIS], n = 352).39 We used the multi-ethnic elastic net prediction model (trained with mixing parameter ) as presented in Mogil et al.39 for our analysis, and we refer this model as PTRS_MESA(monocytes).
Construction of risk scores
Building on the concept of genetically regulated gene expression (GReX) introduced as part of the widely used PrediXcan framework,37 we calculated the PTRS for the individual using the following formula:
where is the GReX of gene for individual , the calculation of is detailed in PrediXcan framework,37 is the estimated effect size of gene , the calculation of is detailed in Summary-PrediXcan (S-PrediXcan),41 and is the total number of genes. The PTRS approach used in the current manuscript was adapted from previous work,36 with the main difference being that the current work leverages summary statistics from published GWASs rather than individual-level data. We applied S-PrediXcan41 using the selected gene expression prediction models with the published lung function GWASs to obtain the estimated effects corresponding to each gene . Finally, the PTRS was inverse-normal transformed in the analysis.
We then applied four methods to select genes for inclusion in the PTRS:
-
(1)
The first method was to take genes within a ± 1-Mb region of 279 variants from previous GWASs11 and then overlap with genes from each of the three PTRS models (“279-1Mb”).
-
(2)
The second method is a variation of the first method (above). We restricted to 261 genes identified in previous GWAS (Table S9 in Shrine’s work11) harboring the 279 variants and then selected genes overlapping with each of the three PTRS models (“279-nearby”).
-
(3)
The third method was to apply different p value thresholds on transcriptome-wide association study (TWAS) results from S-PrediXcan. Nominal, qval, and Bonf represented the p value, q value, and Bonferroni corrected cut-offs at 0.05, respectively (“TWAS-nominal,” “TWAS-qval,” and “TWAS-Bonf”).
-
(4)
The last method was to select genes by regional colocalization probability (RCP). We applied FastEnloc42 on eQTLs and GWASs11 to compute RCP for genes. For eQTLs, we adopted GTEx_V8 eQTL43 for PTRS_GTEx models and MESA eQTLs39 for PTRS_MESA models. The genes were selected first by the range of RCP (from 0.1 to 0.9) and then overlapped with genes from each of the three PTRS models (“Coloc0.1” to “Coloc0.9”).
A summary of the number of genes selected by each method is reported in Table S1.
PRS calculation
To provide a comparison with our proposed PTRS, we incorporated in our study two models that reflect a more standard PRS framework. We further applied inverse normal transformation to the PRS values to standardize the scores.
-
•
PRS_279 model: denotes the previously published genetic risk score that leverages weights for 279 selected variants from previous GWASs11 (“SNPs-279”).
-
•
PRS_(p+t) model: applied pruning and thresholding by PLINK 1.90b --clump and --score,44 a p value and linkage disequilibrium (LD)-driven procedure, to build additional PRS candidates where 1,864 European ancestry samples from MESA who had-whole genome sequence data through TOPMed were used to construct a LD reference panel. For each trait, a range of p values ( and ) and pairwise correlation (0.2, 0.4, 0.6, and 0.8) thresholds were used to create an additional eight PRS candidates (“5e-4_0.2” to “5e-8_0.8”).
Study samples
The training data comprised the participants from eight population/family-based cohorts (the Atherosclerosis Risk in Communities [ARIC] study, the Coronary Artery Risk Development in Young Adults [CARDIA] study, the Cleveland Family Study [CFS], the Cardiovascular Health Study [CHS], the Framingham Heart Study [FHS], the Hispanic Community Health Study/Study of Latinos [HCHS/SOL], the Jackson Heart Study [JHS], and the Multi-Ethnic Study of Atherosclerosis [MESA]). The test data consisted of the participants from two COPD-enriched studies (the Genetic Epidemiology of COPD [COPDGene] study and the Sub-Populations and Intermediate Outcome Measures in COPD Study [SPIROMICS]). For all of the included studies, Institutional Review Boards at each field center approved study protocols, and written informed consent was obtained from all participants. Detailed cohort descriptions are provided in the supplemental methods.
Whole-genome sequence data
Whole-genome sequencing (WGS) in TOPMed had, on average, deep (∼30×) coverage with joint-sample variant calling and variant level quality control in ∼140,000 TOPMed samples for Freeze 8 and ∼159,000 samples for Freeze 9b.45 Analyses in the population/family-based cohorts, as well as COPDGene, used WGS from TOPMed Freeze 8. Study-specific analyses in SPIROMICS used the newer Freeze 9b WGS genotypes as (1) these analyses were carried out at a later stage in our research, and (2) the SPIROMICS WGS data were only available starting from the newer Freeze 9b release. Additional details regarding quality control of genotype data for the present analyses are included in the supplemental methods.
Phenotype definition
The phenotype harmonization of pulmonary function traits (pre-bronchodilator FEV1 and FEV1/FVC ratio) was conducted following the protocol of the NHLBI Pooled Cohorts Study (supplemental methods, Oelsner et al.17). We followed Zhao et al.13 to proceed with phenotype QC and calculate the race/ethnic-specific predicted values of FEV1 using the equations of Hankinson46 that were determined for NHW, AA, and HIS, respectively, COPD cases, and controls were then defined as follows:
-
•
Moderate-to-severe COPD: pre-bronchodilator FEV1 < 80% predicted and FEV1/FVC < 0.7
-
•
Severe COPD: pre-bronchodilator FEV1 < 50% predicted and FEV1/FVC < 0.7
-
•
Controls: pre-bronchodilator FEV1 ≥ 80% predicted and FEV1/FVC ≥ 0.7
Statistical analysis to examine prediction performance
We carried out pooled analyses across self-reported race/ethnic groups for the training data (population/family-based cohorts). For the test data (COPD-enriched studies), analyses were stratified by self-reported race/ethnic group (NHW versus AA) and then meta-analyzed using an inverse-variance weighted fixed effect model. Statistical analyses were conducted using R/GENESIS v.2.21.3,47 and meta-analyses were implemented in R/meta v4.13-0.48
For dichotomous traits, the score with the best prediction accuracy for each set of risk scores corresponding to each model was determined by the maximal AUC. The AUC was calculated using a generalized linear mixed model for association of the dichotomous trait with thescore candidate and including additional covariate adjustment for age, sex, race, study, sequence center, pack-years of smoking, ever versus never smoking, and principal components (PCs) of ancestry. The genetic relationship matrix (GRM) was used to specify the covariance structures of the random effects term in the model. The AUC was calculated by risk score only, and the confidence intervals of AUC were calculated using R/pROC v.1.17.0.1.49
For quantitative traits, we followed Zhao et al.13 and Sofer et al.50 to obtain study-specific variance adjusted residuals as phenotypes for the analyses. More specifically, we applied linear mixed models to obtain study-specific residuals, along with study-specific standard deviations of the residuals. The inverse-normal transformed residuals were scaled by their study-specific standard deviations. The resulting values were used to assess the association with each proposed risk score using a linear mixed model. The linear mixed models included covariate adjustment for age, age-squared, sex, height, height-squared, race, study, sequence center, pack-years of smoking, current smoking, former smoking, PCs of ancestry, and the GRM. Prediction performance of each score was quantified as the proportion of variance explained (%), estimated as 100 × the squared correlation (R2) between the observed phenotypes and the predicted phenotypes by score only.
The GRM of samples and PCs of ancestry for all studies except SPIROMICS were generated on TOPMed Freeze 8 and obtained directly from the TOPMed Data Coordinating Center. For SPIROMICS, the GRM and PCs were based on TOPMed Freeze 9b and were computed by following TOPMed Freeze 8 procedures using R/GENESIS v.2.21.3.47 Analyses in TOPMed Freeze 8 (population/family-based and COPDGene) included adjustment for the first 11 PCs of ancestry, whereas analyses in SPIROMICS included adjustment for the first 4 PCs of ancestry after checking pairwise PC plots.
Smoking interaction
For the best-performing risk-score candidate identified for each model, the smoking × score interaction effects were assessed by adding an interaction term for pack-years of smoking × score in the (generalized) linear mixed models for each of the four traits (moderate-to-severe COPD, severe COPD, FEV1, and FEV1/FVC ratio) on population/family-based cohorts.
Portability analysis
Cross-ethnic portability was defined as the prediction accuracy (AUC) ratio for non-European- versus European-ancestry populations. We applied bootstrapped sampling (i.e., random sampling with replacement) on two COPD-enriched studies to generate 95% confidence intervals of cross-ethnic portability. For each bootstrapped sample of COPD cases and controls, we calculated the cross-ethnic portability. The 95% CIs for portability estimates were then obtained using the percentile method on 10,000 bootstrapped samples, separately for each of the two COPD-enriched studies.
Examination of a combined risk score
To explore the performance of a single risk score that combines PRS and PTRS, we selected one candidate to represent each approach (PRS_279: SNPs-279 and PTRS_GTEx(Lung): 279-nearby) for further investigation. These two risk scores are relevant but provide different levels of genetic risk information. We first examined the interaction between these two scores in the training data (population/family-based cohorts). The interaction was assessed by adding a score interaction term in the same prediction model as for risk-score prediction evaluation. We then explored two schemes to combine two individual risk scores, unweighted sum and weighted sum. The unweighted sum is obtained as the direct sum of the two individual risk scores. The weights in the weighted sum were obtained as the regression coefficients of two individual risk scores in the score interaction model using the population/family-based cohorts. The weights were also applied to COPD-enriched studies to calculate the combined risk score. Finally, we evaluated the predictive performance of the risk scores for each COPD trait (1) using the same prediction model used for our primary analyses as described above, (2) using a baseline set of clinical risk factors alone (age, sex, race, pack-years of smoking), (3) using the risk score alone, and (4) using the combination of clinical risk factors and risk score.
Results
Participant characteristics
Demographic and clinical characteristics of our study samples are summarized in Table 1, which includes 29,381 participants from the eight population/family-based cohorts and 11,771 participants from the COPD-enriched studies. Based on participant self-reported race/ethnicity, 50% and 74% of the participants were categorized as NHW in the population/family-based cohorts and COPD-enriched studies, respectively. The remaining participants represented AA (24% and 26% in the population/family-based and COPD-enriched studies, respectively) and HIS (26% in the population/family-based cohorts).
Table 1.
Characteristics of the study-participants included in the analyses
| Stratum | Study | NHW | AA | HIS | Age, years | Female (%) | Smoking pack-years | FEV1%predicted | FEV1/FVC ratio | Moderate-to-severe COPD | Severe COPD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Population-and family-based | ARIC | 5,717 | 1,458 | – | 62.81 ± 9.69 | 3,998 (56) | 17.28 ± 23.41 | 92 ± 19 | 0.73 ± 0.09 | 1,115 | 199 |
| CARDIA | 1,386 | 1,373 | – | 42.17 ± 6.62 | 1,525 (55) | 0.71 ± 1.58 | 95 ± 14 | 0.79 ± 0.06 | – | – | |
| CFS | 402 | 388 | – | 47.21 ± 16.07 | 432 (55) | 10.29 ± 16.26 | 91 ± 20 | 0.79 ± 0.07 | 64 | 16 | |
| CHS | 2,321 | 312 | – | 78.74 ± 6.09 | 1,574 (60) | 16.08 ± 24.85 | 91 ± 25 | 0.72 ± 0.11 | 500 | 155 | |
| FHS | 3,321 | – | – | 48.86 ± 11.85 | 1,771 (53) | 6.72 ± 15.76 | 96 ± 15 | 0.76 ± 0.07 | 239 | 26 | |
| HCHS/SOL | – | – | 6,750 | 46.65 ± 13.64 | 3,969 (59) | 7.43 ± 15.97 | 92 ± 15 | 0.80 ± 0.07 | 394 | 69 | |
| JHS | – | 2,511 | – | 54.54 ± 12.54 | 1,621 (65) | – | 93 ± 18 | 0.81 ± 0.08 | 123 | 26 | |
| MESA | 1,580 | 983 | 879 | 66.40 ± 9.84 | 2,088 (52) | 10.69 ± 20.90 | 94 ± 18 | 0.75 ± 0.08 | 408 | 52 | |
| Total | 14,727 | 7,025 | 7,629 | – | – | – | – | – | 2,843 | 543 | |
| COPD-enriched | COPDGene | 6,609 | 3,258 | – | 59.55 ± 9.04 | 4,602 (47) | 44.27 ± 24.87 | 73 ± 26 | 0.65 ± 0.16 | 3,981 | 2,022 |
| SPIROMICS | 1,535 | 369 | – | 63.50 ± 9.06 | 886 (47) | 47.60 ± 27.91 | 67 ± 27 | 0.59 ± 0.16 | 1,115 | 547 | |
| Total | 8,144 | 3,627 | – | – | – | – | – | – | 5,096 | 2,569 |
Mean ± standard deviation. ARIC, Atherosclerosis Risk in Communities; CARDIA, Coronary Artery Risk Development in Young Adults; CFS, Cleveland Family Study; CHS, Cardiovascular Health Study; FHS, Framingham Heart Study; HCHS/SOL, Hispanic Community Health Study/Study of Latinos; JSH, Jackson Heart Study; MESA, Multi-Ethnic Study of Atherosclerosis; COPDGene, Genetic Epidemiology of COPD; SPIROMICS, Sub-Populations and Intermediate Outcome Measures in COPD Study; NHW, non-Hispanic White; AA, African American; HIS, Hispanic; FEV1, forced expiratory volume in 1 s; FEV1/FVC ratio, FEV1 ratio to forced vital capacity.
Selection of best-performing risk-score candidate for each model
The overview of study design is shown in Figure 1. We used large-scale GWASs of individuals with European ancestry from the UK Biobank reported by Shrine et al.11 (n = 321,047) to derive both PRS and PTRS candidates for FEV1/FVC ratio and FEV1, respectively. For the two COPD definitions (moderate-to-severe COPD and severe COPD), the risk-score candidates were constructed using GWAS results for FEV1/FVC ratio. Specifically, we derived 42 candidates for PTRS by three different transcriptome reference models (i.e., PTRS_GTEx[Lung], PTRS_GTEx[WB], and PTRS_MESA[monocytes]) and by four different gene selection methods for each transcriptome reference model (i.e., “279-1Mb,” ”279-nearby,” “TWAS-nominal, qval, and Bonf,” and “Coloc0.1 to Coloc0.9”), and 9 candidates for PRS by two models (i.e., PRS_279 and PRS_[p+t]) for each of the complex traits (material and methods). We assessed these candidate scores on the TOPMed population/family-based cohorts (training data) to select the best-performing risk score by maximum AUC for each model and for each trait. Both association strength and cross-ethnic portability of the best risk scores with diseases were then tested in the TOPMed COPD-enriched studies (Figure 1).
Figure 2 shows that the two definitions of COPD (moderate-to-severe COPD and severe COPD) shared the same pattern of the best-performing risk-score candidates for each model. Taking moderate-to-severe COPD for example, PRS_279 had the best prediction accuracy overall (AUC = 0.579 [95% CI: 0.567–0.590]). The best-performing candidate for the PRS pruning and thresholding model was from the accumulation of independently genome-wide significant variants (i.e., PRS_[p+t]: 5e-8_0.2 with AUC = 0.566 [95% CI: 0.555–0.578]). Among our proposed PTRSs, the PTRS with genes near 279 variants had the best AUC for PTRS_GTEx(Lung) model (i.e., PTRS_GTEx[Lung]: 279-nearby with AUC = 0.549 [95% CI: 0.537–0.560]). Among the PTRS_GTEx(WB) model risk scores, the candidate with genes selected using the q value method was the best performing (i.e., PTRS_GTEx[WB]: TWAS-qval with AUC = 0.525 [95% CI: 0.513–0.536]). The PTRS with the second-largest gene size was the best performing for the MESA model (i.e., PTRS_MESA[monocytes]: TWAS-nominal with AUC = 0.537 [95% CI: 0.525–0.548]). The best PRS candidate (PRS_279) has significantly higher AUC than the best PTRS candidate (PTRS_GTEx[Lung]: 279-nearby) for moderate-to-severe COPD (Delong p value = 7.46 × 10−6). The detailed prediction performance of all proposed risk scores for the two COPD traits are shown in Tables S2 and S3.
Figure 2.

Prediction accuracy of all risk-score candidates on multi-ethnic population/family-based cohorts for two COPD traits
AUC, area under the curve, was evaluated by the risk score only. Data are shown as AUC with 95% CI. PRS_279, PRS derived by previously published 279 variants for FEV1/FVC ratio; PRS_(p+t), PRS derived by pruning and thresholding (a range of p value and pairwise correlation thresholds were used to create eight candidates, 5e-4_0.2 to 5e-8_0.8); 279-nearby and 279-1Mb, PTRS derived by genes nearby and within ± 1 Mb region of 279 variants, respectively; TWAS-nominal, TWAS-qval, and TWAS-Bonf, PTRS derived by genes passing TWAS p value threshold of 0.05, q value, and Bonferroni, respectively; Coloc0.1 to Coloc0.9, PTRS derived by genes with regional colocalization probability ranging from 0.1 to 0.9.
The genes included in the best-performing PTRS for two COPD traits may provide additional information to prioritize genes for further investigation of the underlying biological mechanism of COPD (Tables S4–S6). Taking the best-performing PTRS of the GTEx(Lung) model (i.e., PTRS_GTEx[Lung]: 279-nearby) for example, there are 126 genes included in this PTRS that represent a subset of the 261 genes identified based on GWASs of lung function in the UK Biobank.11 Among these, 43 of the genes included in the risk score achieved Bonferroni significance (i.e., TWAS p value < 0.05/126, Table S4). Furthermore, the small number of overlapped genes among the three best-performing PTRS candidates suggested that the best candidate from each PTRS model provided information on a relatively distinct set of genes (Table S1 and Figures S2–S4).
For the two quantitative lung function traits, PRS_279 has overall better prediction accuracy than the other risk scores examined for both traits, and the best-performing PTRS candidate differed by trait (Figure S1). Overall, the risk scores from the model PTRS_GTEx(Lung) presented higher R2 among all PTRS candidates for both traits. The detailed prediction results for all proposed risk scores are shown in Tables S7 and S8, and the genes included for the best-performing score from each PTRS model for two lung function quantitative traits are shown in Tables S9–S13.
PTRS presents stronger association than PRS with COPD in African Americans
Defining subgroups for stratified analysis
As shown in Table 1, the participants in COPD-enriched studies had heavier smoking history than those in TOPMed population/family-based cohorts. Hence, to examine the association strength of best risk scores on COPD-enriched studies, we first examined the impact of pack-years of smoking on the relationship between the proposed risk scores and COPD risk via smoking interaction analysis (material and methods). For each of the four traits (two definitions of COPD and two quantitative traits: FEV1 and FEV1/FVC ratio), at least one selected candidate score showed nominally significant interaction with smoking (i.e., interaction p value < 0.05, Table S14). We then conducted smoking-stratified analyses on COPD-enriched studies to examine the association performance of best risk scores on different smoking strata.
Within smoking strata, we undertook separate analyses for NHW and AA. Due to the limited samples with pack-years of smoking < 20 in SPIROMICS (Table S15), we only applied analyses in COPDGene for the light smokers (i.e., pack-years of smoking < 20) for all four traits. For each of the five risk-score models (i.e., PRS_279, PRS_[p+t], PTRS_GTEx[Lung], PTRS_GTEx[WB], and PTRS_MESA[monocytes]), we selected the best risk scores based on their AUCs in each smoking stratum on population/family-based cohorts and then applied them in analysis of COPD-enriched studies (Tables S16–S19). The meta-analyzed odds ratios for the association of best risk scores with COPD traits on COPD-enriched studies are shown in Figure 3A. Overall, PRS_279 still showed stronger association with both COPD traits in NHW participants from COPD-enriched studies and for each smoking strata (e.g., odds ratio [OR] = 1.57 [95% CI: 1.48–1.67] for moderate-to-severe COPD and OR = 1.66 [95% CI: 1.55–1.79] for severe COPD for smoking pack years ≥ 0). For light smokers (i.e., pack-years of smoking < 20) in AA, PTRS showed either a similar or stronger association than PRS with two COPD traits (OR = 1.33 [95% CI: 1.06–1.68] from PTRS_GTEx[WB] versus 1.33 [95% CI: 1.06–1.68] from PRS_279 for moderate-to-severe COPD, and OR = 1.51 [95% CI: 1.04–2.19] from PTRS_GTEx[WB] versus 1.31 [95% CI: 0.87–1.96] from PRS_279 for severe COPD). For heavy smokers (i.e., pack-years of smoking ≥ 40) in AA, the PTRS presented a stronger association than the PRS for moderate-to-severe COPD (OR = 1.24 [95% CI: 1.08–1.43] from PTRS_GTEx[Lung] versus OR = 1.10 [95% CI: 0.96–1.26] from PRS_279). For the other two lung function traits, FEV1/FVC ratio and FEV1, PRS_279 outperformed in both NHW and AA (Figures S5 and S6).
Figure 3.

Association and cross-ethnic portability of best risk scores with two COPD traits in COPD-enriched studies
The risk-score candidates used in the analyses were based on the best AUC of each smoking stratum on multi-ethnic population/family-based cohorts for both PRS and PTRS.
(A) Association of the best risk scores with two COPD traits. NHW, non-Hispanic Whites; AA, African Americans. Data are shown as meta-analyzed odds ratio with 95% CI.
(B) Cross-ethnic portability of the best risk scores; portability was calculated as the ratio of AUC of AA over NHW. Data are shown as raw portability, and error bars are 95% CIs from 10,000 bootstrapped samples.
PTRS improves cross-ethnic portability of prediction
The noticeably decreased performance of PRS from NHW to AA is shown in Figure 3A. For example, considering the performance of all participants (i.e., pack-years of smoking ≥ 0) for moderate-to-severe COPD, the OR was 1.57 [95% CI: 1.48–1.67] by PRS_279 for NHW, but it dropped to 1.24 [95% CI: 1.14–1.36] for AA (Table S16). To test the cross-ethnic portability of prediction for both PRS and PTRS, we generated 10,000 bootstrapped samples for two COPD-enriched studies (material and methods). By definition of cross-ethnic portability, the reference portability is 1. As shown in Figure 3B, the PTRS models retained overall better portability than that from PRS models for both definitions of COPD. More specifically, we compared the performance of the best portability between PTRS and PRS. For example, in pooled analysis across all smoking strata (i.e., pack-years of smoking ≥ 0) for moderate-to-severe COPD, the PTRS with the best portability was PTRS_GTEx(Lung) model (portability = 1 [95% CI: 0.95–1.05] for COPDGene and portability = 1.05 [95% CI: 0.89–1.19] for SPIROMICS), whereas the PRS with the best portability was the PRS_(p+t) model (portability = 0.95 [95% CI: 0.91–1] for COPDGene and portability = 1 [95% CI: 0.87–1.13] for SPIROMICS). Hence, the gain of portability from PTRS was 5% from both cohorts in this smoking strata for moderate-to-severe COPD, and based on a paired t test comparing the bootstrapped distributions, the PTRS_GTEx(Lung) model has significantly higher portability than that from the PRS_(p+t) model for both cohorts (p < ). We also observed the significantly improved portability from PTRS (gain ranges from 5% to 28%, Tables S16 and S17) for the other three smoking strata for both definitions of COPD.
Comparison of genetic versus smoking-related risk of disease
To aid with practical interpretation, we examined the impact of the combination of genetic risk scores and smoking history on the risk of COPD. These exploratory analyses were conducted on population/family-based cohorts only, as these studies allowed us to examine the population-level risk of disease. Participants were divided into risk categories by the values of both risk scores (quintiles) and pack-years of smoking (< 20 and ≥ 40). The reference group was defined as the participants with low genetic risk (i.e., 1st quintile of genetic risk score) and light smoking (i.e., pack-years of smoking < 20). Comparing the individuals with high genetic risk (i.e., 5th quintile of genetic risk score) and heavy smoking history (i.e., pack-years of smoking ≥ 40) to those in the reference group, the OR by the PRS_279 model was 11.26 (95% CI: 5.94–21.35) for moderate-to-severe COPD and 21.21 (95% CI: 7.93–56.74) for severe COPD (Figure 4A). The impact of smoking history was observed to be greater than genetic risk, as quantified by either the PRS or the PTRS. Taking the results from PRS_279 for example, if an individual has low genetic risk but heavy smoking, then the OR for moderate-to-severe COPD was 4.45 (95% CI: 2.50–7.93). However, for an individual in the light smoking group, even with high genetic risk, the OR was comparably lower at 2.58 (95% CI: 2.14–3.11) for moderate-to-severe COPD (Figure 4A). The same pattern was also observed for severe COPD.
Figure 4.

Risk for two COPD traits by different risk groups and by the combined risk scores
(A) Odds ratios of two COPD traits for different risk groups on multi-ethnic population/family-based cohorts. The reference group was defined as low risk and light smoker; low and high risk are referring to the 1st and the 5th quintile of genetic risk score, respectively; light and heavy smokers are referring to the participants with pack-years of smoking < 20 and ≥ 40, respectively; the risk-score candidates used in the analyses were based on the prediction performance of non-smoking stratum on population/family-based cohorts. Data are shown as odds ratio with 95% CI.
(B) Association of the combined risk scores with two COPD traits. The risk-score candidates, PTRS_GTEx(Lung): 279-nearby and PRS_279: SNPs-279 were used for PTRS and PRS, respectively, in the analyses. Unweighted sum and weighted sum refer to the direct summation and the weighted summation of PTRS and PRS, respectively. Data are shown as odds ratio with 95% CI. For COPD-enriched studies, the odds ratios were meta-analyzed. NHW, non-Hispanic Whites; AA, African Americans; Population_MultiEthnic, multi-ethnic samples in population/family-based cohorts.
Combined PRS and PTRS improves association strength
To explore the performance of a single risk score that borrows information from both PRS and PTRS for COPD, we selected one risk score representing each approach, PRS_279: SNPs-279 and PTRS_GTEx(Lung): 279-nearby, for further examination. These two risk scores are correlated (Pearson’s correlation = 0.27, p < ) but provide different levels of genetic risk information. We first examined the interaction between the two scores in the population/family-based cohorts (material and methods). The significant main effects (i.e., PRS and PTRS effects) and non-significant interactions indicated that two individual risk scores provided independent effects for all four traits (Table S20). Then we applied unweighted- and weighted-sum schemes to combine two individual risk scores into a single score. Figure 4B presents the association results of combined risk score with two COPD traits on both training and test data. In general, the weighted-sum score and PRS showed similar strength of association, and both presented stronger association than unweighted-sum score and PTRS on both NHW dominant training data (population/family-based cohorts) and NHW participants in test data (COPD-enriched studies). The similar performance between the weighted-sum score and the PRS for NHW can be explained by the major contribution from PRS to this combined score (i.e., the weights mainly come from PRS, Table S20). For AA, the unweighted-sum score that equally borrows information from both PRS and PTRS produced noticeable improvement. Taking the AA participants with pack-years of smoking between 20 and 40 for example, the OR was increased from 1.50 (95% CI: 1.24–1.80) by PRS to 1.66 (95% CI: 1.37–2.02) by the unweighted-sum score for severe COPD, which produced 10% increment for OR. The same pattern of improvement was also observed for the two lung traits FEV1/FVC and FEV1 (Figures S7 and S8). For the prediction accuracy evaluated by AUC, the weighted-sum score presented overall outperformance for both COPD traits among risk scores in population/family-based cohorts (Tables S21 and S22). Although the AUC achieved by a baseline model (i.e., AUC based on clinical risk factors) was higher than the AUC for the risk score alone, we observed a trend of lower baseline AUC and high risk-score AUC among heavy smokers compared to those with reduced smoking exposures. Taking moderate-to-severe COPD for example, the baseline AUC dropped from 0.742 (95% CI: 0.729–0.754) for light smokers (i.e., pack-years of smoking < 20) to 0.63 (95% CI: 0.604–0.657) for heavy smokers (i.e., pack-years of smoking ≥ 40), whereas the weighted-sum risk-score AUC increased from 0.592 (95% CI: 0.576–0.608) to 0.599 (95% CI: 0.572–0.625) (Table S21).
Discussion
In the current manuscript, we proposed and applied an integrative framework to quantify genetic risk of COPD and predict quantitative lung function traits. Our proposed polygenic transcriptomic risk score (PTRS) framework, built on the widely used PrediXcan approach used for systematic integration of GWASs with reference eQTL data,39,43 bears a more direct connection to underlying disease biology than standard PRS approaches. Hypothesizing that the underlying biology of complex disease traits is shared across diverse race/ethnic groups, we further anticipated that risk scores constructed under our PTRS framework would have greater portability than the standard PRS. Our application of PTRS to prediction of COPD in African American individuals from COPD-enriched studies demonstrated that our proposed PTRS had better portability for prediction of both moderate-to-severe COPD and severe COPD than the PRS approaches that we examined. Further, examining correlation of our PTRS with a standard PRS, we showed that the two classes of scores are not strongly correlated and thus present independent and complementary information that can be combined.
As the PTRS approaches are restricted primarily to eQTL variants, the number of possible predictors available for construction of these risk scores is relatively constrained in relation to more standard PRS approaches. Thus, we did not hypothesize that the PTRS would show overall stronger predictive performance than comparable PRS approaches. As expected, the PRS approaches showed performance advantages in prediction of COPD risk in individuals with European ancestry. In examining performance specifically among African Americans, we did note a stronger association with COPD for the PTRS compared to PRS, particularly in heavy smokers with 40 or more pack-years of smoking for moderate-to-severe COPD and in light smokers with pack-years of smoking less than 20 for severe COPD. Although our present work did not include a direct comparison to the more recently published COPD PRS18 that is a weighted sum of two individual PRSs for FEV1 and FEV1/FVC and includes more variants not reaching genome-wide significance by lassosum,51 we observed the same pattern as the Moll paper18 that the AUC based on clinical risk factors was higher than the risk-score AUC, but the combined AUC (including both clinical and genetic factors) improved upon each of the separate models. In addition, we observed that the AUC obtained by clinical risk factors alone decreased with increasing smoking history, whereas the AUC achieved by the risk score alone was higher in strata with greater smoking exposures. This result reflects the likely larger effects of the underlying SNPs in the presence of smoking and warrants further investigation. While the Moll PRS18 demonstrated improvements in predictive performance over the Shrine et al.11 risk score for both individuals with European ancestry and individuals with African ancestry, the performance gap between these two ancestry groups was increased (e.g., based on comparison of odds ratios for the respective risk scores observed for COPDGene NHW and AA from the Moll versus Shrine PRS). In our study, the improved association performance of the PTRS in African Americans is concordant with the portability advantages that we also observed for the PTRS. Combined, these results underscore the value of using PTRS approaches to leverage large-scale genomic resources of primarily European ancestry to construct risk scores that can be extended to non-European ancestry populations.
The specific reasons contributing to the particular value of PTRS in improving portability across ancestry groups are not entirely straightforward. While we hypothesized initially that the PTRS may show advantages due to its use of eQTL variants that tie it to biological mechanisms that may be shared across ancestry groups, part of the portability of the PTRS may also stem in part from the methods used to construct the gene expression prediction models. The GTEx prediction models38 incorporated statistical fine-mapped variants in selection of SNPs for gene expression prediction, which may have helped in enriching the resulting predictors for causal variants. While the MESA prediction models were built using the elastic net model without initial selection based on fine mapping, these MESA prediction models were constructed leveraging the diverse and multi-ethnic MESA participants,39 such that the variants ultimately included in the predictors were also enriched for eQTL variants exhibiting shared effects across ancestry groups.
Comparing performance of the PTRS across the different gene expression prediction models used to construct these risk scores, we did not observe a clear trend in terms of which model resulted in better predictive performance overall. Direct comparison of PTRS performance across different gene expression prediction models is not straightforward, in part because the properties of the underlying models from GTEx and MESA differ on multiple levels. Besides the differences in statistical approaches used to develop these prediction models noted earlier, other differences between the GTEx and MESA models include (1) source tissues represented, (2) race/ethnic composition of the underlying studies, with GTEx including roughly 15% non-European-ancestry individuals52 compared to 50% non-European-ancestry individuals in MESA,39 and (3) sample sizes of the underlying models in GTEx lung (n = 444) and whole blood (n = 573) versus MESA monocytes (n = 1,163).38,39 Further, the PTRS constructed using different gene expression prediction models differed markedly in the specific genes included in the final risk scores. While these differences may reflect distinct biology captured by each of the corresponding sources of tissue, we should keep in mind that the sample sizes used to construct the underlying gene expression prediction models were limited. In addition, the quality and disease relevance of the GTEx lung data for application to chronic lung diseases are limited.53 As resources used for eQTL mapping expand in sample size and improve in tissue quality in the future, we expect to gain additional resolution in examining the finer difference in performance among the various PTRSs derived from distinct gene expression prediction models.
In summary, we applied the PTRS framework toward genetic risk prediction of COPD and demonstrated its value in providing biologically interpretable disease risk prediction that is portable across ancestry groups and provides information complementary to the standard polygenic risk scores. A particular strength of our approach is that the PTRS can be constructed using summary statistics from large-scale GWASs and/or TWASs alone, allowing us to leverage the abundant genome-wide summary data from prior GWASs to construct these risk scores. Limitations of our study include our use, for construction of the PTRS, of existing gene expression prediction functions, which themselves rely on limited sample sizes and provide limited ability to compare performance of PTRS approaches across underlying tissue models for gene expression prediction. In future work, we will work to build gene expression prediction models in expanded RNA-seq resources from TOPMed and other sources, allowing us to leverage larger sample sizes for gene expression prediction, while also applying more consistent methods for gene expression prediction to allow more direct comparison across tissues. In addition, our proposed PTRS framework extends naturally to other molecular omics types, and we intend to extend our PTRS approach to leverage proteomics or additional omics as they become more widely available.
While we have demonstrated the value of integrative approaches leveraging eQTLs toward improvement of cross-ancestry portability of risk scores, we further emphasize that constructing more portable risk scores represents just one line of investigation toward achieving equity in risk prediction and personalized medicine. Ultimately, a crucial step toward improving performance of genomic risk prediction for non-European-ancestry groups will be to increase the diversity of participants included and analyzed in genetic studies. Our long-term hope is that our field can make sufficient progress on expanding diverse ancestry resources for genomics, for example from TOPMed, the Population Architecture using Genomics and Epidemiology (PAGE) Consortium,30 and All of Us Research Program,54 such that we can leverage these diverse ancestry resources more directly toward improved prediction in diverse ancestry populations. As there remains a long road ahead toward recruitment, phenotyping, and analysis of diverse ancestry samples for large-scale diverse ancestry genetic studies, we suggest that construction of more portable risk scores will contribute toward improvements in equity in the near future.
Acknowledgments
We dedicate this manuscript to our co-authors, long-time collaborators, and dear friends Dr. Debbie Ann Nickerson and Dr. L. Adrienne Cupples. Debbie was a pioneer in human genomics and advocate for diversity and inclusion in science and research. Adrienne was committed to excellence in statistical methodology and collaborative consortium-based genomic research, with major contributions to teaching and service in biostatistics. This research was supported by NIH/NHLBI R01 HL131565 (X.H. and A.M.); R01 HL153248 (A.M. and M.H.C.); R01 HL135142, R01 HL137927, R01 HL089856, and R01 HL147148 (M.H.C.); and K01-HL129039 (D.Q.). Study-specific acknowledgments are given in the supplemental information. We gratefully acknowledge the cohorts and participants who provided biological samples and data for TOPMed. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the U.S. Department of Health and Human Services. A full list of authors for the NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium is provided at https://www.nhlbiwgs.org/topmed-banner-authorship.
Declaration of interests
In the past three years, E.K.S. and M.H.C. have received institutional grant support from GlaxoSmithKline and Bayer. M.H.C. has received consulting and speaking fees from Illumina and AstraZeneca. B.M.P. serves on the Steering Committee of the Yale Open Data Access project funded by Johnson & Johnson. T.L. is an advisor for Variant Bio, Goldfinch Bio, and GSK. T.L. also has stock in Variant Bio. All other authors have declared no competing interests.
Published: April 5, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.03.007.
Data and code availability
Individual whole-genome sequence data for TOPMed whole genomes are available through dbGaP. The dbGaP accession numbers are: Atherosclerosis Risk in Communities (ARIC) phs001211, Coronary Artery Risk Development in Young Adults (CARDIA) phs001612, Cardiovascular Health Study (CHS) phs001368, Cleveland Family Study (CFS) phs000954, Framingham Heart Study (FHS) phs000974, Hispanic Community Health Study/Study of Latinos (HCHS/SOL) phs001395, Jackson Heart Study (JHS) phs000964, Multi-Ethnic Study of Atherosclerosis (MESA) phs001416, Genetic Epidemiology of COPD (COPDGene) phs000951, and SubPopulations and InteRmediate Outcome Measures in COPD Study (SPIROMICS) phs001927. Data in dbGaP can be downloaded by controlled access with an approved application submitted through dbGaP website. All PrediXcan code used is available in the GitHub repository.
Web resources
PredictDB, https://predictdb.org/
PrediXcan GitHub repository, https://github.com/hakyimlab/PrediXcan
Supplemental information
References
- 1.Heron M. Deaths: Leading Causes for 2016. Natl. Vital Stat. Rep. 2018;67:1–77. [PubMed] [Google Scholar]
- 2.Murphy S.L., Xu J., Kochanek K.D., Arias E. Mortality in the United States, 2017. NCHS Data Brief. 2018;(328):1–8. [PubMed] [Google Scholar]
- 3.Global Health Estimates Life expectancy and leading causes of death and disability. Accessed Jan 18, 2022. URL: https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-leading-causes-of-death.
- 4.Tan W.C., Sin D.D., Bourbeau J., Hernandez P., Chapman K.R., Cowie R., FitzGerald J.M., Marciniuk D.D., Maltais F., Buist A.S., et al. CanCOLD Collaborative Research Group Characteristics of COPD in never-smokers and ever-smokers in the general population: results from the CanCOLD study. Thorax. 2015;70:822–829. doi: 10.1136/thoraxjnl-2015-206938. [DOI] [PubMed] [Google Scholar]
- 5.Smith B.M., Kirby M., Hoffman E.A., Kronmal R.A., Aaron S.D., Allen N.B., Bertoni A., Coxson H.O., Cooper C., Couper D.J., et al. MESA Lung, CanCOLD, and SPIROMICS Investigators Association of Dysanapsis With Chronic Obstructive Pulmonary Disease Among Older Adults. JAMA. 2020;323:2268–2280. doi: 10.1001/jama.2020.6918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silverman E.K., Chapman H.A., Drazen J.M., Weiss S.T., Rosner B., Campbell E.J., O’Donnell W.J., Reilly J.J., Ginns L., Mentzer S., et al. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease. Risk to relatives for airflow obstruction and chronic bronchitis. Am. J. Respir. Crit. Care Med. 1998;157:1770–1778. doi: 10.1164/ajrccm.157.6.9706014. [DOI] [PubMed] [Google Scholar]
- 7.Ingebrigtsen T., Thomsen S.F., Vestbo J., van der Sluis S., Kyvik K.O., Silverman E.K., Svartengren M., Backer V. Genetic influences on Chronic Obstructive Pulmonary Disease - a twin study. Respir. Med. 2010;104:1890–1895. doi: 10.1016/j.rmed.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Zhou J.J., Cho M.H., Castaldi P.J., Hersh C.P., Silverman E.K., Laird N.M. Heritability of chronic obstructive pulmonary disease and related phenotypes in smokers. Am. J. Respir. Crit. Care Med. 2013;188:941–947. doi: 10.1164/rccm.201302-0263OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wain L.V., Shrine N., Artigas M.S., Erzurumluoglu A.M., Noyvert B., Bossini-Castillo L., Obeidat M., Henry A.P., Portelli M.A., Hall R.J., et al. Understanding Society Scientific Group. Geisinger-Regeneron DiscovEHR Collaboration Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat. Genet. 2017;49:416–425. doi: 10.1038/ng.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wyss A.B., Sofer T., Lee M.K., Terzikhan N., Nguyen J.N., Lahousse L., Latourelle J.C., Smith A.V., Bartz T.M., Feitosa M.F., et al. Multiethnic meta-analysis identifies ancestry-specific and cross-ancestry loci for pulmonary function. Nat. Commun. 2018;9:2976. doi: 10.1038/s41467-018-05369-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shrine N., Guyatt A.L., Erzurumluoglu A.M., Jackson V.E., Hobbs B.D., Melbourne C.A., Batini C., Fawcett K.A., Song K., Sakornsakolpat P., et al. Understanding Society Scientific Group New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat. Genet. 2019;51:481–493. doi: 10.1038/s41588-018-0321-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakornsakolpat P., Prokopenko D., Lamontagne M., Reeve N.F., Guyatt A.L., Jackson V.E., Shrine N., Qiao D., Bartz T.M., Kim D.K., et al. SpiroMeta Consortium. International COPD Genetics Consortium Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat. Genet. 2019;51:494–505. doi: 10.1038/s41588-018-0342-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao X., Qiao D., Yang C., Kasela S., Kim W., Ma Y., Shrine N., Batini C., Sofer T., Taliun S.A.G., et al. NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium. TOPMed Lung Working Group Whole genome sequence analysis of pulmonary function and COPD in 19,996 multi-ethnic participants. Nat. Commun. 2020;11:5182. doi: 10.1038/s41467-020-18334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang J., Benyamin B., McEvoy B.P., Gordon S., Henders A.K., Nyholt D.R., Madden P.A., Heath A.C., Martin N.G., Montgomery G.W., et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudbridge F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 2013;9:e1003348. doi: 10.1371/journal.pgen.1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busch R., Hobbs B.D., Zhou J., Castaldi P.J., McGeachie M.J., Hardin M.E., Hawrylkiewicz I., Sliwinski P., Yim J.-J., Kim W.J., et al. National Emphysema Treatment Trial Genetics. Evaluation of COPD Longitudinally to Identify Predictive Surrogate End-Points. International COPD Genetics Network. COPDGene Investigators Genetic Association and Risk Scores in a Chronic Obstructive Pulmonary Disease Meta-analysis of 16,707 Subjects. Am. J. Respir. Cell Mol. Biol. 2017;57:35–46. doi: 10.1165/rcmb.2016-0331OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oelsner E.C., Ortega V.E., Smith B.M., Nguyen J.N., Manichaikul A.W., Hoffman E.A., Guo X., Taylor K.D., Woodruff P.G., Couper D.J., et al. A Genetic Risk Score Associated with Chronic Obstructive Pulmonary Disease Susceptibility and Lung Structure on Computed Tomography. Am. J. Respir. Crit. Care Med. 2019;200:721–731. doi: 10.1164/rccm.201812-2355OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moll M., Sakornsakolpat P., Shrine N., Hobbs B.D., DeMeo D.L., John C., Guyatt A.L., McGeachie M.J., Gharib S.A., Obeidat M., et al. International COPD Genetics Consortium. SpiroMeta Consortium Chronic obstructive pulmonary disease and related phenotypes: polygenic risk scores in population-based and case-control cohorts. Lancet Respir. Med. 2020;8:696–708. doi: 10.1016/S2213-2600(20)30101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khera A.V., Chaffin M., Aragam K.G., Haas M.E., Roselli C., Choi S.H., Natarajan P., Lander E.S., Lubitz S.A., Ellinor P.T., Kathiresan S. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018;50:1219–1224. doi: 10.1038/s41588-018-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knowles J.W., Ashley E.A. Cardiovascular disease: The rise of the genetic risk score. PLoS Med. 2018;15:e1002546. doi: 10.1371/journal.pmed.1002546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharp S.A., Rich S.S., Wood A.R., Jones S.E., Beaumont R.N., Harrison J.W., Schneider D.A., Locke J.M., Tyrrell J., Weedon M.N., et al. Development and Standardization of an Improved Type 1 Diabetes Genetic Risk Score for Use in Newborn Screening and Incident Diagnosis. Diabetes Care. 2019;42:200–207. doi: 10.2337/dc18-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Restuadi R., Garton F.C., Benyamin B., Lin T., Williams K.L., Vinkhuyzen A., van Rheenen W., Zhu Z., Laing N.G., Mather K.A., et al. Polygenic risk score analysis for amyotrophic lateral sclerosis leveraging cognitive performance, educational attainment and schizophrenia. Eur. J. Hum. Genet. 2021 doi: 10.1038/s41431-021-00885-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forrest I.S., Chaudhary K., Paranjpe I., Vy H.M.T., Marquez-Luna C., Rocheleau G., Saha A., Chan L., Van Vleck T., Loos R.J.F., et al. Genome-wide polygenic risk score for retinopathy of type 2 diabetes. Hum. Mol. Genet. 2021;30:952–960. doi: 10.1093/hmg/ddab067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin A.R., Kanai M., Kamatani Y., Okada Y., Neale B.M., Daly M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019;51:584–591. doi: 10.1038/s41588-019-0379-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sirugo G., Williams S.M., Tishkoff S.A. The Missing Diversity in Human Genetic Studies. Cell. 2019;177:26–31. doi: 10.1016/j.cell.2019.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duncan L., Shen H., Gelaye B., Meijsen J., Ressler K., Feldman M., Peterson R., Domingue B. Analysis of polygenic risk score usage and performance in diverse human populations. Nat. Commun. 2019;10:3328. doi: 10.1038/s41467-019-11112-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vilhjálmsson B.J., Yang J., Finucane H.K., Gusev A., Lindström S., Ripke S., Genovese G., Loh P.-R., Bhatia G., Do R., et al. Schizophrenia Working Group of the Psychiatric Genomics Consortium, Discovery, Biology, and Risk of Inherited Variants in Breast Cancer (DRIVE) study Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am. J. Hum. Genet. 2015;97:576–592. doi: 10.1016/j.ajhg.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen C.-Y., Han J., Hunter D.J., Kraft P., Price A.L. Explicit Modeling of Ancestry Improves Polygenic Risk Scores and BLUP Prediction. Genet. Epidemiol. 2015;39:427–438. doi: 10.1002/gepi.21906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Márquez-Luna C., Loh P.-R., Price A.L., South Asian Type 2 Diabetes (SAT2D) Consortium. SIGMA Type 2 Diabetes Consortium Multiethnic polygenic risk scores improve risk prediction in diverse populations. Genet. Epidemiol. 2017;41:811–823. doi: 10.1002/gepi.22083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wojcik G.L., Graff M., Nishimura K.K., Tao R., Haessler J., Gignoux C.R., Highland H.M., Patel Y.M., Sorokin E.P., Avery C.L., et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570:514–518. doi: 10.1038/s41586-019-1310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marigorta U.M., Navarro A. High trans-ethnic replicability of GWAS results implies common causal variants. PLoS Genet. 2013;9:e1003566. doi: 10.1371/journal.pgen.1003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y.R., Keating B.J. Trans-ethnic genome-wide association studies: advantages and challenges of mapping in diverse populations. Genome Med. 2014;6:91. doi: 10.1186/s13073-014-0091-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I., Brown M.A., Yang J. 10 Years of GWAS Discovery: Biology, Function, and Translation. Am. J. Hum. Genet. 2017;101:5–22. doi: 10.1016/j.ajhg.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi H., Burch K.S., Johnson R., Freund M.K., Kichaev G., Mancuso N., Manuel A.M., Dong N., Pasaniuc B. Localizing Components of Shared Transethnic Genetic Architecture of Complex Traits from GWAS Summary Data. Am. J. Hum. Genet. 2020;106:805–817. doi: 10.1016/j.ajhg.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porcu E., Rüeger S., Lepik K., Santoni F.A., Reymond A., Kutalik Z., eQTLGen Consortium. BIOS Consortium Mendelian randomization integrating GWAS and eQTL data reveals genetic determinants of complex and clinical traits. Nat. Commun. 2019;10:3300. doi: 10.1038/s41467-019-10936-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liang Y., Pividori M., Manichaikul A., Palmer A.A., Cox N.J., Wheeler H.E., Im H.K. Polygenic transcriptome risk scores (PTRS) can improve portability of polygenic risk scores across ancestries. Genome Biol. 2022;23:23. doi: 10.1186/s13059-021-02591-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gamazon E.R., Wheeler H.E., Shah K.P., Mozaffari S.V., Aquino-Michaels K., Carroll R.J., Eyler A.E., Denny J.C., Nicolae D.L., Cox N.J., Im H.K., GTEx Consortium A gene-based association method for mapping traits using reference transcriptome data. Nat. Genet. 2015;47:1091–1098. doi: 10.1038/ng.3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barbeira A.N., Bonazzola R., Gamazon E.R., Liang Y., Park Y., Kim-Hellmuth S., Wang G., Jiang Z., Zhou D., Hormozdiari F., et al. GTEx GWAS Working Group. GTEx Consortium Exploiting the GTEx resources to decipher the mechanisms at GWAS loci. Genome Biol. 2021;22:49. doi: 10.1186/s13059-020-02252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mogil L.S., Andaleon A., Badalamenti A., Dickinson S.P., Guo X., Rotter J.I., Johnson W.C., Im H.K., Liu Y., Wheeler H.E. Genetic architecture of gene expression traits across diverse populations. PLoS Genet. 2018;14:e1007586. doi: 10.1371/journal.pgen.1007586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urbut S.M., Wang G., Carbonetto P., Stephens M. Flexible statistical methods for estimating and testing effects in genomic studies with multiple conditions. Nat. Genet. 2019;51:187–195. doi: 10.1038/s41588-018-0268-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barbeira A.N., Dickinson S.P., Bonazzola R., Zheng J., Wheeler H.E., Torres J.M., Torstenson E.S., Shah K.P., Garcia T., Edwards T.L., et al. GTEx Consortium Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nat. Commun. 2018;9:1825. doi: 10.1038/s41467-018-03621-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wen X., Pique-Regi R., Luca F. Integrating molecular QTL data into genome-wide genetic association analysis: Probabilistic assessment of enrichment and colocalization. PLoS Genet. 2017;13:e1006646. doi: 10.1371/journal.pgen.1006646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pividori M., Rajagopal P.S., Barbeira A., Liang Y., Melia O., Bastarache L., Park Y., Consortium G., Wen X., Im H.K., GTEx Consortium PhenomeXcan: Mapping the genome to the phenome through the transcriptome. Sci. Adv. 2020;6:eaba2083. doi: 10.1126/sciadv.aba2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taliun D., Harris D.N., Kessler M.D., Carlson J., Szpiech Z.A., Torres R., Taliun S.A.G., Corvelo A., Gogarten S.M., Kang H.M., et al. NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590:290–299. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hankinson J.L., Odencrantz J.R., Fedan K.B. Spirometric reference values from a sample of the general U.S. population. Am. J. Respir. Crit. Care Med. 1999;159:179–187. doi: 10.1164/ajrccm.159.1.9712108. [DOI] [PubMed] [Google Scholar]
- 47.Gogarten S.M., Sofer T., Chen H., Yu C., Brody J.A., Thornton T.A., Rice K.M., Conomos M.P. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics. 2019;35:5346–5348. doi: 10.1093/bioinformatics/btz567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balduzzi S., Rücker G., Schwarzer G. How to perform a meta-analysis with R: a practical tutorial. Evid. Based Ment. Health. 2019;22:153–160. doi: 10.1136/ebmental-2019-300117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robin X., Turck N., Hainard A., Tiberti N., Lisacek F., Sanchez J.-C., Müller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;12:77. doi: 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sofer T., Zheng X., Gogarten S.M., Laurie C.A., Grinde K., Shaffer J.R., Shungin D., O’Connell J.R., Durazo-Arvizo R.A., Raffield L., et al. NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium A fully adjusted two-stage procedure for rank-normalization in genetic association studies. Genet. Epidemiol. 2019;43:263–275. doi: 10.1002/gepi.22188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mak T.S.H., Porsch R.M., Choi S.W., Zhou X., Sham P.C. Polygenic scores via penalized regression on summary statistics. Genet. Epidemiol. 2017;41:469–480. doi: 10.1002/gepi.22050. [DOI] [PubMed] [Google Scholar]
- 52.GTEx Consortium The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318–1330. doi: 10.1126/science.aaz1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McCall M.N., Illei P.B., Halushka M.K. Complex Sources of Variation in Tissue Expression Data: Analysis of the GTEx Lung Transcriptome. Am. J. Hum. Genet. 2016;99:624–635. doi: 10.1016/j.ajhg.2016.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Denny J.C., Rutter J.L., Goldstein D.B., Philippakis A., Smoller J.W., Jenkins G., Dishman E., All of Us Research Program Investigators The “All of Us” Research Program. N. Engl. J. Med. 2019;381:668–676. doi: 10.1056/NEJMsr1809937. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Individual whole-genome sequence data for TOPMed whole genomes are available through dbGaP. The dbGaP accession numbers are: Atherosclerosis Risk in Communities (ARIC) phs001211, Coronary Artery Risk Development in Young Adults (CARDIA) phs001612, Cardiovascular Health Study (CHS) phs001368, Cleveland Family Study (CFS) phs000954, Framingham Heart Study (FHS) phs000974, Hispanic Community Health Study/Study of Latinos (HCHS/SOL) phs001395, Jackson Heart Study (JHS) phs000964, Multi-Ethnic Study of Atherosclerosis (MESA) phs001416, Genetic Epidemiology of COPD (COPDGene) phs000951, and SubPopulations and InteRmediate Outcome Measures in COPD Study (SPIROMICS) phs001927. Data in dbGaP can be downloaded by controlled access with an approved application submitted through dbGaP website. All PrediXcan code used is available in the GitHub repository.