

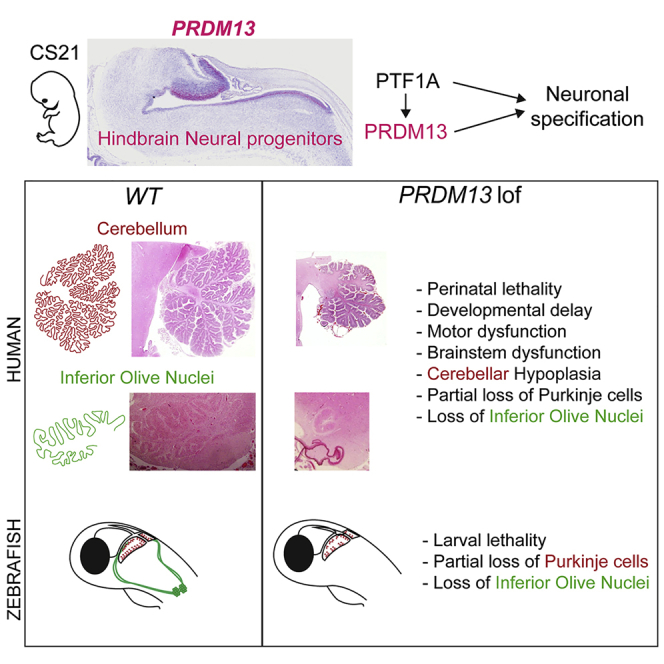
Summary
Pontocerebellar hypoplasias (PCHs) are congenital disorders characterized by hypoplasia or early atrophy of the cerebellum and brainstem, leading to a very limited motor and cognitive development. Although over 20 genes have been shown to be mutated in PCHs, a large proportion of affected individuals remains undiagnosed. We describe four families with children presenting with severe neonatal brainstem dysfunction and pronounced deficits in cognitive and motor development associated with four different bi-allelic mutations in PRDM13, including homozygous truncating variants in the most severely affected individuals. Brain MRI and fetopathological examination revealed a PCH-like phenotype, associated with major hypoplasia of inferior olive nuclei and dysplasia of the dentate nucleus. Notably, histopathological examinations highlighted a sparse and disorganized Purkinje cell layer in the cerebellum. PRDM13 encodes a transcriptional repressor known to be critical for neuronal subtypes specification in the mouse retina and spinal cord but had not been implicated, so far, in hindbrain development. snRNA-seq data mining and in situ hybridization in humans show that PRDM13 is expressed at early stages in the progenitors of the cerebellar ventricular zone, which gives rise to cerebellar GABAergic neurons, including Purkinje cells. We also show that loss of function of prdm13 in zebrafish leads to a reduction in Purkinje cells numbers and a complete absence of the inferior olive nuclei. Altogether our data identified bi-allelic mutations in PRDM13 as causing a olivopontocerebellar hypoplasia syndrome and suggest that early deregulations of the transcriptional control of neuronal fate specification could contribute to a significant number of cases.
Keywords: brainstem, cerebellum, PRDM13, Purkinje cells, olivopontocerebellar hypoplasia, inferior olive nuclei, zebrafish, neurodevelopment, neuronal specification
Graphical abstract
Introduction
Cerebellar hypoplasia (CH) refers to reduced cerebellar volume with a (near-) normal shape based, in vivo, on neuroimaging. It is a relatively common finding associated with a wide range of clinical features.1, 2, 3 Additional neuroimaging findings, such as abnormal size or shape of the pons or the medulla oblongata, are key to categorize CH and better predict the clinical outcome. Early steps of cerebellar development are shared with the brainstem explaining the frequent association of abnormal development of these structures in disorders such as Joubert syndrome (MIM: 213300), rhombencephalosynapsis,4 pontine tegmental cap dysplasia (MIM: 614688), diencephalic–mesencephalic junction dysplasia (MIM: 251280), or pontocerebellar hypoplasia (PCH [MIM: 607596]).5 PCH is described as a group of generally neurodegenerative disorders with prenatal onset, progressive features, and at least 13 subtypes, based on neuropathological, clinical, and MRI criteria.6,7 The prognosis of PCH disorders is poor and most affected individuals die during infancy or childhood. Despite the current effort to identify new genes associated with PCH, a large number of affected individuals remains without genetic diagnosis.6 PCH often results from defects in apparently ubiquitous cellular processes such as tRNA synthesis, mitochondrial, glycosylation, inositol, or purine nucleotide metabolisms.5,7,8 In parallel to the typical PCH presentation, mutations in CASK (MIM: 300172), CHMP1A (MIM: 164010), and TBC1D23 (MIM: 617687) can lead to non-neurodegenerative disorders categorized respectively as PCH-like (or MICPCH [MIM: 300749]), PCH8,9 and PCH1110 disorders. Few cases with mutations in PTF1A (MIM: 607194) can be classified in this category of non-neurodegenerative syndrome, with an imaging pattern similar to PCH,11 although these are characterized by an extremely severe impact on cerebellar development, associated with neonatal diabetes.12 Interestingly, PTF1A is known to play a key role in hindbrain neural stem cell fate specification; whether other cell fate determinants factors contribute to congenital cerebellar disorders remains to be further explored. Indeed, for many key hindbrain stem cell fate determinants (e.g., ATOH1 [MIM: 601461], SKOR2 [MIM: 617138], EN2 [MIM: 131310]), there are no associated and clearly defined Mendelian disorders yet. A better knowledge of the clinical consequences associated with the loss of function of these key genes would improve our understanding of their roles in human cerebellar and whole brain development.
PRDM13 (MIM: 616741), a known target of PTF1A, encodes a transcriptional regulator previously implicated in neuronal specification in the retina and spinal cord.13, 14, 15, 16 However, until recently, no role had been attributed to this factor during cerebellar development. Indeed, Whittaker et al. reported a variant of this gene in three individuals with a hypogonadotropic hypogonadism syndrome associated with cerebellar hypoplasia.17 Here, we report a significantly different syndrome on the basis of eight individuals from four families of different origins associated with loss-of-function mutations of PRDM13. This syndrome associates perinatal lethality with severe brainstem dysfunctions (e.g., feeding and respiratory difficulties, central apnea, bradycardia), together with persistent cerebellar hypoplasia. We also clearly establish that PRDM13 is critically needed for the cerebellar GABAergic lineage differentiation, including Purkinje cells as well as for olivary nuclei genesis.
Material and methods
Patients’ recruitment and investigation
Families 1 and 2 were recruited through the departments of genetics of the Pellegrin and the Necker Enfants Malades Hospitals and the French reference center for cerebellar malformations and congenital diseases at Trousseau Hospital, France. Families 3 and 4 were identified through GeneMatcher.18 Family 3 was referred to the clinical genetics department of university hospitals Bristol. Family 4 was recruited through the Genetic and Developmental Medicine Clinic Sultan Qaboos University Hospital, Oman. Written informed consents have been obtained both from all the participants and the legal representatives of all the children. Procedures followed were in accordance with the ethical standards of the national and institutional responsible committees.
Index cases of the four families have been analyzed via whole-exome sequencing (WES). For family 1, we performed trio WES (proband F.1-1 and both parents) with the Sure Select Human All Exon 58 Mb V6 protocol (Agilent Technologies) to prepare libraries that were sequenced with a HiSeq2500 system (Illumina) at the Imagine institute (Paris, France). Sequences were aligned to the reference human genome hg19 with the Burrows-Wheeler aligner (BWA). Downstream processing was carried out with the Genome Analysis Toolkit (GATK), SAMtools, and Picard. The annotation process was based on the latest release of the Ensembl database, gnomAD 2.1. Variants were annotated and analyzed with the Polyweb software interface designed by the Bioinformatics platform of Imagine-Université de Paris.19 Family 2-1 proband was studied by WES performed in the proband only at Sorbonne Université, Trousseau Hospital, Paris, France, with the following details: SeqCap EZ MedExome capture kit (Roche) and sequence on Illumina NextSeq 500. The BaseSpace cloud computing platform with BWA, GATK Unified Genotyper, and the VariantStudio v.3.0 provided by Illumina was used for analysis. We performed targeted Sanger sequencing in the proband, parents, and affected sibling of family 2 to confirm the presence of the homozygous or heterozygous variants in affected children and parents, respectively. For family 3, WES was performed on both children and their healthy parents by the Exeter Genomics Laboratory (Exeter, UK). Libraries were prepared with the Twist Core Human Exome Kit (Twist Bioscience) and sequenced on the NextSeq 500 or NovaSeq seq platform (Illumina). Reads were aligned to human genome build hg19 and analyzed for sequence variants with a custom-developed analysis tool as previously described.20 Family 4-3 proband was explored with clinical exome at BGI Europe (Copenhagen, Denmark). Exome capture was performed with the Agilent SureSelectXT Human All Exon 50 Mb Kit and sequencing was done on HiSeq2000TM (Illumina). Analysis was performed at Sultan Qaboos University Hospital (Muscat, Oman). An in-house database of 1,562 exomes was used for the filtering out of common population-specific variants.21 Sanger sequencing was used for confirmation of the mutation in the affected individual. The DNA from parents and siblings was not available. Detailed clinical reports are included in supplemental materials.
In situ hybridization with human samples
Tissue collection, processing, and in situ hybridization (ISH) were performed at University College London/HDBR as previously described22 and under the approval of the National Research Ethics Service (NRES). Briefly, tissues were fixed in 4% PFA/10% formalin overnight, embedded and sectioned at 6 microns. ISH was performed with RNAscope 2.5 HD Detection Kit and commercially available probes (Advanced Cell Diagnostics) for PRDM13 and PPIB as a positive control with the references 459851 and 313901, respectively.
Single-cell RNA-sequencing data retrieval
Data re-analyzed as part of this study were retrieved from the Gene Expression Omnibus (GEO) public database under accessions GEO: GSE120372 for mouse embryonic cerebellum23 and GEO: GSE158142 for zebrafish brain single-cell RNA sequencing (scRNA-seq) data24 and from the Human Gene Expression During Development Atlas (Descartes, https://descartes.brotmanbaty.org) for human cerebellum single-nucleotide RNA-sequencing (snRNA-seq) data.25 Mouse scRNA-seq data corresponds to a single developmental time point, embryonic day 13.5 (E13.5; n = 9,353 cells). Zebrafish scRNA-seq data were retrieved as processed R objects (.rds files) for different development stages (24 h post fertilization [hpf] to 8 days post fertilization [dpf]; n = 95,730 cells) and containing, among others, the assigned clusters information at different resolutions as shown in Raj et al.24 Human cerebellum snRNA-seq data were retrieved as sparse gene counts by cell (n = 1,091,999 cells) along with cell-type assignations and gene annotations metadata (Table 1).
Table 1.
Source scRNA-seq and snRNA-seq datasets
| Dataset | Species | Source | Number of single cells |
|---|---|---|---|
| Cerebellum | mouse | GEO: GSE120372 | 9,353 |
| Cerebellum – Purkinje neurons | human | Descartes Atlas | 280,377 |
| Cerebellum – inhibitory interneurons | human | Descartes Atlas | 129,890 |
| Cerebellum – granule neurons | human | Descartes Atlas | 312,674 |
| Cerebellum – astrocytes | human | Descartes Atlas | 268,809 |
| Cerebellum – vascular endothelial | human | Descartes Atlas | 7,349 |
| Cerebellum – unipolar brush | human | Descartes Atlas | 52,646 |
| Cerebellum – microglia | human | Descartes Atlas | 4,428 |
| Cerebellum – oligodendrocytes | human | Descartes Atlas | 16,104 |
| Cerebellum – SLC24A4_PEX5L positive cells | human | Descartes Atlas | 19,722 |
| Head – 24 hpf | zebrafish | GEO: GSE158142 | 9,105 |
| Head – 36 hpf | zebrafish | GEO: GSE158142 | 10,719 |
| Head – 48 hpf | zebrafish | GEO: GSE158142 | 15,558 |
| Brain – 5 dpf | zebrafish | GEO: GSE158142 | 31,659 |
| Brain – 8 dpf | zebrafish | GEO: GSE158142 | 28,689 |
Mouse and human cerebellum data processing
Data were imported into R v4.0.3 creating a Seurat v4.0 object26 from counts. Further data processing followed the standard Seurat workflow for data normalization, dimensionality reduction (principal-component analysis, PCA), and graph-based clustering described in Butler et al.27 Differentially expressed genes for the identified cell subpopulations were determined with Wilcoxon rank-sum tests on genes present in at least 20% of cells in the population of interest, only retaining positive gene markers. Testing was limited to genes that showed, on average, at least 0.2-fold difference (on a log-scale) between the different groups. Finally, genes displaying an adjusted p value inferior to 5% (padj < 0.05) were retained.
Visualization
PRDM13, SOX2, and PTF1A gene expression levels were represented over t-distributed stochastic neighbor embedding (t-SNE) projection plots of distinct single cells at different developmental stages for zebrafish and mouse datasets or over uniform manifold approximation and projection (UMAP) plots for human cerebellum data. Simultaneous visualization of the expression levels of two different genes was performed with the FeaturePlot Seurat function (blend = TRUE parameter).
Zebrafish lines and maintenance
Wild-type AB (ZFIN: ZDB-GENO-960809-7) and prdm13sa16464 (ZFIN: ZDB-ALT-130411-4933) zebrafish were used for the experiments. The prdm13sa16464 line was generated by the Zebrafish Mutation Project (ZMP)28 and obtained from the EZRC center (Karlsruhe). Zebrafish were maintained with standard fish-keeping protocols, and experimental protocols conformed to French and European ethical and animal welfare directives (project authorization from the Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation to M.C.). Genotyping of individuals from the prdm13sa16464 line was performed by PCR with the following allele-specific primer pairs: 5′-GAGCGATACATTTGCCGG-3′, 5′-CTCAGCAGGGGATATCTTCG-3′ (WT allele); 5′-ATGGCCACAAACAACACAAA-3′, 5′-GGTATTTGAATATTCTCCAACAAGAT-3′ (sa16464 allele).
Fresh fin clips from adult breeders or tails from fixed larvae were digested with Proteinase K (200 μg/mL) diluted in PCR buffer (10 mM Tris [pH 8], 2 mM EDTA, 0.2% Triton X-100) for 3 h at 55°C. Proteinase K was inactivated by a 5 min incubation at 95°C. PCR was performed with ReadyMix Taq PCR Reaction Mix (Sigma-Aldrich).
Zebrafish embryos and tissue fixation
Zebrafish embryos and larvae were euthanized with tricaine (0.2% in embryo medium) and fixed overnight at 4°C in a 4% solution of paraformaldehyde (PFA) in PBS. For ISH on larval or juvenile brains (>5 dpf), brains were dissected out in cold PBS after fixation. Following fixation, samples were washed twice in PBS. Whole embryos and larvae were bleached with a peroxide-containing bleaching solution (5% formamide, 0.5× saline sodium citrate [SSC], 3% H2O2) until dark pigments are no longer visible. Samples were either stored in PBS at 4°C (for immunohistochemistry [IHC]) or dehydrated in serial dilutions of methanol/PBS and stored in 100% methanol at −20°C (for ISH).
RNA extraction
Dechorionated 24 hpf zebrafish embryos were euthanized on ice with tricaine (0.2% in embryo medium) and homogenized with a pestle in Trizol solution (Thermofisher, 1 mL Trizol for 50–100 embryos). After a 5 min incubation at RT, chloroform was added (200 μL per mL of Trizol). The Eppendorf tube was shaken for 15 s and centrifuged at 12,000 g for 15 min. The upper phase was transferred to a fresh tube containing 500 μL Isopropanol and incubated at room temperature (RT) for 10 min. The tube was centrifuged at 12,000 g at 4°C for 10 min. After removal of supernatant, the pellet was washed with 70% EtOH and dissolved in 44 μL RNase-free water. Genomic DNA was removed with RNase-free DNase I treatment (NEB): 5 μL of 10× DNase buffer and 1 μL of RNase-free DNase was added to the 44 μL of RNA and the mix was incubated for 30 min at 37°C. To stop the reaction, we added 1 μL of 0.25 mM EDTA and incubated the tube at 65°C for 10 min. RNA concentration was measured with a nanodrop and stored at −80°C.
Cloning
A fragment of zebrafish prdm13 transcript (GenBank: NM_001326454.1) covering the full-length coding sequence was amplified by PCR with the AccuPrime Taq DNA Polymerase System (Invitrogen) with the following primers: 5′-TGGATCCACAGAGCCAACCATGCAAACG-3′ and 5′-TTCTAGATTAGTGTGTCCGTAAAGTGG-3′.
The 1,881 nt-long PCR product was cloned inside the pCR2.1-TOPO plasmid with TOPO TA Cloning Kit (Invitrogen) according to the manufacturer’s instructions. Clone sequences were verified by Sanger sequencing with universal M13 primers.
RNA probes synthesis
For prdm13 RNA probe synthesis, a DNA matrix was first generated by PCR from pCR2.1-prdm13 plasmid with ReadyMix Taq PCR Reaction Mix (Sigma-Aldrich) and M13 primers. For ptf1a RNA probe synthesis, a plasmid containing the full-length ptf1a transcript (pBS-SK-ptf1a(FL)) was kindly donated by Masahiko Hibi (Nagoya University). 10 μg of plasmid DNA was linearized with EcoRI (NEB). DNA matrices were purified with PureLink PCR Micro Kit (Invitrogen) and eluted in 10 μL of RNase-free water. To generate DIG-labeled probes, we used 1 μg linearized plasmid as a template for transcription reaction with T3 or T7 RNA polymerase and DIG RNA Labeling Kit (Roche), according to the manufacturer’s instructions. Transcription reaction was carried out for 3 h at 37°C, and a final incubation with DNase I was performed for removal of template DNA. Unincorporated nucleotides were removed with the ProbeQuant G-50 Micro Columns Kit (GE Healthcare). The RNA probe was stored at −80°C.
Whole-mount ISH
ISH was carried out according to standard protocols.29 Briefly, embryos or dissected brains were rehydrated, washed with PBST, and incubated in hybridization buffer at 65°C for 3 h. Brains were then incubated overnight at 65°C with DIG-labeled RNA probes diluted at 1/100 in hybridization buffer. We washed brains in serial dilutions of hybridization buffer/2×SSC at 65°C to remove excess probe. An immunohistochemistry with anti-DIG antibody coupled to alkaline phosphatase (Roche, 1/5,000 dilution) was then performed. In situ signals were revealed either with NBT/BCIP (Roche) or Fast Red (Sigma) for fluorescent visualization. Following ISH, 2-month-old juvenile brains were sectioned with a vibratome (Leica VT1200) and mounted on microscope slides.
Whole-mount IHC
For whole-mount IHC, zebrafish larvae were permeabilized in acetone at −20°C for 20 min. Samples were washed three times with PBST (0.1% Triton in PBS). Antigen retrieval was then performed through an incubation in HistoVT One (Nacalai Tesque) buffer for 1 h at 65°C. Larvae were washed three times for 5 min each with PBST and once in PBS-DT (0.1% Triton + 1% DMSO + 1% BSA in PBS). Samples were incubated in blocking buffer (5% goat serum in PBS-DT) for at least 1 h at RT and then with primary antibodies (see Table 2) diluted in blocking buffer for 24 h at 4°C. This was followed by six washes with PBST for 15 min. Larvae were incubated overnight at 4°C with secondary antibodies (see Table 2) diluted 1:1,000 in PBS-DT. The brains were then washed six times for 15 min in PBST. Brains were mounted in a glycerol solution (80% glycerol/PBS) for confocal imaging.
Table 2.
Antibodies
| Antibody name | Source | Identifier |
|---|---|---|
| Sheep Anti-Digoxigenin Fab fragments Antibody, AP Conjugated | Sigma-Aldrich | Cat# 11093274910; RRID: AB_514497 |
| Mouse monoclonal (IgG1) Anti-SOX2 antibody [20G5] | Abcam | Cat# ab171380; RRID: AB_2732072 |
| Mouse monoclonal (IgG2b) Anti-HuC/D, Clone 16A11 | Thermo Fisher Scientific | Cat# A-21271; RRID:AB_221448 |
| Rabbit Anti-Calbindin D-28k (for zebrafish) | Swant | Cat# CB-38a; RRID: AB_10000340 |
| Mouse monoclonal Anti-Calbindin-D-28K, Clone CB-955 (for human) | Sigma-Aldrich | Cat# C9848, RRID: AB_476894 |
| Mouse monoclonal (IgG2a) anti-PCNA Antibody, clone PC10 | Santa Cruz Biotechnology | Cat# sc-56; RRID: AB_628110 |
| Mouse polyclonal (IgG1) LHX1 antibody (4F2) | DSHB | Cat# 4F2 s; RRID: AB_531784 |
| Mouse monoclonal (IgG1) anti-Parvalbumin | Millipore | Cat# MAB1572; RRID: AB_11211313 |
| Goat anti-Mouse IgG1-488 | Thermo Fisher Scientific | Cat# A-21121; RRID: AB_2535764 |
| Goat anti-Mouse IgG2a-633 | Thermo Fisher Scientific | Cat# A-21136, RRID: AB_2535775 |
| Goat anti-Mouse IgG2b-633 | Thermo Fisher Scientific | Cat# A-21146, RRID: AB_2535782 |
| Goat anti-Rabbit-546 | Thermo Fisher Scientific | Cat# A-11010, RRID: AB_2534077 |
Image acquisition
Brightfield images were acquired with a stereomicroscope (Leica M165FC) equipped with a color digital camera (Leica DFC450C). Fluorescent images were acquired on confocal microscopes (LSM700) with either a 20× air objective (Plan Apochromat 20×/0.8 M27) or a 40× oil objective (Plan-Apochromat 40×/1.3 Oil M27). 3D renderings were generated with the Imaris software (version 9.7.2, Bitplane). We cropped the 3D image to feature only the cerebellum as our region of interest and contrast was adjusted for each channel. For Sox2 immunostaining signals, background was subtracted with the remove outliers function of the ImageJ image analysis software (version 1.53c; https://fiji.sc/).
Quantification and statistical analyses
3D Images were cropped and then segmented via semi-automatic detection with the Imaris spots function followed by manual curation. We segmented the whole cerebellum to quantify PARV+ Purkinje cells and CALB+ Eurydendroid cells. Data are presented as mean ± 95% confidence interval. Statistical analyses and graphical plots were done with GraphPad Prism version 9.2.0 for Windows (GraphPad Software, San Diego, California, USA, https://www.graphpad.com/). Deviation from Mendelian ratios was tested with Chi-square tests, and pairwise comparisons were carried out with two-tailed unpaired t tests.
Results
Identification of bi-allelic PRDM13 pathogenic variants in families with brainstem dysfunction and cerebellar hypoplasia
In a collaborative effort to identify additional mechanisms underlying PCH and PCH-like disorders,5,19 we recruited a French family of North-African origin with distant consanguinity (family 1) where three siblings presented with cerebellar vermis hypoplasia and brainstem dysgenesis (Figure 1A). For the proband (F.1-1) and sibling F.1-6, fetal brain MRI showed severe cerebellar hypoplasia with abnormal foliation (Figure 1B, Table 3, supplemental materials) and intact supratentorial structures. At birth, they presented with profound hypotonia and feeding difficulties with no sucking abilities. Several episodes of desaturation and altered consciousness related to a potential central origin required oxygenation therapy. The affected child F.1-1 died at the age of 25 months without reaching any developmental milestones and her sibling F.1-6 died at the age of 4 months. During the fourth pregnancy, similar manifestations as the first pregnancy were detected by close ultrasound follow-up and parents opted for a medical termination at 24 weeks of gestation (WG). Fetal pathological examination of F.1-4 further documented the disrupted development of the cerebellum and the brainstem (Figures 2A and 2B; see below), consistent with a type of PCH. Using whole-exome sequencing (WES), we identified a homozygous single base pair deletion in PRDM13 in the three affected children. This variant, c.839del (p.Ala280Glyfs∗21), is predicted to cause a frameshift followed by premature termination after the first zinc finger domain (Figure 1D). PRDM13 encodes a transcription factor with key functions in inhibitory neurons differentiation.13,14 Prdm13 knockout mice display dorso-ventral cell specification defects in the fetal neural tube and die neonatally of unknown reasons.16 Interestingly, a homozygous Prdm13 deletion that removes the C-terminal part of the protein after the first zinc finger domain reflects a null phenotype.16 On the basis of these first observations, we hypothesized that PRDM13 loss of function could cause the neurodevelopmental disorder observed in family 1. In our effort to replicate this finding, we identified five other affected individuals from three additional families (Figure 1, Table 3) with highly comparable clinical phenotypes including cerebellar hypoplasia, brainstem dysfunction, and a diagnosis consistent with a form of PCH (individuals F.2-1, F.2-2, and F.4-3; Figures 1B and 1C). To have a broad view of the phenotypical consequences resulting from PRDM13 mutations, we used a genotype-first approach,18 which led to the identification of families 3 and 4. We used WES systematically to uncover the genetic variants suspected to cause their disorder. For each family, the homozygous variant located in PRDM13 is the only one compliant with autosomal recessive Mendelian inheritance and/or the most likely to disrupt gene function (Supplemental information). Affected individuals of families 2, 3, and 4 were homozygous for variants c.844del (p.Val282Serfs∗19), c.1856A>T (p.His619Leu), and c.800del (p.Gly267Aspfs∗34), respectively (GenBank: NM_021620.3, NP_067633.2; Table 3). All variants are either absent or encountered (at the heterozygous state) with a frequency lower than 0.001% in gnomAD (v2.1.1). The variant identified in family 3 is the only missense variant identified (p.His619Leu), affecting a fully conserved histidine (up to S. cerevisiae, not shown) with a CADD score of 32. This histidine residue (Figure 1D bottom-right, in red) is one of the invariant residues of the Cys2-His2 binding motif.30 Consequently, the functionality of the third zinc finger domain is abrogated by the p.His619Leu variant.
Figure 1.

Bi-allelic mutations in PRDM13 in affected children and fetuses with posterior fossa anomalies
(A) Families with predicted effect of PRDM13 variants and pedigrees. All pathogenic variants are homozygous in affected individuals and segregated as recessive traits.
(B) T2-weighted fetal magnetic resonance images of control individual at 26 WG and affected individuals F.2-1, F.1-1, and F.1-6 at 26 WG, 30 WG, and 33 WG, respectively. Sagittal views are shown on top, and the bottom parts show coronal views for the control individual, F.2-1, and F.1-6 and axial views for F.1-1. The imaging shows reduced cerebellar vermis volume (yellow arrowhead) with abnormal or incomplete foliation and mild tapering of medulla oblongata for F.1-6.
(C) Sagittal T1-weighted (top, for control individual and F.4-3), Sagittal T2-weighted (top, for F.2-2), and coronal T2-weighted (bottom) brain magnetic resonance images of control individual and individuals F.4-3 and F.2-2. For both children, a small and dysmorphic vermis is visible on the sagittal slices and small hemispheres are detected on coronal views.
(D) Identified PRDM13 mutations. Bottom-right, modeling of the third zinc finger domain based on ZFP568 structure (PDB: 5V3J). The mutated histidine residue (in red) is part of the Cys2-His2-binding motif. Blue, zinc ion.
Table 3.
Clinical table
| Family 1 | Family 2 | Family 3 | Family 4 | |||||
|---|---|---|---|---|---|---|---|---|
| Individual identifier | F.1-1 | F.1-4 (fetus) | F.1-6 | F.2-1 (fetus) | F.2-2 | F.3-1 | F.3-2 | F.4-3 |
| Gender (M/F) | F | F | M | F | M | F | F | M |
| Age at last examination, age at death or fetal age | 7 months, died at 25 months | 24 GW | birth, died at 4 months | 31 GW | 2 days, died at 22 days | 4 years 6 months | 11 months | 12 months, died at 16 months |
| Genetic data | ||||||||
| Ethnic origin | Tunisia | Algeria | Pakistan | Arab | ||||
| Consanguinity (yes/no) | yes | yes | no (parents unsure) | yes | ||||
| gDNA (GRCh38/hg38) | chr6(GRCh38): g.99613474del | chr6(GRCh38): g.99613479del | chr6(GRCh38): g.99614491A>T | chr6(GRCh38): g.99613435del | ||||
| cDNA (GenBank: NM_021620.3) | c.839del | c.844del | c.1856A>T | c.800del | ||||
| Protein | p.Ala280Glyfs∗21 | p.Val282Serfs∗19 | p.His619Leu | p.Gly267Aspfs∗34 | ||||
| CADD-PHRED score | N/A | N/A | 32 | NA | ||||
| Prenatal findings | ||||||||
| Prenatal growth retardation | − | − | − | − | − | + | + | − |
| Cerebellar vermis hypoplasia | + | + | + | + | + | not reported | not reported | not reported |
| Growth | ||||||||
| Delivery (weeks) | 39 | 24 | 37 | N/A | 34 | 39 | 37 | 40 |
| Neonatal symptoms | profound hypotonia | N/A | profound hypotonia | N/A | respiratory distress, axial hypotonia | temperature, HR and BP fluctuations; seizures | temperature, HR and BP fluctuations | respiratory distress, recurrent apnea |
| Likely autonomic symptoms | swallowing defect, desaturation, respiratory distress, bradypnea | N/A | swallowing defect, respiratory distress, no cough reflex, bradycardia | N/A | swallowing defect, respiratory distress, recurrent apnea, bradycardia | temperature, HR and BP fluctuations, respiratory distress | temperature, HR and BP fluctuations | respiratory distress, recurrent apnea |
| Weight at last examination (kg) | 7 (−0.8 SD) | N/A | N/K | N/A | N/K | 12.2 at 4 years 6 months (<−2 SD) | 5.0 at 7.7 months (−3 SD) | 5.2 (−4.5 SD) |
| HC at birth (cm) | N/A | N/A | N/K | N/A | 33.5 (+1.3 SD) | N/K | N/K | 34 (−1 SD) |
| HC at last examination (cm) | 42 (−0.9 SD) | N/A | N/K | N/A | N/K | 46.4 at 4 years 6 months (−2.7 SD) | 38.3 at 33 weeks 5 days (−4.7 SD) | 38 (−7 SD) |
| Presence of dysphagia | +, nasogastric tube feeding | N/A | +, nasogastric tube feeding | N/A | +, nasogastric tube feeding | − | − | +, nasogastric tube feeding |
| Developmental history | ||||||||
| Motor (normal/delayed/absent) | absent | N/A | absent | N/A | absent | delayed | delayed | absent |
| Global developmental delay (severity) | severe/profound | N/A | N/A | N/A | N/A | severe | mild | severe |
| Neurological features | ||||||||
| Axial hypotonia | + | N/A | + | N/A | + | + | + | + |
| Distal hypertonia | + | N/A | + | N/A | + | + | + | + |
| Spastic tetraplegia | + | N/A | N/K | N/A | N/K | − | − | + |
| Deep tendon reflexes | normal | N/A | N/K | N/A | N/K | reduced | normal | reduced |
| Epilepsy | ||||||||
| Seizure | − | N/A | − | N/A | − | neonatal period, focal seizures (on levetiracetam) | one seizure, on levetiracetam since | neonatal |
| Brain MRI | ||||||||
| Normal supratentorial brain | + | + | + | + | + | + | + | + |
| Cerebellar hemisphere hypoplasia | + | − | − | + | + | + | + | + |
| Cerebellar vermis hypoplasia | + | + | + | + | + | + | − | + |
| Brainstem hypoplasia | + | + | + | + | − | + | − | + |
| Associated clinical features | ||||||||
| Ophthalmological findings | abnormal ocular movement, suspicion of papillary edema or hypoplasia | N/A | bilateral retinal hemorrhages | N/A | no eye tracking | nystagmus with horizontal gaze, short sighted | − | − |
| Facial dysmorphism | microretrognathia | − | − | − | hypertelorism, epicanthic fold | − | − | hypertrichosis, low anterior hairline, upslanting palpebral fissures, epicanthic folds |
| Oral cavity findings | posterior cleft palate | posterior cleft palate | − | − | − | − | − | − |
| Cardiac abnormalities | − | trabecular ventricular septal defect | ostium secundum atrial septal defect, patent ductus arteriosus | − | N/A | sinus rhythm with occasional marked sinus arrhythmia | − | − |
| Gastrointestinal abnormalities | − | − | gastroesophageal reflux | − | − | − | − | recurrent vomiting, gastroesophageal reflux |
| Respiratory system | oxygen therapy required (nasal canula) | − | oxygen therapy, base of the tongue ptosis, no cough reflex | − | apnea, oxygen therapy required in the context of prematurity | respiratory distress | − | poor respiratory effort, ventilation dependence |
| Other | N/A | mild extremity malpositions | mild feet malpositions | N/A | N/A | postural kyphosis | episodic dystonia | N/A |
Abbreviations: BP, blood pressure; EEG, electroencephalography; HC, head circumference; HR, heart rate; N/A, not applicable; N/K, not known; SD, standard deviation.
Figure 2.

Neuropathological examination of the fetuses F.1-4 and F.2-1 and child F.2-2
(A–F) Posterior view (B) and basal view (A and C–F) of the brain showing cerebellar hypoplasia in affected individuals compared to stage-matched control individuals. Scale bar: 10 mm. Cerebella are outlined with white dashed lines. (B) An incomplete covering of the 4th ventricle by the vermis is visible for the fetus F.1-4.
(G–L) Cross sections of the medulla stained with hematoxylin and eosin (HE) showing a fragmentation or extreme hypoplasia (arrows) of the olivary nuclei in affected individuals. Pyramidal tracts are clearly visible (∗) and inferior olivary nuclei are noted (arrowhead) in control individuals. Scale bar: 500 μm.
(M–R) Horizontal (M) and sagittal (N–R) HE-stained sections showing a dysplasia and fragmentation of the dentate nucleus (arrows) in affected individuals. Arrowheads in control individuals point to the dentate nucleus. Scale bar: 500 μm.
(S–V) Sagittal HE-stained sections at the level of the vermis showing delayed lobulation with absence of tertiary foliation in the cerebellum of F.2-2 and hypoplasia severely affecting the anterior vermis (T and V). Primary fissures are indicated with arrowheads. Scale bar: 1 mm. Purkinje cell cluster heterotopia are visible in the white matter (T, top-left magnification).
(W–AB) Immunostaining of Purkinje cells with calbindin showing a decreased number and delayed maturation of Purkinje cells in the cerebellar cortex of affected individuals. Scale bar: 50 μm
Family 2 is also a French family of North-African origin with consanguinity. They had three pregnancy attempts; the second pregnancy resulted in a fetus who was interrupted for presenting with cerebellar and brainstem hypoplasia (fetus F.2-1, Figure 1B) and the third one resulted in a newborn who died at 22 days with a similar condition and profound hypotonia, inability to eat, and respiratory distress (infant F.2-2, Figure 1C). Family 3 is from the UK, from Pakistani origin, with two affected girls with a milder presentation. The F.3-1 elder sister presented with neonatal hypotonia, bradycardia, and respiratory distress. Brain MRI identified a non-progressive form of PCH. She had recurrent episodes with symptoms of dysautonomia associated with somnolence and then unresponsiveness. Similarly, F.3-2 presented with hypotonia and neonatal autonomic problems, including temperature instability and bradycardia. She has an apparently less severe condition compared to her sister, without severe episodes of altered consciousness and autonomic instability, but she presented with developmental delay, slow weight gain, and cerebellar hemispheres hypoplasia with preserved brainstem on MRI.
Family 4 is a consanguineous family from Oman, with a single affected individual presenting with microcephaly, feeding difficulty, apnea, and hypoventilation and who died at 16 months. Brain MRI showed a dysplasia and a reduced size of the cerebellar vermis and the hemispheres (Figure 1C).
Truncations of PRDM13 are associated with early disruption of cerebellar and brainstem development
Neuropathological examination was performed for fetuses F.1-4 and F.2-1 and the infant F.2-2, covering the stages 24 GW, 31 GW, and 34 GW + 3 postnatal weeks (equivalent to 37 GW) (Figures 2A–2F). These three affected individuals displayed features distinctive of PCH31 with unremarkable supra-tentorial structures: (1) reduced brainstem and cerebellum over total brain weight (3.4% for F.2-1 at 31 GW [−5.2 SD] and 3.7% for F.2-2 at 37 GW [−3.6 SD], Table S1), (2) transverse diameter of the cerebellum below the norm at 31 GW (<5th percentile) and 37 GW (<5th percentile), and (3) delayed/abnormal cerebellar foliation. At the histological level, the three individuals also presented comparable cerebellar and brainstem anomalies. Cerebellar vermis thus showed a hypoplastic but relatively preserved structure at 24 GW (Figures 2A, 2B, 2S, and 2T) and a severe hypoplasia with abnormal foliation at 31 GW and 37 GW (Figures 2C–2F, 2U, and 2V). The dentate nuclei were dysplastic and fragmented for the three affected individuals (Figures 2M–2R). At the level of the brainstem, olivary nuclei were fragmented or extremely hypoplastic (Figures 2G–2L) and pons nuclei appeared smaller (not shown). By contrast, pyramidal tracts were preserved. Histological examination of the cerebellar cortex, with a immunostaining against CALB to label Purkinje cells, revealed the presence of a disorganized Purkinje cell layer at 24 GW and 31 GW (Figures 2W–2Z) and a decreased density of these cells at 31 GW and 37 GW (Figures 2Y–2AB). Additionally, multiple Purkinje cells heterotopia are detected in the white-matter (Figure 2T, top-left inset) for all the affected fetuses and infant.
PRDM13 is expressed in neural stem cells of the hindbrain in mouse and human embryos
During mouse development, Prdm13 was shown to be necessary to balance neuronal fates in the dorsal neural tube and in the retina13,15,16 but no defined role of this factor was described during hindbrain development. Interestingly, the expression of Prdm13 in the neural tube is under the control of Ptf1a.13 Ptf1a is a bHLH (basic helix-loop-helix) proneural transcriptional factor and determines the fate of progenitors of the cerebellar ventricular zone (VZ),32, 33, 34 which give rise to all cerebellar GABAergic neurons (Purkinje cells and inhibitory interneurons) and astrocytes. In addition, Ptf1a is necessary for the development of several brainstem nuclei, including the inferior olivary nucleus (ION).35,36 In order to better appreciate the expression of PRDM13 during central nervous system development, we explored available transcriptomic datasets and performed ISH experiments. Analyzing a published scRNA-seq dataset from the mouse embryonic cerebellum at E13.5,23 we observed that Prdm13 is expressed in cell clusters corresponding to cerebellar progenitors of the VZ (Figure S1A) and co-expressing Ptf1a (Figures S1B and S1C). This confirmed a previous report showing expression of Prdm13 in the mouse cerebellar VZ at E11.5.13 To further demonstrate the link between PRDM13 mutations and hindbrain developmental anomalies, we examined its expression during human brain development. We first exploited publicly available snRNA-seq datasets from fetal cerebellum samples (9–21 post-conception weeks [PCW])25 and observed an enriched expression of PRDM13 in specific cell clusters (Figure 3A). These clusters are located at the crossroad between cell clusters annotated as fully differentiated cells that are Purkinje cells, inhibitory interneurons, and astrocytes. These clusters also show an enriched expression of both PTF1A and the neural stem cell marker SOX2, suggesting they might correspond to VZ progenitors (Figure 3A). To get a comprehensive picture of the spatio-temporal pattern of PRDM13 expression during human brain development, we performed ISH on sections of human embryonic and fetal samples by using the highly-sensitive and specific RNAscope technology. Considering the early expression of PRDM13 seen in the embryonic mouse cerebellum, we studied PRDM13 expression over an extended temporal window from Carnegie stage 13 (CS13, corresponding to ∼32 days post conception [dpc]) to 15 PCW. At CS13, we detected specific expression of PRDM13 in the dorsal neural tube, similarly to the described expression pattern in mice (Figures 3B and 3B″). We also detected PRDM13 transcripts in dorsal ventricular domains of the hindbrain (Figures 3B and 3B′). These expression domains are maintained at CS21 (∼51 dpc) (Figures 3C, 3C″, 3D, and 3D″). At this stage, additional expression sites become apparent, and notably, a strong expression is detected in the cerebellum at the level of the ventricular zone (Figures 3C, 3C′, 3D, and 3D′). This cerebellar expression of PRDM13 is quite transient, as it faints already at 10 PCW (Figure S1D); at this stage PRDM13 expression is maintained only in scattered cells of the subventricular zone (Figure S1D′). Expression of PRDM13 was no longer detected in the cerebellum at 15 PCW (Figure S1E). We also detected expression of PRDM13 in the inner layer of the developing retina (Figures S1F and S1F′), consistent with the described role of Prdm13 in the development of amacrine cells in mice.15 Altogether, our data show that PRDM13 is expressed in human progenitors of the cerebellum and hindbrain at an early neurogenic phase. This result further argues for the implication of this gene in olivopontocerebellar hypoplasia and points to a pathological mechanism involving neuronal fate misspecification. Consistent with the absence of obvious supratentorial anomalies in the individuals investigated in this study, no expression of PRDM13 was detected in the forebrain and the midbrain, except for the hypothalamus (Figures S1G and S1G′).
Figure 3.

PRDM13 expression in neural progenitors in the developing cerebellum and brainstem
(A) Single-nucleus RNA-seq (snRNA-seq) data from human fetal cerebellum (9–21 PCW). snRNA-seq datasets were retrieved from the Human Gene Expression During Development Atlas25 (Descartes, https://descartes.brotmanbaty.org) and analyzed with the Seurat package. Cells are visualized on uniform manifold approximation and projection (UMAP) plots. In the first panel, cells are color-coded according to cell annotations from the Descartes Atlas (PCs, Purkinje cells; UBCs, unipolar brush cells; GCs, granule cells; INs, interneurons; Astros, Astrocytes; OPCs, oligodendrocytes precursors; μglia, microglia; SPs, SLC24A4_PEX5L_positive cells; Endos, vascular endothelial cells). In the three other panels, cells are colored in graded intensities, reflecting the expression levels of PRDM13, PTF1A, and SOX2.
(B–D) Detection of PRDM13 transcripts (red) by RNAscope in situ hybridization on coronal (B and C) or sagittal (D) sections through human embryos at Carnegie stage 13 (B) and 21 (C and D). (B′) and (B″), (C′) and (C″), (D′) and (D′) are higher magnifications of the regions outlined by dotted squares in (B), (C), and (D), respectively. At CS13, specific expression of PRDM13 is detected in the dorsal VZ of the caudal part of the hindbrain (Hb, B′) and in the dorsal VZ of the caudal neural tube (NT, B″). At CS21, PRDM13 starts to be expressed in the primordium of the cerebellum (Cb, C, C′) while being maintained in the medulla oblongata (MO, C, D, D″) and spinal cord (SC, C, C″). In the cerebellum (D′), PRDM13 is more specifically detected in the ventricular zone (VZ, black arrowheads), while it is absent from the rhombic lip (RL, white arrows). Scale bars: 100 μm (B) and 500 μm (C and D).
Homozygous disruption of prdm13 causes hindbrain developmental defects in zebrafish
To validate the implication of PRDM13 in hindbrain development, we took advantage of the zebrafish. Its cerebellar development relies on homologous transcription factors37 and its genome includes a unique ortholog of PRDM13. To further validate this model, we first analyzed the expression of prdm13 during zebrafish brain development by using ISH. At 24 hpf, we detected specific prdm13 expression in rhombomeres 2–7, with a higher intensity in the ventricular zone and in HuC-positive differentiating neurons of rhombomere 7 (r7) (Figures 4A and 4B, right panel). This expression in the developing hindbrain, particularly enriched in r7, was also confirmed by the analysis of available scRNA-seq data from 24 hpf zebrafish embryos24 (Figures S2A and S2B). Of note, dorsal progenitors from r7, expressing ptf1a, were recently shown to give rise to neurons of inferior olivary nuclei.35,38 Additionally, scRNA-seq data revealed expression of zebrafish prdm13 in pharyngeal arch cells at this stage (Figures S2A and S2B). At 48 hpf, we could detect expression of prdm13 in the cerebellar primordium located in the dorsal part of r1 (Figure 4C). The expression of prdm13 in the cerebellar VZ is maintained at larval stages (Figure 4E) and is very similar to the expression of ptf1a (Figure 4F). prdm13 transcripts are located in VZ neural progenitors expressing Sox2 and PCNA and also at least transiently in differentiating cells exiting the VZ (Figures 4C and 4G). Analysis of scRNA-seq data from 5 dpf larval brain also highlighted the expression of prdm13 in cell clusters annotated as “neural progenitors” and co-expressing ptf1a and sox2 (Figures 4H and S2E). The expression of prdm13 in the cerebellar and hindbrain progenitors is maintained in the 2-month-old juvenile zebrafish brain (Figures S2F and S2F′), which is probably related to the maintained neurogenic activity of these regions beyond developmental stages in zebrafish. As in human embryos, we also observed additional expression sites for prdm13 in the developing eye and hypothalamus (Figures 4H and S2E). Altogether, our results demonstrate that prdm13 expression pattern is highly conserved across vertebrates and is consistent with an early role in hindbrain neurogenesis. It also validated the zebrafish as an appropriate model for functional studies of PRDM13-related neurodevelopmental anomalies.
Figure 4.

prdm13 expression during zebrafish hindbrain development
(A) Lateral view of a 3D reconstruction of the zebrafish hindbrain at 24 hpf showing the expression of prdm13 (ISH, magenta) with a whole-mount immunostaining for the neuronal marker HuC (green). Cell nuclei are counterstained with DAPI (blue). A dotted line contours the hindbrain region. Cb, cerebellum; rh1–7, rhombomere 1–7; ov, otic vesicle. Scale bar: 50 μm.
(B) Transverse section through the 3D reconstruction shown in (A) at the level of rhombomere 3 (left panel) and rhombomere 7 (right panel). Scale bar: 50 μm.
(C) Optical z-plane showing prdm13 expression (magenta) together with an immunostaining for Sox2 (blue) and HuC (green) in the cerebellum at 48 hpf. Cell nuclei are counterstained with DAPI (gray). At this stage, prdm13 starts to be expressed in the cerebellar primordium, corresponding to the dorsal part of the first rhombomere, adjacent to the midbrain-hindbrain boundary (MHB). OT, optic tectum. Scale bar: 20 μm.
(D) Schematic representation of the larval zebrafish brain at 6 days post fertilization (dpf), with a magnification on the larval cerebellum. Like in mammals, the zebrafish cerebellum develops from two distinct neurogenic progenitor pools: the rhombic lip (RL, green) and the ventricular zone (VZ, purple). The VZ gives rise to all the GABAergic neuronal populations, including Purkinje neurons, which migrate and align to form the Purkinje cell layer (PCL).
(E and F) Whole-mount ISH on zebrafish larval brains at 6 dpf showing the expression of prdm13 (E) and ptf1a (F) in blue. Note the expression of prdm13 and ptf1a in the cerebellar VZ (black arrowheads) and dorsal hindbrain (black arrows). Cb, cerebellum; OT, optic tectum; Tel, telencephalon; Hyp, hypothalamus; MO: medulla oblongata. Scale bar: 100 μm.
(G) Lateral view of a 3D reconstruction of the zebrafish cerebellum at 6 dpf showing the expression of prdm13 (ISH, magenta) with a whole-mount immunostaining for the proliferation marker PCNA (green). Cell nuclei are counterstained with DAPI (blue). Dotted lines outline the cerebellar primordium. prdm13 transcripts are detected in proliferating progenitors of the VZ (white arrowheads). Scale bar: 10 μm.
(H) t-distributed stochastic neighbor embedding (t-SNE) projection plots of distinct single-cell populations obtained from the zebrafish brain at 5 dpf. The zebrafish single-cell RNA-seq (scRNA-seq) dataset was retrieved from the Gene Expression Omnibus (GEO) public database under accession GEO: GSE158142.24 In the first panel, cells are color-coded according to cluster annotations from the original publication.24 In the three other panels, cells are colored in graded intensities, reflecting the expression levels of prdm13, ptf1a, and sox2. Clusters in which prdm13 expression is enriched are circled with dotted lines. prdm13 expression is enriched in clusters 13, 25, 30, and 38, corresponding to neural progenitors (NPCs) that are also enriched for ptf1a and sox2. Expression of prdm13 is also detected in clusters 1, 34, and 35, which are annotated as hypothalamic clusters (Hyp); 21, which corresponds to amacrine cells (ACs); and 45, which correspond to Müller glia (MG).
We thus analyzed the phenotype of a zebrafish homozygous mutant for prdm13, available from the Zebrafish Mutation Project (sa16464).28 The mutant allele is a point mutation that creates a premature stop codon and leads to a truncation of the Zn fingers domains, comparable to the mutations identified in families 1, 2, and 4 (Figure 5A). Homozygous mutant embryos prdm13sa1646/ sa16464 (hereafter referred to as prdm13−/−), obtained from a cross between heterozygous prdm13sa16464/+ fish (prdm13+/−), survive at least until 7 dpf, when larval lethality begins to be observed. Genotyping individual larvae demonstrated a distortion of the Mendelian distribution at 9 dpf (Chi-square test; p = 0.0002) due to the death of prdm13−/− mutant larvae (Figure 5B). Although prdm13−/− larvae have a roughly normal morphology, we noted that they present an abnormal body curvature and lower jaw morphology (Figure 5C). We then explored their brain phenotype, more particularly the cerebellum and posterior hindbrain. We fluorescently labeled cerebellar Purkinje cells with an antibody against Pvalb7, a validated marker in this species,39 and quantified these cells in 3D reconstructions (Figure 5D). We could show that the number of Purkinje cells was significantly reduced in prdm13−/− larvae when compared to wild-type prdm13+/+ larvae (Figure 5E; n = 5 [prdm13−/−] and n = 6 [prdm13+/+]; unpaired t test p = 0.011). In contrast, other cell populations such as CALB+ Eurydendroid cells (ECs), functionally equivalent of deep cerebellar nuclei in mammals,37 were not affected (Figure 5F; n = 3 [prdm13−/−] and n = 3 [prdm13+/+]; unpaired t test p = 0.7854). We also labeled ION neurons with an antibody against LHX1 in wild-type and mutant larvae (Figure 5G) and observed a total absence of these neurons in prdm13−/− larvae. Altogether, these data indicate that homozygous disruption of prdm13 in zebrafish leads to a late lethality in larvae, associated with a specific reduction in Purkinje cell numbers and an absence of ION neurons (Figure 5H). This phenotype shares striking similarities with the clinical features and neuropathological findings in the affected child and fetuses with bi-allelic PRDM13 mutations, further corroborating their causative role in the pathology.
Figure 5.

Homozygous prdm13 disruption causes reduction in Purkinje neurons numbers and loss of inferior olive nucleus neurons in zebrafish
(A) Zebrafish mutant allele sa16484. The mutant allele is a nonsense single-nucleotide mutation, leading to a truncation of the protein at the level of the first Zn finger domain.
(B) Proportion of embryos of each genotype inside the progeny of a cross between two heterozygous fish from the prdm13sa16484 line at different stages. The number of embryos genotyped per stage is indicated underneath each bar. At 9 dpf, the proportions differ significantly from the expected ratios (Chi-square test; p = 0.0002).
(C) Photographs of wild-type and homozygous mutant larvae from the prdm13sa16484 line at 7 dpf. (C′) and (C″) are higher magnification of the head regions of a wild-type (C′) and homozygous mutant (C″) larva. Homozygous prdm13 mutant larvae display an abnormal body curvature and lower jaw morphology (arrow in C″). Scale bar: 500 μm.
(D) 3D reconstructions of the hindbrain in dorsal view from a wild-type (top panels) and a homozygous mutant (bottom panels) larva at 6 dpf. Larval brains were immunostained for parvalbumin (PARV, green), a marker of Purkinje cells (PCs), and calbindin (CALB, magenta), a marker of some eurydendroid cells (ECs). The panels on the right show a higher magnification of the cerebellum; the last ones show the distribution of PCs (green) and ECs (magenta), as determined by 3D-image segmentation of the cerebellar region. Scale bar: 50 μm,
(E and F) Total number of PARV+ PCs (E) and CALB+ ECs (F) in wild-type and mutant prdm13 larvae. Data are presented as mean ± 95% confidence interval. The number of PCs is significantly reduced in mutant larvae (n = 5 (prdm13+/+) and n = 6 (prdm13−/−); unpaired t test p = 0.011), while no significant changes in EC numbers are observed (n = 3 (prdm13−/−) and n = 3 (prdm13+/+); unpaired t test p = 0.7854).
(G) Representative images of 3D reconstructions of the hindbrain from a wild-type (top panels) and a homozygous mutant (bottom panels) larva at 7 dpf. Larval brains were immunostained for LHX1 (green), which labels inferior olivary nucleus projection neurons (white arrow). The left and middle panels are lateral views and the right panels are ventral views centered on the inferior olivary nuclei (IONs). Note the absence of ION neurons in mutant larvae (four larvae were immunostained for each genotype). Scale bar: 20 μm.
(H) Schematic representation of the phenotype of prdm13 mutant larvae, which present a reduction in the number of Purkinje cells (red) and an absence of ION projection neurons (green), which normally send climbing fibers onto PCs.
Discussion
Several PRDM transcription factors are already known to be involved in central nervous system development and diseases. PRDM8 (MIM: 616639) regulates cadherin-11 to ensure proper neural circuit formation40 and is mutated in progressive myoclonic epilepsy-10 (EPM10 [MIM: 616640]). PRDM12 [MIM: 616458] plays a role in sensory neuron perception and is mutated in hereditary sensory and autonomic neuropathy type VIII (HSAN8 [MIM: 616488]). PRDM15 [MIM: 617692] is mutated in syndromes with neurodevelopmental defects and a progressive nephropathy.41 All these genes play a critical role in the development as illustrated by the embryonic lethality or severe neurodevelopmental phenotypes of their respective knockout mouse mutants. Likewise, the knockout of Prdm13 in mouse, as well as a deletion of its three last Zn finger domains, cause neonatal death, although the cause of this death has not been investigated.16 However, until recently, the only congenital disorders implicating PRDM13 were macular dystrophies. Tandem duplications and single-nucleotide variants in the non-coding region of PRDM13 have thus been reported to cause North Carolina macular dystrophies (NCMD [MIM: 136550]) and other similar autosomal dominant retinal dystrophies.42,43 The implication of PRDM13 in these pathologies is most likely linked to the highly specific expression of PRDM13 in the inner layer of the embryonic human retina that we report in this study and with the role of Prdm13 in the specification of a subset of amacrine cells demonstrated in mice.15 The precise molecular mechanism underlying these PRDM13-associated retinopathies is unknown but is suspected to result from an upregulation of PRDM13.44 This is compatible with our current findings, as we demonstrate that loss-of-function variants cause a very different clinical phenotype. Visual acuity could not be assessed in most of the affected individuals with truncating variants, as they died early during infancy and eye histological examination was not performed. The eldest sister from family 3, carrying a missense mutation and presenting with milder clinical features had a normal fundus exam, although she presented with high myopia. As we show that PRDM13 is expressed in multiple brain regions that are not affected in NCMD, it is also possible that NCMD-causing variants lead to eye-specific deregulations of PRDM13 expression.
During the preparation of this manuscript, Whittaker et al.17 reported a recessive PRDM13 mutation associated with hypogonadotropic hypogonadism syndrome. The three affected individuals are adult or adolescent presenting with scoliosis, sexual development defects, and moderate intellectual deficit. Two of them also present with cerebellar hypoplasia. The authors of this study hypothesized that the mutation identified, a deletion encompassing a splice acceptor site, leads to a truncation of the four Zn finger domains and cause the phenotype. This would be at odds with the lethal phenotype observed in the mouse mutant with a deletion of the Zn finger domains.16 Then, using a different Prdm13 mouse mutant, deleted for the PR domain, Whittaker et al. demonstrated a role for Prdm13 in cerebellar development but without finding any locomotor or Purkinje cell differentiation defect.
Here, we present multiple important differences with the study of Whittaker al. First, we report a significantly different syndrome based on eight individuals from four families of different origins. This syndrome associates perinatal lethality with brainstem dysfunctions (e.g., respiratory and swallowing defects) and/or severe brainstem malformation, together with persistent cerebellar hypoplasia. Consistently with the previously published mouse data, and further demonstrating the critical requirement of the Zn finger domains, three out of the four families carry mutations of PRDM13 that truncate these domains and the last is predicted to disrupt one of them. Using a zebrafish mutant lacking the four Zn finger domains, we further validated the critical need for PRDM13 during brainstem development in vertebrates. Second, we demonstrate that PRDM13 shows a strong and spatio-temporally restricted expression pattern during early human hindbrain development, explaining why Whittaker et al. can barely detect PRDM13 expression in CS23-stage human fetus. Third, correlations of human and zebrafish data from samples with truncation of PRDM13 demonstrate the requirement of this gene for Purkinje cell differentiation. It can be speculated that the clinical differences observed between the two studies come from the specific mutation identified by Whittaker et al. that might alter splicing but still allows for the production of some functional transcripts. However, the exact consequence of this splicing mutation on PRDM13 transcripts remains to be assessed. The partial overlap with our clinical findings, with the presence of cerebellar hypoplasia and global developmental delay, suggests that this variant might be a hypomorphic allele. We note that affected individuals carrying missense variants (i.e., family 3) also present with milder clinical phenotypes in our study. The presence of the three other functional Zn fingers might be sufficient to maintain the functionality of the protein to a certain degree. However, unlike the affected individuals from the aforementioned study, they do not present scoliosis, although postural kyphosis has been detected in the child F.3-1. Clinical follow-up of the very young affected children from family 3 will help to determine whether impaired sexual development, hypogonadism, and progressive scoliosis are consistent clinical features associated with partial loss of function of PRDM13.
Among the clinical features in affected individuals with PRDM13 mutation, some variable features are observed (e.g., cardiac malformation in family 1, see Table 3) and could involve recessive variants other than PRDM13 mutations.45 Differently, epilepsy or seizures have been observed in several members of families 3 and 4. Epilepsy is also present in typical PCH cases that are generally associated with mutations in ubiquitously expressed genes (e.g., genes involved in tRNA processing).7 Here, PRDM13 has a very restricted expression pattern and a direct impact of its mutations in tissues where it is not detected, such as the cerebral cortex, is unlikely. However, a protective role of the cerebellum, through the inhibition of aberrant discharges that lead to cortical seizures, has been suggested46 and so an indirect contribution of the cerebellar malformation to this epilepsy phenotype could be speculated.
Using a meta-analysis of scRNA-seq data as well as a detailed expression study performed during zebrafish hindbrain development, we could define original molecular hypotheses to understand PRDM13’s role in brainstem and cerebellar Purkinje cells development. Several lines of evidence support the idea that these defects result from cell-autonomous neuronal misspecification. Indeed, we show that prdm13 is expressed, together with ptf1a, in progenitors of the cerebellar VZ and dorsal ventricular zone of rhombomere 7. These Ptf1a+ progenitor domains have been demonstrated to generate, respectively, Purkinje cells and ION neurons.32,35,36,38,47 Besides, Ptf1a is required cell autonomously for proper identity specification in neuronal precursors in the retina, spinal cord, cerebellum, and posterior hindbrain.32,35,36,48,49 In the dorsal spinal cord, Ptf1a loss of function thus induces a loss of GABAergic neurons.49 Part of Ptf1a function is mediated by the transcriptional activation of its direct target Prdm13, which in turn inhibits the expression of determinants of the alternative glutamatergic fate, such as Tlx1/3 and Lmx1b.13,16 Accordingly, the Prdm13 loss-of-function phenotype in the spinal cord resembles the one observed in Ptf1a null mutants, albeit less severe, as it affects mostly early-born neurons.16 In the zebrafish cerebellum, we show that the phenotype of prdm13 loss-of-function mutant is comparable but milder to ptf1a mutant.38 This is also the case in humans, as PTF1A loss of function leads to a much more severe cerebellar hypoplasia than the one we observed in presence of PRDM13 mutations. Altogether, these data are in line with the hypothesis that, in the developing cerebellum and ION, PRDM13 plays a cell-autonomous role in neural progenitors fate specification, downstream of PTF1A.
Our findings also suggest that PRDM13 loss of function could impact cerebellar development both directly and indirectly by impairing the development of key afferent pathways of the cerebellum. Indeed, inferior olivary nuclei are the origins of major cerebellar inputs that influence Purkinje cells maturation.50 Interestingly, lesions, size or functional abnormalities of key nuclei of the olivo-cerebellar complex have been observed in sudden infant death syndrome (SIDS).51, 52, 53 Therefore, a defect in these structures could be directly involved in the respiratory symptoms observed in SIDS and in individuals with PRDM13 mutations. Alternatively, other brainstem nuclei could also be affected. Indeed, in mice, Ptf1a loss of function also affects the neuronal composition of brainstem nuclei involved in somatosensory and viscerosensory control circuits.36 Among these nuclei, Ptf1a is required for the proper development of the nucleus of the solitary tract that is a major sensory nucleus of the dorsal medulla oblongata receiving cardiorespiratory afferent inputs among others. Considering the likely involvement of Prdm13 in these developmental processes, its disruption may also impair the development of these nervous centers and thereby explain the various brainstem dysfunctions shared by affected individuals with bi-allelic PRDM13 mutations.
Like the cerebral cortex, the cerebellum is a brain region that has undergone during evolution a strong expansion and complexification in the human lineage. Recent studies in human early embryos showed that cerebellar germinal zones present some human-specific features.54 Notably, as in the cerebral cortex, an expanded subventricular proliferation zone (SVZ) is detected above the VZ in the developing human cerebellum. However, little is known about the diversity of human VZ/SVZ progenitors and the molecular mechanisms involved in this expansion. Our study highlights how defects in the transcriptional regulation of these early human-tailored neurogenic steps can contribute to severe neurodevelopmental disorders of the brainstem and cerebellum. Exploring further this early transcriptional network in a human context55 will be of high interest to understand these pathologies.
Acknowledgments
The project is funded by the Agence Nationale de la Recherche ANR-16-CE12- 0005-01, the Fondation pour la Recherche Médicale FRM-DEQ20160334938, and the patient association CSC “Connaître les syndromes Cérébelleux.” This work was also supported by state funding from the Agence Nationale de la Recherche under “Investissements d’avenir” program (ANR-10-IAHU-01), the Fondation Bettencourt Schueller, and the MSDAvenir Fund (DEVO-DECODE project). The human embryonic and fetal material was provided by the Joint MRC-Wellcome (MR/R006237/1) Human Developmental Biology Resource (https://www.hdbr.org). We would like to thank S. Amat, F. Pelluard, and I. Deryabin for providing clinical information and material; M. Hibi (Nagoya University) for providing the zebrafish ptf1a probe; B. Crespo for technical help; and L. Colleaux for guidance during the course of the project.
Declaration of interests
The authors declare no competing interests.
Published: April 6, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2022.03.010.
Contributor Information
Marion Coolen, Email: marion.coolen@inserm.fr.
Vincent Cantagrel, Email: vincent.cantagrel@inserm.fr.
Data and code availability
This study did not generate datasets or code.
Web resources
dbSNP database, https://www.ncbi.nlm.nih.gov/snp/?cmd=search
Ensembl genome assembly GRCh37, http://grch37.ensembl.org/Homo_sapiens/Info/Index
ESP database, https://evs.gs.washington.edu/EVS/
Gene Expression Omnibus database, https://www.ncbi.nlm.nih.gov/geo/
Human single-cell RNA-seq dataset, Descartes, https://descartes.brotmanbaty.org
Online Mendelian Inheritance in Man, https://www.omim.org
Supplemental information
References
- 1.Accogli A., Addour-Boudrahem N., Srour M. Diagnostic Approach to Cerebellar Hypoplasia. Cerebellum. 2021;20:631–658. doi: 10.1007/s12311-020-01224-5. [DOI] [PubMed] [Google Scholar]
- 2.Poretti A., Boltshauser E., Doherty D. Cerebellar hypoplasia: differential diagnosis and diagnostic approach. Am. J. Med. Genet. C. Semin. Med. Genet. 2014;166C:211–226. doi: 10.1002/ajmg.c.31398. [DOI] [PubMed] [Google Scholar]
- 3.Haldipur P., Millen K.J. What cerebellar malformations tell us about cerebellar development. Neurosci. Lett. 2019;688:14–25. doi: 10.1016/j.neulet.2018.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aldinger K.A., Dempsey J.C., Tully H.M., Grout M.E., Mehaffey M.G., Dobyns W.B., Doherty D. Rhombencephalosynapsis: Fused cerebellum, confused geneticists. Am. J. Med. Genet. C. Semin. Med. Genet. 2018;178:432–439. doi: 10.1002/ajmg.c.31666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ucuncu E., Rajamani K., Wilson M.S.C., Medina-Cano D., Altin N., David P., Barcia G., Lefort N., Banal C., Vasilache-Dangles M.T., et al. MINPP1 prevents intracellular accumulation of the chelator inositol hexakisphosphate and is mutated in Pontocerebellar Hypoplasia. Nat. Commun. 2020;11:6087. doi: 10.1038/s41467-020-19919-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rüsch C.T., Bölsterli B.K., Kottke R., Steinfeld R., Boltshauser E. Pontocerebellar Hypoplasia: a Pattern Recognition Approach. Cerebellum. 2020;19:569–582. doi: 10.1007/s12311-020-01135-5. [DOI] [PubMed] [Google Scholar]
- 7.van Dijk T., Baas F., Barth P.G., Poll-The B.T. What’s new in pontocerebellar hypoplasia? An update on genes and subtypes. Orphanet J. Rare Dis. 2018;13:92. doi: 10.1186/s13023-018-0826-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akizu N., Cantagrel V., Schroth J., Cai N., Vaux K., McCloskey D., Naviaux R.K., Van Vleet J., Fenstermaker A.G., Silhavy J.L., et al. AMPD2 regulates GTP synthesis and is mutated in a potentially treatable neurodegenerative brainstem disorder. Cell. 2013;154:505–517. doi: 10.1016/j.cell.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mochida G.H., Ganesh V.S., de Michelena M.I., Dias H., Atabay K.D., Kathrein K.L., Huang H.T., Hill R.S., Felie J.M., Rakiec D., et al. CHMP1A encodes an essential regulator of BMI1-INK4A in cerebellar development. Nat. Genet. 2012;44:1260–1264. doi: 10.1038/ng.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marin-Valencia I., Gerondopoulos A., Zaki M.S., Ben-Omran T., Almureikhi M., Demir E., Guemez-Gamboa A., Gregor A., Issa M.Y., Appelhof B., et al. Homozygous Mutations in TBC1D23 Lead to a Non-degenerative Form of Pontocerebellar Hypoplasia. Am. J. Hum. Genet. 2017;101:441–450. doi: 10.1016/j.ajhg.2017.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aldinger K.A., Doherty D. The genetics of cerebellar malformations. Semin. Fetal Neonatal Med. 2016;21:321–332. doi: 10.1016/j.siny.2016.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sellick G.S., Barker K.T., Stolte-Dijkstra I., Fleischmann C., Coleman R.J., Garrett C., Gloyn A.L., Edghill E.L., Hattersley A.T., Wellauer P.K., et al. Mutations in PTF1A cause pancreatic and cerebellar agenesis. Nat. Genet. 2004;36:1301–1305. doi: 10.1038/ng1475. [DOI] [PubMed] [Google Scholar]
- 13.Chang J.C., Meredith D.M., Mayer P.R., Borromeo M.D., Lai H.C., Ou Y.H., Johnson J.E. Prdm13 mediates the balance of inhibitory and excitatory neurons in somatosensory circuits. Dev. Cell. 2013;25:182–195. doi: 10.1016/j.devcel.2013.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanotel J., Bessodes N., Thélie A., Hedderich M., Parain K., Van Driessche B., Brandão Kde.O., Kricha S., Jorgensen M.C., Grapin-Botton A., et al. The Prdm13 histone methyltransferase encoding gene is a Ptf1a-Rbpj downstream target that suppresses glutamatergic and promotes GABAergic neuronal fate in the dorsal neural tube. Dev. Biol. 2014;386:340–357. doi: 10.1016/j.ydbio.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 15.Watanabe S., Sanuki R., Sugita Y., Imai W., Yamazaki R., Kozuka T., Ohsuga M., Furukawa T. Prdm13 regulates subtype specification of retinal amacrine interneurons and modulates visual sensitivity. J. Neurosci. 2015;35:8004–8020. doi: 10.1523/JNEUROSCI.0089-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mona B., Uruena A., Kollipara R.K., Ma Z., Borromeo M.D., Chang J.C., Johnson J.E. Repression by PRDM13 is critical for generating precision in neuronal identity. eLife. 2017;6:e25787. doi: 10.7554/eLife.25787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whittaker D.E., Oleari R., Gregory L.C., Le Quesne-Stabej P., Williams H.J., Torpiano J.G., Formosa N., Cachia M.J., Field D., Lettieri A., et al. A recessive PRDM13 mutation results in congenital hypogonadotropic hypogonadism and cerebellar hypoplasia. J. Clin. Invest. 2021;131:e141587. doi: 10.1172/JCI141587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobreira N., Schiettecatte F., Valle D., Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat. 2015;36:928–930. doi: 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chemin J., Siquier-Pernet K., Nicouleau M., Barcia G., Ahmad A., Medina-Cano D., Hanein S., Altin N., Hubert L., Bole-Feysot C., et al. De novo mutation screening in childhood-onset cerebellar atrophy identifies gain-of-function mutations in the CACNA1G calcium channel gene. Brain. 2018;141:1998–2013. doi: 10.1093/brain/awy145. [DOI] [PubMed] [Google Scholar]
- 20.Le Fevre A., Baptista J., Ellard S., Overton T., Oliver A., Gradhand E., Scurr I. Compound heterozygous Pkd1l1 variants in a family with two fetuses affected by heterotaxy and complex Chd. Eur. J. Med. Genet. 2020;63:103657. doi: 10.1016/j.ejmg.2019.04.014. [DOI] [PubMed] [Google Scholar]
- 21.Al-Thihli K., Afting C., Al-Hashmi N., Mohammed M., Sliwinski S., Al Shibli N., Al-Said K., Al-Kasbi G., Al-Kharusi K., Merle U., et al. Deficiency of acyl-CoA synthetase 5 is associated with a severe and treatable failure to thrive of neonatal onset. Clin. Genet. 2021;99:376–383. doi: 10.1111/cge.13883. [DOI] [PubMed] [Google Scholar]
- 22.Aldinger K.A., Thomson Z., Phelps I.G., Haldipur P., Deng M., Timms A.E., Hirano M., Santpere G., Roco C., Rosenberg A.B., et al. Spatial and cell type transcriptional landscape of human cerebellar development. Nat. Neurosci. 2021;24:1163–1175. doi: 10.1038/s41593-021-00872-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wizeman J.W., Guo Q., Wilion E.M., Li J.Y. Specification of diverse cell types during early neurogenesis of the mouse cerebellum. eLife. 2019;8:e42388. doi: 10.7554/eLife.42388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raj B., Farrell J.A., Liu J., El Kholtei J., Carte A.N., Navajas Acedo J., Du L.Y., McKenna A., Relic D., Leslie J.M., et al. Emergence of Neuronal Diversity during Vertebrate Brain Development. Neuron. 2020;108:1058–1074.e6. doi: 10.1016/j.neuron.2020.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao J., O’Day D.R., Pliner H.A., Kingsley P.D., Deng M., Daza R.M., Zager M.A., Aldinger K.A., Blecher-Gonen R., Zhang F., et al. A human cell atlas of fetal gene expression. Science. 2020;370:eaba7721. doi: 10.1126/science.aba7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kettleborough R.N., Busch-Nentwich E.M., Harvey S.A., Dooley C.M., de Bruijn E., van Eeden F., Sealy I., White R.J., Herd C., Nijman I.J., et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature. 2013;496:494–497. doi: 10.1038/nature11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thisse B., Thisse C. In situ hybridization on whole-mount zebrafish embryos and young larvae. Methods Mol. Biol. 2014;1211:53–67. doi: 10.1007/978-1-4939-1459-3_5. [DOI] [PubMed] [Google Scholar]
- 30.Lee M.S., Gippert G.P., Soman K.V., Case D.A., Wright P.E. Three-dimensional solution structure of a single zinc finger DNA-binding domain. Science. 1989;245:635–637. doi: 10.1126/science.2503871. [DOI] [PubMed] [Google Scholar]
- 31.Barth P.G. Pontocerebellar hypoplasias. An overview of a group of inherited neurodegenerative disorders with fetal onset. Brain Dev. 1993;15:411–422. doi: 10.1016/0387-7604(93)90080-r. [DOI] [PubMed] [Google Scholar]
- 32.Hoshino M., Nakamura S., Mori K., Kawauchi T., Terao M., Nishimura Y.V., Fukuda A., Fuse T., Matsuo N., Sone M., et al. Ptf1a, a bHLH transcriptional gene, defines GABAergic neuronal fates in cerebellum. Neuron. 2005;47:201–213. doi: 10.1016/j.neuron.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 33.Pascual M., Abasolo I., Mingorance-Le Meur A., Martínez A., Del Rio J.A., Wright C.V., Real F.X., Soriano E. Cerebellar GABAergic progenitors adopt an external granule cell-like phenotype in the absence of Ptf1a transcription factor expression. Proc. Natl. Acad. Sci. USA. 2007;104:5193–5198. doi: 10.1073/pnas.0605699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Millen K.J., Steshina E.Y., Iskusnykh I.Y., Chizhikov V.V. Transformation of the cerebellum into more ventral brainstem fates causes cerebellar agenesis in the absence of Ptf1a function. Proc. Natl. Acad. Sci. USA. 2014;111:E1777–E1786. doi: 10.1073/pnas.1315024111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamada M., Terao M., Terashima T., Fujiyama T., Kawaguchi Y., Nabeshima Y., Hoshino M. Origin of climbing fiber neurons and their developmental dependence on Ptf1a. J. Neurosci. 2007;27:10924–10934. doi: 10.1523/JNEUROSCI.1423-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iskusnykh I.Y., Steshina E.Y., Chizhikov V.V. Loss of Ptf1a Leads to a Widespread Cell-Fate Misspecification in the Brainstem, Affecting the Development of Somatosensory and Viscerosensory Nuclei. J. Neurosci. 2016;36:2691–2710. doi: 10.1523/JNEUROSCI.2526-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hibi M., Matsuda K., Takeuchi M., Shimizu T., Murakami Y. Evolutionary mechanisms that generate morphology and neural-circuit diversity of the cerebellum. Dev. Growth Differ. 2017;59:228–243. doi: 10.1111/dgd.12349. [DOI] [PubMed] [Google Scholar]
- 38.Itoh T., Takeuchi M., Sakagami M., Asakawa K., Sumiyama K., Kawakami K., Shimizu T., Hibi M. Gsx2 is required for specification of neurons in the inferior olivary nuclei from Ptf1a-expressing neural progenitors in zebrafish. Development. 2020;147:dev190603. doi: 10.1242/dev.190603. [DOI] [PubMed] [Google Scholar]
- 39.Hamling K.R., Tobias Z.J., Weissman T.A. Mapping the development of cerebellar Purkinje cells in zebrafish. Dev. Neurobiol. 2015;75:1174–1188. doi: 10.1002/dneu.22275. [DOI] [PubMed] [Google Scholar]
- 40.Ross S.E., McCord A.E., Jung C., Atan D., Mok S.I., Hemberg M., Kim T.K., Salogiannis J., Hu L., Cohen S., et al. Bhlhb5 and Prdm8 form a repressor complex involved in neuronal circuit assembly. Neuron. 2012;73:292–303. doi: 10.1016/j.neuron.2011.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mzoughi S., Di Tullio F., Low D.H.P., Motofeanu C.M., Ong S.L.M., Wollmann H., Wun C.M., Kruszka P., Muenke M., Hildebrandt F., et al. PRDM15 loss of function links NOTCH and WNT/PCP signaling to patterning defects in holoprosencephaly. Sci. Adv. 2020;6:eaax9852. doi: 10.1126/sciadv.aax9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Small K.W., DeLuca A.P., Whitmore S.S., Rosenberg T., Silva-Garcia R., Udar N., Puech B., Garcia C.A., Rice T.A., Fishman G.A., et al. North Carolina Macular Dystrophy Is Caused by Dysregulation of the Retinal Transcription Factor PRDM13. Ophthalmology. 2016;123:9–18. doi: 10.1016/j.ophtha.2015.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Silva R.S., Arno G., Cipriani V., Pontikos N., Defoort-Dhellemmes S., Kalhoro A., Carss K.J., Raymond F.L., Dhaenens C.M., Jensen H., et al. Unique noncoding variants upstream of PRDM13 are associated with a spectrum of developmental retinal dystrophies including progressive bifocal chorioretinal atrophy. Hum. Mutat. 2019;40:578–587. doi: 10.1002/humu.23715. [DOI] [PubMed] [Google Scholar]
- 44.Manes G., Joly W., Guignard T., Smirnov V., Berthemy S., Bocquet B., Audo I., Zeitz C., Sahel J., Cazevieille C., et al. A novel duplication of PRMD13 causes North Carolina macular dystrophy: overexpression of PRDM13 orthologue in drosophila eye reproduces the human phenotype. Hum. Mol. Genet. 2017;26:4367–4374. doi: 10.1093/hmg/ddx322. [DOI] [PubMed] [Google Scholar]
- 45.Lal D., Neubauer B.A., Toliat M.R., Altmüller J., Thiele H., Nürnberg P., Kamrath C., Schänzer A., Sander T., Hahn A., Nothnagel M. Increased Probability of Co-Occurrence of Two Rare Diseases in Consanguineous Families and Resolution of a Complex Phenotype by Next Generation Sequencing. PLoS ONE. 2016;11:e0146040. doi: 10.1371/journal.pone.0146040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ming X., Prasad N., Thulasi V., Elkins K., Shivamurthy V.K.N. Possible contribution of cerebellar disinhibition in epilepsy. Epilepsy Behav. 2021;118:107944. doi: 10.1016/j.yebeh.2021.107944. [DOI] [PubMed] [Google Scholar]
- 47.Kani S., Bae Y.K., Shimizu T., Tanabe K., Satou C., Parsons M.J., Scott E., Higashijima S., Hibi M. Proneural gene-linked neurogenesis in zebrafish cerebellum. Dev. Biol. 2010;343:1–17. doi: 10.1016/j.ydbio.2010.03.024. [DOI] [PubMed] [Google Scholar]
- 48.Fujitani Y., Fujitani S., Luo H., Qiu F., Burlison J., Long Q., Kawaguchi Y., Edlund H., MacDonald R.J., Furukawa T., et al. Ptf1a determines horizontal and amacrine cell fates during mouse retinal development. Development. 2006;133:4439–4450. doi: 10.1242/dev.02598. [DOI] [PubMed] [Google Scholar]
- 49.Glasgow S.M., Henke R.M., Macdonald R.J., Wright C.V., Johnson J.E. Ptf1a determines GABAergic over glutamatergic neuronal cell fate in the spinal cord dorsal horn. Development. 2005;132:5461–5469. doi: 10.1242/dev.02167. [DOI] [PubMed] [Google Scholar]
- 50.Sotelo C., Dusart I. Intrinsic versus extrinsic determinants during the development of Purkinje cell dendrites. Neuroscience. 2009;162:589–600. doi: 10.1016/j.neuroscience.2008.12.035. [DOI] [PubMed] [Google Scholar]
- 51.Harper R.M. Sudden infant death syndrome: a failure of compensatory cerebellar mechanisms? Pediatr. Res. 2000;48:140–142. doi: 10.1203/00006450-200008000-00004. [DOI] [PubMed] [Google Scholar]
- 52.Bright F.M., Vink R., Byard R.W., Duncan J.R., Krous H.F., Paterson D.S. Abnormalities in substance P neurokinin-1 receptor binding in key brainstem nuclei in sudden infant death syndrome related to prematurity and sex. PLoS ONE. 2017;12:e0184958. doi: 10.1371/journal.pone.0184958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lavezzi A.M., Corna M., Matturri L., Santoro F. Neuropathology of the Guillain-Mollaret Triangle (Dentato-Rubro-Olivary Network) in Sudden Unexplained Perinatal Death and SIDS. Open Neurol. J. 2009;3:48–53. doi: 10.2174/1874205X00903010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haldipur P., Aldinger K.A., Bernardo S., Deng M., Timms A.E., Overman L.M., Winter C., Lisgo S.N., Razavi F., Silvestri E., et al. Spatiotemporal expansion of primary progenitor zones in the developing human cerebellum. Science. 2019;366:454–460. doi: 10.1126/science.aax7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guy B., Zhang J.S., Duncan L.H., Johnston R.J., Jr. Human neural organoids: Models for developmental neurobiology and disease. Dev. Biol. 2021;478:102–121. doi: 10.1016/j.ydbio.2021.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or code.