

Abstract

Room-temperature phosphorescence (RTP) from organic compounds has attracted increasing attention in the field of data security, sensing, and bioimaging. However, realization of RTP with an aggregate induced phosphorescence (AIP) feature via harvesting supersensitive excited charge transfer triplet (3CT) energy under visible light excitation (VLE) in single-component organic systems at ambient conditions remains unfulfilled. Organic donor–acceptor (D–A) based orthogonal structures can therefore be used to harvest the energy of the 3CT state at ambient conditions under VLE. Here we report three phenoxazine–quinoline conjugates (PQ, PQCl, PQBr), in which D and A parts are held in orthogonal orientation around the C–N single bond; PQCl and PQBr are substituted with halogens (Cl, Br) while PQ has no halogen atom. Spectroscopic studies and quantum chemistry calculations combining reference compounds (Phx, QPP) reveal that all the compounds in film at ambient conditions show fluorescence and green-RTP due to (i) radiative decay of both singlet charge transfer (1CT) and triplet CT (3CT) states under VLE, (ii) energetic nondegeneracy of 1CT and 3CT states (1CT– 3CT, 0.17–0.21 eV), and (iii) spatial separation of highest and lowest unoccupied molecular orbitals. Further, we found in a tetrahydrofuran–water mixture (fw = 90%, v/v) that both PQCl (10–5 M) and PQBr (10–5 M) show concentration-dependent AIP with phosphorescence quantum yields (ϕP) of ∼25% and ∼28%, respectively, whereas aggregate induced quenching (ACQ) was observed in PQ. The phosphorescence lifetimes (τP) of the PQCl and PQBr aggregates were shown to be ∼22–62 μs and ∼22–59 μs, respectively. The ϕP of the powder samples is found to be 0.03% (PQ), 15.6% (PQCl), and 13.0% (PQBr), which are significantly lower than that of the aggregates (10–5 M, fw = 90%, v/v). Film (Zeonex, 0.1 wt %) studies revealed that ϕP of PQ (7.1%) is relatively high, while PQCl and PQBr exhibit relatively low ϕP values (PQCl, 9.7%; PQBr, 8.8%), as compared with that of powder samples. In addition, we found in single-crystal X-ray analysis that multiple noncovalent interactions along with halogen···halogen (Cl···Cl) interactions between the neighboring molecules play an important role to stabilize the 3CT caused by increased rigidity of the molecular backbone. This design principle reveals a method to understand nondegeneracy of 1CT and 3CT states, and RTP with a concentration-dependent AIP effect using halogen substituted twisted donor–acceptor conjugates.
Introduction
Single-component organic
luminescent materials (SCOLMs) that can
harness both singlet and triplet excitons have attracted enormous
interest because of their potential application in chemical sensing,1,2 organic light-emitting diodes (OLEDs),3−7 and bioimaging.8,9 It is a long held notion that
fluorescence via radiative decay of an excited singlet state occurs
readily where the relaxation conserves spin.10 However, achieving phosphorescence from the lowest triplet (T1)
state at ambient conditions is a challenging task due to the involvement
of a quantum mechanically forbidden spin flip process.10,11 Therefore, development of SCOLMs that manifest simultaneous radiative
decay of both excited singlet and triplet states, and high photoluminescence
quantum yield (PLQY) are of prime importance. To observe room-temperature
phosphorescence (RTP), heavy-metal based systems (typically iridium)
have been extensively studied.3,10 It has been realized
that these systems show up to 100% internal electroluminescence efficiency
because of the use of noble metals promoting strong spin–orbit
coupling (SOC).3 Since these systems are
expensive, an alternative method with inexpensive organic materials
which can be simply processed and functionalized by a wide variety
of substituents is therefore desirable. To achieve RTP from purely
organic materials, the molecule should undergo a spin flipping process
that occurs via coupling of the magnetic torque with the spin of the
electron.10,11 Thus, the spin–orbit coupling (SOC)
mechanism is crucial to flip the electron’s spin from the singlet
to the triplet state. It is well-known that the magnitude of SOC depends
on the SOC operator which sequentially turns on the nuclear charge.
Hence, the SOC effect will be stronger for the heavy elements as compared
with the lighter elements such as C, N, O, and H.10 Most of the purely organic molecules are made of lighter
elements, which results in poor SOC. For the sake of efficient intersystem
crossing (ISC), heteroatoms such as sulfur and halogens (Cl, Br, I)
are incorporated as they can yield radiationless ISC transition between
the orbitals of different symmetry, that is, 1(π–π*)
to 3(n−π*) and vice versa (El-Sayed rule).12,13 The RTP efficiency of such molecular systems suffers from a slow
rate of ISC that results in a long lifetime of the triplet states.
In addition, the quenching of triplet excitons (i.e., localized and
charge transfer) caused by molecular vibrations and molecular oxygen
becomes another major bottleneck to achieve efficient RTP.14 To this context, achieving RTP from organic
materials also relies on the rate of intersystem crossing (kISC) that depends mainly on the SOC and energy
difference (ΔEST) between the S1
and T1 states which can be expressed as  . It reveals that energetic
close proximity
of the S1 and T1 states is another prerequisite to observe the RTP
feature. To avoid such obstacles, a large number of design principles
based on harnessing the locally excited triplet (3LE) energy,
for example, intra- and/or intermolecular interactions (lone pair···π,
π···π, hydrogen bonding),15−17 aggregates,18−21 host–guest,22,23 deuteration,24 excited states engineering,25 internal heavy chalcogen atom effect,26 and the presence of heavy elements in the molecular backbone,10 have been adopted to reduce the nonradiative
pathways and boost ISC rates at ambient conditions. Recently, a handful
of reports have established that phosphorescence from the 3CT state at ambient conditions can be achieved using charge transfer
complexation,1,27−29 excited-state
hydrogen-bonding effect,30 aggregate induced
phosphorescence (AIP),31,32 and orthogonal donor–acceptor
(D–A) geometries.1,33 On the other hand,
thermally activated delayed fluorescent (TADF) material development
relies on the orthogonal D–A structure with a close proximity
of 3LE, 1CT, and 3CT states which
results in an increased reverse intersystem crossing (RISC) rate by
reducing the energy gap between the 1CT, 3CT,
and 3LE states.34−39 However, most of the reports determine the energy and configuration
of T1 states from a phosphorescence spectrum measured at 77 K.29,40 In addition, 1CT and 3CT states are often
considered degenerate states, assuming the orthogonal D–A geometry
of the TADF molecules.36,37,41 Furthermore, decades of RTP research have shown RTP via ultraviolet
light excitation. Despite this understanding, the structure–property
correlation in CT triplet (3CT) energy harvesting at ambient
conditions remains murky due to (a) inefficient SOC between the energetically
degenerate 1CT and 3CT states owing to the lack
of displacement of the transition orbitals of these states and (b)
the involvement of supersensitive 3CT excitons with longer
migration length as compared with that of 3LE excitons.42−45 Therefore, it is imperative to design new organic light emitters
that unravel the underlying mechanism of RTP with AIP via radiative
decay of 3CT under visible light excitation (VLE) and can
be used in photonics applications.
. It reveals that energetic
close proximity
of the S1 and T1 states is another prerequisite to observe the RTP
feature. To avoid such obstacles, a large number of design principles
based on harnessing the locally excited triplet (3LE) energy,
for example, intra- and/or intermolecular interactions (lone pair···π,
π···π, hydrogen bonding),15−17 aggregates,18−21 host–guest,22,23 deuteration,24 excited states engineering,25 internal heavy chalcogen atom effect,26 and the presence of heavy elements in the molecular backbone,10 have been adopted to reduce the nonradiative
pathways and boost ISC rates at ambient conditions. Recently, a handful
of reports have established that phosphorescence from the 3CT state at ambient conditions can be achieved using charge transfer
complexation,1,27−29 excited-state
hydrogen-bonding effect,30 aggregate induced
phosphorescence (AIP),31,32 and orthogonal donor–acceptor
(D–A) geometries.1,33 On the other hand,
thermally activated delayed fluorescent (TADF) material development
relies on the orthogonal D–A structure with a close proximity
of 3LE, 1CT, and 3CT states which
results in an increased reverse intersystem crossing (RISC) rate by
reducing the energy gap between the 1CT, 3CT,
and 3LE states.34−39 However, most of the reports determine the energy and configuration
of T1 states from a phosphorescence spectrum measured at 77 K.29,40 In addition, 1CT and 3CT states are often
considered degenerate states, assuming the orthogonal D–A geometry
of the TADF molecules.36,37,41 Furthermore, decades of RTP research have shown RTP via ultraviolet
light excitation. Despite this understanding, the structure–property
correlation in CT triplet (3CT) energy harvesting at ambient
conditions remains murky due to (a) inefficient SOC between the energetically
degenerate 1CT and 3CT states owing to the lack
of displacement of the transition orbitals of these states and (b)
the involvement of supersensitive 3CT excitons with longer
migration length as compared with that of 3LE excitons.42−45 Therefore, it is imperative to design new organic light emitters
that unravel the underlying mechanism of RTP with AIP via radiative
decay of 3CT under visible light excitation (VLE) and can
be used in photonics applications.
Inspired by our previous investigations,1,41 donor (phenoxazine) substituents at either the 8 (ortho to the quinolinyl nitrogen atom) or 6,8-positions (ortho and para to the quinolinyl nitrogen atom) of the quinolinyl ring have resulted in orange-red RTP (3CT) via intermolecular CT complexation, and dual emission via TADF and RTP at ambient conditions, respectively. Herein, we report three new phenoxazine–quinoline conjugates (PQ, PQCl, PQBr), where phenoxazine is covalently attached with the halogen (Cl, Br) and/or without halogen substituted quinolinyl fragment via a C–N single bond (Figure 1); the phenoxazine donor is connected at the 6-position (para to the quinolinyl nitrogen atom) of the acceptor fragment. We anticipate that a heavy atom effect caused by halogen substitution would trigger the SOC as well as intermolecular interactions due to the presence of lone pairs of Cl and Br atoms, which may lead to efficient RTP with an AIP feature at ambient conditions. In addition, we imagine that a spatial separation of the highest occupied and the lowest unoccupied molecular orbitals (HOMO, LUMO) may result in a CT absorption band at the ground state and nondegenerate excited CT states (1CT, 3CT) due to donor–acceptor coupling at the excited state, conducive to RTP via radiative decay of 3CT under VLE. Spectroscopic analysis revealed that PQ in solutions exhibits white light emission due to radiative decay of both locally excited (LE) and charge transfer (CT) states. While PQCl and PQBr showed only a predominant CT emission band in solutions. We found in aggregate studies that PQCl and PQBr show concentration-dependent AIP features (PQCl, τ1 = 22.60 μs, τ2 = 62.37 μs; PQBr, τ1 = 22.03 μs, τ2 = 59.15 μs; λex = 425 nm) (quantum yield, ϕP = 25–28%) in the mixture of tetrahydrofuran and water under VLE at ambient conditions, while an aggregate induced quenching (ACQ) effect was observed only for PQ. Further, in the solid state (powder), all the compounds showed fluorescence and green-RTP (PQ: λem = 535 nm, τ1= 18.95 μs, τ2 = 141.21 μs, τ3 = 1.01 ms; PQCl: λem = 529 nm, τ1 = 6.1 ms, τ2 = 1.8 ms; PQBr: λem = 536 nm, τ1 = 0.23 ms, τ2 = 1.8 ms) features with relatively reduced ϕP (PQ, 0.03; PQCl, 15.6; PQBr, 13.0%) as compared with that of aggregates at ambient conditions. The 1CT–3CT gaps are calculated to be ∼0.03–0.1 eV. While ϕP values of all the conjugates in 0.1 wt % films are found to be ∼7–9%, respectively, at ambient conditions. Single crystal X-ray analysis of PQ and PQCl revealed that donor (phenoxazine) and acceptor (quinolinyl part) are held in almost orthogonal fashion. We found in X-structures that Cl···Cl interactions along with a large number of noncovalent interactions (C–H···Cl, C–H···C, C–H···O, π···π) played an important role to rigidify the molecular system and facilitate the ISC process. Thus, we have developed new D–A molecular systems with halogen substitutions which show RTP with AIP characteristics due to radiative decay of the 3CT state at ambient conditions.
Figure 1.

Energy diagram with proposed molecular structures.
Results and Discussion
Single-Crystal X-ray Diffraction (SCXRD) Analysis
SCXRD analyses (Figure 2) of all of the conjugates reveal that the phenoxazine ring and phenyl substituents attached to the C2 and C4 atoms of the quinolinyl moiety deviate from planarity (PQ: −94(1)°, 36(1)° and −126.2(9)°; PQCl: 86.5(3)°, 6.1(2)7° and −52.5(3)°), when viewed along the C11A–N2–C6–C5, N1–C2–C23–C24, and C3–C4–C17–C22 atoms, respectively (Figure S9, Table S1), proving that donor and acceptor units are in almost orthogonal orientation conducive for a low singlet–triplet gap. Interestingly, the phenoxazine donor at the 6-position of the quinolinyl ring (PQ, PQCl) adapts “bent-type” conformation. The donor at the 6-position of the quinolinyl part of both conjugates is folded by 11.29° and 4.36°, respectively, along the N···O vector, ensuring that geometries with folded and nearly planar phenoxazines are present in the solid state. These observations are consistent with recent reports.1,41,46 Further, PQ shows multiple C···C interactions (C9···C14C, C2···C15C, 3.40(1) Å, 3.39(1)Å), one hydrogen-bond (H–B) interaction (C26–H26···O1B, 2.428 Å) and multiple C–H···C interactions (H18–C18···C16B, 2.856 Å; C19B–H19B···C14B, 2.797 Å; C13–H13···C24, 2.852 Å; C7–H7···C2B, 2.66 Å; C27–H27···H16, 3.11 Å) between two neighboring molecules (Figure S10). In addition, a π···π stacking interaction (centroid to centroid, 3.717 Å) between the phenyl ring of the donor fragment and the pyridyl ring of the quinolinyl part of two neighboring molecules is also observed (Figure S10). In PQCl, multiple C–H···C interactions (C28–H28···C13, 2.891 Å; C13–H13···H26, 2.88 Å; C20–H20···C13A, 2.804 Å) along with one hydrogen bond (C8–H8···Cl1, 2.94 Å) between the neighboring molecules were also found (Figure S10). Interestingly, an additional Cl···Cl interaction (3.485 Å) between the two chlorine atoms of two neighboring molecules was also observed (Figure S10). Further, two π···π stacking interactions (centroid to centroid, 3.778 and 4.131 Å) between the donor rings, and phenyl ring of the PhCl fragment and pyridyl ring of the quinolinyl part of two neighboring molecules are also present (Figure S10). We anticipate that these noncovalent intermolecular interactions lead to increase rigidity that reinforces AIP due to stabilization of the 3CT state.
Figure 2.
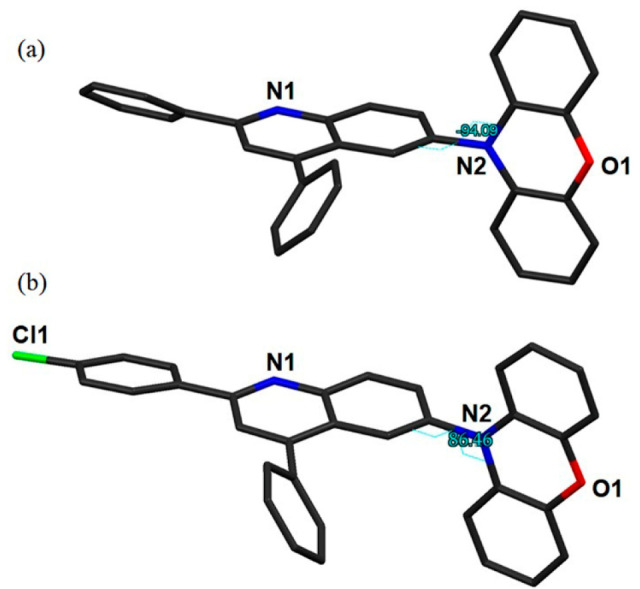
Representative torsions in the (a) PQ and (b) PQCl. H atoms are removed for the sake of clarity.
Quantum Chemistry Analysis
To gain further insight into the excited states of all the conjugates, DFT and TD-DFT47 calculations were performed in Gaussian 09 D.0148 at the M06-2X/6-31G(d,p) level of theory (Figure 3, Figures S11–S13). It should be noted that the crystal geometries of PQ and PQCl were considered for optimization in the gaseous state while the z-matirx was used for optimization of PQBr. The calculations revealed that the HOMOs are predominantly localized on the donor parts, while the LUMOs are distributed over the acceptor units of PQ, PQCl, and PQBr (Figure S11). As shown by the natural transition orbital (NTO)49 analysis in Figure 3, and Figures S12 and S13, the S1 state of PQ, PQCl, and PQBr shows a pronounced CT-excitation character, as the hole and electron wave functions are localized at the donor and the acceptor parts, respectively. In contrast, the T1 state of all the compounds exhibits a hybridized charge transfer (HLCT) character as it consists of a mixture of LE and CT excitons. The S1–T1 gaps were calculated to be 0.08 eV (PQ), 0.03 eV (PQCl), and 0.028 eV (PQBr), respectively. Intriguingly, the calculations predicted a decrease in the S1–T1 gap with halogen substitution. These results promise that the energy of triplet exciton can be harvested via radiative decay of the 3CT state at ambient conditions due to small S1–T1 gaps of all the conjugates.
Figure 3.

The energy diagram of PQCl with natural transition orbitals describing the excitation characters of the 1CT and 3HLCT states (M02X-631G (d,p)).
Absorption and Emission Characteristics in Solutions
Figure 4 and Figure S14 exhibit the normalized ultraviolet–visible (UV–vis) spectra of PQ, PQC, and PQB in the solvents (10–5 M) of disparate polarity. All the compounds exhibit higher energy absorption bands at 265 and 311 nm which can be attributed to the π → π* transition.34,35,50,51 In addition, a broad tail ranging from ∼355 to 500 nm which shows a slight hypsochromic shift upon increasing solvent polarity, was observed. A comparison between the absorption spectra of D (Phx) and A (QPP) fragments with all the conjugates confirms that a higher energy region of the absorption spectra of the D–A conjugates arises due to the arithmetic sum of the D and A absorption spectra, while the appearance of lower energy broad absorption bands (ε = 6100–7100 L·mol–1·cm–1) is due to the charge transfer (CT) caused by D–A torsion (Figure 4), which is conducive for visible light excitation (VLE).
Figure 4.

Absorption (UV–vis) and emission spectra of PQ, PQCl, and PQBr along with donor (Phx) and acceptor (QPP) components in toluene solutions (10 μM) at ambient conditions.
Likewise, PL measurements of all the conjugates were performed in different polar solvents (VLE, λex = 425 nm). Methyl cyclohexane (MCH) solutions (10 μM) of PQ show a local emission (LE) feature at 401, 426, 462, and 482 nm (Figure S15). Interestingly, an increase in solvent polarity (toluene, Tol; tetrahydrofuran, THF; acetonitrile, ACN) leads to a bathochromically shifted new lower energy broad emission band along with the parent LE band which remains almost unaffected (Table 1). In ACN solutions, the dual emission feature (LE, ∼425 nm; CT, ∼598 nm) resulted in a white light emission with Commission Internationale de l’Eclairage (CIE) coordinates of (0.33, 0.35) (Figure S15). To gain more insight, emission measurements of PQ were carried out in THF/ethylene glycol (EtGly) mixture (1.0 μM) (Figure S15). We found that an increasing concentration of EtGly (30%) results in a bathochromic shift of the emission band with reduced intensity. The addition of more EtGly (50–90%) causes a decrease in the intensity of the lower energy broadband emission (∼450–700 nm), while the intensity of the parent LE band (∼405–480 nm) is substantially increased. The dual emission feature suggests that the radiative recombination from LE to the ground state is relatively slow due to D–A torsion, which can significantly compete with the electron transfer that populates the lower-level CT state.34 Further, a close overlap of the steady state spectra of the D part and PQ in toluene clearly indicates that the contribution of the LE band arises from the D component (Figure 4, Figure S15). On the other hand, PQCl and PQBr exhibit only lower energy CT bands that show significant positive solvatochromic effects with increasing solvent polarity as compared with that of PQ (Figure 4, Figure S15). It should be noted that the emission bands of PQCl and PQBr are more bathochromically shifted as compared with that of PQ. These results indicate that the electron withdrawing effect of halogens (Cl, Br) causes a bathochromic shift of the emission bands, proving more CT nature52,53 of the excited states as compared with that of PQ. We found in time-correlated single photon counting (TCSPC) analysis that the lifetime of the LE band remains almost unaltered (τ = 3–4 ns) with increasing polarity of the solvent, while a gradual increase in the lifetime of the lower energy broad emission band (τ = 2.5–18.5 ns) was observed for PQ at RT (Figure S16, Table S3). On the other hand, both PQCl and PQBr showed similar lifetime enhancement with increasing polarity of the solvents (Figure S16, Table S4, Table 1). Further, absolute photoluminescence quantum yield (PLQY) values were found to be ∼3–10% (Table S4). In order to understand the nature of the emission band, temperature-dependent emission studies of all the compounds were performed in MCH (1.0 μM) (Figures S17, S18 and Table S5). At 77 K, both LE and a lower energy broad emission feature (1CT, 580–605 nm) are observed, while the 1CT bands are bathochromically shifted as compared with that of steady state emission bands recorded at RT (Figure S17). These results further substantiate our previous argument for the CT nature of the lower energy broad emission band.
Table 1. Photophysical Parameters of PQ, PQCl and PQBr.
| phosphorescence |
||||||
|---|---|---|---|---|---|---|
| τP (ms) | ϕP (%) | 1CT (eV) | 3CT (eV) | 1CT–3CT (eV) | ||
| Filma | ||||||
| 0.48(0.30) | 0.15(0.6) | 0.5(0.13) | 7.1 | 2.87 | 2.70 | 0.17 |
| 0.31(0.64) | 1.6(0.36) | 9.7 | 2.83 | 2.62 | 0.21 | |
| 0.15(0.62) | 0.6(0.38) | 8.8 | 2.81 | 2.63 | 0.18 | |
| Powder | ||||||
| 0.2(0.1) | 0.15(0.26) | 1.0(0.66) | 0.03 | 2.73 | 2.55 | 0.18 |
| 6.1(0.57) | 1.8(0.43) | 15.6 | 2.51 | 2.48 | 0.03 | |
| 0.2(0.33) | 1.8(0.67) | 13.0 | 2.50 | 2.48 | 0.02 | |
| Aggregated | ||||||
| 0.02(0.36) | 0.06(0.36) | 25 ± 0.3 | ||||
| 0.02(0.73) | 0.06(0.27) | 28 ± 0.8 | ||||
| fluorescence |
||||||
|---|---|---|---|---|---|---|
| samples | λF (nm)b | τF (ns) | ϕF (%) | λP(nm)c | ||
| PQ | 501 | 4.48(0.36) | 16.89(0.6) | 17.0 | 514 | |
| PQCl | 430 | 1.33(0.54) | 5.27(0.46) | 14.9 | 529 | |
| 510 | 3.5 (0.34) | 14.4(0.66) | ||||
| PQBr | 440 | 1.65(0.40) | 5.48(0.60) | 14.8 | 530 | |
| 512 | 5.5(0.25) | 16.4(0.75) | ||||
| PQ | 518 | 9.33(0.44) | 2.78(0.44) | 24.8(0.12) | 2.03 | 535 |
| PQCl | 525 | 4.3(0.57) | 7.2(0.43) | 15.2 | 529 | |
| PQBr | 534 | 4.7(0.11) | 17.9(0.89) | 12.4 | 536 | |
| PQCl | 562 | |||||
| PQBr | 585 | |||||
Zeonex.
λex = 425 nm;
λex = 390 nm,
[M] = 1.0 × 10–5 M (THF)/H2O (fw = 90%).
To understand delayed fluorescence, steady-state emission along with lifetime measurements of all the molecules in MCH solutions were carried out under ambient and degassed conditions at room temperature (RT) (Figure 5a–d, Tables S5–S8). We found in lifetime analysis that no delayed components (2.28–3.26 ns, degassed; 2.14–3.18 ns, ambient) were involved for lower energy emission bands (Tables S5–S8). Further, temperature-dependent (213–298 K) lifetime analysis revealed that a gradual increase in the lifetime values of the short components (prompt fluorescence), ensuring that only radiative decay of the 1CT state is involved for emission at ambient conditions (Figure S18, Tables S5–S8). The phosphorescence measurement (λex = 425 nm, 0.1 ms detector delay) at 77 K shows a broadband emission feature at ∼515 (PQ) nm, ∼530 (PQCl), and ∼535 nm (PQBr), which do not overlap with the energy onsets of the phosphorescence spectra of the D (Phx) and A (QPP) parts of all the molecules (Figure 5d, Figure S19). The 1CT–3CT gaps were calculated to be 0.10–0.22 eV, showing that energetically nondegenerate triplet states are involved for emission, which is in good agreement with the computational analysis discussed above (Figure S20, Figures S11–S13, Table S9).
Figure 5.

Lifetime analysis of (a) PQ, (b) PQCl, and (c) PQBr in different conditions at RT (λex = 425 nm). (d) Phosphorescence spectra (0.1 ms delay) of donor (Phx), acceptor (QPP), PQCl, and PQBr in MCH at 77 K.
Photophysics of Aggregates
In order to understand the aggregation effect on the emission behavior of all the conjugates, aggregate induced emission (AIE) measurements (100 μM) were carried out in THF–water (H2O) mixture at RT under VLE. Figure S21 shows a gradual decrease in the intensity of the emission band of PQ with an increase in concentration of H2O, leading to aggregate induced quenching (ACQ) effect. Interestingly, two other conjugates (PQCl, PQBr) displayed conventional AIE property (Figure 6a,b).16−18 PQCl showed a bathochromic shift (∼625 nm) of the parent broad emission band (∼591 nm) with significant reduction in the intensity of the emission band with a gradual increase in concentration of H2O up to 45% (v/v) to the THF solutions. Further increase in the water content (fw = 90%, v/v) leads to a ∼100-fold increase in emission intensity with a ∼34 nm hypsochromic shift (λem = ∼557 nm). Likewise, PQBr also shows AIE behavior with a ∼55-fold enhancement in emission intensity, and a ∼19 nm blue shift of the emission band (λem = 572 nm). The initial bathochromic shift of PQCl and PQBr with increase in the solvent polarity can be rationalized by the stabilization of the CT excited states, while hypsochromic shifts of the broad emission bands which are observed with higher water content (fw = 70–90%, v/v) are due to aggregation that causes the molecules to come out from the solvent shell, thus weaken the ICT effect.54 We anticipate that halogen substitution (Cl, Br) in the molecular backbone played an important role to achieve AIE in both PQCl and PQBr due to stronger intermolecular interactions caused by free lone pair of electrons of halogens as compared with the parent PQ, thus leading to aggregation.
Figure 6.

Aggregate induced emission (AIE) spectra of (a) PQCl and (b) PQBr with increasing water content in THF (10–6 M) at RT (λex = 425 nm). Aggregate induced phosphorescence (AIP) spectra of (c) PQCl and (d) PQBr aggregates (fw = 90%, THF-H2O) at ambient conditions (0.1 ms delay) (λex = 425 nm). Phosphorescence lifetime of (e) PQCl and (f) PQBr aggregates at ambient conditions (λex = 390 nm).
To explore the aggregate induced phosphorescence (AIP) property, concentration-dependent AIP measurements of PQCl and PQBr were undertaken at fw = 90% in THF under VLE. The phosphorescence measurements of PQCl aggregates (10–6 M, λex = 425 nm, 0.1 ms detector delay) result in an intense broad-RTP peak at ∼562 nm (τ1 = 22.60 μs, τ2 = 62.37 μs; ϕP = 21 ± 0.5%) (Figure 6c,e, Table 1). Similarly, an intense broad-RTP peak at ∼585 nm (τ1 = 22.03 μs, τ2 = 59.15 μs; ϕP = 24 ± 0.4%) was also observed for PQBr (Figure 6d,f, Table 1). Further, AIP measurements (0.1 ms delay) at high concentration (10–5 M) result in a similar AIP feature (fw = 90%, v/v) with increased intensity of the emission bands (ϕP = 25–28%) (Figure S22, Table S10). Interestingly, further increased concentration (10–4 M) leads to a significant reduction in the intensity of the emission bands (ϕP = 12 ± 0.3–16 ± 0.5%) while keeping the same water content (fw = 90%, v/v), suggesting quenching of AIP due to increased concentration (Figure S22, Table S10). Further, the transmission electron microscopy (TEM) measurements revealed that both PQCl and PQBr form aggregates (fw = 90%, v/v) with dimensions of ca. 400 and 80 nm, respectively (Figures S24, S25); aggregates are amorphous in nature, as no clear diffraction patterns are observed (Figures S24, S25). These observations indicate that AIP features of both molecules are activated due to (a) efficient ISC (El-Sayed rule) and (b) intermolecular interactions caused by halogen substitution in the molecular backbone.
Photophysics in Films
To gain further insight on the emission features of PQ, PQCl, and PQBr, photophysical studies were performed in a rigid matrix (Zeonex, 0.1 wt %). Steady state measurements of all the conjugates show both LE and CT emission bands (PQ: 410, 501 nm. PQCl: 430, 510 nm. PQBr: 440, 512 nm) with PLQY values of ∼23–26%, respectively (Figure 7a,b, Table 1). Further, TCSPC analyses of both 1LE and 1CT show only nanosecond component (PQ: λem = 410 nm, τ1 = 5.55, τ2 = 17.42; λem = 501 nm; τ1= 4.48 ns, τ2 = 16.89 ns. PQCl: λem = 430 nm, τ1 = 1.33 ns, τ2 = 5.27 ns; λem = 510 nm, τ1 = 3.5 ns, τ2 = 14.4 ns. PQBr: λem = 440 nm, τ1 = 1.65 ns, τ2 = 5.48 ns; λem = 512 nm, τ1 = 5.5 ns, τ2 = 16.4 ns) at ambient conditions (Figure S26). A comparison between the steady state emission spectra of D component as well as D–A conjugates revealed that the 1LE bands of the compounds result from donor local emissions (Figure S27). We anticipate that the radiative recombination from 1LE to the ground state is relatively slow due to rigidity of the host, which can significantly compete with the electron transfer that populates the lower-level 1CT state.34 On the other hand, phosphorescence measurements (λex = 425 nm, 0.1 ms delay) showed green-RTP (3CT, broad) peaks at 514 (PQ: τ1 = 48.28 μs, τ2 = 152.45 μs, τ3 = 525.94 μs), 529 nm (PQCl: τ1 = 313 μs, τ2 = 1.6 ms), and 530 nm (PQBr: τ1 = 148 μs, τ2 = 569 μs), which are red-shifted (∼10–19 nm) as compared with the steady state emission spectra recorded at ambient conditions (Figure 7a,b, Table 1, Figures S26a, S28). Moreover, a significant increase in the lifetimes under deoxygenated conditions at RT confirms the involvement of triplet state (3CT) (Figure S27). Furthermore, comparison of the energy onsets of the phosphorescence spectra of all compounds along with their D and A parts shows that lower energy 3CT emission bands result from D–A coupling at the excited state (Figure S29), which is fully consistent with the emission measured in MCH (Figure 5d). Phosphorescence quantum yield (ϕP) values were found to be ∼7–9% (Table 1). The CT–3CT gaps were calculated from the difference between the energy onsets of the steady state emission band recorded at RT and the phosphorescence spectrum recorded at 77 K, and found to be 0.17–0.21 eV (Figure S30, Table S11). Similar emission dynamics were also observed in bis[2-(di(phenyl)phosphino)-phenyl]ether oxide (DPEPO) (0.1 wt %), except a significant bathochromic shift of the lower energy broad emission bands (520, 530, 541 nm) was measured due to increased polarity of the host material (Figures S31, S32).35 The phosphorescence lifetimes of the conjugates (PQ: τ1 = 64 ms, τ2= 130.3 ms. PQCl: τ1 = 84.39 ms, τ2= 200.56 ms. PQBr: τ1 = 8.46 ms, τ2= 37.71 ms) further ensure that the 3CT state contributes for RTP of all the conjugates at ambient conditions (Figures S31, S32). The longer lifetimes of the triplet states in DPEPO as compared with the Zeonex films can be explained by the stabilization of the triplet excitons caused by a more polar host. Taking all together, we believe that all the conjugates (aggregates/films/powder) exhibit fluorescence and RTP via radiative decay of 1LE (donor part), 1CT (D–A), and 3CT (D–A), respectively, at ambient conditions.
Figure 7.

Steady state and RTP spectra (0.1 ms detector delay) of (a) PQCl and (b) PQBr at ambient conditions in Zeonex films (0.1 wt %) (λex = 425 nm). Steady state and RTP (0.1 ms detector delay) spectra of (c) PQCl and (d) PQBr in powder (λex = 425 nm). Inset shows RTP decays (λex = 390 nm) of PQCl and PQBr in Zeonex films as well as powder at ambient conditions.
Photophysics in Solid State
The steady state PL measurements of all the conjugates were also carried out in solid state (powder) under VLE. The emission measurements (λex = 425 nm) of powder samples show a broad green emission band at ∼518 nm (PQ: τ1 = 9.33 ns, τ2 = 2.78 ns, τ3 = 24.79 ns), 525 nm (PQCl: τ1= 4.3 ns, τ2 = 7.2 ns), and 534 nm (PQBr: τ1 = 4.7 ns, τ2 = 17.9 ns) at RT (Figure 7c,d, Figures S33, S34, Table 1). It should be noted that no LE emission bands are observed for all the compounds. Thus, the disappearance of locally excited emission from the 1LE state (donor unit) can be explained by the predominant population of the 1CT state caused by the aggregation effect. Likewise, phosphorescence measurements were also performed at ambient conditions (0.1 ms delay). Phosphorescence spectra (λex = 425 nm) of all the conjugates exhibit a broad green-RTP at 535 nm (PQ: τ1 = 18.95 μs, τ2 = 145.21 μs, τ3 = 1.01 ms), 529 nm (PQCl: τ1 = 6.1 ms, τ2 = 1.8 ms), and 536 nm (PQBr: τ1 = 0.23 ms, τ2 = 1.8 ms) which is red-shifted as compared with the steady state emission bands recorded at RT (Figures S33, S34). Absolute PLQY values were found to be 2.1% (PQ), 30.8% (PQCl), and 25.4% (PQBr) (Table 1, Tables S12–S14). The ϕP values were calculated to be 0.03% (PQ), 15.6% (PQCl), and 13.0% (PQBr) (Table 1, Tables S12–S14). When we compare the phosphorescence spectra of powder samples with the AIP spectra (ϕP = 25%, PQCl; ϕP = 28%, PQBr) (aggregation studies discussed above, fw = 90%, c = 10–5 M), we found that the ϕP values of powder samples are relatively smaller than that of aggregate samples, while energy onsets of the phosphorescence bands remained almost the same (Figure S35, Table S15), indicating that the powder samples suffer from a significant quenching effect on PL. When we compared the TEM analysis of the aggregates (fw = 90%, v/v) and powder samples, surprisingly, we found that both powder samples (PQCl, PQBr) showed a clear diffraction pattern along with increased average size of the particles (Figures S36–S38), indicating that powder samples are crystalline in nature, which is therefore responsible for the observation of reduced PL in the powder state. These results clearly indicate that all the conjugates are capable of harnessing singlet and triplet energy via fluorescence (1CT) and RTP (3CT) at ambient conditions, which is consistent with the PL analysis in films. The high ϕP values of PQCl and PQBr indicate that both efficient ISC and AIP mechanisms are activated due to the presence of a lone pair of electrons of halogen atoms (Cl, Br) (El-Sayed rule) in the molecular backbone.12,13 While a low PLQY and ϕP values (without halogen substitution) suggest that the ACQ effect played an important role in the emission characteristics of PQ. Such difference in emission characteristics of PQCl/PQBr with PQ can be further explained by the stabilization of the triplet state (3CT) due to reduced nonradiative channels caused by increased rigidity via intermolecular interactions induced by halogens.
Discussion
We demonstrate RTP with AIP/ACQ features from three D–A molecular systems in which the donor is held in almost orthogonal orientation around the D–A single bond. The donor and acceptor coupling at the ground state has sufficient driving force to obtain a CT absorption band at the ground state that can be used for visible light excitation. By substituting the acceptor part (quinolinyl unit) with electron withdrawing halogens (Cl, Br), an alteration of the photophysical property (ACQ to AIP) occurred while energetic nondegeneracy of the triplet states (1CT, 3CT) remained the same. Moreover, a decrease in the phosphorescence lifetime was found in PQCl and PQBr as compared with the parent molecule (PQ) due to the halogen substitution of the molecules. In addition, halogen substitution in such systems leads to AIP via radiative decay of the 3CT state at ambient conditions. The low 1CT–3CT gap is also involved to show efficient AIP. The relatively high phosphorescence quantum yield of ∼12–15% (powder) and ∼25–28% (aggregates) as compared with that of films (∼7–9%, 0.1 wt %) recorded at ambient conditions highlights the fundamental process of aggregation caused by the presence of a lone pair of electrons of the halogens. We hypothesize that heavy halogens (Cl, Br) in the molecular backbone offer (a) efficient ISC (El-Sayed rule) from 1CT to 3CT state and (b) stabilization of the 3CT state caused by intermolecular interactions (Cl···Cl, C–H···C, π···π, C–H···O) to sustain RTP with AIP characteristics in both PQCl and PQBr at ambient conditions. In addition, the orthogonal D–A structure leads to a controlled low energy gap (1CT–3CT = 0.17–0.21 eV) due to spatial separation of HOMO and LUMO. Accordingly, this approach paves a new way to achieve RTP with AIP and facilitate charge transfer triplet energy harvesting. From these results, the photophysics of such molecules is anticipated to become more deeply explored for the understanding of RTP materials.
Conclusion
In summary, three donor–acceptor based phenoxazine–quinoline conjugates with and/or without halogen substitution have been successfully synthesized. We have shown that all molecular systems show RTP via radiative decay of charge transfer triplet states under visible light excitation at ambient conditions. The presence of heavy halogens (Cl, Br) in the molecular backbone of PQCl and PQBr resulted in concentration-dependent aggregate-induced phosphorescence and led to a high phosphorescence quantum yield (∼25–28%, aggregates) as compared with the PQ (∼0.03%), which showed an aggregate caused quenching effect due to the absence of a halogen atom. The aggregate induced phosphorescence feature of both PQCl and PQBr with decreased phosphorescence lifetimes as compared with that of PQ can be explained by (a) noncovalent interactions (for example, Cl···Cl, C–H···Cl, C–H···C, π···π) which stabilize the triplet excitons due to increased rigidity of the molecular backbone and (b) increased SOC caused by heavy halogens. The design principle may enrich our understanding of RTP from charge transfer triplet state under visible light excitation. Further modification of these systems considering visible light excitation (for bioimaging) and triplet charge transfer state (for photocatalysis) are underway in our laboratory.
Experimental Section
Synthesis and Characterization
The final compound (PQ) was synthesized by the reaction between phenoxazine and 4-fluoro-nitrobenzene, followed by a reduction of the nitrofunction to the amine intermediates, and treated with ethynylbenzene and benzaldehyde in the presence of a catalytic amount of iron(III)-trifluoromethanesulfonate in oxygenated conditions. Similarly, PQCl and PQBr were synthesized using the above procedure except using 4-chlorobenzaldehyde and 4-bromobenzaldehyde separately. The reference compound (QPP) was synthesized by following a similar methodology using aniline, benzaldehyde, and ethynylbenzene to afford QPP (Scheme S1, Supporting Information).1,34,50,51 To check the purity of the target compounds, high performance liquid chromatography (HPLC) of the vacuum sublimated samples was used (Figures S6–S8). All the compounds were characterized by NMR spectroscopy, high resolution mass spectrometry (HRMS), and X-ray analysis (see the Supporting Information).
Photophysical Measurements
Absorption measurements were performed using a Cary 8454 UV–vis instrument from Agilent Technologies. Steady-state emission, phosphorescence, and lifetime analysis of the samples were recorded on HORIBA Fluorolog-3 and Edinburgh FLS980 spectrometers (see section S1 in the Supporting Information). All the phosphorescence spectra were recorded using the detector delay of more than 0.5 ms. The measurement details can be found in the reported literature.1,34,50,51 Temperature-dependent PL measurements were performed using cryostat (JANIS CS204SE-FMX-1AL). High performance liquid chromatography (HPLC) experiments were carried out on a Waters Alliance system (Milford, MA) consisting of e2695 separation module and a 2998 photodiode-array detector.
Acknowledgments
D.R. is grateful to the Science & Engineering Research Board (SERB) (File No: EEQ/2020/000029), DST. S.K., S.D., and M.U. thank SNU for fellowship. We thankfully acknowledge the use of the MAGUS supercomputing system at SNU.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.2c01909.
Crystallographic data for PQ (CIF)
Crystallographic data for PQCl (CIF)
Experimental procedures, NMR spectra of all compounds, high performance liquid chromatography, single crystal X-ray diffraction analysis, UV–vis absorption spectra, steady-state emission, time correlated single photon counting measurement, phosphorescence spectra, quantum yields, rate parameters, and computational details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bhattacharjee I.; Acharya N.; Karmakar S.; Ray D. Room-Temperature Orange-Red Phosphorescence by Way of Intermolecular Charge Transfer in Single-Component Phenoxazine–Quinoline Conjugates and Chemical Sensing. J. Phys. Chem. C 2018, 122, 21589–21597. 10.1021/acs.jpcc.8b06171. [DOI] [Google Scholar]
- Yu X.; Liang W.; Huang Q.; Wu W.; Chruma J. J.; Yang C. Room-Temperature Phosphorescent Y-Cyclodextrin-Cucurbit [6] Uril-Cowheeled [4] Rotaxanes for Specific Sensing of Tryptophan. Chem. Commun. 2019, 55, 3156–3159. 10.1039/C9CC00097F. [DOI] [PubMed] [Google Scholar]
- Baldo M. A.; O’Brien D. F.; You Y.; Shoustikov A.; Sibley S.; Thompson M. E.; Forrest S. R. Highly Efficient Phosphorescent Emission from Organic Electroluminescent Devices. Nature 1998, 395, 151–154. 10.1038/25954. [DOI] [Google Scholar]
- Köhler A.; Bässler H. Triplet States in Organic Semiconductors. Mater. Sci. Eng. R Rep. 2009, 66, 71–109. 10.1016/j.mser.2009.09.001. [DOI] [Google Scholar]
- Liu Y.; Li C.; Ren Z.; Yan S.; Bryce M. R. All-Organic Thermally Activated Delayed Fluorescence Materials for Organic Light-Emitting Diodes. Nat. Rev. Mater. 2018, 3, 18020. 10.1038/natrevmats.2018.20. [DOI] [Google Scholar]
- Reineke S. Phosphorescence Meets Its Match. Nat. Photonics 2014, 8, 269–270. 10.1038/nphoton.2014.78. [DOI] [Google Scholar]
- Uoyama H.; Goushi K.; Shizu K.; Nomura H.; Adachi C. Highly Efficient Organic Light-Emitting Diodes from Delayed Fluorescence. Nature 2012, 492, 234–238. 10.1038/nature11687. [DOI] [PubMed] [Google Scholar]
- Yang J.; Zhang Y.; Wu X.; Dai W.; Chen D.; Shi J.; Tong B.; Peng Q.; Xie H.; Cai Z.; Dong Y.; Zhang X. Rational Design of Pyrrole Derivatives with Aggregation-Induced Phosphorescence Characteristics for Time-Resolved and Two-Photon Luminescence Imaging. Nat. Commun. 2021, 12, 4883. 10.1038/s41467-021-25174-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Palmer G. M.; Dewhirst M. W.; Fraser C. L. A Dual-Emissive-Materials Design Concept Enables Tumour Hypoxia Imaging. Nat. Mater. 2009, 8, 747–751. 10.1038/nmat2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turro N. J.; Ramamurthy V.; Ramamurthy V.; Scaiano J. C.. Principles of Molecular Photochemistry: An Introduction; University Science Books, 2009. [Google Scholar]
- Penfold T. J.; Gindensperger E.; Daniel C.; Marian C. M. Spin-Vibronic Mechanism for Intersystem Crossing. Chem. Rev. 2018, 118, 6975–7025. 10.1021/acs.chemrev.7b00617. [DOI] [PubMed] [Google Scholar]
- El-Sayed M. Spin-Orbit Coupling and the Radiationless Processes in Nitrogen Heterocyclics. J. Chem. Phys. 1963, 38, 2834–2838. 10.1063/1.1733610. [DOI] [Google Scholar]
- Lower S.; El-Sayed M. The Triplet State and Molecular Electronic Processes in Organic Molecules. Chem. Rev. 1966, 66, 199–241. 10.1021/cr60240a004. [DOI] [Google Scholar]
- Hirata S. Intrinsic Analysis of Radiative and Room-Temperature Nonradiative Processes Based on Triplet State Intramolecular Vibrations of Heavy Atom-Free Conjugated Molecules toward Efficient Persistent Room-Temperature Phosphorescence. J. Phys. Chem. Lett. 2018, 9, 4251–4259. 10.1021/acs.jpclett.8b01711. [DOI] [PubMed] [Google Scholar]
- Acharya N.; Dey S.; Deka R.; Ray D. Molecular-Level Understanding of Dual-RTP Via Host-Sensitized Multiple Triplet-to-Triplet Energy Transfers and Data Security Application. ACS Omega 2022, 7, 3722–3730. 10.1021/acsomega.1c06390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia H.; Bhattacharjee I.; Ray D. Biluminescence Via Fluorescence and Persistent Phosphorescence in Amorphous Organic Donor (D4)–Acceptor (A) Conjugates and Application in Data Security Protection. J. Phys. Chem. Lett. 2018, 9, 3808–3813. 10.1021/acs.jpclett.8b01551. [DOI] [PubMed] [Google Scholar]
- Bhatia H.; Dey S.; Ray D. Effect of π···π Interactions of Donor Rings on Persistent Room-Temperature Phosphorescence in D4–A Conjugates and Data Security Application. ACS Omega 2021, 6, 3858–3865. 10.1021/acsomega.0c05666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia H.; Ray D. Use of Dimeric Excited States of the Donors in D4-A Systems for Accessing White Light Emission, Afterglow, and Invisible Security Ink. J. Phys. Chem. C 2019, 123, 22104–22113. 10.1021/acs.jpcc.9b07762. [DOI] [Google Scholar]
- Ma H.; Lv A.; Fu L.; Wang S.; An Z.; Shi H.; Huang W. Room-Temperature Phosphorescence in Metal-Free Organic Materials. Ann. Phys. 2019, 531, 1800482. 10.1002/andp.201800482. [DOI] [PubMed] [Google Scholar]
- Yang Z.; Mao Z.; Zhang X.; Ou D.; Mu Y.; Zhang Y.; Zhao C.; Liu S.; Chi Z.; Xu J.; Wu Y.-C.; Lu P.-Y.; Lien A.; Bryce M. R. Intermolecular Electronic Coupling of Organic Units for Efficient Persistent Room-Temperature Phosphorescence. Angew. Chem., Int. Ed. 2016, 55, 2181–2185. 10.1002/anie.201509224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W.; He Z.; Tang B. Z. Room-Temperature Phosphorescence from Organic Aggregates. Nat. Rev. Mater. 2020, 5, 869–885. 10.1038/s41578-020-0223-z. [DOI] [Google Scholar]
- Bhattacharjee I.; Hayashi K.; Hirata S. Key of Suppressed Triplet Nonradiative Transition-Dependent Chemical Backbone for Spatial Self-Tunable Afterglow. J. Am. Chem. Soc. Au 2021, 1, 945–954. 10.1021/jacsau.1c00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata S.; Totani K.; Zhang J.; Yamashita T.; Kaji H.; Marder S. R.; Watanabe T.; Adachi C. Efficient Persistent Room Temperature Phosphorescence in Organic Amorphous Materials under Ambient Conditions. Adv. Funct. Mater. 2013, 23, 3386–3397. 10.1002/adfm.201203706. [DOI] [Google Scholar]
- Bhattacharjee I.; Hirata S. Highly Efficient Persistent Room-Temperature Phosphorescence from Heavy Atom-Free Molecules Triggered by Hidden Long Phosphorescent Antenna. Adv. Mater. 2020, 32, 2001348. 10.1002/adma.202001348. [DOI] [PubMed] [Google Scholar]
- Higginbotham H. F.; Okazaki M.; de Silva P.; Minakata S.; Takeda Y.; Data P. Heavy-Atom-Free Room-Temperature Phosphorescent Organic Light-Emitting Diodes Enabled by Excited States Engineering. ACS Appl. Mater. Interfaces 2021, 13, 2899–2907. 10.1021/acsami.0c17295. [DOI] [PubMed] [Google Scholar]
- Goto S.; Nitta Y.; Decarli N. O.; de Sousa L. E.; Stachelek P.; Tohnai N.; Minakata S.; de Silva P.; Data P.; Takeda Y. Revealing the Internal Heavy Chalcogen Atom Effect on the Photophysics of the Dibenzo [a, J] Phenazine-Cored Donor–Acceptor–Donor Triad. J. Mater. Chem. C 2021, 9, 13942–13953. 10.1039/D1TC02635F. [DOI] [Google Scholar]
- D’Agostino S.; Grepioni F.; Braga D.; Ventura B. Tipping the Balance with the Aid of Stoichiometry: Room Temperature Phosphorescence Versus Fluorescence in Organic Cocrystals. Cryst. Growth Des. 2015, 15, 2039–2045. 10.1021/acs.cgd.5b00226. [DOI] [Google Scholar]
- Gao H. Y.; Zhao X. R.; Wang H.; Pang X.; Jin W. J. Phosphorescent Cocrystals Assembled by 1, 4-Diiodotetrafluorobenzene and Fluorene and Its Heterocyclic Analogues Based on C–I···π Halogen Bonding. Cryst. Growth Des. 2012, 12, 4377–4387. 10.1021/cg300515a. [DOI] [Google Scholar]
- Iwata S.; Tanaka J.; Nagakura S. Phosphorescence of the Charge-Transfer Triplet States of Some Molecular Complexes. J. Chem. Phys. 1967, 47, 2203–2209. 10.1063/1.1703292. [DOI] [Google Scholar]
- Liu Z.-Y.; Hu J.-W.; Huang C.-H.; Huang T.-H.; Chen D.-G.; Ho S.-Y.; Chen K.-Y.; Li E. Y.; Chou P.-T. Sulfur-Based Intramolecular Hydrogen-Bond: Excited-State Hydrogen-Bond On/Off Switch with Dual Room-Temperature Phosphorescence. J. Am. Chem. Soc. 2019, 141, 9885–9894. 10.1021/jacs.9b02765. [DOI] [PubMed] [Google Scholar]
- Wang T.; Su X.; Zhang X.; Nie X.; Huang L.; Zhang X.; Sun X.; Luo Y.; Zhang G. Aggregation-Induced Dual-Phosphorescence from Organic Molecules for Nondoped Light-Emitting Diodes. Adv. Mater. 2019, 31, 1904273. 10.1002/adma.201904273. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Sharman E.; Yang L.; Jiang J.; Zhang G. Aggregation-Induced Enhancement of Molecular Phosphorescence Lifetime: A First-Principle Study. J. Phys. Chem. C 2018, 122, 25796–25803. 10.1021/acs.jpcc.8b07087. [DOI] [Google Scholar]
- Huang R.; Kukhta N. A.; Ward J. S.; Danos A.; Batsanov A. S.; Bryce M. R.; Dias F. B. Balancing Charge-Transfer Strength and Triplet States for Deep-Blue Thermally Activated Delayed Fluorescence with an Unconventional Electron Rich Dibenzothiophene Acceptor. J. Mater. Chem. C 2019, 7, 13224–13234. 10.1039/C9TC02175B. [DOI] [Google Scholar]
- Acharya N.; Hasan M.; Dey S.; Lo S.-C.; Namdas E. B.; Ray D. Phenothiazine–Quinoline Conjugates Realizing Intrinsic Thermally Activated Delayed Fluorescence and Room-Temperature Phosphorescence: Understanding the Mechanism and Electroluminescence Devices. Adv. Photonics Res. 2021, 2, 2000201. 10.1002/adpr.202000201. [DOI] [Google Scholar]
- Bhatia H.; Ray D. Asymmetric-Donor (D2D2•)–Acceptor (A) Conjugates for Simultaneously Accessing Intrinsic Blue-RTP and Blue-TADF. Mater. Adv. 2020, 1, 1858–1865. 10.1039/D0MA00382D. [DOI] [Google Scholar]
- Dos Santos P. L.; Ward J. S.; Congrave D. G.; Batsanov A. S.; Eng J.; Stacey J. E.; Penfold T. J.; Monkman A. P.; Bryce M. R. Triazatruxene: A Rigid Central Donor Unit for a D–A3 Thermally Activated Delayed Fluorescence Material Exhibiting Sub-Microsecond Reverse Intersystem Crossing and Unity Quantum Yield Via Multiple Singlet-Triplet State Pairs. Adv. Sci. 2018, 5, 1700989. 10.1002/advs.201700989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherington M. K.; Gibson J.; Higginbotham H. F.; Penfold T. J.; Monkman A. P. Revealing the Spin-Vibronic Coupling Mechanism of Thermally Activated Delayed Fluorescence. Nat. Commun. 2016, 7, 13680. 10.1038/ncomms13680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. U.; Park I. S.; Chan C.-Y.; Tanaka M.; Tsuchiya Y.; Nakanotani H.; Adachi C. Nanosecond-Time-Scale Delayed Fluorescence Molecule for Deep-Blue OLEDs with Small Efficiency Rolloff. Nat. Commun. 2020, 11, 1765. 10.1038/s41467-020-15558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y.; Nakagawa H.; Matsumoto S.; Wakisaka Y.; Kaji H. Organic Light Emitters Exhibiting Very Fast Reverse Intersystem Crossing. Nat. Photonics 2020, 14, 643–649. 10.1038/s41566-020-0667-0. [DOI] [Google Scholar]
- Woo S.-J.; Kim Y.-H.; Kim J.-J. Dihedral Angle Distribution of Thermally Activated Delayed Fluorescence Molecules in Solids Induces Dual Phosphorescence from Charge-Transfer and Local Triplet States. Chem. Mater. 2021, 33, 5618–5630. 10.1021/acs.chemmater.1c01011. [DOI] [Google Scholar]
- Dey S.; Hasan M.; Shukla A.; Acharya N.; Upadhyay M.; Lo S.-C.; Namdas E. B.; Ray D. Thermally Activated Delayed Fluorescence and Room-Temperature Phosphorescence in Asymmetric Phenoxazine-Quinoline (D2–A) Conjugates and Dual Electroluminescence. J. Phys. Chem. C 2022, 126, 5649–5657. 10.1021/acs.jpcc.2c00512. [DOI] [Google Scholar]
- Luppi B. T.; Majak D.; Gupta M.; Rivard E.; Shankar K. Triplet Excitons: Improving Exciton Diffusion Length for Enhanced Organic Photovoltaics. J. Mater. Chem. A 2019, 7, 2445–2463. 10.1039/C8TA10037C. [DOI] [Google Scholar]
- Mahato P.; Monguzzi A.; Yanai N.; Yamada T.; Kimizuka N. Fast and Long-Range Triplet Exciton Diffusion in Metal-Organic Frameworks for Photon Upconversion at Ultralow Excitation Power. Nat. Mater. 2015, 14, 924–930. 10.1038/nmat4366. [DOI] [PubMed] [Google Scholar]
- Mikhnenko O. V.; Blom P. W.; Nguyen T.-Q. Exciton Diffusion in Organic Semiconductors. Energy Environ. Sci. 2015, 8, 1867–1888. 10.1039/C5EE00925A. [DOI] [Google Scholar]
- Narushima K.; Kiyota Y.; Mori T.; Hirata S.; Vacha M. Suppressed Triplet Exciton Diffusion Due to Small Orbital Overlap as a Key Design Factor for Ultralong-Lived Room-Temperature Phosphorescence in Molecular Crystals. Adv. Mater. 2019, 31, 1807268. 10.1002/adma.201807268. [DOI] [PubMed] [Google Scholar]
- Ward J. S.; Nobuyasu R. S.; Fox M. A.; Batsanov A. S.; Santos J.; Dias F. B.; Bryce M. R. Bond Rotations and Heteroatom Effects in Donor–Acceptor–Donor Molecules: Implications for Thermally Activated Delayed Fluorescence and Room Temperature Phosphorescence. J. Org. Chem. 2018, 83, 14431–14442. 10.1021/acs.joc.8b02187. [DOI] [PubMed] [Google Scholar]
- Runge E.; Gross E. K. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997. 10.1103/PhysRevLett.52.997. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas O.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, rev. D. 01; Gaussian, Inc.: Wallingford CT, 2009.
- Martin R. L. Natural Transition Orbitals. J. Chem. Phys. 2003, 118, 4775–4777. 10.1063/1.1558471. [DOI] [Google Scholar]
- Bhattacharjee I.; Acharya N.; Bhatia H.; Ray D. Dual Emission through Thermally Activated Delayed Fluorescence and Room-Temperature Phosphorescence, and Their Thermal Enhancement Via Solid-State Structural Change in a Carbazole-Quinoline Conjugate. J. Phys. Chem. Lett. 2018, 9, 2733–2738. 10.1021/acs.jpclett.8b00937. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee I.; Acharya N.; Ray D. Thermally Activated Delayed Fluorescence and Room-Temperature Phosphorescence in Naphthyl Appended Carbazole–Quinoline Conjugates, and Their Mechanical Regulation. Chem. Commun. 2019, 55, 1899–1902. 10.1039/C8CC09220F. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee I.; Ghosh N.; Raina A.; Dasgupta J.; Ray D. Conformational Switching Via an Intramolecular H-Bond Modulates the Fluorescence Lifetime in a Novel Coumarin–Imidazole Conjugate. Phys. Chem. Chem. Phys. 2018, 20, 6060–6072. 10.1039/C7CP07274K. [DOI] [PubMed] [Google Scholar]
- Karmakar S.; Ray D. Synthesis, Optical Properties, Acid-Base Vapochromism and Anti-Counterfeiting of Novel K-Extended Pyridine Fused Coumarins. J. Lumin. 2020, 223, 117229. 10.1016/j.jlumin.2020.117229. [DOI] [Google Scholar]
- Mei J.; Leung N. L.; Kwok R. T.; Lam J. W.; Tang B. Z. Aggregation-Induced Emission: Together We Shine, United We Soar!. Chem. Rev. 2015, 115, 11718–11940. 10.1021/acs.chemrev.5b00263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.