

Summary
Desmoplasia around pancreatic lesions is a barrier for immune cells and a hallmark of developing and established pancreatic cancer. However, the contribution of the innate immune system to this process is ill-defined. Using the KC mouse model and primary cells in vitro, we show that alternatively activated macrophages (AAM) crosstalk with pancreatic lesion cells and pancreatic stellate cells (PSCs) to mediate fibrosis and progression of lesions. TGFβ1 secreted by AAM not only drives activation of quiescent PSCs but also in activated PSCs upregulates expression of TIMP1, a factor previously shown as crucial in fibrosis. Once activated, PSCs auto-stimulate proliferation via CXCL12. Furthermore, we found that TIMP1/CD63 signaling mediates PanIN lesion growth and TGFβ1 contributes to a cadherin switch and drives structural collapse of lesions, indicating a potential progression step. Taken together, our data indicate TGFβ1 produced by Ym1+ AAM as a major driver of processes that initiate the development of pancreatic cancer.
Subject areas: Microenvironment, Immunology, Cancer
Graphical abstract
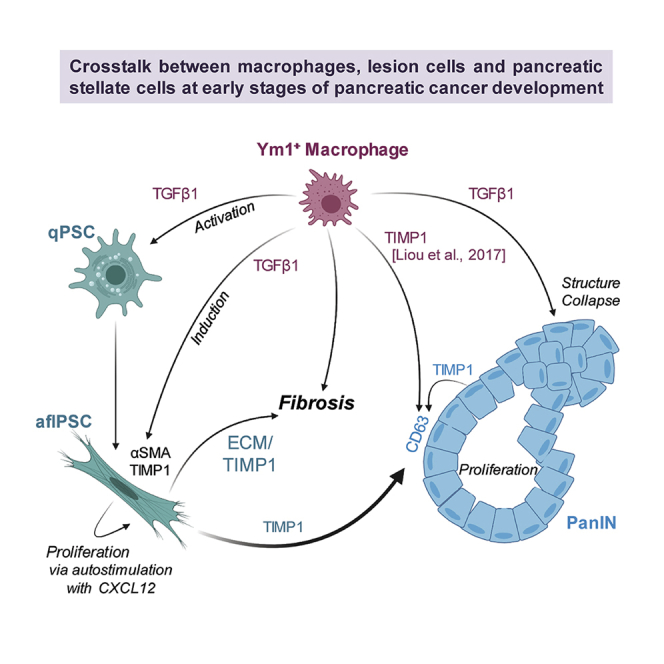
For a Figure360 author presentation of this figure, see https://doi.org/10.1016/j.isci.2022.104327
Highlights
-
•
Ym1+ macrophages are the main drivers of fibrosis in developing mouse PDA
-
•
Ym1+ macrophages are the primary producers of TGFβ1 and TIMP1
-
•
TGFβ1 activates quiescent PSCs and induces them to produce αSMA
-
•
TGFβ1 can cause a collapsed PanIN structure via an EMT-like event
Microenvironment; Immunology; Cancer
Introduction
The desmoplastic reaction is the growth of fibrous tissue around pancreatic lesions and the resulting deposition of stromal collagen and extracellular matrix (ECM) provides a protective barrier for these structures ((Ozdemir et al., 2014; Provenzano et al., 2012); reviewed in (Perez et al., 2021)). Fibrogenesis occurs in response to pancreatic inflammation in pancreatitis and during development of pancreatic cancer, and was linked to activation of quiescent, tissue-resident pancreatic stellate cells (qPSCs) (reviewed in (Apte et al., 2015)). Upon activation, qPSCs undergo a change in morphology to a myofibroblast-like phenotype (Bachem et al., 1998). These activated myofibroblast-like PSCs (aflPSCs) express alpha smooth muscle actin (αSMA) and have a central role in producing the collagen-rich stroma around early premalignant lesions and established tumors (Apte et al., 1999, 2004, 2013)). Therefore, both, presence of αSMA+ cells and collagen deposition can serve as a bona-fide marker for the fibrosis surrounding pancreatic lesions (Shi et al., 2014).
PSCs regulate the ECM by producing both matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) (Phillips et al., 2003), and TIMP1 is increased during pancreatic cancer progression (D'Costa et al., 2017). TIMP1 is best known as an inhibitor of soluble MMPs, and therefore promotes fibrosis and collagen deposition by preventing their ECM-degrading functions (Iredale et al., 1996; Yoshiji et al., 2000). However, TIMP1 also exerts signaling functions via activation of its receptor CD63 (Jung et al., 2006).
A population of alternatively activated macrophages (AAM), best characterized by expression of Chil3 (encodes Ym1), Arg1, Fizz1, IL1rn, and Il10, can stimulate the growth of early pancreatic lesions (low-grade PanIN) via secretion of CCL2, IL-1ra, and other factors (Bastea et al., 2019; Liou et al., 2017). Recent work suggests that these Ym1+ AAM at the earliest stages of tumor development do not originate from expansion of a tissue-resident population (Pandey et al., 2021), but rather from infiltrating inflammatory macrophages after a polarization switch that is mediated by IL-13 secretion from PanIN (Liou et al., 2017). Although a depletion of Ym1+ macrophages (i.e. via neutralization of IL-13) in the p48cre;LSL-KrasG12D (KC) animal model suggested that fibrosis is orchestrated by these cells (Bastea et al., 2019; Liou et al., 2017), their exact contribution to this process is still ill-defined. For example, it is unclear if they secrete factors that activate PSC directly and drive further proliferation of activated PSC, or if they act indirectly through regulating other cell types in the lesion microenvironment.
Here, we used primary PSCs, primary Ym1+ macrophages (peritoneal or bone-marrow-derived), as well as PanIN organoids to determine this crosstalk. We found that by providing critical signaling through TGFβ1 and TIMP1, Ym1+ macrophages have crucial roles in driving fibrosis and progression of early lesions and thus the initiation of pancreatic ductal adenocarcinoma (PDA).
Results
The depletion of Ym1+ macrophages decreases αSMA+ fibroblasts and fibrosis around low-grade (LG) PanIN lesions
Alternatively activated (M2), Ym1+ macrophages have important roles in the initiation of pancreatic cancer in the p48cre;LSL-KrasG12D (KC) animal model (Liou et al., 2013, 2015, 2017). Depletion of this macrophage population can be obtained by neutralization of IL-13 (Figure 1A (Liou et al., 2017)), which results in a loss of fibrosis around PanIN lesions (Figure 1B), specifically due to a significant decrease in αSMA+ fibroblasts of more than 2-fold (Figure 1C), which also is reflected in a 2-fold decrease in collagen deposition (Figure 1D). While this suggests that Ym1+ macrophages control fibrosis at early stages of tumor development, the underlying crosstalk between cell types remains unclear. By using primary cells and PanIN organoids, we here investigated how Ym1+ macrophages crosstalk with the two other major cell types in precancerous lesion areas, αSMA+ fibroblasts and PanIN cells, to obtain this effect.
Figure 1.

Depletion of Ym1+ macrophages results in a decrease in fibrosis around precancerous lesions
(A-D) Three-week-old p48cre;LSL-KrasG12D or control mice were treated with mIL-13 neutralizing antibody or isotype matching control antibody for 7 weeks.
(A) Pancreas tissue samples were analyzed for Ym1+ macrophages using IHC. Shown are representative images (left side; scale bars indicate 50 μm), as well as a quantification analyses of the fold change of Ym1+ cells in whole pancreas sections from n = 3 mice per group. For statistical analysis, data were transformed via arcsine transformation before a t test was performed. Error bars indicate standard deviation. The p-value (p = 0.0290) indicates statistical significance.
(B) Pancreas tissue samples from indicated groups were stained with H&E and representative pictures are shown. The scale bar indicates 50 μm.
(C and D) Pancreas tissue samples were analyzed for expression of αSMA (marker for aflPSC) using IHC (C) or for collagen deposition using a Trichrome staining method (D). Shown are representative images (left side; scale bars indicate 50 μm), as well as quantification analyses of the fold change in signal in whole pancreas sections from n = 3 mice per group. For statistical analysis, data were transformed via arcsine transformation before a t-test was performed. Error bars indicate standard deviation. The p-values (p = 0.0252 in C and p = 0.0316 in D) indicate statistical significance.
Pancreatic Ym1+ macrophages produce TGFβ1
Previously, it was shown that in KC mice, inflammatory macrophages (IM) infiltrate the pancreas to induce acinar-to-ductal metaplasia (ADM) (Liou et al., 2015). After progression to low-grade PanIN, lesions produce IL-13 to induce a polarization switch to alternatively activated macrophages (AAM). A characteristic marker for this AAM subpopulation is Ym1 (encoded by Chil3), but they also express Arg1, Fizz1, IL1rn, and Il10 (Bastea et al., 2019). Because the origin of infiltrated macrophages is still under discussion, in our studies, we analyzed both, bone-marrow-derived macrophages (BMDM) and peritoneal macrophages (PM), and found that when acquiring M2 polarization, both express Ym1 (Figure 2A and 2B) and additional markers CD206/Mrc1, Trem2, and C1qb (Figure S1A) that recently have been shown to be characteristic for this pancreas-relevant population (Chen et al., 2021; Kemp et al., 2021), as well as decreased expression of the M1 marker Cd38 (Figure S1B). In addition, Ym1+ macrophages from both sources also express TGFβ1 (Figures 2A, 2B, and S1C), which is activated during fibrosis and has a known function in wound healing and tissue repair (reviewed in (Huet et al., 2019)). Analysis of pancreatic tissue surrounding PanIN lesions confirmed that Ym1+ macrophages produce TGFβ1 in situ, as shown with a TGFβ1 (Y369) antibody, which recognizes the TGFβ1 proprotein (Figure 2C). Moreover, the depletion of this macrophage population led to an approximately 90% decrease in TGFβ1-producing cells (Figures 2D and 2E), indicating that Ym1+ macrophages indeed are the source for this cytokine in the abnormal pancreas areas.
Figure 2.

Pancreatic Ym1+ macrophages produce TGFβ1
(A and B) Macrophages were isolated from murine bone marrow (BMDM) or peritoneal cavity (PM) and polarized using 10 ng/mL LPS and 20 ng/mL IFNγ (M1) or 20 ng/mL IL-4 (M2). qPCR analysis indicates Ym1+ M2 macrophages produce Tgfb1/TGFβ1. Error bars represent the standard deviation and using the t-test, p-values indicate statistical significance.
(C) IF-IHC for Ym1 and TGFβ1(Y369) with nuclei stained by DAPI. Subsequently, an H&E staining was done. Scale bar is 50 μm.
(D and E) Pancreas tissue samples were examined for expression of TGFβ1(Y369). Representative images are shown with 50 μm scale bars (D). Quantification of TGFβ1(Y369) was done on whole tissue slide area of n = 5 control or n = 3 Ym1+ macrophage-depleted tissues. Error bars represent the standard deviation and using the t-test, p-values indicate statistical significance.
TGFβ1 activates quiescent PSCs and increases SMA expression in activated, fibroblast-like PSCs
We next investigated if TGFβ1 affects pancreatic stellate cells, either in their quiescent state (qPSC) or once they are activated αSMA-positive fibroblast-like cells (aflPSCs). Freshly isolated primary PSCs, when stimulated with TGFβ1, within 2 days, undergo a transformation to a myofibroblast-like phenotype (Figures 3A and S2A) and upregulate αSMA, collagen type I α1, fibronectin, desmin, and periostin, all aflPSC-relevant markers (Figures 3B and S2B). Additionally, the expression of αSMA in activated pancreatic stellate cells is further increased with TGFβ1 (Figure 3C). It should be noted that neither TGFβ1 nor supernatant from either M2 (AA) macrophages, PanIN organoids, or PanIN organoids stimulated with M2 macrophage-conditioned media was capable of driving further proliferation of the aflPSC population (Figures S2C–S2E). This suggests that proliferation of aflPSCs most likely is mediated by autocrine/paracrine signaling. To test this, we performed a cytokine array on media conditioned by aflPSCs, in which we found M-CSF low and CCL2 (JE/MCP-1), CXCL12 (SDF-1), and TIMP1 highly abundant (Figure 3D). While CCL2 and M-CSF rather have roles in monocyte/macrophage recruitment and survival (Boring et al., 1997; Lu et al., 1998; Stanley et al., 1997), CXCL12 previously was implicated in hepatic stellate cell proliferation (Hong et al., 2009). Indeed, blocking antibodies for CXCL12 led to a significant decrease in proliferation of aflPSCs (Figure 3E), suggesting that they auto-stimulate through this chemokine.
Figure 3.

TGFβ1 activates quiescent PSCs and increases SMA and TIMP1 expression in activated, αSMA-positive, fibroblast-like PSCs
(A) Brightfield images of freshly isolated qPSCs treated with TGFβ1 or vehicle control for 48 h. Scale bar indicates 100 μm.
(B) qPCR analysis of aflPSC markers in freshly isolated PSCs treated with TGFβ1 or vehicle control for 48 h. Error bars represent the standard deviation. The t-test indicates statistical significance.
(C): Acta2/αSMA mRNA expression in response to treatment of aflPSCs with TGFβ1. Error bars represent the standard deviation, and the t-test indicates statistical significance.
(D) Array to detect cytokines secreted by aflPSCs.
(E) Proliferation assay for aflPSCs treated with CXCL12 neutralizing antibody (NAB) or isotype control. Error bars represent the standard deviation, and the t-test indicates statistical significance.
TIMP1 expression in activated PSCs is mediated by TGFβ1 signaling and contributes to growth of LG-PanIN lesions
Besides the previously mentioned factors, we also detected high levels of TIMP1 in media conditioned by aflPSC (Figure 3D). But unlike TGFβ1, TIMP1 failed to activate freshly isolated quiescent pancreatic stellate cells (Figure 4A), nor did it contribute to aflPSC proliferation (Figure S3A). A comparison of TIMP1 mRNA expression showed that all three major cell types in the pancreatic lesion microenvironment produce TIMP1; however, aflPSCs show highest expression of this factor (Figure 4B). This was confirmed by an ISH for TIMP1 mRNA in tissue of KC mice, which showed that stromal components (fibroblasts) around lesions indeed are major producers of TIMP1 (Figure 4C). TIMP1 expression in aflPSCs is further enhanced by the macrophage-secreted factor TGFβ1 (Figure 4D). Similarly, TGFβ1 increases TIMP1 expression in freshly isolated PSCs (Figure S3B). Moreover, depletion of Ym1+ macrophages in KC pancreata completely abolishes TIMP1 expression in fibroblasts surrounding lesions (Figure 4E). Moreover, after depletion of Ym1+ macrophages, the remaining SMA-positive fibroblasts show no expression of TIMP1 (Figure 4F), suggesting that TIMP1 expression in these fibroblasts most likely is regulated by AAM.
Figure 4.

TIMP1 expression is enhanced by TGFβ1 signaling in aflPSCs
(A) Brightfield images of PSCs treated with recombinant TIMP1 or vehicle control. The scale bar indicates 100 μm.
(B) qPCR analysis for Timp1 in PanIN cells, aflPSCs, Ym1+ BMDM, and Ym1+ PM. Statistical significance determined by the t-test and error bars represent the standard deviation.
(C) In situ hybridization (ISH) for Timp1 mRNA in brown combined with IHC for Ym1 protein in pink on KC mouse tissue. Scale bar indicates 100 μm.
(D) qPCR analysis of Timp1 in aflPSCs treated with TGFβ1 or vehicle. Error bars represent standard deviation and the t-test was used to determine statistical significance.
(E) IHC for TIMP1 protein (brown) in control or Ym1+ macrophage-depleted KC tissue. Scale bars indicate 50 μm.
(F) Pancreas tissue from p48cre;LSL-KrasG12D mice treated with mIL-13 neutralizing antibody or isotype matching control antibody was stained for SMA (red) and TIMP1 (green). Nuclei were visualized using DAPI and scale bars indicate 50 μm.
Extracellular matrix metalloproteinases (MMPs) such as MMP9 and their specific inhibitors such as TIMP1 regulate matrix metabolism and are biomarkers of fibrosis (Lachowski et al., 2019; Phillips et al., 2003). However, TIMP1 also can have signaling function through the surface receptor CD63 (Jung et al., 2006). We found this receptor highly expressed in PanIN cells of pancreata of KC mice as well as in PanIN organoids (Figures 5A, 5B and S3C). Stimulation of PanIN organoids with TGFβ1 did not enhance CD63 expression, indicating that it is regulated independently of this factor (Figure 5C). We then determined the effects of TIMP1/CD63 signaling in PanIN lesions. Treatment of PanIN organoids with recombinant TIMP1 over 4 days significantly increased their growth as measured by determining the ductal area (Figures 5D and 5E), indicating a growth-promoting effect. TIMP1-induced increases in ductal area were reduced in presence of a CD63 neutralizing antibody, indicating that TIMP1 indeed induces its effects on PanIN lesion growth via its receptor CD63 (Figure 5F).
Figure 5.

TIMP1 increases growth of LG-PanINs via its receptor CD63
(A) Representative picture of an IHC for CD63 (brown) in normal acinar, stromal, and lesion areas of KC tissue. Scale bars indicate 50 μm.
(B) Quantification of CD63 expression in acinar, stromal, and lesion areas (n = 15 each). Points are graphed in a box and whiskers plot, where the whiskers extend to the minimum and maximum and the midline of the box indicates the median. Statistical significance was determined by a one-way ANOVA(p < 0.0001) followed by t-tests, p-values noted in figure.
(C) qPCR for TGFβ1 or vehicle-treated PanIN organoids. Error bars represent the standard deviation and statistical significance was determined by the t-test.
(D) TIMP1 increases the growth of duct-like structures. PanIN cells were seeded on top of Matrigel and treated with vehicle control or TIMP1 (50 ng/mL) for 4 days. Pictures show a representative area of single cells seeded at day 0 or ductal structures developed at day 4. The scale bar indicates 50 μm.
(E) Quantification of areas of ductal structures in control (n = 68) or TIMP1 (n = 63)-stimulated organoids on day 4. For statistical analysis of data, a t-test was performed. The p-value (p = 0.0043) indicates statistical significance. The line indicates the median.
(F) Quantification of areas of ductal structures in TIMP1-stimulated organoids that were incubated with 1 μg/mL control (IgG) or CD63 neutralization antibody (NOVUS, NBP2-42225SS). On day 4, size of ducts was analyzed (n = 276 for control; n = 404 for CD63-NAB). For statistical analysis of data, a t-test was performed. The p-value (p < 0.0001) indicates statistical significance. The line indicates the median.
Additional functions of TGFβ1-TIMP1 signaling on PanIN lesion growth
While TIMP1 regulates PanIN lesion cell growth via CD63, TGFβ1 has a possible role in further progression toward a more invasive phenotype. We found that established PanIN organoids, after treatment with TGFβ1 alter their phenotype from well-organized duct-like structures to collapsed, disorganized tumor-like structures (Figure 6A). This goes along with a decrease in expression of ZO-1 and E-cadherin, and an increase in N-cadherin, suggesting an EMT event (Figures 6B and S4). Such collapsed areas can be detected in some PanIN areas in KC mice, and cells in these areas stain positive for nuclear SMAD4, an indicator for TGFβ1 activity (Figure 6C) and EMT (Whittle et al., 2015). Moreover, an analysis of tissue for N-Cadherin expression in collapsed versus non-collapsed PanIN areas in KC tissue showed that it correlated with SMAD4 expression (Figure 6D).
Figure 6.

TGFβ1 drives EMT-like structural changes in PanIN Cells
(A) PanIN organoids were treated with vehicle control or TGFβ1 three days after plating on top of Matrigel, and then imaged on days one and five post treatment. Scale bars indicate 100 μm.
(B) qPCR analysis for Cdh1 (e-cadherin) and Cdh2 (n-cadherin) in vehicle or TGFβ1-treated PanIN organoids. Error bars indicate standard deviation and the t-test was used to determine statistical significance.
(C) Left side: IHC for SMAD4 (brown) in KC mouse tissue. Images show collapsed PanIN structures (top, arrows) and non-collapsed PanIN structures (bottom image). Scale bars indicate 100 μm. Right side: Quantification of SMAD4 expression in non-collapsed (n = 46) and collapsed PanIN (n = 45) areas. Points are graphed in a box and whiskers plot, where the whiskers extend to the minimum and maximum and the midline of the box indicates the median. Statistical significance was determined by a t-test.
(D) Tissue from KC mice was stained for N-cadherin (pink) and SMAD4 (green). DAPI was used to visualize nuclei. The scale bars indicate 50 μm.
Taken together, our data indicate that Ym1+ macrophages in the initiation phase of pancreatic cancer regulate fibrosis, lesion growth, and progression mainly through TGFβ1. TGFβ1 not only activates quiescent PSCs but also increases expression of αSMA and TIMP1 in activated PSCs. While TIMP1 has a role in fibrosis as well as PanIN cell proliferation via its receptor CD63, TGFβ1 impacts the morphology of PanIN cells by mediating a collapse to a disorganized structure, which may lead to progression.
Discussion
Fibrosis is an event occurring early in pancreatic cancer development, and in established tumors can be a key barrier for efficient treatment of PDA (Hessmann et al., 2018; Olive et al., 2009). This desmoplastic reaction is regulated via the three major cell types in lesion/tumor areas: lesion cells, macrophages, and fibroblasts (Storz and Crawford, 2020). PDA cancer-associated fibroblasts (CAFs) can have different origins (Manoukian et al., 2021), but one major source is pancreas-resident quiescent stellate cells (qPSCs) (Apte et al., 2004; Bachem et al., 2005; Ohlund et al., 2017). Recent in vivo lineage tracing using Fabp4-driven Cre indicated that only a portion of CAFs arise from PSCs, but that myCAFs with PSC origins have a tumor-promoting role (Helms et al., 2021). In vitro, primary PSCs show plasticity and can differentiate into both, inflammatory iCAFs, which generate an immunosuppressive microenvironment, and myofibroblast-like myCAFs (Ohlund et al., 2017). Using primary cells in in vitro assays, we here show that fibroblasts at low-grade (LG) PanINs can originate from qPSCs. Specifically, we uncover TGFβ1-and TIMP1-driven crosstalk signaling between AAMs, PSCs, and PanIN lesions that regulate fibrosis and lesion growth.
Macrophages contribute to pancreatic malignancy at several levels. While inflammatory macrophages (IM) can induce ADM and drive formation of low-grade lesions (Liou et al., 2013, 2015), alternatively activated Ym1+ macrophages regulate fibrosis and contribute to PanIN growth (Liou et al., 2017). The polarization switch between IMs and AAMs in the lesion microenvironment is mediated by IL-13 which is mainly produced by DCLK1+ PanIN cells (Liou et al., 2017). We now provide mechanistic insight of how depletion of Ym1+ AAMs leads to a decrease in fibrosis around precancerous lesions, as shown by decrease in SMA+ cells and collagen deposition (Figure 1). We found that Ym1+ AAMs are the primary producers of TGFβ1 (Figure 2) and provide evidence that TGFβ1 activates qPSCs (Figures 3A and 3B) and induces aflPSCs to increase expression of αSMA (Figure 3C) and TIMP1 (Figure 4D), thereby contributing to fibrosis.
An interesting finding is that neither TGFβ1, nor AAM-conditioned or PanIN organoid-conditioned media increased proliferation of PSCs once they were activated (Figure S2). Instead, we found that aflPSCs regulated their proliferation via autostimulation through production of CXCL12 (Figures 3D and 3E), a known regulator of fibrosis in lung (Li et al., 2020; Phillips et al., 2004; Xu et al., 2007) and liver (Akcora et al., 2018). CXCL12 also has been shown to decrease CD8+ T cells in tumor areas (Ene-Obong et al., 2013; Garg et al., 2018). This suggests a potential opportunity for therapeutical intervention, for example by using neutralizing antibodies for CXCL12 or its receptors CXCR4 and CXCR7.
One mechanism of how aflPSCs control fibrosis at LG lesions is via upregulation of TIMP1, an inhibitor of soluble MMPs to prevent ECM degradation and to promote fibrosis and collagen deposition (Iredale et al., 1996; Yoshiji et al., 2000). Out of the three major cell types in the lesion microenvironment, aflPSCs are the main producers of TIMP1, and TIMP1 expression is even further enhanced with TGFβ1 (Figures 4B-4D). Because TGFβ1 is mainly produced by Ym1+ AAMs (Figure 2), the depletion of this cell type in vivo abolished presence of TIMP1 in the lesion microenvironment (Figures 4E and 4F), as well as collagen deposition and fibrosis (Figure 1). The latter may be due to additional effects on matrix metalloproteinase activity, since TGFβ can also decrease MMP expression in PSCs (Tjomsland et al., 2016). Our data also show that TIMP1, via activation of its receptor CD63, has an additional function as a stimulator of PanIN lesion growth (Figures 5D-5F). This is in line with work showing that TIMP1 induces hyperproliferation of KRASG12D-transformed pancreatic ductal cells (Botta et al., 2013). Moreover, we found that TGFβ1 can cause a collapsed PanIN structure possibly via an EMT-like event (Figures 6A–6D and S4), similar as observed in the KPC mouse model (Whittle et al., 2015).
In preclinical animal models, multiple tumor-promoting functions have been assigned to TIMP1. For example, chemoresistance to gemcitabine can be reverted by the downregulation of TIMP1 (Tan et al., 2021). Moreover, TIMP1 produced in pancreatic premalignant lesions can activate hepatic stellate cells and create a metastatic niche in the liver (Grunwald et al., 2016). Consequently, for human pancreatic adenocarcinoma, TIMP1 in serum has been identified as a diagnostic and prognostic biomarker (Jenkinson et al., 2015; Poruk et al., 2013), and high expression of TIMP1 in pancreatic tumors correlates with shorter patient survival (Tan et al., 2021).
The stroma and fibrosis around pancreatic lesions has been shown to sequester T cells and block them from reaching tumor cells (Ene-Obong et al., 2013), but also to prevent efficient delivery of gemcitabine to tumor cells (Hessmann et al., 2018; Olive et al., 2009). However, because different subtypes of fibroblasts can have tumor restraining or tumor-promoting roles, specific populations need to be defined and targeted. This becomes evident by approaches to target αSMA+ cells (Chen et al., 2021; Ozdemir et al., 2014) or sonic hedgehog (SHH) (Lee et al., 2014; Rhim et al., 2014), which both resulted in more rapid tumor progression and decreased survival in mice. Likewise, high stromal density in patients has been associated with increased survival (Bever et al., 2015; Torphy et al., 2018). In contrast, recent reports show that decreased patient survival is associated with high levels of type I collagen (Elahi-Gedwillo et al., 2019), and mouse studies using antifibrotic therapies showed promising results (Elahi-Gedwillo et al., 2019; Ernsting et al., 2015).
Our data now suggest that Ym1+ AAMs are the main drivers of fibrogenesis in mouse PDA (Figure 1 and (Bastea et al., 2019; Liou et al., 2017)) through TGFβ1 and TIMP1 (Graphical abstract). An additional effect of targeting this population may be an increase in infiltrating activated T cells to KPC syngeneic tumors (Bastea et al., 2019). Therefore, our data suggest targeting AAMs for therapeutic benefit.
Limitations of the study
While our data are strengthened by using primary cells, it needs to be noted that Ym1+ is a mouse-specific AAM marker and the equivalent human AAM population is not defined, yet. Nevertheless, for human PDA, survival data indicate presence of AAMs and TGFβ1 are each predictive of decreased survival (Liu et al., 2020), and activated stroma with a macrophage-associated gene signature also indicates poorer survival (Moffitt et al., 2015). Thus, our data are in line with literature supporting therapeutic targeting of AAMs.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-CD63 | Sigma-Aldrich | Cat# SAB5700799; RRID:AB_2904550 |
| Rabbit monoclonal anti-CD63 (clone EPR5702) | Abcam | Cat# ab134045; RRID:AB_2800495 |
| Mouse monoclonal anti-CD63 (clone H5C6) | Novus | Cat# NBP2-42225; RRID:AB_2884028 |
| Mouse monoclonal anti-E-cadherin | BD Biosciences | Cat# 610,181; RRID:AB_397580 |
| Rabbit monoclonal anti-GAPDH (clone D16H11) | Cell Signaling Technology | Cat# 5174; RRID:AB_10622025 |
| Mouse monoclonal anti-mIL-13-IgG | InvivoGen | Cat# mabg-mil13; RRID:AB_2722583 |
| Mouse monoclonal IgG1 isotype control | R&D Systems | Cat# MAB002; RRID:AB_357344 |
| Rabbit monoclonal anti-N-cadherin (clone EPR1791-4) | Abcam | Cat# ab76011; RRID:AB_1310479 |
| Sheep polyclonal anti-N-cadherin | R&D Systems | Cat# AF6426; RRID: AB_10718850 |
| Rabbit polyclonal anti-Periostin | Abcam | Cat# ab14041; RRID:AB_2299859 |
| Rabbit polyclonal anti-Smad4 | Cell Signaling Technology | Cat# 9515; RRID:AB_2193344 |
| Rabbit polyclonal anti-Alpha smooth muscle actin | Abcam | Cat# ab5694, RRID:AB_2223021 |
| Goat polyclonal anti-TIMP1 | R&D Systems | Cat# AF980; RRID:AB_355759 |
| Rabbit polyclonal anti-Y369-TGFβ1 | Boster Biological Technology | Cat# A00019Y369; RRID:AB_2904551 |
| Goat polyclonal anti-Ym1 | R&D Systems | Cat# AF2446; RRID:AB_2079008 |
| Rabbit polyclonal anti-Ym1 | Stem Cell Technologies | Cat# 60130; RRID:AB_2868482 |
| Mouse monoclonal anti-ZO1 (clone 1A12) | Invitrogen | Cat# 33-9100; RRID:AB_2533147 |
| Chemicals, peptides, and recombinant proteins | ||
| Recombinant murine IL4 | PeproTech | Cat# 214-14 |
| Recombinant murine IFNɣ | PeproTech | Cat# 315-05 |
| Recombinant human TGFβ1 | PeproTech | Cat# 100-21C |
| Recombinant mouse TIMP1 | R&D Systems | Cat# 980-MT |
| Critical commercial assays | ||
| CyQUANT cell proliferation assay | Invitrogen | Cat# C7026 |
| High Capacity cDNA RT Kit | Applied Biosystems | Cat# 4368814 |
| Proteome Profiler Mouse Cytokine Array Kit, Panel A | R&D Systems | Cat# ARY006 |
| miRNeasy Mini Kit | Qiagen | Cat# 217004 |
| RNeasy Plus Mini Kit | Qiagen | Cat# 74134 |
| RNAscope® Assay 2.5 HD Reagent Kit-Brown | Advanced Cell Diagnostics, Inc. | Cat# 322300 |
| Experimental models: Cell lines | ||
| Mouse: Primary peritoneal macrophages | Method: Zhang et al. (2008); Bastea et al. (2019); Pandey et al. (2021) | N/A |
| Mouse: Primary bone marrow-derived macrophages | Method: Zhang et al. (2008); Pandey et al. (2021) | N/A |
| Mouse: Primary pancreatic stellate cells | Method: Bastea et al. (2019) | N/A |
| Mouse: Primary PanIN organoids | Method: Agbunag et al. (2006); Liou et al. (2017) | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: Ptf1a/p48cre/+ and LSL-KrasG12D/+ mouse strains | Mayo Clinic, described in Liou et al. (2015) | N/A |
| Oligonucleotides | ||
| qPCR primer/probe: Gapdh | Thermo Fisher Scientific | Mm99999915_g1 |
| qPCR primer/probe: Acta2/SMA | Thermo Fisher Scientific | Mm00725412_s1 |
| qPCR primer/probe: C1qb | Thermo Fisher Scientific | Mm01179619_m1 |
| qPCR primer/probe: Cd38 | Thermo Fisher Scientific | Mm01220906_m1 |
| qPCR primer/probe: Cd63 | Thermo Fisher Scientific | Mm07294134_g1 |
| qPCR primer/probe: Cdh1/e-cadherin | Thermo Fisher Scientific | Mm01247357_m1 |
| qPCR primer/probe: Cdh2/n-cadherin | Thermo Fisher Scientific | Mm01162497_m1 |
| qPCR primer/probe: Chil3/Ym1 | Thermo Fisher Scientific | Mm00657889_mH |
| qPCR primer/probe: Col1a1 | Thermo Fisher Scientific | Mm00801666_g1 |
| qPCR primer/probe: Des/Desmin | Thermo Fisher Scientific | Mm00802453_m1 |
| qPCR primer/probe: Fn1/Fibronectin | Thermo Fisher Scientific | Mm01256744_m1 |
| qPCR primer/probe: Mrc1/CD206 | Thermo Fisher Scientific | Mm01329362_m1 |
| qPCR primer/probe: Tgfb1 | Thermo Fisher Scientific | Mm01178820_m1 |
| qPCR primer/probe: Timp1 | Thermo Fisher Scientific | Mm01341361_m1 |
| qPCR primer/probe: Trem2 | Thermo Fisher Scientific | Mm04209424_g1 |
| Software and algorithms | ||
| ImageJ | Schneider et al. (2012) | RRID:SCR_003070; https://imagej.nih.gov/ij/ |
| Aperio ImageScope v12.4.3 | Leica Biosystems | RRID:SCR_020993 |
| Aperio ImageScope Positive Pixel Algorithm | Leica Biosystems | https://www.leicabiosystems.com/us/analyze/ihc/aperio-positive-pixel-count-algorithm/ |
| GraphPad Prism v.9.2.0 | GraphPad Software | RRID:SCR_002798 |
| CaseViewer software v2.3.0.99276 | 3DHISTECH | RRID:SCR_017654 |
| Other | ||
| Aperio AT2 Digital Scanner | Leica Biosystems | RRID:SCR_021256 |
| Aperio FL Slide Scanner | Leica Biosystems | RRID:SCR_022191 |
| EVOS XL Core Microscope | Thermo Fisher Scientific | RRID:SCR_022190; Cat# AMEX1000 |
| Olympus IX71 Microscope | Olympus Corporation | RRID:SCR_022185 |
| Pannoramic 250 Flash III | 3DHISTECH | RRID:SCR_022184 |
| QuantStudio 7 Flex Real-Time PCR System | Applied Biosystems | RRID:SCR_020245 |
| Collagenase | Sigma-Aldrich | Cat# C0130 |
| 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI) | Sigma-Aldrich | Cat# D8417 |
| Lipopolysaccharides from E. coli | Sigma-Aldrich | Cat# L4391 |
| Matrigel | Corning Inc. | Cat# 354234 |
| Nycoprep Universal 60% solution (Nycodenz solution) | Accurate Chemical & Scientific Corporation | Cat# AN1106865 |
| Oil Red O | Sigma-Aldrich | Cat# 00625 |
| RNAscope in situ hybridization probe: Timp1 | Advanced Cell Diagnostics, Inc. | Cat# 316841 |
| Taqman Fast Mix 2x | Applied Biosystems | Cat# 4352042 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Peter Storz (storz.peter@mayo.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mouse lines
Ptf1a/p48cre/+ and LSL-KrasG12D/+ mouse strains and genotyping of mice have been described previously (Liou et al., 2015). Male and female mice were randomly distributed between treatment groups. Age of mice is noted for each experiment in the method details. All animal experiments were performed in accordance with relevant institutional and national guidelines and regulations and were approved by the Mayo Clinic IACUC committee.
Primary macrophages and pancreatic stellate cells
All primary mouse cells were cultured at 37°C with 5% CO2. Murine peritoneal-derived macrophages (PMs) were cultured in RPMI +10% FBS +1% Pen/Strep and bone marrow-derived macrophages (BMs) were cultured in DMEM/F-12 + 10% FBS +100 U/mL of macrophage colony stimulating factor (PeproTech, Cranbury, NJ). Primary murine pancreatic stellate cells were cultured in DMEM HG + 10% FBS +1% Pen/Strep. For proliferation assays and cytokine arrays, aflPSCs were cultured in serum-free media and while this decreased proliferation, as expected, no morphological changes or signs of stress were seen.
Culture of PanIN organoids
PanIN organoids were generated by seeding primary duct-like cells isolated from pancreata of 6 weeks old Pdx1Cre/+;KrasG12D/+ mice as previously described (Agbunag et al., 2006; Liou et al., 2017). Culture media was composed of DMEM/F-12 media (Sigma-Aldrich, St. Louis, MO) containing 5% FBS, 25 μg/mL bovine pituitary extract (Gibco/Thermo Scientific, Waltham, MA), 20 ng/mL EGF, 0.1 mg/mL soybean trypsin inhibitor type I (AMRESCO, Solon, OH), 5 mg/mL D-glucose (Sigma-Aldrich), 1.22 mg/mL nicotinamide (Sigma-Aldrich), 5 nM triiodo-L-thyronine (Sigma-Aldrich), 1 μM dexamethasone (Sigma-Aldrich), 100 ng/mL cholera toxin (Sigma-Aldrich), 5 mL/L insulin-transferrin-selenium (Corning, Corning, NY) and 100 U/mL penicillin/streptomycin (Gibco/Thermo Scientific) in a 37°C incubator supplemented with 5% CO2. For organoid culture, single cells were seeded on Matrigel (200 μL/well of a 24 well plate) and then stimulated with recombinant proteins or vehicle control, as indicated in the figure legends. The area of ductal structures formed was determined using ImageJ software (Schneider et al., 2012).
Method details
Antibodies and reagents
All antibodies and reagents are identified in the Key resources table. Additional information on dilution of all antibodies used for Western blotting, immunohistochemistry and immunofluorescence are provided in Table S1.
Depletion of Ym1+ macrophages in vivo
To deplete Ym1+ macrophages in vivo, 3 week old p48cre;LSL-KrasG12D mice were intraperitoneally (IP) injected with mIL-13 neutralizing antibody (or isotype control; both described in (Liou et al., 2017) at a dose of 1 mg/kg every other day for 7 weeks.
Primary macrophage isolation and polarization
Peritoneal macrophages (PM): Non-transgenic mice at 10-20 weeks of age were injected with 2-3 mL of 5% aged thioglycollate. Five days later, mice were euthanized and peritoneal macrophages were collected, as described in detail in (Pandey et al., 2021; Zhang et al., 2008). Briefly, 5-7 mL cold RPM1-1640 containing 10% FBS and 1% Penicillin-Streptomycin was injected into the peritoneum (21G needle) and after massaging the abdomen, the peritoneal wash was drawn back up into the syringe and transferred to a 50 mL tube. This step was repeated, and the cells were centrifuged (37 x g, 10m, 4°C). After collection, macrophages were plated onto 10 cm dishes, and once completely adhered, plates were washed 2-3 times to remove non-adherent cells. Fresh media was added, and cells were allowed to acclimatize overnight before further experimental manipulation. Bone marrow-derived macrophages (BMDM): The collection of BMDM from femurs of non-transgenic mice occurred as described in detail in (Pandey et al., 2021; Zhang et al., 2008). Briefly, the femur was cut and a syringe with a 26 ½ G needle was used to push 1 mL of PBS through the middle of the bone, causing the bone marrow to drop into a 50 mL tube. An additional 2-3 mL of PBS was pushed through to collect any remaining cells. Cells were then centrifuged at 500 x g for 10 min at room temperature. After isolation, 4 × 105 cells were added to 10 cm dishes in 10 mL macrophage complete medium. On days 3 and 5, 5 mL of medium was replaced with fresh complete medium. On day 7, the cells were polarized. Polarization of peritoneal or bone marrow-derived macrophages to a Ym1+ alternatively-activated phenotype was obtained by stimulation with IL-4 for 24 h as previously described (Liou et al., 2017).
Isolation of quiescent PSCs and activation
Quiescent PSCs were isolated from pancreata of 5-12 months old Ptf1a/p48cre/+;LSL-KrasG12D/+; mice as previously described (Bastea et al., 2019). Briefly, the pancreatic tissue was washed with PBS, minced, and digested for 1.5 h (37°C, 220 rpm) in HBSS +2 mg/mL collagenase +200 units DNase I (Sigma-Aldrich). The digestion was stopped and washed twice with cold HBSS +5% Fetal Bovine Serum (FBS). Following the washes, the cell pellet was resuspended in 5% FBS in HBSS and filtered through 500 and 105 μm nylon meshes, respectively. The cells were washed and resuspended in cold HBSS +0.3% BSA then mixed with 30% (w/v) Nycodenz + HBSS solution. The cell suspension was carefully layered under an HBSS +0.3% BSA solution and centrifuged at 1400 g, 4°C for 20 min. The cells were collected from the thin white band above the Nycodenz interface, washed, resuspended in DMEM HG + 10% FBS +1% Pen/Strep and transferred to a cell culture plate. After 48 h, the medium was changed daily. To obtain activated fibroblast-like PSC (aflPSC) cells were grown to confluency and then passaged as described in (Apte et al., 1998; Sparmann et al., 2004).
RNA extraction and quantitative PCR
RNA extraction was performed with the RNeasy Plus Mini Kit and miRNeasy Mini Kit, and cDNA was prepared using the High-Capacity cDNA RT Kit. TaqMan Fast Mix 2x was used to prepare qPCR reactions with the following TaqMan primer/probe sets: Gapdh (Mm99999915_g1), Acta2/SMA (Mm00725412_s1), C1qb (Mm01179619_m1), Cd38 (Mm01220906_m1), Cd63 (Mm07294134_g1), Cdh1/e-cadherin (Mm01247357_m1), Cdh2/n-cadherin (Mm01162497_m1), Chil3/Ym1 (Mm00657889_+mH), Col1a1 (Mm00801666_g1), Des/Desmin (Mm00802453_m1), Fn1/Fibronectin (Mm01256744_m1), Mrc1/CD206 (Mm01329362_m1), Tgfb1 (Mm01178820_m1), Timp1 (Mm01341361_m1), and Trem2 (Mm04209424_g1). Samples were run on a QuantStudio 7 Flex Real-Time PCR System. For analyses all CT values were normalized to Gapdh and fold changes were calculated using the ΔΔCT method.
Cytokine arrays
Indicated cells were starved for 48 h and supernatants were analyzed for a panel of 40 cytokines using a mouse cytokine array (Proteome Profiler Mouse Cytokine Array Kit Panel A).
Immunofluorescence (IF) and cell imaging
For IF, cells were washed in PBS, fixed in 4% paraformaldehyde in PBS for 15 min at 37°C, and washed again in PBS. Cells were then permeabilized using 0.1% Triton X-100 in PBS for 2 min at room temperature (RT) and then incubated with 100 mM glycine in PBS (2 min, RT) before blocking with 3% BSA, 0.05% Tween in PBS (30 min, RT) and incubating with primary antibodies (as listed in Table S1) overnight at 4°C. Cells were then washed in PBS before incubating with Alexa Fluor secondary antibodies (1:800; Invitrogen, Waltham, MA) and DAPI (0.25 μg/mL) for 2 h at RT. Cells were then washed and imaged. Brightfield and fluorescent images were taken using an Olympus IX71 or EVOS XL Core.
Oil red O staining
For Oil Red O staining, cells were fixed in 10% formalin for 1h at RT, washed with PBS, and then stained with Oil Red O working solution for 10 min at RT on rocker. Then cells were washed with PBS and imaged using an Olympus IX71 microscope. To obtain an Oil Red O working solution, first a stock solution (5 mg/mL in 100% isopropanol) was made and then the working solution was prepared just before use (3 parts oil red stock and 2 parts water; mixed, incubated for 10 min, and passed through a 0.2 μm filter).
Immunohistochemistry (IHC)
Slides were deparaffinized and rehydrated as previously described (Liou et al., 2016). Antigen retrieval was performed with 10 mM sodium citrate buffer (pH 6.0), and samples were then treated with H2O2 (3%, 5 min), washed with 0.5% Tween 20/PBS, and blocked (5 min, RT) with Protein Block Serum-Free Solution (Agilent Dako, Santa Clara, CA). For DAB immunohistochemistry, primary antibodies (as listed in Table S1) were diluted in Antibody Diluent Background Reducing Solution (DAKO) and visualized with the EnVision Plus Anti-Rabbit Labeled Polymer Kit (Agilent Dako). H&E and Trichrome staining (Masson Trichrome Stain Kit from Sigma-Aldrich) were performed as previously described (Pandey et al., 2021). For fluorescent immunohistochemistry (IF-IHC), slides were incubated with indicated primary antibodies (as listed in Table S1) in Antibody Diluent Background Reducing solution at 4°C overnight. After three washes with 0.05% Tween 20/PBS, Alexa Fluor 488 or 647 labeled secondary antibodies (Invitrogen) were added (1:500, RT) for 1 h with DAPI (0.5 μg/mL). LabVision PermaFluor (Thermo Fisher Scientific) was used as mounting medium. Brightfield images were captured using an Aperio ScanScope XT scanner and ImageScope software and fluorescent images were captured using a Pannoramic 250 Flash III Scanner and CaseViewer software.
sRNAscope in situ hybridization (ISH) combined with IHC
In situ hybridization (ISH) was performed using RNAscope® Assay 2.5 HD Reagent Kit-Brown (Advanced Cell Diagnostics [ACD], Hayward, CA) using the ACD mouse target probe Timp1 (NM_001044384.1, region 2-772), exactly as described in (Liou et al., 2017). The kit was used according to the manufacturer’s instructions, with an 8-min target retrieval and 15-min protease plus incubation. The mRNA signal was detected with DAB. To continue with IHC, ISH tissue samples were blocked with Protein Block Serum-Free Solution (Agilent Dako) for 30 min at RT, and then incubated overnight at 4 °C with indicated antibody diluted in Antibody Diluent Background Reducing Solution (Agilent Dako). After three washes (TBS), the slides were incubated with Rabbit AP-Polymer (Biocare Medical, Concord, CA) for 30 min at RT, rinsed with TBS (3 times), treated with Warp Red Chromogen Kit (Biocare Medical), counterstained with hematoxylin, dehydrated and mounted. Images were captured using the ScanScope XT scanner and ImageScope software.
Proliferation assays
For proliferation assays, the CyQUANT Cell Proliferation Assay kit (Invitrogen) was used per the manufacturer’s instructions and plates were read on a Synergy HT plate reader (BioTek, Winooski, VT). Cells were plated and allowed to attach for 6 h before changing the media to serum-free media and beginning treatment with TGFβ1 (50 ng/mL), TIMP1 (50 ng/mL), CXCL12 neutralizing antibody (1 μg/mL), vehicle control, or isotype control (1 μg/mL). For proliferation assays with conditioned media, cells were starved overnight before replacing media with conditioned or control media. To create conditioned media, media sat on cells for 24 h before being collected.
Western blotting
For Western blotting, cells were washed two times with ice-cold PBS (140 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4, pH 7.2) and lysed with lysis buffer (50 mM Tris-HCl, pH 7.4, 1% Triton X-100, 150 mM NaCl, 5 mM EDTA, pH 7.4) plus protease inhibitor cocktail (PIC; Sigma-Aldrich) for 30 min on ice. After addition of 2x Laemmli buffer, samples were subjected to SDS-PAGE, transferred to nitrocellulose membrane, and proteins of interest were detected using indicated specific primary antibodies (Table S1) and HRP-conjugated secondary antibodies.
Quantification and statistical analysis
All experiments (cell biological and biochemical) shown have been performed at least 3 times. If not stated otherwise in the figure legends, for quantification analyses in all animal experiments, pancreatic samples from n = 3 mice have been used. IHC data was quantified by manual counting of positive cells or by using the Aperio Positive Pixel Count Algorithm. Error bars represent ±SD. If not stated otherwise in the figure legends, p values were acquired with the unpaired Student’s t test with Welch’s correction using Graph Pad software (GraphPad Inc., La Jolla, CA). When pancreatic areas were compared, the data were transformed via arcsine transformation (ASIN(SQRT(proportion of abnormal tissue area to total tissue area))∗180/PI) before a t test was performed. p < 0.05 was considered statistically significant.
Acknowledgments
This work was supported by NIH grant CA229560. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The graphical abstract was created with BioRender.com. All authors have no conflict of interest. We thank Dr. Yan Asmann (Associate Professor of BioMedical Informatics, Mayo Clinic) for advice on the statistical analyses, and our colleagues in the Department for Cancer Biology for helpful discussions. We also thank the Champions For Hope (Funk-Zitiello Foundation) and the Gary Chartrand Foundation for supporting these studies.
Author contributions
Conceived and designed the experiments: AKFM, GYL, LIB, HRD, PS. Performed the experiments: AKFM, LIB, HRD, GYL, BHE. Analyzed the data: GYL, HRD, LIB, AKFM, PS. Wrote the paper: AKFM, PS.
Declaration of interests
The authors declare no competing interests.
Published: May 20, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104327.
Supplemental information
Data and code availability
Data reported in this paper will be shared by the Lead author upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.
References
- Agbunag C., Lee K.E., Buontempo S., Bar-Sagi D. Pancreatic duct epithelial cell isolation and cultivation in two-dimensional and three-dimensional culture systems. Methods Enzymol. 2006;407:703–710. doi: 10.1016/s0076-6879(05)07055-2. [DOI] [PubMed] [Google Scholar]
- Akcora B.O., Storm G., Bansal R. Inhibition of canonical WNT signaling pathway by beta-catenin/CBP inhibitor ICG-001 ameliorates liver fibrosis in vivo through suppression of stromal CXCL12. Biochim. Biophys. Acta Mol. Basis Dis. 2018;1864:804–818. doi: 10.1016/j.bbadis.2017.12.001. [DOI] [PubMed] [Google Scholar]
- Apte M., Pirola R.C., Wilson J.S. Pancreatic stellate cell: physiologic role, role in fibrosis and cancer. Curr. Opin. Gastroenterol. 2015;31:416–423. doi: 10.1097/mog.0000000000000196. [DOI] [PubMed] [Google Scholar]
- Apte M.V., Haber P.S., Applegate T.L., Norton I.D., McCaughan G.W., Korsten M.A., Pirola R.C., Wilson J.S. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43:128–133. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte M.V., Haber P.S., Darby S.J., Rodgers S.C., McCaughan G.W., Korsten M.A., Pirola R.C., Wilson J.S. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999;44:534–541. doi: 10.1136/gut.44.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte M.V., Park S., Phillips P.A., Santucci N., Goldstein D., Kumar R.K., Ramm G.A., Buchler M., Friess H., McCarroll J.A., et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas. 2004;29:179–187. doi: 10.1097/00006676-200410000-00002. [DOI] [PubMed] [Google Scholar]
- Apte M.V., Wilson J.S., Lugea A., Pandol S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology. 2013;144:1210–1219. doi: 10.1053/j.gastro.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachem M.G., Schneider E., Gross H., Weidenbach H., Schmid R.M., Menke A., Siech M., Beger H., Grunert A., Adler G. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115:421–432. doi: 10.1016/s0016-5085(98)70209-4. [DOI] [PubMed] [Google Scholar]
- Bachem M.G., Schunemann M., Ramadani M., Siech M., Beger H., Buck A., Zhou S., Schmid-Kotsas A., Adler G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology. 2005;128:907–921. doi: 10.1053/j.gastro.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Bastea L.I., Liou G.Y., Pandey V., Fleming A.K., von Roemeling C.A., Doeppler H., Li Z., Qiu Y., Edenfield B., Copland J.A., et al. Pomalidomide alters pancreatic macrophage populations to generate an immune-responsive environment at precancerous and cancerous lesions. Cancer Res. 2019;79:1535–1548. doi: 10.1158/0008-5472.can-18-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bever K.M., Sugar E.A., Bigelow E., Sharma R., Laheru D., Wolfgang C.L., Jaffee E.M., Anders R.A., De Jesus-Acosta A., Zheng L. The prognostic value of stroma in pancreatic cancer in patients receiving adjuvant therapy. HPB. 2015;17:292–298. doi: 10.1111/hpb.12334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boring L., Gosling J., Chensue S.W., Kunkel S.L., Farese R.V., Jr., Broxmeyer H.E., Charo I.F. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Invest. 1997;100:2552–2561. doi: 10.1172/jci119798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botta G.P., Reichert M., Reginato M.J., Heeg S., Rustgi A.K., Lelkes P.I. ERK2-regulated TIMP1 induces hyperproliferation of K-Ras(G12D)-transformed pancreatic ductal cells. Neoplasia. 2013;15:359–372. doi: 10.1593/neo.121708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Kim J., Yang S., Wang H., Wu C.J., Sugimoto H., LeBleu V.S., Kalluri R. Type I collagen deletion in αSMA+ myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell. 2021;39:548–565.e6. doi: 10.1016/j.ccell.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Costa Z., Jones K., Azad A., van Stiphout R., Lim S.Y., Gomes A.L., Kinchesh P., Smart S.C., Gillies McKenna W., Buffa F.M., et al. Gemcitabine-induced TIMP1 attenuates therapy response and promotes tumor growth and liver metastasis in pancreatic cancer. Cancer Res. 2017;77:5952–5962. doi: 10.1158/0008-5472.can-16-2833. [DOI] [PubMed] [Google Scholar]
- Elahi-Gedwillo K.Y., Carlson M., Zettervall J., Provenzano P.P. Antifibrotic therapy disrupts stromal barriers and modulates the immune landscape in pancreatic ductal adenocarcinoma. Cancer Res. 2019;79:372–386. doi: 10.1158/0008-5472.can-18-1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ene-Obong A., Clear A.J., Watt J., Wang J., Fatah R., Riches J.C., Marshall J.F., Chin-Aleong J., Chelala C., Gribben J.G., et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145:1121–1132. doi: 10.1053/j.gastro.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernsting M.J., Hoang B., Lohse I., Undzys E., Cao P., Do T., Gill B., Pintilie M., Hedley D., Li S.D. Targeting of metastasis-promoting tumor-associated fibroblasts and modulation of pancreatic tumor-associated stroma with a carboxymethylcellulose-docetaxel nanoparticle. J. Control Release. 2015;206:122–130. doi: 10.1016/j.jconrel.2015.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg B., Giri B., Modi S., Sethi V., Castro I., Umland O., Ban Y., Lavania S., Dawra R., Banerjee S., et al. NFκB in pancreatic stellate cells reduces infiltration of tumors by cytotoxic T cells and killing of cancer cells, via up-regulation of CXCL12. Gastroenterology. 2018;155:880–891.e8. doi: 10.1053/j.gastro.2018.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwald B., Harant V., Schaten S., Fruhschutz M., Spallek R., Hochst B., Stutzer K., Berchtold S., Erkan M., Prokopchuk O., et al. Pancreatic premalignant lesions secrete tissue inhibitor of metalloproteinases-1, which activates hepatic stellate cells via CD63 signaling to create a premetastatic niche in the liver. Gastroenterology. 2016;151:1011–1024.e7. doi: 10.1053/j.gastro.2016.07.043. [DOI] [PubMed] [Google Scholar]
- Helms E.J., Berry M.W., Chaw R.C., DuFort C.C., Sun D., Onate M.K., Oon C., Bhattacharyya S., Sanford-Crane H., Horton W., et al. Mesenchymal lineage heterogeneity underlies non-redundant functions of pancreatic cancer-associated fibroblasts. Cancer Discov. 2021;12:484–501. doi: 10.1158/2159-8290.CD-21-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessmann E., Patzak M.S., Klein L., Chen N., Kari V., Ramu I., Bapiro T.E., Frese K.K., Gopinathan A., Richards F.M., et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut. 2018;67:497–507. doi: 10.1136/gutjnl-2016-311954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F., Tuyama A., Lee T.F., Loke J., Agarwal R., Cheng X., Garg A., Fiel M.I., Schwartz M., Walewski J., et al. Hepatic stellate cells express functional CXCR4: role in stromal cell-derived factor-1α-mediated stellate cell activation. Hepatology. 2009;49:2055–2067. doi: 10.1002/hep.22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huet E., Jaroz C., Nguyen H.Q., Belkacemi Y., de la Taille A., Stavrinides V., Whitaker H. Stroma in normal and cancer wound healing. FEBS J. 2019;286:2909–2920. doi: 10.1111/febs.14842. [DOI] [PubMed] [Google Scholar]
- Iredale J.P., Benyon R.C., Arthur M.J., Ferris W.F., Alcolado R., Winwood P.J., Clark N., Murphy G. Tissue inhibitor of metalloproteinase-1 messenger RNA expression is enhanced relative to interstitial collagenase messenger RNA in experimental liver injury and fibrosis. Hepatology. 1996;24:176–184. doi: 10.1002/hep.510240129. [DOI] [PubMed] [Google Scholar]
- Jenkinson C., Elliott V., Menon U., Apostolidou S., Fourkala O.E., Gentry-Maharaj A., Pereira S.P., Jacobs I., Cox T.F., Greenhalf W., et al. Evaluation in pre-diagnosis samples discounts ICAM-1 and TIMP-1 as biomarkers for earlier diagnosis of pancreatic cancer. J. Proteomics. 2015;113:400–402. doi: 10.1016/j.jprot.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Jung K.K., Liu X.W., Chirco R., Fridman R., Kim H.R.C. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 2006;25:3934–3942. doi: 10.1038/sj.emboj.7601281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp S.B., Steele N.G., Carpenter E.S., Donahue K.L., Bushnell G.G., Morris A.H., The S., Orbach S.M., Sirihorachai V.R., Nwosu Z.C., et al. Pancreatic cancer is marked by complement-high blood monocytes and tumor-associated macrophages. Life Sci. Alliance. 2021;4 doi: 10.26508/lsa.202000935. e202000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachowski D., Cortes E., Rice A., Pinato D., Rombouts K., Del Rio Hernandez A. Matrix stiffness modulates the activity of MMP-9 and TIMP-1 in hepatic stellate cells to perpetuate fibrosis. Sci. Rep. 2019;9:7299. doi: 10.1038/s41598-019-43759-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.J., Perera R.M., Wang H., Wu D.C., Liu X.S., Han S., Fitamant J., Jones P.D., Ghanta K.S., Kawano S., et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc. Natl. Acad. Sci. U S A. 2014;111:E3091–E3100. doi: 10.1073/pnas.1411679111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Xu X., Geng J., Wan X., Dai H. The autocrine CXCR4/CXCL12 axis contributes to lung fibrosis through modulation of lung fibroblast activity. Exp. Ther. Med. 2020;19:1844–1854. doi: 10.3892/etm.2020.8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou G.Y., Bastea L., Fleming A., Doppler H., Edenfield B.H., Dawson D.W., Zhang L., Bardeesy N., Storz P. The presence of interleukin-13 at pancreatic ADM/PanIN lesions alters macrophage populations and mediates pancreatic tumorigenesis. Cell Rep. 2017;19:1322–1333. doi: 10.1016/j.celrep.2017.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou G.Y., Doppler H., DelGiorno K.E., Zhang L., Leitges M., Crawford H.C., Murphy M.P., Storz P. Mutant KRas-induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016;14:2325–2336. doi: 10.1016/j.celrep.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou G.Y., Doppler H., Necela B., Edenfield B., Zhang L., Dawson D.W., Storz P. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015;5:52–63. doi: 10.1158/2159-8290.cd-14-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou G.Y., Doppler H., Necela B., Krishna M., Crawford H.C., Raimondo M., Storz P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-κB and MMPs. J. Cell Biol. 2013;202:563–577. doi: 10.1083/jcb.201301001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Wu H., Li Y., Zhang R., Kleeff J., Zhang X., Cui M., Liu J., Li T., Gao J., et al. Combined blockade of TGf-β1 and GM-CSF improves chemotherapeutic effects for pancreatic cancer by modulating tumor microenvironment. Cancer Immunol. Immunother. 2020;69:1477–1492. doi: 10.1007/s00262-020-02542-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B., Rutledge B.J., Gu L., Fiorillo J., Lukacs N.W., Kunkel S.L., North R., Gerard C., Rollins B.J. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med. 1998;187:601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoukian P., Bijlsma M., van Laarhoven H. The cellular origins of cancer-associated fibroblasts and their opposing contributions to pancreatic cancer growth. Front. Cell Dev. Biol. 2021;9:743907. doi: 10.3389/fcell.2021.743907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt R.A., Marayati R., Flate E.L., Volmar K.E., Loeza S.G., Hoadley K.A., Rashid N.U., Williams L.A., Eaton S.C., Chung A.H., et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015;47:1168–1178. doi: 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlund D., Handly-Santana A., Biffi G., Elyada E., Almeida A.S., Ponz-Sarvise M., Corbo V., Oni T.E., Hearn S.A., Lee E.J., et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017;214:579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive K.P., Jacobetz M.A., Davidson C.J., Gopinathan A., McIntyre D., Honess D., Madhu B., Goldgraben M.A., Caldwell M.E., Allard D., et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir B.C., Pentcheva-Hoang T., Carstens J.L., Zheng X., Wu C.C., Simpson T.R., Laklai H., Sugimoto H., Kahlert C., Novitskiy S.V., et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey V., Fleming-Martinez A., Bastea L., Doeppler H.R., Eisenhauer J., Le T., Edenfield B., Storz P. CXCL10/CXCR3 signaling contributes to an inflammatory microenvironment and its blockade enhances progression of murine pancreatic precancerous lesions. Elife. 2021;10:e60646. doi: 10.7554/elife.60646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez V.M., Kearney J.F., Yeh J.J. The PDAC extracellular matrix: a review of the ECM protein composition, tumor cell interaction, and therapeutic strategies. Front. Oncol. 2021;11:751311. doi: 10.3389/fonc.2021.751311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips P.A., McCarroll J.A., Park S., Wu M.J., Pirola R., Korsten M., Wilson J.S., Apte M.V. Rat pancreatic stellate cells secrete matrix metalloproteinases: implications for extracellular matrix turnover. Gut. 2003;52:275–282. doi: 10.1136/gut.52.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R.J., Burdick M.D., Hong K., Lutz M.A., Murray L.A., Xue Y.Y., Belperio J.A., Keane M.P., Strieter R.M. Circulating fibrocytes traffic to the lungs in response to CXCL12 and mediate fibrosis. J. Clin. Invest. 2004;114:438–446. doi: 10.1172/jci200420997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poruk K.E., Firpo M.A., Scaife C.L., Adler D.G., Emerson L.L., Boucher K.M., Mulvihill S.J. Serum osteopontin and tissue inhibitor of metalloproteinase 1 as diagnostic and prognostic biomarkers for pancreatic adenocarcinoma. Pancreas. 2013;42:193–197. doi: 10.1097/mpa.0b013e31825e354d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano P.P., Cuevas C., Chang A.E., Goel V.K., Von Hoff D.D., Hingorani S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim A.D., Oberstein P.E., Thomas D.H., Mirek E.T., Palermo C.F., Sastra S.A., Dekleva E.N., Saunders T., Becerra C.P., Tattersall I.W., et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C., Washington M.K., Chaturvedi R., Drosos Y., Revetta F.L., Weaver C.J., Buzhardt E., Yull F.E., Blackwell T.S., Sosa-Pineda B., et al. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab. Invest. 2014;94:409–421. doi: 10.1038/labinvest.2014.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparmann G., Hohenadl C., Tornoe J., Jaster R., Fitzner B., Koczan D., Thiesen H.J., Glass A., Winder D., Liebe S., Emmrich J. Generation and characterization of immortalized rat pancreatic stellate cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;287:G211–G219. doi: 10.1152/ajpgi.00347.2003. [DOI] [PubMed] [Google Scholar]
- Stanley E.R., Berg K.L., Einstein D.B., Lee P.S., Pixley F.J., Wang Y., Yeung Y.G. Biology and action of colony--stimulating factor-1. Mol. Reprod. Dev. 1997;46:4–10. doi: 10.1002/(SICI)1098-2795(199701)46:1<4::AID-MRD2>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Storz P., Crawford H.C. Carcinogenesis of pancreatic ductal adenocarcinoma. Gastroenterology. 2020;158:2072–2081. doi: 10.1053/j.gastro.2020.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y., Li X., Tian Z., Chen S., Zou J., Lian G., Chen S., Huang K., Chen Y. TIMP1 down-regulation enhances gemcitabine sensitivity and reverses chemoresistance in pancreatic cancer. Biochem. Pharmacol. 2021;189:114085. doi: 10.1016/j.bcp.2020.114085. [DOI] [PubMed] [Google Scholar]
- Tjomsland V., Pomianowska E., Aasrum M., Sandnes D., Verbeke C.S., Gladhaug I.P. Profile of MMP and TIMP expression in human pancreatic stellate cells: regulation by IL-1α and TGFβ and implications for migration of pancreatic cancer cells. Neoplasia. 2016;18:447–456. doi: 10.1016/j.neo.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torphy R.J., Wang Z., True-Yasaki A., Volmar K.E., Rashid N., Yeh B., Anderson J.M., Johansen J.S., Hollingsworth M.A., Yeh J.J., Collisson E.A. Stromal content is correlated with tissue site, contrast retention, and survival in pancreatic adenocarcinoma. JCO Precis Oncol. 2018;2018 doi: 10.1200/po.17.00121. PO.17.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle M.C., Izeradjene K., Rani P.G., Feng L., Carlson M.A., DelGiorno K.E., Wood L.D., Goggins M., Hruban R.H., Chang A.E., et al. RUNX3 controls a metastatic switch in pancreatic ductal adenocarcinoma. Cell. 2015;161:1345–1360. doi: 10.1016/j.cell.2015.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Mora A., Shim H., Stecenko A., Brigham K.L., Rojas M. Role of the SDF-1/CXCR4 axis in the pathogenesis of lung injury and fibrosis. Am. J. Respir. Cell Mol. Biol. 2007;37:291–299. doi: 10.1165/rcmb.2006-0187oc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiji H., Kuriyama S., Miyamoto Y., Thorgeirsson U.P., Gomez D.E., Kawata M., Yoshii J., Ikenaka Y., Noguchi R., Tsujinoue H., et al. Tissue inhibitor of metalloproteinases-1 promotes liver fibrosis development in a transgenic mouse model. Hepatology. 2000;32:1248–1254. doi: 10.1053/jhep.2000.20521. [DOI] [PubMed] [Google Scholar]
- Zhang X., Goncalves R., Mosser D.M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. 2008 doi: 10.1002/0471142735.im1401s83. Chapter 83, Unit 14 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the Lead author upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead contact upon request.