

Abstract
Sporadic inclusion body myositis (IBM) is the most common acquired muscle disease in adults over age 50, yet it remains unclear whether the disease is primarily driven by T cell–mediated autoimmunity. IBM muscle biopsies display nuclear clearance and cytoplasmic aggregation of TDP-43 in muscle cells, a pathologic finding observed initially in neurodegenerative diseases, where nuclear loss of TDP-43 in neurons causes aberrant RNA splicing. Here, we show that loss of TDP-43–mediated splicing repression, as determined by inclusion of cryptic exons, occurs in skeletal muscle of subjects with IBM. Of 119 muscle biopsies tested, RT-PCR–mediated detection of cryptic exon inclusion was able to diagnose IBM with 84% sensitivity and 99% specificity. To determine the role of T cells in pathogenesis, we generated a xenograft model by transplanting human IBM muscle into the hindlimb of immunodeficient mice. Xenografts from subjects with IBM displayed robust regeneration of human myofibers and recapitulated both inflammatory and degenerative features of the disease. Myofibers in IBM xenografts showed invasion by human, oligoclonal CD8+ T cells and exhibited MHC-I up-regulation, rimmed vacuoles, mitochondrial pathology, p62-positive inclusions, and nuclear clearance and cytoplasmic aggregation of TDP-43, associated with cryptic exon inclusion. Reduction of human T cells within IBM xenografts by treating mice intraperitoneally with anti-CD3 (OKT3) suppressed MHC-I up-regulation. However, rimmed vacuoles and loss of TDP-43 function persisted. These data suggest that T cell depletion does not alter muscle degenerative pathology in IBM.
INTRODUCTION
Sporadic inclusion body myositis (IBM) causes progressive muscle atrophy, weakness, and disability, with a median of 14 years between symptom onset and wheelchair dependence (1, 2). The insidious progression of weakness and atrophy makes the disease difficult to diagnose, with a median time from onset to diagnosis of about 5 years (3, 4). Clinically, subjects with IBM exhibit a distinct pattern of muscle weakness with finger flexor and quadriceps muscles most severely affected. Diagnosis is confirmed by muscle biopsy, which shows characteristic features including ubiquitinated protein inclusions, rimmed vacuoles, mitochondrial pathology, and intense endomysial inflammation (5). Although many clinical trials using immunomodulatory drugs have been performed in IBM, as well as trials of drugs targeting proteostasis or the myostatin pathway, none have proven successful, and there remains no established therapy for the disease (2).
The combination of inflammatory and degenerative pathological features has led to considerable debate as to the primary cause of the disease (2, 6–8). The presence of major histocompatibility complex class I (MHC-I) up-regulation within myofibers that are invaded by highly differentiated cytotoxic CD8+ T cells, the association of IBM with other autoimmune disorders and specific human leukocyte antigen haplotypes, and the presence of autoantibodies in sera from subjects with IBM support an autoimmune trigger for the disease (9–12). The lack of clinical efficacy of immunosuppression therapies, however, suggests that endomysial inflammation may not be required for disease progression. Alternatively, IBM pathology may be driven by a T cell population refractory to immunosuppressive therapy (9, 13).
A competing idea hypothesizes that IBM is primarily a degenerative disease, analogous to that of neurodegenerative disease. This view is supported by ultrastructural characterization of inclusions in myofibers of IBM biopsies, which form amyloid-like fibrils and tubulofilaments analogous to those observed in the brain of Alzheimer’s disease (AD) (14). Intracellular amyloid accumulation is observed in IBM muscle, and overexpression of β-amyloid (Aβ) precursor protein (APP) in human myoblasts or mouse muscle causes toxicity (15, 16). More recently, nuclear loss and cytoplasmic inclusions of transactivation response element DNA binding protein 43 (TDP-43) have been observed in myofibers of subjects with IBM (17–19), a pathological feature initially identified in amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (20). By binding to the uracil and guanine (UG)-rich repeats of their target RNAs to repress splicing of nonconserved cryptic exons in a transcriptome-wide manner (21–23), TDP-43 serves to protect cells by ensuring the proper formation of the transcriptome (24). TDP-43 is recognized as the founding member of a growing family of RNA binding proteins that serve this critical cellular function (25). Recent studies in human disease support the view that loss of TDP-43 splicing repression underlies neuronal loss in neurodegenerative diseases (24, 26, 27). Although a pathological link between IBM pathogenesis and age-dependent neurodegeneration exists, it remains unclear whether loss of TDP-43 splicing repression occurs in skeletal muscle of subjects with IBM.
The lack of IBM laboratory models that recapitulate both the inflammatory and degenerative features of the disease is a primary reason IBM pathogenesis remains elusive. Donor-derived xenograft models have been effectively used to develop laboratory models and treatments for malignancies, as well as personalized therapies (28, 29). Xenografts have recently been used to study genetic disorders of skeletal muscle, and whole muscle xenografts have been successfully used to model facioscapulohumeral muscular dystrophy (FSHD) and test potential therapies (30, 31). Here, we determine whether human skeletal muscle xenografts can be used to model sporadic, late-onset, inflammatory muscle diseases, such as IBM. Aging is known to diminish the regenerative capacity of skeletal muscle, resulting in loss of muscle mass and strength observed in elderly populations (32). In addition, in vitro studies have shown myoblasts from donors with IBM proliferate at a slower rate than age-matched controls and undergo premature senescence (33). Furthermore, skeletal muscle regeneration is highly regulated by the immune system, and chronic inflammation has been hypothesized to negatively influence muscle regeneration (34, 35). Conversely, it has been reported that muscle from subjects with IBM exhibits increased numbers of satellite cells and regenerating fibers in comparison to age-matched controls, as well as up-regulation of the myogenic regulatory factor myogenin, which drives differentiation of satellite cells (36). Because xenograft formation requires successful myofiber regeneration, this approach allows us to test the ability of IBM muscle to regenerate in vivo and develop a laboratory model for IBM.
Here, we show that the detection of cryptic exons in muscle biopsies is a sensitive approach to identify TDP-43 pathology in the muscle from donors with IBM. Cryptic exons can be detected in IBM xenografts, which recapitulate both inflammatory and degenerative pathological features of the human disease. We used this model to carry out mechanistic studies to better understand disease pathogenesis. By treating mice containing IBM xenografts with a monoclonal CD3 antibody (OKT3), we specifically depleted human T cells from xenografts to assess their role in IBM pathophysiology. Our data demonstrate in this model that T cells are required for MHC-I up-regulation in muscle cells but not for rimmed vacuole formation and loss of TDP-43 function.
RESULTS
Detection of TDP-43 cryptic exons in human muscle from subjects with IBM
Of many proteins that form aggregates within IBM muscle cells, cytoplasmic accumulations of TDP-43 and p62/sequestosome 1 have been suggested to have high specificity for IBM (17, 18). In ALS-FTD, AD, and other neurodegenerative diseases termed TDP-43 proteinopathies (37), nuclear loss of TDP-43 is more frequent than cytoplasmic aggregation, and assays for loss of function are more sensitive for detecting TDP-43 pathology as compared to immunohistochemical stains (24, 38). TDP-43 functions in pre-mRNA splicing to repress the incorporation of “cryptic” exons, and detection of cryptic exons in neurodegenerative disease tissue using RNA sequencing (RNA-seq) and reverse transcription polymerase chain reaction (RT-PCR) has been shown to be a sensitive functional assay for TDP-43 pathology (24, 39). Cryptic exon incorporation can be detected in hippocampal samples from subjects with AD lacking TDP-43 aggregation (38). Whether loss of TDP-43 splicing repression occurs in skeletal muscle of subjects with IBM remains to be determined. Because nonconserved cryptic exons are species- and cell type–specific, we first sought to identify human skeletal muscle-associated TDP-43 cryptic exon targets (24, 39).
To identify TDP-43 cryptic exons from muscle cells, we used previously characterized small interfering RNAs (siRNAs) (24) to deplete TDP-43 from healthy human myoblasts and performed RNA-seq analysis (Fig. 1A). We identified a set of RNA targets in which nonconserved cryptic exons were flanked by “UG” repeats, the known binding site of TDP-43 (fig. S1). In addition to cassette exons, we identified a subset of nonstandard cryptic exons that could be categorized as alternative transcriptional start sites, premature polyadenylation sites, or expansions of conserved canonical exons (Fig. 1B and table S1). These results confirm that TDP-43 represses nonconserved cryptic exons in human myoblasts.
Fig. 1. Cryptic exon detection is a sensitive and specific assay for TDP-43 pathology in IBM biopsies.
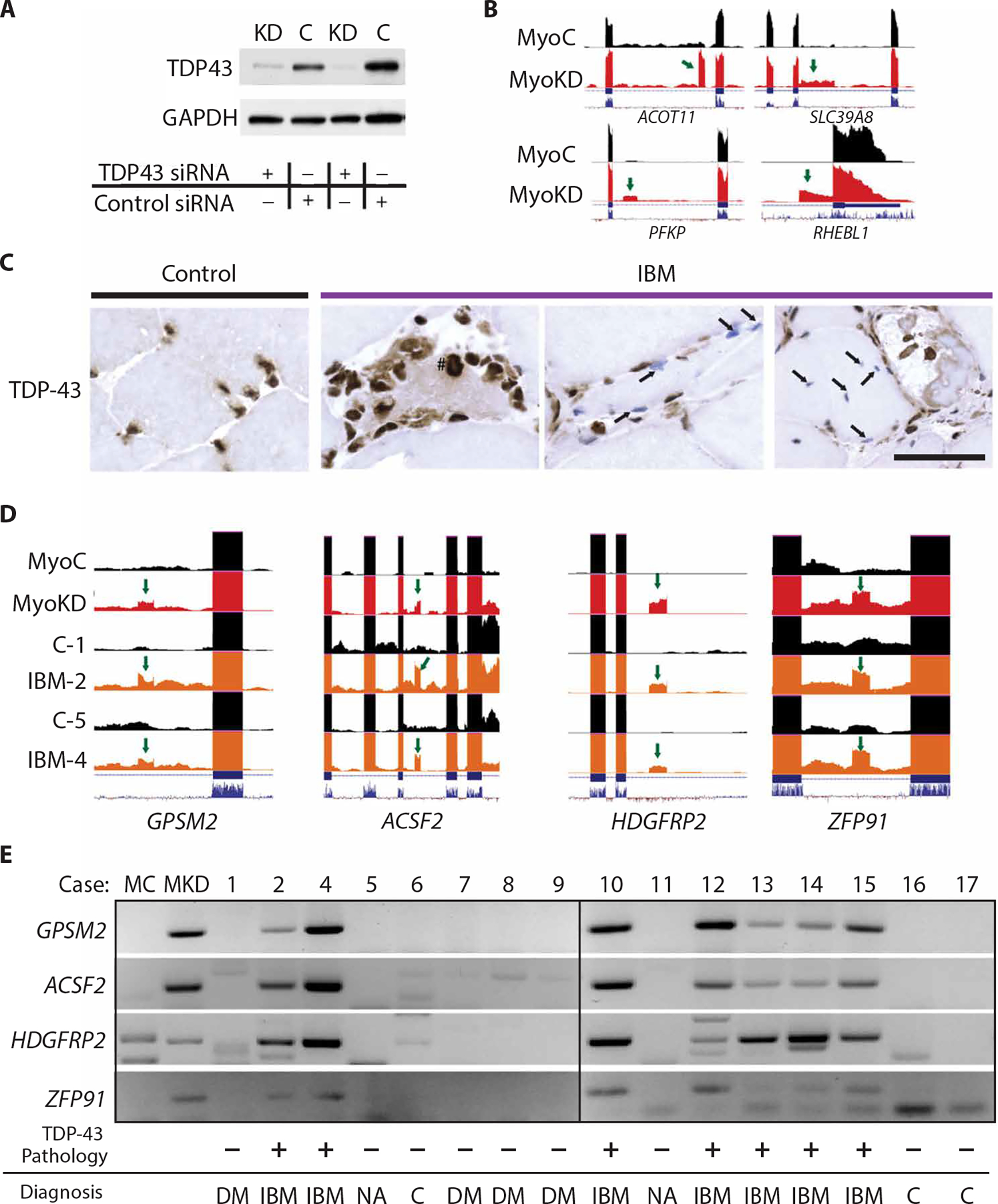
(A) TDP-43 Western blot in myoblasts treated with TDP43 siRNA [knockdown (KD)] compared to control siRNA (C). (B) Visualization of cryptic exons (green arrows) in myoblast cells with TDP-43 knockdown (MyoKD) compared to control (MyoC) for TDP-43 target genes ACOT11, SLC39A8, PFKP, and RHEBL1. ACOT11, acyl-CoA thioesterase 11; SLC39A8, solute carrier family 39 member 8; PFKP, phosphofructokinase, platelet; RHEBL1, RHEB (ras homolog, mechanistic target of rapamycin kinase binding) like 1. (C) Immunohistochemical TDP-43 staining of muscle sections showing accumulation of TDP-43 in the cytoplasm (#) or nuclear clearing (arrows). Scale bar, 50 μm. (D) Visualization of cryptic exons (green arrow) in control myoblasts (MyoC), myoblasts with TDP-43 knockdown (MyoKD), or subject muscle biopsies [control (C) and IBM]; numbers indicate cases (table S2). (E) Representative gel showing cryptic exon expression from TDP-43 target genes GPSM2, ACSF2, HDGFRP2, and ZFP91 in skeletal muscle biopsies from IBM and control biopsies (DM, dermatomyositis; NA, neurogenic atrophy; C, normal muscle or mild nonspecific features).
If loss of TDP-43 function occurs in IBM, then there should be an incorporation of these nonconserved cryptic exons in IBM skeletal muscles. To test this hypothesis, we first confirmed the diagnosis from muscle biopsies of a cohort of cases that exhibit pathological hallmarks of IBM, including rimmed vacuoles, endomysial inflammation, and p62 and TDP-43–positive protein aggregates (table S2). As shown previously (40), besides the abnormal cytoplasmic accumulation of TDP-43, we also found clearance of TDP-43 from nuclei in otherwise morphologically normal myofibers (Fig. 1C).
Total RNA from biopsies of quadriceps muscle from two subjects with IBM along with two controls were extracted and subjected to RNA-seq analysis (table S2). We found several nonconserved cryptic exons in both IBM cases but not in controls (Fig. 1D). Analysis of the frequency that a cryptic exon is incorporated into mRNA transcripts [percent spliced-in (PSI)] revealed that 5 to 17% of G protein signaling modulator 2 (GPSM2), acyl-CoA synthetase family member 2 (ACSF2), hepatoma-derived growth factor–related protein 2 (HDGFRP2) (or HDGFL2), or zinc finger protein 91 (ZFP91) transcripts in biopsies from subjects with IBM showed incorporation of cryptic exons, whereas cryptic exons of GPSM2, ACSF2, and HDGFRP2 were not detected in control donor biopsies (table S3). ACSF2 showed the highest amount of incorporation with an average PSI of 13.6%. To determine whether the incorporation of cryptic exons can be readily detected in muscle from subjects with IBM, total RNA was extracted from rectus femoris muscle biopsies of age- and sex-matched IBM [meeting the European Neuromuscular Centre (ENMC) 2011 clinically defined diagnostic criteria] and control donors (n = 16, table S2) and subjected to RT-PCR analysis. We selected a panel of four RNA targets (GPSM2, ACSF2, HDGFRP2, and ZFP91) for our analysis. Sequence and alignment validation of cryptic exon RT-PCR products were performed for the RNA targets (figs. S2 and S3). Cryptic exons of all four RNA targets were detected in all seven IBM cases, but not in any of the nine controls (Fig. 1E and table S4). Together, these results establish that loss of TDP-43 function occurs in biopsies from subjects with IBM.
To determine the sensitivity and specificity of cryptic exon detection for the diagnosis of IBM in a large cohort of myositis center subjects, we assayed muscle biopsies from an additional 103 subjects (table S4), including 30 donors used in generating the xenograft model (see below). Of 119 total donors, 36 of 44 IBM and only 1 of 75 controls were positive for cryptic exons (P < 0.0001 by Fisher’s exact test), resulting in a sensitivity of 84% and specificity of 99% for diagnosis of IBM. These data show that detection of TDP-43 cryptic exons serves as a useful assay for IBM diagnosis among subjects with myositis.
IBM xenografts regenerate robustly in immunodeficient mice
To determine whether human skeletal muscle xenografts transplanted into immunodeficient mice can successfully engraft and recapitulate key features of human muscle pathology, we recruited subjects at the time of diagnostic open muscle biopsy at the Johns Hopkins Outpatient Center (table S5). Subjects were classified as IBM (n = 15) if they met ENMC 2011 diagnostic criteria or as a control (n = 12) if they were more than 40 years of age and IBM was not considered to be a possible diagnosis at follow-up (3). The IBM biopsies display characteristic features of the disease, including endomysial inflammation, invasion of myofibers by CD3+ T cells, cytochrome c oxidase (COX)–deficient fibers, and/or rimmed vacuoles (table S5). The control biopsies were taken from subjects with weakness, elevated muscle enzymes, and/or muscle pain and typically showed mild abnormalities such as perivascular inflammation, mild myopathic features such as mild necrosis or internalized nuclei, mild neurogenic atrophy, and/or mitochondrial abnormalities, although 5 of 12 were pathologically normal. The sex ratio and disease duration of the subjects were not significantly different between groups (P > 0.05); however, the population of subjects with IBM was older than the control group (average age 70.1 versus 61.2 years, P = 0.04) (table S6).
Human skeletal muscle biopsy specimens obtained from both subjects with IBM and controls were dissected and transplanted into nonobese diabetic–Rag1nullIL2rγnull (NRG) mice lacking the ability to generate mature B or T cells and innate lymphoid cells, including natural killer cells (fig. S4A) (30, 41). In this model, the mature human myofibers cut during the biopsy degenerate and are replaced by newly regenerated myofibers, derived from transplanted satellite cells that are revascularized and reinnervated by the mouse host (30, 41). At 1 month after xenotransplantation, all myofibers in IBM and control xenografts are positive for embryonic myosin heavy chain (eMHC), a marker of newly regenerated myofibers (fig. S4, B and C) (30, 41). At 4 months after xenotransplantation, myofibers within IBM xenografts and control xenografts successfully regenerate (Fig. 2, A to E). There is no difference in the number of regenerated fibers or the fraction of the xenograft composed of myofibers (fiber fraction) between the two groups (Fig. 2, B and C), and the percentage of eMHC–positive fibers is unchanged (Fig. 2D), indicating that the process of myofiber maturation, as assessed by the turnover of eMHC, is unaltered (42). In comparison to control, myofibers of IBM xenografts show a significant increase in the median cross-sectional area (369 versus 249 μm2, P = 0.021) (Fig. 2E). These data indicate that muscle from IBM subjects is capable of robust regeneration within a mouse host, allowing the establishment of a mouse model of sporadic IBM.
Fig. 2. IBM xenografts regenerate robustly in NRG mice.
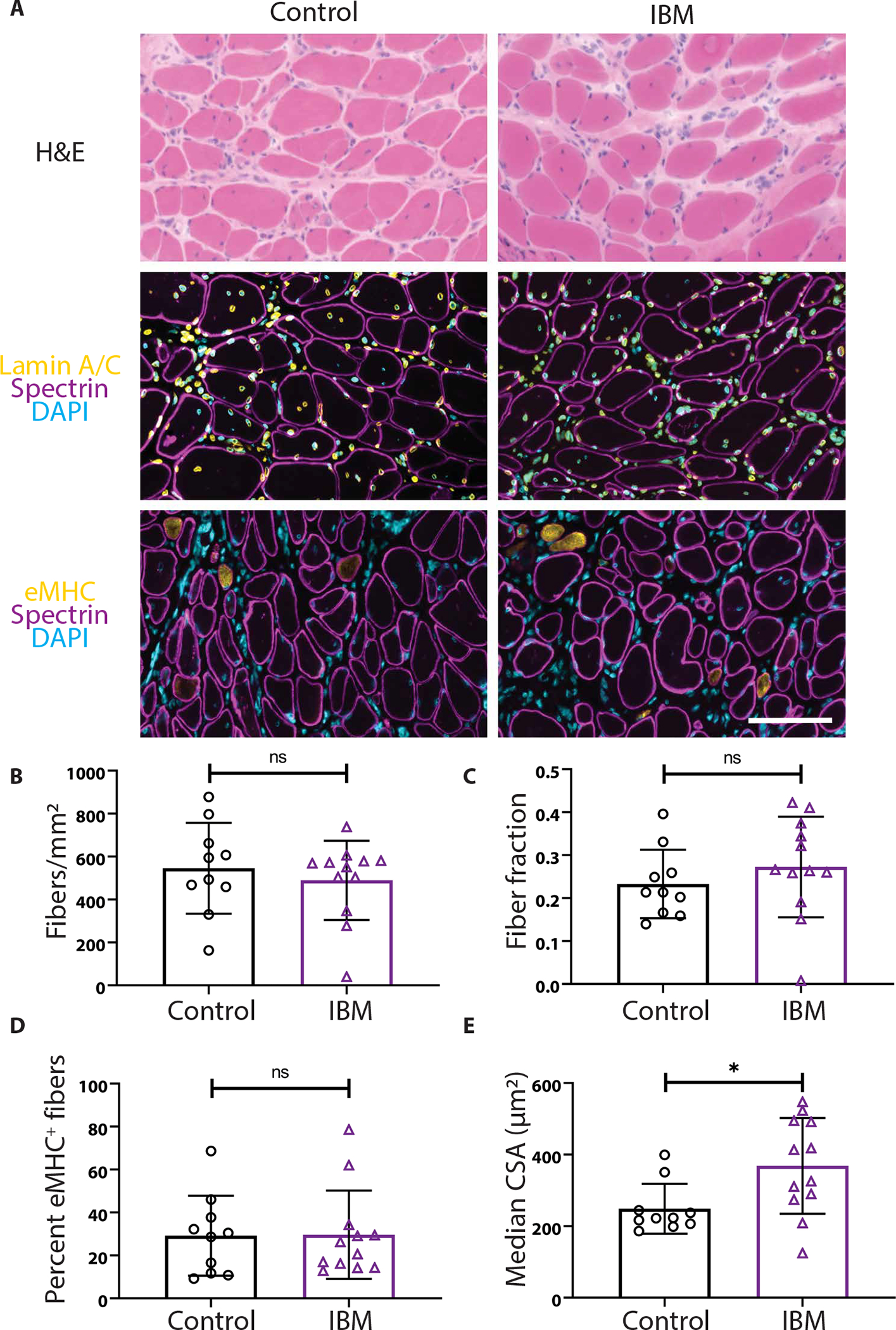
(A) Representative images of 4-month control and IBM xenografts stained with hematoxylin and eosin (H&E), human spectrin (magenta), human lamin A/C (yellow), embryonic myosin heavy chain (eMHC) (yellow), and 4′,6-diamidino-2-phenylindole (DAPI) (cyan). Scale bar, 100 μm. (B to D) The number of fibers per xenograft area (B), the fraction of the xenograft covered by myofibers (C), and the percentage of eMHC+ regenerating fibers are similar between control and IBM xenografts (D). (E) The median cross-sectional area (CSA) of myofibers within the xenografts (*P < 0.05, Mann-Whitney test). For all graphs, each point denotes one subject (control, n = 10; IBM, n = 12).
IBM xenografts recapitulate pathological features of the human disease
To determine whether newly formed IBM muscle xenografts exhibit features of IBM pathology present in the original biopsy, we first examined 4-month xenografts for rimmed vacuoles, a pathologic hallmark of IBM. Rimmed vacuoles are frequently seen in IBM xenografts as judged by Gomori trichrome (GT) histological stain, whereas they are rarely observed in controls (0.54% versus 0.05% of fibers; P = 0.007) (Fig. 3, A and B, and fig. S5, A to C). The only control xenograft that showed rimmed vacuoles was from a subject diagnosed with a vacuolar myopathy based on muscle biopsy (case 27) (fig. S5A). These findings demonstrate that regenerated myofibers in muscle biopsies from subjects with IBM develop rimmed vacuoles within a mouse host, indicating that this pathology is likely intrinsic to IBM muscle rather than resulting from a circulating factor.
Fig. 3. Rimmed Vacuoles and TDP-43 pathology are observed in IBM xenografts.
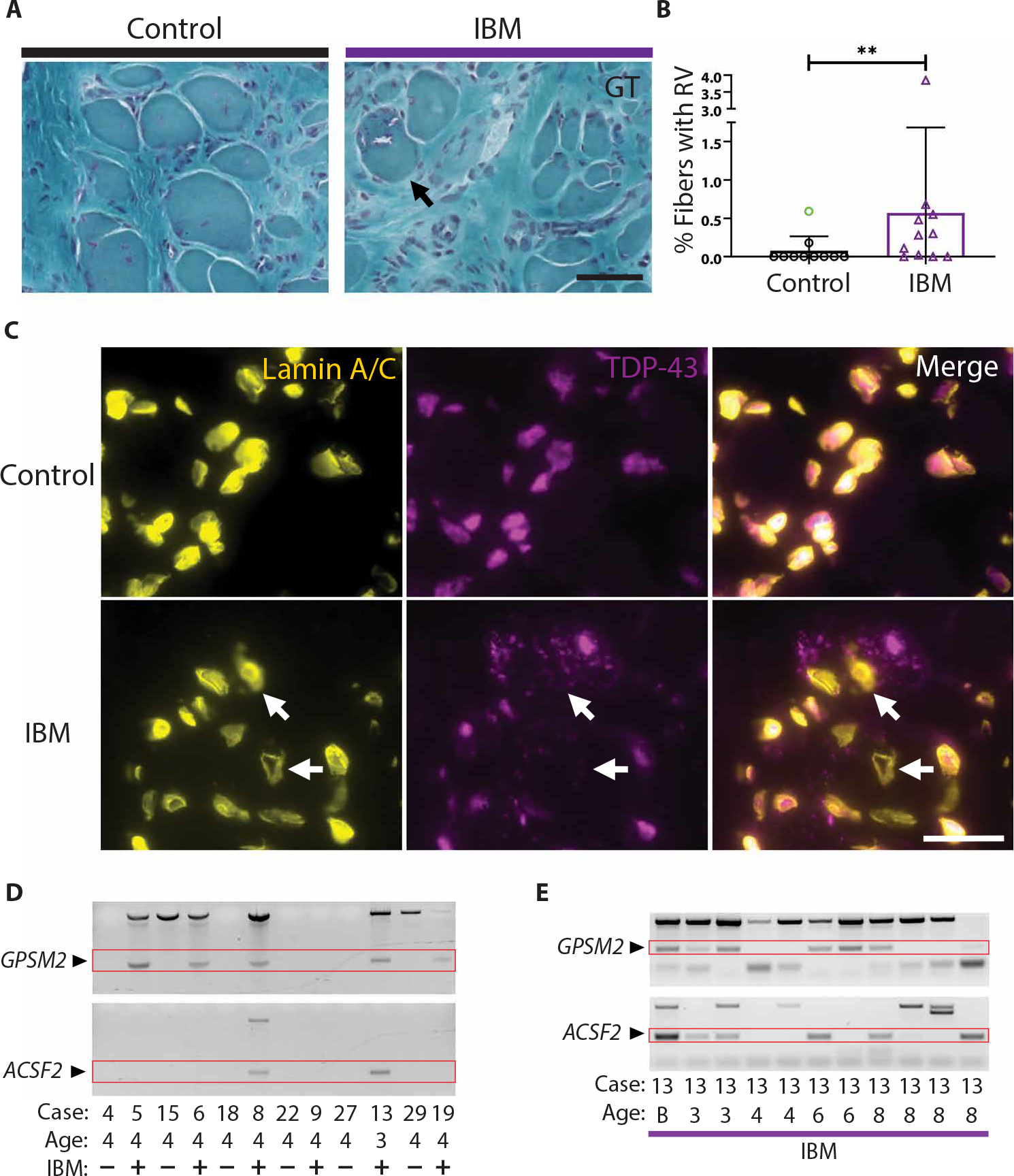
(A) Representative images from GT stains of 4-month control and IBM xenografts. Examples of rimmed vacuoles (arrow) are exhibited by the IBM xenograft. Scale bar, 50 μm. (B) Quantification of the percentage of fibers with rimmed vacuoles (RV) in each group; each point denotes one subject (control, n = 10; IBM, n = 11). The control sample highlighted in green indicates the case 27 subject diagnosed with a vacuolar myopathy and was excluded from statistical analysis. Mann-Whitney test was used to determine significance (**P < 0.01). (C) TDP-43 aggregates and examples of nuclear clearing (arrows) in 4-month xenografts. Scale bar, 25 μm. (D) Cryptic exon expression from GPSM2 and ACSF2 in 3- and 4- month IBM xenografts and controls. (E) Cryptic exon expression from TDP-43 target genes GPSM2 and ACSF2 detected in IBM xenografts ranging from 3 to 8 months (B, subject biopsy).
To assess IBM xenografts for pathologic protein aggregation, we first performed immunostaining for p62 and observed robust p62-positive aggregates at 4-month time points in the majority of IBM xenografts (n = 4 of 5), but in only one control xenograft (n = 1 of 5) with a diagnosis of dermatomyositis (fig. S6A). We next looked for TDP-43 pathology as evidenced by cytoplasmic accumulation and/or nuclear loss of TDP-43. Nuclear clearance and cytoplasmic aggregation of TDP-43 are observed in 4-month IBM (n = 5 of 5), but not in control (n = 0 of 5), xenografts (P = 0.008 using Fisher’s exact test; Fig. 3C). Furthermore, we reliably detect cryptic exon inclusion from TDP-43 targets GPSM2 (n = 17 of 40) and ACSF2 (n = 11 of 34) within IBM, but not within control, xenografts from 2 to 10 months (GPSM2: P < 0.0001; ACSF2: P = 0.0013 using Fisher’s exact test; Fig. 3, D and E, and table S7). Cryptic exon incorporation in IBM xenografts corresponds to that of the subject biopsies (only human biopsies showing the inclusion of cryptic exons result in xenografts with cryptic exon incorporation as shown by IBM cases 9 and 33; Fig. 3D and tables S4 and S7). The examination of the inclusion of cryptic exons longitudinally within IBM (cases 8 and 13) reveals that the inclusion of cryptic exons can be detected at all time points examined: 3 months (2 of 2), 4 months (1 of 4), 6 months (3 of 4), 8 months (3 of 7), and 10 months (3 of 3) (Fig. 3E and table S7); about 50% (19 of 40) of IBM xenografts shows incorporation of cryptic exons regardless of time point, compared with only 3% (1 of 34) of control xenografts. Together, these data show that TDP-43 nuclear function is impaired in IBM xenografts, as is observed in muscle biopsies from subjects with IBM.
After determining that IBM xenografts display characteristic degenerative pathology including rimmed vacuoles and loss-of-TDP-43 function, we investigated whether the human immune cells transferred via the xenograft surgery persist within regenerated xenografts. Most control xenografts (n = 22 of 27) recapitulate features of normal muscle tissue including MHC-I staining highlighting capillaries and absence of CD3+ T cells (Fig. 4A, left). However, some control xenografts (n = 5 of 27) and most IBM xenografts (n = 22 of 33) reveal sarcoplasmic up-regulation of MHC-I and the presence of CD3+ T cells (P = 0.0002 using Fisher’s exact test; Fig. 4A, middle and right). Multiple human immune cells are present within both control and IBM xenografts, including helper (CD4+) and cytotoxic (CD8+) T cells, B cells (CD20+), macrophages (CD68+), and plasma cells (CD138+) (fig. S7). Despite the presence of CD3+ T cells in both IBM and control xenografts, T cell invasion of non-necrotic fibers is only observed in IBM xenografts (n = 0 of 27 control xenografts and n = 4 of 33 IBM xenografts examined, P = 0.13 using Fisher’s exact test; Fig. 4B). T cells are not observed in regions of mouse muscle adjacent to the human xenograft, arguing against a graft-versus-host response. Furthermore, IBM xenografts show significantly higher numbers of CD3+ T cells than control xenografts (842 versus 195 per square millimeter, P = 0.017) (Fig. 4C), a finding that is also observed in the original muscle biopsies.
Fig. 4. IBM xenografts recapitulate pathological features of human disease.
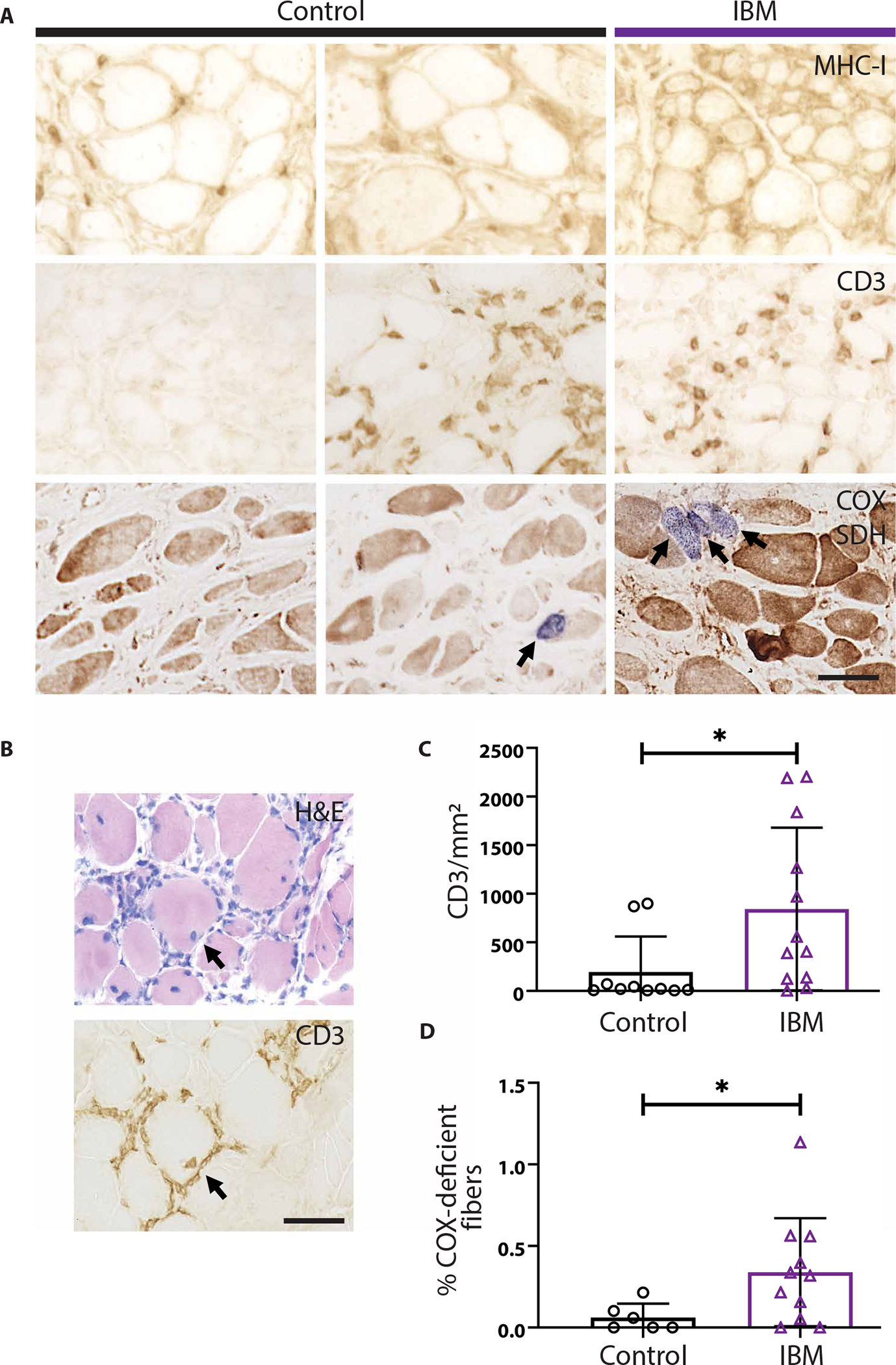
(A) Representative images from MHC-I, CD3, and dual COX/SDH (COX-negative, SDH dark fibers stain blue) stains of 4-month control and IBM xenografts. (B) Serial sections of a 4-month IBM xenograft stained with H&E and anti-CD3 showing an example of T cell invasion of a non-necrotic fiber. (C) Quantification of the number of CD3+ T cells per xenograft area in 4-month collections. Each point denotes one subject (control, n = 10; IBM, n = 12). (D) Quantification of the percentage of COX-deficient fibers in each group; each point denotes one subject (control, n = 6; IBM, n = 11). Mann-Whitney test was used to determine significance (*P < 0.05). Scale bars, 50 μm.
Most muscle biopsies from subjects with IBM exhibit mitochondrial pathology, characterized by an increase in the number of COX-deficient fibers, an accumulation of abnormal mitochondria, and mitochondrial DNA deletions (43, 44). To assess IBM xenografts for mitochondrial pathology, we carried out dual COX and succinate dehydrogenase (SDH) histological stains on 4-month control and IBM xenografts (Fig. 4A). In comparison to control xenografts, we found that the percentage of COX-deficient fibers was significantly increased in IBM xenografts (0.34% versus 0.06% of fibers, P = 0.04) (Fig. 4D). Collectively, these results indicate that this xenograft model of IBM successfully recapitulates key features of both degenerative and inflammatory pathology.
T cells in IBM xenografts share clones and immunophenotypes with the original IBM biopsy
The majority of IBM muscle biopsies show oligoclonal populations of T cells, consistent with the hypothesis that they proliferate in response to unknown antigens (45, 46). In peripheral blood mononuclear cells (PBMCs) and muscle from subjects with IBM, oligoclonal populations of CD8+ T cells persist over time and exhibit a highly differentiated, cytotoxic T cell phenotype, as indicated by loss of CD28 and gain of CD57 and killer cell lectin like receptor G1 (KLRG1) expression (9, 13, 47, 48). Flow cytometry analysis of PBMCs demonstrates that patients with IBM have significantly more CD8+ T cells that are CD57+ (45% versus 11%, P = 0.016, Mann-Whitney test) and KLRG1+ (67% versus 38%, P = 0.032) in comparison to control patients (fig. S8). We found that IBM xenografts contain both CD57+ and KLRG1+ cells, suggesting that immune cells within xenografts recapitulate immunophenotypes found within the muscle from subjects with IBM (Fig. 5A). To better characterize the human immune cells within IBM xenografts, we performed flow cytometry on immune cells isolated from 4-month xenografts (n = 4), as well as PBMCs isolated from the same subjects at the time of biopsy (n = 2) (Fig. 5, B to D). IBM xenografts show a predominance of CD4+ T cells and large populations of CD57+KLRG1+CD8+ T cells, similar to what is seen in PBMCs from subjects with IBM (Fig. 5, C and D) (9, 13). Together, these data indicate that the majority of CD8+ T cells in 4-month IBM xenografts contain markers of highly differentiated cytotoxic T cells as seen in IBM biopsies and peripheral blood.
Fig. 5. T cells in IBM xenografts are phenotypically similar to subjects with IBM.
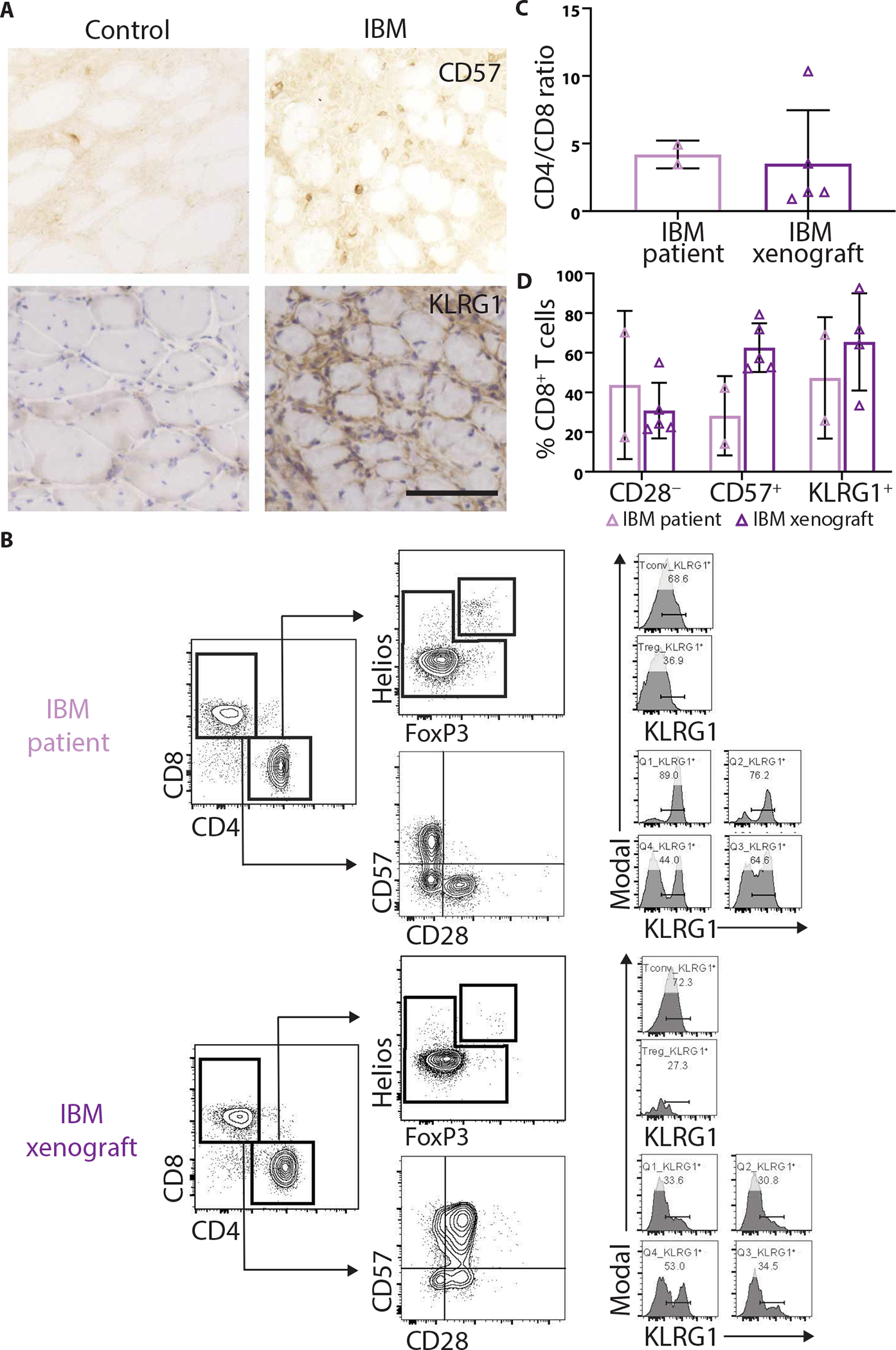
(A) Representative CD57 and KLRG1 stains of control and IBM xenografts. Scale bar, 100 μm. (B) Representative flow cytometry plots of PBMCs from a subject with IBM (top) and a 4-month xenograft (bottom) generated from the same subject. Treg, regulatory T cells; Tconv, conventional T cells. Quantification of (C) CD4/CD8 ratio and (D) percent of CD8+ T cells that are CD28−, CD57+, and KLRG1+ to evaluate T cell phenotypes in PBMCs and xenografts.
To explore whether the T cells in IBM xenografts are oligoclonal and share clones in common with the original muscle biopsy, we examined matched biopsy and xenograft T cell receptor (TCR) repertoires using the multiplex PCR-based approach Framework Region 3 AmplifiKation sequencing (49). This analysis revealed that T cells within IBM xenografts are more clonally restricted than the original IBM biopsies (P = 0.027, Mann-Whitney test; Fig. 6A). In addition, as compared to control xenografts, IBM xenografts show an increased richness or number of unique clones (P = 0.0014, Mann-Whitney test; Fig. 6B). Using a Morisita-Horn index to compare the similarity of clones between biopsies and xenografts, we found that T cell clones within a biopsy from a subject and its corresponding xenografts are more similar to each other than to T cell clones from other subjects with IBM or xenografts (Fig. 6C). Last, the proportion of T cell clones was compared between biopsies from subjects with IBM and corresponding xenografts at multiple time points (Fig. 6D). Multiple xenograft cases showed T cell clones that persist in xenografts across the 1-week to 8-month collections. Together, these data demonstrate that T cells in IBM xenografts are composed of an enriched subset of clones also detected in the original muscle biopsy.
Fig. 6. T cells in IBM xenografts show clonality and persistence.
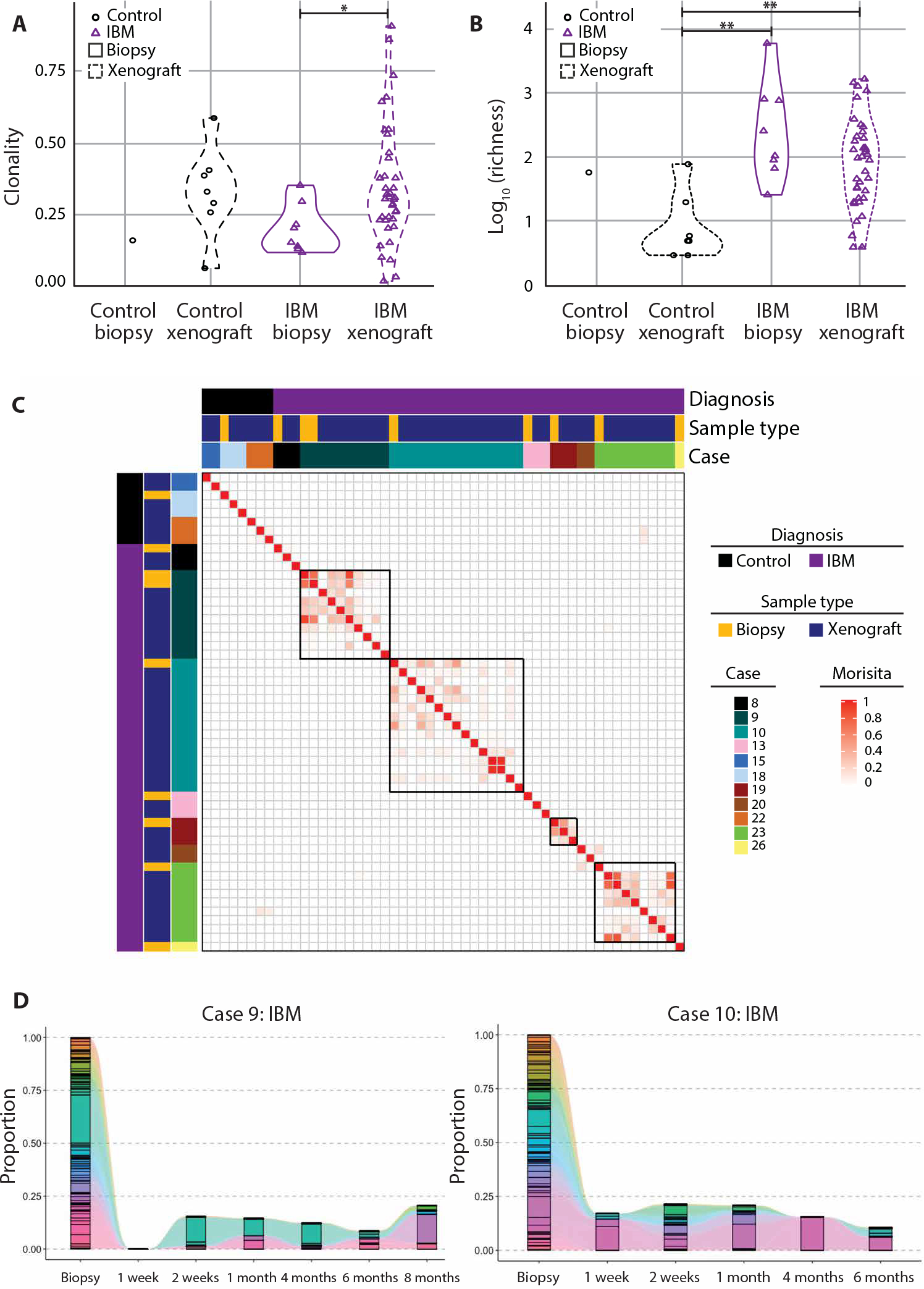
TCR sequencing was performed to determine clonality (A) and the number of unique clonotype (richness) (B) in both control and IBM biopsies and xenografts (control biopsy, n = 1; control xenograft, n = 7; IBM biopsy, n = 8; IBM xenograft, n = 38). Mann-Whitney test was used to determine significance (*P < 0.05; **P < 0.01). (C) Heatmap displaying the Morisita-Horn index between biopsies and xenografts. (D) The proportion of TCR clones was compared between biopsies from subjects with IBM and corresponding xenografts at multiple time points as shown for two IBM xenograft cases. Each color represents a unique clonotype.
Intraperitoneal delivery of OKT3 depletes T cells in IBM xenografts
A central question in IBM is whether inflammation drives pathology or is secondary to muscle degeneration. Recently, highly differentiated effector CD8+ KLRG1+ T cells present in IBM muscle have been suggested to be refractory to conventional immunotherapy, and targeted approaches to deplete this subpopulation of T cells are in therapeutic development for IBM (9, 13). Monoclonal antibodies can be used to deplete specific immune cells via induction of apoptosis or by antibody-dependent cell-mediated cytotoxicity (50). Murine monoclonal anti-CD3 antibody (OKT3) recognizes a nonpolymorphic subunit of the human TCR: CD3ε (51). OKT3 has been used clinically to treat transplant rejection and has also been delivered intraperitoneally in xenograft models to ablate human T cells in mice engrafted with human hematopoietic cells (52). We hypothesized that treating xenografted mice with OKT3 would specifically eliminate T cells within grafts and that this would enable us to study the effect of T cells on aspects of IBM pathology.
All four biopsies from subjects with IBM (cases 23, 26, 36, and 42) used in OKT3 experiments met ENMC clinically defined IBM criteria and showed marked endomysial inflammation with T cell invasion of non-necrotic fibers and mitochondrial pathology (table S5 and fig. S9). Consistent with previous studies showing that about 20% of subjects with typical clinical features of IBM do not exhibit rimmed vacuoles (53, 54), one of the four IBM cases (case 36) did not show rimmed vacuoles. Xenografted mice were treated weekly via intraperitoneal injection with OKT3 (10 mg/kg), as this treatment regimen has been shown to effectively ablate human T cells in vivo (52). Whereas IBM xenograft mice treated with vehicle alone had an average of 741 CD3+ T cells/mm2, OKT3 reduced this number to an average of 93 CD3+ T cells/mm2 at 2 months (P = 0.03) and 31 CD3+ T cells/mm2 at 4 months (P < 0.0001) (Fig. 7, A and B). Although treatment with OKT3 was highly effective in depleting T cells, myofiber regeneration was unchanged between control and treatment groups (Fig. 7, C to E). In addition, OKT3 treatment did not affect the proportion of eMHC+ fibers at 4 months (Fig. 7F), indicating that the process of myofiber maturation is unchanged (42). In healthy muscle, the inflammatory response to muscle injury is a highly complex and coordinated process involving cells from both the innate and adaptive immune systems (35). These data demonstrate that T cells within IBM xenografts do not affect myofiber regeneration in our model.
Fig. 7. OKT3 treatment ablates T cells from IBM xenografts but does not affect myofiber regeneration.
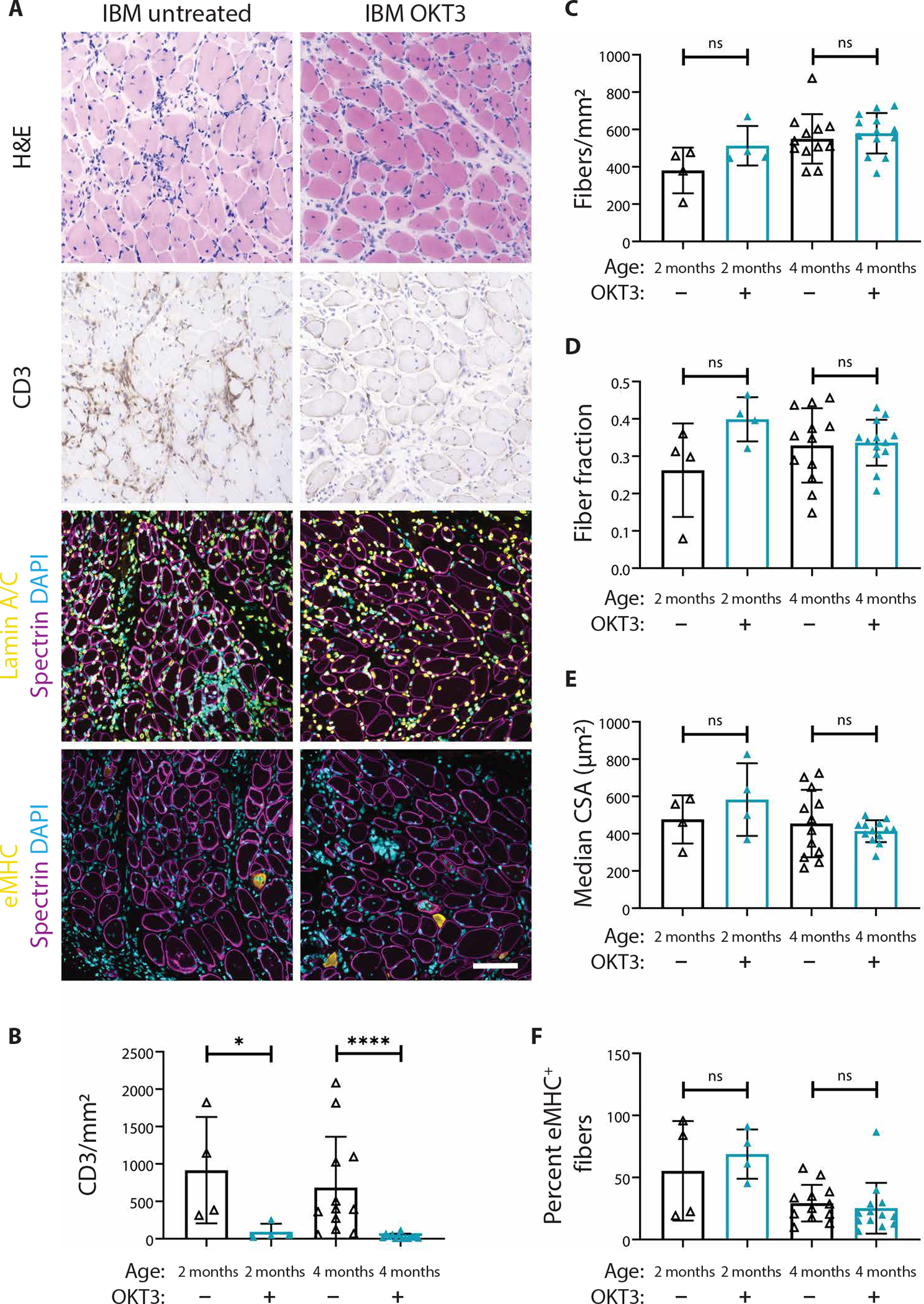
(A) Representative H&E, CD3, lamin A/C (yellow), spectrin (magenta), eMHC (yellow), and DAPI stains of 4-month untreated and OKT3-treated IBM xenografts. Scale bar, 100 μm. Quantification of the number of CD3+ T cells over the xenograft area (B), the number of fibers over the xenograft area (C), fiber fraction (D), median fiber CSA (E), and percentage of eMHC+ fibers (F) are shown. For all graphs, each point denotes one xenograft (2-month untreated, n = 4; 2-month OKT3, n = 4; 4-month untreated, n = 12; and 4-month OKT3, n = 13). Xenografts were obtained from four subjects with IBM (cases 23, 26, 36, and 42). Mann-Whitney U test was used to test for significance (*P < 0.05; ****P < 0.0001).
Depletion of T cells in IBM xenografts does not affect rimmed vacuoles or inclusion of TDP-43 cryptic exons
As xenografts from mice treated with OKT3 or vehicle showed comparable regeneration, these samples are ideal to address the question of how T cells influence aspects of IBM pathology. As expected, given the marked depletion of T cells, OKT3 treatment successfully reduced both the percentage of fibers showing MHC-I up-regulation (15.4 versus 67.1; P = 0.016) and the number of KLRG1+ cells (7.3 versus 26.3; P = 0.002) within xenografts at 4 months (Fig. 8, A to C). However, OKT3 treatment did not significantly reduce the number of COX-deficient fibers (from 0.37 to 0.19%, P = 0.12) (fig. S10, A and B). We found that the number of COX-deficient fibers was significantly correlated to the number of CD3+ T cells in IBM but not in control xenografts (control R2: 0.03; IBM R2: 0.2; P = 0.02) (fig. S10C), consistent with previous work demonstrating positive correlations between the number of COX-deficient muscle fibers and the severity of inflammation in biopsies from subjects with IBM (55). The amelioration of MHC-I up-regulation within IBM myofibers demonstrates that depletion of T cells reduces inflammatory changes within muscle cells.
Fig. 8. OKT3 depletion of T cells reduces inflammation but not rimmed vacuoles or p62 pathology.
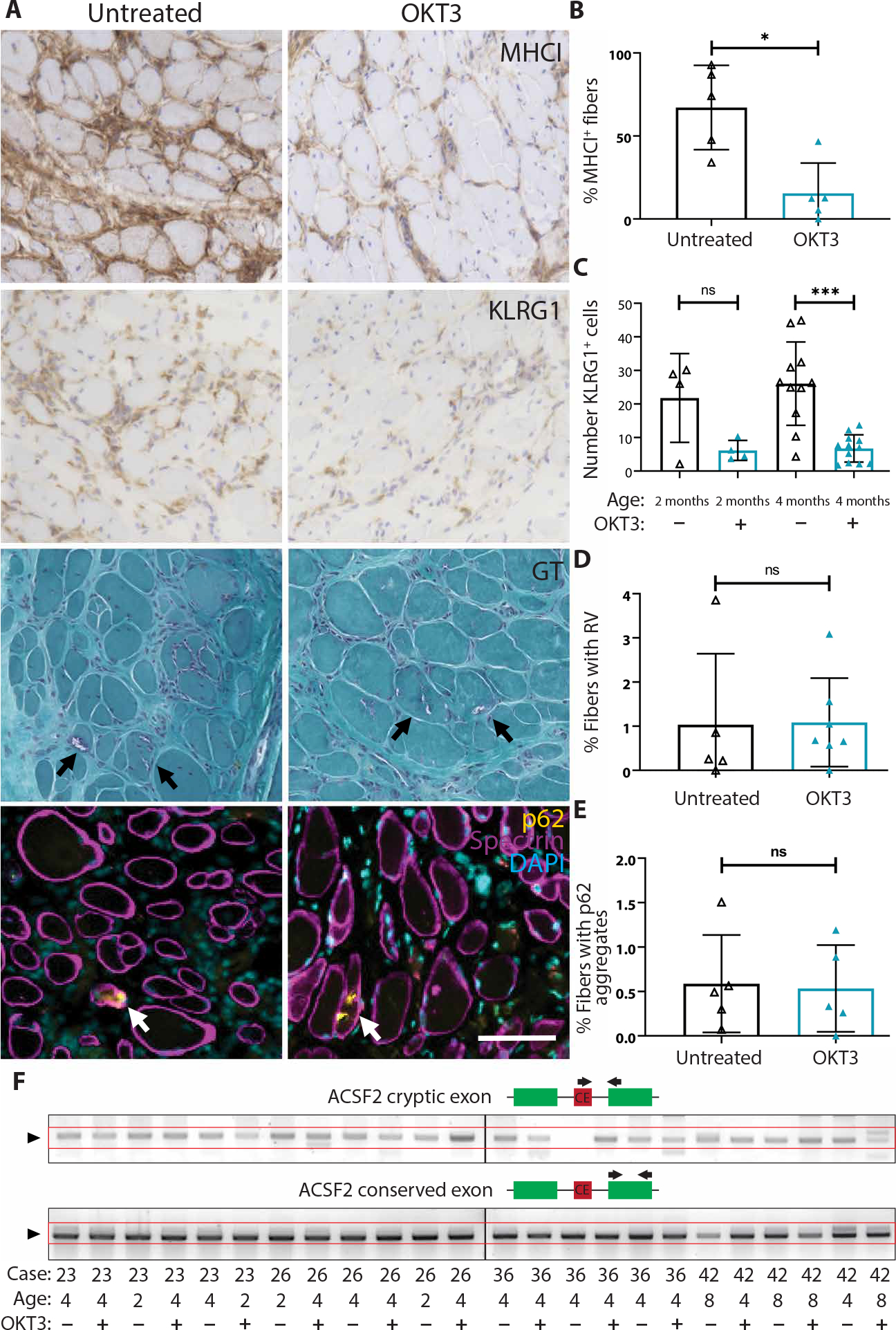
(A) Representative images from MHC-I, KLRG1, GT histological stains, and fluorescent costaining of p62 (yellow), spectrin (magenta), and DAPI in untreated and OKT3-treated 4-month IBM xenografts. Rimmed vacuoles and p62 aggregates are indicated by arrows. Scale bar, 100 μm. (B) Quantification of the percentage of MHC-I–positive fibers in untreated and OKT3-treated xenografts at 4 months (untreated, n = 5; OKT3, n = 5). Mann-Whitney test was used to determine significance (*P < 0.05). (C) Quantification of the number of KLRG1+ cells per xenograft cross section in untreated and OKT3-treated xenografts at both 2-month (untreated, n = 4; OKT3, n = 4) and 4-month (untreated, n = 11; OKT3, n = 12) time points. Mann-Whitney test was used to determine significance (***P < 0.001). (D) Quantification of the percentage of myofibers containing rimmed vacuoles (RV) in untreated and OKT3-treated xenografts at 4 months (untreated, n = 5; OKT3, n = 7). Mann-Whitney test was used to determine significance (xenograft samples from case 36 were excluded from this analysis as the subject biopsy did not display RVs). (E) Quantification of the percentage of fibers containing p62 aggregates in 4-month untreated (n = 5) and OKT3 (n = 5) treated xenografts. Mann-Whitney test was used to determine significance. (F) Expression from the ACSF2 cryptic exon compared to an ACSF2 conserved exon in 2-, 4-, and 8-month untreated and OKT3-treated IBM xenografts. The box diagrams show the primer design strategy for each PCR reaction. Top: One primer (black arrow) is designed to the cryptic exon (red), and the other is designed to a conserved exon (green) to detect cryptic exon (CE) expression. Bottom: Both primers are designed to a conserved exon to determine conserved exon expression as a control for total ACSF2 message.
Given that T cell depletion reduced inflammatory changes in IBM xenografts, we next determined whether OKT3 affected degenerative muscle pathology. As determined by GT staining, OKT3 treatment did not affect the percentage of myofibers containing rimmed vacuoles (Fig. 8, A and D). Similarly, p62 immunostaining revealed no difference (P = 0.84) in the percentage of fibers showing p62-positive aggregates between untreated and OKT3-treated 4-month xenografts (Fig. 8, A and E). In addition, ACSF2, GPSM2, and/or HDGFRP2 cryptic exons were detected in OKT3-treated IBM xenografts at 2-, 4-, and 8-month time points at similar frequencies as controls (P > 0.999 at each time point) (Fig. 8F and table S8). These findings demonstrate that rimmed vacuole pathology and TDP-43 dysfunction persist in IBM xenografts depleted of T cells.
DISCUSSION
The intertwined degenerative and inflammatory pathological features have fueled the debate underlying the pathogenesis of IBM and have impeded the generation of laboratory models. Although IBM muscle biopsies exhibit nuclear clearance and cytoplasmic aggregation of TDP-43, it has not been clear whether loss of nuclear TDP-43 function occurs in this disease. Evidence that loss of TDP-43 function is a common cellular pathogenic mechanism in neurodegenerative disease exhibiting TDP-43 pathology would support such a view. Recent evidence strongly supports the view that loss of TDP-43 splicing repression triggers neurodegeneration: (i) TDP-43 splicing repression is compromised in brains of subjects with ALS-FTD and AD (24, 38), (ii) TDP-43 nuclear depletion in neurons has been reported at the presymptomatic stage in a subject with C9orf72-linked ALS-FTD (56), and (iii) ALS-linked mutant TDP-43 fails to repress nonconserved cryptic exons independent of TDP-43 cytoplasmic aggregation (26, 27) and facilitates the formation of RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells (57). Our demonstration of TDP-43 cryptic exons in muscle from subjects with IBM is consistent with the notion that nuclear depletion of TDP-43 represents an early contributor to IBM pathogenesis. Although many different immunohistochemical assays and combinations of clinical and pathological features have been suggested to have high sensitivity and specificity for the diagnosis of IBM, the PCR-based cryptic exon detection assay that we report here demonstrates high sensitivity (84%) and specificity (99%) for IBM diagnosis in a large myositis cohort (119 subjects: IBM, n = 44; control, n = 75). Because the incorporation of cryptic exons that are spliced in-frame likely encode previously unidentified epitopes (neoantigens), we hypothesize that such neoantigens may contribute to the autoimmune response in IBM. If confirmed in additional cohorts, then the detection of these neoantigens in serum or muscle has potential as functional biomarkers for clinical applications.
Our study shows that skeletal muscle xenografts provide a valuable approach to model acquired muscle diseases such as IBM. A variety of laboratory models have been developed for various forms of hereditary inclusion body myopathy (hIBM), such as transgenic (58, 59) or knockin (60) models of IBMPFD caused by mutations in valosin-containing protein (VCP). However, despite the fact that hIBMs share degenerative pathological features with IBM (TDP-43 pathology, protein aggregates, and rimmed vacuoles), they are clinically distinct from IBM in that subjects with hIBM have an earlier age of onset and different patterns of muscle involvement, and, in stark contrast with IBM, muscle biopsies typically lack inflammation.
In addition, other animal models have been developed to recapitulate specific features of IBM. For example, transgenic approaches that drive Aβ expression in skeletal muscle result in a vacuolar myopathy. However, these models also fail to show inflammatory pathology, and the potential contribution of Aβ and APP in the pathogenesis of IBM remains controversial (61, 62). Although transgenic mice that conditionally overexpress MHC-I show myofiber degeneration, they lack other aspects of IBM pathology (63). Thus, in contrast to the xenograft model described here, existing mouse models of IBM recapitulate some aspects of IBM pathology, but none show the full spectrum of pathological features. These xenografts can recapitulate the complex genetic and epigenetic abnormalities that exist in human disease that may never be reproducible in other animal models, and xenografts form a complete in vivo system for modeling disease and developing new therapies.
Our data show that muscle from subjects with IBM robustly regenerates in immunodeficient mice to form skeletal muscle xenografts despite the presence of an inflammatory milieu, and the characteristic degenerative pathological features of IBM are recapitulated in this xenograft model. Most IBM xenografts display robust rimmed vacuoles at 4 months, and nuclear clearance of TDP-43 leading to missplicing and incorporation of cryptic exons is detected in the majority of IBM xenograft cases, indicating a loss of TDP-43 function in our model. Because all mature myofibers cut during the biopsy procedure are replaced with myofibers that form de novo from satellite cells by 4 months, our data indicate that rimmed vacuole formation and TDP-43 pathology are intrinsic to muscle from subjects with IBM and do not require factors circulating within the blood. TDP-43 is thought to function in normal muscle regeneration through the formation of cytoplasmic, amyloid-like “myo-granules” associated with sarcomeric mRNAs (64). These myo-granules form in healthy muscle after injury and are readily cleared as myofibers mature. However, purified myo-granules can seed the formation of amyloid-like fibrils in vitro and therefore may lead to the formation of stable aggregates that may drive disease pathology (65), an idea that can now be tested in our IBM xenograft model.
In addition to these degenerative features, IBM xenografts also show elevation of MHC-I, intense endomysial inflammation, and oligoclonal expansion of CD8+ T cells that express markers of highly differentiated cytotoxic T cells including CD57 and KLRG1. Persistence of these T cells and evidence of invasion of non-necrotic myofibers in IBM xenografts strongly suggest ongoing antigen stimulation by newly forming myofibers. In contrast, there is no evidence of graft-versus-host disease, as human T cells do not invade surrounding mouse muscle, and the expanded T cell clones in xenografts mirror those of the original IBM muscle biopsy.
Using a monoclonal CD3 antibody (OKT3) (52), we successfully depleted 96% of T cells from IBM xenografts. This treatment reduced MHC-I expression in myofibers, demonstrating that T cells are required for MHC-I up-regulation. T cell burden correlates with the number of COX-deficient fibers in IBM xenografts, as has been identified in IBM biopsies, suggesting that T cells may directly contribute to mitochondrial pathology in IBM. Such a view is consistent with in vitro studies showing primary myotubes cultured with proinflammatory cytokines such as interferon-γ and interleukin-1β led to a decrease in amount of COX protein (66).
Although OKT3 treatment substantially ameliorated inflammatory changes in IBM xenografts, degenerative pathological features including rimmed vacuoles and loss of TDP-43 function persist. This finding may explain why subjects with IBM do not respond to immunosuppressive treatment. However, we cannot exclude the possibility that T cells induce these degenerative features during early myofiber regeneration, before OKT3-mediated ablation. Furthermore, the low number of subjects with IBM used in these studies (n = 4), the variability in IBM pathology, the short period of treatment (4 months), and low numbers of fibers exhibiting degenerative pathology in xenografts preclude us from excluding the possibility that OKT3 treatment might have an effect on myofiber degeneration or atrophy.
In addition to the caveats mentioned above, this xenograft model has other limitations. Typical functional or behavioral studies used to assess muscle weakness will be challenging to apply to this model because the human xenograft is adjacent to mouse muscle. Because pathologic analysis requires sacrificing the entire xenograft, longitudinal studies to demonstrate alterations of IBM pathology over time require large numbers of xenografts. Although single fiber contractility of xenografts can be assessed in vitro (30), the lack of in vivo assays to assess functional outcomes would preclude mechanistic interpretation of pathological features of IBM responsible for muscle weakness, limiting the translatability of this model to predict clinical benefit. In addition, the number of xenografts that could be obtained from each individual subject was limited by the size of the research piece obtained at biopsy. Given the variability and heterogeneity of IBM pathology, a large number of xenografts may need to be performed in preclinical studies to demonstrate statistically significant improvements in IBM pathology.
Nonetheless, this xenograft model of IBM has the advantage of exhibiting both degenerative and inflammatory features. Our data are most consistent with a model in which loss of TDP-43 function and rimmed vacuole formation in IBM occur independently or upstream of T cell infiltration. These findings support the view that IBM should be considered within the spectrum of TDP-43 proteinopathy, along with ALS, FTD, and other neurodegenerative diseases exhibiting TDP-43 pathology.
MATERIALS AND METHODS
Study design
The objective of this study was to characterize the pathological features of IBM recapitulated in a xenograft model and use that model to conduct mechanistic studies of disease pathology. Our rationale behind developing a xenograft model of IBM arose from the fact that xenografts have been effectively used to develop animal models of FSHD and malignancies, and we hypothesized that this approach would closely replicate this complex human disease in comparison to traditional laboratory models.
All use of research specimens from human subjects was approved by the Johns Hopkins Institutional Review Board (IRB00235256 and IRB00072691). Subjects with suspected muscle disease scheduled for a diagnostic muscle biopsy provided informed consent before providing an extra muscle sample for use in xenograft surgeries. All IBM cases met ENMC 2011 criteria for clinically defined or probable IBM (3). Control samples were selected from non-IBM subjects scheduled for a diagnostic muscle biopsy. Subject samples with excessive fibroadipose replacement or in poor condition were excluded. Sample size calculations and subject randomization were not performed. Because IBM is classified as an orphan disease (a disease that affects less than 200,000 Americans), our aim was to consent as many IBM and control subjects as possible following our inclusion and exclusion criteria. Xenograft procedures were performed as previously described (41). All animal experiments were approved by the Johns Hopkins University Institutional Animal Care and Use Committee (protocol number: MO20M09).
Our experimental goals were to (i) study the expression of TDP-43 cryptic exons in muscle from subjects with IBM and their potential utility as biomarkers, (ii) identify the degenerative and inflammatory features of IBM found in this xenograft model, and (iii) determine whether removal of human T cells from IBM xenografts would influence these pathological features. Xenograft collection time points were determined prospectively on the basis of xenograft regeneration. Xenografts were considered mature at 4 months on the basis of the expression of eMHC, and unless noted in the figure legends, experiments were performed on samples 4 months after xenotransplantation. We performed all fiber morphology and pathological analyses on the entire frozen section and quantified three muscle sections per sample (technical replicates), and the value reported is an average of these three counts. Pathological quantifications (TDP-43 nuclear clearing, p62 aggregation, and rimmed vacuoles) were performed by an independent investigator after sample blinding. The investigator carrying out the staining protocols and microscopy would provide the blinded investigator deidentified image files to analyze.
For xenografted mice treated with OKT3 (10 mg/kg) (Fisher Scientific, 50561956), OKT3 diluted in sterile PBS was injected intraperitoneally immediately after the xenograft surgery and once weekly until xenograft collection was performed. Control “untreated” mice in OKT3 experiments were injected with sterile PBS after the same treatment regimen. Animal numbers for OKT3 experiments were determined by previous experience with xenograft models of FSHD and availability of subjects with IBM (30, 31).
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 8.3.1 (GraphPad software, La Jolla, CA, USA, www.graphpad.com). For non-normally distributed data (Shapiro-Wilk test, P < 0.05), the nonparametric Mann-Whitney test was used to determine significance between two groups, or Fisher’s exact test where noted. Data are presented as means ± SD unless otherwise indicated in the figure legends. Significance markers on figures are from post hoc analysis (ns, not significant; *P < 0.05, **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.00001) with values of P < 0.05 considered significant unless otherwise noted in the figure legends.
Supplementary Material
Acknowledgments:
We thank the staff of the Next Generation Sequencing Center (JHMI) for RNA-seq service and the Johns Hopkins NINDS Multiphoton Imaging Core (NS050274) for technical expertise and providing imaging equipment.
Funding:
This work was supported, in part, by grants from the NIH (R01-NS095969 to P.C.W., R01-NS082563 to T.E.L., R01-NS120060 to S.A.V., and R01-AR076390 to T.E.L. and P.C.W.), Maryland Technology Development Corporation to P.C.W., Muscular Dystrophy Association 630399 to T.E.L., Johns Hopkins Discovery Award to T.E.L., Myositis UK Speed Funding Award 132406 to K.A.B., Intramural Research Program of NIAMS to A.L.M., and Peter and Carmen Lucia Buck Foundation to T.E.L. and P.C.W.
Footnotes
Competing interests: T.E.L. has served as a consultant for AAVogen, Abata Therapeutics, Acceleron Pharma, Novartis, Kezar Life Sciences, and Orphazyme.
SUPPLEMENTARY MATERIALS
Data and materials availability:
Processed data from TCR sequencing experiments are available on VDJServer (http://VDJServer.org) with UUID no. 2551710644631825940-242 ac114-0001-012. Sequencing data from human myoblasts were deposited in SRA under the accession SRP341966. Individual-specific sequencing data were deposited in dbGaP under the accession phs002663.v1. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.
REFERENCES AND NOTES
- 1.Benveniste O, Guiguet M, Freebody J, Dubourg O, Squier W, Maisonobe T, Stojkovic T, Leite MI, Allenbach Y, Herson S, Brady S, Eymard B, Hilton-Jones D, Long-term observational study of sporadic inclusion body myositis. Brain 134, 3176–3184 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Greenberg SA, Inclusion body myositis: Clinical features and pathogenesis. Nat. Rev. Rheumatol. 15, 257–272 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Rose MR; ENMC IBM Working Group, 188th ENMC international workshop: Inclusion body myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul. Disord. 23, 1044–1055 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Needham M, Corbett A, Day T, Christiansen F, Fabian V, Mastaglia FL, Prevalence of sporadic inclusion body myositis and factors contributing to delayed diagnosis. J. Clin. Neurosci. 15, 1350–1353 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Catalán M, Selva-O'Callaghan A, Grau JM, Diagnosis and classification of sporadic inclusion body myositis (sIBM). Autoimmun. Rev 13, 363–366 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Benveniste O, Stenzel W, Hilton D, Marco J, Olivier S, Engelen BGMV, Amyloid deposits and inflammatory infiltrates in sporadic inclusion body myositis: The inflammatory egg comes before the degenerative chicken. Acta Neuropathol. 129, 611–624 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weihl CC, Mammen AL, Sporadic inclusion body myositis—A myodegenerative disease or an inflammatory myopathy. Neuropathol. Appl. Neurobiol. 43, 82–91 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Keller CW, Schmidt J, Lünemann JD, Immune and myodegenerative pathomechanisms in inclusion body myositis. Ann. Clin. Transl. Neurol. 4, 422–445 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenberg SA, Pinkus JL, Kong SW, Baecher-Allan C, Amato AA, Dorfman DM, Highly differentiated cytotoxic T cells in inclusion body myositis. Brain 142, 2590–2604 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Rothwell S, Cooper RG, Lundberg IE, Gregersen PK, Hanna MG, Machado PM, Herbert MK, Pruijn GJM, Lilleker JB, Roberts M, Bowes J, Seldin MF, Vencovsky J, Danko K, Limaye V, Selva-O'Callaghan A, Platt H, Molberg Ø, Benveniste O, Radstake TRDJ, Doria A, De Bleecker J, De Paepe B, Gieger C, Meitinger T, Winkelmann J, Amos CI, Ollier WE, Padyukov L, Lee AT, Lamb JA, Chinoy H, Denton C, Gheorghe K, Hilton-Jones D, Kiely P, Mann H, Immune-array analysis in sporadic inclusion body myositis reveals HLA–DRB1 amino acid heterogeneity across the myositis spectrum. Arthritis Rheum. 69, 1090–1099 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larman HB, Salajegheh M, Nazareno R, Lam T, Sauld J, Steen H, Kong SW, Pinkus JL, Amato AA, Elledge SJ, Greenberg SA, Cytosolic 5′-nucleotidase 1A autoimmunity in sporadic inclusion body myositis. Ann. Neurol. 73, 408–418 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Pluk H, Van Hoeve BJA, Van Dooren SHJ, Stammen-Vogelzangs J, Van Der Heijden A, Schelhaas HJ, Verbeek MM, Badrising UA, Arnardottir S, Gheorghe K, Lundberg IE, Boelens WC, Van Engelen BG, Pruijn GJM, Autoantibodies to cytosolic 5′-nucleotidase 1A in inclusion body myositis. Ann. Neurol. 73, 397–407 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Greenberg SA, Pinkus JL, Amato AA, Kristensen T, Dorfman DM, Association of inclusion body myositis with T cell large granular lymphocytic leukaemia. Brain 139, 1348–1360 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Askanas V, Engel WK, Bilak M, Alvarez RB, Selkoe DJ, Twisted tubulofilaments of inclusion body myositis muscle resemble paired helical filaments of Alzheimer brain and contain hyperphosphorylated tau. Am. J. Pathol. 144, 177–187 (1994). [PMC free article] [PubMed] [Google Scholar]
- 15.Askanas V, McFerrin J, Baque S, Alvarez RB, Sarkozi E, Engel WK, Transfer of β-amyloid precursor protein gene using adenovirus vector causes mitochondrial abnormalities in cultured normal human muscle. Proc. Natl. Acad. Sci. U.S.A. 93, 1314–1319 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugarman MC, Yamasaki TR, Oddo S, Echegoyen JC, Murphy MP, Golde TE, Jannatipour M, Leissring MA, Laferla FM, Inclusion body myositis-like phenotype induced by transgenic overexpression of β-APP in skeletal muscle. Proc. Natl. Acad. Sci. 99, 6334–6339 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dubourg O, Wanschitz J, Maisonobe T, Béhin A, Allenbach Y, Herson S, Benveniste O, Diagnostic value of markers of muscle degeneration in sporadic inclusion body myositis. Acta Myologica 30, 103–108 (2011). [PMC free article] [PubMed] [Google Scholar]
- 18.Hiniker A, Daniels BH, Lee HS, Margeta M, Comparative utility of LC3, p62 and TDP-43 immunohistochemistry in differentiation of inclusion body myositis from polymyositis and related inflammatory myopathies. Acta Neuropathol. Commun. 1–29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenberg SA, Theories of the pathogenesis of inclusion body myositis. Curr. Rheumatol. Rep 12, 221–228 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY, Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Lukavsky PJ, Daujotyte D, Tollervey JR, Ule J, Stuani C, Buratti E, Baralle FE, Damberger FF, Allain FH-T, Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20, 1443–1449 (2013). [DOI] [PubMed] [Google Scholar]
- 22.Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW, Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 14, 459–468 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V, Patani R, Chandran S, Rot G, Zupan B, Shaw CE, Ule J, Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 14, 452–458 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ling JP, Pletnikova O, Troncoso JC, Wong PC, TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science 349, 650–655 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ling JP, Chhabra R, Merran JD, Schaughency PM, Wheelan SJ, Corden JL, Wong PC, PTBP1 and PTBP2 repress nonconserved cryptic exons. Cell Rep. 17, 104–113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klim JR, Williams LA, Limone F, Juan IGS, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, Moccia R, Cassel SH, Chen K, Wainger BJ, Woolf CJ, Eggan K, ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 22, 167–179 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, Ohkubo T, Rodriguez M, Lin N, Wu D, Bennett CF, Rigo F, Da Cruz S, Ravits J, Lagier-Tourenne C, Cleveland DW, Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 22, 180–190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubio-Viqueira B, Hidalgo M, Direct in vivo xenograft tumor model for predicting chemotherapeutic drug response in cancer patients. Clin. Pharmacol. Ther. 85, 217–221 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Izumchenko E, Paz K, Ciznadija D, Sloma I, Katz A, Vasquez-Dunddel D, Ben-Zvi I, Stebbing J, McGuire W, Harris W, Maki R, Gaya A, Bedi A, Zacharoulis S, Ravi R, Wexler LH, Hoque MO, Rodriguez-Galindo C, Pass H, Peled N, Davies A, Morris R, Hidalgo M, Sidransky D, Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 28, 2595–2605 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, King OD, Rahimov F, Jones TI, Ward CW, Kerr JP, Liu N, Emerson CP Jr., Kunkel LM, Partridge TA, Wagner KR, Human skeletal muscle xenograft as a new preclinical model for muscle disorders. Hum. Mol. Genet. 23, 3180–3188 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen JC, King OD, Zhang Y, Clayton NP, Spencer C, Wentworth BM, Emerson CP Jr., Wagner KR, Morpholino-mediated knockdown of DUX4 toward facioscapulohumeral muscular dystrophy therapeutics. Mol. Ther. 24, 1405–1411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suetta C, Frandsen U, Mackey AL, Jensen L, Hvid LG, Bayer ML, Petersson SJ, Schroder HD, Andersen JL, Aagaard P, Schjerling P, Kjaer M, Ageing is associated with diminished muscle re-growth and myogenic precursor cell expansion early after immobility-induced atrophy in human skeletal muscle. J. Physiol. 591, 3789–3804 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morosetti R, Broccolini A, Sancricca C, Gliubizzi C, Gidaro T, Tonali PA, Ricci E, Mirabella M, Increased aging in primary muscle cultures of sporadic inclusion-body myositis. Neurobiol. Aging 31, 1205–1214 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Loell I, Lundberg IE, Can muscle regeneration fail in chronic inflammation: A weakness in inflammatory myopathies? J. Intern. Med. 269, 243–257 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Tidball JG, Regulation of muscle growth and regeneration by the immune system. Nat. Rev. Immunol. 17, 165–178 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wanschitz JV, Dubourg O, Lacene E, Fischer MB, Höftberger R, Budka H, Romero NB, Eymard B, Herson S, Butler-Browne GS, Voit T, Benveniste O, Expression of myogenic regulatory factors and myo-endothelial remodeling in sporadic inclusion body myositis. Neuromuscul. Disord. 23, 75–83 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cortese A, Plagnol V, Brady S, Simone R, Lashley T, Acevedo-Arozena A, De Silva R, Greensmith L, Holton J, Hanna MG, Fisher EMC, Fratta P, Widespread RNA metabolism impairment in sporadic inclusion body myositis TDP43-proteinopathy. Neurobiol. Aging 35, 1491–1498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun M, Bell W, LaClair KD, Ling JP, Han H, Kageyama Y, Pletnikova O, Troncoso JC, Wong PC, Chen LL, Cryptic exon incorporation occurs in Alzheimer's brain lacking TDP-43 inclusion but exhibiting nuclear clearance of TDP-43. Acta Neuropathol. 133, 923–931 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jeong YH, Ling JP, Lin SZ, Donde AN, Braunstein KE, Majounie E, Traynor BJ, LaClair KD, Lloyd TE, Wong PC, Tdp-43 cryptic exons are highly variable between cell types. Mol. Neurodegener. 12, 13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salajegheh M, Pinkus JL, Taylor JP, Amato AA, Nazareno R, Baloh RH, Greenberg SA, Sarcoplasmic redistribution of nuclear TDP-43 in inclusion body myositis. Muscle Nerve 40, 19–31 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Britson KA, Black AD, Wagner KR, Lloyd TE, Performing human skeletal muscle xenografts in immunodeficient mice. J. Vis. Exp 151, (2019). [DOI] [PubMed] [Google Scholar]
- 42.Schiaffino S, Rossi AC, Smerdu V, Leinwand LA, Reggiani C, Developmental myosins: Expression patterns and functional significance. Skelet. Muscle 5, 22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oldfors A, Moslemi AR, Jonasson L, Ohlsson M, Kollberg G, Lindberg C, Mitochondrial abnormalities in inclusion-body myositis. Neurology 66, S49–S55 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Lindgren U, Roos S, Hedberg Oldfors C, Moslemi AR, Lindberg C, Oldfors A, Mitochondrial pathology in inclusion body myositis. Neuromuscul. Disord. 25, 281–288 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Fyhr IM, Moslemi AR, Mosavi AA, Lindberg C, Tarkowski A, Oldfors A, Oligoclonal expansion of muscle infiltrating T cells in inclusion body myositis. J. Neuroimmunol. 79, 185–189 (1997). [DOI] [PubMed] [Google Scholar]
- 46.Salajegheh M, Rakocevic G, Raju R, Shatunov A, Goldfarb LG, Dalakas MC, T cell receptor profiling in muscle and blood lymphocytes in sporadic inclusion body myositis. Neurology 69, 1672–1679 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Müntzing K, Lindberg C, Moslemi AR, Oldfors A, Inclusion body myositis: Clonal expansions of muscle-infiltrating T cells persist over time. Scand. J. Immunol. 58, 195–200 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Pandya JM, Fasth AE, Zong M, Arnardottir S, Dani L, Lindroos E, Malmstrom V, Lundberg IE, Expanded T cell receptor Vβ-restricted T cells from patients with sporadic inclusion body myositis are proinflammatory and cytotoxic CD28null T cells. Arthritis Rheum. 62, 3457–3466 (2010). [DOI] [PubMed] [Google Scholar]
- 49.Montagne JM, Zheng XA, Pinal-Fernandez I, Milisenda JC, Christopher-Stine L, Lloyd TE, Mammen AL, Larman HB, Ultra-efficient sequencing of T Cell receptor repertoires reveals shared responses in muscle from patients with myositis. EBioMedicine 59, 102972 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lo Nigro C, Macagno M, Sangiolo D, Bertolaccini L, Aglietta M, Merlano MC, NK-mediated antibody-dependent cell-mediated cytotoxicity in solid tumors: Biological evidence and clinical perspectives. Ann. Transl. Med. 7, 105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kung P, Goldstein G, Reinherz EL, Schlossman SF, Monoclonal antibodies defining distinctive human T cell surface antigens. Science 206, 347–349 (1979). [DOI] [PubMed] [Google Scholar]
- 52.Wunderlich M, Brooks RA, Panchal R, Rhyasen GW, Danet-Desnoyers G, Mulloy JC, OKT3 prevents xenogeneic GVHD and allows reliable xenograft initiation from unfractionated human hematopoietic tissues. Blood 123, e134–e144 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chahin N, Engel AG, Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology 70, 418–424 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Ikenaga C, Kubota A, Kadoya M, Taira K, Uchio N, Hida A, Maeda MH, Nagashima Y, Ishiura H, Kaida K, Goto J, Tsuji S, Shimizu J, Clinicopathologic features of myositis patients with CD8-MHC-1 complex pathology. Neurology 89, 1060–1068 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Rygiel KA, Miller J, Grady JP, Rocha MC, Taylor RW, Turnbull DM, Mitochondrial and inflammatory changes in sporadic inclusion body myositis. Neuropathol. Appl. Neurobiol. 41, 288–303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vatsavayai SC, Yoon SJ, Gardner RC, Gendron TF, Vargas JN, Trujillo A, Pribadi M, Phillips JJ, Gaus SE, Hixson JD, Garcia PA, Rabinovici GD, Coppola G, Geschwind DH, Petrucelli L, Miller BL, Seeley WW, Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain 139, 3202–3216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu H, Lu S, Gasior K, Singh D, Vazquez-Sanchez S, Tapia O, Toprani D, Beccari MS, Yates JR 3rd, Da Cruz S, Newby JM, Lafarga M, Gladfelter AS, Villa E, Cleveland DW, HSP70 chaperones RNA-free TDP-43 into anisotropic intranuclear liquid spherical shells. Science 371, eabb4309 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weihl CC, Miller SE, Hanson PI, Pestronk A, Transgenic expression of inclusion body myopathy associated mutant p97/VCP causes weakness and ubiquitinated protein inclusions in mice. Hum. Mol. Genet. 16, 919–928 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Custer SK, Neumann M, Lu H, Wright AC, Taylor JP, Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum. Mol. Genet. 19, 1741–1755 (2010). [DOI] [PubMed] [Google Scholar]
- 60.Nalbandian A, Llewellyn KJ, Badadani M, Yin HZ, Nguyen C, Katheria V, Watts G, Mukherjee J, Vesa J, Caiozzo V, Mozaffar T, Weiss JH, Kimonis VE, A progressive translational mouse model of human valosin-containing protein disease: The VCPR155H/+ mouse. Muscle Nerve 47, 260–270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greenberg SA, How citation distortions create unfounded authority: Analysis of a citation network. BMJ 339, b2680 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fergusson D, Inappropriate referencing in research. BMJ 339, b2049 (2009). [DOI] [PubMed] [Google Scholar]
- 63.Nagaraju K, Raben N, Loeffler L, Parker T, Rochon PJ, Lee E, Danning C, Wada R, Thompson C, Bahtiyar G, Craft J, Hooft van Huijsduijnen R, Plotz P, Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc. Natl. Acad. Sci. 97, 9209–9214 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vogler TO, Wheeler JR, Nguyen ED, Hughes MP, Britson KA, Lester E, Rao B, Betta ND, Whitney ON, Ewachiw TE, Gomes E, Shorter J, Lloyd TE, Eisenberg DS, Taylor JP, Johnson AM, Olwin BB, Parker R, TDP-43 and RNA form amyloid-like myo-granules in regenerating muscle. Nature 563, 508–513 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cutler AA, Ewachiw TE, Corbet GA, Parker R, Olwin BB, Myo-granules connect physiology and pathophysiology. J. Exp. Neurosci. 13, 117906951984215 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chinoy H, Lilleker JB, in BMC Rheumatol (2019), vol. 3, pp. 1–56. [Google Scholar]
- 67.Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, Chudakov DM, MiXCR: Software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Shannon CE, The mathematical theory of communication. 1963. MD Comput. 14, 306–317 (1997). [PubMed] [Google Scholar]
- 69.Villalta SA, Rosenthal W, Martinez L, Kaur A, Sparwasser T, Tidball JG, Margeta M, Spencer MJ, Bluestone JA, Regulatory T cells suppress muscle inflammation and injury in muscular dystrophy. Sci. Transl. Med. 6, 258ra142 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mayeuf-Louchart A, Hardy D, Thorel Q, Roux P, Gueniot L, Briand D, Mazeraud A, Bougle A, Shorte SL, Staels B, Chretien F, Duez H, Danckaert A, MuscleJ: A high-content analysis method to study skeletal muscle with a new Fiji tool. Skelet. Muscle 8, 25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed data from TCR sequencing experiments are available on VDJServer (http://VDJServer.org) with UUID no. 2551710644631825940-242 ac114-0001-012. Sequencing data from human myoblasts were deposited in SRA under the accession SRP341966. Individual-specific sequencing data were deposited in dbGaP under the accession phs002663.v1. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials.