

Abstract
Escalated innate immunity plays a critical role in SARS-CoV-2 pathology; however, the molecular mechanism is incompletely understood. Thus, we aim to characterize the molecular mechanism by which SARS-CoV-2 Spike protein advances human macrophage (Mϴ) inflammatory and glycolytic phenotypes and uncover novel therapeutic strategies. We found that human Mϴs exposed to Spike protein activate IRAK4 phosphorylation. Blockade of IRAK4 in Spike protein-stimulated Mϴs nullifies signaling of IRAK4, AKT, and baseline p38 without affecting ERK and NF-κB activation. Intriguingly, IRAK4 inhibitor (IRAK4i) rescues the SARS-CoV-2-induced cytotoxic effect in ACE2+HEK 293 cells. Moreover, the inflammatory reprogramming of Mϴs by Spike protein was blunted by IRAK4i through IRF5 and IRF7, along with the reduction of monokines, IL-6, IL-8, TNFα, and CCL2. Notably, in Spike protein-stimulated Mϴs, suppression of the inflammatory markers by IRAK4i was coupled with the rebalancing of oxidative phosphorylation over metabolic activity. This metabolic adaptation promoted by IRAK4i in Spike protein-activated Mϴs was shown to be in part through constraining PFKBF3, HIF1α, cMYC, LDHA, lactate expression, and reversal of citrate and succinate buildup. IRAK4 knockdown could comparably impair Spike protein-enhanced inflammatory and metabolic imprints in human Mϴs as those treated with ACE2, TLR2, and TLR7 siRNA. Extending these results, in murine models, where human SARS-CoV-2 Spike protein was not recognized by mouse ACE2, TLRs were responsible for the inflammatory and glycolytic responses instigated by Spike protein and were dysregulated by IRAK4i therapy. In conclusion, IRAK4i may be a promising strategy for severe COVID-19 patients by counter-regulating ACE2 and TLR-mediated Mϴ hyperactivation.
Graphical abstract
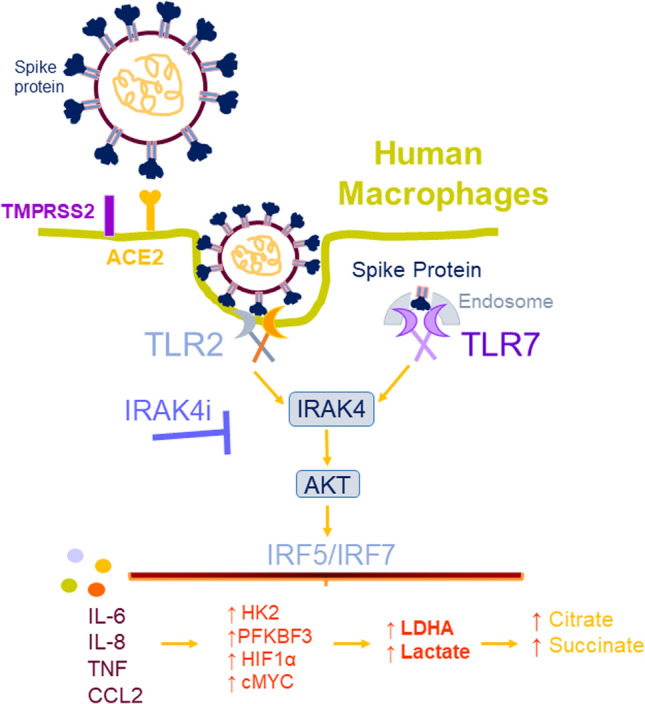
IRAK4i therapy counteracts Mϴ inflammatory and glycolytic reprogramming triggered by Spike protein. This study illustrates that SARS-CoV-2 Spike protein activates IRAK4 signaling via ACE2 as well as TLR2 and TLR7 sensing in human Mϴs. Remarkably, IRAK4i treatment can dysregulate both ACE-dependent and independent (via TLR sensing) SARS-CoV-2 Spike protein-activated inflammatory and metabolic imprints.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00018-022-04329-8.
Keywords: SARS-COV-2 Spike protein, IRAK4 inhibitor, Macrophages, TLRs, Glycolysis, Mitochondrial oxidative phosphorylation
Introduction
The emergence of the 2019 strain of coronavirus has resulted in profound morbidity and mortality worldwide. Severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) is a novel enveloped RNA virus that is associated with circulating hypercytokinemia and lymphopenia, in addition to substantial mononuclear cell infiltration in the lungs, heart, spleen, and lymph nodes in patients with severe disease [1–3]. These manifestations are observed as a result of excessive innate response and restrained adaptive immune defense leading to tissue damage in SARS-CoV-2-infected patients.
Several studies have characterized the peripheral blood transcriptome profile of COVID-19 patients at different disease stages compared to healthy donors [4, 5]. It was shown that the number of the classical CD14+ IL-1β+ monocytes, as well as IFN-activated monocytes, were expanded in COVID-19 patients relative to controls [6]. The classical CD14+ monocytes displayed amplified levels of IL-1β, JUN, FOS, JUNB, KLF6, CCL4 and CXCR4 in SARS-CoV-2-infected patients compared to the control group [7]. An independent study substantiated that the classical monocytes in COVID-19 peripheral blood mononuclear cells (PBMCs) were the main source of inflammatory monokines including IL-1β, IL-18, CCL2, CXCL8, and TNFα [4]. These authors also observed that the classical monocyte markers (S100A8, S100A9, and S100A12) dominated the nonclassical counterparts (C1AQ, C1BQ, LSTB1) in critical compared to mild/moderate COVID-19 patients [4]. In contrast, IFNγ, CXCL8, and CXCL9 had comparable expression levels in moderate and critical COVID-19 patients [4]. Others have shown that myeloid cell reprogramming by SARS-CoV-2 infection is partially due to hypermetabolic activity [8, 9].
The receptor-binding domain (RBD) of Spike protein binds to ACE2 to facilitate SARS-CoV-2 entry [10–13]. Recent findings demonstrated that coronavirus envelope protein can directly interact with TLR2 [14]. Others have shown that the Spike protein but not the envelope protein is responsible for sensing and activation of TLR2 in human and murine Mϴs [15]. Conversely, the S1 subunit of Spike protein was shown to be responsible for advancing inflammation in connection with TLR4 but not TLR2 [16]. These studies suggest that TLRs may be accountable for SARS-CoV-2 pathology via different structural proteins. However, the question remains whether the SARS-CoV-2 virus can utilize multiple strategies to enter cells and trigger the host cell’s immune response.
Interestingly, the expression of TLR1, TLR4, TLR5, TLR8, and TLR9 was associated with the severity of COVID-19 in patients with severe and critical disease relative to controls [5, 14]. Further, TLR7 transcription levels were elevated in patients with moderate COVID-19, while MyD88 and TLR2 transcriptomes were potentiated in all analyzed patients compared to healthy controls [14]. Consistently, activation of NLRP3-inflammasome and escalation of IL-1β secretion were aligned with COVID-19 severity [17, 18]. TLR and IL-1β bind to MyD88 and recruit Interleukin-1 receptor-activated kinase 4/2 into the Toll–IL-1 receptor (TIR) domain. Moreover, IRAK4 is recruited with other partners including IRAK1, TRAF6, and IRF5 to form the myddosome complex which can be triggered by TLRs [19]. Consequently, human and murine cells with IRAK4 deficiency are nonresponsive to TLR or IL-1β signaling [20, 21].
In this study, we sought to characterize the mechanism of Spike protein-instigated pathology and strategies that may subside its function. We exhibit that in human Mϴs, Spike protein phosphorylates IRAK4 and AKT which is counteracted by IRAK4i. Remarkably, IRAK4i disrupts Spike protein-induced pathogenesis by restoring oxidative phosphorylation via suppression of citrate and succinate buildup as well as downregulating the expression of inflammatory monokines and glycolytic intermediates. Similar to IRAK4 siRNA, the inflammatory and metabolic reprogramming exacerbated by Spike protein in human Mϴs were reversed by ACE2, TLR2, and TLR7 knockdown. Our findings indicate that in human Mϴs, IRAK4 inhibition impedes Spike protein signaling through ACE2 in addition to its potentiated TLR sensing.
Results
IRAK4i impairs spike protein-induced AKT signaling in myeloid cells and nullifies SARS-CoV-2 infection
Myeloid cells are the first line of defense in SARS-CoV-2 infection, we therefore, characterized the signaling pathways that were activated by Spike protein in these cells. In these studies, the dose of Spike protein was titrated based on its EC50 value (29.1 nM) in monocyte-differentiated Mϴs by TNFα quantification (Suppl. 6A) and earlier studies [22]. We found that human myeloid cells exposed to Spike protein lead to activation of IRAK4 and AKT phosphorylation, while p38, ERK, and NF-κB signaling were not implicated in this process (Fig. 1A, Suppl. 2D, Suppl. 6). We further authenticated that similar to R837, Spike protein expands IRAK4 phosphorylation by twofold, whereas the pan IRAK4 levels were unaffected in human myeloid cells (Suppl. 6B). Interestingly, IRAK4i represses Spike protein-enhanced AKT and IRAK4 signaling as well as baseline p38 phosphorylation in human myeloid cells (Fig. 1B).
Fig. 1.

IRAK4 signaling is triggered by SARS-CoV-2 Spike protein in human myeloid cells and inhibition of IRAK4 prevents SARS-CoV-2-mediated viral infection in hACE2 + 293 T cells. Human myeloid cells were untreated or stimulated with Spike Protein (30 nM; ExonBio) for 0–30 min (A). In a different set of studies, human myeloid cells were either untreated or activated with Spike protein (Exonbio; 30 nM) in the presence of DMSO (D) or IRAK4i (1 µM, PF06650833, Sigma #PZ0327) for 30 min (B). Lysates from both studies were examined for IRAK4, AKT1, p38, ERK phosphorylation, or IKBα degradation (1:1000, Cell Signaling) and actin (1:3000, Santa Cruz), n = 3. hACE2 + HEK 293 T cells were either uninfected or infected with SARS-CoV-2, (Isolate USA-WA1/2020, BEI) in the presence of IRAK4i (1 µM, PF06650833). Cytopathic effect (CPE) was imaged (C) at 48 h post-infection and quantified using ATPLite luminescence assay (D), n = 3. E Negatively selected human monocytes were either untreated (PBS) or treated with IRAK4i (1 µM, PF06650833) for 18 h before staining with FITC conjugated CD14 (Biolegend) and APC labeled ACE2 (R&D Systems) staining for 1 h, n = 10. F Human monocyte-differentiated macrophages seeded on a coverslip were either untreated (PBS) or treated with IRAK4i (1 µM) for 18 h. Thereafter, cells were fixed in 10% formalin and stained with WGA (5 µg/ml, Biotium), ACE2 (1:200 dilution; Cell Signaling), CD14 (1:100 dilution; Proteintech), and a representative image is shown, n = 3. The data are shown as mean ± SEM, *p < 0.05, **p < 0.01. Significant differences were determined by one-way ANOVA followed by Tukey’s multiple comparisons or unpaired student’s T-test
Next, the efficacy of IRAK4i therapy was evaluated on SARS-CoV-2-mediated infection and cytopathic effect (CPE) in hACE2+ HEK 293 T cells. At the dose of 1 µM, IRAK4i did not display any cytotoxic effect in hACE2+ HEK 293 T cells. However, SARS-CoV-2 infection in hACE2+HEK 293 T cells was constrained by 85% through IRAK4i therapy (Fig. 1C, D). Interestingly, we found that while IRAK4i interferes with cell death mediated by SARS-CoV-2 infection in hACE2+ HEK 293 T cells, this compound alone is inconsequential on ACE2 internalization in the absence of Spike protein (Fig. 1E, F). Collectively, in human Mϴs, IRAK4i interferes with Spike protein downstream signaling including IRAK4 and AKT activation.
IRAK4i dysregulates spike protein-enhanced IRF5/IRF7 transcription and inflammatory response in human Mϴs
Several other groups have exhibited that Mϴ inflammatory reprogramming is accompanied by specific IRF transcriptional regulation [22, 23]. In this study, we examined the inflammatory imprint of Spike protein in human monocyte-derived Mϴs and delineated the mechanism by which IRAK4i intercepts this phenomenon. Interestingly, Spike protein induction of IRF5 and IRF7 was 80–70% nullified by IRAK4i, this was followed by a 50% reduction of IRF1/8/9 in human monocyte-derived Mϴs (Fig. 2A). Transcription of IL-1β (130x), IL-6 (60x), IL-8 (34x), and CCL2 (9x), but not CCL5, was highly potentiated by Spike protein and was ablated by IRAK4i therapy (70–95%) in human monocyte-derived Mϴs (Fig. 2B). Likewise, type I (α, β) IFN and II (γ) responses amplified by Spike protein were diminished by IRAK4i (Fig. 2C, D), Suppl. 3A). Extending these findings, secretion of IL-8 was upregulated by Spike protein exposure trailed by CCL2, TNFα, and IL-6 in human monocyte-derived Mϴs (Fig. 2E–H). Nevertheless, Spike protein-induced IL-6 production levels were more potently reduced (90%) by IRAK4i relative to other monokines (40–50%). Taken together, IRAK4i reverses Spike protein-mediated inflammatory phenotype especially by disrupting IL-6 expression via IRF5 and IRF7 suppression in human monocyte-derived Mϴs.
Fig. 2.

IRAK4i therapy disrupts SARS-CoV-2 Spike protein-mediated inflammatory imprints in human Mϴs. Human monocytes-differentiated Mϴs were pretreated with DMSO (D) or IRAK4i (1 µM, PF06650833) before treating the cells with PBS or Spike protein (30 nM) for 8 h. Cells were utilized to determine the transcription of IRFs (A) or monokines including IL-1β, TNFα, IL-6, IL-8, CCL2, CCL5 (B), IFNα, and IFNβ (C–D) were quantified by real-time RT-PCR, n = 6. Concurrently, IL-6, IL-8, TNFα, and CCL2 production (E–H) were measured in the conditioned media by ELISA, n = 6. The data are shown as mean ± SEM, *p < 0.05, **p < 0.01. Significant differences were determined by one-way ANOVA followed by Tukey’s or Šídák’s multiple comparison test
IRAK4i transposes spike protein-induced metabolic reconfiguration in Mϴs
Next, experiments were performed to determine the impact of Spike protein on Mϴ immunometabolism and IRAK4i ability to counteract its function. Surprisingly, while glycolytic activity measured by extracellular acidification rate (ECAR) was amplified in Mϴs and the oxygen consumption rate (OCR) was reduced in response to Spike protein, only downregulation of OCR was rescued by IRAK4i (Fig. 3A–E). Nevertheless, expression of the glycolysis master regulators, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB)3, hexokinase (HK)2, HIF1α, cMYC, LDHA, and lactate was amplified by Spike protein in human Mϴs and abrogated via IRAK4i, while pyruvate levels were unchanged (Fig. 3B–D), Suppl. 3D). Spike protein-triggered Mϴ glycolysis was also accompanied by transcriptional downregulation of non-oxidative PPP intermediates, CARKL which was restored by IRAK4i to the baseline level (Suppl. 3F). Remarkably, Spike protein inhibitory effect on oxidative phosphorylation was linked to citrate and succinate buildup in human monocyte-derived Mϴs which was counteracted by IRAK4i (Fig. 3F, G) In short, IRAK4i shifts the Spike protein-induced glycolytic Mϴs to oxidative cells by impairing the upregulation of metabolic intermediates and accumulation of citrate or succinate.
Fig. 3.

IRAK4i counter-regulates glycolysis potentiated by SARS-CoV-2 Spike protein in Mϴs and rebalances oxidative phosphorylation. Glycolytic capacity (ECAR) and oxygen consumption (OCR) were tested in RAW 264.7 cells (5 × 103 cells/well) treated with PBS, Spike Protein (300 nM; ExonBio) with or without IRAK4i (10 µM) using the XF Cell Mito Stress Test kit (103,015–100; Agilent Technologies) as per manufacturer’s instructions. Cells were pre-conditioned with the stimuli in 0% FBS/DMEM for 24 h before ECAR (A) and OCR (E) evaluation, n = 5. Human monocytes-differentiated Mϴs were pretreated with DMSO (D) or IRAK4i (1 µM, PF06650833) before treating the cells with PBS or Spike protein (30 nM) for 8 h. Cells were utilized to determine the transcription of glycolytic intermediates by real-time RT-PCR (B) and the protein expression by Western blot analysis (C) and conditioned media was employed to quantify levels of Lactate (D), Citrate (F), and Succinate (G) by colorimetric assay, n = 5–6. The data are shown as mean ± SEM, *p < 0.05, **p < 0.01. Significant differences were determined by one-way ANOVA followed by Šídák’s multiple comparison test
Spike protein-elevated inflammatory and metabolic imprints are abrogated by TLR2, TLR7, and ACE2 knockdown in Mϴs
Since IRAK4 signaling is required for innate immune responses escalated by cell surface or endosomal TLRs, we asked if TLR2 or TLR7 signaling is involved in Spike protein-instigated Mϴ pathology. We found that Spike protein-expanded IL-6, TNFα, IL-8, or IL-1β transcription and/or production were nullified by inhibitors or knockdown of TLR2 or TLR7 in Mϴs relative to counterpart controls (Figs. 4A–C, 5A–C, Suppl. 4–5). In line with these observations, inhibition or knockdown of TLR2 or TLR7 blunted Spike protein transcriptional upregulation of metabolic markers, HIF1α, cMYC, LDHA in Mϴs compared to the control group (Figs. 4D–F, 5D–F). To further substantiate the specificity of TLR2i and TLR7i compounds, their impact was examined on LPS-elevated inflammatory and metabolic phenotypes in human monocyte-differentiated Mϴs. LPS signals through TLR2/TLR4 pathways, hence blockade of TLR2 impairs LPS-induced TNFα and lactate secretion, while dysregulation of TLR7 was ineffective on these manifestations (Suppl. 6G-H).
Fig. 4.

Spike protein-induced inflammatory and metabolic responses are dysregulated by TLR2 and TLR7 inhibitors. Human monocytes, differentiated into Mϴs were either untreated (PBS) or treated with Spike protein (30 nM; ExonBio) for 8 h in the presence of DMSO, TLR2i (oxPAPC, InvivoGen, 30 µg/ml), or TLR7i (A151 InvivoGen, 1 µM). Conditioned media was utilized for quantifying monokines such as IL-6, TNFα, and CCL2 (A–C) protein levels by ELISA as well as employing the harvested cells to evaluate glycolytic metabolite including HIF1α, cMYC, and LDHA (D–F) transcriptional regulation by real-time RT-PCR, n = 3. The data are shown as mean ± SEM, *p < 0.05, **p < 0.01. Significant differences were determined by one-way ANOVA followed by Bonferroni’s or Šídák’s multiple comparison test
Fig. 5.

Spike protein-potentiated inflammatory and metabolic markers were impaired by ACE2, IRAK4, TLR2, and TLR7 knockdown in Mϴs. Human monocyte-differentiated Mϴs were transfected with ACE2, IRAK4, TLR2, and TLR7 specific and nonspecific control siRNA (Santa Cruz Biotechnologies) at a final concentration of 100 nM using Lipofectamine 3000 (Thermo Fisher) following the manufacturer’s instruction. The transfected cells were utilized following 48 h of transfection and were either untreated or stimulated with Spike protein (30 nM) for 8 h. Conditioned media was utilized for quantifying monokines such as IL-6, TNFα, and CCL2 (A–C) secretion by ELISA as well as employing harvested cells to characterize glycolytic metabolite mRNA expression including HIF1α, cMYC, and LDHA (D–F) by real-time RT-PCR, n = 3. The data are shown as mean ± SEM, *p < 0.05 compared to PBS, **p < 0.01 compared to PBS, #p < 0.05 relative to Spike protein, and ##p < 0.01 relative to Spike protein. Significant differences were determined by one-way ANOVA followed by Šídák’s multiple comparison test
Subsequently, experiments were performed to substantiate the specificity of IRAK4i and to compare its action with ACE2 ablation in Spike protein-activated Mϴs. Similar to the blockade of TLR2 and TLR7, the knockdown of IRAK4 and ACE2 could effectively restrain Spike protein-potentiated inflammatory monokines and glycolytic metabolites, HIF1α, cMYC, LDHA in Mϴs (Figs. 4D–F, 5D–F). In short, our results suggest that dysregulation of ACE2 or TLR2 and TLR7 function can intercept Spike protein-induced pathology in Mϴs. Hence, in cells that express ACE2, IRAK4 inhibition blocks Spike protein-induced inflammatory profile through 2 distinct pathways that are dependent on ACE or TLR-mediated function. These findings are in line with IRAK4i therapy rescuing ACE2+ HEK 293 T cells from SARS-CoV-2-induced cell death.
IRAK4i reverses spike protein-induced inflammatory and metabolic landscape in vivo
SARS-CoV-2 does not cause infection in the upper and lower airways as the human and mouse ACE2 are not homologous [24], and the murine ACE2 is not recognized by the human SARS-CoV-2 Spike protein [25]. Hence, we asked if the inflammatory response can be locally triggered by Spike protein in the peritoneal cavity and whether this function can be negated by IRAK4i or TLR blockade. Interestingly, we found that in IRAK4i treated mice and TLR7-/- animals, IL-1β transcription and secretion as well as IL-6, CCL2, and LDHA mRNA levels were compromised in response to systemic injection of Spike protein (Fig. 6A–D, F). Yet, treatment with IRAK4i showed a greater potency by suppressing several Spike protein-activated monokines that were unchanged by TLR7 deficiency including CCL5, TNFα, and MIP2 (Fig. 6E, G–H). In conclusion, in the absence of ACE2 ligation, Spike protein's modest inflammatory phenotype can be effectively ablated by IRAK4i in part through TLRs.
Fig. 6.

IRAK4i therapy reverses Spike protein-induced inflammatory and metabolic landscape in vivo. Wild-type C57BL6 and/or TLR7-/- mice were pretreated with IRAK4i (10 mg/kg BW only in WT) or untreated for 18 h before receiving Spike protein (2 µg, ExonBio) i.p. injection for 24 h. The peritoneal gavage and the exudate cells were harvested. Cells were utilized to quantify the expression of IL-1β, IL-6, CCL2, LDHA (A–D) n = 5, by real-time RT-PCR, and peritoneal gavage was employed to determine protein levels of CCL5, IL-1β, TNFα, and MIP2 (E–H), n = 6–8. The data are shown as mean ± SEM, *p < 0.05, **p < 0.01. Significant differences were determined by one-way ANOVA followed by Šídák’s multiple comparison test
Discussion
The current study uncovers that IRAK4 dysregulation obstructs the SARS-CoV-2-induced cytotoxic effect in ACE2+ HEK 293 T cells. Although IRAK4i therapy alone does not interfere with ACE2 internalization, it can effectively transpose Spike protein-driven inflammatory and metabolic reprogramming in human Mϴs. IRAK4i intercepts the metabolic activity potentiated by Spike protein via HIF1α, cMYC, LDHA, and lactate downregulation. While Spike protein-mediated suppression of oxidative phosphorylation is counter-regulated by IRAK4i through constraining citrate and succinate buildup in Mϴs. In murine models, where human SARS-CoV-2 Spike protein is unable to internalize mouse ACE2, its inflammatory and metabolic imprints are deregulated by IRAK4i therapy in part through TLR sensing. Our results indicate that blockade of IRAK4 signaling interferes with Spike protein-driven pathology through ACE2 dependent and independent strategies that include TLR deactivation (Graphical abstract).
A growing body of evidence has established that SARS-CoV-2’s, Envelope, and Spike protein advance inflammation in murine myeloid cells through NF-κB activation [14–16]. Others have elucidated that SARS-CoV-2’s Envelope or Spike protein S1 subunit is involved in ERK and JNK signaling in murine Mϴs [14, 16]. Although we were unable to show ERK involvement, a strong and time-dependent IRAK4, and AKT signaling was exhibited in human Mϴs exposed to Spike protein, which was impaired by IRAK4 inhibition. Notably, NF-κB activation was transient and sporadic in Spike protein-stimulated human Mϴs and was unaffected by IRAK4i.
ACE2 is the main port of SARS-CoV-2 Spike protein entry into the host cells [10–13]. The contact between ACE2 and Spike protein is facilitated by its RBD; hence, viral entry can be prevented by therapeutics that disengage this interaction [10, 26, 27]. More recent studies have identified alternative receptors that allow SARS-CoV-2 to access the host cells. Lectins, CD147 glycoprotein, and neuropilin (NRP1) were identified as the additional port of entry, however, only NRP1 was shown to bind to Spike protein S1 [13]. Despite IRAK4i’s ability to reverse cell death in ACE2+HEK 293 T cells infected with SARS-CoV-2, this compound alone was inconsequential on ACE2 expression and internalization in Mϴs. These findings imply that in ACE2+HEK 293 T cells and human ACE2+ Mϴs, IRAK4i disrupts IRAK4 signaling potentiated by SARS-CoV-2 Spike protein interaction with ACE2.
Earlier studies have revealed that transcriptomes of innate immunity and TLR signaling are closely connected to disease severity in COVID-19 PBMCs [5]. While MYD88 and TLR2 expression were exponentially elevated in patients with moderate to critical disease, TLR7 levels were upregulated in those with moderate disease compared to healthy controls [14]. Since TLR expression was distinctly upregulated in COVID-19 patients we asked whether blockade of a common signaling molecule, IRAK4, may effectively mitigate disease pathology expanded by Spike protein.
The SARS-CoV-2-induced inflammatory phenotype has been associated with the massive influx of monocytes that exacerbate lung injury and promote acute respiratory distress syndrome (ARDS) [28]. Moreover, monocytes recruited to the inflamed tissues are reconfigured into inflammatory cells. We found that in human Mϴs, amplification of IRF5 and IRF7 was instrumental in Spike protein-induced inflammatory landscape and was potently negated by IRAK4i. Extending these observations, IRF7 transcription was increased in COVID-19 patients with moderate disease compared to healthy individuals [5]. Similarly, transcriptional upregulation of IRF5 and IRF7 observed in TLR7-activated RA Mϴs was markedly reversed by IRAK4i [29]. A recent review has postulated that SARS-CoV-2 infection advances Mϴ metabolic remodeling in part through IRF5 activation [19]. Hence, IRF5 may be a critical link between the so-called “metabolic inflammation” and the amplified monokine profile in COVID-19 that can be disrupted by IRAK4i.
Additionally, IRAK4i counteracted Spike protein Mϴ inflammatory reprogramming that was accompanied by the expansion of IL-6, IL-8, TNFα, and CCL2 at transcriptional and translational levels. Corroborating these findings, exposure of human PBMCs to Envelope protein contributed to an increase in IL-6 and TNFα transcription [14], which was shown to be TLR2 dependent and TLR7 independent using deficient mice compared to control animals. On the contrary, our data indicate that akin to IRAK4 blockade, inhibition, and knockdown of TLR2 or TLR7 abrogate Spike protein-induced Mϴ inflammatory and metabolic rewiring.
Enriched levels of lactate dehydrogenase (LDHA) and lactate are linked to disease severity and hypermetabolic activity in COVID-19 patients [5]. Glucose can dose-dependently enhance viral load, ACE2, and IL-1β expression in SARS-CoV-2-infected monocytes, thus upregulated blood glucose levels are a major risk factor for these patients [8]. Spike protein misregulates ECAR over OCR by increasing glycolytic intermediates including, HK2, PFKBF3, HIF1α, cMYC, LDHA, and lactate in circulating monocyte-differentiated Mϴs. Notably, IRAK4i can more effectively reverse Spike protein-induced glycolytic reprogramming in Mϴs compared to Tofacitinib in part by impairing HIF1α transcription [22]. Nonetheless, both IRAK4i and Tofacitinib therapies rebalance OCR through a different mechanism of action [22]. IRAK4i advances mitochondrial oxidative phosphorylation by repressing citrate and succinate buildup, whereas Tofacitinib restores PPARγ transcription. We also showed that knockdown of ACE2, TLR2, or TLR7 counteracts Spike protein-potentiated glycolytic reprogramming in Mϴs by downregulating transcription of cMYC, LDHA, and HIF1α. In line with our data, inhibition of glucose uptake or HIF1α activity dysregulated SARS-CoV-2 replication and monocyte inflammatory response [8]. It was also observed that SARS-CoV-2-induced mitochondrial ROS and HIF1α stability were responsible for IL-1β transcription and inflammatory response in monocytes [8].
Human ACE2 transgenic mice are required for SARS-CoV-2 infection in the lungs [24]. Yet, a modest local inflammation can be triggered by Spike protein in the absence of human ACE2 expression in vivo, suggesting that alternative strategies may function in parallel to ACE2. To delineate the influence of IRAK4 and TLR7 signaling on Spike protein-modulated inflammatory phenotype in vivo, wild type, or TLR7-/- mice were systemically injected with Spike protein in the presence or absence of IRAK4i. Interestingly, Spike protein administration activated peritoneal IL-1β secretion that was impeded by IRAK4i and TLR7 deficiency, while CCL5, TNFα, and MIP2 production were unaffected by TLR7-/- mice. In contrast, the modest inflammatory and glycolytic imprints accentuated by local Spike protein injection were strongly intercepted by IRAK4i treatment. Distinct from the in vivo findings, IL-1β production was unaltered in Spike protein stimulated monocyte-differentiated Mϴs, perhaps due to low pro-IL-1β or caspase1 expression. However, downregulation of IL-1β production by IRAK4i in response to Spike protein in vivo administration may indicate that both TLR and IL-1βR function can be nullified by this compound. A recent elegant study exhibits that in humanized K18-hACE-2 mice, treatment with IL-1 receptor antagonist rescues SARS-CoV-2-mediated lung infection [30], substantiating the significance of IL-1β in COVID-19 pathogenesis.
Supporting IRAK4i efficiency in counter-regulation of Spike protein-induced macrophage activation syndrome, others have shown that TLR2 sensing is involved in SARS-CoV-2 amplified infection [14, 15]. Intriguingly, the significance of IRAK4 signaling in SARS-CoV-2 infection was shown in an independent study where IRAK4 deficiency leads to resistance to COVID-19 through impaired IFNα2, CXCL10/IP-10, and IL-6 secretion from blood myeloid cells [31]. Supporting this notion, IRAK4 was phosphorylated following SARS-CoV-2 infection in Vero E6 cells [32] which was shown to be abrogated by Capmatinib an IRAK4 inhibitor [33]. More importantly, IRAK4i (PF-06650833) is currently in phase 2 clinical trial for hospitalized patients with COVID-19 pneumonia that suffer from exuberant inflammation.
Spike-human ACE2 interaction is considered the canonical pathway of SARS-CoV-2 entry into cells and initiation of signals, which mirrors our human data. However, our animal data most likely represents an ACE2 independent mechanism as the mice do not express human ACE2 and thus have a low affinity for the Spike protein. As recent studies have shown, treatment with ACE2 inhibitor (MLN-4760) did not abrogate the inflammatory phenotype triggered by Spike protein in human myeloid cells [15, 34]. In agreement with this notion, others have validated that TLR7-MyD88 signaling is critical for SARS-CoV-2-induced IFN secretion, while TLR2 is responsible for the IL-6 response in dendritic cells [35].
The current study uncovers that inhibition of IRAK4 signaling can reverse Spike protein-induction of inflammatory and metabolic profiles in human Mϴs that express ACE2 as well as in vivo models where ACE2 is absent, highlighting its potential use for treating patients with severe COVID-19.
Materials and methods
Human myeloid cells
Studies were approved by the University of Illinois at Chicago (UIC) Institutional Ethics Review Board and all donors gave informed written consent. Human mononuclear cells were isolated by Histopaque gradient centrifugation and monocytes were isolated from human peripheral blood (PB) using a negative selection kit according to the manufacturer’s instruction (StemCell Technology) [36–43].
Western blot analysis
Human myeloid cells were untreated or stimulated with Spike Protein (30 nM; ExonBio) for 0–40 min before cells were harvested. In a different set of studies, human myeloid cells were either untreated or activated with Spike protein (Exonbio; 30 nM) in the presence of DMSO (D) or IRAK4i (1 µM, PF06650833, Sigma #PZ0327) [23] for 30 min. Lysates from both studies were examined for IRAK4, AKT1, p38, ERK phosphorylation, or IKBα degradation (1:1000, Cell Signaling) and actin equal loading (1:3000, Santa Cruz). For glycolytic metabolites, human myeloid cells were untreated or stimulated with Spike protein (30 nM) with DMSO or IRAK4i (1 µM, PF06650833) for 8 h. Lysates were probed for HK2, PFKBF3, (1:1000, Cell Signaling), HIF1α (1:1000, Santa Cruz), cMYC (1:1000, Novus Biologicals), and actin (1:3000, Santa Cruz). Raw Western blots and their quantification are shown in Suppl-2.
Flow cytometry
Negatively selected human monocytes were either untreated (PBS) or treated with IRAK4i (1 µM, PF06650833, Sigma #PZ0327) [23] for 18 h before staining with FITC conjugated CD14 (Biolegend) and APC labeled ACE2 (R&D Systems) staining for 1 h. Zombie Violet PB450 was used to exclude the dead cells. The gating strategy is shown in Suppl-1.
Immunofluorescence visualization
Human monocyte-differentiated macrophages seeded on a coverslip were either untreated (0 h) or treated with IRAK4i (1 µM) for 18 h. Thereafter, cells were fixed in 10% formalin and stained with WGA (5 µg/ml, Biotium), ACE2 (1:200 dilution; Cell Signaling), CD14 (1:100 dilution; Proteintech), and a representative image is shown.
CPE assay using SARS-CoV-2 infection assay in hACE2-293 T cells
hACE2-293 T cells were either uninfected or infected with SARS-CoV-2, (Isolate USA-WA1/2020, BEI) in the presence or absence of IRAK4i (1 µM, PF06650833, Sigma #PZ0327) [23] and the assay was performed in triplicates. Cytopathic effect (CPE) was imaged at 48 h post-infection and %CPE was quantified using ATPLite luminescence screening. For quantification purposes, the medium was removed and following the addition of Cell Titer Glo assay reagent (Promega Inc.) for 15 min, luminescence was measured [44]. The virus-induced cytotoxicity % was calculated as follows: 100 × [1–(X–MIN)/(MAX–MIN)]. X = Mean RLU of triplicates; Min = Mean RLU of infected control wells with the virus; Max = Mean RLU of uninfected wells without virus. These experiments were performed in collaboration with Drs. Kumar and Prabhakar in a BCL3 certified facility. Dr. Prabhakar’s laboratory was responsible for the virus propagation experiments which were performed under BSL3 containment with the approval of the University of Illinois at Chicago Institutional Biosafety Committee.
Use of inhibitors and/or knockdown cells
Human monocytes were differentiated into Mϴs for 3 days in 10% RPMI. On day 4, Mϴs were either untreated (PBS) or treated with Spike protein (30 nM; ExonBio) for 8 h in the presence of DMSO, TLR2i (oxPAPC, InvivoGen, 30 µg/ml) [14, 45] or TLR7i (A151 InvivoGen, 1 µM) [23, 46] and IRAK4i (1 µM, PF06650833, Sigma #PZ0327)[23]. Cells were harvested in Trizol for mRNA quantification and conditioned media was collected for ELISA. Human monocyte-differentiated Mϴs were transfected with ACE2, IRAK4, TLR2, and TLR7 specific and nonspecific control siRNA (Santa Cruz Biotechnologies) at a final concentration of 100 nM using Lipofectamine 3000 (Thermo Fisher) complying with the manufacturer’s instruction. The transfected cells were utilized following 48 h of transfection and were either untreated or stimulated with Spike protein (30 nM) for 8 h.
Seahorse assay
Glycolytic capacity (ECAR) and oxygen consumption (OCR) were tested in RAW 264.7 cells (5 × 103 cells/well) treated with PBS, Spike Protein (300 nM; ExonBio) with or without IRAK4i (10 µM) using the XF Cell Mito Stress Test kit (103,015–100; Agilent Technologies) as per manufacturer’s instructions. Cells were pre-conditioned with the stimuli in 0% FBS/DMEM for 24 h before ECAR and OCR evaluation [22, 47].
Spike protein immune response in vivo
Next, we examined if the inflammatory response can be activated by local Spike protein administration and whether this function can be negated by IRAK4i or TLR blockade. For this purpose, wild-type C57BL6 and/or TLR7-/- mice were pretreated and/or untreated with IRAK4i (10 mg/kg BW only in WT) for 18 h before receiving Spike protein (2 µg, ExonBio) i.p. injection for 24 h. Thereafter, peritoneal gavage and peritoneal exudate cells were harvested.
Statistical analysis
For comparison among multiple groups, one-way ANOVA followed by Tukey's, Bonferroni’s, or Šídák’s multiple comparison test was employed, using Graph Pad Prism8 software. The data were also analyzed using a two-tailed Student’s T test for paired or unpaired comparisons between two groups. Values of p < 0.05 were considered significant.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file1 The gating strategy is shown for unstained and stained myeloid cells for determining the % CD14+ ACE2+ cells (TIF 201 KB)
Supplementary file2 Raw Western blots are shown in Suppl. 2A, Suppl. 2B, and Suppl. 2C for Figs. 1A, 1B, and 3C respectively, and Western blot density was analyzed by Image J in Suppl. 2D and 2E for Figs. 1B and 3C, n=3 (TIF 175 KB)
Supplementary file3 Human monocyte-differentiated Mϴs were pretreated with DMSO (D) or IRAK4i (1 µM, PF06650833) before treating the cells with PBS or Spike protein (30 nM) for 8h. The harvested conditioned media was utilized for quantifying IFNγ, IL-1β, CCL5, or Pyruvate protein levels by ELISA (A-C), n=6 or colorimetric assay (D), n=3. Cells were utilized to determine the transcriptional regulation of HK2 and CARKL (E-F), n=6. The data are shown as mean ± SEM, *p<0.05, **p<0.01. Significant differences were determined by one-way ANOVA followed by Šídák’s multiple comparison test (TIF 81 KB)
Supplementary file4 Human monocytes, differentiated into Mϴs were either untreated (PBS) or treated with Spike protein (30 nM; ExonBio) for 8h in the presence of DMSO, TLR2i (oxPAPC, InvivoGen, 30 µg/ml), or TLR7i (A151 InvivoGen, 1 µM). The harvested cells were used to quantify IL-1β (A), IL-8 (B), TNFα (C), and CCL2 (D) mRNA expression, n=3. The data are shown as mean ± SEM, *p<0.05, **p<0.01. Significant differences were determined by one-way ANOVA followed by Bonferroni’s, Šídák’s, or Holm-Šídák’s multiple comparison test (TIF 65 KB)
Supplementary file5 Human monocyte-differentiated Mϴs were transfected with ACE2, IRAK4, TLR2, and TLR7 specific and nonspecific control siRNA (Santa Cruz Biotechnologies) at a final concentration of 100 nM using Lipofectamine 3000 (Thermo Fisher). The transfected cells were utilized following 48h of transfection and expression of the targeted genes was evaluated by real-time RT-PCR (A-D), n=3. Next, the control and knockdown cells were untreated or stimulated with Spike protein (30 nM) for 8h to determine IL-1β, IL-8, TNFα, and HK2 transcriptional expression (E-H), n=5. The data are shown as mean ± SEM, *p<0.05, **p<0.01. Significant differences were determined by one-way ANOVA followed by Šídák’s multiple comparison test or unpaired Student’s T-test (TIF 93 KB)
Supplementary file6 (A) The EC50 value was determined for Spike Protein (0, 5, 25, and 50 nM) in monocyte-differentiated Mϴs by measuring TNFα production by ELISA. (B) Human myeloid cells were untreated (PBS) or stimulated with Spike Protein (30 nM; ExonBio) and R837 (10 µg/ml) for 30min and protein levels of pIRAK4 or IRAK4 (Cell Signaling) were determined by flow cytometry. (C) pIRAK4 was quantified by ImageJ and normalized to total housekeeping protein (actin) (n=5). (D) Human myeloid cells were untreated or stimulated with Spike Protein (30 nM; ExonBio) for 0-30 min and probed for pAKT (n=3), p-p38 (n=3), and corresponding pan antibodies (Cell Signaling).(E, F) Protein concentration of pAKT and p-p38 was quantified by ImageJ and normalized to total housekeeping protein (actin). (G, H) Human monocyte-differentiated Mϴs were stimulated with LPS (100 ng/ml) in the presence or absence of TLR2i (30 µg/ml; InvivoGen) and TLR7i (1 µM; InvivoGen) for 24h and secretion of TNFα or lactate was quantified. The data are shown as mean ± SEM, *p<0.05, **p<0.01. Significant differences were determined by one-way ANOVA followed by Tukey’s multiple comparison test (TIF 168 KB)
Acknowledgements
The authors thank Balaji B. Ganesh, Director of the Flow Cytometry Core at the University of Illinois at Chicago for his excellent scientific advice.
Author contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Shahrara had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study conception and design. SU and SS. Acquisition of data. SU, KP, AM, PK, MVV, RKZ, and SS. Analysis and interpretation of data. SU, KP, AM, PK, BP, MVV, RR, MA, HJC, RKZ, JR, and SS. Providing crucial reagents. MA and HJC.
Funding
This work was supported in part by awards from the Department of Veteran’s Affairs MERIT Award BX002286, the National Institutes of Health NIH AI147697, the National Psoriasis Foundation (NPF), Pfizer Investigator-Initiated Research (IIR) Program and Chicago Biomedical Consortium (CBC) Accelerator Award.
Data availability
All findings are exhibited in the paper and the material and data are available for transparency.
Code availability
Not applicable.
Declarations
Conflict of interest
The authors have declared that no commercial or financial conflict of interest exists.
Ethical approval
All peripheral blood (PB) was collected in accordance with our protocol approved by the University of Illinois at Chicago Institutional Ethics Review Board. Furthermore, all animal studies were approved by the University of Illinois at Chicago Animal Care and Use Committee following the legal requirements and guidelines of the state of Illinois in the USA and NIH. Dr. Prabhakar’s laboratory was responsible for the virus propagation experiments which were performed under BSL3 containment with approval of the University of Illinois at Chicago Institutional Biosafety Committee.
Consent to publication
All authors have been involved in writing the manuscript and consented to publication.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, Liu S, Zhao P, Liu H, Zhu L, Tai Y, Bai C, Gao T, Song J, Xia P, Dong J, Zhao J, Wang FS. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8(4):420–422. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, Collaboration HAS, UK, COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20:355–362. doi: 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vanderbeke L, Van Mol P, Van Herck Y, De Smet F, Humblet-Baron S, Martinod K, Antoranz A, Arijs I, Boeckx B, Bosisio FM, Casaer M, Dauwe D, De Wever W, Dooms C, Dreesen E, Emmaneel A, Filtjens J, Gouwy M, Gunst J, Hermans G, Jansen S, Lagrou K, Liston A, Lorent N, Meersseman P, Mercier T, Neyts J, Odent J, Panovska D, Penttila PA, Pollet E, Proost P, Qian J, Quintelier K, Raes J, Rex S, Saeys Y, Sprooten J, Tejpar S, Testelmans D, Thevissen K, Van Buyten T, Vandenhaute J, Van Gassen S, Velasquez Pereira LC, Vos R, Weynand B, Wilmer A, Yserbyt J, Garg AD, Matthys P, Wouters C, Lambrechts D, Wauters E, Wauters J. Monocyte-driven atypical cytokine storm and aberrant neutrophil activation as key mediators of COVID-19 disease severity. Nat Commun. 2021;12(1):4117. doi: 10.1038/s41467-021-24360-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, Pere H, Charbit B, Bondet V, Chenevier-Gobeaux C, Breillat P, Carlier N, Gauzit R, Morbieu C, Pene F, Marin N, Roche N, Szwebel TA, Merkling SH, Treluyer JM, Veyer D, Mouthon L, Blanc C, Tharaux PL, Rozenberg F, Fischer A, Duffy D, Rieux-Laucat F, Kerneis S, Terrier B. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang D, Guo R, Lei L, Liu H, Wang Y, Wang Y, Qian H, Dai T, Zhang T, Lai Y, Wang J, Liu Z, Chen T, He A, O'Dwyer M, Hu J. Frontline Science: COVID-19 infection induces readily detectable morphologic and inflammation-related phenotypic changes in peripheral blood monocytes. J Leukoc Biol. 2021;109(1):13–22. doi: 10.1002/JLB.4HI0720-470R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wen W, Su W, Tang H, Le W, Zhang X, Zheng Y, Liu X, Xie L, Li J, Ye J, Dong L, Cui X, Miao Y, Wang D, Dong J, Xiao C, Chen W, Wang H. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell Discov. 2020;6(1):31. doi: 10.1038/s41421-020-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, Prodonoff JS, Carregari VC, de Biagi Junior CAO, Crunfli F, Jimenez Restrepo JL, Vendramini PH, Reis-de-Oliveira G, Bispo Dos Santos K, Toledo-Teixeira DA, Parise PL, Martini MC, Marques RE, Carmo HR, Borin A, Coimbra LD, Boldrini VO, Brunetti NS, Vieira AS, Mansour E, Ulaf RG, Bernardes AF, Nunes TA, Ribeiro LC, Palma AC, Agrela MV, Moretti ML, Sposito AC, Pereira FB, Velloso LA, Vinolo MAR, Damasio A, Proenca-Modena JL, Carvalho RF, Mori MA, Martins-de-Souza D, Nakaya HI, Farias AS, Moraes-Vieira PM. Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1alpha/glycolysis-dependent axis. Cell Metab. 2020 doi: 10.1016/j.cmet.2020.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bojkova D, Klann K, Koch B, Widera M, Krause D, Ciesek S, Cinatl J, Munch C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature. 2020;583:469–472. doi: 10.1038/s41586-020-2332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, Zhang Q, Shi X, Wang Q, Zhang L, Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581(7807):215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 11.Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z, Lu G, Qiao C, Hu Y, Yuen KY, Wang Q, Zhou H, Yan J, Qi J. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181(4):894–904 e899. doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson CB, Farzan M, Chen B, Choe H. Mechanisms of SARS-CoV-2 entry into cells. Nat Rev Mol Cell Biol. 2021;23:3–20. doi: 10.1038/s41580-021-00418-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, Jonsson CB, Kanneganti TD. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol. 2021;22(7):829–838. doi: 10.1038/s41590-021-00937-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan S, Shafiei MS, Longoria C, Schoggins JW, Savani RC, Zaki H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-kappaB pathway. Elife. 2021;10:e68563. doi: 10.7554/eLife.68563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shirato K, Kizaki T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon. 2021;7(2):e06187. doi: 10.1016/j.heliyon.2021.e06187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodrigues TS, de Sa KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L, Goncalves AV, Perucello DB, Andrade WA, Castro R, Veras FP, Toller-Kawahisa JE, Nascimento DC, de Lima MHF, Silva CMS, Caetite DB, Martins RB, Castro IA, Pontelli MC, de Barros FC, do Amaral NB, Giannini MC, Bonjorno LP, Lopes MIF, Santana RC, Vilar FC, Auxiliadora-Martins M, Luppino-Assad R, Almeida SCL, de Oliveira FR, Batah SS, Siyuan L, Benatti MN, Cunha TM, Alves-Filho JC, Cunha FQ, Cunha LD, Frantz FG, Kohlsdorf T, Fabro AT, Arruda E, de Oliveira RDR, Louzada-Junior P, Zamboni DS. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J Exp Med. 2021 doi: 10.1084/jem.20201707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bryant C. COVID-19 stokes inflammasomes. J Exp Med. 2021 doi: 10.1084/jem.20202413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stoy N. Involvement of Interleukin-1 Receptor-Associated Kinase 4 and Interferon Regulatory Factor 5 in the Immunopathogenesis of SARS-CoV-2 Infection: Implications for the Treatment of COVID-19. Front Immunol. 2021;12:638446. doi: 10.3389/fimmu.2021.638446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Suzuki N, Suzuki S, Duncan GS, Millar DG, Wada T, Mirtsos C, Takada H, Wakeham A, Itie A, Li S, Penninger JM, Wesche H, Ohashi PS, Mak TW, Yeh WC. Severe impairment of interleukin-1 and Toll-like receptor signalling in mice lacking IRAK-4. Nature. 2002;416(6882):750–756. doi: 10.1038/nature736. [DOI] [PubMed] [Google Scholar]
- 21.De Nardo D, Balka KR, Cardona Gloria Y, Rao VR, Latz E, Masters SL. Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll-like receptor signaling. J Biol Chem. 2018;293(39):15195–15207. doi: 10.1074/jbc.RA118.003314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palasiewicz K, Umar S, Romay B, Zomorrodi RK, Shahrara S. Tofacitinib therapy intercepts macrophage metabolic reprogramming instigated by SARS-CoV-2 Spike protein. Eur J Immunol. 2021;51:2330–2340. doi: 10.1002/eji.202049159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Umar S, Palasiewicz K, Van Raemdonck K, Volin MV, Romay B, Amin MA, Zomorrodi RK, Arami S, Gonzalez M, Rao V, Zanotti B, Fox DA, Sweiss N, Shahrara S. IRAK4 inhibition: a promising strategy for treating RA joint inflammation and bone erosion. Cell Mol Immunol. 2021;18(9):2199–2210. doi: 10.1038/s41423-020-0433-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bao L, Deng W, Huang B, Gao H, Liu J, Ren L, Wei Q, Yu P, Xu Y, Qi F, Qu Y, Li F, Lv Q, Wang W, Xue J, Gong S, Liu M, Wang G, Wang S, Song Z, Zhao L, Liu P, Zhao L, Ye F, Wang H, Zhou W, Zhu N, Zhen W, Yu H, Zhang X, Guo L, Chen L, Wang C, Wang Y, Wang X, Xiao Y, Sun Q, Liu H, Zhu F, Ma C, Yan L, Yang M, Han J, Xu W, Tan W, Peng X, Jin Q, Wu G, Qin C. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature. 2020;583:830–833. doi: 10.1038/s41586-020-2312-y. [DOI] [PubMed] [Google Scholar]
- 25.Leist SR, Dinnon KH, 3rd, Schafer A, Tse LV, Okuda K, Hou YJ, West A, Edwards CE, Sanders W, Fritch EJ, Gully KL, Scobey T, Brown AJ, Sheahan TP, Moorman NJ, Boucher RC, Gralinski LE, Montgomery SA, Baric RS. A mouse-adapted SARS-CoV-2 induces acute lung injury and mortality in standard laboratory mice. Cell. 2020;183(4):1070–1085 e1012. doi: 10.1016/j.cell.2020.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A, Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581(7807):221–224. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tong P, Gautam A, Windsor IW, Travers M, Chen Y, Garcia N, Whiteman NB, McKay LGA, Storm N, Malsick LE, Honko AN, Lelis FJN, Habibi S, Jenni S, Cai Y, Rennick LJ, Duprex WP, McCarthy KR, Lavine CL, Zuo T, Lin J, Zuiani A, Feldman J, MacDonald EA, Hauser BM, Griffths A, Seaman MS, Schmidt AG, Chen B, Neuberg D, Bajic G, Harrison SC, Wesemann DR. Memory B cell repertoire for recognition of evolving SARS-CoV-2 spike. Cell. 2021;184(19):4969–4980 e4915. doi: 10.1016/j.cell.2021.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, Guo L, Yang J, Wang C, Jiang S, Yang D, Zhang G, Li H, Chen F, Xu Y, Chen M, Gao Z, Yang J, Dong J, Liu B, Zhang X, Wang W, He K, Jin Q, Li M, Wang J. Heightened innate immune responses in the respiratory tract of COVID-19 patients. Cell Host Microbe. 2020;27(6):883–890 e882. doi: 10.1016/j.chom.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Umar S, Palasiewicz K, Volin MV, Zanotti B, Al-Awqati M, Sweiss N, Shahrara S. IRAK4 inhibitor mitigates joint inflammation by rebalancing metabolism malfunction in RA macrophages and fibroblasts. Life Sci. 2021;287:120114. doi: 10.1016/j.lfs.2021.120114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiong S, Zhang L, Richner JM, Class J, Rehman J, Malik AB. Interleukin-1RA mitigates SARS-CoV-2-induced inflammatory lung vascular leakage and mortality in humanized K18-hACE-2 Mice. Arterioscler Thromb Vasc Biol. 2021;41:2773–2785. doi: 10.1161/ATVBAHA.121.316925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Onodi F, Bonnet-Madin L, Meertens L, Karpf L, Poirot J, Zhang SY, Picard C, Puel A, Jouanguy E, Zhang Q, Le Goff J, Molina JM, Delaugerre C, Casanova JL, Amara A, Soumelis V. SARS-CoV-2 induces human plasmacytoid predendritic cell diversification via UNC93B and IRAK4. J Exp Med. 2021;5;218(4):e20201387. doi: 10.1084/jem.20201387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu S, Zhu L, Xie G, Mok BW-Y, Yang Z, Deng S, Lau S-Y, Chen P, Wang P, Chen H, Cai Z. Potential antiviral target for SARS-CoV-2: a key early responsive kinase during viral entry. CCS Chemistry. 2022;4(1):112–121. [Google Scholar]
- 33.Sugiyama MG, Cui H, Redka DS, Karimzadeh M, Rujas E, Maan H, Hayat S, Cheung K, Misra R, McPhee JB, Viirre RD, Haller A, Botelho RJ, Karshafian R, Sabatinos SA, Fairn GD, Madani Tonekaboni SA, Windemuth A, Julien JP, Shahani V, MacKinnon SS, Wang B, Antonescu CN. Multiscale interactome analysis coupled with off-target drug predictions reveals drug repurposing candidates for human coronavirus disease. Sci Rep. 2021;11(1):23315. doi: 10.1038/s41598-021-02432-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sawa T, Akaike T. What triggers inflammation in COVID-19? Elife. 2022;11:e76231. doi: 10.7554/eLife.76231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van der Sluis RM, Cham LB, Gris-Oliver A, Gammelgaard KR, Pedersen JG, Idorn M, Ahmadov U, Hernandez SS, Cemalovic E, Godsk SH, Thyrsted J, Gunst JD, Nielsen SD, Jorgensen JJ, Bjerg TW, Laustsen A, Reinert LS, Olagnier D, Bak RO, Kjolby M, Holm CK, Tolstrup M, Paludan SR, Kristensen LS, Sogaard OS, Jakobsen MR (2022) TLR2 and TLR7 mediate distinct immunopathological and antiviral plasmacytoid dendritic cell responses to SARS-CoV-2 infection. EMBO J 16;41(10):e109622. 10.15252/embj.2021109622 [DOI] [PMC free article] [PubMed]
- 36.Chamberlain ND, Kim SJ, Vila OM, Volin MV, Volkov S, Pope RM, Arami S, Mandelin AM, 2nd, Shahrara S. Ligation of TLR7 by rheumatoid arthritis synovial fluid single strand RNA induces transcription of TNFalpha in monocytes. Ann Rheum Dis. 2013;72(3):418–426. doi: 10.1136/annrheumdis-2011-201203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Z, Kim SJ, Chamberlain ND, Pickens SR, Volin MV, Volkov S, Arami S, Christman JW, Prabhakar BS, Swedler W, Mehta A, Sweiss N, Shahrara S. The novel role of IL-7 ligation to IL-7 receptor in myeloid cells of rheumatoid arthritis and collagen-induced arthritis. J Immunol. 2013;190(10):5256–5266. doi: 10.4049/jimmunol.1201675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen Z, Kim SJ, Essani AB, Volin MV, Vila OM, Swedler W, Arami S, Volkov S, Sardin LV, Sweiss N, Shahrara S. Characterising the expression and function of CCL28 and its corresponding receptor, CCR10. RA pathogenesis Ann Rheum Dis. 2015;74(10):1898–1906. doi: 10.1136/annrheumdis-2013-204530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim SJ, Chen Z, Chamberlain ND, Essani AB, Volin MV, Amin MA, Volkov S, Gravallese EM, Arami S, Swedler W, Lane NE, Mehta A, Sweiss N, Shahrara S. Ligation of TLR5 promotes myeloid cell infiltration and differentiation into mature osteoclasts in rheumatoid arthritis and experimental arthritis. J Immunol. 2014;193(8):3902–3913. doi: 10.4049/jimmunol.1302998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim SJ, Chen Z, Essani AB, Elshabrawy HA, Volin MV, Volkov S, Swedler W, Arami S, Sweiss N, Shahrara S. Identification of a novel toll-like receptor 7 endogenous ligand in rheumatoid arthritis synovial fluid that can provoke arthritic joint inflammation. Arthritis Rheumatol. 2016;68(5):1099–1110. doi: 10.1002/art.39544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim SJ, Chen Z, Essani AB, Elshabrawy HA, Volin MV, Fantuzzi G, McInnes IB, Baker JF, Finn P, Kondos G, Volkov S, Swedler W, Arami S, Sweiss N, Shahrara S. Differential impact of obesity on the pathogenesis of RA or preclinical models is contingent on the disease status. Ann Rheum Dis. 2017;76(4):731–739. doi: 10.1136/annrheumdis-2016-209206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim SJ, Chang HJ, Volin MV, Umar S, Van Raemdonck K, Chevalier A, Palasiewicz K, Christman JW, Volkov S, Arami S, Maz M, Mehta A, Zomorrodi RK, Fox DA, Sweiss N, Shahrara S. Macrophages are the primary effector cells in IL-7-induced arthritis. Cell Mol Immunol. 2019;17:728–740. doi: 10.1038/s41423-019-0235-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Raemdonck K, Umar S, Palasiewicz K, Volkov S, Volin MV, Arami S, Chang HJ, Zanotti B, Sweiss N, Shahrara S. CCL21/CCR7 signaling in macrophages promotes joint inflammation and Th17-mediated osteoclast formation in rheumatoid arthritis. Cell Mol Life Sci. 2019;77:1387–1399. doi: 10.1007/s00018-019-03235-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Neerukonda SN, Vassell R, Herrup R, Liu S, Wang T, Takeda K, Yang Y, Lin TL, Wang W, Weiss CD. Establishment of a well-characterized SARS-CoV-2 lentiviral pseudovirus neutralization assay using 293T cells with stable expression of ACE2 and TMPRSS2. PLoS ONE. 2021;16(3):e0248348. doi: 10.1371/journal.pone.0248348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von Schlieffen E, Oskolkova OV, Schabbauer G, Gruber F, Bluml S, Genest M, Kadl A, Marsik C, Knapp S, Chow J, Leitinger N, Binder BR, Bochkov VN. Multi-hit inhibition of circulating and cell-associated components of the toll-like receptor 4 pathway by oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2009;29(3):356–362. doi: 10.1161/ATVBAHA.108.173799. [DOI] [PubMed] [Google Scholar]
- 46.Kim SJ, Chen Z, Essani AB, Elshabrawy HA, Volin MV, Volkov S, Swedler W, Arami S, Sweiss N, Shahrara S. Identification of a novel TLR7 endogenous ligand in RA synovial fluid that can provoke arthritic joint inflammation. Arthritis Rheumatol. 2016 doi: 10.1002/art.39544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Raemdonck K, Umar S, Palasiewicz K, Volin MV, Elshabrawy HA, Romay B, Tetali C, Ahmed A, Amin MA, Zomorrodi RK, Sweiss N, Shahrara S. IL-34 reprograms glycolytic and osteoclastic RA macrophages via Syndecan-1 and M-CSFR. Arthritis Rheumatol. 2021;73:2003–2014. doi: 10.1002/art.41792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file1 The gating strategy is shown for unstained and stained myeloid cells for determining the % CD14+ ACE2+ cells (TIF 201 KB)
Supplementary file2 Raw Western blots are shown in Suppl. 2A, Suppl. 2B, and Suppl. 2C for Figs. 1A, 1B, and 3C respectively, and Western blot density was analyzed by Image J in Suppl. 2D and 2E for Figs. 1B and 3C, n=3 (TIF 175 KB)
Supplementary file3 Human monocyte-differentiated Mϴs were pretreated with DMSO (D) or IRAK4i (1 µM, PF06650833) before treating the cells with PBS or Spike protein (30 nM) for 8h. The harvested conditioned media was utilized for quantifying IFNγ, IL-1β, CCL5, or Pyruvate protein levels by ELISA (A-C), n=6 or colorimetric assay (D), n=3. Cells were utilized to determine the transcriptional regulation of HK2 and CARKL (E-F), n=6. The data are shown as mean ± SEM, *p<0.05, **p<0.01. Significant differences were determined by one-way ANOVA followed by Šídák’s multiple comparison test (TIF 81 KB)
Supplementary file4 Human monocytes, differentiated into Mϴs were either untreated (PBS) or treated with Spike protein (30 nM; ExonBio) for 8h in the presence of DMSO, TLR2i (oxPAPC, InvivoGen, 30 µg/ml), or TLR7i (A151 InvivoGen, 1 µM). The harvested cells were used to quantify IL-1β (A), IL-8 (B), TNFα (C), and CCL2 (D) mRNA expression, n=3. The data are shown as mean ± SEM, *p<0.05, **p<0.01. Significant differences were determined by one-way ANOVA followed by Bonferroni’s, Šídák’s, or Holm-Šídák’s multiple comparison test (TIF 65 KB)
Supplementary file5 Human monocyte-differentiated Mϴs were transfected with ACE2, IRAK4, TLR2, and TLR7 specific and nonspecific control siRNA (Santa Cruz Biotechnologies) at a final concentration of 100 nM using Lipofectamine 3000 (Thermo Fisher). The transfected cells were utilized following 48h of transfection and expression of the targeted genes was evaluated by real-time RT-PCR (A-D), n=3. Next, the control and knockdown cells were untreated or stimulated with Spike protein (30 nM) for 8h to determine IL-1β, IL-8, TNFα, and HK2 transcriptional expression (E-H), n=5. The data are shown as mean ± SEM, *p<0.05, **p<0.01. Significant differences were determined by one-way ANOVA followed by Šídák’s multiple comparison test or unpaired Student’s T-test (TIF 93 KB)
Supplementary file6 (A) The EC50 value was determined for Spike Protein (0, 5, 25, and 50 nM) in monocyte-differentiated Mϴs by measuring TNFα production by ELISA. (B) Human myeloid cells were untreated (PBS) or stimulated with Spike Protein (30 nM; ExonBio) and R837 (10 µg/ml) for 30min and protein levels of pIRAK4 or IRAK4 (Cell Signaling) were determined by flow cytometry. (C) pIRAK4 was quantified by ImageJ and normalized to total housekeeping protein (actin) (n=5). (D) Human myeloid cells were untreated or stimulated with Spike Protein (30 nM; ExonBio) for 0-30 min and probed for pAKT (n=3), p-p38 (n=3), and corresponding pan antibodies (Cell Signaling).(E, F) Protein concentration of pAKT and p-p38 was quantified by ImageJ and normalized to total housekeeping protein (actin). (G, H) Human monocyte-differentiated Mϴs were stimulated with LPS (100 ng/ml) in the presence or absence of TLR2i (30 µg/ml; InvivoGen) and TLR7i (1 µM; InvivoGen) for 24h and secretion of TNFα or lactate was quantified. The data are shown as mean ± SEM, *p<0.05, **p<0.01. Significant differences were determined by one-way ANOVA followed by Tukey’s multiple comparison test (TIF 168 KB)
Data Availability Statement
All findings are exhibited in the paper and the material and data are available for transparency.
Not applicable.