

Abstract
Purpose:
Mendelian disease genomic research has undergone a massive transformation over the past decade. With increasing availability of exome and genome sequencing, the role of Mendelian research has expanded beyond data collection, sequencing, and analysis to worldwide data sharing and collaboration.
Methods:
Over the past 10 years, the National Institutes of Health–supported Centers for Mendelian Genomics (CMGs) have played a major role in this research and clinical evolution.
Results:
We highlight the cumulative gene discoveries facilitated by the program, biomedical research leveraged by the approach, and the larger impact on the research community. Beyond generating a list of gene-phenotype relationships and participating in widespread data sharing, the CMGs have created resources, tools, and training for the larger community to foster understanding of genes and genome variation. The CMGs have participated in a wide range of data sharing activities, including deposition of all eligible CMG data into the Analysis, Visualization, and Informatics Lab-space (AnVIL), sharing candidate genes through the Matchmaker Exchange and the CMG website, and sharing variants in Genotypes to Mendelian Phenotypes (Geno2MP) and VariantMatcher.
Conclusion:
The work is far from complete; strengthening communication between research and clinical realms, continued development and sharing of knowledge and tools, and improving access to richly characterized data sets are all required to diagnose the remaining molecularly undiagnosed patients.
Keywords: Centers for Mendelian Genomics (CMG), Data sharing, Mendelian conditions, Novel gene-disease discovery, Rare disease tools
Introduction
The completion of the first human reference genome in 2001, complemented by the rapid development of next-generation sequencing (NGS), brought about a paradigm shift in the field of human genetics and Mendelian disease research. Candidate gene sequencing, positional cloning, and physical mapping of genes were rapidly replaced when NGS technologies enabled routine examination of all annotated human coding regions in the assayable genome, ie, the exome, in a single analysis. By 2010, it was clear that NGS methods, particularly exome sequencing (ES), offered a robust approach to identify candidate novel disease genes and molecular diagnoses.1–5 With these advancements, it was now possible to study Mendelian conditions in a rapid and cost-effective manner. High-throughput sequencing approaches lent themselves to coordination through national programs, allowing for efficient generation of high-quality genomic data analyzed through rigorous sustainable pipelines. Effectively finding causes for the rarest of diseases requires expanding the reach of genomic research through training more clinicians and researchers in genomic analysis and a robust infrastructure that supports data sharing and interdisciplinary collaborative research.
With a focus on identifying the causative variants in the genes responsible for all Mendelian phenotypes, several national and international efforts were quickly developed and included collaborative efforts such as the FORGE Canada Consortium (now referred to as the Care4Rare Canada Consortium),6 Deciphering Developmental Disorders in the UK study,7 Undiagnosed Diseases Network in the United States,8 and the International Rare Diseases Research Consortium.9 In 2011, the National Human Genome Research Institute (NHGRI) within the United States National Institutes of Health (NIH) established the Centers for Mendelian Genomics (CMGs), with additional support from the National Heart, Lung, and Blood Institute and, later, the National Eye Institute. The need for common infrastructure, workflows, and methods development across all disease areas provided the rationale for a centralized CMG structure that could support national and international collaborative efforts with clinicians and researchers who might not otherwise have the necessary resources or environments to engage in genomic research, thus truly opening the door to the study of all Mendelian conditions. The goal of the CMG program was to accelerate the identification of genetic variation underlying Mendelian conditions leveraging genome-wide NGS technologies and to disseminate discoveries, approaches, and technology broadly to drive discovery worldwide.10 The consortium initially consisted of 3 centers11: Baylor-Hopkins CMG, University of Washington CMG, and Yale CMG. A fourth center, Broad Institute of MIT and Harvard CMG, was added during the second phase12 of the consortium. Now in the final year of funding, we highlight our achievements through discoveries, data sharing, tools, and impact on the community as well as reflect on the remaining challenges and lessons learned from a decade of gene discovery. We have learned much from the CMGs, and although the CMG program has come to an end, a new NHGRI-funded program, the Genomics Research to Elucidate the Genetics of Rare Diseases Consortium (GREGoR), launched in 2021. GREGoR’s mandate is to sequence, identify, and validate disease-contributing genes and variants in families for whom current approaches have failed to find a molecular diagnosis, essentially developing approaches to solve the unsolved Mendelian disease cases and families.
The Importance of Collaboration in Mendelian Gene Discovery
The rarity and diversity of Mendelian diseases require that we cast a broad net across the human population to meet our goal of complete enumeration of Mendelian disease genes. Accordingly, the 4 CMGs collaborated directly with 2283 researchers during the last decade. Through extended collaborations and publications, the CMGs worked with 11,771 researchers across 2773 institutions in 90 countries. Building this vast collaborative network required global outreach efforts, including advertisements in journals, seminars at conferences, educational courses, boots on the ground–style recruitment, and word of mouth from collaborators. Collaborators brought DNA samples from deeply phenotyped, unsolved families affected by rare disease who consented for genomic studies and data sharing; CMGs provided sequencing, data processing, and data sharing, and analysis was done in collaboration through joint distributive research efforts. Although collaboration styles varied based on preferences of individual CMGs and collaborators, often CMGs would perform initial genomic analyses to prioritize a short list of high priority candidate variants for each proband/family that could be discussed in consultation with the collaborator. Collaborators typically pursued the variants of interest to gather additional cases, performed functional studies, synthesized the science, and wrote the findings, often with resources and support from CMGs.
Most successful discoveries required collaboration that extended beyond a single CMG and collaborator. In an analysis of the CMG’s gene discovery publications, we found that >90% involved more than 1 institution contributing cases to the publication, highlighting the fundamental role of collaboration in solving Mendelian disease. These connections are often made via data-sharing platforms and activities, which have become a critical component to gene discovery.13
In 10 years, CMGs have contributed to 961 manuscripts by providing sequencing data, analysis, methods development, and training in genomic analysis. CMGs have generated 75,573 exomes, 3876 genomes, 714 transcriptomes through RNA sequencing (RNA-seq), and 385 methylation arrays across 28,991 families (Figure 1). The general strategy was to sequence trios or probands that had prior gene panel testing to exclude known genetic causes of disease, although as exome prices fell over the course of the program, this became less pragmatic, particularly for samples from individuals with limited health care access.
Figure 1. Overview of the Centers for Mendelian Genomics (CMGs) by numbers.
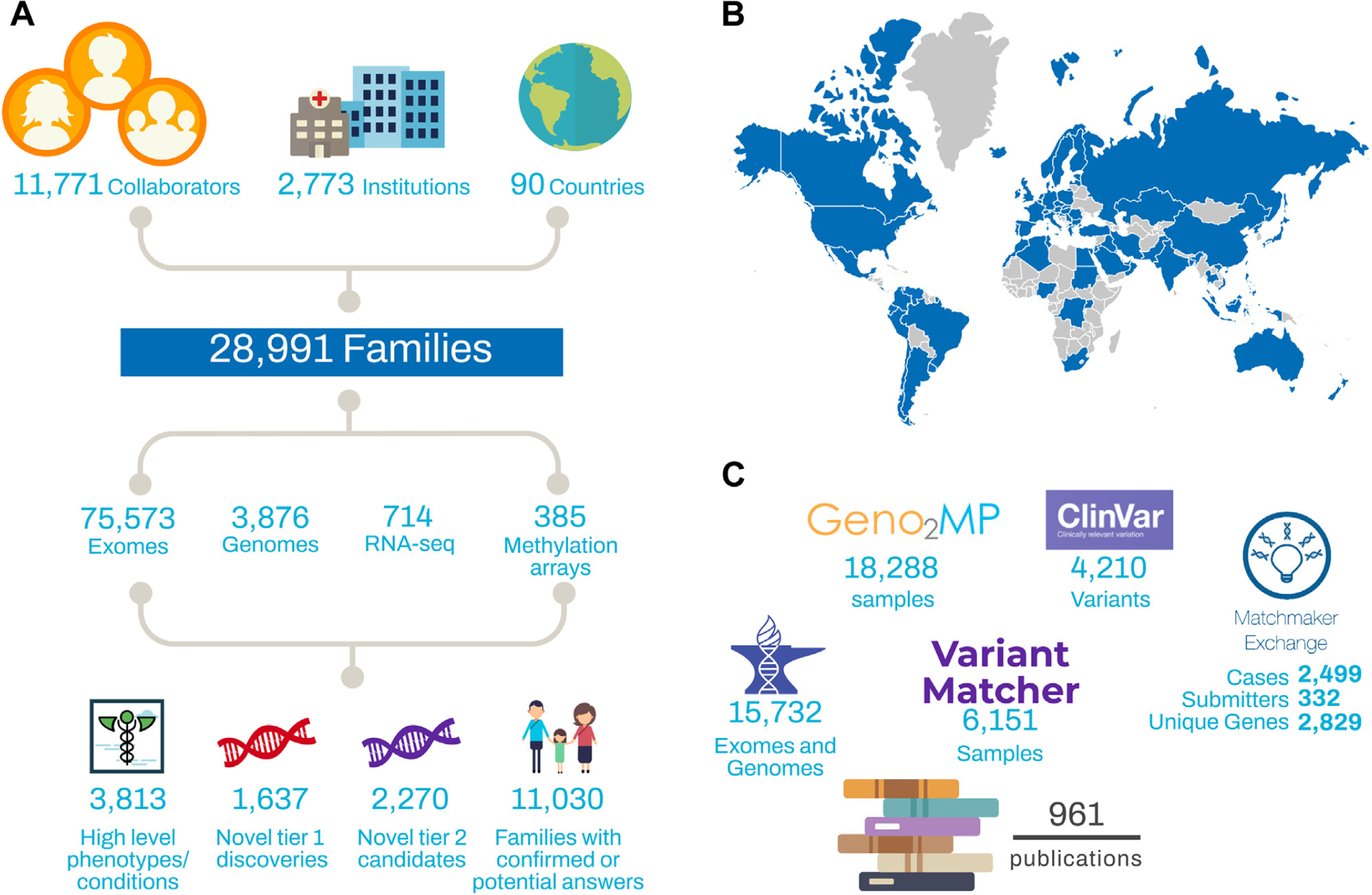
(A) A high-level summary of activities performed by the CMGs, including the number of collaborators, number of kindreds, volume of testing performed, and discovery rates. (B) Map of CMG collaborators. Blue indicates that the CMGs collaborated with at least 1 researcher in that country (based on country listing in PubMed affiliations). (C) Data-sharing metrics for the CMGs.
Track record in discovering gene-disease relationships
Remarkably, the rate of CMG discovery has continued on a fairly constant trajectory over the decade of the program.11,12 The CMGs have contributed to the identification of 1637 new disease-gene relationships (172/year) and 2270 candidates (239/year) over the course of the program (Table 1). Details of each novel gene-disease relationship are provided in Supplemental Table 1. This continued rate of discovery tells us that there are still more Mendelian genes to be found and that ongoing sequencing efforts should continue to be fruitful.
Table 1.
Breakdown of CMG findings by category
| Category of Findings | Gene-Disease Entities, n | Families, n | Unique Genes, n | Publications |
|---|---|---|---|---|
| Novel gene-disease discoveries (tier 1) | 1637 | 3387 | 1286 | 379 |
| Novel gene-disease candidates (tier 2) | 2270 | 2505 | 1936 | 75 |
| Known gene-disease relationships | 2584 | 5138 | 1598 | 163 |
| Total | 6089 | 10,349 | 3846a | 533a |
Tier 1 discoveries are defined as multiple affected kindreds identified for a gene-disease relationship or very strong functional data supporting the relationship. Tier 2 candidates are strong enough to enter the gene in the Matchmaker Exchange, often having some data in the literature implicating the gene for the phenotype, but further data are needed, such as additional unrelated probands with an overlapping phenotype, model organism, or other functional studies. Known gene-disease relationships are distinguished from novel phenotypes using OMIM’s Phenotype MIM IDs.
CMG, Center for Mendelian Genomics.
Total for unique genes and publications is not equal to the sum of the rows above because genes can have multiple disease associations, with varying certainty of associations, and publications can be duplicated across categories.
There are intriguing observations and interesting trends in the success of gene-disease discovery efforts across organ systems, although we note the caveat that efforts have not been applied evenly. Much effort has focused on neuro-developmental conditions and syndromic disorders where there has been a continued high rate of gene-disease discovery (Figure 2). For cases where only a single system was impacted, disorders of blood and blood-forming tissues, metabolism/homeostasis, the immune system, integument, and the nervous system have the highest rates of novel findings. When multiple systems are involved, involvement of the skeletal system or connective tissue has the highest rate of findings (including known gene-disease relationships, novel gene-disease relationship discoveries, and candidates). Skeletal abnormalities or involvement of the immune system had the highest novel discovery rate. For ear, eye, kidney, connective tissue, and muscle phenotypes, some new genes have been identified, but many cases were solved with variants in genes that have known gene-phenotype relationships in OMIM. Solving many of these cases required additional data types, including RNA-seq or targeted splicing assays, long-read sequencing, and methylation analysis.14–17
Figure 2. Solve and discovery rates by high-level Human Phenotype Ontology (HPO) category.
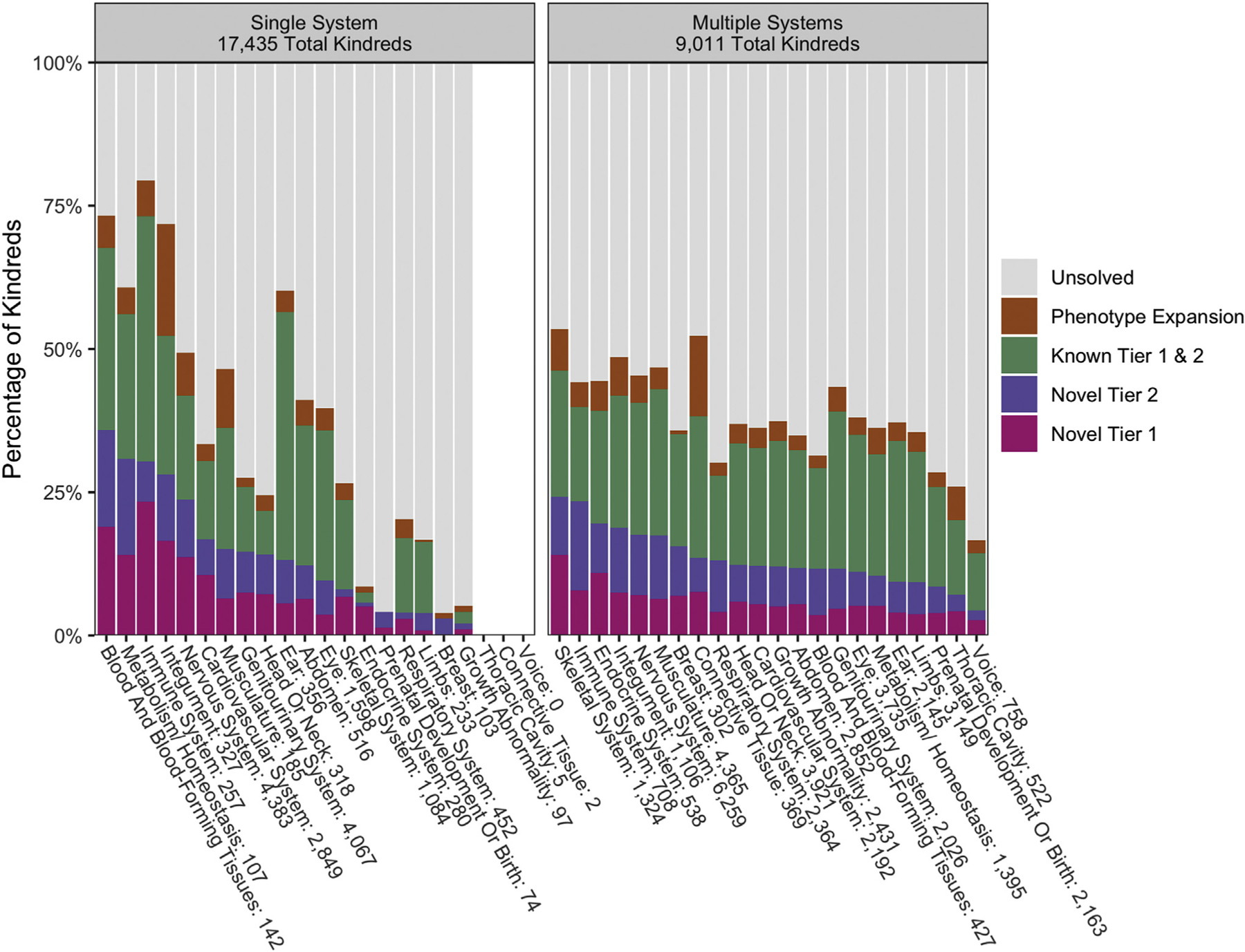
Kindreds were categorized as having phenotypes in 1 HPO high-level system or multiple high-level systems. From there, the solve and discovery types were analyzed for each system. There were 494 kindreds in our Center for Mendelian Genomics (CMG) cohort with no HPO terms available, and therefore they were not able to be included in this analysis. Systems with fewer than 10 kindreds were noted but excluded.
The CMGs have contributed to the discovery of 778 phenotypic expansions associated with previously established disease genes. Such discoveries represent an important contribution to both the research and clinical fields because the full phenotypic spectrum of a Mendelian disease, or the set of phenotypes associated with a genomic locus, may not be fully revealed at the time of the initial disease-gene discovery. Indeed, approximately 961 genes, or 24% of genes implicated in Mendelian disease, now have more than 1 phenotype associated with the gene.18,19 The rate of disease-gene discovery continues to outpace that of phenotype expansion, highlighting the continued need for dedicated research programs. Many of the CMG discoveries are also uncovering molecular and biological processes that can inform therapeutic intervention or management, ranging from existing medications known to impact a particular metabolic pathway or ion channel, to avoidance of certain medications, to novel molecular interventions.20–24
Impact on the Rare Disease Community
To advance discovery of the underlying basis of Mendelian disease, interactions with clinical laboratories are important, both to relay discoveries more rapidly and efficiently than the peer review publication process allows and to access primary evidence from patient testing. However, navigating the clinical research boundaries can be challenging. Many genes discovered and published through traditional research still lack the evidence required to definitively establish a role in disease and enable the molecular diagnosis of patient symptomatology.25,26 Including genes with limited evidence on clinical testing panels increases the number of variants of uncertain significance on patient’s test results without increasing yield, which has led many labs to set a threshold of evidence for genes to be included on clinical test reports. Giving patients uncertain results can distract them from pursuing other causes of disease yet, in other circumstances, empower them to engage in the research process. Thus, researchers, clinical labs, and patients must work together to share primary evidence, build the clinical genomic knowledgebase, and ensure rapid translation of discoveries to patient care.
Thus far, the CMGs have contributed to 419 publications describing novel gene-disease discoveries and candidate gene-disease relationships, whereas another 163 articles have added to the understanding of known gene-disease relationships. To date, 23.2% (379) of the 1637 novel gene-disease discoveries have been published in peer-reviewed journals. Many of the CMG discoveries and candidates are still in the process of gathering cases and functional data or going through the peer review publication process.
Although the primary goal of CMGs was gene discovery, their impact on the genetic community goes beyond establishing or clarifying gene-disease relationships. Through community outreach, training, and data sharing, the CMGs provided students, clinician-scientists, and investigators with the training and tools to discover new Mendelian disease genes. The CMGs provided educational and networking opportunities by hosting in-person courses attended by over 300 analysts and researchers, including the Mendelian Data Analysis Workshop (University of Washington), Interpreting Genomes for Rare Disease (Broad), and McKusick Short Course in Human and Mammalian Genetics (Baylor-Hopkins). The CMGs have enabled researchers and clinicians investigating rare Mendelian diseases around the world to access gene discovery techniques, including those in countries where access to research opportunities is limited, such as the Democratic Republic of the Congo,27 South Africa, Kenya, Egypt,28,29 Iraq,30 Chile,31,32 Turkey,33–35 and Lithuania. For some, this has involved training opportunities within a CMG-affiliated laboratory, whereas for others, learning happened through collaborative meetings to discuss analysis results on teleconferences.
To better understand the impact of the CMG program, we surveyed CMG collaborators in early 2021. A total of 206 responses were collected, including collaborators from basic research (37%), clinical research (33%), clinical practice (24%), and diagnostic laboratories (6%). Most were senior investigators with >10 years of experience (63%), although junior faculty with <5 years (9%) and 5–10 years of experience (20%) were also well represented, whereas some were not in faculty positions (8%). Overall, 70% reported starting new collaborations because of their work with a CMG, and 76% were more likely to share data after working with CMGs. Most collaborators (69%) clinically confirm and return results identified through the CMG to the rare disease families, but several groups were unable to because of insufficient resources.
Sharing Data to Improve Rare Disease Diagnosis
Given the genetic heterogeneity and rarity of Mendelian disease and the diversity of our species, rapid and international data sharing is critical to build sufficient evidence to unambiguously identify novel gene-disease relationships. Many of the discoveries made by the CMGs were only possible because of collaborations, which were necessary for obtaining enough samples to convincingly associate a gene with a given phenotype. Data sharing is another key element in helping researchers with related cases find each other and work together.
Approaches that share only the results or top candidates from sequencing studies are inadequate to maximize discovery rates, because not all pathogenic variants are equally recognizable. Mendelian disease investigators therefore should design the infrastructure of each study in such a way that enables easy sharing of both the genomic data and accompanying phenotype and other metadata.
The CMGs have committed to rapid and extensive data sharing (Table 2), developing new platforms accessible to the global community and requiring participation by all CMG collaborators to the benefit of other researchers and patients with rare diseases. The CMGs share data using a variety of tools that support 1 of 3 primary modes of sharing: connecting 2 parties that each have a predefined candidate gene regardless of whether the variants in those genes are the same or different (two-sided matching); allowing one party to query another party’s primary data for any variant or certain types of variants (predicted loss-of-function variants, biallelic rare variants, etc.) in a particular candidate gene (one-sided matching); or allowing credentialed researchers who apply for access to the data to directly analyze cohorts of read-level sequence data (shared via CRAM files) and detailed metadata in structured files to make discoveries in the absence of any identified gene candidates (zero-sided matching). By participating in multiple types of data sharing, CMG-sequenced data is made accessible to the broadest possible range of researchers and use-cases across the rare disease community.
Table 2.
CMG Data Sharing
| Type of Data Shared | CMG Website | Matchmaker Exchange | ClinVar | Geno2MP | Variant Matcher | AnVIL |
|---|---|---|---|---|---|---|
| Type of data sharing (2-sided, 1-sided, 0-sided) | 2 | 2 | 2 | 1 | 1 | 0 |
| BAM/CRAM files | ✓ | |||||
| Gene name | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| Variant information | Optional | ✓ | ✓ | ✓ | ✓ | |
| High-level phenotype | ✓ | ✓ | ✓ | ✓ | ||
| HPO terms | Optional | ✓ | ✓ | ✓ | ||
| Other | Variant classification and supporting evidence | See Supplemental Table 3 |
A breakdown for the various data-sharing activities in which the CMGs participated. Each column is a different data-sharing platform, and the rows are the types of data that the CMGs could share.
AnVIL, Analysis, Visualization, and Informatics Lab-space; CMG, Center for Mendelian Genomics; HPO, Human Phenotype Ontology.
The CMGs submitted that file or data type to the platform.
Sharing for two-sided matching (both parties have gene candidates for their cases)
Two-sided matching historically occurred through parties listing and/or searching for genes of interest on websites, using search engines such as Google as the matcher, or emailing inquiries to colleagues. This method can be error prone and time consuming and lead to either overly inclusive or limited results. However, websites such as ClinVar or mendelian.org allow for data to be shared quickly and openly to anyone with internet access. More recently, informatics have been used to create genomics match-making tools that allow matching based on gene and/or phenotype. The CMGs have contributed to both website-based and informatics-enabled matching.
CMG website
The coordinating center of the NHGRI Genome Sequencing Program has created a website to disseminate information arising from the CMG program (http://mendelian.org/). This site hosts a searchable list of phenotypes under investigation, including novel gene-phenotype relationships (http://mendelian.org/phenotypes-genes), and a list of all publications that acknowledge CMG support.
Matchmaker Exchange
Matchmaker Exchange (MME)13 plays a critical role in facilitating the aggregation of individual cases with variants in a given candidate gene. MME is a federated network designed to connect databases of gene candidates and phenotypic data via a common application programming interface. It allows for computational ranking of matches by phenotype and enables collaborators to connect for follow-up and more detailed manual comparisons of phenotypes.36 In addition to committing to sharing candidate genes to MME, the CMGs have also contributed to this platform by building 3 of the 8 current nodes (Gene-Matcher,37 MyGene2,38 and seqr39; Table 3).
Table 3.
CMG gene candidate sharing through MME
| Cases in MME |
Submitters |
Unique Genes |
||||||
|---|---|---|---|---|---|---|---|---|
| MME Node | CMG Developer | CMG Submitter | CMG | Non-CMG | CMG | Non-CMG | CMG | Non-CMG |
| GeneMatcher | BH | BH | 343 | 55,210 | 99 | 11,790 | 817 | 12,964 |
| Yale | 270 | 0 | 36 | 0 | 276 | 0 | ||
| MyGene2 | UW | UW | 871 | 599 | 119 | 125 | 635 | 563 |
| seqr | Broad | Broad | 1015 | 70 | 78 | 13 | 1101 | 81 |
This is a summary of the data submitted to MME through CMG-developed nodes. The number of cases, submitters, and unique genes that were contributed to MME are broken down into CMG and non-CMG cases because non-CMG participants are still able to use the CMG-developed nodes to share on MME.
BH, Baylor-Hopkins; CMG, Center for Mendelian Genomics; MME, Matchmaker Exchange; UW, University of Washington.
ClinVar
In addition to facilitating novel gene-disease relationships and increasing solve rates, data generated by the CMGs can be valuable for many other clinical and research uses, including clinical molecular diagnosis, characterizing the natural history of disease, and providing the foundational data to catalyze interventional and disease mitigation therapeutic strategies. To support these efforts, the CMGs have committed to sharing all published variants and supporting evidence to ClinVar, an open-source database that collects and reports variant and phenotype relationships. Since the inception of the program, the CMGs have together submitted over 4200 variants, along with supporting evidence summaries, to ClinVar. The center-specific names and pages can be found in Supplemental Table 2.
Sharing for one-sided matching (only one party has a gene candidate and wishes to query other primary data sets to find a match that was not previously recognized by the other party)
Although two-sided matching is helpful when both parties share a gene or phenotype of interest, many researchers have cases that do not yet have an identified candidate gene. In addition to these unsolved cases, there remain many candidate genes without matches, highlighting the need to search databases of patients with rare diseases for additional copies of specific candidate variants or other similar potentially deleterious variants within the candidate genes. The CMGs have created 2 databases to support one-sided variant matching. These databases also help to exclude candidates, because the variant of interest may be found in unaffected relatives or well-phenotyped individuals with unrelated phenotypes with documented absence of the phenotype in question. Nevertheless, caution must be exercised in exclusion based on possible nonpenetrance.
The Genotypes to Mendelian Phenotypes (Geno2MP) browser was created by the University of Washington CMG to facilitate new gene discovery efforts and prioritization of candidate variants. It contains aggregate genotype data from >19,000 samples (>15 million rare variants with <2.5% frequency in gnomAD) sequenced by the University of Washington, Broad, and Yale CMGs (https://geno2mp.gs.washington.edu/).40 Each rare variant is linked to de-identified phenotypic information about the affected individuals and unaffected relatives who carry the variant. Genotypes to Mendelian Phenotypes users may contact original submitters of the samples through the website and provide details about their own cases to pursue potential gene discovery matches.
VariantMatcher, created by the Baylor-Hopkins CMG, allows users to query rare (<1% allele frequency), non-synonymous variants from over 6151 samples sequenced by the Baylor-Hopkins CMG for specific variants of interest. Users of the site must register and be approved by site administrators. If phenotypic features are submitted in combination with the variant in the query, the phenotype from the matched entry will be shared in a simultaneous email to the submitter and the matching investigator. If a match is not made, the queried coordinates with submitted phenotypes can be stored for future matching.
Sharing for zero-sided matching (querying larger, combined data sets to identify novel gene-disease candidates in the absence of any prior identified gene candidate)
Novel methods and aggregation of larger data sets will be needed to solve cases that remain unsolved after thorough analysis by the CMGs. In the initial years of the program, CRAM files from the CMGs were shared through dbGaP (NIH Database of Genotypes and Phenotypes), but access for analysis required download and storage. The center-specific study accession numbers can be found in Supplemental Table 2. As genomic data grows in scale, the community needs better solutions.
NHGRI’s Genomic Data Science Analysis, Visualization, and Informatics Lab-space (AnVIL) is a cloud-based environment where both data and tools can coexist, thereby improving logistics for the wider community to be able to share and access them together. AnVIL is assembling the most commonly used tools and pipelines to support genomic analysis and make them available on the AnVIL platform. In addition, investigators can add their own tools to the platform. There are currently a number of workflows important in rare disease analysis set up in AnVIL, including germline variant calling with GATK, RNA-seq processing, and mitochondrial variant calling. The seqr analysis platform is on AnVIL, and any researcher can request that a jointly called vcf located within an AnVIL workspace be loaded in seqr. By creating an environment to share tools and pipelines, the application of analysis methods to data sets will be facilitated, allowing for comparison of the performance of approaches to each other to help develop best practices. The introduction of novel tools and methods will be immediately leveraged and readily testable. This will ultimately allow authorized researchers anywhere in the world to explore their own hypotheses using CMG data and a constantly evolving set of analysis tools.
CMG data sharing has transitioned to AnVIL, where >60 terabytes of data are available alongside other common and rare disease data sets. As of this publication, the CMGs have deposited over 15,025 exomes and 707 genomes to 39 AnVIL workspaces (https://anvilproject.org/data). In addition to the raw sequencing data, the CMGs have uploaded accompanying metadata for each sample, including sample-, subject-, family-, discovery-, and sequence-level information (see Supplemental Table 3 for file formats and data dictionary).
Although the tools and workflows are open access for anyone logging into the interface, AnVIL data sets have 3 types of data access: open access, controlled access, and consortium access. To learn more about the types of AnVIL access and examples, see Supplemental Table 4. All CMG data sets are controlled access, meaning researchers must request access through dbGaP and obtain permission for use, consistent with the subjects’ informed consent (eg, General Research Use, disease-specific research and clinical care). Access is regulated in accordance with NIH policy and full details can be found at https://anvilproject.org/data/requesting-data-access.
Development of Tools and Improved Methods
The CMGs have developed the open-source analysis tools PhenoDB and seqr (Pais L, Snow H, Weisburd B, et al. seqr: a web-based analysis and collaboration tool for rare disease genomics. https://doi.org/10.1101/2021.10.27.21265326) for filtering and prioritizing variants in individuals or families with Mendelian disease.41 These platforms enable streamlined analysis of exome or genome sequence data through incorporation of in silico predictions and population and disease databases, as well as integration with other external databases to facilitate review of novel candidates (eg, GeneCards, Mouse Genome Informatics). Clinical data as structured Human Phenotype Ontology (HPO) terms and pedigree data can also be recorded, enabling coupling of genetic and phenotypic data. These web-based tools facilitate collaboration by providing a platform for researchers from disparate locations to work as a team on analyses. PhenoDB and seqr continue to be periodically updated and are available for free download and implementation, thereby providing broadly useful resources for Mendelian disease research.
The CMGs have also developed or contributed to methods to reanalyze exome data from unsolved cases using semiautomated pipelines and updated annotations. This yielded confirmed or potential genetic diagnoses in up to one third of unsolved cases, mostly within disease genes published after initial analysis.42,43 Although the pace of disease-gene discovery emphasizes the importance of periodic reanalysis, it is the automation of these processes that will meet the challenge of a continued accrual of unsolved cases. CMG investigators have also developed and applied gene-centric analyses to identify candidate disease genes in exome-negative cohorts, such as burden tests, to identify genes enriched in deleterious rare variants across cases with the same phenotype44,45 and a phenotype-agnostic method that prioritizes genes most likely to underlie Mendelian disease.46 Reanalysis methods for more complicated patterns of inheritance led to the development of a two-locus genome-wide test that enables detection of digenic inheritance in exome data.47
CMG investigators have successfully applied sequencing approaches beyond the exome to identify or validate causal variant(s), including genome, RNA, bisulfite DNA methylation sequencing, and long-read genome sequencing, showing their utility in cases where ES fails to find a molecular diagnosis. Examples include utilizing genome sequencing to identify pathogenic structural variants missed by exome, such as the homozygous inversion in QDPR detected in a patient with dihydropteridine reductase deficiency;48 applying RNA-seq to identify genes with aberrant expression and/or splicing, including an intronic variant in trans with a missense in muscle disease gene DES that resulted in a pseudoexon insertion and allelic imbalance49; using bisulfite sequencing to identify gene silencing epivariation, such as the characterization of aberrant hypermethylation associated with a pathogenic repeat expansion in the XYLT1 promoter region17; and the application of long-read genome sequencing to characterize a complex genomic rearrangement involving an inverted triplication flanked by duplications in a proband with Temple syndrome.50
Data Sharing Empowers and Expedites Solving Rare Disease
There are a number of unsolved syndromes that have perplexed the clinical genetics community for decades.51 Although collectively the CMGs have sequenced over 28,991 families, each individual CMG often has only a handful of cases of a given phenotype. We formed the CMG Data Analysis working group to share cohorts across the CMGs to increase power to solving these challenging phenotypes. For our pilot project focusing on Dubowitz syndrome, we built a cohort of 20 individuals from 16 clinically diagnosed families. It would have been very challenging to collect this many families with a rare condition without access to an international network of researchers and clinicians. On analysis, no two cases shared the same genetic diagnosis and no specific pathways were identified. A collaboration was then established with the Canadian Care4Rare Program, which had been building a similar cohort. Only after combining the CMG and Care4Rare cohorts (31 individuals from 27 families) was the group able to recognize that the diagnosis of Dubowitz syndrome was not pointing to a single disorder but a collection of disorders with overlapping phenotypic features, highlighting the benefit of data aggregation for these studies. Overall, we found that the Dubowitz syndrome phenotype has extensive locus heterogeneity rather than a single gene etiology. Diagnoses were made for a number of recently molecularly defined and phenotypically similar conditions with growth restriction, microcephaly, and developmental delay, with a molecular diagnosis made in 13 of 27 families (48%) or strong candidate variants in known and candidate disease genes identified in an additional 7 of 27 families (26%).52 This experience highlights the need to continue to work across national and international rare disease programs to build larger cohorts for rare conditions, both to discover unifying genetic causes and, as in the case of Dubowitz syndrome, to provide evidence refuting that a cluster of features represents a single syndrome.
Model organism data are an important component of gene discovery. Even when multiple families with overlapping phenotypes have had a variant identified in the same gene, establishing a causal gene-disease relationship can be difficult. As part of the effort to clarify these relationships, the CMGs formed a collaboration with the Knock-Out Mouse Project to help prioritize genes that may be important for human disease (https://www.komp.org). We shared candidate genes after a short embargo period, and results were shared through the International Mouse Phenotype Consortium database. Several gene discoveries, including TONSL for skeletal dysplasia and FAM92A for limb and digit anomalies, were supported by this collaborative mechanism.53,54
Future Goals of Mendelian Rare Disease Genomics
As the CMG program comes to a close, additional work is still needed to identify the genetic basis of Mendelian disease for many families. Hundreds of novel gene-disease candidates were identified, but many still lack sufficient data to confirm or refute the relationship. Over half of the families sequenced remain unsolved despite having phenotypes that are strongly suggestive of a Mendelian cause. There are also thousands of genes predicted by human or model organism data to result in a human phenotype when disrupted that have not yet been linked to a human phenotype.55,56 The mission started by the CMGs remains far from finished, but the data and tools developed will continue to support the ongoing research efforts in this area.
New bioinformatic methods and resources are needed to help address the limitations of current analytical approaches and variant detection, and interpreting rare variation in the genome is a major barrier for achieving genetic diagnoses. Telomere-to-telomere genome assembly and the use of a pan-genome human reference sequence representing diverse ancestries will improve variant interpretation57 (Nurk S, Koren S, Rhie A, et al. The complete sequence of a human genome. https://doi.org/10.1101/2021.05.26.445798); however, it will also require new analytical pipelines (Kaminow B, Ballouz S, Gillis J, Dobin A. Virtue as the mean: panhuman consensus genome significantly improves the accuracy of RNA-seq analyses. https://doi.org/10.1101/2020.12.22.423111), including support for graph-based representation typically used for pan-genome data.58 Improved annotation of enhancers and promoter regions, which benefit by leveraging data from the Encyclopedia of DNA Elements project, can further facilitate variant interpretation, particularly for genome analyses.59,60
Efforts to resolve variants of uncertain significance, particularly missense variant alleles, are needed. With progress in recent years for deep mutational scanning, there is now an international focus through the Atlas of Variant Effect Alliance, which aims to systematically determine the impact of variants in functionally important genomic regions.61 The application of artificial intelligence methods for predicting the functional effect of variants also offers an avenue for improving in silico predictions of pathogenicity.62,63 Much of our knowledge about gene and transcript expression patterns relies on adult tissue; the Developmental Genotype-Tissue Expression (dGTEx) and Pediatric Cell Atlas initiatives will facilitate interpretation for developmental disorders.64
Population data sets and analytical tools that aid the interpretation of long-read sequencing and methylation data in patients with rare diseases will be needed to help realize the diagnostic utility of these technologies. Furthermore, the integration of methylation, structural, and RNA-seq data into variant analytical tools will ultimately be required to streamline analysis for cases remaining unsolved after ES, particularly for compound heterozygotes where only one of the variants will be detected and prioritized by exome or genome.
Although CMGs strived to include collaborators and cases from around the world (Figure 1B), there is still progress to be made on the diversity of both the participating researchers we collaborate with and the genetic ancestry of the cases sequenced. A total of 73% of collaborators reported their ancestry to be White/European, and the majority (58%) were male. Additionally, 67% percent of the cases sequenced were also White/European. To make good on the promise of genomics, a much greater fraction of the world’s populations must be involved. Research programs must push to expand the diversity of the research workforce as well as the families and individuals studied.
Debate continues regarding models for funding rare disease research, either distributing funds to individual rare disease investigators working in their own fields or centralizing funding for centers that can build infrastructure used by many investigators. The success of the CMG collaborators in discoveries, publications, data sharing, and subsequent research funding highlights the power of centralized funding for centers. The CMG structure has allowed for sequencing and analysis of invaluable samples to be performed in centers of excellence by teams of experts, and more than that, it has facilitated investigators of the same rare disease around the world to connect and collaborate. This approach offers cost efficiency by distributing shared infrastructure across large numbers of investigators and enabling better data sharing and cohort aggregation for increased statistical power. There are also limitations. Participants were not directly recruited by the CMGs, which can make collecting detailed phenotypes more challenging; recontact/reconsent for follow-up studies was typically not feasible; and reviewing the data-sharing language in the hundreds of submitting collaborator consent forms required substantial overhead. Additionally, because the CMG data set consisted of a highly heterogeneous set of phenotypes, large-scale analyses such as enrichment analyses were not practical. In this decade of gene discovery by the CMGs, there has been substantial progress made, but much remains to be done. As routine genomic analysis becomes more successful, and approaching a 40% diagnosis rate in clinical diagnostic labs becomes more attainable,65 the cases that remain unsolved are increasingly challenging. Undiscovered diseases are ultra rare, the functional impact of variation is difficult to determine, and causality is hard to prove, particularly for variants with incomplete penetrance and complex genetic architectures. Gene discovery rates remain steady, highlighting the continued need for national and international programs in rare disease genomic analysis, including the recently funded GREGoR Consortium (https://gregorconsortium.org).
Supplementary Material
Acknowledgments
We extend our gratitude and respect to our friend and colleague, Dr. Deborah A. Nickerson, who passed away in December of 2021. She was a leader in human genomics research, a passionate advocate for trainees and women in science, and contributed to hundreds of discoveries that have set the stage for precision medicine. The Baylor-Hopkins Center for Mendelian Genomics, Broad Institute of MIT and Harvard Center for Mendelian Genomics, University of Washington Center for Mendelian Genomics, and Yale Center for Mendelian Genomics were funded by the National Human Genome Research Institute (NHGRI) awards UM1HG006542, UM1HG008900, UM1HG006493, and UM1HG006504, respectively. Funds were also provided under the National Heart, Lung, and Blood Institute under the Trans-Omics for Precision Medicine Program and the National Eye Institute. The GSP Coordinating Center (U24HG008956) contributed to cross-program scientific initiatives and provided logistical and general study coordination. Aspects of this work were funded by NHGRI K08HG008986 (J.E.P.), NHGRI R01HG009141 (H.L.R. and D.G.M.), the National Institute of Neurological Disorders and Stroke R35NS105078 (J.R.L.), and National Health and Medical Research Council Early Career Fellowship 1159456 (N.J.L.). The Centers for Mendelian Genomics would like to thank all of our collaborators from around the world as well as the families and individuals who contributed their data to this study.
Members of the Centers for Mendelian Genomics Consortium
A full list of members and their affiliations appears in the Supplement.
Marcia Adams, François Aguet, Gulsen Akay, Peter Anderson, Corina Antonescu, Harindra M. Arachchi, Mehmed M. Atik, Christina A. Austin-Tse, Larry Babb, Tamara J. Bacus, Vahid Bahrambeigi, Suganthi Balasubramanian, Yavuz Bayram, Arthur L. Beaudet, Christine R. Beck, John W. Belmont, Jennifer E. Below, Kaya Bilguvar, Corinne D. Boehm, Eric Boerwinkle, Philip M. Boone, Sara J. Bowne, Harrison Brand, Kati J. Buckingham, Alicia B. Byrne, Daniel Calame, Ian M. Campbell, Xiaolong Cao, Claudia M.B. Carvalho, Varuna Chander, Jaime Chang, Katherine R. Chao, Ivan K. Chinn, Declan Clarke, Ryan L. Collins, Beryl Cummings, Zain Dardas, Moez Dawood, Kayla Delano, Stephanie P. DiTroia, Harshavardhan Doddapaneni, Haowei Du, Renqian Du, Ruizhi Duan, Mohammad Eldomery, Christine M. Eng, Eleina England, Emily Evangelista, Selin Everett, Jawid Fatih, Adam Felsenfeld, Laurent C. Francioli, Christian D. Frazar, Jack Fu, Emmanuel Gamarra, Tomasz Gambin, Weiniu Gan, Mira Gandhi, Vijay S. Ganesh, Kiran V. Garimella, Laura D. Gauthier, Danielle Giroux, Claudia Gonzaga-Jauregui, Julia K. Goodrich, William W. Gordon, Sean Griffith, Christopher M. Grochowski, Shen Gu, Sanna Gudmundsson, Stacey J. Hall, Adam Hansen, Tamar Harel, Arif O. Harmanci, Isabella Herman, Kurt Hetrick, Hadia Hijazi, Martha Horike-Pyne, Elvin Hsu, Jianhong Hu, Yongqing Huang, Jameson R. Hurless, Steve Jahl, Gail P. Jarvik, Yunyun Jiang, Eric Johanson, Angad Jolly, Ender Karaca, Michael Khayat, James Knight, J. Thomas Kolar, Sushant Kumar, Seema Lalani, Kristen M. Laricchia, Kathryn E. Larkin, Suzanne M. Leal, Gabrielle Lemire, Richard A. Lewis, He Li, Hua Ling, Rachel B. Lipson, Pengfei Liu, Alysia Kern Lovgren, Francesc López-Giráldez, Melissa P. MacMillan, Brian E. Mangilog, Stacy Mano, Dana Marafi, Beth Marosy, Jamie L. Marshall, Renan Martin, Colby T. Marvin, Michelle Mawhinney, Sean McGee, Daniel J. McGoldrick, Michelle Mehaffey, Betselote Mekonnen, Xiaolu Meng, Tadahiro Mitani, Christina Y. Miyake, David Mohr, Shaine Morris, Thomas E. Mullen, David R. Murdock, Mullai Murugan, Donna M. Muzny, Ben Myers, Juanita Neira, Kevin K. Nguyen, Patrick M. Nielsen, Natalie Nudelman, Emily O’Heir, Melanie C. O’Leary, Chrissie Ongaco, Jordan Orange, Ikeoluwa A. Osei-Owusu, Ingrid S. Paine, Lynn S. Pais, Justin Paschall, Karynne Patterson, Davut Pehlivan, Benjamin Pelle, Samantha Penney, Jorge Perez de Acha Chavez, Emma Pierce-Hoffman, Cecilia M. Poli, Jaya Punetha, Aparna Radhakrishnan, Matthew A. Richardson, Eliete Rodrigues, Gwendolin T. Roote, Jill A. Rosenfeld, Erica L. Ryke, Aniko Sabo, Alice Sanchez, Isabelle Schrauwen, Daryl A. Scott, Fritz Sedlazeck, Jillian Serrano, Chad A. Shaw, Tameka Shelford, Kathryn M. Shively, Moriel Singer-Berk, Joshua D. Smith, Hana Snow, Grace Snyder, Matthew Solomonson, Rachel G. Son, Xiaofei Song, Pawel Stankiewicz, Taylorlyn Stephan, V. Reid Sutton, Abigail Sveden, Diana Cornejo Sánchez, Monica Tackett, Michael Talkowski, Machiko S. Threlkeld, Grace Tiao, Miriam S. Udler, Laura Vail, Zaheer Valivullah, Elise Valkanas, Grace E. VanNoy, Qingbo S. Wang, Gao Wang, Lu Wang, Michael F. Wangler, Nicholas A. Watts, Ben Weisburd, Jeffrey M. Weiss, Marsha M. Wheeler, Janson J. White, Clara E. Williamson, Michael W. Wilson, Wojciech Wiszniewski, Marjorie A. Withers, Dane Witmer, Lauren Witzgall, Elizabeth Wohler, Monica H. Wojcik, Isaac Wong, Jordan C. Wood, Nan Wu, Jinchuan Xing, Yaping Yang, Qian Yi, Bo Yuan, Jordan E. Zeiger, Chaofan Zhang, Peng Zhang, Yan Zhang, Xiaohong Zhang, Yeting Zhang, Shifa Zhang, Huda Zoghbi, Igna van den Veyver
Consortium members received funding as follows:
Alicia B. Byrne was supported by the Australian Government Research Training Program Scholarship, the Australian Genomics Health Alliance & NHMRC (GNT1113531), and the Maurice de Rohan International Scholarship. Laurent C. Francioli was supported by the Swiss National Science Foundation (Advanced Post-doc.Mobility 177853). Vijay S. Ganesh was supported by NIH/NHGRI T32HG010464. Sanna Gudmundsson was supported by the Knut and Alice Wallenberg Foundation. Miriam S. Udler was supported by NIH/NIDDK K23DK114551. Monica H. Wojcik was supported by NIH/ NICHD K23HD102589 and by an Early Career Award from the Thrasher Research Fund.
Additional Information
The online version of this article (https://doi.org/10.1016/j.gim.2021.12.005) contains supplementary material, which is available to authorized users.
Footnotes
Ethics Declaration
Informed consent was obtained by collaborators for all participants in studies across the Centers for Mendelian Genomics (CMGs), and individual-level data, including genomics and clinical data, were de-identified and coded by our collaborators before submission to the CMGs. The participants’ samples used for this study were obtained from multiple institutions, and each CMG (Baylor-Hopkins, Baylor College of Medicine Institutional Review Board [IRB] and Johns Hopkins Medicine IRB; Broad Institute of MIT and Harvard, Mass General Brigham IRB; University of Washington, University of Washington IRB; Yale, Yale University IRB) was responsible for submitting to their own IRB to receive local approval. There was no central IRB for this consortium; however, the main IRB for this publication is Broad Institute of MIT and Harvard, Mass General Brigham IRB.
Conflict of Interest
Baylor College of Medicine and Miraca Holdings Inc have formed a joint venture with shared ownership and governance of Baylor Genetics, formerly the Baylor Miraca Genetics Laboratories, which performs clinical ES and chromosomal microarray analysis for genome-wide detection of copy number variants. J.R.L. serves on the Scientific Advisory Board of Baylor Genetics. J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, and is a coinventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. H.L.R. receives funding from Illumina to support rare disease gene discovery and diagnosis. Consortium author conflicts of interest are listed in the Supplement. All other authors have no disclosures relevant to the manuscript.
Data Availability
Candidate genes identified by the Centers for Mendelian Genomics (CMGs) have been submitted to Matchmaker Exchange (https://www.matchmakerexchange.org/). Variant classifications have been submitted to ClinVar. Deidentified and coded genomic and phenotype data have been shared on the National Human Genome Research Institute Analysis, Visualization, and Informatics Lab-space platform. Data access requests can be made per instructions here: https://anvilproject.org/learn/accessing-data/requesting-data-access#accessing-controlled-access-data. ClinVar submissions for each CMG can be found at Baylor-Hopkins CMG, Lupski Lab, https://www.ncbi.nlm.nih.gov/clinvar/submitters/505572; Baylor-Hopkins CMG (Johns Hopkins University), https://www.ncbi.nlm.nih.gov/clinvar/submitters/505755; Broad Institute Rare Disease Group (Broad Institute), https://www.ncbi.nlm.nih.gov/clinvar/submitters/506627; University of Washington Center for Mendelian Genomics (University of Washington), https://www.ncbi.nlm.nih.gov/clinvar/submitters/505516; and Yale CMG (Yale University) https://www.ncbi.nlm.nih.gov/clinvar/submitters/506150.
References
- 1.Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot–Marie–Tooth neuropathy. N Engl J Med 2010;362(13):1181–1191. 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bainbridge MN, Wiszniewski W, Murdock DR, et al. Whole-genome sequencing for optimized patient management. Sci Transl Med 2011;3(87):87re3. 10.1126/scitranslmed.3002243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature 2009;461(7261):272–276. 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a Mendelian disorder. Nat Genet 2010;42(1):30–35. 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Biesecker LG. Exome sequencing makes medical genomics a reality. Nat Genet 2010;42(1):13–14. 10.1038/ng0110-13. [DOI] [PubMed] [Google Scholar]
- 6.Beaulieu CL, Majewski J, Schwartzentruber J, et al. FORGE Canada Consortium: outcomes of a 2-year national rare-disease gene-discovery project. Am J Hum Genet 2014;94(6):809–817. 10.1016/j.ajhg.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright CF, McRae JF, Clayton S, et al. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet Med 2018;20(10):1216–1223. 10.1038/gim.2017.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Splinter K, Adams DR, Bacino CA, et al. Effect of genetic diagnosis on patients with previously undiagnosed disease. N Engl J Med 2018;379(22):2131–2139. 10.1056/NEJMoa1714458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Austin CP, Cutillo CM, Lau LPL, et al. Future of rare diseases research 2017–2027: an IRDiRC perspective. Clin Transl Sci 2018;11(1): 21–27. 10.1111/cts.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bamshad MJ, Shendure JA, Valle D, et al. The Centers for Mendelian Genomics: a new large-scale initiative to identify the genes underlying rare Mendelian conditions. Am J Med Genet A 2012;158A(7): 1523–1525. 10.1002/ajmg.a.35470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chong JX, Buckingham KJ, Jhangiani SN, et al. The genetic basis of Mendelian phenotypes: discoveries, challenges, and opportunities. Am J Hum Genet 2015;97(2):199–215. 10.1016/j.ajhg.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Posey JE, O’Donnell-Luria AH, Chong JX, et al. Insights into genetics, human biology and disease gleaned from family based genomic studies. Genet Med 2019;21(4):798–812. 10.1038/s41436-018-0408-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azzariti DR, Hamosh A. Genomic data sharing for novel Mendelian disease gene discovery: the Matchmaker Exchange. Annu Rev Genomics Hum Genet 2020;21:305–326. 10.1146/annurevgenom-083118-014915. [DOI] [PubMed] [Google Scholar]
- 14.Bryen SJ, Joshi H, Evesson FJ, et al. Pathogenic abnormal splicing due to intronic deletions that induce biophysical space constraint for spliceosome assembly. Am J Hum Genet 2019;105(3):573–587. 10.1016/j.ajhg.2019.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bryen SJ, Ewans LJ, Pinner J, et al. Recurrent TTN metatranscript-only c.39974–11T>G splice variant associated with autosomal recessive arthrogryposis multiplex congenita and myopathy. Hum Mutat 2020;41(2):403–411. 10.1002/humu.23938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wahlster L, Verboon JM, Ludwig LS, et al. Familial thrombocytopenia due to a complex structural variant resulting in a WAC-ANKRD26 fusion transcript. J Exp Med 2021;218(6):e20210444. 10.1084/jem.20210444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.LaCroix AJ, Stabley D, Sahraoui R, et al. GGC repeat expansion and exon 1 methylation of XYLT1 is a common pathogenic variant in Baratela-Scott syndrome. Am J Hum Genet 2019;104(1):35–44. 10.1016/j.ajhg.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mendelian traits by the numbers Centers for Mendelian Genomics. http://mendelian.org/mendelian-traits-numbers. Accessed September 9, 2021. [Google Scholar]
- 19.Amberger JS, Bocchini CA, Scott AF, Hamosh A. OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res 2019;47(D1):D1038–D1043. 10.1093/nar/gky1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee-Barber J, English TE, Britton JF, et al. Apparent acetaminophen toxicity in a patient with transaldolase deficiency. JIMD Rep 2019;44:9–15. 10.1007/8904_2018_116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hanczko R, Fernandez DR, Doherty E, et al. Prevention of hepatocarcinogenesis and increased susceptibility to acetaminophen-induced liver failure in transaldolase-deficient mice by N-acetylcysteine. J Clin Invest 2009;119(6):1546–1557. 10.1172/JCI35722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donkervoort S, Mohassel P, Laugwitz L, et al. Biallelic loss of function variants in SYT2 cause a treatable congenital onset presynaptic myasthenic syndrome. Am J Med Genet A 2020;182(10):2272–2283. 10.1002/ajmg.a.61765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Karnebeek CDM, Ramos RJ, Wen XY, et al. Bi-allelic GOT2 mutations cause a treatable malate-aspartate shuttle-related encephalopathy. Am J Hum Genet 2019;105(3):534–548. 10.1016/j.ajhg.2019.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marafi D, Mitani T, Isikay S, et al. Biallelic GRM7 variants cause epilepsy, microcephaly, and cerebral atrophy. Ann Clin Transl Neurol 2020;7(5):610–627. 10.1002/acn3.51003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bean LJH, Funke B, Carlston CM, et al. Diagnostic gene sequencing panels: from design to report-a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet Med 2020;22(3):453–461. 10.1038/s41436-019-0666-z. [DOI] [PubMed] [Google Scholar]
- 26.Strande NT, Riggs ER, Buchanan AH, et al. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the Clinical Genome Resource. Am J Hum Genet 2017;100(6):895–906. 10.1016/j.ajhg.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lumaka A, Race V, Peeters H, et al. A comprehensive clinical and genetic study in 127 patients with ID in Kinshasa, DR Congo. Am J Med Genet A 2018;176(9):1897–1909. 10.1002/ajmg.a.40382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saad AK, Marafi D, Mitani T, et al. Neurodevelopmental disorder in an Egyptian family with a biallelic ALKBH8 variant. Am J Med Genet A 2021;185(4):1288–1293. 10.1002/ajmg.a.62100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saad AK, Marafi D, Mitani T, et al. Biallelic in-frame deletion in TRAPPC4 in a family with developmental delay and cerebellar atrophy. Brain 2020;143(10):e83. 10.1093/brain/awaa256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duan R, Saadi NW, Grochowski CM, et al. A novel homozygous SLC13A5 whole-gene deletion generated by Alu/Alu-mediated rearrangement in an Iraqi family with epileptic encephalopathy. Am J Med Genet A 2021;185(7):1972–1980. 10.1002/ajmg.a.62192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Contreras JL, Ladino MA, Aránguiz K, et al. Immune dysregulation mimicking systemic lupus erythematosus in a patient with lysinuric protein intolerance: case report and review of the literature. Front Pediatr 2021;9:673957. 10.3389/fped.2021.673957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aird A, Lagos M, Vargas-Hernández A, et al. Novel heterozygous mutation in NFKB2 is associated with early onset CVID and a functional defect in NK cells complicated by disseminated CMV infection and severe nephrotic syndrome. Front Pediatr 2019;7:303. 10.3389/fped.2019.00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karaca E, Harel T, Pehlivan D, et al. Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron 2015;88(3):499–513. 10.1016/j.neuron.2015.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bayram Y, Karaca E, Coban Akdemir Z, et al. Molecular etiology of arthrogryposis in multiple families of mostly Turkish origin. J Clin Invest 2016;126(2):762–778. 10.1172/JCI84457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pehlivan D, Bayram Y, Gunes N, et al. The genomics of arthrogryposis, a complex trait: candidate genes and further evidence for oligogenic inheritance. Am J Hum Genet 2019;105(1):132–150. 10.1016/j.ajhg.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sobreira NLM, Arachchi H, Buske OJ, et al. Matchmaker Exchange. Curr Protoc Hum Genet 2017;95:9.31.1–9.31.15. 10.1002/cphg.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat 2015;36(10):928–930. 10.1002/humu.22844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Website aims to accelerate gene discovery, diagnosis, treatment: MyGene2.org fosters open sharing among families, researchers, and clinicians. Am J Med Genet A 2016;170(6):1388–1389. 10.1002/ajmg.a.37746. [DOI] [PubMed] [Google Scholar]
- 39.Arachchi H, Wojcik MH, Weisburd B, et al. matchbox: an open-source tool for patient matching via the Matchmaker Exchange. Hum Mutat 2018;39(12):1827–1834. 10.1002/humu.23655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD:predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47(D1):D886–D894. 10.1093/nar/gky1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sobreira N, Schiettecatte F, Boehm C, Valle D, Hamosh A. New tools for Mendelian disease gene identification: PhenoDB variant analysis module; and GeneMatcher, a web-based tool for linking investigators with an interest in the same gene. Hum Mutat 2015;36(4):425–431. 10.1002/humu.22769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu P, Meng L, Normand EA, et al. Reanalysis of clinical exome sequencing data. N Engl J Med 2019;380(25):2478–2480. 10.1056/NEJMc1812033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmitz-Abe K, Li Q, Rosen SM, et al. Unique bioinformatic approach and comprehensive reanalysis improve diagnostic yield of clinical exomes. Eur J Hum Genet 2019;27(9):1398–1405. 10.1038/s41431-019-0401-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo H, Bettella E, Marcogliese PC, et al. Disruptive mutations in TANC2 define a neurodevelopmental syndrome associated with psychiatric disorders. Nat Commun 2019;10(1):4679. 10.1038/s41467-019-12435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang M, Chun J, Genovese G, et al. Contributions of rare gene variants to familial and sporadic FSGS. J Am Soc Nephrol 2019;30(9):1625–1640. 10.1681/ASN.2019020152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansen AW, Murugan M, Li H, et al. A genocentric approach to discovery of Mendelian disorders. Am J Hum Genet 2019;105(5):974–986. 10.1016/j.ajhg.2019.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kerner G, Bouaziz M, Cobat A, et al. A genome-wide case-only test for the detection of digenic inheritance in human exomes. Proc Natl Acad Sci U S A 2020;117(32):19367–19375. 10.1073/pnas.1920650117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lilleväli H, Pajusalu S, Wojcik MH, et al. Genome sequencing identifies a homozygous inversion disrupting QDPR as a cause for dihydropteridine reductase deficiency. Mol Genet Genomic Med 2020;8(4):e1154. 10.1002/mgg3.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mohammadi P, Castel SE, Cummings BB, et al. Genetic regulatory variation in populations informs transcriptome analysis in rare disease. Science 2019;366(6463):351–356. 10.1126/science.aay0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carvalho CMB, Coban-Akdemir Z, Hijazi H, et al. Interchromosomal template-switching as a novel molecular mechanism for imprinting perturbations associated with Temple syndrome. Genome Med 2019;11(1):25. 10.1186/s13073-019-0633-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boycott KM, Dyment DA, Innes AM. Unsolved recognizable patterns of human malformation: challenges and opportunities. Am J Med Genet C Semin Med Genet 2018;178(4):382–386. 10.1002/ajmg.c.31665. [DOI] [PubMed] [Google Scholar]
- 52.Dyment DA, O’Donnell-Luria A, Agrawal PB, et al. Alternative genomic diagnoses for individuals with a clinical diagnosis of Dubowitz syndrome. Am J Med Genet A 2021;185(1):119–133. 10.1002/ajmg.a.61926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burrage LC, Reynolds JJ, Baratang NV, et al. Bi-allelic variants in TONSL cause SPONASTRIME dysplasia and a spectrum of skeletal dysplasia phenotypes. Am J Hum Genet 2019;104(3):422–438. 10.1016/j.ajhg.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schrauwen I, Giese AP, Aziz A, et al. FAM92A underlies nonsyndromic postaxial polydactyly in humans and an abnormal limb and digit skeletal phenotype in mice. J Bone Miner Res 2019;34(2):375–386. 10.1002/jbmr.3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bamshad MJ, Nickerson DA, Chong JX. Mendelian gene discovery: fast and furious with no end in sight. Am J Hum Genet 2019;105(3):448–455. 10.1016/j.ajhg.2019.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581(7809):434–443. Published correction appears in Nature. 2021;590(7846):E53. 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Logsdon GA, Vollger MR, Hsieh P, et al. The structure, function and evolution of a complete human chromosome 8. Nature 2021; 593(7857):101–107. 10.1038/s41586-021-03420-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lappalainen T, Scott AJ, Brandt M, Hall IM. Genomic analysis in the age of human genome sequencing. Cell 2019;177(1):70–84. 10.1016/j.cell.2019.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sethi A, Gu M, Gumusgoz E, et al. Supervised enhancer prediction with epigenetic pattern recognition and targeted validation. Nat Methods 2020;17(8):807–814. 10.1038/s41592-020-0907-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sisu C, Muir P, Frankish A, et al. Transcriptional activity and strain-specific history of mouse pseudogenes. Nat Commun 2020;11 (1):3695. 10.1038/s41467-020-17157-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Esposito D, Weile J, Shendure J, et al. MaveDB: an open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol 2019;20(1):223. 10.1186/s13059-019-1845-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sundaram L, Gao H, Padigepati SR, et al. Predicting the clinical impact of human mutation with deep neural networks. Nat Genet 2018;50(8):1161–1170. Published correction appears in Nat Genet. 2019;51(2):364 10.1038/s41588-018-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell 2019;176(3):535–548.e24. 10.1016/j.cell.2018.12.015. [DOI] [PubMed] [Google Scholar]
- 64.Taylor DM, Aronow BJ, Tan K, et al. The pediatric cell atlas: defining the growth phase of human development at single-cell resolution. Dev Cell 2019;49(1):10–29. 10.1016/j.devcel.2019.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Clark MM, Stark Z, Farnaes L, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom Med 2018;3:16. 10.1038/s41525-018-0053-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Candidate genes identified by the Centers for Mendelian Genomics (CMGs) have been submitted to Matchmaker Exchange (https://www.matchmakerexchange.org/). Variant classifications have been submitted to ClinVar. Deidentified and coded genomic and phenotype data have been shared on the National Human Genome Research Institute Analysis, Visualization, and Informatics Lab-space platform. Data access requests can be made per instructions here: https://anvilproject.org/learn/accessing-data/requesting-data-access#accessing-controlled-access-data. ClinVar submissions for each CMG can be found at Baylor-Hopkins CMG, Lupski Lab, https://www.ncbi.nlm.nih.gov/clinvar/submitters/505572; Baylor-Hopkins CMG (Johns Hopkins University), https://www.ncbi.nlm.nih.gov/clinvar/submitters/505755; Broad Institute Rare Disease Group (Broad Institute), https://www.ncbi.nlm.nih.gov/clinvar/submitters/506627; University of Washington Center for Mendelian Genomics (University of Washington), https://www.ncbi.nlm.nih.gov/clinvar/submitters/505516; and Yale CMG (Yale University) https://www.ncbi.nlm.nih.gov/clinvar/submitters/506150.