

Abstract
Metastatic castration resistant (mCRPC) prostate cancer has been largely resistant to immunotherapy. Natural killer (NK) cells are cytotoxic lymphocytes that detect and kill transformed cells without prior sensitization, and their infiltration into prostate tumors corresponds with an increased overall survival among patients with mCRPC. We sought to harness this knowledge to develop an approach to NK-cell based immunotherapy for mCRPC. We engineered an NK cell line (NK-92MI) to express CD64, the sole human high-affinity IgG Fcγ receptor (FcγR1), and bound these cells with antibodies to provide interchangeable tumor-targeting elements. NK-92MICD64 cells were evaluated for cell-activation mechanisms and antibody-dependent cell-mediated cytotoxicity (ADCC). A combination of monoclonal antibodies was used to target the prostate tumor antigen tumor-associated calcium signal transducer 2 (TROP2) and the cancer-associated fibroblast marker fibroblast activation protein alpha (FAP). We found that CD64, which is normally expressed by myeloid cells and associates with the adaptor molecule FcRγ, can be expressed by NK-92MI cells and mediate ADCC through an association with CD3ζ. Cytotoxicity from the combination approach was two-fold higher than that by NK-92MICD64 cells with either mAb alone bound to them, and seven-fold higher than NK-92MICD64 cells alone at an effector–target cell ratio of 20:1. The cytotoxic effect was lost when using isotype control antibodies, indicating a selective targeting mechanism. The combination approach demonstrated efficacy in vivo as well and significantly reduced tumor growth compared with the saline control. This combination therapy presents a potential approach for treating mCRPC and could improve immunotherapy response.
Keywords: Prostate cancer, natural killer cell, immunotherapy, combination therapy, tumor microenvironment
Introduction
Metastatic castration resistant prostate cancer (mCRPC) is uniformly lethal with 5-year survival rates remaining at 30%. Innovative treatments for patients with the disease are urgently needed and efforts are being focused on developing immunotherapies [1]. The FDA-approved autologous cell–based vaccine Sipuleucel-T, which stimulates T-cell response against the prostate tumor antigen prostatic acid phosphatase, improved median overall survival (OS) by 4.1 months, indicating that prostate cancer was a good candidate for cancer immunotherapy [2]. Current immunotherapy clinical trials for mCRPC have focused on evaluating checkpoint inhibitors, however, they have had modest outcomes and none have been approved [3–5]. The lack of immunotherapeutic efficacy in mCRPC is due, in part, to the immunosuppressive tumor microenvironment (TME) that promotes the survival of the cancer cells and progression of the disease [6]. Cancer-associated fibroblasts are the major cell type in the TME that regulate growth and metastasis of epithelial cancers, including prostate cancer [7]. Cancer-associated fibroblast paracrine signaling and mechanical stress contribute to tumor survival, inflammation, immunosuppression, and therapeutic resistance, and drive tumor invasion [8, 9]. Fibroblast activation protein alpha (FAP) is a transmembrane serine protease highly expressed by cancer-associated fibroblasts that contributes to extracellular matrix remodeling and pro-tumor signal transduction [10]. Immunotherapies that target malignant cells alone may not be enough to elicit an immune response capable of overcoming pro-tumorigenic functions of the TME. In contrast, an immunotherapeutic approach that targets both the tumor stroma and malignant cells has been shown effective in multiple cancer models [11, 12].
Natural killer (NK) cells are innate, cytotoxic lymphocytes that detect and kill stressed, virus-infected, and transformed cells without prior sensitization [13]. Infiltration of NK cells in prostate tumors corresponds with an increased OS among patients with mCRPC [14], which suggests the potential to exploit NK cells for the treatment of prostate cancer. A key antitumor effector function of human NK cells is antibody-dependent cell-mediated cytotoxicity (ADCC), which is mediated through the IgG Fcγ receptor CD16A (FcγRIIIA) [15]. Several clinically successful antitumor monoclonal antibodies (mAbs) mediate ADCC [16]; however, attributes of CD16A, such as a low-to-moderate affinity for IgG and rapid downregulation by a disintegrin and metalloproteinase-17 (ADAM17) may limit the efficacy of mAb therapies [17].
CD64 (FcγR1), the sole functional high affinity FcγR in humans, binds to IgG isotypes IgG1 and IgG3, like CD16A; however, CD64 binds monomeric IgG with an affinity ~30-fold higher than CD16A [18]. CD64 is expressed by myeloid cells including monocytes, macrophages and neutrophils, but not lymphocytes, including NK cells [19]. Unlike CD16A, CD64 expression is not downregulated by ADAM17 upon activation [20]. CD16A non-covalently associates with the signaling adaptor FcRγ and CD3ζ as either homodimers or heterodimers [21, 22], whereas CD64 associates with FcRγ, because myeloid cells lack expression of CD3ζ [23]. These immunoreceptor tyrosine-based activation motif (ITAM)-containing signaling adaptors trigger downstream signaling cascades for cell activation.
In this study, we stably expressed human CD64 in the NK-92MI human NK cell line that lacks expression of endogenous FcγRs and synthesizes IL2 to promote cell proliferation [24]. These cells are referred to herein as NK-92MICD64 cells. Our analyses revealed that CD64 surface levels can be modulated by CD3ζ expression. Furthermore, we observed that CD64 induces ADCC and that this was facilitated through CD3ζ signaling. The antitumor activity of these genetically modified NK cells was tested against the TROP2-expressing prostate cancer cell line, DU145, and the FAP-expressing prostate stromal cell line, hPrCSC-44. Effector-cell function was enhanced with the addition of TROP2-or FAP-targeted antibodies. Therapeutic efficacy of the combination therapy was also demonstrated in a prostate tumor stroma xenograft model. Overall, these data suggest the therapeutic potential of the engineered NK cells in combination with antibodies targeting both the tumor stroma and malignant cells, a strategy that could provide urgently needed immunotherapies for patients with mCRPC.
Methods
Cell Culture
DU145 (#HTB-81) and HEK293T (#CRL-11268) cell lines were purchased from ATCC in 2019 and maintained in DMEM (Gibco, catalog no. 11965092) supplemented with 10% FBS (Gibco, catalog no. 16140071). hPrCSC-44 cells were obtained from Dr. W. Nathaniel Brennen (Johns Hopkins Medicine, Baltimore, MD) and maintained in RoosterNourish-MSC media (RoosterBio, catalog no. KT-001). Human NK-92MI (#CRL-2408) cells were purchased from ATCC in 2018 and maintained in MEM-α media without nucleosides (Gibco catalogue no. 12561056, Gibco) supplemented with 12.5% Fetal Bovine Serum (Gibco, catalog no. 16140071), 12.5% Horse Serum (Gibco, catalog no. 26050088), and 0.1 nM 2-mercaptoethanol (Sigma-Aldrich, catalog no. M6250). Jurkat (#TIB-152) cells were obtained from ATCC in 2009 and maintained in RPMI-1640 (Gibco, catalog no. 11875093). Gryphon Ampho cells (293T cells stably expressing gag, pol and env genes) were purchased from Allele Biotechnology in 2019 and were maintained DMEM (Gibco, catalog no. 11965092) supplemented with 10% FBS (Gibco, catalog no. 16140071). All cell experiments were performed within 4 months of thawing cell lines from frozen cell stocks. Cell lines were authenticated using short-tandem repeat analysis and routinely monitored for mycoplasma contamination prior to our studies.
Generation of CD64, FcRγ-chain, and CD3ζ-chain expression constructs
Fresh peripheral blood of healthy blood donors was purchased from the Innovative Blood Resources (St. Paul, MN). Total RNA was isolated from two human peripheral blood leukocytes using TRIzol total RNA isolation reagent (ThermoFisher Scientific, catalog no. 10296010). Peripheral blood cDNA synthesized with the SuperScript First-Strand Synthesis System for RT-PCR kit (ThermoFisher Scientific, catalog no. 11904018) was used in RT-PCR for expression construct generation. Bam HI-flanked full-length CD64 cDNA (1,169 bps) was amplified using the forward primer 5’-CTC TAG ACT GCC GGA TCC GGA GAC AAC ATG TGG TTC TTG ACA ACT CT-3’ and the reverse primer 5’-TCG AAT TTA AAT GGA TCC CTA CGT GGC CCC CTG GGG CTC CTT-3’ (underlined nucleotides indicate Bam HI restriction sites). The RT-PCR reaction was performed with 2 μl of cDNA, 200 nM of each primer, 200 μM of dNTPs, 2.0 mM of MgSO4, and 1 U of Platinum Taq DNA Polymerase High Fidelity (ThermoFisher Scientific, catalog no. 11304011) in a 25 μl reaction volume. The ABI Veriti 96-well Thermal Cycler was used for the RT-PCR reaction starting with 94 ºC for 3 min, 35 cycles of denaturing at 94 ºC for 30 s, annealing at 56 ºC for 45 s, extension at 68 ºC for 1 min and 30 s with a final extension at 72 ºC for 7 min. In-Fusion HD Cloning Kit (Takara Bio USA, catalog no. 638917) was used to insert the CD64 cDNA into the bi-cistronic retroviral expression vector pBMN-I-GFP (Addgene Plasmid# 1736; deposited by Dr. Garry Nolan) at the Bam HI restriction enzyme site. Pseudo-retrovirus was generated as previously described [25]. Briefly, approximately 24 hours before transfection, AmphoPhoenix cells were seeded in a 60-mm cell culture dish. AmphoPhoenix cells were incubated overnight at 37 ºC, 5% CO2 to reach about 85% confluence. For transfection of AmphoPhoenix cells, the complexes of 5 μg CD64-pBMN-I-GFP plasmid DNA and 10 μl Lipofectamine 2000 Tranfection Reagent (ThermoFisher Sceintific, catalog no. 11668019) waer prepared according to the vendor’s instructions. The DNA-Lipofectamine 2000 complexes were added to AmphoPhoenix cells in the dish and subsequently mixed with culture medium by rocking back and forth gently. The medium containing DNA-Lipofectamine 2000 complexes was removed from the culture dish and 5 ml of fresh culture medium was added after 12–16 hours of incubation. The cell culture medium containing pseudo-retrovirus particles was harvested after additional 48-hour incubation. The harvested cell culture supernatant was centrifuged at 2500 RPM (1282 x g) for 3 min at 4 ºC to remove cell debris and filtered through 0.45 μm filter before cell transduction.
For NK-92MI cell transduction, 1 ml of filtered cell culture supernatant containing virus particles was mixed with 4 ml of NK-92MI cell culture medium and 5 μg/m polybrene (Sigma-Aldrich, catalog no. TR-1003) to form an infection cocktail. To infect NK-92MI cells, 5×105 NK-92MI cell were mixed with 5 ml of the infection cocktail and seed into one well of a 6-well plate. The same volume of fresh medium was added one day after infection. Two days after infection, the transduced NK92-MI cells were stained with APC-conjugated anti-CD64 (BioLegend, catalog no. 305014) and examined by flow cytometric analysis to determine CD64 expression. The NK-92MI cells were sorted by flow cytometry for a homogeneous CD64 expression population.
Kpn I/Bam HI-flanked full-length CD64 cDNA was amplified using the forward primer 5’-TAA CGG GGT ACC GGA GAC AAC ATG TGG TTC TTG ACA ACT-3’ (underlined nucleotides indicate the Kpn I restriction site) and the reverse primer 5’-CCG GGA TCC CTA CGT GGC CCC CTG GGG CTC CTT-3’ (underlined nucleotides indicate the Bam HI restriction site). Bam HI/EcoR I-flanked full-length FcRγ chain cDNA (285 bps) was amplified with the sense (5’-CGC GGA TCC CAG CCC AAG ATG ATT CCA GCA GTG GTC-3’) (underlined nucleotides indicate the Bam HI restriction site) and anti-sense primer (5’-CCG GAA TTC CTA AAG CTA CTG TGG TGG TTT CTC ATG-3’ (the underlined nucleotides indicate the EcoR I restriction site). The human FcRγ cDNA was cloned into the pcDNA3.1 expression vector (ThermoFisher Scientific, catalog no. V79020). Similarly, Kpn I/EcoR I-flanked full-length CD3ζ chain cDNA (546 bps) was amplified with the sense primer (5’-CGG GGT ACC TCT GAG GGA AAG GAC AAG ATG A-3’) (underlined nucleotides indicate the Kpn I restriction site) and anti-sense primer (5’-CCG GAA TTC GGT GAA ATC CCC TGC CTG TTA-3’ (underlined nucleotides indicate the EcoR I restriction site). The human CD3ζ cDNA was cloned into the pcDNA3.1 expression vector. Expression constructs were transfected into HEK-293T cells for transient expression analysis using the TransIT-293 transfection reagent (Mirus Bio, catalog no. MIR 2704). Successful transfection was confirmed via flow cytometric analysis.
Immunoprecipitation (IP) and Western Blotting
Cells were lysed in sodium dodecyl sulfate (SDS) lysis buffer (1% SDS, 50 mM Tris-Cl, 10 mM Ethylenediaminetetraacetic acid (EDTA), protease inhibitor cocktail (MilliporeSigma, catalog no. P8340) and phosphatase inhibitor cocktail 2 (MilliporeSigma, catalog no. P5726)) for Western blot, or digitonin IP lysis buffer (1% digitonin, 20 mM Tris-Cl, 135 mM NaCl, protease and phosphatase cocktail inhibitors) for IP. SDS lysates were sonicated and centrifuged at 12,000 ×g to pellet insoluble material. Digitonin lysates were rocked gently for 30 min at 4°C and centrifuged at 10,000 ×g to remove insoluble cell debris.
Protein concentrations were quantified using a Bicinchoninic acid assay (Pierce Biotechnology, catalog no. 23225). 50 μg of protein (1X Laemmli sample buffer (BIO-RAD, catalog no. 161-0747), 0.1M DTT) was resolved by SDS-PAGE and transferred to a nitrocellulose membrane. Blots were blocked in Intercept TBS blocking buffer (LI-COR, catalog no. 927-60001) and incubated with primary antibodies and Quick Western IRDye® 680RD detection reagent (LI-COR, catalog no. 926-69100) overnight at 4°C. Blots were visualized using an Odyssey infrared imager (LI-COR). Primary antibodies used were anti-CD64 (Abcam, catalog no. ab134073), anti-FcRγ subunit (EMD Millipore, catalog no. 06-727), anti-CD3ζ (Santa Cruz Biotechnology, catalog no. sc-1239), and anti-β-actin (Sigma-Aldrich, catalog no. A5316).
For IP, lysates were incubated for 1 hr with human serum IgG (Sigma-Aldrich, catalog no. I2511-10MG) at 4°C to block non-specific interactions between CD64 and IP antibodies. CD64 or isotype matched control antibodies (Bethyl Laboratories, catalog no. catalog no. P120-101) were immobilized to Pierce Direct IP Kit immunoprecipitation beads (Pierce Biotechnology, catalog no. 26148) following the manufacturer’s directions. Protein was eluted at 100°C using SDS elution buffer containing 0.1M DTT, and samples were resolved by SDS-PAGE for Western blot as described above.
CRISPR/Cas9 knock-out of CD3ζ
Single guide RNAs (sgRNA) were designed using Synthego’s Knockout Guide Design tool (https://design.synthego.com/#). Alt-R CRISPR-Cas9 sgRNAs were purchased from IDT (Integrated DNA Technologies) and contain both tracrRNA and crRNA sequences; CD247 (CD3ζ) guide RNA sequence: AGCAGAGUUUGGGAUCCAGC. Cas9 recombinant protein was purchased from IDT (catalog no. 1081059). CRISPR/Cas9 ribonucleoprotein (RNP) complexes were assembled by mixing 100 pmol of guide RNA and 30 pmol of Cas9. Five million NK-92MI cells were resuspended in primary cell P3 buffer (Lonza, catalog no. PBP3-00675) with the Cas9 RNP complexes and nucleofected using the Amaxa 4D Nucleofector program CA-137. Knockout of CD3ζ was confirmed via Western blot (anti-CD3ζ, Santa Cruz Biotechnology, catalog no. sc-1239), flow cytometry, and Inference of CRISPR Edits (ICE) analysis. For ICE analysis, DNA was isolated from NK-92MI cells using Qiagen’s DNEasy kit (QIAGEN, catalog no. 69506) and PCR with the SsoAdvanced Universal SYBR Green Supermix (BIO-RAD, catalog no. 1725270) was used to amplify a 467 bp region that contained the gRNA cut site for CD3ζ (forward primer 5’-CCTTGTTCCTGAGAGTGAAG-3’, reverse primer 5’-CCAAATAAATCATGACACGGAG-3’). PCR products were submitted to UMN Genomics for Sanger sequencing with the sequencing primer 5’-GATGTGTTCTCGTCACCTT-3’. Sequence results were then uploaded to Synthego’s ICE analysis tool to confirm gene ablation (https://ice.synthego.com/#).
Antibody Expression
An in-house murine naïve single-chain variable fragment (scFv) antibody phage display library with a diversity of 1x109 was used to identify clones against recombinant human FAP (R&D Systems, catalog no. 3715-SE-010). Anti-TROP2 was generated as previously described [26]. The heavy and light chain variable domains of the anti-FAP (B12) and anti-TROP2 scFv sequences were cloned separately into the pFUSEss human IgG1 expression vectors pFUSEss-CHIg-hG1 (InvivoGen, catalog no. pfusess-hchg1) and pFUSE2ss-CLIg-hk (InvivoGen, catalog no. pfuse2ss-hclk). Anti-FAP light chain primers (forward primer 5’-AAAACGAATTCGGATATTGTGATCAGTCTCCATCCTCC −3’, reverse primer 5’-AAAACGTACGGCGTTTGATTTCCAGCTTGGTGC −3’) and heavy chain primers (forward primer 5’-AAAACGAATTCGGAAGTGATGCGAGTCTGGG-3’, reverse primer 5’-AAAACGTAGCCGAGGAGACTGTGAGAGTGGTGC-3’). Anti-TROP2 light chain primers (forward primer 5’-GCCGCGAATTCGGACATCCAGCTGACCCAAAGCCACAAGTTCATG-3’, reverse primer 5’-CCGCGCGTACGACGCTTCAGCTCCAGCTTGGTACCCGCGC-3’) and heavy chain primers (forward primer 5’-GCCGCGAATTCGCAAGTGCAACTGGGTGAAAGCGGTCCGG-3’, reverse primer 5’-GCCGCGCTAGCGCTGCTAACGGTCACGGTGGTACCTTGGCC-3’). The vectors were co-transfected into HEK293T cells using the calcium carbonate method. The cell supernatant was collected after 72 hours and purified using a 5 mL HiTrap Protein A HP column (GE Healthcare) following the manufacturer’s guidelines. Eluted antibody was collected, and the purity was analyzed by reduced and nonreduced SDS-PAGE and visualized using GelCode Blue Safe Protein Stain (ThermoFisher Scientific, catalog no. PI24596). Protein concentration was measured based on absorbance at 280 nm using a NanoDrop One UV-Vis Spectrophotometer (Thermo-Fisher).
Cell Staining and IFNγ Quantification
To measure antibody capture by CD64, NK-92MICD64 cells were incubated with the generated anti-TROP2 or anti-FAP (B12) (5 μg/mL) in serum free MEM-α media (SFM) without nucleosides (Gibco, catalog no. 12561056) supplemented with 0.1 nM 2-mercaptoethanol (Sigma-Aldrich, catalog no. M6250) for 2 hours at 37°C and 5% CO2. Effector cells were washed once with SFM, and stained with APC-conjugated donkey anti-human IgG secondary mAb (1:200, catalog no. 709-136-14, Jackson Immunoresearch) for 30 minutes on ice. Cells were fixed with 1% paraformaldehyde, resuspended in FACS buffer, and analyzed on a BD LSR II flow cytometer. For IFNγ quantification, NK-92MICD64 cells were preincubated in SFM for 2 hours at 37°C and 5% CO2. Effector cells were combined with DU145 or hPrSCS-44 target cells at an E:T ratio of 1:1 (1.5x105 effector and 1.5x105 target cells, respectively) and incubated with or without anti-TROP2 or B12 (5 μg/mL) in SFM for 2 hours at 37°C and 5% CO2. After incubation, the cells were centrifuged and cell supernatants were collected for IFNγ determination using the human IFNγ flex set (cat. #560111, BD Biosciences) and a BD LSR II flow cytometer. Analysis was performed using FlowJo software.
Flow cytometric analysis was performed on FACSCelesta and BD LSR II instruments using BD FACSDiva Software (BD Biosciences) or the Beckman Coulter Cytoflex flow cytometer. CD64 expression was determined using the human anti-CD64 clone 10.1 (BioLegend, catalog no. 305014). To analyze CD3ζ expression, cells were fixed and permeabilized using the Tonbo Foxp3/Transcription Factor Staining Buffer Kit (Tonbo, TNB-0607-KIT) and stained with anti-CD247(BioLegend, catalog no. 644106). For controls, fluorescence minus one was used as well as appropriate isotype-matched antibodies. An FSC-A/SSC-A plot was used to set an electronic gate on cell populations, and an FSC-A/FSC-H plot was used to set an electronic gate on single cells. To distinguish live vs. dead cells, 7AAD was used as per the manufacturer’s instructions (Biolegend, catalog no. 420403). Analysis was performed using FlowJo software.
Cytotoxicity Assays
ADCC was determined using the DELFIA EuTDA cytotoxicity assay (cat. #AD0116, Perkin-Elmer) according to the manufacturer’s instructions and performed as previously described [20]. Cytotoxicity for each sample is represented as % specific release and was calculated using the following formula:
Conventional ADCC: CD3z KO and NK-92MICD64 cells were preincubated in SFM for 2 hours at 37°C and 5% CO2. For the monotherapy, effector (CD3z KO or NK-92MICD64) and target cells (DU145 or hPrCSC-44) were combined at the indicated E:T ratios in SFM with or without anti-TROP2 or B12 (5 ug/mL). For the combination therapy, effector cells (NK-92MICD64) were combined with a mixture of target cells (1:1; 4x103 hPrCSC-44: 4x103 DU145 cells/well) at the indicated E:T ratios in SFM with or without anti-TROP2 and B12 (2.5 ug/mL each).
Bound mAb ADCC: NK-92MICD64 cells were preincubated with or without anti-TROP2 or B12 (5 ug/mL) in SFM for 2 hours at 37°C and 5% CO2 then washed once with SFM. For the monotherapy, effector and target cells (DU145 or hPrCSC-44) were combined at the indicated E:T ratios. The experiment was repeated with NK-92MICD64 cells preincubated with or without a human IgG isotype control (5 ug/mL) in SFM for 2 hours at 37°C and 5% CO2. For the combination therapy, effector cells (1:1; anti-TROP2: B12) were combined with a mixture of target cells (1:1; 4x103 hPrCSC-44: 4x103DU145 cells/well) at the indicated E:T ratios.
Animal Studies
Animal studies were approved by the University of Minnesota Institutional Animal Care and Use Committee (IACUC). Three-to-four-week-old NOD/scid/IL-2 common γ chain −/− (NSG) mice were purchased from Jackson Labs (cat. #005557). Tumors were implanted by subcutaneous injection of a mixture of 2x106 hPrCSC-44 and 1x106 DU145 cells suspended in 100 μL of 1:1 PBS and Matrigel (Corning, catalog no. 356234) into the rear flanks of mice. NK-92MICD64 cells were irradiated (10 gray) before use as a treatment. Randomized mice (n=4/arm) with tumor volumes 100 – 200 mm3 were assigned to the following treatment arms: saline; NK-92MICD64 cells alone; i.p. injection of anti-TROP2 and B12 (6 mg/kg of each mAb) 24 hours before adoptive NK-92MICD64 cell therapy or adoptive NK-92MICD64 cell therapy with bound anti-TROP2 and B12 (1:1; anti-TROP2: B12). Treatment (1x107 cells/mouse) was administered peritumorally once a week for four weeks. Tumor volumes were measured two to three times a week until tumors reached 1500 mm3. For IHC, mice (n=3/arm) were treated with the aforementioned arms. For the ADCC combination, mice were injected i.p. with anti-TROP2 and B12 (6 mg/kg of each mAb) every 2 days. After 7 days, the tumors were excised and fixed for IHC analysis.
Immunohistochemistry (IHC)
IHC was performed on formalin-fixed paraffin-embedded tissue sections using (1:400 dilution) anti-human cleaved caspase 3 (cCasp3) (Catalog no. 9661L, Cell Signaling). Unstained sections (4 μm thick) were deparaffinized and rehydrated using standard methods. Antigen retrieval and staining were performed as previously described [27]. Slides were incubated in secondary biotinylated goat anti-rabbit IgG (10 μL/mL; Vector, BA-1000) diluted in 10% blocking solution (Peroxidase, Biocare Medical, catalog no. PX968)/90% TBST. Detection of the antibody complexes was by the avidin–biotin immunoperoxidase method. Images of cCasp3-stained tumor tissue were collected using an Axiolab 5 microscope (ZEISS). Images (n=3/mouse) were collected for each treatment group (n=3 mice/group). Image J was used to remove background color and the total image was measured for mean grey value.
Statistical Analysis
All statistical analyses were performed using Prism GraphPad software. Comparison between two groups was done using one-way ANOVA and followed by Tukey multiple comparisons test. Statistical significance of tumor growth was determined using a two-way ANOVA and corrected for multiple comparisons using Sidak hypothesis testing. Survival curves were constructed using the Kaplan–Meier method and statistical analysis of survival was performed using a log-rank (Mantel-Cox) test. IHC staining was quantified as mean intensity. Mean intensity = 255 – mean grey value. Results are depicted as mean ± SEM. The symbols used to represent the P values were as follows; ns, P > 0.05; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
Results
NK-92MICD64 cells utilize CD3ζ to induce ADCC
NK-92MI cells are derived from NK-92 cells, which do not express endogenous FcγRs but can mediate ADCC through recombinant CD16A [28]. We transduced NK-92MI cells to express recombinant human CD64 at high levels (Fig. 1A). CD64 is normally expressed by myeloid cells in which its signaling is mediated by its association with the signaling adapter FcRγ [29]. We investigated whether recombinant CD64 can associate with CD3ζ in NK-92MI cells. As shown in Fig. 1B, CD3ζ was observed to co-immunoprecipitate with CD64, indicating their non-covalent association.
Figure 1. CD64 in transduced NK-92MI cells associates with the signaling adaptor CD3ζ.
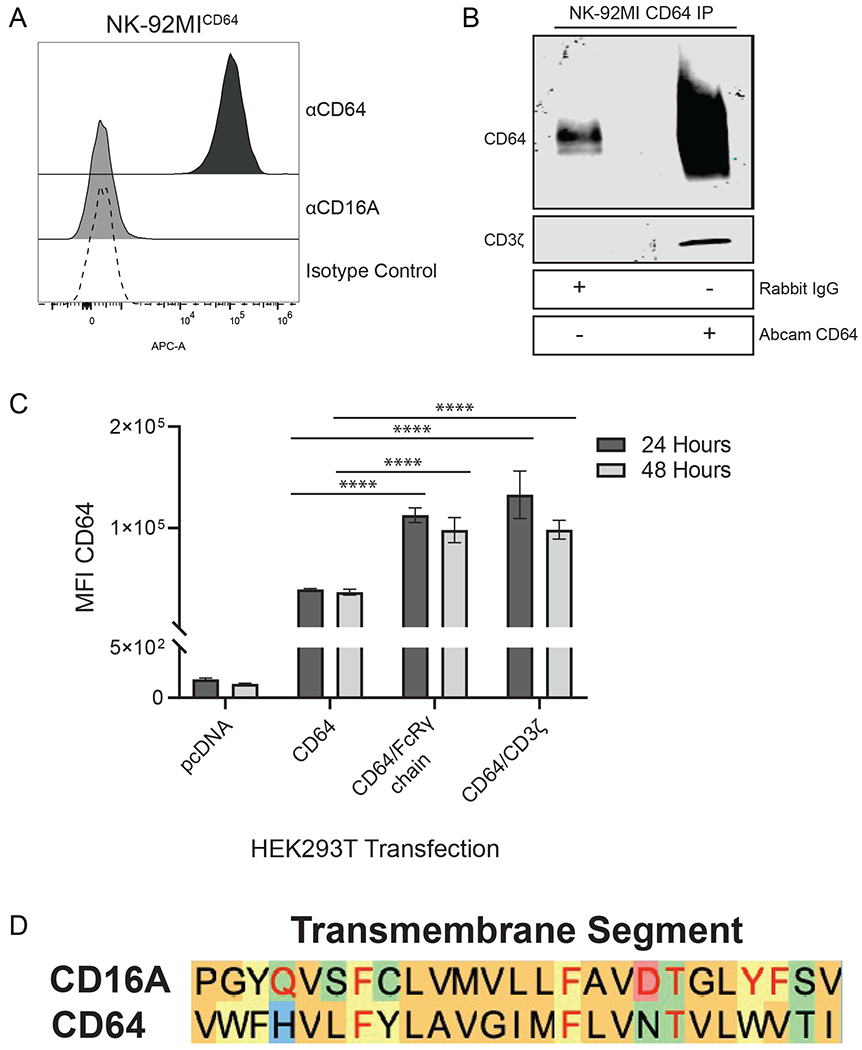
A. CD64-transduced NK-92MI cells were stained with anti-CD64, anti-CD16, or isotype matched control antibodies and analyzed via flow cytometry. Histogram is a representative of data from 3 independent experiments. B. Lysates from NK-92MICD64 cells were immunoprecipitated with either a rabbit isotype matched control antibody or anti-CD64 and immunoblotted for CD64 and CD3ζ. Blot is a representative of 3 independent experiments. C. HEK293T cells were transiently transfected with pcDNA3.1 vector, CD64 expression construct, CD64 plus FcRγ expression constructs, or CD64 plus CD3ζ expression constructs. Cell surface expression of CD64 was determined by flow cytometry. Data are represented as mean ± SEM; n = 3 independent experiments; ****, P ≤ 0.0001 by ANOVA and Tukey’s post hoc for multiple comparisons. D. Amino acid alignment of the predicted human CD16A and human CD64 transmembrane regions. The amino acid sequences of the Fc receptor transmembrane regions are based on a previous study [50]. The color of the shaded letters represents groupings based on side chain properties. The red letters in the CD16A sequence represent amino acids critical for CD3ζ association [51]. The red letters in the CD64 sequence represent identical amino acids.
It is well established that CD16A expression on the cell surface requires the co-expression of either FcRγ or CD3ζ [21, 30, 31]. In contrast, CD64 surface expression is not dependent on FcRγ, but its level of expression at the cell surface can be increased with co-expression of FcRγ [23]. We examined the effects of FcRγ and CD3ζ co-expression on the cell surface levels of CD64. This was performed in HEK-293T cells, which lack endogenous CD64, FcRγ, and CD3ζ. HEK-293T cells transfected with CD64 cDNA alone expressed low levels of CD64, whereas its expression was markedly increased with the co-transfection of either FcRγ or CD3ζ (Fig. 1C). Taken together, we demonstrate that CD64 can be expressed in NK cells and that it can associate with the signaling adaptor CD3ζ when available. Moreover, alignment of the amino acid sequences of the transmembrane regions of CD16A and CD64 revealed a high degree of homology and the conservation of several residues that are critical for CD16A association with CD3ζ (Fig. 1D).
To assess whether CD3ζ affects CD64 function, we used CRISPR/Cas9 genome editing to knockout CD3ζ from NK-92MI cells. NK-92MI cells were transfected with a single CD3ζ CRISPR guide RNA plus Cas9 RNP or Cas9 alone as a control. The genotype of CRISPR edited NK-92MI cells was determined via Sanger sequencing. Short electropherograms of the sequence area near the sgRNA binding site were used to determine the number of indels at the site of excision and subsequently scored for knockout using ICE analysis (Fig. 2A). This revealed that 97% of the sequences in the CRISPR/cas9 sgRNA transfected sequences contained a frameshift mutation. CD3ζ knockout in NK-92MI cells was confirmed by Western blotting and flow cytometry, which both demonstrated a dramatic reduction in CD3ζ expression (Fig. 2B and 2C).
Figure 2. Generation of a CD3ζ KO NK-92MI cell line.
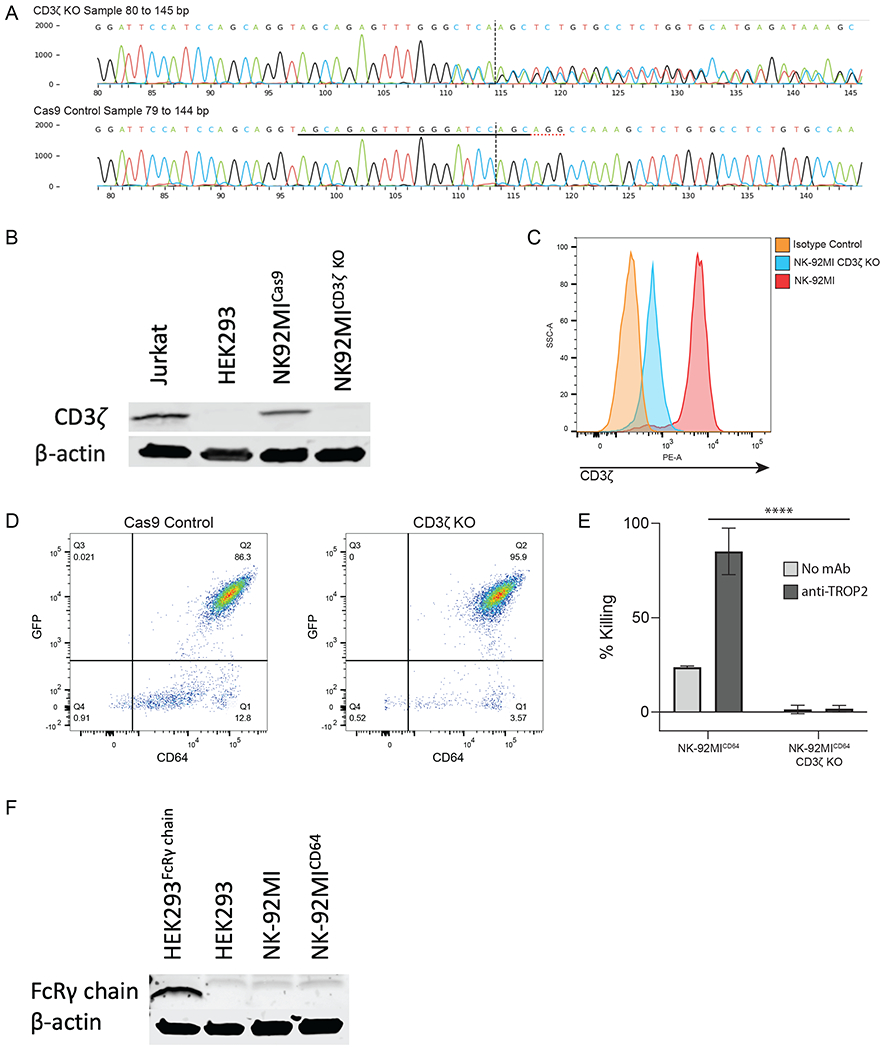
A. Sequencing electropherogram of NK-92MICD3ζ KO cells. The black underline indicates the sgRNA sequence, the red dotted underline indicates the protospacer adjacent motif sequence, and the vertical dotted line shows the cut site of Cas9. B. Western blot analysis of CD3ζ chain in NK-92MI control cells or NK-92MICD3ζ KO cells. Lysates from Jurkat and HEK293 cells were used as a positive and negative control, respectively. Equal protein loading was confirmed via β-actin. Blot is representative of 3 independent experiments. C. Flow cytometric analysis of CD3ζ chain expression in NK-92MI or NK-92MICD3ζ KO cells, n = 1. D. Flow cytometric analyses of transduced NK-92MI control cells or NK-92MICD3ζ KO cells showing equivalent expression of CD64 and GFP, n = 1. E. NK-92MICD64 or NK-92MICD3ζ KO cells were combined with DU145 target cells for ADCC. (Data are represented as mean ± SEM; n = 3 independent experiments; ****, P ≤ 0.0001 by Student’s t-test). F. Western blot analysis of FcRγ chain in NK-92MI and NK-92MICD64 cells. Lysates from HEK293 cells expressing FcRγ chain and HEK293 cells were used as a positive and negative control, respectively. Equal protein loading was confirmed via β-actin. Blot is representative of 3 independent experiments.
Next, we evaluated the cytotoxic capacity of NK-92MICD64 cells lacking CD3ζ using a conventional ADCC assay. For these studies, CD3ζ knockout and Cas9-transfected control NK-92MI cells were transduced with equal amounts of viral supernatant to express CD64 and sorted to match cell surface expression levels of CD64 (Fig. 2D). A bicistronic retroviral vector was used for transduction that expresses GFP in a proportional manner, and its levels were equivalent for the NK-92MICD64/CD3ζ KO and NK-92MICD64 transductants (Fig. 2D). Each transductant was then incubated with the TROP2-positive prostate cancer cell line DU145 in the presence or absence of an anti-TROP2 mAb. As shown in Fig. 2E, high levels of DU145 cell cytotoxicity occurred by NK-92MICD64 cells in the presence but not absence of anti-TROP2. However, NK-92MICD64/CD3ζ KO cells demonstrated highly diminished levels of cytotoxicity in the presence of anti-TROP2 (Fig. 2E), indicating that CD3ζ had a key role in CD64 induction of ADCC. These findings show that CD64 in NK-92MI cells can utilize CD3ζ as a signaling adaptor. It was interesting to us that ADCC by NK-92MICD64/CD3ζ KO cells was so highly ablated, as we presumed that FcRγ in these cells would mediate signaling as well. However, we were unable to detect FcRγ expression by Western blotting, suggesting that its levels are very low in NK-92MI cells (Fig. 2F).
ADCC efficiency of NK-92MICD64 cells against prostate cancer and stromal cells
Having elucidated the functional activity of CD64 in NK-92MI cells and its activation mechanism, we evaluated the potential of the engineered cell therapy and ADCC-inducing mAbs as a strategy for treating mCRPC. The effector function of NK-92MICD64 cells was first tested against DU145, a human prostate cancer cell line that expresses TROP2, and hPrCSC-44, a human prostate cancer–associated fibroblast cell line that expresses FAP. Previously developed therapeutic mAbs against TROP2 [26] and FAP [32, 33] were used in this study. The FAP-specific mAb is known as B12. The anti-TROP2 and B12 were shown to induce ADCC by the NK-92MICD64 cells. Killing levels were measured using the Delfia EuTDA cell cytotoxicity assay with data represented as percent specific release (or % killing). A concentration-dependent effect of anti-TROP2 and B12 on ADCC was observed, with highest concentration the most potent (Fig. 3A). A cytotoxicity assay with different effector-target (E:T) ratios at the highest concentration of mAbs was performed (Fig. 3B). NK-92MICD64 cells had significantly higher DU145-cell killing efficiencies in the presence of anti-TROP2 than in its absence at different E:T ratios (83.28% versus 8.67% at 20:1; 66.6% versus 3.88% at 10:1; and 44.83% versus 7.37% at 5:1 E:T). Similarly, NK-92MICD64 cells demonstrated significant hPrCSC-44-cell killing efficiencies in the presence of B12 mAb compared with its absence at different E:T ratios (77.6% versus 8.44% at 20:1; and 52.4% versus 9.35% at 10:1). Next, as a negative control, cytotoxicity by NK-92MICD64 cells was measured using the highest concentration of anti-TROP2 in the presence of hPrCSC-44 cells, which do not express TROP2, or B12 mAb in the presence of DU145 cells, which do not express FAP (Fig. 3C). No ADCC was observed under these conditions. Finally, IFNγ production was measured by flow cytometry to evaluate the function of NK-92MICD64 cells after incubation with or without anti-TROP2 or B12 mAb (Fig. 3D). IFNγ levels increased by 20-fold in the presence of target cells and anti-TROP2 and by 4-fold in the presence of B12 mAb, respectively. These data suggest that NK-92MICD64-cell effector functions against cancer and associated stromal cells can be enhanced in the presence of anti-TROP2 or B12 mAb in vitro.
Figure 3. NK-92MICD64 cells are functional against prostate tumor cell lines.
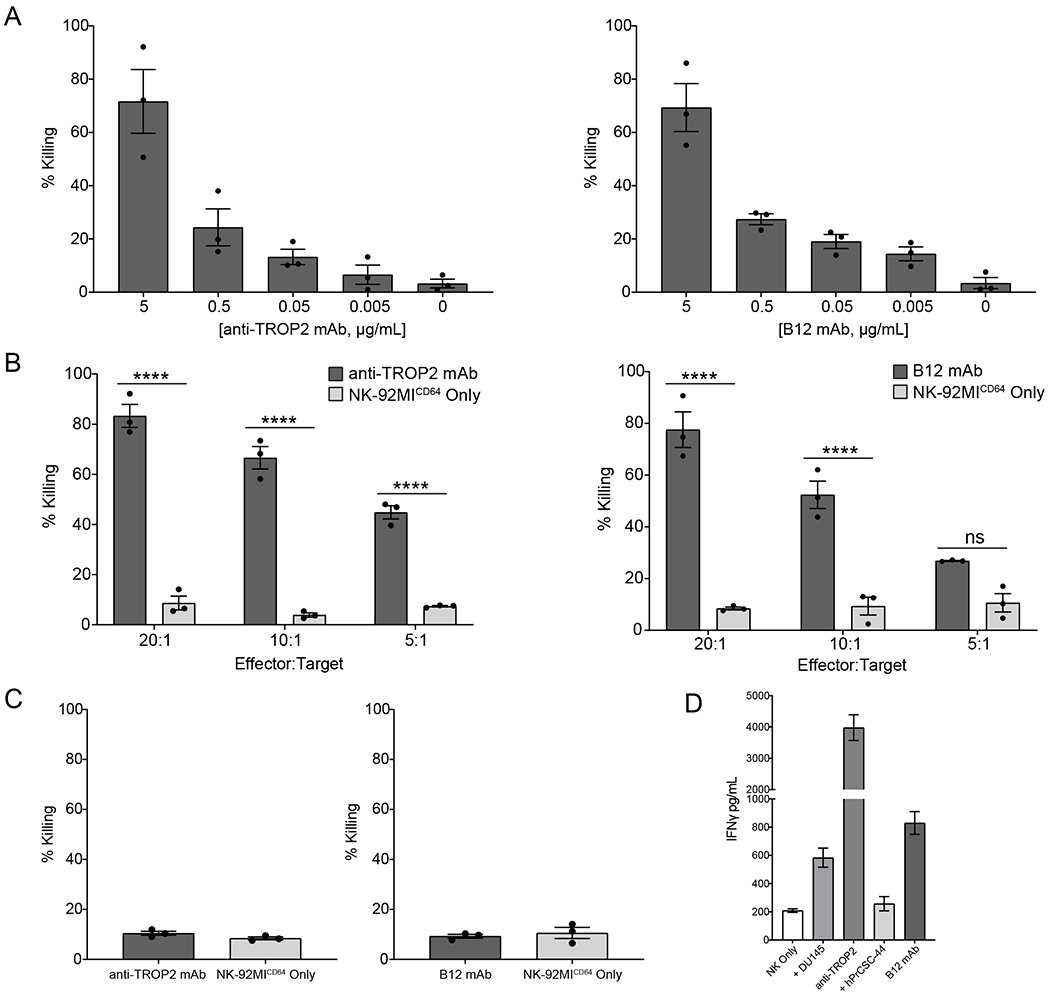
A. NK-92MICD64 cells were combined with DU145 or hPrCSC-44 target cells at an E:T ratio of 20:1 (1.6x105: 8x103 cells/well). Anti-TROP2 and B12 mAb were titrated in to the DU145 and hPrCSC-44 target cell cultures, respectively, and incubated for 2 hours. Data are represented as mean ± SEM of n=3 technical replicates; n = 3 independent experiments. B. NK-92MICD64 cells were combined with DU145 or hPrCSC-44 cells at the indicated E:T ratios (1 = 8x103 cells/well) with anti-TROP2 or B12 mAb (5 μg/mL), respectively, and incubated for 2 hours. Data are represented as mean ± SEM of n=3 technical replicates; n = 3 independent experiments; ****, P ≤ 0.0001 by ANOVA and Tukey’s post hoc for multiple comparisons. C. NK-92MICD64 cells were combined with DU145 or hPrCSC-44 target cells at an E:T ratio of 20:1 (1.6x105: 8x103 cells/well) in the presence or absence of the opposing antibody (5 μg/mL), so B12 mAb and anti-TROP2, respectively, and incubated for 2 hours. This represented a target-null cell assay. There was no difference in cell killing. Data are represented as mean ± SEM of n=3 technical replicates; n = 3 independent experiments. D. NK-92MICD64 cells were incubated alone, or at an E:T ratio of 1:1 (1.5x105 cells) in the presence or absence of anti-TROP2 or B12 mAb (5 μg/mL) for 2 hours. Cell culture supernatant was collected and IFNγ levels were quantified using a bead-based assay. Data are represented as mean ± SEM of n = 3 technical replicates; n = 2 independent experiments.
Soluble therapeutic mAb can be captured by CD64 for target cell killing
Because CD64 binds IgG with high affinity and can stably bind soluble antibody [34], we evaluated the killing capacity of NK-92MICD64 cells. The capture of anti-TROP2 and B12 mAb by CD64 on NK-92MICD64 cells in SFM was detected by flow cytometry using an APC-conjugated secondary mAb specific for human IgG1 (Fig. 4A). More than 95% of the cells incubated with either anti-TROP2 or B12 mAb were positively stained compared with NK-92MICD64 cells stained with secondary mAb alone, indicating that CD64 on NK-92MICD64 can stably bind both mAbs. We next determined the mAb-directed killing effect using the Delfia EuTDA cell cytotoxicity assay (Fig. 4B). NK-92MICD64 cells with bound anti-TROP2 (anti-TROP2-NK-92MICD64) or B12 mAb (B12-NK-92MICD64) were incubated with their respective target cells at the indicated E:T ratios. Compared with the NK-92MICD64 cells alone, the anti-TROP2-NK-92MICD64 cells had significantly higher cytotoxicity at different E:T ratios (86.94% versus 8.67 at 20:1; and 64.63% versus 3.88% at 10:1). B12-NK-92MICD64 cell cytotoxicity was also significantly higher than the NK-92MICD64 cells alone (67.53% versus 8.44% at 20:1; and 36.97% versus 9.35% at 10:1). NK-92MICD64 bound with human IgG1 isotype control had basal levels of cytotoxicity that were not significantly different from the untreated NK-92MICD64 cells for both cell lines (Fig. 4C). In conclusion, anti-TROP2 or B12 mAb stably bound to CD64 on cells can effectively direct the NK-92MICD64 cells to prostate cancer and stromal target cells, resulting in antigen-selective cytotoxicity.
Figure 4. CD64 can capture soluble therapeutic mAb to induce cell killing.
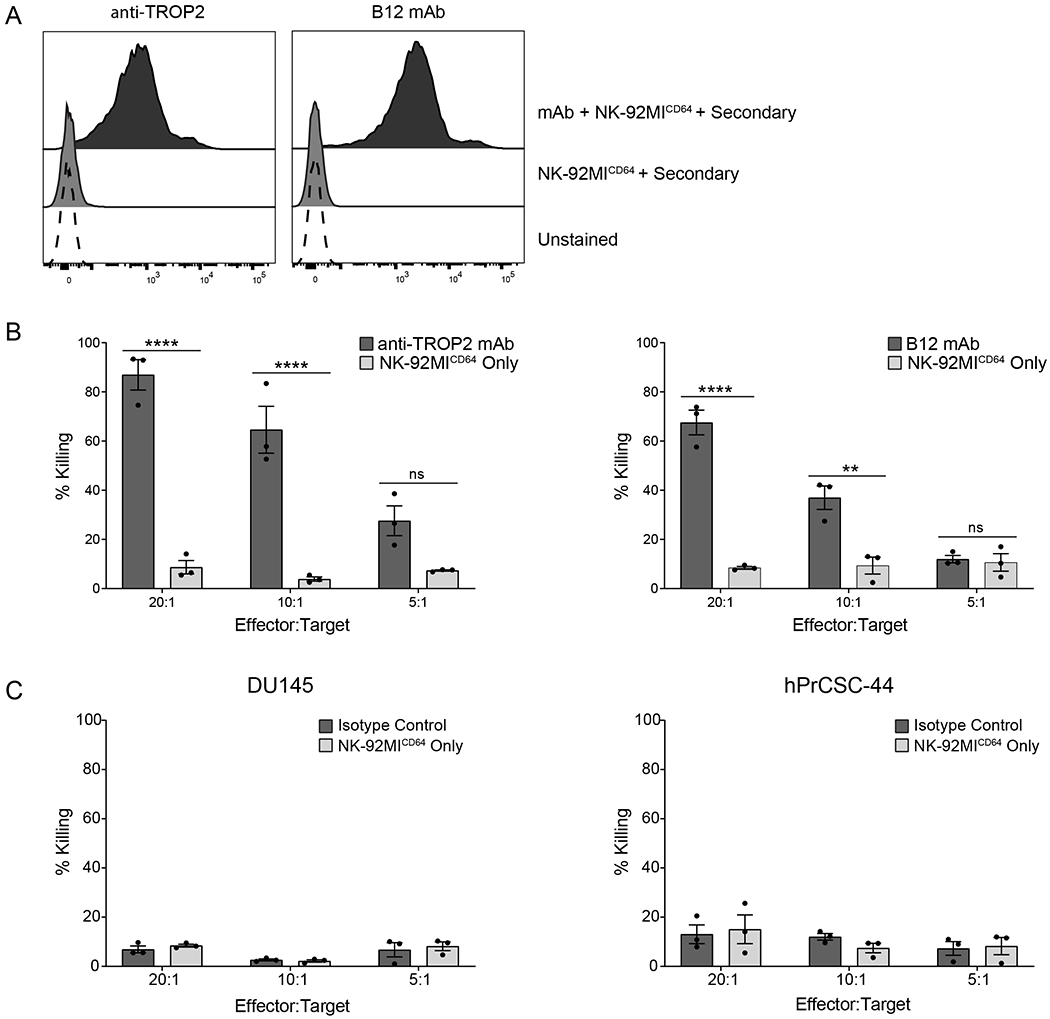
A. NK-92MICD64 cells preincubated with or without anti-TROP2 or B12 mAb (5 μg/mL) in SFM were stained with an APC-conjugated anti-human IgG secondary fab and analyzed by flow cytometry. Over 95% of NK-92MICD64 cells incubated with therapeutic mAb were positively stained compared with unstained NK-92MICD64 cells. Histogram is a representative of data from 2 independent experiments. B. mAb-directed killing effect was measured using the Delfia EuTDA cell cytotoxicity assay. Anti-TROP2-NK-92MICD64 cell killing of DU145 cells was significantly higher than NK-92MICD64 cells alone at the indicated E:T ratios (1 = 8x103 cells/well) after 2 hours incubation. The experiment was repeated with B12-NK-92MICD64 cells cultured with hPrCSC-44 target cell and yielded similar results. Data are represented as mean ± SEM of n=3 technical replicates; n = 3 independent experiments; **, P ≤ 0.01; ****, P ≤ 0.0001 by ANOVA and Tukey’s post hoc for multiple comparisons. C. The experiment was repeated but NK-92MICD64 cells were preincubated with or without an isotype control mAb. Effector cells were combined with DU145 or hPrCSC-44 target cells at the indicated E:T ratios (1 = 8x103 cells/well) and incubated for 2 hours. There was no difference in cell killing between the groups. Data are represented as mean ± SEM of n=3 technical replicates; n = 3 independent experiments.
Combination of two mAbs with NK-92MICD64 cell therapy approach enhances ADCC
With the demonstrated success of the conventional ADCC and bound mAb NK-92MICD64 cell monotherapies, we evaluated the efficacy of anti-TROP2 and B12 mAb as a mixable therapeutic approach. Previous studies have shown a therapeutic effect from combination therapies that target both the tumor stroma and malignant cells compared with either monotherapy alone [12, 35, 36]. To fabricate the prostate TME in an in vitro model, we combined DU145 and hPrCSC-44 target cells at a 1:1 ratio. First, the anti-TROP2 and B12 mAb combination was tested for conventional ADCC induction with the NK-92MICD64 cells (Fig. 5A). We measured potency of target cell killing following co-culture with the NK-92MICD64 cells in the presence of both mAbs compared with NK-92MICD64 cells alone at each E:T ratio. The combination therapy demonstrated enhanced cytotoxicity and % killing in the presence of both mAbs was significantly higher compared to co-culture with NK-92MICD64 cells alone at different E:T ratios (74.83% versus 10.23% at 20:1; 59.3% versus 6.53% at 10:1; and 38.5% versus 4.03% at 5:1). Next, we tested the bound mAb combination therapy (Fig. 5B). The anti-TROP2-NK-92MICD64 and B12-NK-92MICD64 cell combination therapy exhibited significantly higher cell killing compared with NK-92MICD64 cells alone (75.36% versus 10.23% at 20:1; and 40.00% versus 6.53% at 10:1). These data suggest that treatment with NK-92MICD64 cells in combination with anti-TROP2 and B12 mAb by the conventional or bound ADCC mechanism could be effective at targeting prostate tumors.
Figure 5. NK-92MICD64 cell therapy in combination with TROP2-and FAP-targeted antibodies demonstrates additive antitumor effect in vitro.
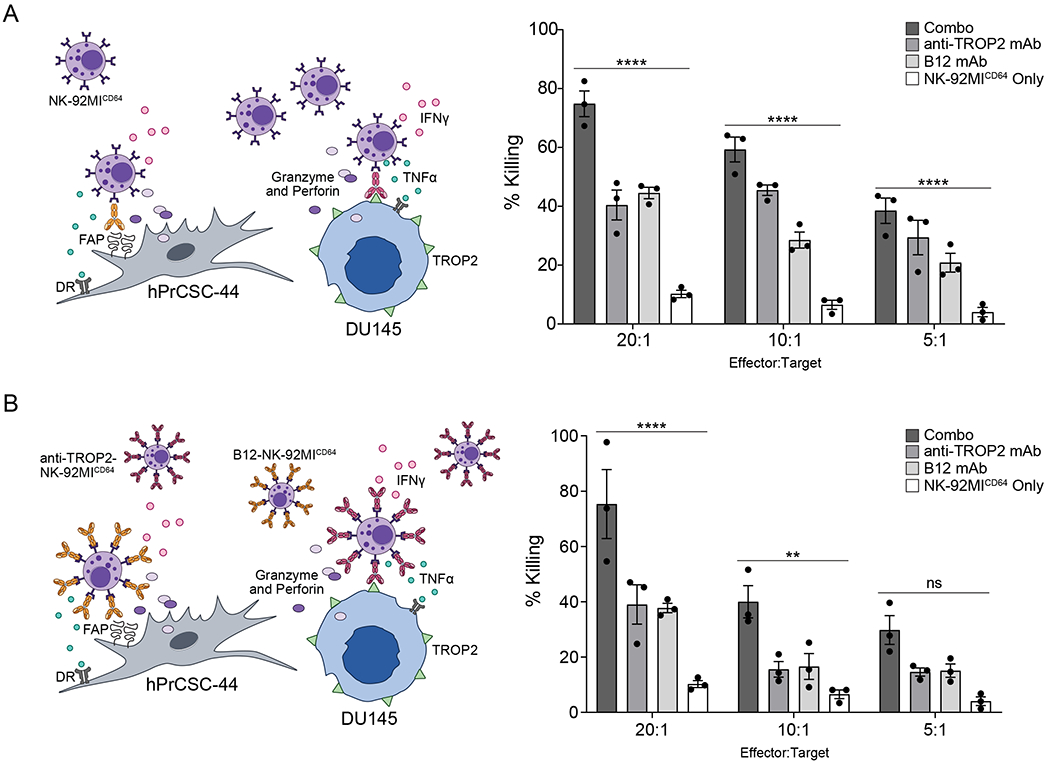
A. anti-TROP2 and B12 mAb combination mediates ADCC. NK-92MICD64 cells were combined with target cells (1:1; hPrCSC-44: DU145; 8x103 cells total/well) at the indicated E:T ratios with or without the antibody combination (2.5 μg/mL each) or with the antibodies individually (2.5 μg/mL) and incubated for 2 hours. Cell killing was significantly higher with both antibodies compared with NK-92MICD64 cells alone. Data are represented as mean ± SEM of n=3 technical replicates; n = 3 independent experiments; ****, P ≤ 0.0001 ANOVA and Tukey’s post hoc for multiple comparisons. B. anti-TROP2-NK-92MICD64 and B12-NK-92MICD64 combination enhances cell killing. NK-92MICD64 cells were preincubated with anti-TROP2 or B12 mAb (5 μg/mL) separately and effector cells (1:1; anti-TROP2: B12 mAb) were combined with target cells (1:1; hPrCSC-44: DU145; 8 x 103 cells total/well) at the indicated E:T ratios and incubated for 2 hours. Cell killing was significantly higher with the combination therapy compared with NK-92MICD64 cells alone. Data are represented as mean ± SEM of n=3 technical replicates; n = 3 independent experiments; **, P ≤ 0.01; ****, P ≤ 0.0001 by ANOVA and Tukey’s post hoc for multiple comparisons.
Adoptive NK-92MICD64 cell transfer with therapeutic mAbs controls tumor growth and prolongs survival
Therapeutic efficacy of the NK-92MICD64 cells in combination with anti-TROP2 and B12 mAb was evaluated in a prostate cancer xenograft mouse model (Fig. 6A). NSG mice bearing subcutaneous DU145 and hPrCSC-44 xenografts were randomly assigned to the treatment arms: saline; adoptive NK-92MICD64 cell transfer; intraperitoneal (i.p.) injection of anti-TROP2 and B12 mAb 24 hours before adoptive NK-92MICD64 cell transfer; and adoptive anti-TROP2-NK-92MICD64 and B12-NK-92MICD64 cell transfer. Treatment was administered peritumorally once per week for four weeks and tumor volumes were measured up to 1500 mm3. Robust antitumor activity in the animals treated with either combination therapy was observed (Fig. 6B). At day 26 post-treatment, mice receiving the ADCC or bound mAb combination therapies had significantly reduced tumor growth by 72.1% (P=0.0013) and 75.5% (P=0.0016) compared with the saline control. Furthermore, treatment with either combination therapy significantly improved overall survival (Fig. 6C). Median survival was 26 days for the saline control versus 40 days for the conventional ADCC combination (P=0.0069) and 48 days for the bound mAb combination (P=0.0069). Tumor-bearing mice treated once in each of the previously mentioned arms had tumors excised after 7 days. IHC staining with anti-human cleaved caspase 3 (cCasp3) was used to measure apoptosis in the tumor tissue (Fig. 6D). Tumors treated with either of the combination therapies had significantly higher mean intensity indicating increased cCasp3 compared to the saline control (Fig. 6E). In conclusion, adoptive NK-92MICD64 cell transfer with antibodies targeting the tumor stroma and malignant cells is effective in a prostate tumor model.
Figure 6. Adoptive NK-92MICD64 cell therapy with combination tumor-targeted antibodies reduces tumor growth and prolongs survival in mice.
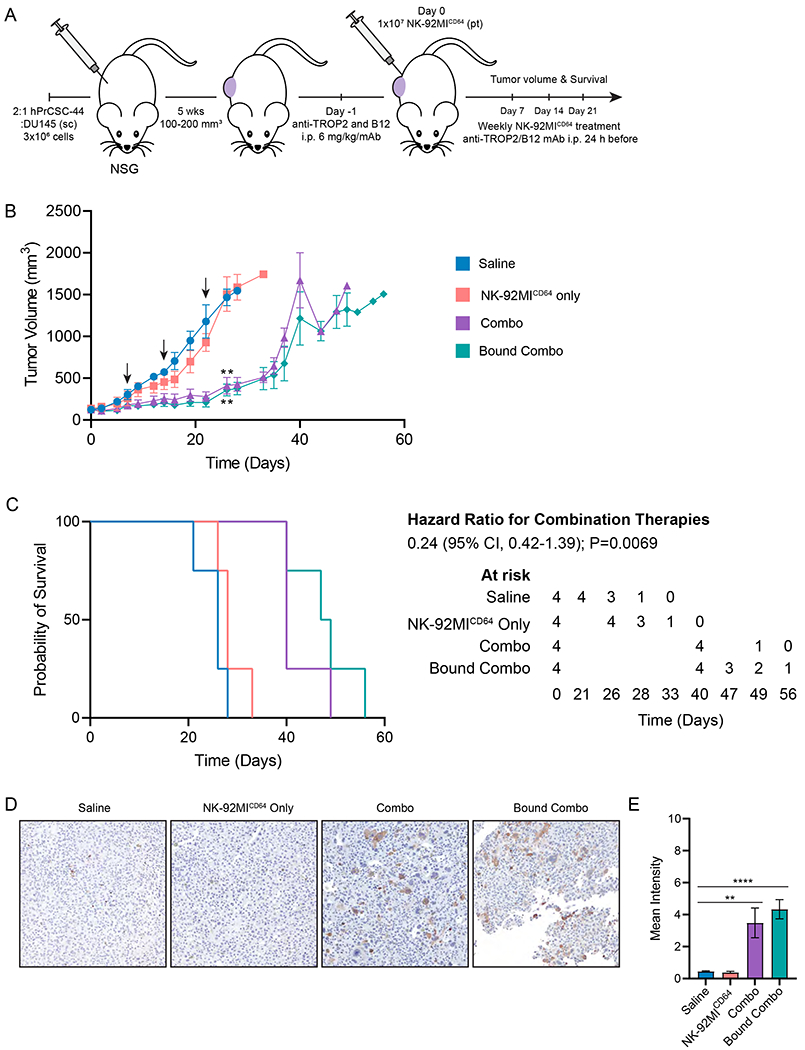
A. Schematic of experimental design. A 2:1 mixture of hPrCSC-44: DU145 cells (3x106) were implanted subcutaneously in the flank of NSG mice. Tumors were allowed to grow until tumor volumes measured 100–200 mm3 and mice (n=4/arm) were randomly assigned to the following treatment arms: Saline; NK-92MICD64 cells alone; i.p. injection of anti-TROP2 and B12 mAb 24 hours before adoptive NK-92MICD64 cell transfer or; adoptive NK-92MICD64 cell transfer with bound anti-TROP2 and B12 mAb. Treatment was administered peritumorally once per week for four weeks. B. Tumor volumes were measured two-to-three times per week until tumors reached 1500 mm3. Conventional ADCC and bound mAb combination therapies demonstrated significant tumor control compared with the saline arm at 26 days. Black arrows signify treatment administration. n=4/group; n=1 independent experiment; **, P ≤ 0.01, ANOVA mixed methods test. C. Kaplan-Meier survival curve for mice treated with NK-92MICD64 cells in combination with therapeutic antibodies compared with control mice. NK-92MICD64 cells in combination with anti-TROP2 and B12 mAb treated mice survived significantly longer than the controls. n=4/group; n=1 independent experiment; ** P=0.0069 by log-rank Mantel-Cox test). D. Ex vivo evaluation of combination adoptive NK-92MICD64 cell therapy. Representative images of IHC staining with a secondary anti-human cleaved caspase 3 (cCasp3). Mice bearing hPrCSC-22:DU145 subcutaneous xenografts (n = 3/arm) were treated with one dose and tumors were excised and fixed 7 days after treatment. Tumor tissue from mice treated with the combination or bound antibody therapy showed stronger staining for cCasp3 compared to the saline control. E. Quantification of IHC staining for cCasp3 in tumor tissue. Data is represented as mean intensity ± SEM of n = 3 images/mouse/group; n=1 independent experiment; **, P≤0.01; ****, P≤0.0001 by ANOVA and Tukey’s post hoc for multiple comparisons.
Discussion
NK cells utilize the low affinity Fc receptor CD16A to kill antibody-opsonized tumor cells via ADCC. In humans, CD16A has two known allelic variants, CD16A-176V and CD16A-176F, with CD16A-176V having a higher affinity for IgG (~2–3 fold). In clinical trials with tumor-targeting mAbs, patients homozygous for CD16A-176V have improved outcomes compared with homozygous CD16A-176F or heterozygous patients [37], suggesting that increasing the binding affinity of CD16A for IgG mAbs may improve the efficacy of antibody therapy. One common approach to achieve higher affinity interactions is to modify the Fc region of the therapeutic mAb; however, this requires modification and optimization of each individual antibody. Another approach that has been explored is to increase the Ab binding affinity of CD16A. We have previously generated a recombinant FcγR consisting of the extracellular region of CD64 and the transmembrane and cytoplasmic regions of CD16A to retain its signaling process for NK-cell activation [20]. Here, we show that intact CD64 can be expressed in an NK cell line and induce potent ADCC. We further demonstrate that like CD16A, CD64 can utilize the CD3ζ signaling adaptor for ADCC. It is thus conceivable that CD64 could be expressed in T cells as well to induce ADCC, which has been done for CD16A [38]. Of note, the alpha chains of CD16A and CD64 differ and mediate distinct signaling mechanisms [39, 40], and therefore it will be interesting to determine in future studies how these regions may differentially signal in NK cells.
Our combination approach introduces what we believe to be a new strategy for treating mCRPC, which, so far, has been largely resistant to immunotherapy. The engineered NK-92MICD64 cells showed enhanced effector function against prostate cancer and stromal cell lines in the presence of TROP2-and FAP-targeted antibodies, suggesting the therapeutic potential of NK cell therapies for prostate cancer. Dual targeting of stromal and cancer cells by the combination approach demonstrated potent cytotoxicity in vitro compared with NK cells alone when tested in our prostate tumor stroma model. Using the bound mAb method, CD64 captured the soluble therapeutic mAbs and effectively directed cell killing. Finally, treatment with the engineered NK-92MICD64 cells in combination with the therapeutic mAbs led to tumor control and prolonged survival of mice in a subcutaneous xenograft model, further indicating the clinical potential of the combination approach.
A patient’s response to immunotherapy depends on a number of factors including tumor heterogeneity, previous treatment history, T-cell infiltration, mutation burden, TME-driven immunosuppression, tissue hypoxia, and density of the stromal network [1]. Although prostate cancer has a low tumor mutation burden and is considered to be immunologically “cold” [41, 42], several preclinical studies show that pharmacological targeting of the TME improves immunotherapy efficacy, suggesting that prostate cancer could be sensitized to immunotherapy [43–45]. Targeting both the tumor stroma and prostate cancer cells could be a key strategy to overcome the limitations of current therapeutics given the central role of the TME in survival and progression of the disease. Recent studies demonstrating the high genomic heterogeneity found in prostate cancer patients indicate the importance of identifying patient-specific molecular signatures to guide immunotherapy strategies [46, 47]. The localization and upregulated expression of stromal biomarkers and tumor antigens, including FAP and TROP2, facilitate the development of targeted therapies. FAP is highly expressed by cancer-associated fibroblasts in mCRPC across genomic subtypes and metastatic lesion sites [27] and TROP2 is overexpressed in several epithelial tumors including mCRPC [48].
Several preclinical studies have reported a therapeutic effect from targeting FAP and various tumor antigens using combinations of vaccines, antibody-drug conjugates, and chemotherapies [11, 36, 49]. Few studies, however, have used a combination of antigen-targeting cell therapies. Adoptive transfer of a combination of FAP-specific and EphA2-specific CAR T cells enhances antitumor activity and prolongs survival in a lung cancer mouse model [12]. In addition, a phase I clinical trial for Nectin4-and FAP-targeted CAR T cells in solid tumors is currently ongoing (ClinicalTrials.gov Identifier: NCT03932565). Our approach is the first to our knowledge that uses engineered NK cells and a combination of antibodies to simultaneously target solid tumor cells and stromal cells. With this strategy, patients can be pre-treated with therapeutic antibodies before administration of the engineered cells. Additionally, CD64 can also be used as a docking platform where therapeutic antibodies are bound before treatment to direct NK cells to varied tumor antigens. Antibody attachment to CD64 thus has the advantage of dynamic tumor antigen targeting, whereas the FDA-approved CARs are restricted to a specific tumor antigen.
The NK-92 cell platform is a well-characterized human NK cell line that has received US FDA approval for testing in cancer patients. Genetically modified NK-92 cells expressing the CD16A-176V variant (referred to as haNK) are currently being investigated in clinical trials (ClinicalTrials.gov Identifier: NCT03387085, NCT03387111, NCT03586869). Advantages of NK-92 cells include unlimited clonal expansion for achieving clinical doses. However, it will be interesting to engineer other NK-cell platforms with CD64, including peripheral blood NK cells, cord blood NK cells, or induced pluripotent stem cell (iPSC)-derived NK cells that do not require irradiation prior to infusion.
Synopsis:
The data in this study suggest targeting the tumor stroma and malignant cells with engineered natural killer cells and therapeutic antibodies could overcome immunotherapy resistance in prostate cancer and result in a next-generation targeted therapy approach.
Acknowledgements
Financial Support:
This work was supported by NIH/NCI T32CA009138 (to H.M. Hintz), DOD Idea Award W81XWH-19-1-0243 (to A.M. LeBeau), a 2018 Prostate Cancer Foundation Challenge Award (to A.M. LeBeau), a 2013 Prostate Cancer Foundation Young Investigator Award (to A.M. LeBeau), NIH R01s CA237272, CA233562, and CA245922 (to A.M. LeBeau) NIH R01CA203348 (to B. Walcheck), NIH R21AI149395 (to J. Wu), Howard Hughes Medical Institute and Burroughs Wellcome Fund Medical Research Fellowship (to K.M. Snyder), and University of Minnesota Medical School intramural grant for studying CRISPR in NK cells (to G.D. Hart). The authors would also like to thank Colleen Forster and the Biorepository and Laboratory Services Division at the University of Minnesota (Minneapolis, MN) for their histology and pathology support.
Footnotes
Conflict of Interest Statement: A.M.L. and H.M.H are listed as inventors on the patent application, 20200246383 (Compounds binding to fibroblast activation protein alpha and methods of making and using). Applicant: University of Minnesota. Monoclonal antibody targeting FAP. J.W. and B.W. are inventors on the patent application, WO2019084388A1 (Recombinant immune cells, methods of making, and methods of use). Human CD64 described in the patent application has been exclusively licensed. All other authors declare no conflicts.
References
- 1.Cha HR, Lee JH, and Ponnazhagan S. Revisiting immunotherapy: A focus on prostate cancer. Cancer Res 2020;80:1615–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-t immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363:411–22. [DOI] [PubMed] [Google Scholar]
- 3.Venturini NJ and Drake CG. Immunotherapy for prostate cancer. Cold Spring Harb Perspect Med 2019;9:a030627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, et al. Randomized, double-blind, phase iii trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol 2017;35:40–47. [DOI] [PubMed] [Google Scholar]
- 5.Powles T, Fizazi K, Gillessen S, Drake CG, Rathkopf DE, Narayanan S, et al. A phase iii trial comparing atezolizumab with enzalutamide vs enzalutamide alone in patients with metastatic castration-resistant prostate cancer (mcrpc). J Clin Oncol 2017;35:TPS5090–TPS90. [Google Scholar]
- 6.Valkenburg KC, De Groot AE, and Pienta KJ. Targeting the tumour stroma to improve cancer therapy. Nat Rev Clin Oncol 2018;15:366–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen X and Song E. Turning foes to friends: Targeting cancer-associated fibroblasts. Nature Reviews Drug Discovery 2019;18:99–115. [DOI] [PubMed] [Google Scholar]
- 8.Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol 2017;216:3799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ziani L, Chouaib S, and Thiery J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front Immunol 2018;9:414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brennen WN, Isaacs JT, and Denmeade SR. Rationale behind targeting fibroblast activation protein-expressing carcinoma-associated fibroblasts as a novel chemotherapeutic strategy. Mol Cancer Ther 2012;11:257–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fabre M, Ferrer C, Domínguez-Hormaetxe S, Bockorny B, Murias L, Seifert O, et al. Omtx705, a novel fap-targeting adc demonstrates activity in chemotherapy and pembrolizumab-resistant solid tumor models. Clin Cancer Res 2020;26:3420–30. [DOI] [PubMed] [Google Scholar]
- 12.Kakarla S, Chow KK, Mata M, Shaffer DR, Song X-T, Wu M-F, et al. Antitumor effects of chimeric receptor engineered human t cells directed to tumor stroma. Mol Ther 2013;21:1611–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abel AM, Yang C, Thakar MS, and Malarkannan S. Natural killer cells: Development, maturation, and clinical utilization. Front Immunol 2018;9: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasero C, Gravis G, Granjeaud S, Guerin M, Thomassin-Piana J, Rocchi P, et al. Highly effective nk cells are associated with good prognosis in patients with metastatic prostate cancer. Oncotarget 2015;6:14360–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zahavi D and Weiner L. Monoclonal antibodies in cancer therapy. Antibodies (Basel) 2020;9: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ochoa MC, Minute L, Rodriguez I, Garasa S, Perez-Ruiz E, Inoges S, et al. Antibody-dependent cell cytotoxicity: Immunotherapy strategies enhancing effector nk cells. Immunol Cell Biol 2017;95:347–55. [DOI] [PubMed] [Google Scholar]
- 17.Wu J, Mishra HK, and Walcheck B. Role of adam17 as a regulatory checkpoint of cd16a in nk cells and as a potential target for cancer immunotherapy. J Leukoc Biol 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human fcgamma receptors and their polymorphic variants for human igg subclasses. Blood 2009;113:3716–25. [DOI] [PubMed] [Google Scholar]
- 19.Bruhns P Properties of mouse and human igg receptors and their contribution to disease models. Blood 2012;119:5640–49. [DOI] [PubMed] [Google Scholar]
- 20.Snyder KM, Hullsiek R, Mishra HK, Mendez DC, Li Y, Rogich A, et al. Expression of a recombinant high affinity igg fc receptor by engineered nk cells as a docking platform for therapeutic mabs to target cancer cells. Front Immunol 2018;9:2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lanier LL, Yu G, and Phillips JH. Co-association of cd3 zeta with a receptor (cd16) for igg fc on human natural killer cells. Nature 1989;342:803–5. [DOI] [PubMed] [Google Scholar]
- 22.Letourneur O, Kennedy IC, Brini AT, Ortaldo JR, O’shea JJ, and Kinet JP. Characterization of the family of dimers associated with fc receptors (fc epsilon ri and fc gamma riii). J Immunol 1991;147:2652–6. [PubMed] [Google Scholar]
- 23.Van Vugt MJ, Heijnen IA, Capel PJ, Park SY, Ra C, Saito T, et al. Fcr gamma-chain is essential for both surface expression and function of human fc gamma ri (cd64) in vivo. Blood 1996;87:3593–9. [PubMed] [Google Scholar]
- 24.Chen Y, You F, Jiang L, Li J, Zhu X, Bao Y, et al. Gene-modified nk-92mi cells expressing a chimeric cd16-bb-zeta or cd64-bb-zeta receptor exhibit enhanced cancer-killing ability in combination with therapeutic antibody. Oncotarget 2017;8:37128–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jing Y, Ni Z, Wu J, Higgins L, Markowski TW, Kaufman DS, et al. Identification of an adam17 cleavage region in human cd16 (fcgammariii) and the engineering of a non-cleavable version of the receptor in nk cells. PLoS One 2015;10:e0121788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Strop P, Tran TT, Dorywalska M, Delaria K, Dushin R, Wong OK, et al. Rn927c, a site-specific trop-2 antibody-drug conjugate (adc) with enhanced stability, is highly efficacious in preclinical solid tumor models. Mol Cancer Ther 2016;15:2698–708. [DOI] [PubMed] [Google Scholar]
- 27.Hintz HM, Gallant JP, Vander Griend DJ, Coleman IM, Nelson PS, and Lebeau AM. Imaging fibroblast activation protein alpha improves diagnosis of metastatic prostate cancer with positron emission tomography. Clin Cancer Res 2020;26:4882–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Binyamin L, Alpaugh RK, Hughes TL, Lutz CT, Campbell KS, and Weiner LM. Blocking nk cell inhibitory self-recognition promotes antibody-dependent cellular cytotoxicity in a model of anti-lymphoma therapy. 2008;180:6392–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ernst LK, Duchemin AM, and Anderson CL. Association of the high-affinity receptor for igg (fc gamma ri) with the gamma subunit of the ige receptor. Proceedings of the National Academy of Sciences 1993;90:6023–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hibbs ML, Selvaraj P, Carpen O, Springer TA, Kuster H, Jouvin MH, et al. Mechanisms for regulating expression of membrane isoforms of fc gamma riii (cd16). Science 1989;246:1608–11. [DOI] [PubMed] [Google Scholar]
- 31.Kurosaki T and Ravetch JV. A single amino acid in the glycosyl phosphatidylinositol attachment domain determines the membrane topology of fc gamma riii. Nature 1989;342:805–7. [DOI] [PubMed] [Google Scholar]
- 32.Hintz HM, Cowan A, Shapovalova M, and Lebeau A. Development of a cross-reactive monoclonal antibody for detecting the tumor stroma. Bioconjug Chem 2019; [DOI] [PubMed] [Google Scholar]
- 33.Hintz HM, Gallant JP, Vander Griend DJ, Coleman IM, Nelson PS, and Lebeau AM. Imaging fibroblast activation protein alpha improves diagnosis of metastatic prostate cancer with positron emission tomography. Clin Cancer Res 2020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nimmerjahn F and Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol 2008;8:34–47. [DOI] [PubMed] [Google Scholar]
- 35.Duperret EK, Trautz A, Ammons D, Perales-Puchalt A, Wise MC, Yan J, et al. Alteration of the tumor stroma using a consensus DNA vaccine targeting fibroblast activation protein (fap) synergizes with antitumor vaccine therapy in mice. Clin Cancer Res 2018;24:1190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loeffler M, Kruger JA, Niethammer AG, and Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest 2006;116:1955–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamura K, Shimizu C, Hojo T, Akashi-Tanaka S, Kinoshita T, Yonemori K, et al. Fcγr2a and 3a polymorphisms predict clinical outcome of trastuzumab in both neoadjuvant and metastatic settings in patients with her2-positive breast cancer. Ann Oncol 2011;22:1302–07. [DOI] [PubMed] [Google Scholar]
- 38.Ollier J, Vivien R, Vié H, and Clémenceau B. Transfection of fcγriiia (cd16) alone can be sufficient to enable human αβtcr t lymphocytes to mediate antibody-dependent cellular cytotoxicity. ImmunoHorizons 2017;1:63–70. [DOI] [PubMed] [Google Scholar]
- 39.Li X, Baskin JG, Mangan EK, Su K, Gibson AW, Ji C, et al. The unique cytoplasmic domain of human fcgammariiia regulates receptor-mediated function. J Immunol 2012;189:4284–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Edberg JC, Qin H, Gibson AW, Yee AM, Redecha PB, Indik ZK, et al. The cy domain of the fcgamma ria alpha-chain (cd64) alters gamma-chain tyrosine-based signaling and phagocytosis. J Biol Chem 2002;277:41287–93. [DOI] [PubMed] [Google Scholar]
- 41.Maleki Vareki S High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. Journal for ImmunoTherapy of Cancer 2018;6: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Castle JC, Uduman M, Pabla S, Stein RB, and Buell JS. Mutation-derived neoantigens for cancer immunotherapy. Front Immunol 2019;10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jayaprakash P, Ai M, Liu A, Budhani P, Bartkowiak T, Sheng J, et al. Targeted hypoxia reduction restores t cell infiltration and sensitizes prostate cancer to immunotherapy. The Journal of Clinical Investigation 2018;128:5137–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou J, Yang T, Liu L, and Lu B. Chemotherapy oxaliplatin sensitizes prostate cancer to immune checkpoint blockade therapies via stimulating tumor immunogenicity. Mol Med Report 2017;16:2868–74. [DOI] [PubMed] [Google Scholar]
- 45.Chiappinelli KB, Zahnow CA, Ahuja N, and Baylin SB. Combining epigenetic and immunotherapy to combat cancer. Cancer Res 2016;76:1683–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lovf M, Zhao S, Axcrona U, Johannessen B, Bakken AC, Carm KT, et al. Multifocal primary prostate cancer exhibits high degree of genomic heterogeneity. Eur Urol 2019;75:498–505. [DOI] [PubMed] [Google Scholar]
- 47.Lu Z, Williamson SR, Carskadon S, Arachchige PD, Dhamdhere G, Schultz DS, et al. Clonal evaluation of early onset prostate cancer by expression profiling of erg, spink1, etv1, and etv4 on whole-mount radical prostatectomy tissue. Prostate 2020;80:38–50. [DOI] [PubMed] [Google Scholar]
- 48.Trerotola M, Jernigan DL, Liu Q, Siddiqui J, Fatatis A, and Languino LR. Trop-2 promotes prostate cancer metastasis by modulating 1 integrin functions. 2013;73:3155–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gottschalk S, Yu F, Ji M, Kakarla S, and Song XT. A vaccine that co-targets tumor cells and cancer associated fibroblasts results in enhanced antitumor activity by inducing antigen spreading. PLoS One 2013;8:e82658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zidovetzki R, Rost B, Armstrong DL, and Pecht I. Transmembrane domains in the functions of fc receptors. Biophys Chem 2003;100:555–75. [DOI] [PubMed] [Google Scholar]
- 51.Blázquez-Moreno A, Park S, Im W, Call MJ, Call ME, and Reyburn HT. Transmembrane features governing fc receptor cd16a assembly with cd16a signaling adaptor molecules. Proceedings of the National Academy of Sciences 2017;114:E5645–E54. [DOI] [PMC free article] [PubMed] [Google Scholar]