

Abstract
Further clinical development of PF74, a lead compound targeting HIV-1 capsid, is impeded by low antiviral activity and inferior metabolic stability. By modifying the benzene (region I) and indole of PF74, we identified two potent compounds (7m and 7u) with significantly improved metabolic stability. Compared to PF74, 7u displayed greater metabolic stability in human liver microsomes (HLMs) with half-life (t1/2) 109-fold that of PF74. Moreover, mechanism of action (MOA) studies demonstrated that 7m and 7u effectively mirrored the MOA of compounds that interact within the PF74 interprotomer pocket, showing direct and robust interactions with recombinant CA, and 7u displaying antiviral effects in both the early and late stages of HIV-1 replication. Furthermore, MD simulation corroborated that 7u was bound to the PF74 binding site, and the results of the online molinspiration software predicted that 7m and 7u had desirable physicochemical properties. Unexpectedly, this series of compounds exhibited better antiviral activity than PF74 against HIV-2, represented by compound 7m whose anti-HIV-2 activity was almost 5 times increased potency over PF74. Therefore, we have rationally redesigned the PF74 chemotype to inhibitors with novel structures and enhanced metabolic stability in this study. We hope that these new compounds can serve as a blueprint for developing a new generation of HIV treatment regimens.
Keywords: HIV-1, Capsid inhibitor, Phenylalanine, Benzothiazole, Metabolic stability
1. Introduction
Acquired immunodeficiency syndrome (AIDS) caused by human immunodeficiency virus (HIV) is a destructive infectious disease that seriously threatens human life [1]. HIV is comprised of two genotypes, namely HIV-1 and HIV-2, in which HIV-1 represents the primary pathogenic pathogen and results in thousands of death every year [2]. HIV-2 is mainly prevalent in West Africa and induces lower pathogenicity than HIV-1 [3]. However, the risk of HIV-2 infection is increasing and cases have been found in the United States, Europe, South Africa, India and China, which should also be paid more attention [4]. Today, combination antiretroviral therapy (cART) significantly improves the longevity and life quality of patients [5], whereas HIV cannot be eliminated from the body due to its latency, and therefore requires lifelong drug treatment [6,7]. The long-term use of antiviral agents against the error prone replication of HIV inevitably leads to complicated drug resistance and various serious side effects [8–10]. Therefore, there is an urgent and challenging need to identify anti-HIV agents with new and as yet unexplored mechanisms of action (MOA) to increase the available treatment options for patients who have developed resistance to other clinically used drugs.
HIV-1 capsid protein (CA) is an essential structural protein that plays critical structural and regulatory functions within the HIV-1 lifecycle. HIV-1 CA’s structural role is to form a protective shell within the infectious virion that encloses the viral genome and key enzymes [11]. CA’s normal assembly and structural stability are critical to the infectivity of the virus, making CA a novel target for anti-HIV-1 therapies with druggable potential [12,13]. The protective shell formed by CA is a fullerene-shaped mature capsid core composed of approximately 250 CA hexamers and exactly 12 CA pentamers [14,15]. The individual building block of these higher-order structures, the CA monomer, is highly helical, consisting of an N terminal domain (NTD, residues 1 to 145) and a C terminal domain (CTD, residues 150 to 231) with a flexible linker in-between [16,17]. In addition to its structural role, HIV-1 CA plays critical regulatory roles in the early stage of HIV-1 replication, including participating in processes such as uncoating, reverse transcription, and integration [18–21]. In the early stage, once mature HIV-1 particle enters the cell by binding to the host cell CD4 receptor, CXCR4 or CCR5 co-receptor, CA will interact with many cellular factors, including cleavage and polyadenylation specific factor 6 (CPSF6), nucleoporin 153 kDa, (NUP153), nucleoporin 358 (NUP358), cyclophilin A (CypA), TRIM5α, MxB, Sec24C and so on [22–29]. These interactions promote the completion of the uncoating process and regulate the critical steps of downstream reverse transcription, pre-integration complex (PIC) entry into the nucleus, and selection of integration site [30,31]. Moreover, during these processes, the CA shell also serves to evade innate immune surveillance. Given these many and essential roles that CA performs, it has been identified as a very attractive drug target [32,33].
To date, several classes of CA inhibitors have been reported [34], according to their binding sites, CA inhibitors can be divided into NTD-targeted, CTD-targeted, and NTD-CTD interface-targeted groups [35–38]. Due to the crucial role of the NTD-CTD interface in CA assembling, perturbation of this interface can affect CA inherent flexibility, rearrange the hydration layer, and subsequently destroy viral particle integrity. PF74 has received significant attention as the first compound to be structurally described in a co-crystal structure with the CA NTD–CTD interface [39]. Still, it failed to enter clinical development due to its insufficient antiviral activity and metabolic instability. However, Gilead Sciences designed the highly potent GS-6207 by multi-round derivatization of PF74 and is now in the clinical phase [40–42] (Fig. 1). However, GS-6207 also has many drawbacks, with the most notable being its complicated synthesis, large molecular weight, poor solubility, and drug resisance [43]. Therefore, the challenge remains to discover new structural types of HIV-1 CA inhibitors that can be seamlessly integrated into current cART regimens.
Fig. 1.
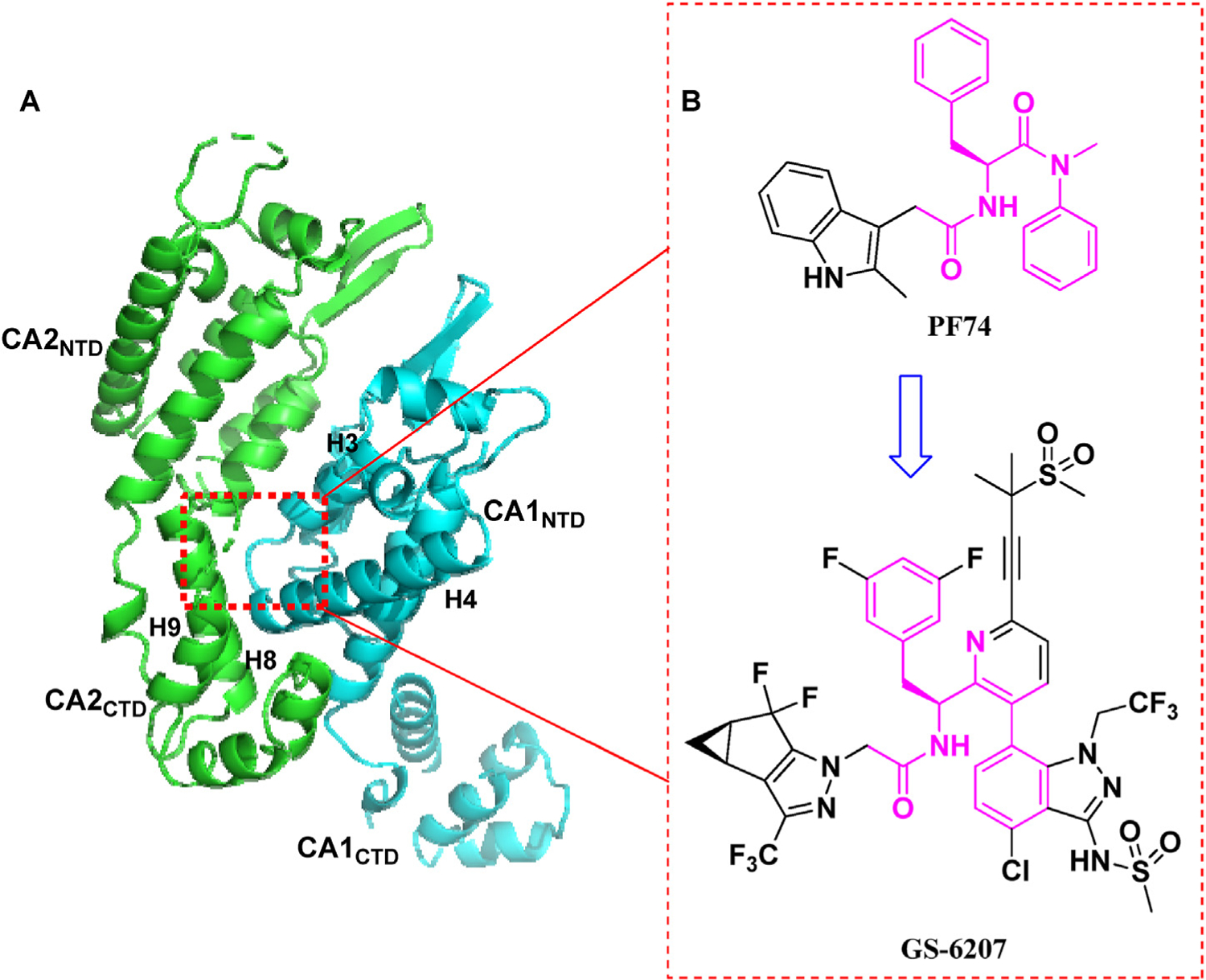
(A) The highly helical structure of CA dimer (PDB code: 5HGL) and a critical ligand-binding pocket (NTD-CTD interface, red dotted line). The pocket formed around H3 and H4 of the CA1NTD (blue), and H8 and H9 of the adjacent CA2CTD (green) accommodates the binding of multiple ligands, including host factors CPSF6 and NUP153, and small molecules PF74 and GS-6207; (B) The chemical structures of PF74 and GS-6207. The figure is generated in PyMOL (www.pymol.org).
To improve the antiviral activity and metabolic stability of PF74, our previous efforts involved replacing the indole moiety with a 1,2,3-triazole ring or benzenesulfonamide moiety and obtained several compounds with enhanced activity but only marginally improved microsomal stability [44–46]. Therefore, although a significant step forward in the redevelopment of HIV-1 CA inhibitors, room for optimization remains. Assessment of the PF74-CA complex structure reveals ample space within the NTD-CTD interfacial pocket that could be exploited in the further modification of PF74. In order to identify novel CA inhibitors with improved drug-like properties and promising activities, our current work focuses on employing a scaffold-hopping strategy to modify the electron-rich benzene ring (region I) and indole ring (region II, Fig. 2) of PF74 [47]. Our design strategy includes the addition of new groups that could make additional contacts within the binding site with key residues, such as Tyr130, Tyr169, Arg173 and Gln176. Furthermore, the susceptibility to oxidative metabolism could be effectively mitigated by replacing the electron-rich groups. Specifically, the electron-deficient benzothiazole moiety is used to replace the benzene ring in region I, while in region II, the phenylamide group serves as a linker, and the aromatic ring bearing different substituents or aliphatic substituents are conjugated. When R is a substituted aromatic ring, we further explored the effects of various substituents on the antiviral activity, including halogen substitution, amino substitution at different positions, 4-CH3, 4-CH3O, 4-CH3CO, 4-CN substitution and so on.
Fig. 2.
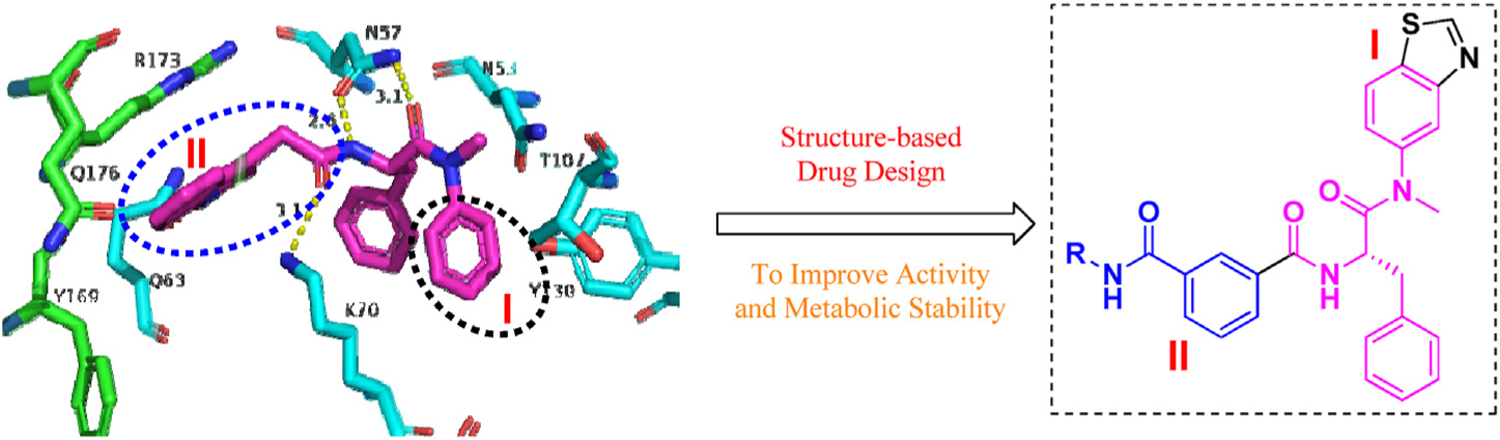
Binding modes of PF74 (purple, PDB code: 5HGL) and design principle of novel HIV-1 CA inhibitors. The figure is generated in PyMOL (www.pymol.org).
Herein we report the design, synthesis and biological evaluation of a series of phenylalanine derivatives with a benzothiazole moiety as HIV-1 CA inhibitors. The target compounds were screened for their antiviral activities in MT-4 cells. Surface plasmon resonance (SPR) direct interaction assays, action stage determination and molecular dynamics (MD) simulation, were performed to provide mechanistic understanding in to the mode of inhibition of the target compounds. Furthermore, the stability assays of representative compounds 7m and 7u were also implemented in the presence of human liver microsomes and human plasma, respectively. Finally, we predicted the physicochemical properties of 7m and 7u using Molinspiration Cheminformatics free web services (https://www.molinspiration.com, Slovensky Grob, Slovakia).
2. Results and discussion
2.1. Chemistry
The target compounds were prepared via a concise synthetic route as outlined in Scheme 1. Commercially available benzo[d] thiazol-5-amine (1) was treated with paraformaldehyde, sodium methoxide and sodium borohydride in methanol to give 2. Then, 2 was treated with N-(tert-butoxycarbonyl)-L-phenylalanine and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluoro phosphate (PyBOP) in N,N-diisopropylethyl-amine (DIEA) and dichloromethane (DCM) to afford 3, followed by the removal of tert-butyloxycarbonyl (Boc) group, afforded the free amine 4. The acylation of 4 with 3-methoxycarbonylbenzoic acid in dichloromethane solution led to the key intermediate 5. 6 was achieved by hydrolysis of 5 using lithium hydroxide. The simultaneous amide condensation reaction of 6 with various substituted aromatic amines or aliphatic amines to get the target compounds 7(a-t). Compounds 7(u-w) were obtained by the removal of Boc group of 7(r-t).
Scheme 1.
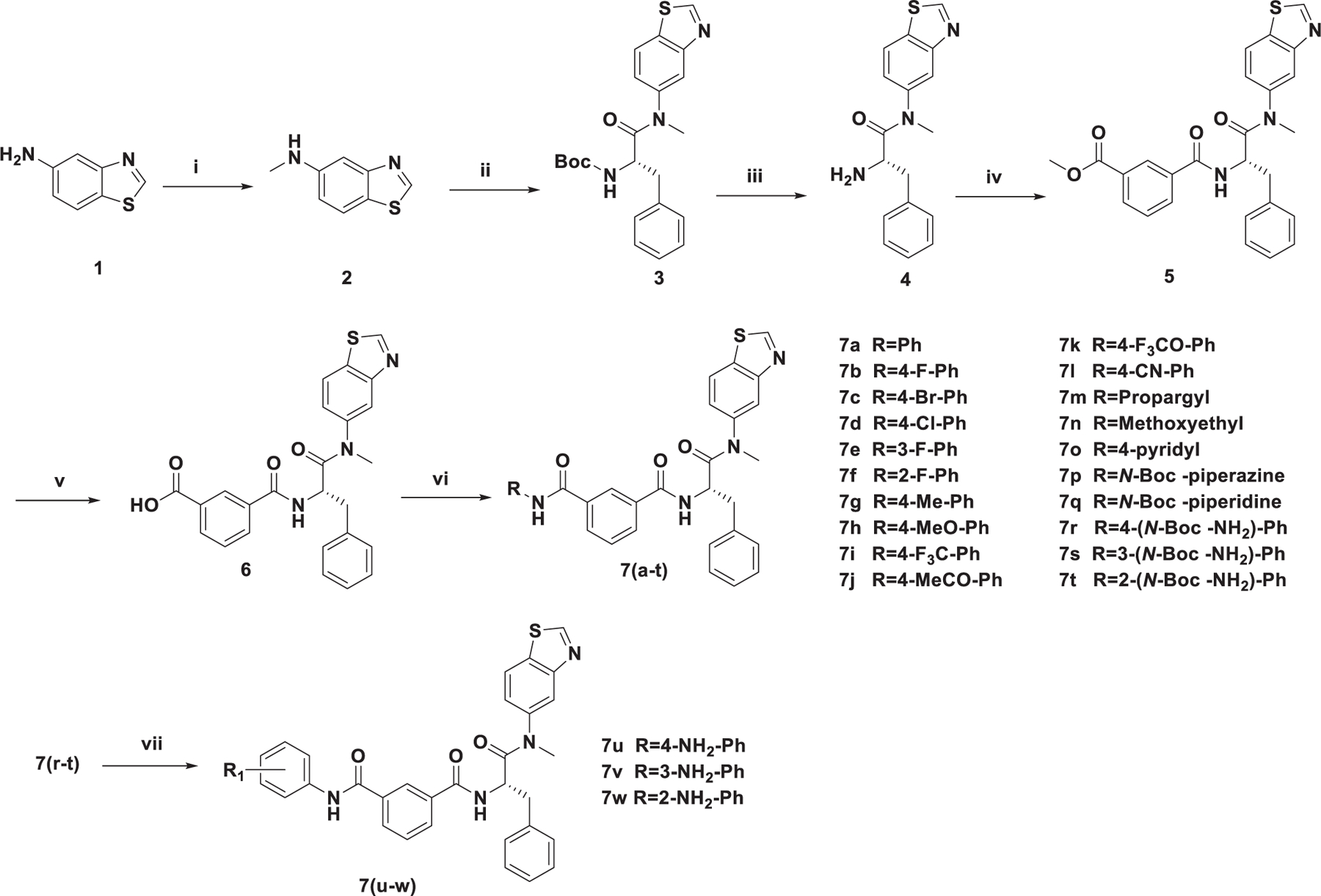
The synthetic route of target compounds. Reagents and conditions: (i) Paraformaldehyde macromolecule, CH3ONa, CH3OH, 60 °C, NaBH4, r.t. Yield: 79%; (ii) N-Boc-L-Phe, PyBOP, DIEA, DCM, 0 °C to r.t. Yield: 76%; (iii) TFA, DCM, r.t. Yield: 79%; (iv) 3-methoxycarbonylbenzoic acid, HATU, DIEA, DCM, 0 °C to r.t. Yield: 61%; (v) LiOH, THF, H2O, r.t. Yield: 77%; (vi) Substituted aromatic amine or substituted aliphatic amine, HATU, DIEA, DCM, 0 °C to r.t. Yield: 65%–82%; (vii) TFA, DCM, r.t. Yield: 76%–77%.
2.2. Antiviral activity against HIV replication in MT-4 cells and structure-activity relationship (SAR) analysis
All the newly synthesized compounds were evaluated for their antiviral activity and cytotoxicity against wild-type (WT) HIV-1 (IIIB) and HIV-2 (ROD) in MT-4 cells. PF74 was included for comparison. The values of EC50 (50% effective concentration), CC50 (50% cytotoxic concentration), and SI (selectivity index, CC50/EC50 ratio) of the target compounds were displayed in Table 1.
Table 1.
Antiviral activity against HIV replication in MT-4 cells.

| |||||||
|---|---|---|---|---|---|---|---|
| Compounds | R | EC50a(μM) |
CC50c (μM) | SId |
|||
| IIIB | ROD | ratiob | IIIB | ROD | |||
| 7a | Ph | 22.37 ± 9.81 | 2.40 ± 0.26 | 9.32 | >234.01 | >10.46 | >97.50 |
| 7b | 4-F-Ph | 31.80 ± 6.34 | 3.66 ± 2.99 | 8.69 | 54.01 ± 2.48 | 1.70 | 14.76 |
| 7c | 4-Br-Ph | ≥185.61 | 6.21 ± 0.95 | >29.89 | >204.22 | N.D.e | >32.89 |
| 7d | 4-Cl-Ph | >220.02 | 3.85 ± 1.00 | >57.15 | >220.02 | N.D.e | >57.15 |
| 7e | 3-F-Ph | >226.38 | 2.84 ± 0.20 | >79.71 | >226.39 | N.D.e | >79.71 |
| 7f | 2-F-Ph | 21.88 ± 5.89 | 2.01 ± 0.18 | 10.88 | 66.38 ± 13.31 | 3.03 | 33.02 |
| 7g | 4-CH3-Ph | 41.15 ± 8.83 | 3.30 ± 1.64 | 12.47 | >228.02 | >5.54 | >69.10 |
| 7h | 4-CH3O-Ph | ≥204.92 | 3.72 ± 1.17 | 55.09 | >221.56 | ≤1.08 | >59.56 |
| 7i | 4-F3C-Ph | ≥39.49 | >88.22 | N.D.e | 88.22 ± 73.35 | ≤2.23 | <1 |
| 7j | 4-CH3CO-Ph | >164.95 | 19.18 ± 7.19 | 8.60 | 164.95 ± 26.33 | <1 | 8.60 |
| 7k | 4-F3CO-Ph | 34.89 ± 1.23 | ≥31.08 | ≤1.12 | 118.13 ± 56.20 | 3.39 | ≤3.80 |
| 7l | 4-CN-Ph | 13.20 ± 2.52 | 6.83 ± 1.23 | 1.93 | >223.55 | >16.94 | >32.73 |
| 7m | Propargyl | 5.02 ± 2.02 | 0.85 ± 0.48 | 5.91 | 133.61 ± 9.71 | 26.62 | 157.19 |
| 7n | Methoxyethyl | 15.69 ± 0.50 | 2.42 ± 0.60 | 6.48 | 203.38 ± 27.68 | 12.96 | 84.04 |
| 7o | 4-pyridyl | ≥41.41 | 3.51 ± 0.67 | 11.80 | 37.63 ± 7.19 | <0.91 | 10.72 |
| 7p | N-Boc-piperazine | 13.50 ± 0.29 | 2.68 ± 0.49 | 5.04 | 93.90 ± 19.78 | 6.96 | 35.04 |
| 7q | N-Boc-piperidine | >29.32 | 2.78 ± 0.19 | 10.55 | 29.32 ± 2.88 | <1 | 10.55 |
| 7r | 4-(N-BoceNH2)–Ph | >2.23 | >2.23 | N.D.e | 2.23 ± 1.11 | <1 | <1 |
| 7s | 3-(N-BoceNH2)–Ph | >192.53 | 28.28 ± 19.78 | 6.81 | >192.53 | N.D.e | >6.81 |
| 7t | 2-(N-BoceNH2)–Ph | 138.96 ± 25.43 | 2.46 ± 0.11 | 56.49 | >192.53 | >1.39 | >78.26 |
| 7u | 4-NH2-Ph | 3.57 ± 0.27 | 2.93 ± 0.33 | 1.22 | 57.36 ± 17.88 | 16.07 | 19.58 |
| 7v | 3-NH2-Ph | 23.71 ± 10.83 | 1.55 ± 0.58 | 15.30 | 133.60 ± 59.03 | 5.63 | 86.19 |
| 7w | 2-NH2-Ph | >25.37 | 1.84 ± 0.35 | 13.79 | 25.37 ± 0.76 | <1 | 13.79 |
| PF74 | 0.75 ± 0.33 | 4.16 ± 2.02 | 0.18 | 32.27 ± 2.94 | 43.03 | 7.76 | |
EC50: concentration of compound required to achieve 50% protection of MT-4 cell cultures against HIV-1-induced cytotoxicity, as determined by the MTT method.
EC50(IIIB)/EC50(ROD) ratio.
CC50: concentration required to reduce the viability of mock-infected cell cultures by 50%, as determined by the MTT method.
SI: selectivity index, the ratio of CC50/EC50.
N.D.: not determined.
Anti-HIV activities of the LA-series were depicted in Table 1. The biochemical evaluation of synthesized compounds against HIV-1 allowed a description of the structure-activity relationship (SAR). More than half of the compounds exhibited moderate activity against HIV-1 in MT-4 cells with EC50 values ranging from 3.57 μM to 41.15 μM, in which 7u (EC50 = 3.57 ± 0.27 μM) and 7m (EC50 = 5.02 ± 2.02 μM) were considered to be most potent HIV-1 inhibitors. Derivatives bearing halogenated aniline lost antiviral activity, whereas compounds substituted by amino-substituted aniline (7u, EC50 = 3.57 ± 0.27 μM) and cyano-substituted aniline (7l, EC50 = 13.20 ± 2.52 μM) displayed good antiviral activity. Meanwhile, the same substituent in different positions would also fluctuate with antiviral activity. Generally speaking, there was a rule of 4-substituted > 3-substituted > 2-substituted, such as 7u (4-NH2, EC50 = 3.57 ± 0.27 μM) > 7v (3-NH2, EC50 = 23.71 ± 10.83 μM) > 7w (2-NH2, EC50 > 25.37 μM). Indeed, the replacement of R with aliphatic substitution showed an overall improvement of the inhibition power of this series of compounds as determined by the comparison of the EC50 values of aromatic substituted derivatives.
Interestingly, 16 compounds conferred better anti-HIV-2 potency than PF74 (EC50 = 4.16 ± 2.02 μM). Among the compounds, 7m and 7v were found the most dominant ones of the series against HIV-2, with EC50 values of 0.85 ± 0.48 μM and 1.55 ± 0.58 μM, respectively. Taking the unsubstituted benzene compound 7a (EC50 = 2.40 ± 0.26 μM) as a reference, a general SAR observation was that when R was an aromatic substituent: substitution with 2-F (7f, EC50 = 2.01 ± 0.18 μM), 3-NH2 (7v, EC50 = 1.55 ± 0.58 μM) and 2-NH2 (7w, EC50 = 1.84 ± 0.35 μM) increased the antiviral activity, while other substitutions did not improve the antiviral activity profile. From the para-substituted benzene derivatives, substitution on the benzene ring with 4-NH2 (7u, EC50 = 2.93 ± 0.33 μM), 4-CH3 (7g, EC50 = 3.30 ± 1.64 μM), 4-F (7b, EC50 = 3.66 ± 2.99 μM), 4-CH3O (7h, EC50 = 3.72 ± 1.17 μM), 4-Cl (7d, EC50 = 3.85 ± 1.00 μM) contributed to much more enhanced antiviral activity than PF74. The activity order of para halogen substitutions was F (7b) >Cl (7d) >Br (7c, EC50 = 6.21 ± 0.95 μM), which was positively related to the electronegativity of substituents. Besides, the hydrogen-bond donor (NH2, 7u, 7v, 7w) was beneficial to antiviral activity. The position of substituents also had great influence on the antiviral activity, such as 7v (3-NH2, EC50 = 1.55 ± 0.58 μM) > 7w (2-NH2, EC50 = 1.84 ± 0.35 μM) > 7u (4-NH2, EC50 = 2.93 ± 0.33 μM), and 7f (2-F, EC50 = 2.01 ± 0.18 μM) > 7e (3-F, EC50 = 2.84 ± 0.20 μM) > 7b (4-F, EC50 = 3.66 ± 2.99 μM). Additionally, rules of antiviral activity were consistent with that of HIV-1 when R was aliphatic substitution.
Overall, the 23 synthesized analogs in this work displayed moderate anti-HIV-1 activities and potent anti-HIV-2 activities, and many compounds could selectively inhibit HIV-2. Also, most of the compounds had higher safety profile compared to PF74, as indicated by their CC50 values. The SAR studies demonstrated that different substituents and the position of substituents were of utmost importance on antiviral activity. Considering that HIV-1 is more infectious and lethal than HIV-2 worldwide, we choose further to explore the MOA of representative compounds on HIV-1.
2.3. Demonstration of the retention of target specificity - surface plasmon resonance (SPR) analysis of compound-HIV-1 CA interactions
Since several compounds demonstrated moderate antiviral activities, we then sought to determine whether the target compounds retained target specificity to the HIV-1 CA protein. In this work, we selected two different CA protein constructs, the CA monomer and the disulphide-stabilized CA hexamer, to determine their binding affinity with the highest potency compounds, 7m and 7u. The SPR-based method had previously been reported [48,49] and PF74 was used as an in-line control for comparison. The SPR binding results were shown in Fig. 3, Fig. 4 and Table 2.
Fig. 3.
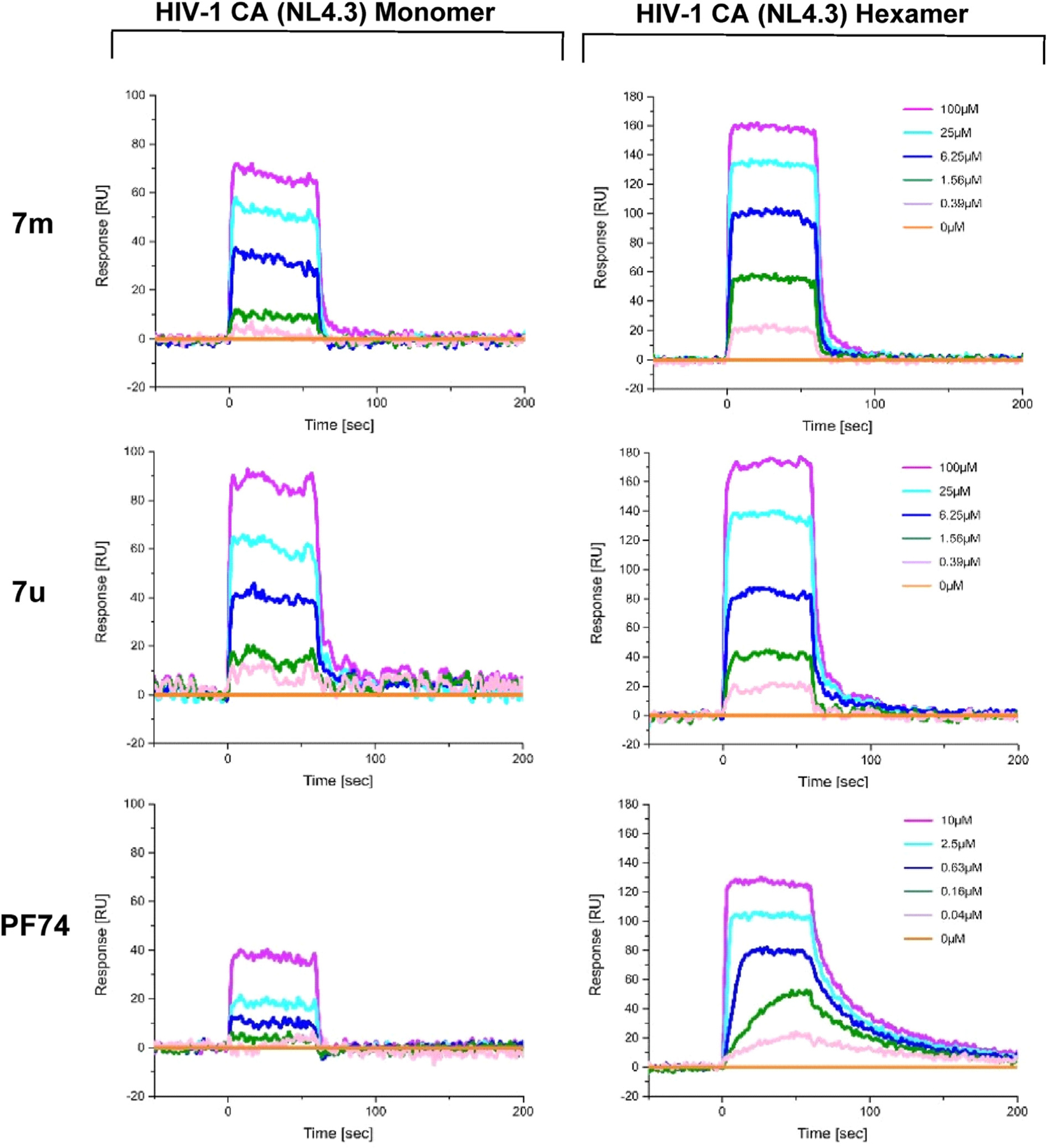
SPR sensorgrams of 7m and 7u binding to two variants of the CA protein (monomer and disulfide-stabilized hexamer) with PF74 as the reference.
Fig. 4.
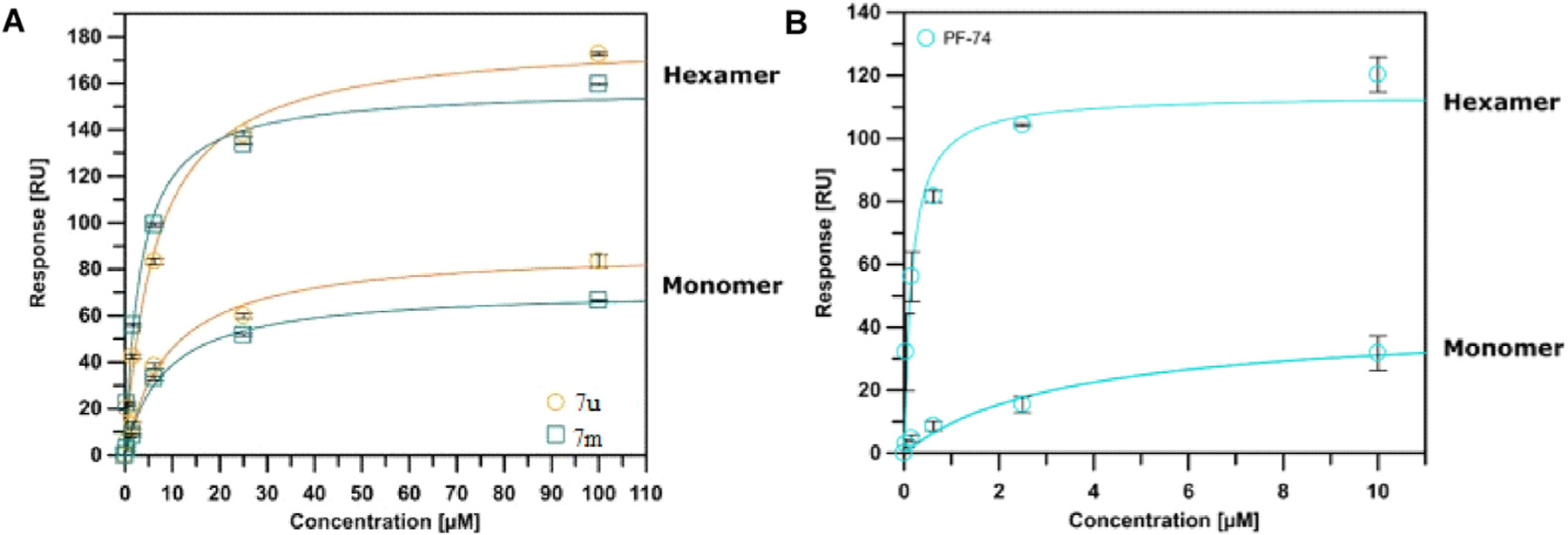
SPR isotherms of 7m and 7u (A) binding to two variants of the CA protein (monomer and disulfide-stabilized hexamer) with PF74 (B) as the reference. Isotherms are an average of 3 replicates with error bars indicating the standard error of the mean (SEM).
Table 2.
SPR results of 7m, 7u and PF74 binding to monomeric and hexameric CA constructs.
| Compounds | KDa (μM) |
Koffa(s−1) |
Ratiob | ||
|---|---|---|---|---|---|
| Monomer | Hexamer | Monomer | Hexamer | ||
| 7m | 8.393 ± 1.085 | 3.218 ± 0.485 | 0.374 ± 0.095 | 0.200 ± 0.003 | 2.608 |
| 7u | 9.306 ± 1.469 | 6.647 ± 1.098 | 0.210 ± 0.080 | 0.148 ± 0.026 | 1.400 |
| PF74 | 3.410 ± 1.310 | 0.159 ± 0.041 | N.D.c | 0.018 ± 0.00039 | 21.446 |
All values represent the average response from at least 3 replicates. Error bars represent standard deviation
..
N.D.: not determined.
The SPR analysis demonstrated that compounds 7m and 7u interact with both of the CA constructs used. Interestingly, the interaction of 7m and 7u to the monomer and hexamer mirrored the preference of the parental compound PF74, displaying a preference for the hexamer. Moreover, the calculated affinities of the compounds were comparable to the EC50 values derived from the fully infectious viral assay. Finally, the ability of compounds 7m and 7u to interact with both oligomeric forms of the HIV-1 CA, indicated that these compounds would be expected to retain the dual-stage inhibition characteristic of PF74.
2.4. Determination of the action stage of 7u
The above SPR results showed that these inhibitors demonstrated high affinities with CA proteins, characterizing their target engagement. Given that CA exerts essential roles in both early and late stages, we then sought to determine which stage(s) of the HIV-1 lifecycle can be inhibited by these inhibitors. Therefore, the modular nature of the single-round infection assay was harnessed to determine whether the new compounds, as indicated by the SPR results, would retain the dual, early and late-stage inhibition signature of PF74 and other compounds that interact within the interprotomer pocket of HIV-1 CA hexamers. In the single-round infection assay, the infective HIV-1 particles were generated recombinantly in 293T cells and subsequently utilized to infect U87.CD4.CCR5 cells allowing for separation of the assessment of early and late stages of the HIV-1 lifecycle [48]. The most potent representative compound 7u was chosen for this analysis.
As shown in Fig. 5, 7u could function at both the early and late stages of the HIV-1 life cycle, but it had a greater effect in the early-stage assay. Although the KD difference for the monomer and hexamer was not significant, the off-rate signature of 7u, as could be seen from the sensorgram (Fig. 3 and Table 2), indicated a slower off-rate for 7u for the monomeric CA protein as compared to PF74. The off-rate of PF74 from the monomeric CA was so fast that no accurate off-rate determination was achievable. The slower off-rate of 7u could explain the stronger late-stage effect as compared to PF74 when both compounds are used at 10-fold their EC50 values. Combining the above SPR data that the binding interaction of 7u to the hexamer was tighter than that two the monomer, and that these compounds were designed to the interprotomer pocket, we could speculate that the MOA of 7u in the spreading antiviral assay would be dominated by an early-stage effect, and by a multimodal mechanism, similar to that of PF74 [50].
Fig. 5.
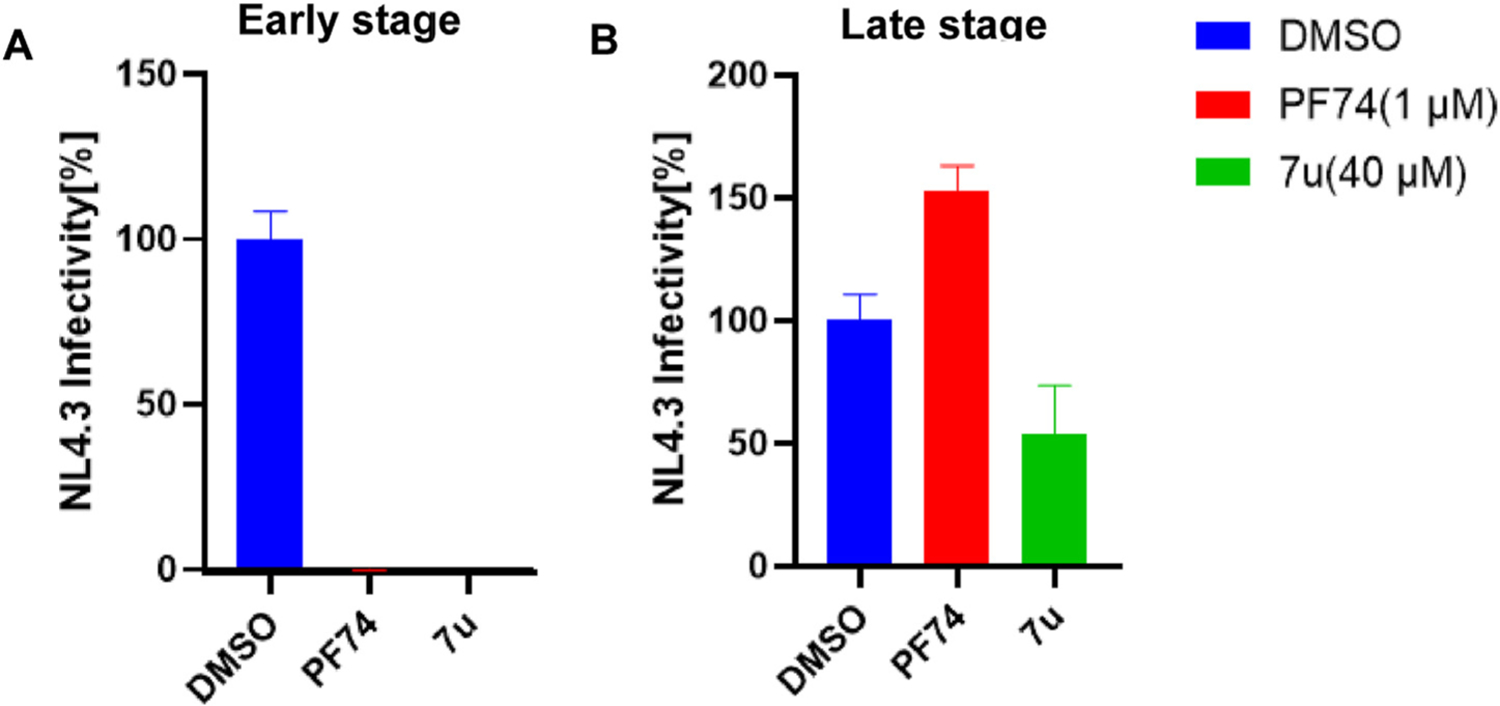
Results of single-round infection assay. (A) Early stage; (B) late stage. Infections are an average of 3 replicates with error bars indicating SEM.
We also want to clarify that PF74 under the concentrations used in our assay has no late-stage effect but has a late-stage impact at higher concentrations [13] (>10 μM, see reference 13).
2.5. Molecular dynamics (MD) simulation on 7u
The most active HIV-1 CA inhibitor in this series was selected to explore its binding to the pocket. Fig. 6A showed the root mean square deviation (RMSD) of HIV-1 CA amino acids from the first frame of the 500 ns MD simulation. The figure showed that the protein had slightly deviated from the X-ray structure and formed few but similar conformations. Deviated amino acids were investigated by calculating the root mean square fluctuation (RMSF) of amino acids (Fig. 6B). Most amino acids had deviated from the X-ray structure; however, mostly, they had slight deviation. This indicated that the HIV-1 CA visited different similar conformations during MD simulation. Thus, the presence of an ensemble of conformation could result in different binding modes of 7u. To investigate the binding of 7u to the binding site, its RMSD was calculated and represented in Fig. 6C. The figure showed that 7u had deviated from the docked conformer and stabilized in different conformers.
Fig. 6.
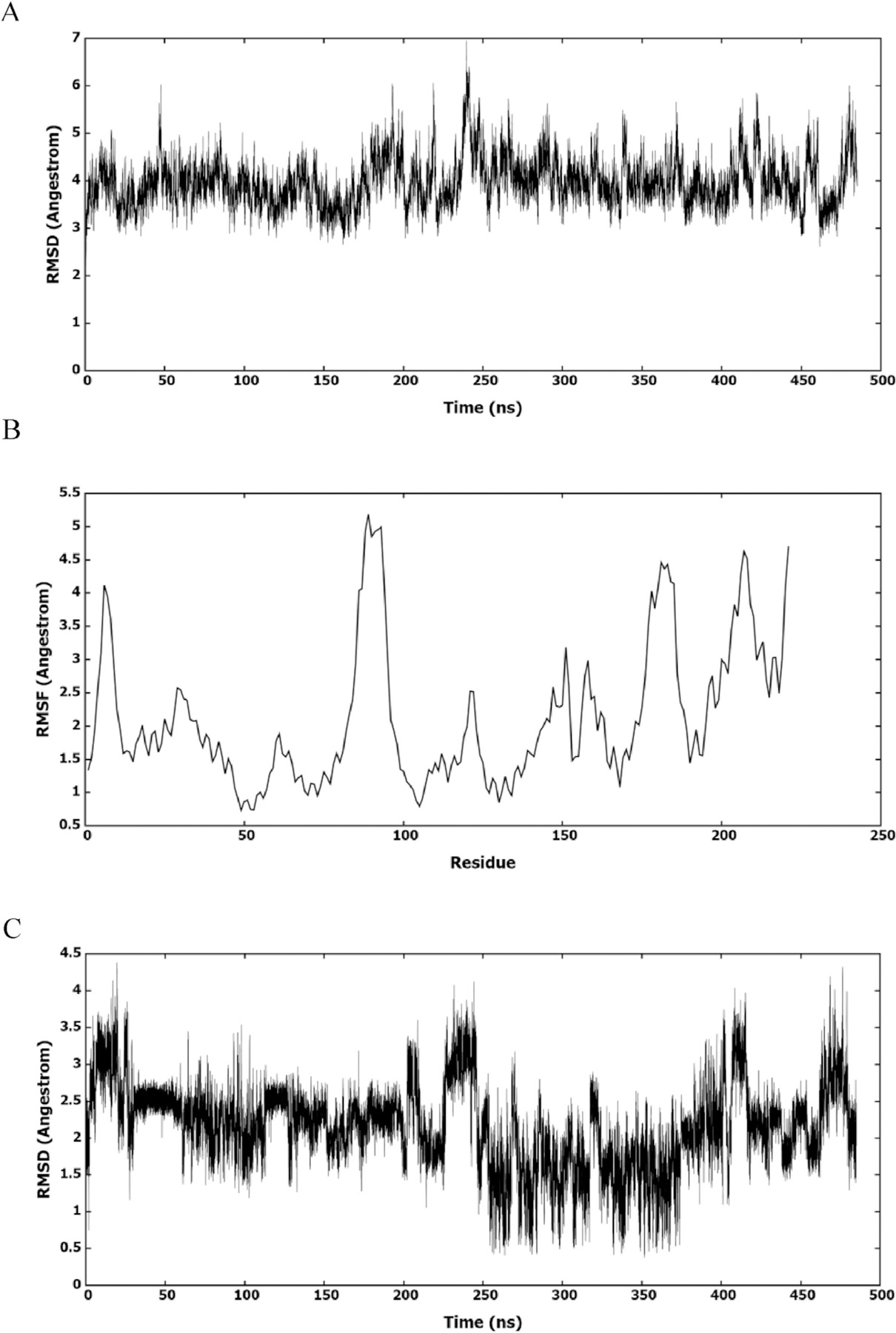
(A) RMSD of amino acids of HIV-1 CA monomer in reference to the first frame of the MD simulation. (B) RMSF of the backbone Cα atoms for amino acids of HIV-1 CA monomer. (C) RMSD (heavy atoms) of the bound 7u.
To find the conformations of HIV-1 CA and 7u, the whole trajectory was clustered. The MD trajectory was clustered based on 7u to investigate its interactions with the binding site. The clustering procedure returned 5 clusters with one most populated. This also showed that the deviation from the X-ray structure was slight and within limited conformational space. Fig. 7 showed the representative structure of the most populated cluster. The conformation of the HIV-1 CA representative structure of the most populated cluster was similar to that of the X-ray structure, and it had its binding site at the same location as the X-ray structure.
Fig. 7.
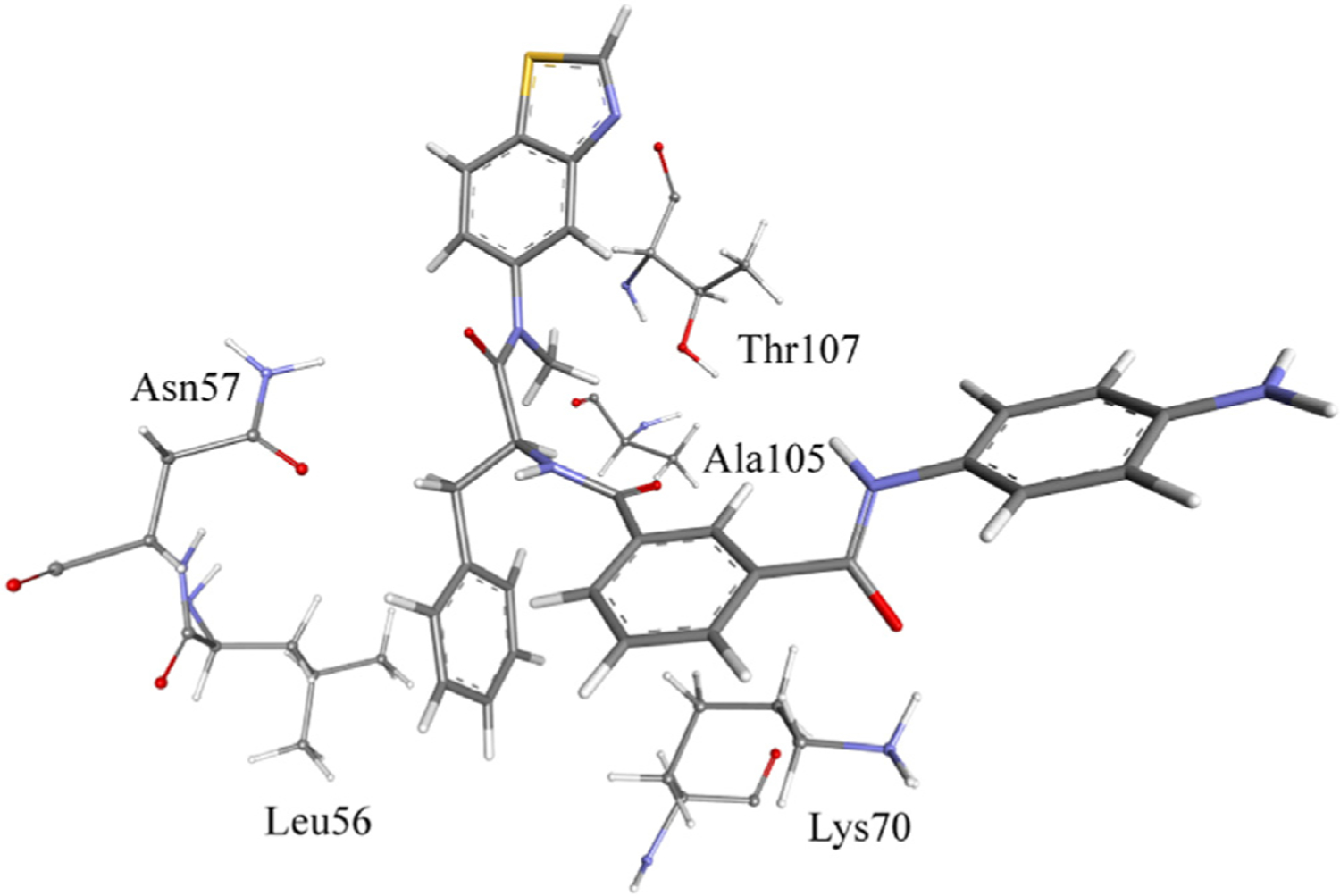
Binding of 7u to HIV-1 CA in the most populated cluster.
The bonding forces between HIV-1 CA and 7u were investigated in the most populated cluster (Fig. 7). Thr107 had aliphatic hydrogen bonding by its side-chain OH group to the methyl group of the N-methylamide of 7u. However, Ala105 had this bonding force by its backbone carbonyl group. Also, Thr107 could form hydrophobic interactions by its Ha with the benzothiazole system. Leu56 formed hydrophobic interactions with the terminal benzene ring of 7u. On the other hand, Asn57 (NH2) formed a hydrogen bond with the carbonyl oxygen of 7u, as shown in the figure. Indeed, according to the hydrogen bond analysis, Asn57 formed this hydrogen bond for 40% of the MD simulation time. Also, Asn57 formed a hydrogen bond through its side chain carbonyl oxygen with the nitrogen amide of 7u for 14% of the simulation time, as shown in Fig. 7. The 7u carbonyl group formed a hydrogen bond with Lys70, as shown in Fig. 7. However, according to the hydrogen bond analysis, this bond was formed for only 2.0% of the simulation time. This could be due to the slight fluctuation of Lys70, which made it out of the hydrogen bond distance. Results of MD simulation analysis revealed that 7u and PF74 bind to the same site. However, the antiviral activity of 7u was lower than that of PF74, maybe since the newly introduced aniline group did not maintain the original interactions with the NTD-CTD interface.
2.6. Metabolic stabilities in the presence of human liver microsomes and human plasma
Metabolic stability is reflected by the absorption, distribution, metabolism, and excretion (ADME) property that significantly impacts drug bioavailability. A serious disadvantage of PF74 is the poor metabolic stability. Since most metabolic enzymes are found in the liver, measuring liver microsomes’ in vitro metabolic stability can reliably predict oral drug bioavailability of drugs. Low stability in liver microsomes, as signified by the short half-life (t1/2), indicates excessive first-pass liver metabolism and poor oral bioavailability [51]. To evaluate the metabolic stability of our analogs, we tested the two most potent antiviral compounds (7m and 7u), as well as PF74 in human liver microsomes (HLMs). Testosterone, diclofenac, and propranolol were used as a control with moderate metabolic stability. As shown in Table 3, PF74 was rapidly metabolized with a half-life (t1/2) of 0.5 min, while half-life (t1/2) of 7m and 7u was 25.7 min and 54.6 min, respectively. Remarkably, 7u demonstrated hugely improved metabolic stability in HLMs (a 109-fold increase over PF74), and CLint(liver) of 7m and 7u was also much lower than that of PF74.
Table 3.
Metabolic stability assay in human liver microsomes.
| Sample | HLM (Final concentration of 0.5 mg protein/mL) |
|||||
|---|---|---|---|---|---|---|
| R2a | T1/2b (min) | CLint(mic)c (µL/min/mg) | CLint(liver)d (mL/min/kg) | Remaining (T = 60min) | Remaining (NCFe = 60min) | |
| 7m | 0.9055 | 25.7 | 53.9 | 48.5 | 17.0% | 100.1% |
| 7u | 0.9028 | 54.6 | 25.4 | 22.8 | 42.2% | 92.4% |
| PF74 | 1.0000 | 0.5 | 2862.5 | 2576.2 | 0.0% | 112.6% |
| Testosterone | 0.9982 | 16.7 | 82.8 | 74.5 | 7.9% | 90.7% |
| Diclofenac | 0.9947 | 3.7 | 372.0 | 334.8 | 0.0% | 96.7% |
| Propafenone | 0.9350 | 5.0 | 279.5 | 251.5 | 0.0% | 93.6% |
R2 is the correlation coefficient of the linear regression for the determination of the kinetic constant.
T1/2 is the half-life, and CLint(mic) is the intrinsic clearance.
CLint(mic) = (0.693/half-life)/mg microsome protein per mL.
CLint(liver) = CLint(mic) × mg microsomal protein/g liver weight × g liver weight/kg body weight.
NCF: no cofactor. No NADPH regenerating system was added to the NCF sample (replaced by buffer) during the 60 min incubation. If the remaining amount is less than 60%, then a non-NADPH dependent reaction occurs.
Oral drugs enter the systemic circulation after being metabolized by the liver to exert their pharmacological activity. Thus, the plasma stability of the compound is also a key factor in evaluating its druggability. Next, the representative compounds 7m and 7u and PF74 were tested for human plasma stability. Propantheline bromide was tested for control. As shown in Fig. 8, 7m and 7u remained intact after incubation for 120 min, and they also showed better stability than PF74.
Fig. 8.
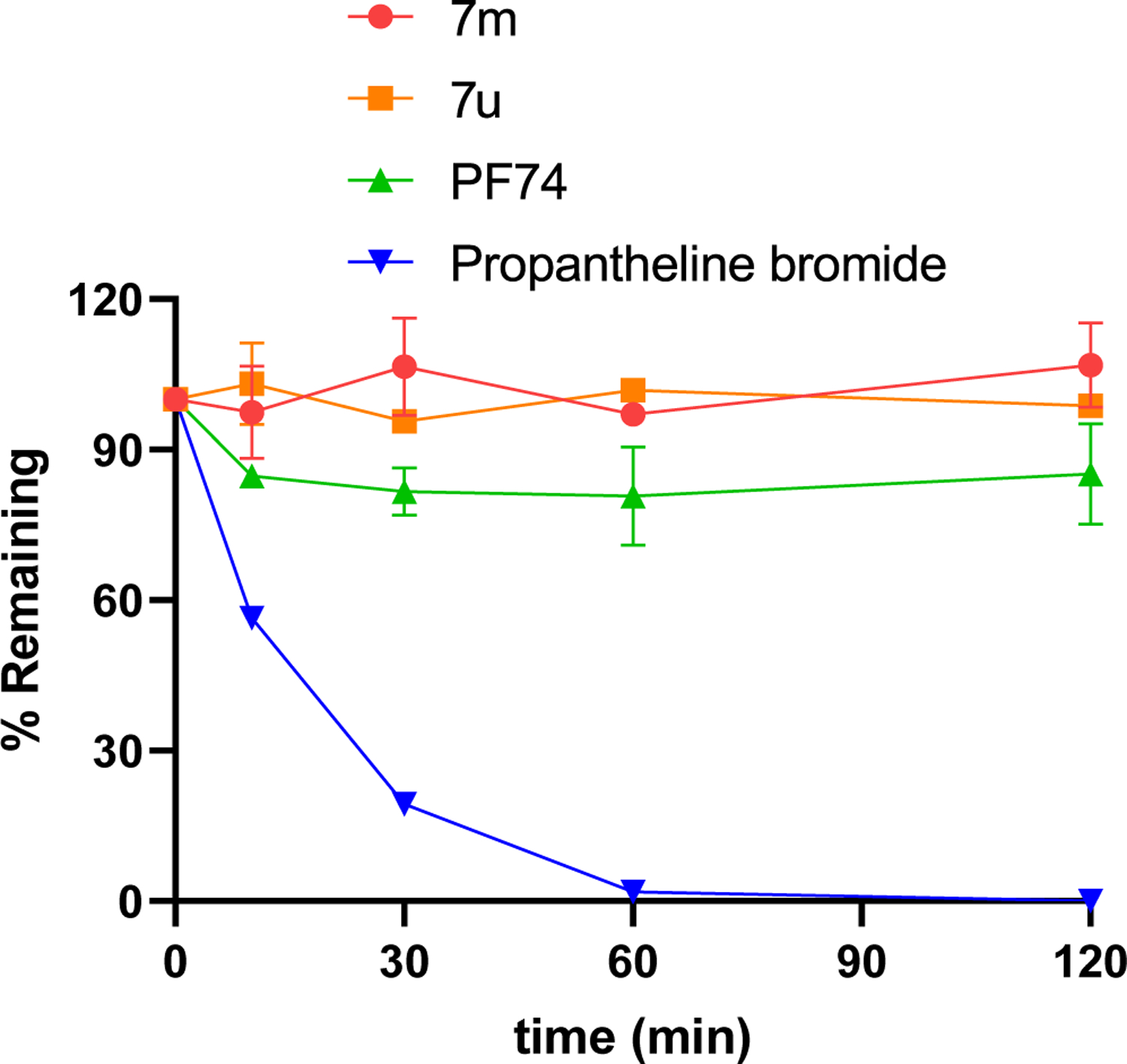
Result summary of human plasma stability assay. Experiments were performed in triplicate. % remaining = 100 × (PAR at appointed incubation time/PAR at time T0). PAR is the peak area ratio of a test compound to the internal standard. Accuracy should be within 80–120% of the indicated value.
For both the aniline in region I and indole ring in region II are electron-rich groups, they are easy to be oxidized and metabolized. Therefore, the two compounds with significant improvement on metabolic stability compared with PF74 may be due to the introduction of the benzothiazole and the change of indole to electron-deficient groups playing roles in blocking metabolic sites.
2.7. In silico prediction of physicochemical properties
According to the classical definition for drug evaluation, a potential drug candidate should own favorable drug-likeness features. Therefore, to further guide subsequent structural optimization and preliminary drug-like properties evaluation, some significant physicochemical properties of compounds 7m, 7u and PF74 were predicted comprehensively by using free online molinspiration software (http://www.molinspiration.com/), including molecular weight (MW), hydrogen bond acceptors (nON), hydrogen bond donors (nOHNH), rotatable bonds (nrotb), topological polar surface area (TPSA), miLogP, nviolations(nViol), natoms and molecular volume (MV). As displayed in Table 4, 7m and PF74 were in full compliance with the “Lipinski’s rule of five” [52] and 7u showed a slight violation regarding molecular weight, indicating that these compounds might have good absorption or cell permeability properties. Therefore, 7m and 7u had achieved a well balance between moderate efficacy and desirable physicochemical properties as same as PF74.
Table 4.
Predicted physicochemical properties calculated by using Molinspiration software for 7m, 7u and PF74a.
| Compounds | MW(Da) | nON | nOHNH | nrotb | TPSA(A2) | miLogP | nViol | natoms | MV(m3/mol) |
|---|---|---|---|---|---|---|---|---|---|
| Accepted range | <500 | <10 | <5 | ≤10 | <140 | <5 | – | – | |
| 7m | 496.59 | 7 | 2 | 8 | 91.40 | 3.71 | 0 | 36 | 441.20 |
| 7u | 549.66 | 8 | 4 | 8 | 117.42 | 4.32 | 1 | 40 | 484.83 |
| PF74 | 425.53 | 5 | 2 | 7 | 65.20 | 4.43 | 0 | 32 | 402.55 |
Using free on-line software (http://www.molinspiration.com/); MW = molecular weight; nON = no. of hydrogen bond acceptors; nOHNH = no. of hydrogen bond donors; nrotb = no. of rotatable bonds; TPSA = topological polar surface area; miLogP = molinspiration predicted LogP; nViol = number of violations; natoms = no. of atoms; MV = molar volume.
3. Conclusions
HIV remains one of the fatal diseases in the world, requiring new classes of agents with unique mechanistic preferences to overcome current drug resistance. This study has designed, synthesized, and evaluated a novel series of phenylalanine derivatives bearing benzothiazole moiety as HIV-1 capsid inhibitors. Compounds 7m and 7u were found to be the most potent inhibitors. Additionally, the newly synthesized compounds revealed an outstanding antiviral activity profile against HIV-2, represented by compound 7m whose anti-HIV-2 activity was 4.89-fold better than PF74 and most compounds could selectively inhibit HIV-2, providing valuable lead compounds as promising HIV-2 inhibitors. Herein, this paper discussed the MOA of representative compounds only on HIV-1. Firstly, the binding assay based on SPR showed that 7m and 7u could bind to recombinant CA tightly. The single-round infection assay displayed that 7u possessed a dual-stage inhibition profile with the more potent inhibition in the early stage of HIV-1 replication than the late stage. Considering the above results, we could now speculate that 7u shared a similar mode of action to PF74, stabilizing the CA hexamer and interfering with the disassembly of CA and also preventing interaction with host cell factors that utilize the same binding site. The results of MD simulation on 7u showed that it could bind to the PF74 binding site. Remarkably, 7u conferred up to ~109-fold higher half-life (t1/2) than PF74 in HLMs, which is a significant breakthrough considering the poor metabolic stability of PF74. Finally, the silico prediction suggested that 7m and 7u balanced moderate efficacy and desirable physicochemical properties. In summary, these data indicated that 7u could be a potentially viable antiviral lead compound.
In the follow-up work, to find CA inhibitors with enhanced potency and better drug-like properties, we will further verify the effect of phenylalanine derivatives containing benzothiazole moiety on metabolic stability.
4. Experimental
4.1. Chemistry
1H NMR and 13C NMR spectra were recorded on a Bruker AV-400 spectrometer using solvents as indicated (DMSO-d6). Chemical shifts were reported in δ values (ppm) with tetramethylsilane (TMS) as the internal reference, and J values were reported in hertz (Hz). Melting points (mp) were determined on a micro melting point apparatus. TLC was performed on Silica Gel GF254 for TLC (Merck), and spots were visualized by iodine vapor or irradiation with UV light (λ = 254 nm, or λ = 365 nm). Flash column chromatography was performed on a column packed with Silica Gel 60 (200–300 mesh). Thin-layer chromatography was performed on precoated HUANGHAI_HSGF254, 0.15–0.2 μM TLC plates. Solvents were of reagent grade and were purified and dried by standard methods when necessary. The concentration of the reaction solutions involved the use of a rotary evaporator at reduced pressure. The solvents of dichloromethane, TEA, methanol, etc., were obtained from Sinopharm Chemical Reagent Co., Ltd. (SCRC), AR grade. The key reactants, including benzo[d]thiazol-5-amine, N-Boc-L-phenylalanine, 3-methoxycarbonylbenzoic acid, etc., were purchased from Bide Pharmatech Co. Ltd. The purity of final representative compounds was checked by HPLC and was >95%.
4.1.1. N-methylbenzo[d]thiazol-5-amine (2)
A solution of 1,3-benzothiazol-5-amine (1, 0.50 g, 3.33 mMol) in 20 mL of methanol was slowly added to paraformaldehyde (0.22 g, 6.66 mMol) and sodium methoxide (0.90 g, 16.64 mMol), and the mixture was stirred for 16 h at 60 °C. When the temperature was gradually reduced to room temperature, NaBH4 (0.31 g, 8.32 mMol) was added, and the mixture was continually stirred at room temperature for 16 h (monitored by TLC). The reaction solution was filtered, and the solvent was evaporated under reduced pressure, 30 mL of water was added, and the mixture was extracted with ethyl acetate (3 × 30 mL). Then, the combined organic layer was washed with saturated sodium chloride solution (3 × 20 mL), dried over anhydrous Na2SO4, filtered, under reduced pressure to afford crude product, which was purified by flash column chromatography (EA:PE = 1:8 + 1% TEA) to obtain intermediate 2 as a yellow oil with the yield of 78.6%. 1H NMR (400 MHz, DMSO-d6): δ 9.20 (s, 1H, CH), 7.77 (d, J = 8.7 Hz, 1H, NH), 7.08 (d, J = 2.2 Hz, 1H, Ph-H), 6.83 (dd, J = 8.7, 2.2 Hz, 1H, Ph-H), 5.92 (d, J = 5.2 Hz, 1H, Ph-H), 2.75 (d, J = 5.1 Hz, 3H, CH3). ESI-MS: m/z 165.00 [M+1]+. C8H8N2S (164.04).
4.1.2. Tert-butyl (S)-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (3)
To a solution of N-tert-butoxycarbonyl-L-phenylalanine (1.94 g, 7.31 mMol) in 30 mL of dichloromethane, PyBOP was added (4.75 g, 9.13 mMol) at 0 °C, and the mixture was stirred for 0.5 h. Subsequently, intermediate 2 (1.00 g, 6.09 mMol) and DIEA (2.83 g, 21.87 mMol) was added to the mixture and then stirred at room temperature for another 3 h (monitored by TLC). The resulting mixture was evaporated under reduced pressure, and the residue was initially washed by 1 N HCl and extracted with ethyl acetate (3 × 20 mL). Then, the combined organic layer was washed with saturated sodium bicarbonate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford the corresponding crude product, which was purified by flash column chromatography (EA:PE = 1:4) to obtain intermediate 3 as a yellow solid with the yield of 76.0%. 1H NMR (400 MHz, DMSO-d6): δ 9.50 (s, 1H, CH), 8.27 (d, J = 8.6 Hz, 1H, Ph-H), 7.99 (s, 1H, Ph-H), 7.38 (d, J = 8.6 Hz, 1H, NH), 7.17 (d, J = 8.1 Hz, 1H, Ph-H), 7.09 (d, J = 7.2 Hz, 3H, Ph-H), 6.73 (d, J = 7.0 Hz, 2H), 4.18 (q, J = 7.3 Hz, 1H, CH), 3.24 (s, 3H, NCH3), 2.84 (dd, J = 13.7, 4.5 Hz, 1H, PhCH), 2.65 (dd, J = 13.6, 9.7 Hz, 1H, PhCH), 1.31 (s, 9H, C(CH3)3). ESI-MS: m/z 411.87 [M+H]+, 433.99 [M+Na]+.C22H25N3O3S (411.16).
4.1.3. (S)-2-amino-N-(benzo[d]thiazol-5-yl)-N-methyl-3-phenylpropanamide (4)
Trifluoroacetic acid (12.15 mMol) was added dropwise to intermediate 3 (1.00 g, 2.43 mMol) in 20 mL of dichloromethane and stirred at room temperature for 1 h (monitored by TLC). Then, the resulting mixture solution was alkalized to pH~7 with saturated sodium bicarbonate solution and then extracted with dichloromethane (30 mL). Then, the combined organic layer was washed with saturated sodium bicarbonate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford corresponding crude product 4 as a yellow oil with the yield of 79.3%. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H, CH), 9.01 (d, J = 7.6 Hz, 1H, NH), 8.28 (d, J = 8.5 Hz, 1H, NH), 8.03 (d, J = 7.6 Hz, 1H, Ph-H), 7.58 (t, J = 7.7 Hz, 1H, Ph-H), 7.47 (d, J = 8.5 Hz, 1H, Ph-H), 7.09 (d, J = 5.3 Hz, 3H, Ph-H), 6.90–6.77 (m, 2H, Ph-H), 4.67 (q, J = 7.4, 6.9 Hz, 1H, CH), 3.26 (s, 3H, NCH3), 3.08–2.92 (m, 2H, PhCH2). ESI-MS: m/z 312.02 [M+H]+. C17H17N3OS (311.11).
4.1.4. Methyl (S)-3-((1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamoyl)benzoate (5)
To a solution of 3-methoxycarbonylbenzoic acid (0.48 g, 2.67 mMol) in 20 mL dichloromethane, HATU was added (1.27 g, 3.33 mMol) at 0 °C, and the mixture was stirred for 0.5 h. Subsequently, intermediate 4 (0.69 g, 2.22 mMol) and DIEA (0.57 g, 4.44 mMol) were added to the mixture and then stirred at room temperature for another 3 h (monitored by TLC). The resulting mixture was evaporated under reduced pressure, and the residue was initially washed by 1 N HCl and extracted with ethyl acetate (3 × 20 mL). Then, the combined organic layer was washed with saturated sodium bicarbonate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford the corresponding crude product, which was purified by flash column chromatography (EA:PE = 1:3) to obtain intermediate 5 as a yellow solid with the yield of 60.8%. 1H NMR (400 MHz, DMSO-d6) δ 9.50 (s, 1H, CH), 9.03 (d, J = 7.6 Hz, 1H, NH), 8.42 (s, 1H, Ph-H), 8.27 (d, J = 8.5 Hz, 1H, Ph-H), 8.15–8.02 (m, 3H, Ph-H), 7.61 (t, J = 7.8 Hz, 1H, Ph-H), 7.46 (d, J = 8.6 Hz, 1H, Ph-H), 7.09 (d, J = 5.1 Hz, 3H, Ph-H), 6.90–6.77 (m, 2H, Ph-H), 4.66 (d, J = 6.5 Hz, 1H, CH), 3.90 (s, 3H, OCH3), 3.26 (s, 3H, NCH3), 3.10–2.92 (m, 2H, PhCH2). ESI-MS: m/z 474.02 [M+H]+. C26H23N3O4S (473.14).
4.1.5. (S)-3-((1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl) carbamoyl)benzoic acid (6)
To a solution of intermediate 5 (0.63 g, 1.33 mMol) in 10 mL of tetrahydrofuran, 10 mL of water was added, and lithium hydroxide (63.7 mg, 2.66 mMol) at room temperature, and the mixture was stirred for 2 h (monitored by TLC). The tetrahydrofuran was evaporated under reduced pressure, and then the resulting mixture solution was alkalized to pH~3 with 2 N HCl, and the white solid 6 was precipitated with a yield of 76.9%. 1H NMR (400 MHz, DMSO-d6) δ 13.16 (s, 1H, COOH), 9.50 (s, 1H, CH), 9.01 (d, J = 7.2 Hz, 1H, NH), 8.43 (s, 1H, Ph-H), 8.28 (d, J = 8.4 Hz, 1H, Ph-H), 8.05 (dd, J = 14.7, 7.5 Hz, 3H, Ph-H), 7.58 (t, J = 7.7 Hz, 1H, Ph-H), 7.47 (d, J = 7.5 Hz, 1H, Ph-H), 7.09 (s, 3H, Ph-H), 6.91–6.76 (m, 2H, Ph-H), 4.67 (q, J = 7.4 Hz, 1H, CH), 3.26 (s, 3H, NCH3), 3.01 (dt, J = 15.0, 7.7 Hz, 2H, PhCH2). ESI-MS: m/z 460.05 [M+1]+. C25H21N3O4S (459.13).
4.1.6. General procedure for the synthesis of 7(a–t)
A solution of intermediate 6 (0.10 g, 0.22 mMol) in 20 mL dichloromethane was added HATU (0.12 g, 0.33 mMol) at 0 °C, and the mixture was stirred for 0.5 h. Subsequently, the differently substituted amino derivatives (0.26 mMol) and DIEA (0.08 g, 0.65 mMol) were added to the mixture and then stirred at room temperature for another 3 h (monitored by TLC). The resulting mixture was evaporated under reduced pressure, and the residue was initially washed by 1 N HCl and extracted with ethyl acetate (3 × 20 mL). Then, the combined organic layer was washed with saturated sodium bicarbonate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford a corresponding crude product, purified by flash column chromatography to afford products 7(a–t).
4.1.6.1. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-phe nylisophthalamide (7a).
White solid, yield: 81%. Mp: 102–103 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.39 (s, 1H, NH), 9.52 (s, 1H, CH), 8.96 (d, J = 7.6 Hz, 1H, NH), 8.41 (s, 1H, Ph-H), 8.30 (d, J = 8.5 Hz, 1H, Ph-H), 8.19–8.05 (m, 2H, Ph-H), 8.02 (d, J = 7.8 Hz, 1H, Ph-H), 7.80 (d, J = 8.0 Hz, 2H, Ph-H), 7.62 (t, J = 7.7 Hz, 1H, Ph-H), 7.49 (d, J = 8.4 Hz, 1H, Ph-H), 7.41–7.34 (m, 2H, Ph-H), 7.12 (q, J = 6.2, 5.1 Hz, 4H, Ph-H), 6.86 (d, J = 4.9 Hz, 2H, Ph-H), 4.68 (d, J = 8.2 Hz, 1H, CH), 3.34 (d, J = 1.5 Hz, 3H, NCH3), 3.02 (qd, J = 13.7, 7.0 Hz, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.67 (C=O), 166.31 (C=O), 165.62 (C=O), 158.58, 154.21, 141.81,139.56, 138.36, 135.65, 134.37, 133.57, 130.95, 130.82, 129.21, 129.14, 128.85, 128.61, 127.45, 126.91, 125.69, 124.26, 123.84, 122.62, 120.83, 53.24, 38.05, 37.00. HRMS m/z C31H26N4O3S: Calcd. 534.1726, Found 535.1802 [M+H]+. HPLC purity: 96.09%.
4.1.6.2. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(4-fluorophenyl)isophthalamide (7b).
White solid, yield: 80%. Mp: 111–112 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.45 (s, 1H, NH), 9.52 (s, 1H, CH), 8.96 (d, J = 7.6 Hz, 1H, NH), 8.41 (s, 1H), 8.29 (d, J = 8.5 Hz, 1H, Ph-H), 8.16–8.05 (m, 2H, Ph-H), 8.02 (d, J = 7.8 Hz, 1H, Ph-H), 7.82 (dd, J = 8.7, 5.0 Hz, 2H, Ph-H), 7.62 (t, J = 7.7 Hz, 1H, Ph-H), 7.49 (d, J = 8.4 Hz, 1H, Ph-H), 7.22 (t, J = 8.7 Hz, 2H, Ph-H), 7.11 (d, J = 5.2 Hz, 3H, Ph-H), 6.97–6.76 (m, 2H, Ph-H), 4.69 (s, 1H, CH), 3.28 (s, 3H, NH3), 3.02 (qd, J = 13.8, 7.1 Hz, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.66 (C=O), 166.30 (C=O), 165.53 (C=O), 158.86 (d, 1JC-F = 240.4 Hz), 158.56e158.42 (m), 154.20, 141.81, 138.31, 135.91, 135.47, 134.43, 133.55, 130.88 (d, 3JC-F = 5.3 Hz), 129.21, 128.86, 128.60, 127.41, 126.91, 125.65, 123.80, 122.75, 122.67, 122.58, 115.70 (d, 2JC-F = 22.2 Hz), 53.20, 38.07, 37.09. HRMS m/z C31H25FN4O3S: Calcd. 552.1631, Found 553.1702 [M+H]+. HPLC purity: 97.91%.
4.1.6.3. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(4-bromophenyl)isophthalamide (7c).
White solid, yield: 76%. Mp: 103–105 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.52 (s, 1H, NH), 9.52 (s, 1H, CH), 8.96 (d, J = 7.6 Hz, 1H, NH), 8.41 (s, 1H, Ph-H), 8.29 (d, J = 8.5 Hz, 1H, Ph-H), 8.18e7.96 (m, 3H, Ph-H), 7.83–7.76 (m, 2H, Ph-H), 7.63 (t, J = 7.8 Hz, 1H, Ph-H), 7.59–7.54 (m, 2H, Ph-H), 7.49 (d, J = 8.3 Hz, 1H, Ph-H), 7.10 (d, J = 5.4 Hz, 3H, Ph-H), 6.85 (d, J = 6.3 Hz, 2H, Ph-H), 4.69 (s, 1H, CH), 3.28 (s, 3H, NCH3), 3.03 (td, J = 16.4, 15.2, 7.4 Hz, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.66 (C=O), 166.28 (C=O), 165.73 (C=O), 158.51, 154.18, 141.79, 138.93, 138.30, 135.36, 134.42, 133.56, 131.96, 130.97, 129.21, 129.13, 128.90, 128.60, 127.46, 126.91, 125.65, 123.81, 122.74, 122.59, 115.98, 53.21, 38.06, 37.06. HRMS m/z C31H25BrN4O3S: Calcd.612.0831, Found 613.0906 [M+H]+. HPLC purity: 97.81%.
4.1.6.4. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(4-chlorophenyl)isophthalamide (7d).
White solid, yield: 75%. Mp: 134–137 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.52 (s, 1H, NH), 9.52 (s, 1H, CH), 8.96 (d, J = 7.6 Hz, 1H, NH), 8.41 (s, 1H, Ph-H), 8.29 (d, J = 8.5 Hz, 1H, Ph-H), 8.05 (dd, J = 20.4, 8.0 Hz, 3H, Ph-H), 7.85 (d, J = 8.4 Hz, 2H, Ph-H), 7.63 (t, J = 7.8 Hz, 1H, Ph-H), 7.44 (d, J = 8.3 Hz, 3H, Ph-H), 7.11 (s, 3H, Ph-H), 6.85 (d, J = 6.3 Hz, 2H, Ph-H), 4.68 (q, J = 7.3 Hz, 1H, CH), 3.28 (s, 3H, NCH3), 3.15–2.90 (m, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.66 (C=O), 166.27 (C=O), 165.71 (C=O), 158.55, 154.20, 141.80, 138.54, 138.34, 135.37, 134.41, 133.57, 130.98, 130.96, 129.21, 129.06, 128.90, 128.61, 127.87, 127.47, 126.91, 125.67, 123.83, 122.61, 122.34, 53.23, 38.05, 37.02. HRMS m/z C31H25ClN4O3S: Calcd.568.1336, Found 569.1413 [M+H]+. HPLC purity: 98.95%.
4.1.6.5. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(3-fluorophenyl)isophthalamide (7e).
White solid, yield: 65%. Mp: 104–105 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.58 (s, 1H, NH), 9.52 (s, 1H, CH), 8.97 (d, J = 7.6 Hz, 1H, NH), 8.42 (s, 1H, Ph-H), 8.30 (d, J = 8.5 Hz, 1H, Ph-H), 8.07 (dt, J = 17.9, 7.8 Hz, 3H, Ph-H), 7.78 (dd, J = 11.8, 2.2 Hz, 1H, Ph-H), 7.62 (dd, J = 19.7, 8.1 Hz, 2H, Ph-H), 7.42 (q, J = 7.8 Hz, 2H, Ph-H), 7.11 (s, 3H, Ph-H), 6.96 (t, J = 8.5 Hz, 1H, Ph-H), 6.85 (d, J = 6.3 Hz, 2H, Ph-H), 4.76–4.60 (m, 1H, CH), 3.28 (s, 3H, NCH3), 3.02 (qd, J = 13.7, 7.2 Hz, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.66 (C=O), 166.26 (C=O), 165.89 (C=O), 162.55 (d, 1JC-F = 240.9 Hz), 158.56, 154.21, 141.81, 141.39, 141.28, 138.35, 135.30, 134.43, 133.57, 131.03, 130.77 (d, 3JC-F = 9.3 Hz), 129.21, 128.92, 128.61, 127.50, 126.91, 125.67, 123.83, 122.61, 116.48 (d, 3JC-F = 2.7 Hz), 110.71 (d, 2JC-F = 21.1 Hz), 107.46 (d, 2JC-F = 26.4 Hz), 53.25, 38.06, 37.01. HRMS m/z C31H25FN4O3S: Calcd.552.1631, Found 553.1707 [M+H]+, 575.1517 [M+23]+. HPLC purity: 98.66%.
4.1.6.6. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(2-fluorophenyl)isophthalamide (7f).
White solid, yield: 82%. Mp: 105–107 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.29 (s, 1H, NH), 9.55 (s, 1H, CH), 8.99 (d, J = 7.6 Hz, 1H, NH), 8.48 (s, 1H, Ph-H), 8.33 (d, J = 8.5 Hz, 1H, Ph-H), 8.14 (d, J = 7.4 Hz, 2H, Ph-H), 8.06 (d, J = 7.8 Hz, 1H, Ph-H), 7.68 (q, J = 8.4 Hz, 2H, Ph-H), 7.52 (d, J = 8.4 Hz, 1H, Ph-H), 7.38–7.27 (m, 3H, Ph-H), 7.15 (d, J = 5.1 Hz, 3H, Ph-H), 6.90 (d, J = 6.2 Hz, 2H, Ph-H), 4.73 (d, J = 7.2 Hz, 1H, CH), 3.32 (s, 3H, NCH3), 3.14–2.96 (m, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.64 (C=O), 166.27 (C=O), 165.51 (C=O), 158.54, 156.22 (d, 1JC-F = 246.9 Hz), 154.21, 141.79, 138.32, 134.52 (d, 2JC-F = 12.9 Hz), 133.57, 131.08 (d, 3JC-F = 6.3 Hz), 131.03, 129.22, 128.94, 128.61, 127.54 (d, 2JC-F = 8.0 Hz), 127.44, 126.92, 126.18, 126.06, 125.67, 124.82 (d, 3JC-F = 3.5 Hz), 123.83, 122.60, 116.44, 116.24, 53.19, 38.06, 37.09. HRMS m/z C31H25FN4O3S: Calcd.552.1631, Found 553.1709 [M+H]+, 575.1509 [M+23]+. HPLC purity: 95.95%.
4.1.6.7. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(p-tolyl)isophthalamide (7g).
White solid, yield: 81%. Mp: 132–134 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.30 (s, 1H, NH), 9.51 (s, 1H, CH), 8.94 (d, J = 7.5 Hz, 1H, NH), 8.40 (s, 1H, Ph-H), 8.29 (d, J = 8.5 Hz, 1H, Ph-H), 8.18–8.04 (m, 2H Ph-H), 8.00 (d, J = 7.8 Hz, 1H, Ph-H), 7.68 (d, J = 8.0 Hz, 2H, Ph-H), 7.61 (t, J = 7.8 Hz, 1H, Ph-H), 7.55–7.42 (m, 1H, Ph-H), 7.18 (d, J = 8.0 Hz, 2H, Ph-H), 7.11 (d, J = 5.4 Hz, 3H, Ph-H), 6.85 (d, J = 6.3 Hz, 2H, Ph-H), 4.69 (s, 1H, CH), 3.28 (s, 3H, NCH3), 3.12–2.93 (m, 2H, PhCH2), 2.30 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6): δ 171.66 (C=O), 166.33 (C=O), 165.40 (C=O), 158.55, 154.20, 141.80, 138.35, 137.03, 135.72, 134.36, 133.56, 133.22, 130.89, 130.73, 129.52, 129.21, 128.82, 128.60, 127.39, 126.91, 125.68, 123.83, 122.61, 120.85, 53.21, 38.05, 37.03, 20.98. HRMS m/z C32H28N4O3S: Calcd.548.1882, Found 549.1957 [M+H]+. HPLC purity: 97.81%.
4.1.6.8. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(4-methoxyphenyl)isophthalamide (7h).
White solid, yield: 80%. Mp: 143–144 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.27 (s, 1H, NH), 9.52 (s, 1H, CH), 8.95 (d, J = 7.6 Hz, 1H, NH), 8.40 (s, 1H, Ph-H), 8.30 (d, J = 8.5 Hz, 1H, Ph-H), 8.17–7.93 (m, 3H, Ph-H), 7.79–7.67 (m, 2H, Ph-H), 7.61 (t, J = 7.7 Hz, 1H, Ph-H), 7.55–7.42 (m, 1H, Ph-H), 7.11 (d, J = 5.2 Hz, 3H, Ph-H), 7.04–6.92 (m, 2H, Ph-H), 6.85 (d, J = 4.5 Hz, 2H, Ph-H), 4.69 (t, J = 7.6 Hz, 1H, CH), 3.76 (d, J = 1.5 Hz, 3H, NCH3), 3.28 (s, 3H, OCH3), 3.01 (qd, J = 14.6, 14.0, 7.5 Hz, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.67 (C=O), 166.35 (C=O), 165.16 (C=O), 158.51, 156.13, 154.20, 141.81, 138.33, 135.72, 134.38, 133.55, 132.61, 130.82, 130.65, 129.21, 128.80, 128.60, 127.33, 126.90, 125.66, 123.80, 122.59, 122.46, 114.28, 55.67, 53.20, 38.06, 37.07. HRMS m/z C32H28N4O4S: Calcd.564.1831, Found 565.1905 [M+H]+, 587.1723 [M+23]+. HPLC purity: 98.87%.
4.1.6.9. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(4-(trifluoromethyl)phenyl)isophthalamide (7i).
White solid, yield: 79%. Mp: 134–136 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.74 (s, 1H, NH), 9.52 (s, 1H, CH), 8.98 (d, J = 7.6 Hz, 1H, NH), 8.43 (s, 1H, Ph-H), 8.30 (d, J = 8.6 Hz, 1H, Ph-H), 8.07 (dd, J = 23.9, 8.2 Hz, 5H, Ph-H), 7.76 (d, J = 8.4 Hz, 2H, Ph-H), 7.64 (t, J = 7.8 Hz, 1H, Ph-H), 7.49 (s, 1H, Ph-H), 7.11 (s, 3H, Ph-H), 6.85 (d, J = 6.3 Hz, 2H, Ph-H), 4.68 (s, 1H, CH), 3.28 (s, 3H, NCH3), 3.11–2.89 (m, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.65 (C=O), 166.23 (C=O), 166.11 (C=O), 165.07, 158.50, 154.21, 143.21, 141.81, 138.32, 135.17, 134.48, 133.55, 131.11 (d, 2JC-F = 6.4 Hz), 129.22, 128.93, 128.60, 127.58, 126.91, 126.48, 126.43 (d, 3JC-F = 3.8 Hz), 126.22, 124.73 (d, 1JC-F = 186.2 Hz), 124.41, 124.09, 123.52, 53.22, 38.06, 37.08. HRMS m/z C32H25F3N4O3S: Calcd. 602.1599, Found 603.1676 [M+H]+. HPLC purity: 96.38%.
4.1.6.10. (S)–N1-(4-acetylphenyl)-N3-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-ph enylpropan-2-yl)isophthalamide (7j).
White solid, yield: 74%. Mp: 103–104 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.71 (s, 1H, NH), 9.51 (s, 1H, CH), 8.97 (d, J = 7.6 Hz, 1H, NH), 8.43 (s, 1H, Ph-H), 8.30 (d, J = 8.5 Hz, 1H, Ph-H), 8.10 (d, J = 7.5 Hz, 2H, Ph-H), 7.99 (q, J = 8.6 Hz, 5H, Ph-H), 7.64 (t, J = 7.8 Hz, 1H, Ph-H), 7.49 (d, J = 8.5 Hz, 1H, Ph-H), 7.11 (d, J = 5.2 Hz, 3H, Ph-H), 6.85 (d, J = 6.2 Hz, 2H, Ph-H), 4.69 (d, J = 7.0 Hz, 1H, CH), 3.28 (s, 3H, NCH3), 3.14–2.93 (m, 2H, PhCH2), 2.57 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6): δ 197.11 (C=O), 171.65 (C=O), 166.24 (C=O), 166.06 (C=O), 158.59, 154.20, 143.97, 141.78, 138.34, 135.24, 134.40, 133.57, 132.60, 131.13, 129.83, 129.20, 128.95, 128.62, 127.60, 126.92, 125.67, 123.85, 122.62, 119.95, 53.26, 38.05, 36.97, 26.97. HRMS m/z C33H28N4O4S: Calcd. 576.1831, Found 577.1899 [M+H]+. HPLC purity: 99.16%.
4.1.6.11. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(4-(trifluoromethoxy)phenyl)isophthalamide (7k).
White solid, yield: 71%. Mp: 112–113 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.58 (s, 1H, NH), 9.52 (s, 1H, CH), 8.97 (d, J = 7.6 Hz, 1H, NH), 8.42 (s, 1H, Ph-H), 8.29 (d, J = 8.5 Hz, 1H, Ph-H), 8.09 (d, J = 8.3 Hz, 2H, Ph-H), 8.04 (d, J = 7.8 Hz, 1H, Ph-H), 7.98–7.86 (m, 2H, Ph-H), 7.63 (t, J = 7.9 Hz, 1H, Ph-H), 7.49 (d, J = 8.4 Hz, 1H, Ph-H), 7.40 (d, J = 8.5 Hz, 2H, Ph-H), 7.11 (d, J = 5.3 Hz, 3H, Ph-H), 6.85 (d, J = 6.3 Hz, 2H, Ph-H), 4.69 (d, J = 7.4 Hz, 1H, CH), 3.28 (s, 3H, NCH3), 3.14–2.90 (m, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.66 (C=O), 166.27 (C=O), 165.76 (C=O), 158.55, 154.20, 144.44, 141.80, 138.77, 138.35, 135.31, 134.43, 133.58, 131.00, 129.21, 128.90, 128.61, 127.51, 126.91, 125.67, 124.75 (d, 1JC-F = 184.7 Hz), 122.62, 122.18, 122.09, 121.99, 121.91, 53.25, 38.05, 37.01. HRMS m/z C32H25F3N4O4S: Calcd. 618.1549, Found 619.1623 [M+H]+. HPLC purity: 99.06%.
4.1.6.12. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(4-cyanophenyl)isophthalamide (7l).
White solid, yield: 68%. Mp: 171–172 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.78 (s, 1H, NH), 9.50 (s, 1H, CH), 8.96 (d, J = 7.5 Hz, 1H, NH), 8.42 (s, 1H, Ph-H), 8.28 (d, J = 8.6 Hz, 1H, Ph-H), 8.09 (d, J = 7.8 Hz, 2H, Ph-H), 8.01 (dd, J = 8.8, 1.7 Hz, 3H, Ph-H), 7.86–7.82 (m, 2H, Ph-H), 7.64 (t, J = 7.8 Hz, 1H, Ph-H), 7.47 (d, J = 8.5 Hz, 1H, Ph-H), 7.10 (d, J = 5.5 Hz, 3H, Ph-H), 6.85 (d, J = 6.3 Hz, 2H, Ph-H), 4.69 (t, J = 8.4 Hz, 1H, CH), 3.27 (s, 3H, NCH3), 3.11–2.93 (m, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.64 (C=O), 166.24 (C=O), 166.19 (C=O), 158.54, 154.20, 143.88, 141.80, 138.33, 135.04, 134.47, 133.65, 133.56, 131.26, 131.14, 129.21, 128.98, 128.61, 127.63, 126.91, 125.65, 123.83, 122.61, 120.72, 119.52, 105.99, 53.25, 38.06, 37.02. HRMS m/z C32H25N5O3S: Calcd. 559.1678, Found 560.1749 [M+H]+. HPLC purity: 99.35%.
4.1.6.13. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(prop-2-yn-1-yl)isophthalamide (7m).
White solid, yield: 76%. Mp: 134–135 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.51 (s, 1H, CH), 9.04 (t, J = 5.6 Hz, 1H, NH), 8.89 (d, J = 7.6 Hz, 1H, NH), 8.42–8.22 (m, 2H, Ph-H), 8.08 (s, 1H, Ph-H), 7.97 (t, J = 7.6 Hz, 2H, Ph-H), 7.56 (t, J = 7.7 Hz, 1H, Ph-H), 7.47 (d, J = 8.1 Hz, 1H, Ph-H), 7.11 (d, J = 4.9 Hz, 3H, Ph-H), 6.97–6.72 (m, 2H, Ph-H), 4.67 (d, J = 7.0 Hz, 1H, CH), 4.09 (dd, J = 5.7, 2.4 Hz, 2H, CH2), 3.27 (s, 3H, NCH3), 3.16 (s, 1H, C≡CH), 3.08–2.91 (m, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.62 (C=O), 166.28 (C=O), 166.01 (C=O), 158.54, 154.19, 141.78, 138.30, 134.44, 134.39, 133.55, 130.73, 130.50, 129.21, 128.84, 128.61, 127.10, 126.91, 125.66, 123.82, 122.58, 81.67, 73.43, 53.13, 38.05, 37.10, 29.05. HRMS m/z C28H24N4O3S: Calcd. 496.1569, Found 497.1639 [M+H]+, 519.1444 [M+23]+. HPLC purity: 97.95%.
4.1.6.14. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(2-methoxyethyl)isophthalamide (7n).
White solid, yield: 80%. Mp: 105–106 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.50 (s, 1H, CH), 8.85 (d, J = 7.6 Hz, 1H, NH), 8.60 (d, J = 5.7 Hz, 1H, NH), 8.28 (d, J = 14.2 Hz, 2H, Ph-H), 8.07 (s, 1H, Ph-H), 7.95 (dd, J = 13.1, 7.7 Hz, 2H, Ph-H), 7.54 (t, J = 7.7 Hz, 1H, Ph-H), 7.46 (d, J = 8.3 Hz, 1H, Ph-H), 7.10 (d, J = 5.2 Hz, 3H, Ph-H), 6.84 (d, J = 6.2 Hz, 2H, Ph-H), 4.67 (d, J = 7.4 Hz, 1H, CH), 3.47 (t, J = 5.1 Hz, 4H, CH2 × 2), 3.31–3.21 (m, 6H, NCH3, OCH3), 3.09–2.91 (m, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.65 (C=O), 166.38 (C=O), 166.30 (C=O), 158.52, 154.19, 141.80, 138.32, 135.02, 134.31, 133.55, 130.49, 130.43, 129.21, 128.71, 128.60, 126.97, 126.90, 125.65, 123.81, 122.59, 70.93, 58.41, 53.13, 39.54, 38.05, 37.11. HRMS m/z C28H28N4O4S: Calcd. 516.1831, Found 517.1899 [M+H]+. HPLC purity: 99.27%.
4.1.6.15. (S)–N1-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-N3-(pyridin-4-yl)isophthalamide (7o).
White solid, yield: 80%. Mp: 143–145 oC. 1H NMR (400 MHz DMSO-d6): δ 10.75 (s, 1H, NH), 9.51 (s, 1H, CH), 8.97 (d, J = 7.5 Hz, 1H, NH), 8.51 (d, J = 5.4 Hz, 2H, Ph-H), 8.43 (s, 1H, Ph-H), 8.29 (d, J = 8.5 Hz, 1H, Ph-H), 8.08 (dd, J = 16.3, 7.5 Hz, 3H, Ph-H), 7.82 (d, J = 5.5 Hz, 2H, Ph-H), 7.65 (t, J = 7.8 Hz, 1H, Ph-H), 7.48 (d, J = 8.5 Hz, 1H, Ph-H), 7.11 (d, J = 5.3 Hz, 3H, Ph-H), 6.86 (d, J = 6.2 Hz, 2H, Ph-H), 4.70 (t, J = 7.6 Hz, 1H, CH), 3.28 (s, 3H, NCH3), 3.02 (dq, J = 23.0, 12.8, 10.9 Hz, 2H, PhCH2). 13C NMR(100 MHz, DMSO-d6): δ 171.64 (C=O), 166.55 (C=O), 166.19 (C=O), 158.54, 154.20, 150.79, 146.37, 141.80, 138.33, 134.90, 134.48, 133.57, 131.33, 131.15, 129.21, 128.99, 128.61, 127.64, 126.92, 125.66, 123.82, 122.60, 114.53, 53.25, 38.06, 37.04. HRMS m/z C30H25N5O3S: Calcd. 535.1678, Found 536.1745 [M+H]+. HPLC purity: 99.01%.
4.1.6.16. tert-butyl(S)-4-(3-((1-(benzo[d]thiazol-5-yl(methyl) amino)-1-oxo-3-phenylpropan −2-yl)carbamoyl)benzoyl)piperazine-1-carboxylate (7p).
White solid, yield: 77%. Mp: 152–153 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.51 (s, 1H, CH), 8.89 (d, J = 7.7 Hz, 1H, NH), 8.30 (d, J = 8.5 Hz, 1H, Ph-H), 8.11 (s, 1H, Ph-H), 7.98–7.81 (m, 2H, Ph-H), 7.66–7.42 (m, 3H, Ph-H), 7.09 (d, J = 5.3 Hz, 3H, Ph-H), 6.82 (d, J = 6.3 Hz, 2H, Ph-H), 4.65 (d, J = 9.4 Hz, 1H, CH), 3.62 (s, 8H, CH2 × 4), 3.27 (s, 3H, NCH3), 3.01 (td, J = 16.9, 15.4, 7.5 Hz, 2H, PhCH2), 1.42 (s, 9H, C(CH3)3). 13C NMR (100 MHz, DMSO-d6): δ 171.63 (C=O), 169.06 (C=O), 166.04 (C=O), 158.55, 154.30 (C=O), 154.20, 141.79, 138.40, 136.17, 134.23, 133.56, 130.29, 129.18, 129.01, 128.56, 126.86, 126.44, 125.67, 123.84, 122.59, 79.72, 53.20, 38.72, 38.06, 36.90, 28.49. HRMS m/z C34H37N5O5S: Calcd. 627.2515, Found 628.2588 [M+H]+, 650.2403 [M+23]+. HPLC purity: 99.86%.
4.1.6.17. tert-butyl(S)-4-(3-((1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phenylpropan −2-yl)carbamoyl)benzamido)piperidine-1-carboxylate (7q).
White solid, yield: 73%. Mp: 138–140 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.51 (s, 1H, CH), 8.89 (d, J = 7.5 Hz, 1H, NH), 8.41 (d, J = 7.8 Hz, 1H, NH), 8.33–8.22 (m, 2H, Ph-H), 8.10 (s, 1H, Ph-H), 7.95 (t, J = 8.0 Hz, 2H, Ph-H), 7.51 (dt, J = 17.0, 8.1 Hz, 2H, Ph-H), 7.16–7.04 (m, 3H, Ph-H), 6.90–6.79 (m, 2H, Ph-H), 4.65 (q, J = 6.8, 5.3 Hz, 1H, CH), 3.97 (dd, J = 14.5, 9.2 Hz, 3H, CH2, CH), 3.27 (s, 3H, NCH3), 3.07–2.80 (m, 4H, CH2 × 2), 1.80 (dd, J = 13.1, 4.0 Hz, 2H, CH2), 1.47 (dd, J = 12.1, 4.2 Hz, 2H, CH2), 1.42 (s, 9H, C(CH3)3). 13C NMR (100 MHz, DMSO-d6): δ 171.69 (C=O), 166.50 (C=O), 165.61 (C=O), 158.59 (C=O), 154.41, 154.19, 141.80, 138.38, 135.25, 134.27, 133.57, 130.62, 130.37, 129.19, 128.60, 127.15, 126.90, 125.67, 123.84, 122.61, 79.12, 53.25, 47.05, 38.03, 36.93, 31.76, 28.56. HRMS m/z C35H39N5O5S: Calcd. 641.2672, Found 642.2741 [M+H]+, 664.2534 [M+23]+. HPLC purity: 99.71%.
4.1.6.18. tert-butyl(S)-(4-(3-((1-(benzo[d]thiazol-5-yl(methyl) amino)-1-oxo-3-phenylpropa n −2-yl)carbamoyl)benzamido)phenyl) carbamate (7r).
White solid, yield: 76%. Mp: 162–163 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.29 (s, 1H, NH), 9.52 (s, 1H, NH), 9.32 (s, 1H, CH), 8.94 (d, J = 7.6 Hz, 1H, NH), 8.40 (s, 1H, Ph-H), 8.30 (d, J = 8.5 Hz, 1H, Ph-H), 8.17–8.03 (m, 2H, Ph-H), 8.00 (d, J = 7.8 Hz, 1H, Ph-H), 7.67 (d, J = 8.5 Hz, 2H, Ph-H), 7.60 (t, J = 7.8 Hz, 1H, Ph-H), 7.44 (d, J = 8.6 Hz, 3H, Ph-H), 7.10 (d, J = 5.2 Hz, 3H, Ph-H), 6.85 (d, J = 6.4 Hz, 2H, Ph-H), 4.68 (d, J = 6.8 Hz, 1H, CH), 3.28 (s, 3H, NCH3), 3.01 (qd, J = 14.0, 7.3 Hz, 2H, PhCH2), 1.49 (s, 9H, C(CH3)3). 13C NMR (100 MHz, DMSO-d6): δ 171.65 (C=O), 166.34 (C=O), 165.21 (C=O), 158.48 (C=O), 154.20, 153.32, 141.82, 138.32, 135.99, 135.69, 134.38, 133.95, 133.56, 130.83, 130.68, 129.22, 128.79, 128.60, 127.34, 126.90, 125.65, 123.80, 122.60, 121.42, 118.84, 79.38, 53.18, 38.71, 38.06, 28.64. HRMS m/z C36H35N5O5S: Calcd. 649.2359, Found 650.2421 [M+H]+, 672.2222 [M+23]+. HPLC purity: 97.93%.
4.1.6.19. tert-butyl(S)-(3-(3-((1-(benzo[d]thiazol-5-yl(methyl) amino)-1-oxo-3-phenylpropa n-2-yl)carbamoyl)benzamido)phenyl) carbamate (7s).
White solid, yield: 72%. Mp: 151–152 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.36 (s, 1H, NH), 9.52 (s, 1H, NH), 9.41 (s, 1H, CH), 8.94 (d, J = 7.6 Hz, 1H, NH), 8.39 (s, 1H, Ph-H), 8.30 (d, J = 8.5 Hz, 1H, Ph-H), 8.17–8.05 (m, 2H, Ph-H), 8.06–7.94 (m, 2H, Ph-H), 7.60 (t, J = 7.8 Hz, 1H, Ph-H), 7.47 (dd, J = 18.2, 8.3 Hz, 2H, Ph-H), 7.23 (t, J = 8.1 Hz, 1H, Ph-H), 7.12 (t, J = 8.4 Hz, 4H, Ph-H), 6.97–6.77 (m, 2H, Ph-H), 4.68 (d, J = 7.2 Hz, 1H, CH), 3.28 (s, 3H, NCH3), 3.15–2.92 (m, 2H, PhCH2), 1.49 (s, 9H, C(CH3)3). 13C NMR (100 MHz, DMSO-d6): δ 171.66 (C=O), 166.33 (C=O), 165.52 (C=O), 158.55 (C=O), 154.20, 153.23, 141.80, 140.28, 139.81, 138.34, 135.63, 134.31, 133.56, 130.97, 130.79, 129.22, 129.07, 128.78, 128.61, 127.47, 126.91, 125.68, 123.83, 122.61, 114.93, 114.46, 111.09, 79.44, 53.20, 38.05, 37.05, 28.63. HRMS m/z C36H35N5O5S: Calcd. 649.2359,Found 650.2453 [M+H]+. HPLC purity: 99.34%.
4.1.6.20. tert-butyl(S)-(2-(3-((1-(benzo[d]thiazol-5-yl(methyl) amino)-1-oxo-3-phenylpropa n-2-yl)carbamoyl)benzamido)phenyl) carbamate (7t).
White solid, yield: 69%. Mp: 104–105 °C.1H NMR (400 MHz, DMSO-d6): δ 9.93 (s, 1H, NH), 9.51 (s, 1H, NH), 8.98 (d, J = 7.6 Hz, 1H, NH), 8.69 (s, 1H, CH), 8.45 (s, 1H, Ph-H), 8.29 (d, J = 8.5 Hz, 1H, Ph-H), 8.06 (dd, J = 20.3, 7.6 Hz, 3H, Ph-H), 7.67–7.44 (m, 4H, Ph-H), 7.26–7.08 (m, 5H, Ph-H), 6.86 (d, J = 6.2 Hz, 2H, Ph-H), 4.69 (d, J = 6.8 Hz, 1H, CH), 3.28 (s, 3H, NCH3), 3.10–2.92 (m, 2H, PhCH2), 1.44 (s, 9H, C(CH3)3). 13C NMR (100 MHz, DMSO-d6): δ 171.64 (C=O), 166.24 (C=O), 165.48 (C=O), 158.55 (C=O), 154.21, 153.82, 141.79, 138.34, 135.01, 134.47, 132.50, 131.01, 130.91, 129.93, 129.22, 128.92, 128.61, 127.47, 126.91, 126.71, 126.22, 125.65, 124.44, 124.19, 123.82, 80.07, 53.22, 38.05, 37.05, 28.51. HRMS m/z C36H35N5O5S: Calcd. 649.2359, Found 650.2421 [M+H]+, 672.2249 [M+23]+. HPLC purity: 98.40%.
4.1.7. General procedure for the synthesis of 7(u–w)
Trifluoroacetic acid (0.77 mMol) was added dropwise to the target compounds 7(r–t) (0.10 g, 0.15 mMol) in 20 mL of dichloromethane and stirred at room temperature for 1 h (monitored by TLC). Then, the resulting mixture solution was alkalized to pH ~7 with saturated sodium bicarbonate solution and then extracted with dichloromethane (30 mL). Then, the combined organic layer was washed with saturated sodium bicarbonate (3 × 20 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford a corresponding crude product, purified by flash column chromatography to afford products 7(u–w).
4.1.7.1. (S)eN1-(4-aminophenyl)-N3-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phe nylpropan-2-yl)isophthalamide (7u).
White solid, yield: 77%. Mp: 131e132 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.98 (s, 1H, NH), 9.50 (s, 1H, CH), 8.88 (d, J = 7.6 Hz, 1H, NH), 8.36 (s, 1H, Ph-H), 8.28 (d, J = 8.5 Hz, 1H, Ph-H), 8.14–7.91 (m, 3H, Ph-H), 7.57 (t, J = 7.8 Hz, 1H, Ph-H), 7.47 (d, J = 8.5 Hz, 1H, Ph-H), 7.39 (d, J = 8.2 Hz, 2H, Ph-H), 7.10 (s, 3H, Ph-H), 6.85 (s, 2H, Ph-H), 6.67–6.50 (m, 2H, Ph-H), 4.94 (s, 2H, NH2), 4.68 (s, 1H, CH), 3.27 (s, 3H, NCH3), 3.00 (dt, J = 23.5, 13.5 Hz, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.67 (C=O), 166.39 (C=O), 164.71 (C=O), 158.54, 154.21, 145.79, 141.81, 138.34, 136.01, 134.31, 133.40, 130.70, 130.40, 129.22, 128.73, 128.60, 128.50, 127.21, 126.91, 125.68, 123.82, 122.66, 118.61, 114.18, 53.17, 38.06, 37.07. HRMS m/z C31H27N5O3S: Calcd. 549.1835, Found 550.1912 [M+H]+. HPLC purity: 99.28%.
4.1.7.2. (S)–N1-(3-aminophenyl)-N3-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phe nylpropan-2-yl)isophthalamide (7v).
White solid, yield: 76%. Mp: 129e130 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.05 (s, 1H, NH), 9.50 (s, 1H, CH), 8.89 (d, J = 7.6 Hz, 1H, NH), 8.35 (s, 1H, Ph-H), 8.28 (d, J = 8.5 Hz, 1H, Ph-H), 8.13–7.93 (m, 3H, Ph-H), 7.58 (t, J = 7.8 Hz, 1H, Ph-H), 7.47 (d, J = 8.4 Hz, 1H, Ph-H), 7.23–7.06 (m, 4H, Ph-H), 6.98 (t, J = 7.9 Hz, 1H, Ph-H), 6.87 (t, J = 8.9 Hz, 3H, Ph-H), 6.34 (d, J = 7.9 Hz, 1H, Ph-H), 5.09 (s, 2H, NH2), 4.68 (d, J = 7.0 Hz, 1H, CH), 3.27 (s, 3H, NCH3), 3.13–2.91 (m, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.67 (C=O), 166.35 (C=O), 165.36 (C=O), 158.52, 154.20, 149.45, 141.82, 140.12, 138.33, 135.99, 134.31, 133.56, 130.88, 130.60, 129.31, 129.22, 128.76, 128.61, 127.37, 126.91, 125.67, 123.82, 122.61, 110.37, 108.83, 106.57, 53.18, 38.06, 37.09. HRMS m/z C31H27N5O3S: Calcd. 549.1835, Found 550.1904 [M+H]+. HPLC purity: 99.50%.
4.1.7.3. (S)eN1-(2-aminophenyl)-N3-(1-(benzo[d]thiazol-5-yl(methyl)amino)-1-oxo-3-phe nylpropan-2-yl)isophthalamide (7w).
White solid, yield: 77%. Mp: 169–171 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.76 (s, 1H, NH), 9.52 (s, 1H, CH), 8.92 (d, J = 7.6 Hz, 1H, NH), 8.45 (s, 1H, Ph-H), 8.29 (d, J = 8.6 Hz, 1H, Ph-H), 8.11 (d, J = 9.4 Hz, 2H, Ph-H), 8.00 (d, J = 7.8 Hz, 1H, Ph-H), 7.60 (t, J = 7.8 Hz, 1H, Ph-H), 7.48 (d, J = 8.5 Hz, 1H, Ph-H), 7.20 (d, J = 7.9 Hz, 1H, Ph-H), 7.11 (d, J = 5.1 Hz, 3H, Ph-H), 7.00 (t, J = 7.7 Hz, 1H, Ph-H), 6.86 (d, J = 6.2 Hz, 2H, Ph-H), 6.81 (d, J = 8.0 Hz, 1H, Ph-H), 6.62 (t, J = 7.6 Hz, 1H, Ph-H), 4.96 (s, 2H, NH2), 4.69 (d, J = 7.2 Hz, 1H, CH), 3.28 (s, 3H, NCH3), 3.13–2.90 (m, 2H, PhCH2). 13C NMR (100 MHz, DMSO-d6): δ 171.66 (C=O), 166.32 (C=O), 165.53 (C=O), 158.50, 154.19, 142.51, 141.80, 138.28, 135.25, 134.29, 133.57, 131.05, 130.74, 129.23, 128.77, 128.62, 127.51, 127.19, 127.11, 126.92, 125.65, 124.19, 123.80, 122.58, 117.61, 117.17, 53.12, 38.07, 37.18. HRMS m/z C31H27N5O3S: Calcd. 549.1835, Found 550.1909 [M+H]+. HPLC purity: 99.51%.
4.2. In vitro anti-HIV assay in MT-4 cells
Evaluation of the antiviral activity of the compounds against HIV in MT-4 cells was performed using the MTT assay as described below. Stock solutions (10 × final concentration) of test compounds were added in 25 μL volumes to two series of triplicate wells to allow simultaneous evaluation of their effects on mock- and HIV-infected cells at the beginning of each experiment. Serial 5-fold dilutions of test compounds were made directly inflat-bottomed 96-well microtiter trays using a Biomek 3000 robot (Beckman Instruments, Fullerton, CA). Untreated HIV- and mock-infected cell samples were included as controls. HIV stock (50 μL) at 100–300 CCID50 (50% cell culture infectious doses) or a culture medium was added to either the infected or mock-infected wells of the microtiter tray. Mock-infected cells were used to evaluate the effects of test compounds on uninfected cells to assess the test compounds’ cytotoxicity. Exponentially growing MT-4 cells were centrifuged for 5 min at 220 g, and the supernatant was discarded. The MT-4 cells were resuspended at 6 × 105 cells/mL, and 50 μL volumes were transferred to the microtiter tray wells. Five days after infection, the viability of mock- and HIV-infected cells was examined spectrophotometrically using the MTT assay. The MTT assay is based on the reduction of yellow coloured 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Acros Organics) by mitochondrial dehydrogenase activity in metabolically active cells to a blue-purple formazan that can be measured spectrophotometrically. The absorbances were read in an eight-channel computer-controlled photometer (Infinite M1000, Tecan), at two wavelengths (540 and 690 nm). All data were calculated using the median absorbance value of three wells. The 50% cytotoxic concentration (CC50) was defined as the concentration of the test compound that reduced the absorbance (OD540) of the mock-infected control sample by 50%. The concentration achieving 50% protection against the cytopathic effect of the virus in infected cells was defined as the 50% effective concentration (EC50).
4.3. Binding to CA proteins analysis via surface plasmon resonance (SPR)
All binding assays were performed on a ProteOn XPR36 SPR Protein Interaction Array System (Bio-Rad Laboratories, Hercules, CA). The instrument temperature was set at 25 °C for all kinetic analyses. ProteOn GLH sensor chips were preconditioned with two short pulses each (10 s) of 50 mM NaOH, 100 mM HCl, and 0.5% sodium dodecyl sulfide. Then the system was equilibrated with PBS-T buffer (20 mM sodium phosphate, 150 mM NaCl, and 0.005% polysorbate 20, pH 7.4). The surface of a GLH sensor chip was activated with a 1:100 dilution of a 1:1 mixture of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (0.2 M) and sulfo-N-hydroxysuccinimide (0.05 M). Immediately after chip activation, the HIV-1 NL4–3 capsid protein constructs, purified as in Xu et al. [53], were prepared at a concentration of 100 mg/mL in 10 mM sodium acetate, pH 5.0 and injected across ligand flow channels for 5 min at a flow rate of 30 μL/min. Then, after unreacted protein had been washed out, excess active ester groups on the sensor surface were capped by a 5 min injection of 1 M ethanolamine HCl (pH 8.0) at a flow rate of 5 μL/min. A reference surface was similarly created by immobilizing a non-specific protein (IgG b12 anti-HIV-1 gp120; was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Anti-HIV-1 gp120 Monoclonal (IgG1 b12) from Dr. Dennis Burton and Carlos Barbas) and was used as a background to correct non-specific binding. To prepare a compound for direct binding analysis, compound stock solutions, along with 100% DMSO, and totalling 30 mL was made to a final volume of 1 mL by addition of sample preparation buffer (PBS, pH 7.4). Preparation of analyte in this manner ensured that the concentration of DMSO was matched with that of running buffer with 3% DMSO. Serial dilutions were then prepared in the running buffer (PBS, 3% DMSO, 0.005% polysorbate 20, pH 7.4) and injected at a flow rate of 100 μL/min, for a 1 min association phase, followed by up to a 5 min dissociation phase using the “one-shot kinetics” capability of the Proteon instrument [54]. Data were analyzed using the ProteOn Manager Software version 3.0 (Bio-Rad). The responses from the reference flow cell were subtracted to account for the nonspecific binding and injection artifacts. Experimental data were fitted to a simple 1:1 binding model (where applied). The average kinetic (dissociation [kd] rates) and equilibrium parameters generated from 3 replicates were used to define the off-rates and equilibrium dissociation constant (KD).
4.4. Action stage determination of 7u
Please refer to our previously published research for methodology [44].
4.5. Molecular dynamics simulation
To keep consistency for MD simulation of different series of CA HIV-1 monomer inhibitors, we used the same procedure for docking, MD simulation and its analysis, please refer to our previously published research for methodology [44].
4.6. Metabolic stability in human liver microsomes
Details of the analytical method and raw data are given in the Supporting Information.
4.7. Human plasma stability assay
The pooled frozen plasma was thawed in a water bath at 37 °C before the experiment. Plasma was centrifuged at 4000 rpm for 5 min, and the clots were removed, if any. The pH was adjusted to 7.4 ± 0.1, if required. To obtain the compounds, 1 mM intermediate solution was prepared by diluting 10 μL of the stock solution with 90 μL of DMSO; 1 mM intermediate of positive control Propantheline was prepared by diluting 10 μL of the stock solution with 90 μL of ultrapure water. Then, 100 μM dosing solution was prepared by diluting 20 μL of the intermediate solution (1 mM) with 180 μL of 45% MeOH/H2O, and 98 μL of blank plasma was spiked with 2 μL of dosing solution (100 μM) to achieve 2 μM of the final concentration in triplicate. Samples were incubated at 37 °C in a water bath. At each time point (0, 10, 30, 60, and 120 min), 400 μL of stop solution (200 ng/mL tolbutamide and 200 ng/mL labetalol in 50% ACN/MeOH) was added to precipitate protein and mixed thoroughly. Sample plates were centrifuged at 4000 rpm for 10 min. An aliquot of supernatant (50 mL) was transferred from each well and mixed with 100 μL of ultra-pure water. The samples were shaken at 800 rpm for about 10 min before submitting them to LC-MS/MS analysis.
Supplementary Material
Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (NSFC Nos. 82173677, 81773574), the Shandong Provincial Key research and development project (No. 2019JZZY021011), Science Foundation for Outstanding Young Scholars of Shandong Province (ZR2020JQ31), Foreign cultural and educational experts Project (GXL20200015001), Qilu Young Scholars Program of Shandong University, the Taishan Scholar Program at Shandong Province and NIH/NIAID grant R01AI150491 (Cocklin, PI, Salvino, CoeI).
Footnotes
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejmech.2021.113903.
References
- [1].De Clercq E, Antivirals: past, present and future, Biochem. Pharmacol 85 (2013) 727–744. [DOI] [PubMed] [Google Scholar]
- [2].Ponnan SM, Vidyavijayan KK, Thiruvengadam K, Hilda JN, Mathayan M, Murugavel KG, Hanna LE, Role of circulating T follicular helper cells and stem-like memory CD4(+) T cells in the pathogenesis of HIV-2 infection and disease progression, Front. Immunol 12 (2021) 666388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dang Z, Lai W, Qian K, Ho P, Lee KH, Chen CH, Huang L, Betulinic acid derivatives as human immunodeficiency virus type 2 (HIV-2) inhibitors, J. Med. Chem 52 (2009) 7887–7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Smith RA, Wu VH, Zavala CG, Raugi DN, Ba S, Seydi M, Gottlieb GS, University of Washington-Dakar HIVSG, Vitro antiviral activity of cabotegravir against HIV-2, Antimicrob. Agents Chemother 62 (2018) e01299–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Margolis DM, Hazuda DJ, Combined approaches for HIV cure, Curr. Opin. HIV AIDS 8 (2013) 230–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Davenport MP, Khoury DS, Cromer D, Lewin SR, Kelleher AD, Kent SJ, Functional cure of HIV: the scale of the challenge, Nat. Rev. Immunol 19 (2019) 45–54. [DOI] [PubMed] [Google Scholar]
- [7].Chong H, Xue J, Zhu Y, Cong Z, Chen T, Guo Y, et al. , Design of novel HIV-1/2 fusion inhibitors with high therapeutic efficacy in rhesus monkey models, J. Virol 92 (2018) e00775–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Feinstein MJ, Achenbach CJ, Stone NJ, Lloyd-Jones DM, A systematic review of the usefulness of statin therapy in HIV-infected patients, Am. J. Cardiol 115 (2015) 1760–1766. [DOI] [PubMed] [Google Scholar]
- [9].Zhan P, Pannecouque C, De Clercq E, Liu X, Anti-HIV drug discovery and development: current innovations and future trends, J. Med. Chem 59 (2016) 2849–2878. [DOI] [PubMed] [Google Scholar]
- [10].Du J, Guo J, Kang D, Li Z, Wang G, Wu J, Zhang Z, Fang H, Hou X, Huang Z, Li G, Lu X, Liu X, Ouyang L, Rao L, Zhan P, Zhang X, Zhang Y, New techniques and strategies in drug discovery, Chin. Chem. Lett 31 (2020) 1695–1708. [Google Scholar]
- [11].Sun L, Zhang X, Xu S, Huang T, Song S, Cherukupalli S, Zhan P, Liu X, An insight on medicinal aspects of novel HIV-1 capsid protein inhibitors, Eur. J. Med. Chem 217 (2021) 113380. [DOI] [PubMed] [Google Scholar]
- [12].Engelman AN, Singh PK, Cellular and molecular mechanisms of HIV-1 integration targeting, Cell. Mol. Life Sci 75 (2018) 2491–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Blair WS, Pickford C, Irving SL, Brown DG, Anderson M, Bazin R, Cao J, Ciaramella G, Isaacson J, Jackson L, Hunt R, Kjerrstrom A, Nieman JA, Patick AK, Perros M, Scott AD, Whitby K, Wu H, Butler SL, HIV capsid is a tractable target for small molecule therapeutic intervention, PLoS Pathog 6 (2010), e1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Engelman AN, HIV capsid and integration targeting, Viruses 13 (2021) 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ganser-Pornillos BK, Cheng A, Yeager M, Structure of full-length HIV-1 CA: a model for the mature capsid lattice, Cell 131 (2007) 70–79. [DOI] [PubMed] [Google Scholar]
- [16].Pornillos O, Ganser-Pornillos BK, Kelly BN, Hua Y, Whitby FG, Stout CD, Sundquist WI, Hill CP, Yeager M, X-ray structures of the hexameric building block of the HIV capsid, Cell 137 (2009) 1282–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wagner JM, Zadrozny KK, Chrustowicz J, Purdy MD, Yeager M, Ganser-Pornillos BK, Pornillos O, Crystal structure of an HIV assembly and maturation switch, Elife 5 (2016), e17063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Le Sage V, Mouland AJ, Valiente-Echeverría F, Roles of HIV-1 capsid in viral replication and immune evasion, Virus Res 193 (2014) 116–129. [DOI] [PubMed] [Google Scholar]
- [19].Campbell EM, Hope TJ, HIV-1 capsid: the multifaceted key player in HIV-1 infection, Nat. Rev. Microbiol 13 (2015) 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rossi E, Meuser ME, Cunanan CJ, Cocklin S, Structure, function, and interactions of the HIV-1 capsid protein, Life 11 (2021) 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Novikova M, Zhang Y, Freed EO, Peng K, Multiple roles of HIV-1 capsid during the virus replication cycle, Virol. Sin 34 (2019) 119–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chen B, HIV capsid assembly, mechanism, and structure, Biochemistry 55 (2016) 2539–2552. [DOI] [PubMed] [Google Scholar]
- [23].Ning J, Zhong Z, Fischer DK, Harris G, Watkins SC, Ambrose Z, Zhang P, Truncated CPSF6 forms higher-order complexes that bind and disrupt HIV-1 capsid, J. Virol 92 (2018) e00368–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Saito A, Henning MS, Serrao E, Dubose BN, Teng S, Huang J, Li X, Saito N, Roy SP, Siddiqui MA, Ahn J, Tsuji M, Hatziioannou T, Engelman AN, Yamashita M, Capsid-CPSF6 interaction is dispensable for HIV-1 replication in primary cells but is selected during virus passage in vivo, J. Virol 90 (2016) 6918–6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Matreyek KA, Yücel SS, Li X, Engelman A, Nucleoporin NUP153 phenylalanine-glycine motifs engage a common binding pocket within the HIV-1 capsid protein to mediate lentiviral infectivity, PLoS Pathog 9 (2013), e1003693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Meehan AM, Saenz DT, Guevera R, Morrison JH, Peretz M, Fadel HJ, Hamada M, van Deursen J, Poeschla EM, A cyclophilin homology domain-independent role for Nup358 in HIV-1 infection, PLoS Pathog 10 (2014), e1003969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim K, Dauphin A, Komurlu S, McCauley SM, Yurkovetskiy L, Carbone C, Diehl WE, Strambio-De-Castillia C, Campbell EM, Luban J, Cyclophilin A protects HIV-1 from restriction by human TRIM5a, Nat. Microbiol 4 (2019) 2044–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fricke T, White TE, Schulte B, de Souza Aranha Vieira DA, Dharan A, Campbell EM, Brandariz-Nuñez A, Diaz-Griffero F, MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1, Retrovirology 11 (2014) 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Rebensburg SV, Wei G, Larue RC, Lindenberger J, Francis AC, Annamalai AS, Morrison J, Shkriabai N, Huang SW, KewalRamani V, Poeschla EM, Melikyan GB, Kvaratskhelia M, Sec24C is an HIV-1 host dependency factor crucial for virus replication, Nat. Microbiol 6 (2021) 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Blanco-Rodriguez G, Gazi A, Monel B, Frabetti S, Scoca V, Mueller F, Schwartz O, Krijnse-Locker J, Charneau P, Di Nunzio F, Remodeling of the core leads HIV-1 preintegration complex into the nucleus of human lymphocytes, J. Virol 94 (2020) e00135–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Burdick RC, Li C, Munshi M, Rawson JMO, Nagashima K, Hu WS, Pathak VK, HIV-1 uncoats in the nucleus near sites of integration, Proc. Natl. Acad. Sci. U. S. A 117 (2020) 5486–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ, HIV-1 evades innate immune recognition through specific cofactor recruitment, Nature 503 (2013) 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Dick A, Cocklin S, Recent advances in HIV-1 gag inhibitor design and development, Molecules 25 (2020) 1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Thenin-Houssier S, Valente ST, HIV-1 capsid inhibitors as antiretroviral agents, Curr. HIV Res 14 (2016) 270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Carnes SK, Sheehan JH, Aiken C, Inhibitors of the HIV-1 capsid, a target of opportunity, Curr. Opin. HIV AIDS 13 (2018) 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhang J, Liu X, De Clercq E, Capsid (CA) protein as a novel drug target: recent progress in the research of HIV-1 CA inhibitors, Mini Rev. Med. Chem 9 (2009) 510–518. [DOI] [PubMed] [Google Scholar]
- [37].Kortagere S, Xu JP, Mankowski MK, Ptak RG, Cocklin S, Structure-activity relationships of a novel capsid targeted inhibitor of HIV-1 replication, J. Chem. Inf. Model 54 (2014) 3080–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Kortagere S, Madani N, Mankowski MK, Schön A, Zentner I, Swaminathan G, Princiotto A, Anthony K, Oza A, Sierra LJ, Passic SR, Wang X, Jones DM, Stavale E, Krebs FC, Martín-García J, Freire E, Ptak RG, Sodroski J, Cocklin S, Smith AB 3rd, Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid, J. Virol 86 (2012) 8472–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shi J, Zhou J, Halambage UD, Shah VB, Burse MJ, Wu H, Blair WS, Butler SL, Aiken C, Compensatory substitutions in the HIV-1 capsid reduce the fitness cost associated with resistance to a capsid-targeting small-molecule inhibitor, J. Virol 89 (2015) 208–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Margot N, Ram R, Rhee M, Callebaut C, Absence of lenacapavir (GS-6207) phenotypic resistance in HIV gag cleavage site mutants and in isolates with resistance to existing drug classes, Antimicrob. Agents Chemother 65 (2021) e02057–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Singh K, Gallazzi F, Hill KJ, Burke DH, Lange MJ, Quinn TP, Neogi U, Sönnerborg A, GS-CA compounds: first-in-class HIV-1 capsid inhibitors covering multiple grounds, Front. Microbiol 10 (2019) 1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Link JO, Rhee MS, Tse WC, Zheng J, Somoza JR, Rowe W, Begley R, Chiu A, Mulato A, Hansen D, Singer E, Tsai LK, Bam RA, Chou CH, Canales E, Brizgys G, Zhang JR, Li J, Graupe M, Morganelli P, Liu Q, Wu Q, Halcomb RL, Saito RD, Schroeder SD, Lazerwith SE, Bondy S, Jin D, Hung M, Novikov N, Liu X, Villasenor AG, Cannizzaro CE, Hu EY, Anderson RL, Appleby TC, Lu B, Mwangi J, Liclican A, Niedziela-Majka A, Papalia GA, Wong MH, Leavitt SA, Xu Y, Koditek D, Stepan GJ, Yu H, Pagratis N, Clancy S, Ahmadyar S, Cai TZ, Sellers S, Wolckenhauer SA, Ling J, Callebaut C, Margot N, Ram RR, Liu YP, Hyland R, Sinclair GI, Ruane PJ, Crofoot GE, McDonald CK, Brainard DM, Lad L, Swaminathan S, Sundquist WI, Sakowicz R, Chester AE, Lee WE, Daar ES, Yant SR, Cihlar T, Clinical targeting of HIV capsid protein with a long-acting small molecule, Nature 584 (2020) 614–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Marcelin AG, Charpentier C, Jary A, Perrier M, Margot N, Callebaut C, Calvez V, Descamps D, Frequency of capsid substitutions associated with GS-6207 in vitro resistance in HIV-1 from antiretroviral-naive and -experienced patients, J. Antimicrob. Chemother 75 (2020) 1588–1590. [DOI] [PubMed] [Google Scholar]
- [44].Wu G, Zalloum WA, Meuser ME, Jing L, Kang D, Chen CH, Tian Y, Zhang F, Cocklin S, Lee KH, Liu X, Zhan P, Discovery of phenylalanine derivatives as potent HIV-1 capsid inhibitors from click chemistry-based compound library, Eur. J. Med. Chem 158 (2018) 478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sun L, Huang T, Dick A, Meuser ME, Zalloum WA, Chen CH, Ding X, Gao P, Cocklin S, Lee KH, Zhan P, Liu X, Design, synthesis and structure-activity relationships of 4-phenyl-1H-1,2,3-triazole phenylalanine derivatives as novel HIV-1 capsid inhibitors with promising antiviral activities, Eur. J. Med. Chem 190 (2020) 112085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sun L, Dick A, Meuser ME, Huang T, Zalloum WA, Chen CH, Cherukupalli S, Xu S, Ding X, Gao P, Kang D, De Clercq E, Pannecouque C, Cocklin S, Lee KH, Liu X, Zhan P, Design, synthesis, and mechanism study of benzenesulfonamide-containing phenylalanine derivatives as novel HIV-1 capsid inhibitors with improved antiviral activities, J. Med. Chem 63 (2020) 4790–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Xu S, Sun L, Huang B, Liu X, Zhan P, Medicinal chemistry strategies of targeting HIV-1 capsid protein for antiviral treatment, Future Med. Chem 12 (2020) 1281–1284. [DOI] [PubMed] [Google Scholar]
- [48].Xu JP, Francis AC, Meuser ME, Mankowski M, Ptak RG, Rashad AA, Melikyan GB, Cocklin S, Exploring modifications of an HIV-1 capsid inhibitor: design, synthesis, and mechanism of action, J. Drug Des. Res 5 (2018) 1070. [PMC free article] [PubMed] [Google Scholar]
- [49].Meuser ME, Reddy PAN, Dick A, Maurancy JM, Salvino JM, Cocklin S, Rapid optimization of the metabolic stability of a human immunodeficiency virus type-1 capsid inhibitor using a multistep computational workflow, J. Med. Chem 64 (2021) 3747–3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Saito A, Ferhadian D, Sowd GA, Serrao E, Shi J, Halambage UD, Teng S, Soto J, Siddiqui MA, Engelman AN, Aiken C, Yamashita M, Roles of capsid-interacting host factors in multimodal inhibition of HIV-1 by PF74, J. Virol 90 (2016) 5808–5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Wang L, Casey MC, Vernekar SKV, Sahani RL, Kirby KA, Du H, Zhang H, Tedbury PR, Xie J, Sarafianos SG, Wang Z, Novel PF74-like small molecules targeting the HIV-1 capsid protein: balance of potency and metabolic stability, Acta Pharm. Sin. B 11 (2021) 810–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Deliv. Rev 46 (2001) 3–26. [DOI] [PubMed] [Google Scholar]
- [53].Xu JP, Branson JD, Lawrence R, Cocklin S, Identification of a small molecule HIV-1 inhibitor that targets the capsid hexamer, Bioorg. Med. Chem. Lett 26 (2016) 824–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bravman T, Bronner V, Lavie K, Notcovich A, Papalia GA, Myszka DG, Exploring “one-shot” kinetics and small molecule analysis using the ProteOn XPR36 array biosensor, Anal. Biochem 358 (2006) 281–288. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.