

Abstract
The world was ambushed in 2019 by the COVID-19 virus which affected the health, economy, and lifestyle of individuals worldwide. One way of combating such a public health concern is by using appropriate, rapid, and unbiased diagnostic tools for quick detection of infected people. However, a current dearth of bioinformatics tools necessitates modeling studies to help diagnose COVID-19 cases. Molecular-based methods such as the real-time reverse transcription polymerase chain reaction (rRT-PCR) for detecting COVID-19 is time consuming and prone to contamination. Modern bioinformatics tools have made it possible to create large databases of protein sequences of various diseases, apply data mining techniques, and accurately diagnose diseases. However, the current sequence alignment tools that use these databases are not able to detect novel COVID-19 viral sequences due to high sequence dissimilarity. The objective of this study, therefore, was to develop models that can accurately classify COVID-19 viral sequences rapidly using protein vectors generated by neural word embedding technique. Five machine learning models; K nearest neighbor regression (KNN), support vector machine (SVM), random forest (RF), Linear discriminant analysis (LDA), and Logistic regression were developed using datasets from the National Center for Biotechnology. Our results suggest, the RF model performed better than all other models on the training dataset with 99% accuracy score and 99.5% accuracy on the testing dataset. The implication of this study is that, rapid detection of the COVID-19 virus in suspected cases could potentially save lives as less time will be needed to ascertain the status of a patient.
Keywords: COVID-19, Natural language processing, Word vectors, Receiver operator characteristic curve, Continuousbag-of-words, Artificial intelligence
Introduction
The world was ambushed in 2019 by the Corona virus disease 2019 (COVID-19) which affected the health, economy, and lifestyle of individuals worldwide. First discovered in Wuhan, Hubei Province, China in late 2019, COVID-19 has spread worldwide and subsequently been declared as a global pandemic by the World Health Organization on March 11, 2020 [1]. At the time of writing this paper, the total confirmed cases of COVID-19 stood at 298,915,721 with 5,469,303 confirmed deaths worldwide [2]. COVID-19 is a novel infectious disease caused by severe acute respiratory syndrome coronavirus-2 (SARS-COV-2), a new type of virus family that had not yet been detected in humans [3]. The viral disease is transmitted through person-to-person contact through respiratory droplets generated by breathing, sneezing, coughing etc. as well as direct contact with an infected person [3]. As a result of the widespread ubiquitous nature of the COVID-19 virus, scientists and clinicians are researching new technologies to screen infected patients at various stages of the viral infection [4].
The heightened interest of researchers on the COVID-19 virus is in part due to the discovery and publishing of the genetic sequence of the virus on January 11, 2020 [5]. The current standard diagnostic testing method for COVID-19 is the real-time reverse transcription polymerase chain reaction (rRT-PCR) which detects the virus’ genetic material and the antigen test which detects specific proteins in the virus [6]. According to Long et al. [7], it takes about 5–6 hours to obtain results from rRT-PCR. In addition, the average accuracy of the rRt-PCR has been reported to be 60–70% [7] which means some tests may turn out to be false positives. Nasopharyngeal swabs are commonly taken to the lab before the swabs are then investigated using PCR which could take longer in places where PCR is not readily available. Although the rRT-PCR test for COVID-19 has been largely successful, sometimes, there can be a 12–48-hour lag in reporting the results to individuals. The apparent lag in getting test results prolongs the time in which an asymptomatic person may inadvertently transmit the disease to others. Rani et al. [8] also argue that doctors spend time studying COVID-19 test reports which can be time-consuming. In addition, molecular-based methods such as rRT-PCR and microarrays are time consuming and can lead to contamination of sequences [9].
Modern bioinformatics tools have made it possible to create large databases of protein sequences of various diseases [10, 11], apply data mining techniques, and accurately diagnose diseases [12, 13]. However, current sequence alignment methods which depend on databases to align sequences are not able to detect novel sequences due to the high dissimilarities among the queried sequences [14]. In such cases, there is a risk of pronouncing a potential COVID-19 case as a negative when the patient is actually infected with the virus (false negative) [14]. There is therefore a need for the development of alternative data-driven models which can help in the quick identification COVID-19 using modern machine learning techniques.
Despite the recent discovery of different COVID-19 vaccines, Forni et al. [15] argues that the development of the vaccines in a short period of time inherently suggests that the long-term efficacy and the side effects of the vaccines is still not known. We now know that the BioNTech/Pfizer vaccine is 95% efficacious after two doses and apparently safe [16]. However, Shen et al. [17] raises the challenge of equitable distribution of the COVID vaccine which underscores the uphill battle ahead to fight COVID-19. Most people in developing countries have yet to receive a single dose of the vaccine and must rely solely on implementing public health practices to prevent spread of COVID-19. The prompt and early detection of the COVID-19 virus using artificial intelligence is still an evolving area that needs more attention.
Review of relevant studies
Recently, there has been a surge in the use of machine learning and artificial intelligence (AI) to either screen, predict or forecast the occurrence of COVID-19 [4]. In this section, we briefly discuss studies that used laboratory test results as a basis for detecting COVID-19 using various machine learning techniques. We also briefly outline studies that applied machine learning methods to protein sequences to predict the presence or absence of COVID-19. Dutta and Bandyopadhyay [18] determined the feasibility of using machine learning methods to evaluate the prediction accuracy of confirmed negative, released, and death cases of COVID-19. Khanday et al. [19] used supervised machine learning techniques (LR, Multinomial Naïve Bayes, SVM, DT, bagging, AdaBoost, RF classifier, and stochastic gradient boosting) to classify textural clinical reports of COVID-19 patients. The classification was done to separate the cases into four different categories COVID-19, SARS, and acute respiratory distress syndrome (ARDS). Their results suggest that logistic regression and multinomial Naïve Bayesian classifiers gave the best performance results with 94% precision, 96% recall, 95% F1 score and 96.2% accuracy. Aljame et al. [20] obtained 5644 data samples with 559 confirmed COVID-19 cases from the Albert Einstein Hospital in Brazil. The authors used classical methods (extra trees, random forest, and logistic regression) to predict the presence of COVID-19. They then increased the performance by applying XGBoost algorithm to the prediction results from the classical methods. Overall, the authors reported that the improved ensemble model achieved an accuracy of 99.88%, area under the curve (AUC) of 99.38%, a sensitivity of 98.72%, and a specificity of 99.99%. Brinati et al. [21] used machine learning classification algorithms (decision tree, extremely randomized trees, K-nearest neighbor, logistic regression, naïve Bayes, random forest, and support vector regression) to develop models to detect COVID-19 from routine blood examinations of 279 patients. The best performing model was a modified version of the random forest model called the three-way random forest classifier. The authors report that the three-way random forest model achieved an accuracy of 86%, sensitivity of 95%, and a specificity of 75%. Turabieh and Karaa [22] predicted the presence of COVID-19 by using blood test results of 5644 patient. The authors combined wrapper feature selection algorithm with convolutional neural network, decision trees, K-nearest neighbor, and na¨ıve bayes. The authors obtained a best accuracy of 76% when they combined a wrapper feature selection method based called a binary genetic algorithm with convolutional neural network.
Deep learning models have also been applied to help diagnose COVID-19 [23, 24]. Alakus and Turkoglu [23] developed and evaluated clinical predictive models to determine COVID-19 infection using 111 laboratory findings from 5644 patients. The algorithms employed in their study included: artificial neural networks (ANN), convolutional neural networks (CNN), long-short term memory (LSTM), recurrent neural networks (RNN), CNNLSTM, and CNNRNN. Their results suggest the CNNLSTM model attained the best accuracy, recall, and AUC values of 92.3%, 93.68%, and 90.0% respectively. Göreke et al. [24] created a feature group based on laboratory findings which were used to design a hybrid classifier based on deep learning architecture (ANN, CNN, and RNN) to detect COVID-19. The authors report that RNN attained the best accuracy of 94.95%, F1-score of 94.98%, precision of 94.98%, and recall of 94.98%. An ensemble-based model called Deep Forest was developed by Aljame [6] by combining three classifiers: extra tree, XGBoost, and LightGBM. Their proposed model achieved an accuracy of 99.5%, a sensitivity of 95.3%, and a specificity of 99.96%.
Some studies also focused on developing machine learning models by finding the degree of similarity of a COVID-19 genome against a given genomic sequence [8, 25–27]. Rani et al. [8] developed a deep learning model to find similarities between a given genome and a COVID-19 genome and detecting the presence of COVID-19 in humans using data from the National Centre for Biotechnology Information. The authors employed Convolutional Neural Networks (CNN) and Long-Short-Term-Memory (LSTM) for improving the accuracy of classification and similarity score prediction. The authors claimed their CNN model was efficient at detecting genome sequences of COVID-19 in a host genome with 99.27% accuracy. Jamshidi et al. [25] reviewed studies that delineated the application of Generative Adversial Networks (GANs) in detecting COVID-19 genome in a human genome sample. Cleemput et al. [26] developed a bioinformatics based tool “Genome Detective Coronavirus Typing Tool” which was capable of identifying genomic sequences of COVID-19 with 87.5% accuracy. Similarly, Arslan [27] developed a method that distinguishes human COVID-19 genome from bat SARS-CoV-like coronavirus. The authors applied classical machine learning techniques like support vector machine (SVM), K-nearest neighbor (KNN), decision trees (DT), random forests (RF), Adaptive Boosting (AdaBoost), and multi-layer perceptron (MLP) on 1000 genome sequences of COVID-19 and 615 genome sequences of other types of human coronavirus. They report that their KNN model attained an accuracy of 99.2% in detecting COVID-19 genomes. While using laboratory test results and genomic sequences to predict COVID-19 cases have been largely ubiquitous, few studies focus on using the protein sequences of samples from COVID-19 cases to predict the presence or absence of COVID-19 [12, 13, 28].
Rationale for application of word vectors
Protein sequences are represented by a continuous string of letters arranged in the order of their amino acid monomers, and this information can be translated to determine properties of the protein such as the shape, thus, the application of word vectors for preprocessing [29]. Word vectors are numerical representations of words in a low dimensional space. These vectors are generated using a one-layer neural network with hyperparameters such as context windows, vector dimension, etc. Different techniques for vectorizing words include skip-gram, continuous-bag-of-words (CBOW), Global vectors (GloVe), FastText [30]. The former two language models are collectively known as Word2vec [29]. Skip-gram predicts the context words (surrounding words) given the target word within a predefined context window size, whereas CBOW predicts the context words given a target word [31]. GloVe model uses the probability of word co-occurrence within a context window and claims that ratios of the co-occurrence probabilities of words can distinctively indicate the relatedness of words [30]. Like the skip-gram, it predicts the context words given a target word. Different tasks such as word similarity, analogy, and other downstream processing usage have yielded varied results using these different language models. Therefore, there is no clear advantage of using a particular language model. Word embedding is preferred to one-hot encoding because of its ability to provide expressive representation of word relations instead of zeros and ones in one-hot encoding of words [32]. Also, one-hot-encoding is computationally expensive due to the curse of dimensionality [33]. Ubiquitous successful application of word embedding models in biology and bioinformatics have been reported in literature. Chen et al. [34] used the GloVe model to represent protein sequences and subsequently used it to predict self-interacting proteins. Also, word embedding models have been used in down streaming prediction of antimicrobial peptides [35] and identifying substrate specificities of transporters [36].
Purpose of study
The main goal of this machine learning study was to use trained data to elicit some underlying patterns in big data, build models using the trained data, and make predictions based on an optimized line of best fit [37]. To the best of our knowledge, no study has implemented continuous-bag-of-words to generate vectors of the COVID-19 viral sequences and used the vectors as input features to build models used for prediction. By using modern data science tools like machine learning, models can be built to accurately predict if a viral sequence can cause COVID-19 or not. The objective of this study therefore was to develop models that can rapidly and accurately classify a viral sequence as COVID-19 positive or negative based on its similarity to labelled COVID-19 data. Here, we exploited different machine learning algorithms, namely, support vector machine, K-nearest neighbor, random forest, linear discriminant analysis, and logistic regression to assist in identifying COVID-19 viral sequences. We first created k-mers of the protein sequences and subsequently vectorized them using continuous-bag-of-words neural embedding technique to create the input features building the model.
Materials and methods
Description of the database
Protein sequence data of COVID-19 virus was obtained from a platform (NCBI Virus) of the National Center for Biotechnology [10]. The “NCBI virus” is an integrative platform designed to support retrieval, display, and analysis of large viral sequences [10]. Non-COVID-19 virus sequences were also obtained from the NCBI taxonomy database [38]. The main purpose of the NCBI taxonomy database is to aggregate information on organism names and classifications for every sequence in the protein sequence databases of the International Nucleotide Sequence Database collaboration [38].
Preprocessing
Sequences were obtained to build a corpus which resulted in about 10,533 unique (words). We created trigrams from each sequence, and each trigram was considered as a word. The trigrams consisted of continuous sliding windows of each of the sequences. The trigrams were trained using the continuous bag of words (CBOW) neural model [29]. Window sizes of , , were used to train the corpus at a vector dimension of 200. Context window size of was also found to be suitable, and, thus, chosen.
Five hundred COVID-19 viral sequences and non-COVID-19 viral sequences considered as positive and negative datasets, respectively, were used as training datasets. CD-Hit was used to reduce redundancy to make sure no two sequences overlap more than 90% (percent identity) [39]. The vectors of trigrams for each sequence used for training the models were obtained from the trained CBOW model and added to construct features equal to the vector dimension of 200. The word vectors were standardized to range between 0 and 1. After this, principal component analysis was used to reduce redundancy in the dataset [40, 41]. Principal component analysis led to the reduction of features from 200 to 10 based on the resulting cumulative explained variance.
Model training using cross-validation
Logistic regression (LR), random forest (RF), support vector machine (SVM), K-nearest neighbor (KNN), linear discriminant (LD) models were trained on the data. A ten-fold cross-validation was used in training and evaluation of the models [42]. N-1 folds were used to train the models and evaluated on the remaining folds iteratively. To test the performance of the model on unseen data, we divided the data into 70% for training and 30% for testing. Here we report the evaluated results (training) and the test results of all models. Figure 1 depicts a summary of the steps that were followed in implementing the machine learning algorithms.
Fig. 1.

A summary of the methodology we adopted in this paper
Model evaluation
Generally, four statistical criteria were used to assess how well the models developed performed on data that was used to train the models and on data that was unseen by the models. Three statistical criteria (Accuracy, Precision, Recall, and F1 scores) were used to compare the performance of the models [43].
, , ,
TP true positive, FP false positives, TN true negative, FN false negatives, F1 F1 score.
Machine learning techniques
K-nearest neighbor
K-Nearest neighbor (KNN) is one of the simplest non-parametric learning algorithms that is easy to implement. KNN has been referred to as a “lazy” learning algorithm since it does not yield a function previously but yields the closest “K” records of the training data set [44]. KNN predicts a target class of an unseen datapoint by comparing it to 'K' similar cases in the input training dataset. To implement the KNN classifier to an unknown sample and a training dataset, a value of K is chosen and the distances of unknown cases to all cases in the training set is computed. In calculating the distance between an observation to the nearest neighbors, the Euclidean distance equation can be used. Cases in the training set with the shortest distances to the unknown observation are then used to predict the class of the unknown observation [45]. A value for K which is a hyperparameter of the algorithm that can be tuned is chosen carefully as a low value can lead to overfitting of the data while a larger K value can lead to a higher bias [46]. It is common to use cross-validation to choose an optimal value of K that reduces the number of errors while increasing accuracy.
Support vector machine
Support vector machine (SVM) is a supervised algorithm that classifies cases by finding a separator. With an appropriate non-linear mapping to a high-dimensional feature space, data points which will otherwise not be linearly separable can be separated by a hyperplane [47]. Noble [48] argues that to understand and implement SVM, one needs to first understand what the separating hyperplane is, the maximum margin hyperplane, the soft margin, and the kernel function. In simple terms, to implement SVM, first, an optimized hyperplane is found by minimizing a regularized cost function [49]. The hyperplane is then learned using the data set aside for training the algorithm making sure the hyperplane maximizes the margin between the classified data. The best hyperplane is one that results in the largest separation of the two classes being classified. Once an optimized linear function has been obtained, new predictions are classified by feeding in unknown data to this optimized linear function. An important decision that must be made during the training of the SVM model is the type of kernel function to use. Some popular kernel functions that can be used include linear, polynomial, radial basis functions and sigmoid functions.
Random forest
The Random Forest (RF) classifier algorithm is a supervised method of classifying input variables based on constructing multiple decision trees during model training and outputting a class. According to Breiman [50], RF refers to a combination of tree predictors in a way that each tree is constructed based on random independent sampling of vectors with the same distribution from the trees in the forest. The RF algorithm addresses the problem of overfitting of training data sets which is common with decisions when they become complex [51]. The method of using multiple trees to make a prediction is known as bootstrap aggregation (bagging) [52]. According to Hastie et al. [53], to generate a prediction from a random forest, you have to first create a random bootstrap sample from the original dataset with replacement, create a decision tree using the bootstrapped data but at each node of the tree, randomly select a subset of predictors to obtain the best split from the subset. Finally, from the output RF trees, a new prediction is made by choosing the class that had the majority votes. Two of the most important parameters to specify during training is the number of decision trees to be grown and the number of predictors at each node of a decision tree [50].
Linear discriminant analysis
Linear discriminant analysis (LDA) is a dimension reduction technique method whereby an optimal transformation that maximizes class separability is found [54]. While many techniques exist for classification of data, principal component analysis (PCA) and LDA are the most used techniques for data classification and dimensionality reduction [55]. Dimensional reduction techniques like LDA are mostly used to reduce dimensions by removing redundant features and transforming those features into lower dimensions [56]. LDA has been prominently used in bankruptcy prediction and facial recognition [57]. According to Tharwat et al. [56], to implement LDA, the separability between different classes is first calculated, also known as the between-class variance or between-class matrix. The distance between the mean and the samples of each class are then calculated (within-class variation). Finally, a lower dimensional space which maximizes the between-class variance and minimizes the within class variance is then calculated.
Logistic regression
Logistic regression (LR) is a classification algorithm for categorical variables. In LR, the aim of developing a model is to describe the relationship of many independent variables to a dichotomous dependent variable i.e. a variable that takes two possible outcomes [58]. LR models binary response variables and tries to fit an equation to that data [59]. According to Kleinbaum et al. [58], LR is by far the most popular modeling procedure for analyzing epidemiological data when the measure of an illness results in two outcomes. In LR, the independent variable should be continuous but if categorical then they should be converted to indicator variables.
Results and discussion
Model evaluation aimed to quantify the ability of the models developed to accurately generalize to new sequence data that was not used to train the models. Four evaluation metrics were used in this classification study (accuracy, precision, recall, and F1 score). Figure 2
Fig. 2.
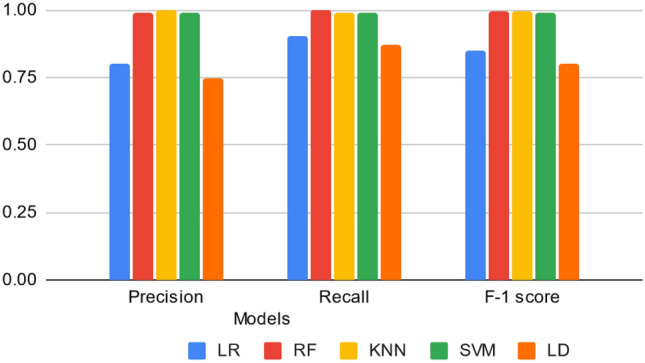
Average precision, recall, and F-1 score of the model after 10-fold cross-validation for the five models: Logistic Regression (LR), Random Forest (RF), K-Nearest Neighbor (KNN), Support Vector Machine (SVM), and Linear Discriminant Analysis (LDA).
Table 1 presents the result of the training and test accuracy of the five models. Test accuracy here refers to the ratio of the correctly predicted COVID-19 positive viral sequence to the total predictions made on the test set [60]. Prediction on the test dataset was done after 10-fold cross validations and the model was used to predict the sequences. Random forest (RF) produced the highest average train accuracy of 0.99 while the LDA model resulted in the lowest accuracy among all the models. Also, the RF model achieved 99.5% accuracy on the test viral sequences. Dutta & Bandyopadhyay [18] investigated the feasibility of using machine learning methods to evaluate the prediction accuracy of confirmed, negative, released, and death cases of COVID-19. Their study applied a Long short-term memory (LSTM), a Gated Recurrent Unit (GRU), and a combined LSTM-GRU frameworks to predict the COVID-19 cases. Their study revealed that the combined LSTM-GRU based Recurrent Neural Network model produced the best prediction accuracy with 0.87 on confirmed cases. Our model’s accuracy was, thus, 12% higher than the accuracy achieved in their study. In another study by Khanday et al. [19], logistic regression and multinomial Naïve Bayesian algorithms achieved an accuracy of 96.2% which is lower than the accuracy achieved by the RF model in our study. Afify and Zanaty [13] were able to achieve an accuracy of 100% when Linear Regression, KNN, and SVM were used to classify human protein sequences of COVID-19 using data from 27 countries. Mohammed et al. [61] applied decision trees, logistic regression, naive bayes, support vector machine, and artificial neural network to develop modals to predict COVID-19 cases for epidemiological data in Mexico. Their results suggest decision trees had the highest prediction accuracy with 94.99%. Our best model’s accuracy was higher than the best model prediction accuracy in [61]. We did not use decision trees for our study because decision trees have been shown to overfit to training data especially when they become too complex [51]. Random forest performed best because of its ensemble nature whereby multiple decision trees are constructed during model training and the average prediction of the decision trees gives the class of a new observation [51].
Table 1.
Accuracy of different machine learning techniques in detecting COVID-19 viral sequences
| Models | Train accuracy | Test accuracy |
|---|---|---|
| Logistic regression | 0.856 (0.022535) | 0.85 |
| Linear discriminant analysis | 0.841 (0.028532) | 0.80 |
| K-nearest neighbor | 0.984 (0.012562) | 0.995 |
| Random forest | 0.990 (0.007500) | 0.995 |
| Support vector regression | 0.973 (0.024238) | 0.99 |
Values in bold are highest accuracy in each column. Numbers in parenthesis are the standard deviations after 10-fold cross validation
Receiver operating characteristic (ROC) curves for the five models were created by plotting the true positive rate (sensitivity) on the y-axis against the false-positive rate (1-specificity) on the x-axis (Fig. 3). Sensitivity was formally defined as the proportion of positives which are correctly identified (the probability of a positive test) while specificity represented the proportion of negatives correctly identified (probability of a negative test) [62]. The area under the curve (AUC) for each model is displayed in each of the graphs. The higher the AUC value, the better the model. Model accuracies correlate with AUC values, that is, models that achieved high accuracies also achieved high AUC values. The use of protein vectors generated using neural word embedding technique word2vec (continuous bag-of-words) to represent and extract features has resulted in a high accuracy in detecting COVID-19 viral sequences. The ability of word embeddings to efficiently represent characteristic relations among words through the building of low dimensional vectors could be attributed to the relative higher performance of models in this study.
Fig. 3.

Receiver operating characteristic curves for the machine learning models after 10-fold cross-validation. The models: a Logistic regression, b Linear discriminant analysis, c K-nearest neighbor, d Random Forest, and e Support vector machine is presented with their respective area under curve values
Conclusions
With the enormous amounts of genetic sequence data generated on a regular basis due to the advancement in whole-genome sequencing, there exists an opportunity for researchers to transform these data into useful insights. In this study, we apply popular machine learning classification algorithms to develop models that accurately classify a viral protein sequence as COVID-19 positive or negative based on its similarity to labelled COVID-19 data using a readily available data (NCBI Virus) from the National Center for Biotechnology Information. The protein sequences were sliced into k-mers and subsequently vectors were generated and used as input data into training the models. The non-linear models, random forest and k-nearest neighbors outperformed the linear model (linear discriminant analysis). Thus, a non-linear relationship was established between the generated protein vectors and their respective labels. Specifically, the RF model performed better than all the other models on the training dataset with 99% accuracy score and 99.5% accuracy on the testing dataset while the KNN model resulted in perfect precision that produced no false positive result. While KNN models are relatively easy to implement, they can be computationally expensive especially when the data involved is large. Both KNN and RF models are also known to perform well when there is no known relationship in a dataset. Thus, both KNN and RF were better suited to learning patterns in the viral protein sequences and using those patterns to accurately predict the status of the protein sequences. While machine learning models on their own may not be a panacea, when used to complement actual laboratory diagnostic techniques, health practitioners can increase the certainty of laboratory test results while rapidly diagnosing COVID-19. Future studies could explore the application of clustering and deep learning techniques to the protein sequence data to predict COVID-19 viral sequences.
Funding
This research received no external funding.
Declarations
Conflict of interest
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Toby A. Adjuik, Email: adjuiktoby@gmail.com
Daniel Ananey-Obiri, Email: dananeyobiri@aggies.ncat.edu.
References
- 1.WHO (2020) WHO Director-General’s opening remarks at the media briefing on COVID-19. https://doi.org/https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020
- 2.WHO (2020) WHO coronavirus disease (COVID-19) dashboard. https://doi.org/https://covid19.who.int/
- 3.Yadav M, Perumal M, Srinivas M. Analysis on novel coronavirus (COVID-19) using machine learning methods. Chaos Solitons Fract. 2020;139:110050. doi: 10.1016/j.chaos.2020.110050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lalmuanawma S, Hussain J, Chhakchhuak L. Applications of machine learning and artificial intelligence for Covid-19 (SARS-CoV-2) pandemic: a review. Chaos Solitons Fract. 2020;139:110059. doi: 10.1016/j.chaos.2020.110059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le TT, Andreadakis Z, Kumar A, Roman RG, Tollefsen S, Saville M, Mayhew S. The COVID-19 vaccine development landscape. Nat Rev Drug Discov. 2020;19:305–306. doi: 10.1038/d41573-020-00151-8. [DOI] [PubMed] [Google Scholar]
- 6.Aljame M, Imtiaz A, Ahmad I, Mohammed A (2021) Deep forest model for diagnosing COVID-19 from 10.21203/rs.3.rs-567774/v1. Routine blood tests [DOI] [PMC free article] [PubMed]
- 7.Long C, Xu H, Shen Q, Zhang X, Fan B, Wang C, Zeng B, Li Z, Li X, Li H. Diagnosis of the Coronavirus disease (COVID-19): rRT-PCR or CT? Eur J Radiol. 2020;126:108961. doi: 10.1016/j.ejrad.2020.108961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rani G, Oza MG, Dhaka VS, Pradhan N, Verma S, Rodrigues JJ. Applying deep learning for genome detection of coronavirus. Res Sq. 2020 doi: 10.21203/rs.3.rs-93564/v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, Walker AW. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014 doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hatcher EL, Zhdanov SA, Bao Y, Blinkova O, Nawrocki EP, Ostapchuck Y, Schäffer AA, Brister JR. Virus Variation Resource–improved response to emergent viral outbreaks. Nucleic Acids Res. 2017;45:D482–D490. doi: 10.1093/nar/gkw1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoch CL, Ciufo S, Domrachev M, Hotton CL, Kannan S, Khovanskaya R, Leipe D, Mcveigh R, O’Neill K, Robbertse B (2020) NCBI Taxonomy: a comprehensive update on curation, resources and tools. Database 2020 [DOI] [PMC free article] [PubMed]
- 12.Dey L, Chakraborty S, Mukhopadhyay A. Machine learning techniques for sequence-based prediction of viral–host interactions between SARS-CoV-2 and human proteins. Biomed J. 2020 doi: 10.1016/j.bj.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Afify HM, Zanaty MS. Computational Predictions for Protein Sequences of COVID-19 virus via. Mach Learn Algorithm. 2020 doi: 10.21203/rs.3.rs-34004/v2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chowdhury AS, Call DR, Broschat SL. Antimicrobial resistance prediction for gram-negative bacteria via game theory-based feature evaluation. Sci Rep. 2019 doi: 10.1038/s41598-019-50686-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Forni G, Mantovani A. COVID-19 vaccines: where we stand and challenges ahead. Cell Death Differ. 2021;28:626–639. doi: 10.1038/s41418-020-00720-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chagla Z. The BNT162b2 (BioNTech/Pfizer) vaccine had 95% efficacy against COVID-19 ≥ 7 days after the 2nd dose. Ann Intern Med. 2021;174:JC15. doi: 10.7326/ACPJ202102160-015. [DOI] [PubMed] [Google Scholar]
- 17.Shen AK, Hughes IR, DeWald E, Rosenbaum S, Pisani A, Orenstein WJ. Ensuring equitable access to COVID-19 vaccines in the US: current system challenges and opportunities: analysis examines ensuring equitable access to COVID-19 vaccines. Health Affairs. 2021 doi: 10.1377/hlthaff.2020.01554. [DOI] [PubMed] [Google Scholar]
- 18.Dutta S, Bandyopadhyay SK. Machine learning approach for confirmation of covid-19 cases: positive, negative, death and release. Mol Biol. 2020 doi: 10.1101/2020.03.25.20043505. [DOI] [Google Scholar]
- 19.Khanday AMUD, Rabani ST, Khan QR, Rouf N, Mohi Ud Din M. Machine learning based approaches for detecting COVID-19 using clinical text data. Int J Inf Technol. 2020;12:731–739. doi: 10.1007/s41870-020-00495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aljame M, Ahmad I, Imtiaz A, Mohammed A. Ensemble learning model for diagnosing COVID-19 from routine blood tests. Inf Med Unlocked. 2020;21:100449. doi: 10.1016/j.imu.2020.100449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brinati D, Campagner A, Ferrari D, Locatelli M, Banfi G, Cabitza F. Detection of COVID-19 infection from routine blood exams with machine learning: a feasibility study. J Med Syst. 2020 doi: 10.1007/s10916-020-01597-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turabieh H, Ben Abdessalem Karaa W (2021) Predicting the existence of COVID-19 using machine learning based on laboratory findings. IEEE
- 23.Alakus TB, Turkoglu I. Comparison of deep learning approaches to predict COVID-19 infection. Chaos Solitons Fract. 2020;140:110120. doi: 10.1016/j.chaos.2020.110120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Göreke V, Sarı V, Kockanat S. A novel classifier architecture based on deep neural network for COVID-19 detection using laboratory findings. Appl Soft Comput. 2021;106:107329. doi: 10.1016/j.asoc.2021.107329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jamshidi M, Lalbakhsh A, Talla J, Peroutka Z, Hadjilooei F, Lalbakhsh P, Jamshidi M, Spada LL, Mirmozafari M, Dehghani M, Sabet A, Roshani S, Roshani S, Bayat-Makou N, Mohamadzade B, Malek Z, Jamshidi A, Kiani S, Hashemi-Dezaki H, Mohyuddin W. Artificial intelligence and COVID-19: deep learning approaches for diagnosis and treatment. IEEE Access. 2020;8:109581–109595. doi: 10.1109/access.2020.3001973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cleemput S, Dumon W, Fonseca V, Abdool Karim W, Giovanetti M, Alcantara LC, Deforche K, De Oliveira T. Genome detective coronavirus typing tool for rapid identification and characterization of novel coronavirus genomes. Bioinformatics. 2020;36:3552–3555. doi: 10.1093/bioinformatics/btaa145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arslan H. COVID-19 prediction based on genome similarity of human SARS-CoV-2 and bat SARS-CoV-like coronavirus. Comput Ind Eng. 2021;161:107666. doi: 10.1016/j.cie.2021.107666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ren J, Song K, Deng C, Ahlgren NA, Fuhrman JA, Li Y, Xie X, Poplin R, Sun F. Identifying viruses from metagenomic data using deep learning. Quant Biol. 2020;8:64–77. doi: 10.1007/s40484-019-0187-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mikolov T, Chen K, Corrado G, Dean J (2013) Efficient estimation of word representations in vector space. ArXiv Pre-Print Serv
- 30.Pennington J, Socher R, Manning CD (2014) Glove: global vectors for word representation, pp 1532–1543
- 31.Shi T, Liu Z (2014) Linking GloVe with word2vec.ArXiv PreprArXiv14115595
- 32.Dahouda MK, Joe I. A deep-learned embedding technique for categorical features encoding. IEEE Access. 2021;9:114381–114391. doi: 10.1109/ACCESS.2021.3104357. [DOI] [Google Scholar]
- 33.Cerda P, Varoquaux G, Kégl B. Similarity encoding for learning with dirty categorical variables. Mach Learn. 2018;107:1477–1494. doi: 10.1007/s10994-018-5724-2. [DOI] [Google Scholar]
- 34.Chen Y, Zhang W, Cheng A. Global Vectors Representation of Protein Sequences and Its Application for Predicting Self-Interacting Proteins with Multi-Grained Cascade Forest Model. Genes. 2019;10:924. doi: 10.3390/genes10110924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamid M-N, Friedberg I. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks. Bioinformatics. 2019;35:2009–2016. doi: 10.1093/bioinformatics/bty937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ho Q-T, Phan D-V, Ou Y-Y. Using word embedding technique to efficiently represent protein sequences for identifying substrate specificities of transporters. Anal Biochem. 2019;577:73–81. doi: 10.1016/j.ab.2019.04.011. [DOI] [PubMed] [Google Scholar]
- 37.Min S, Lee B, Yoon S. Deep learning in bioinformatics. Brief Bioinform. 2016 doi: 10.1093/bib/bbw068. [DOI] [PubMed] [Google Scholar]
- 38.Schoch CL, Ciufo S, Domrachev M, Hotton CL, Kannan S, Khovanskaya R, Leipe D, Mcveigh R, O’Neill K, Robbertse B (2020) NCBI taxonomy: a comprehensive update on curation, resources and tools. Database 2020 [DOI] [PMC free article] [PubMed]
- 39.Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT Suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. doi: 10.1093/bioinformatics/btq003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jolliffe IT. Principal components in regression analysis. New York: Springer; 2002. pp. 167–198. [Google Scholar]
- 41.Ringnér M. What is principal component analysis? Nat Biotechnol. 2008;26:303–304. doi: 10.1038/nbt0308-303. [DOI] [PubMed] [Google Scholar]
- 42.Vabalas A, Gowen E, Poliakoff E, Casson AJ. Machine learning algorithm validation with a limited sample size. PLoS ONE. 2019;14:e0224365. doi: 10.1371/journal.pone.0224365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hossin M, Sulaiman MN. A review on evaluation metrics for data classification evaluations. Int J Data Min Knowl Manage Process. 2015;5:1. doi: 10.5121/ijdkp.2015.5201. [DOI] [Google Scholar]
- 44.Alkhatib K, Najadat H, Hmeidi I, Shatnawi MKA. Stock price prediction using k-nearest neighbor (kNN) algorithm. Int J Bus Humanit Technol. 2013;3:32–44. [Google Scholar]
- 45.Imandoust SB, Bolandraftar M. Application of k-nearest neighbor (knn) approach for predicting economic events: theoretical background. Int J Eng Res Appl. 2013;3:605–610. [Google Scholar]
- 46.Altman NS. An introduction to kernel and nearest-neighbor nonparametric regression. Am Stat. 1992;46:175–185. doi: 10.1080/00031305.1992.10475879. [DOI] [Google Scholar]
- 47.Larrañaga P, Calvo B, Santana R, Bielza C, Galdiano J, Inza I, Lozano JA, Armañanzas R, Santafé G, Pérez A, Robles V. Machine learning in bioinformatics. Brief Bioinf. 2006;7:86–112. doi: 10.1093/bib/bbk007. [DOI] [PubMed] [Google Scholar]
- 48.Noble WS. What is a support vector machine? Nat Biotechnol. 2006;24:1565–1567. doi: 10.1038/nbt1206-1565. [DOI] [PubMed] [Google Scholar]
- 49.Cortes C, Vapnik V. Support-vector networks. Mach Learn. 1995;20:273–297. doi: 10.1007/BF00994018. [DOI] [Google Scholar]
- 50.Breiman L. Random forests. Mach Learn. 2001;45:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 51.Ho TK (1995) Random decision forests. IEEE, pp 278–282
- 52.Liaw A, Wiener M. Classification and regression by random forest. R News. 2002;2:18–22. [Google Scholar]
- 53.Hastie T, Tibshirani R, Friedman J. The elements of statistical learning: data mining, inference, and prediction. New York: Springer Science & Business Media; 2009. [Google Scholar]
- 54.Park CH, Park H. A comparison of generalized linear discriminant analysis algorithms. Pattern Recogn. 2008;41:1083–1097. doi: 10.1016/j.patcog.2007.07.022. [DOI] [Google Scholar]
- 55.Balakrishnama S, Ganapathiraju A. Linear discriminant analysis-a brief tutorial. Inst Signal Inf Process. 1998;18:1–8. [Google Scholar]
- 56.Tharwat A, Gaber T, Ibrahim A, Hassanien AE. Linear discriminant analysis: a detailed tutorial. AI Commun. 2017;30:169–190. doi: 10.3233/AIC-170729. [DOI] [Google Scholar]
- 57.Xiaozhou Y (2020) Linear discriminant analysis, explained. https://doi.org/https://towardsdatascience.com/linear-discriminant-analysis-explained-f88be6c1e00b
- 58.Kleinbaum DG, Dietz K, Gail M, Klein M, Klein M. Logistic regression. New York: Springer; 2002. [Google Scholar]
- 59.Ananey-Obiri D, Sarku E. Predicting the presence of heart diseases using comparative data mining and machine learning algorithms. Int J Comput Appl. 2019;975:8887. [Google Scholar]
- 60.Géron A (2017) Hands-on machine learning with Scikit-learn and tensor flow: Concepts, tools, and techniques to build intelligent systems (N. Tache Ed. 1st ed.). Sebastopol, CA , USA: O’Reilly Media, Inc.
- 61.Muhammad LJ, Algehyne EA, Usman SS, Ahmad A, Chakraborty C, Mohammed IA. Supervised machine learning models for prediction of covid-19 infection using epidemiology dataset. SN Comput Sci. 2021 doi: 10.1007/s42979-020-00394-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Carter JV, Pan J, Rai SN, Galandiuk S. ROC-ing along: evaluation and interpretation of receiver operating characteristic curves. Surgery. 2016;159:1638–1645. doi: 10.1016/j.surg.2015.12.029. [DOI] [PubMed] [Google Scholar]