

Abstract
As one of two highly conserved cellular degradation systems, autophagy plays a critical role in regulation of protein, lipid, and organelle quality control and cellular homeostasis. This evolutionarily conserved pathway singles out intracellular substrates for elimination via encapsulation within a double-membrane vesicle and delivery to the lysosome for degradation. Multiple cancers disrupt normal regulation of autophagy and hijack its degradative ability to remodel their proteome, reprogram their metabolism and adapt to environmental challenges, making the autophagy-lysosome system a prime target for anti-cancer interventions. Here we discuss the roles of autophagy in tumor progression, including cancer-specific mechanisms of autophagy regulation and the contribution of tumor and host autophagy in metabolic regulation, immune evasion and malignancy. We further discuss emerging proteomics-based approaches for systematic profiling of autophagosome-lysosome composition and contents. Together, these approaches are uncovering new features and functions of autophagy, leading to more effective strategies for targeting this pathway in cancer.
ETOC blurb
Autophagy plays a critical role in regulation of metabolic homeostasis and cellular quality control. Hernandez et al. review how these essential functions of autophagy contribute to cancer progression and stress adaptation. They further discuss emerging mass spectrometry-based approaches for systematic profiling of autophagosome-lysosome composition and contents in health and disease.
INTRODUCTION
Macroautophagy (hereafter referred to as autophagy) is a highly conserved cellular catabolic process that occurs at a basal level in virtually all cells and is further increased under conditions of stress including nutrient starvation, organelle damage and abnormal protein accumulation. Studies in budding yeast first defined a core network of genes required for autophagy (Tsukada and Osumi, 1993), and earned Yoshinori Osumi the Nobel prize in 2016. Most of these proteins were subsequently shown to be conserved in mammals (Galluzzi et al., 2017). The process of autophagy is a multistep pathway involving several protein complexes, accessory proteins and membranes, which coordinate to generate a double membrane vesicle that encapsulates intracellular substrates (referred to as ‘cargo’) and delivers them to the lysosome for degradation (Mizushima, 2020). Under basal conditions autophagy serves to degrade damaged cellular constituents and recycle nutrients to maintain the metabolic and energetic state of the cell. These key features of autophagy are especially advantageous to cancer cells, such as those harboring activating mutations in the Ras and Braf oncogenes (Guo et al., 2011; Lock et al., 2011; Perera et al., 2015; Rao et al., 2014; Strohecker et al., 2013; Yang et al., 2011), which display elevated levels of basal autophagy to maintain metabolic homeostasis, proteome remodeling and adaptation to environmental stress. Therefore, a detailed molecular understanding of each step along the autophagy pathway is necessary for developing effective strategies to modulate pathway activity in the context of disease. Development of genetically engineered mouse (GEM) models of cancer have helped to uncover critical roles for autophagy in tumor cells, as well as host organs and supporting cells within the tumor microenvironment in promoting tumor growth. Likewise, recent studies have also demonstrated an important role for tumor cell autophagy in promoting escape from immune cell detection. Moreover, improvements in autophagosome and lysosome isolation coupled to mass-spectrometry based proteomic profiling techniques have shed light on the specificity of autophagic cargo selection under different nutritional, environmental and diseased states. Together, these studies highlight how autophagy interfaces with multiple aspects of malignant disease.
In this review we discuss how autophagy works differently in cancer cells compared to normal cells, enabling protein quality control and metabolic reprogramming under the exceptionally challenging conditions under which tumors thrive. Finally, we discuss recent advances contributing to the emerging role of tumor autophagy in immune evasion and host autophagy in the regulation of tumor growth.
AUTOPHAGY IS A MULTI-STEP PROCESS
The mammalian autophagy process can be subdivided into several steps involving phagophore formation and elongation, maturation and fusion with lysosomes (Figure 1). Activation of the constitutively associated Unc-51-like kinase (ULK) complex consisting of ULK family kinase, ATG13, ATG101 and focal adhesion kinase interacting protein 200 kDa (FIP200), is the initiating event that triggers the autophagy cascade (Galluzzi et al., 2017). Once activated, ULK1 kinase activates a second complex consisting of the lipid kinase VPS34, Beclin1, VPS15, ATG14L and p150 leading to stimulation of VPS34 kinase activity and generation of phosphatidylinositol 3-phosphate (PI3P) on membranes derived most commonly from the endoplasmic reticulum (ER) (Yang et al., 2021) as well as ER-Golgi sites (Ge et al., 2017; Kumar et al., 2021).
Figure 1: The autophagy pathway.
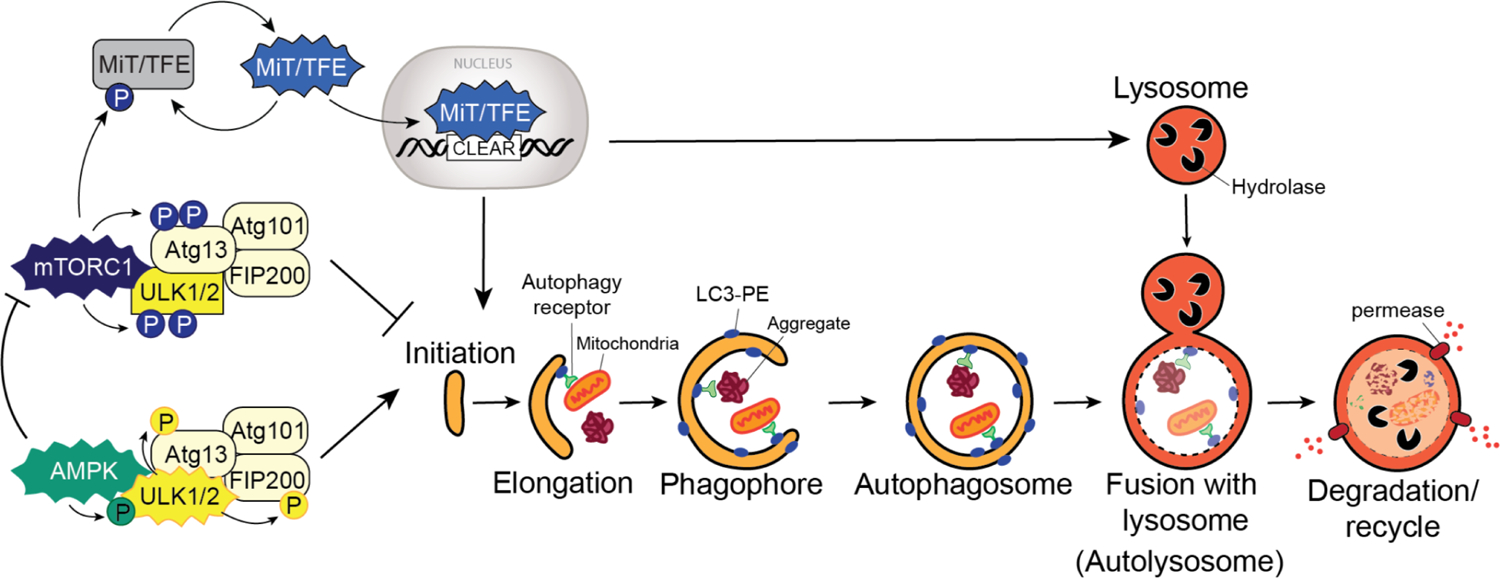
Autophagy is a multi-step process, and its activation is dependent on key upstream regulators. Under nutrient replete conditions, mTORC1 is active and phosphorylates ULK1/2 and Atg13 to block autophagy initiation. mTORC1 also phosphorylated MiT/TFE transcription factors leading to their cytoplasmic retention. Under low nutrient conditions, mTORC1 is inactivated relieving the negative regulation on the pathway. MiT/TFE factors enter the nucleus, recognize CLEAR elements present in the promoters of autophagy and lysosome genes and promote the coordinate transcriptional upregulation of these genes. Similarly, low ATP levels triggers AMPK activation, which phosphorylates ULK1/2 promoting its kinase activity. ULK1/2 in turn phosphorylates Atg13 and FIP200 promoting pathway initiation. AMPK also phosphorylates the mTORC1 subunit, Raptor, leading to suppression of mTORC1 activity. Cancer cells can bypass some, if not all, of these regulator circuits to maintain heighted activation of autophagy, independent of nutrient status or mTORC1/AMPK activity. Capture of intracellular “cargo” (eg. mitochondria, protein aggregates) is mediated by autophagy receptors which bind simultaneously to the cargo and to lipidated LC3 (LC3-II) associated with the growing autophagosome. Autophagosome fusion with lysosomes leads to hydrolase dependent degradation of cargo followed by efflux of degradation products via lysosomal permeases.
Elongation of the growing autophagosome (AP) membrane is mediated by two parallel ubiquitin (Ub)-like conjugation systems that serve to attach a protein - an ATG8 family member (LC3s/GABARAPs) - to a lipid – phosphatidylethanolamine (PE). First, ATG7 and ATG10, which function as E1- and E2-like enzymes respectively, facilitate conjugation of ATG12 to ATG5 to generate the ATG5-ATG12 complex. This complex is subsequently non-covalently conjugated to ATG16L1 to form the ATG5-ATG12-ATG16L complex that displays E3 ligase activity towards LC3 (and GABARAP) family members (Walczak and Martens, 2013). LC3 itself is first modified by the cysteine protease ATG4 to generate its soluble form (LC3-I) which is subsequently conjugated to PE by the ATG5-ATG12-ATG16L complex.
The lipidated form of LC3 (LC3-II) is a critical component of the growing AP and functions as a docking site for autophagy cargo receptors that deliver autophagic cargo to the AP (Figure 1). These cargo receptors, including sequestosome 1 (p62/SQSTM1) and neighbor of BRCA1 (NBR1), harbor LC3 interacting region/s (LIR) which facilitate binding to LC3 while the presence of Ub binding domains enables simultaneous recognition of ubiquitylated cargo proteins and organelles destined for autophagic capture and lysosomal degradation (Lamark and Johansen, 2021). Autophagy receptors can be soluble or membrane bound proteins and provide the selectivity to eliminate specific cellular components (Lamark and Johansen, 2021). For example, ER membrane bound autophagy receptors, including FAM134B and RTN3, facilitate remodeling of the ER in response to, or recovery from, ER stress. Moreover, the ability of autophagy receptors to heterodimerize with each other may also provide additional cargo selectivity (Clausen et al., 2010; Kirkin et al., 2009; Waters et al., 2009).
ATG9-associated vesicles serve as seeds for phagophore formation (Chang et al., 2021), and ATG9 was recently described as a phospholipid scramblase that participates in phagophore expansion (Matoba et al., 2020). Similarly, direct lipid transfer from donor organelles was also recently shown to be mediated by ATG2 – which funnels lipids, presumably in a unidirectional manner, from a donor membrane, most likely the ER, towards the growing AP (Osawa et al., 2019; Valverde et al., 2019). Locally produced phospholipids generated at the site of AP generation and maturation may be important for ensuring efficient AP growth. For instance, newly synthesized lipids were shown to be directly integrated into the growing AP (Andrejeva et al., 2020). Moreover, the lipid demand for generation of autophagic membrane, particularly under stress conditions which trigger the biogenesis of hundreds of autophagic vesicles, likely requires changes in cellular lipid synthesis and catabolism rates in order to provide the necessary lipid content to support AP growth (Melia et al., 2020). How lipids are harvested and transported to the expanding phagophore and how this process is coupled to the metabolic state of the cell remains largely unknown.
Once the AP membrane is sealed the mature vesicle must dock and fuse with lysosomes to deliver its cargo for degradation. This step is regulated by soluble NSF attachment protein receptors (SNARE) present on both the AP and the lysosome, Rab GTPases, and the homotypic fusion and protein sorting (HOPS) complex, which is thought to mediate lysosome-autophagosome tethering, while SNARE proteins mediate fusion (Nakamura and Yoshimori, 2017). Resident lysosomal hydrolases in turn degrade incoming cargo material which is either stored or recycled via efflux through dedicated lysosome membrane transporters and channels (Xu and Ren, 2015) (Figure 1). Together the autophagy-lysosome process ensures delivery, degradation and recycling of diverse intracellular material, and efficient flux through this pathway helps to maintain cellular health.
Recent studies have revealed that ATG8 proteins can be incorporated into additional single-membrane vesicles, including phagosomes, macropinosomes, endosomes and entotic vesicles which ultimately fuse with lysosomes (Nieto-Torres et al., 2021). Formation of these vesicles is independent of the ULK1 initiation machinery but requires the Ub-like ATG5 and ATG7 conjugation machinery that function during canonical autophagy. LC3 positive phagosomes generated via the LC3-associated phagocytosis (LAP) pathway enable highly phagocytic cells to engulf extracellular fungal and bacterial pathogens or apoptotic bodies. LC3 is also recruited to macropinosomes (Florey et al., 2011) and to transport vesicles during the process of entosis, which involves the engulfment of live cells (Florey and Overholtzer, 2019). Additional non-canonical functions for ATG8s include its involvement in LC3-associated endocytosis (LANDO) (Heckmann et al., 2019) and incorporation in a subset of extracellular vesicles (EVs) that mediate loading and exocytosis of RNA-binding proteins and small non-coding RNAs in a process known as LC3-dependnet EV loading and secretion (LDELS) (Leidal et al., 2020). LC3 is likely incorporated into these vesicular carriers after they are fully formed. Therefore, unlike canonical autophagosome formation, LC3 is unlikely to contribute to the biogenesis of LAP-, LANDO-, and LDELS-associated structures. Instead, LC3 on these carriers may enable recruitment of proteins that enable transport to and fusion with lysosomes, or efficient secretion, however this remains to be determined. Studies exploring the physiological relevance and molecular mechanisms mediating non-canonical autophagy related processes in the context of cancer may uncover additional vulnerabilities that could be targeted in combination with canonical autophagy.
REGULATION OF AUTOPHAGY IN NORMAL AND CANCER CELLS
Activation of autophagy in response to acute nutrient stress is primarily mediated by AMP-activated protein kinase (AMPK) (Trefts and Shaw, 2021) and the mechanistic target of rapamycin complex 1 (mTORC1) protein kinase (Shin and Zoncu, 2020). Activated mTORC1 has a pivotal role in suppression of autophagy via phosphorylation of key autophagy regulators, ULK1 and ATG13 (Hosokawa et al., 2009). Under growth factor or nutrient deprivation, mTORC1 becomes inactivated, leading to initiation of autophagy (Shin and Zoncu, 2020). Moreover, under energy-starved (low ATP) conditions caused by glucose withdrawal or mitochondrial dysfunction, AMPK becomes activated and promotes autophagosome formation and elongation through phosphorylation of ULK1, the type III PI3K Vps34 and Beclin1. AMPK also aids autophagy initiation by negatively regulating mTORC1 (Trefts and Shaw, 2021) (Figure 1).
In addition to kinase dependent acute regulation of autophagy initiation, transcriptional activation mediated by members of the MiT/TFE family of master transcription factors (TFE3, TFE3, MITF) enable prolonged pathway activation through coordinated upregulation of autophagy and lysosome genes (Sardiello et al., 2009; Settembre et al., 2011). MiT/TFE factors are also negatively regulated by mTORC1 phosphorylation, which leads to their cytoplasmic retention, thereby limiting autophagy and lysosome gene expression under conditions in which mTORC1 is active (Settembre et al., 2012) (Figure 1). Paradoxically, cancer cells which display increased autophagy and lysosome biogenesis, often do so under conditions in which mTORC1 activity is also high (Perera et al., 2015), suggesting that bypass mechanisms must be in place to sustain simultaneous activation of pro-growth and quality control programs. This can be achieved through upregulation and constitutive nuclear localization of MiT/TFE factors (Di Malta et al., 2017; Perera et al., 2015; Ploper et al., 2015), AMPK activity (Eichner et al., 2019), amplification, mutation or translocation of MiT/TFE genes, leading to their constitutive activation (Perera et al., 2019) or loss of upstream regulators of mTORC1-dependent MiT/TFE phosphorylation such as Folliculin (Napolitano et al., 2020) and tuberous sclerosis (TSC) (Alesi et al., 2021). Each of these conditions, leads to hyper-activation of MiT/TFE factors and increased autophagy and lysosome biogenesis, which in turn drive tumorigenesis.
In addition to their canonical role in nutrient deprivation induced autophagy, the MiT/TFE factors have been shown to respond to a broad range of cellular stressors (Martina and Puertollano, 2017). These include aneuploidy (Santaguida et al., 2015), DNA damage (Brady et al., 2018), mitochondrial stress (Nezich et al., 2015; Wang et al., 2020) and ER stress (Martina et al., 2016) – all of which are conditions associated with tumorigenesis. For example, alterations in ER homeostasis due to accumulation of un- or mis-folded proteins, triggers the unfolded protein response (UPR), which serves to restore homeostasis. TFEB and TFE3 are activated in response to ER stress and translocate into the nucleus in a PERK-dependent and mTORC1-independent manner. Targets of ER-stress induced by TFEB/TFE3 activity include autophagy and lysosome genes in addition to key regulators of ER homeostasis, including the ATF4 transcription factor (Martina et al., 2016). Thus, MiT/TFE activation induced ER-stress serves to restore cellular homeostasis and may function as an additional safety mechanism to ensure ER function in highly proliferative cancer cells. Likewise, induction of mitophagy upon mitochondrial damage was shown to be TFEB dependent (Nezich et al., 2015). Mitochondrial stress induced a PINK/Parkin-dependent, and mTORC1 independent, activation of TFEB. Importantly, depletion of all MiT/TFE family members led to an inability to eliminate damaged mitochondria due to defective autophagy (Nezich et al., 2015). Whether broader stress induced activation of MiT/TFE factors contributes to cellular adaptation and growth of cancer cells under austere cellular and microenvironmental conditions, remains to be determined. Moreover, analysis of the wider transcriptional program regulated by MiT/TFE factors, may uncover complementary pathways that integrate with autophagy and lysosome induction, providing important insight into how these factors support the growth of different cancer types at different stages.
ROLE OF AUTOPHAGY IN CANCER: LESSONS LEARNED FROM MOUSE MODELS
In normal cells, baseline levels of autophagy serve to maintain homeostasis via removal of toxic or damaged proteins and organelles, suppression of reactive oxygen species (ROS), DNA damage, tissue damage and inflammation. Accordingly, autophagy helps to prevent accumulation of chronic cellular damage that could promote transition to a cancerous state (Kenific and Debnath, 2015). Consistent with this idea, genetic suppression of autophagy genes such as ATG5, ATG7, FIP200 in combination with activation of tumor-initiating mutations leads to an increase in premalignant lesions in GEM models of cancer (Guo et al., 2011; Wei et al., 2011; Yang et al., 2014; Yang et al., 2011). In tissues susceptible to chronic damage and inflammation, such as the liver, deletion of Atg5 or Atg7 alone gives rise to spontaneous benign hepatomas (Takamura et al., 2011). In all cases, the lesions which develop remain benign, suggesting that autophagy is required for tumors to progress to a malignant state. More recent studies in a GEM model of pancreatic cancer, showed that in the absence of initiating oncogenic mutations, autophagy suppression within the pancreatic epithelia does not lead to metaplasia or growth of benign lesions (Yang et al., 2018). This suggests that benign tumor growth following autophagy inhibition likely only occurs in the presence of additional oncogenic insults within tumor cells in most tissues.
In contrast to early-stage disease, genetic or pharmacological inhibition of autophagy in tumor cells in models of advanced disease leads to a significant block in tumor growth. These include GEM models of melanoma (Xie et al., 2015), breast (Huo et al., 2013; Wei et al., 2011), lung (Guo et al., 2011; Guo et al., 2013; Karsli-Uzunbas et al., 2014), brain (Gammoh et al., 2016; Shchors et al., 2015), prostate (Santanam et al., 2016) and pancreas (Yang et al., 2018; Yang et al., 2014; Yang et al., 2011). Most of these studies incorporate tumor specific deletion of Atg5 or Atg7. Given that non-canonical autophagy pathways (eg. LAP, LANDO, LDELS described above) also depend on these critical ATG proteins, further studies in cancer models may help to determine the relative contribution of canonical vs non-canonical functions to tumor growth. Nevertheless, it is clear that diverse cancers benefit from and are dependent on functional autophagy for their growth, survival, and malignant progression. Taken together with the infrequency of mutations in essential autophagy genes in human cancers (Lebovitz et al., 2015), this pathway likely serves a pro-tumorigenic function in human tumors.
DEFINING AUTOPHAGY DEPENDENT CARGOS
Autophagy receptors and target selection
The function of autophagy is closely associated with the identity of the substrates that are targeted for degradation. It is also dependent on the upstream stimuli that triggers pathway activation, which in turn may define substrate selectivity. For instance, starvation-induced autophagy may capture distinct cargos that directly restore nutrient balance and may differ from baseline autophagy in its cargo specificity, function, and regulation (discussed later). Similarly, molecular, and biochemical differences between autophagosomes associated with basal autophagy versus stress induced autophagy may exist but remain poorly defined (Figure 2).
Figure 2: Factors influencing cargo selection.

Multiple factors influence how cargo is selected for autophagy mediated degradation. The nature of the upstream stimuli and its duration (acute versus prolonged) can dictate the types of cargo selected for capture via autophagy. Posttranslational modifications of the cargo (eg. Ub-dependent versus -independent) can influence autophagy receptor engagement. Some autophagy receptors and cargos are uniquely associated with a particular cellular location (eg. mitochondria versus ER). The abundance or condition of the cargo (healthy, damaged, aggregated, modified) can influence the efficiency and rate of capture. Autophagy receptors themselves can be modified by posttranslational modifications or participate as oligomers which can influence the efficiency and specificity of cargo capture.
Perhaps the best studied mechanisms for targeted degradation of cellular constituents involves the engagement of a class of molecules known as selective autophagy receptors (Lamark and Johansen, 2021). These receptors link their bound cargo to the growing AP membrane via simultaneous binding to LC3 proteins. Recent advances in the identification of a growing list of autophagy cargo receptors have highlighted how autophagy controls the regulated turnover of diverse and complex substrates, including proteins, aggregates, organelles and pathogens (Galluzzi et al., 2017). For example, autophagy receptors mediate targeted degradation of mitochondria (mitophagy), ER (ER-phagy), glycogen (glycophagy), lipid droplets (lipophagy), peroxisomes (pexophagy) and several additional intracellular and pathogenic macromolecules (Lamark and Johansen, 2021).
Two different classes of autophagy receptors exist based on whether they recognize cargo via a Ub dependent or independent mechanism (Khaminets et al., 2016). For instance, autophagy receptors containing Ub-interacting motifs (UIM) utilize their UIM and a LC3-interacting region (LIR), to bind to ubiquitylated cargo and LC3, respectively, within the AP (Khaminets et al., 2016). In contrast, several autophagy receptors, such as the membrane bound ER-phagy receptors do not contain UIMs and directly bind to LC3 (Khaminets et al., 2016). Similarly, recognition of lipids, sugars and the iron storage protein ferritin, occurs in a Ub independent manner by distinct autophagy receptors (Mancias and Kimmelman, 2016).
Some of these autophagy receptors play especially important roles in the context of cancer. For example, the nuclear receptor co-activator 4 (NCOA4) was identified as a Ub-independent adaptor for autophagy mediated degradation of ferritin – a process referred to as ferritinophagy (Dowdle et al., 2014; Mancias et al., 2014). By combining autophagosome isolation from Pancreatic ductal adenocarcinoma (PDA) cells with mass-spectrometry based proteomics, Mancias and colleagues found that NCOA4 was enriched in autophagosomes. NCOA4 also directly binds to ferritin heavy chain 1 (FTH1) subunit and is necessary for delivery of ferritin to autophagosomes and its subsequent degradation in the lysosome. Accordingly, NCOA4-deficient cells were unable to degrade ferritin leading to a decrease in intracellular bioavailable iron. NCOA4 itself is regulated via post-translational modification by the E3 Ub ligase HERC2, which targets it for proteosomal degradation under iron replete conditions, when ferritinophagy is no longer required (Mancias et al., 2014). Thus, the discovery and regulation of NCOA4 in the context of ferritinophagy, uncovered a mechanism for autophagy dependent maintenance of cellular iron supply. The relatively high levels of NCOA4 and ferritinophagy in PDA cells further suggests that cancer cells may have a higher requirement for iron storage and/or utilization (Torti and Torti, 2020).
Additional forms of selective autophagy, such as mitophagy (discussed later), similarly contribute to metabolic and organelle health in cancer cells. For instance, aberrant expression of ER-phagy receptors FAM134B and SEC62 were detected in several cancers (Hubner and Dikic, 2020; Islam et al., 2019; Zhang et al., 2019). Additional mechanistic studies will help to establish the role of ER-phagy in cancer progression and whether induction of this selective form of autophagy-dependent ER remodeling is linked to alterations in ER homeostasis, stress adaptation or environmental cues. Other forms of selective autophagy have been implicated in regulation of signaling complexes (Sandilands et al., 2011; Sandilands et al., 2012), turnover of focal adhesions (Kenific et al., 2016), downregulation of antigen presentation machinery (Yamamoto et al., 2020) and response to hypoxia (Daskalaki et al., 2018; Liu et al., 2012) in cancer cells.
Autophagosome content profiling
The development of methods to identify autophagosome substrates and associated proteins has been the focus of studies spanning several decades – one of the first being the establishment of an autophagosome purification method using density gradient fractionation (Stromhaug et al., 1998). Subsequent studies coupled subcellular fractionation, with two-dimensional electrophoresis and liquid chromatography tandem mass spectrometry (LC-MS/MS) to identify and quantify intralumenal cargo proteins and autophagosome membrane associated proteins (Dengjel et al., 2012; Gao et al., 2010; Overbye et al., 2007) (Box 1). These studies identified several proteins enriched in autophagosome fractions predicted to have functional roles in cargo recognition, modification of membrane lipids, and establishing autophagosome shape. Cargos were predominantly cytoplasm-derived and represented a diverse array of proteins. An interesting observation from these studies was the relative specificity of autophagosome associated proteins to a given upstream stimuli. For example, proteomics analysis of autophagosomes isolated from cells exposed to either amino acid (AA) starvation or treatment with rapamycin (an inhibitor of mTORC1 which mimics AA starvation) identified only a small cohort of common targets with the vast majority being unique to each condition (Dengjel et al., 2012). Similarly, whole cell proteomic profiling revealed stimuli specific autophagic substrate clearance in response to acute AA deprivation versus treatment with rapamycin (Mejlvang et al., 2018). These results suggest that seemingly analogous conditions can trigger significantly different outcomes, either due to qualitative differences in pathway activation and duration or due to integration with parallel trafficking routes in the cell.
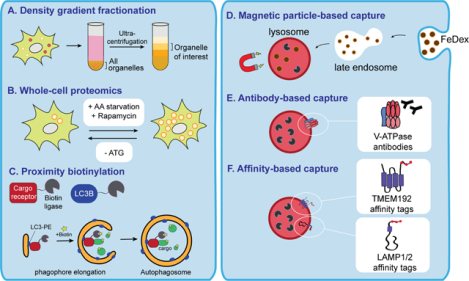
Box 1: Methods for autophagosome and lysosome composition and content profiling.
A. Early techniques to isolate autophagosomes and lysosome relied on density gradient fractionation-based approaches. Cells or tissues were homogenized and centrifuged at low speed to pellet intact cells, nuclei, and debris. Organelles located within the supernatant were concentrated and subjected to density gradient separation. Ultracentrifugation of the sample leads to migration of distinct organelles to distinct bands (colored), which can be individually isolated for biochemical or mass spectrometry-based analysis.
B. Identification of autophagy dependent cargo proteins can be determined via whole cell proteomics analysis of cells treated with autophagy inducing agents (Rapamycin), or stimuli (nutrient starvation). Under these conditions, increased autophagic degradation would lead to decreased abundance of select proteins species. Control experiments in which these treatments are combined with acute lysosomal blockade would restore the levels of autophagy targeted substrates. Likewise, proteins which accumulate following starvation or Rapamycin treatment in ATG null cells would represent autophagy cargos.
C. Proximity labeling (PL) using engineered enzymes such as peroxidases [engineered ascorbate peroxidase 2 (APEX2)], biotin ligases (BioID) or TurboID, conjugated to LC3 or to autophagy receptors. Upon addition of biotin-phenol/HO2 or biotin, proteins in close proximity to the enzyme are biotinylated, which are subsequently enriched using streptavidin conjugated beads. PL using autophagy receptors combined with autophagosome purification enables autophagosome content profiling. PL using LC3 enables broader analysis of LC3 interacting proteins associated with canonical and non-canonical autophagy pathways. Both techniques may identify putative regulators and interacting proteins that modulate pathway activity.
D. Strategies to isolate lysosomes take advantage of the fact that the lysosome is the final destination of several plasma membrane-derived internalization pathways. For example, lysosome isolation can be achieved following uptake of iron-dextran (FeDex) or related particles, followed by magnetic separation from lysed cells. The eluted samples can then be analyzed by mass spectrometry-based proteomics.
E. Immunoprecipitation of intact lysosome can be achieved using antibody-based capture from lysed cells. For example, antibodies specific to subunits of the lysosomal V-ATPase have been used to capture and isolate lysosomes from cultured cells. Following elution from the capture beads, lysosomal proteins can be analyzed by mass spectrometry-based proteomics.
F. Stable ectopic expression of lysosomal membrane proteins (LAMP1/2, TMEM192) conjugated to affinity handles (HA, FLAG, Biotin) enables rapid and efficient immunoprecipitation of intact lysosomes. Following mechanical disruption of cells, lysosomes are affinity enriched on beads containing immobilized streptavidin or antibodies against HA or FLAG. Lysosomal elutes can subsequently be analyzed using biochemical or mass spectrometry-based approaches. The advantages of this technique include its relative speed, ability to generate high purity samples and its independence from uptake of exogenous particles prior to organelle separation. Drawbacks include the need for stable expression of the exogenous construct which may not be possible in some cell types such as primary cells.
Several studies have revealed broader proteomic changes associated with autophagy induction or suppression (Kristensen et al., 2008; Mathew et al., 2014; Mejlvang et al., 2018; Zhang et al., 2016) (Box 1). Analysis of protein half-lives in autophagy-competent and deficient fibroblasts under basal conditions using SILAC and mass spectrometry uncovered two important findings: i) that autophagy is largely responsible for turnover of a significant fraction of the proteome, and ii) the existence of clear biases in autophagic target selection (Mathew et al., 2014; Zhang et al., 2016). Time dependent analysis of changes in protein abundance following AA starvation have also uncovered preference for autophagic degradation of certain cellular proteins. For instance, cytosolic proteins were noted to be the first cohort degraded during early stages of starvation induced autophagy while organelle derived proteins and protein complexes are targeted at later time points (Kristensen et al., 2008; Mathew et al., 2014). Therefore, in addition to substrate preference, targeting and degradation of cellular proteins appears to occur in a time-dependent, ordered manner under starvation conditions. These studies contributed to a general shift in perception of autophagy from a bulk non-selective process, to one that is highly selective in terms of substrate preference and timing of capture and clearance.
Targeted elimination of cellular proteins by autophagy plays increasingly recognized roles in cancer growth and stress adaptation. For instance, proteomic analysis of autophagy-functional versus -deficient Ras-transformed cells showed that proteins selectively targeted for starvation induced degradation included those associated with the innate immune response and the interferon response (Mathew et al., 2014). An advantage of specifically eliminating tumor derived inflammatory mediators upon starvation may be to subvert premature cell death and maintain cell viability under nutrient limiting conditions. In contrast, proteins involved in vesicle trafficking or general stress response pathways were resistant to autophagic degradation in response to starvation, thereby ensuring efficient activation of these protective pathways (Mathew et al., 2014). Constituents of the splicesome and ribosome were also shown to be largely protected against autophagy mediated degradation under baseline or starvation induced conditions (Mathew et al., 2014; Zhang et al., 2016). Recent studies utilizing global proteomics to measure protein translation and degradation rates, have also suggested that ribosomal subunits are not subject to significant autophagic turnover in response to starvation (An and Harper, 2018; An et al., 2020). However, it is possible that certain disease states may nevertheless trigger autophagy dependent ribosome turnover in response to nutrient stress (Wyant et al., 2018), and future studies across cell types and conditions may help to clarify a potential role for autophagy in ribosome turnover.
How specificity for select protein substrates is established remains an active area of current investigation. Similarly, the mechanisms in place to protect specific classes of proteins against autophagic capture under basal conditions (Dengjel et al., 2012; Zhang et al., 2016) or following starvation (Mathew et al., 2014) is an important question in the field. It is possible that ubiquitylation or other protein modifications (Rigbolt et al., 2014), differences in overall abundance, presence of specific interacting partners (Behrends et al., 2010), and cellular location may impact recognition and/or targeting of select protein subsets under basal and stress induced conditions in different cell types (Figure 3).
Figure 3: Tumor cell autonomous functions of autophagy in cancer.
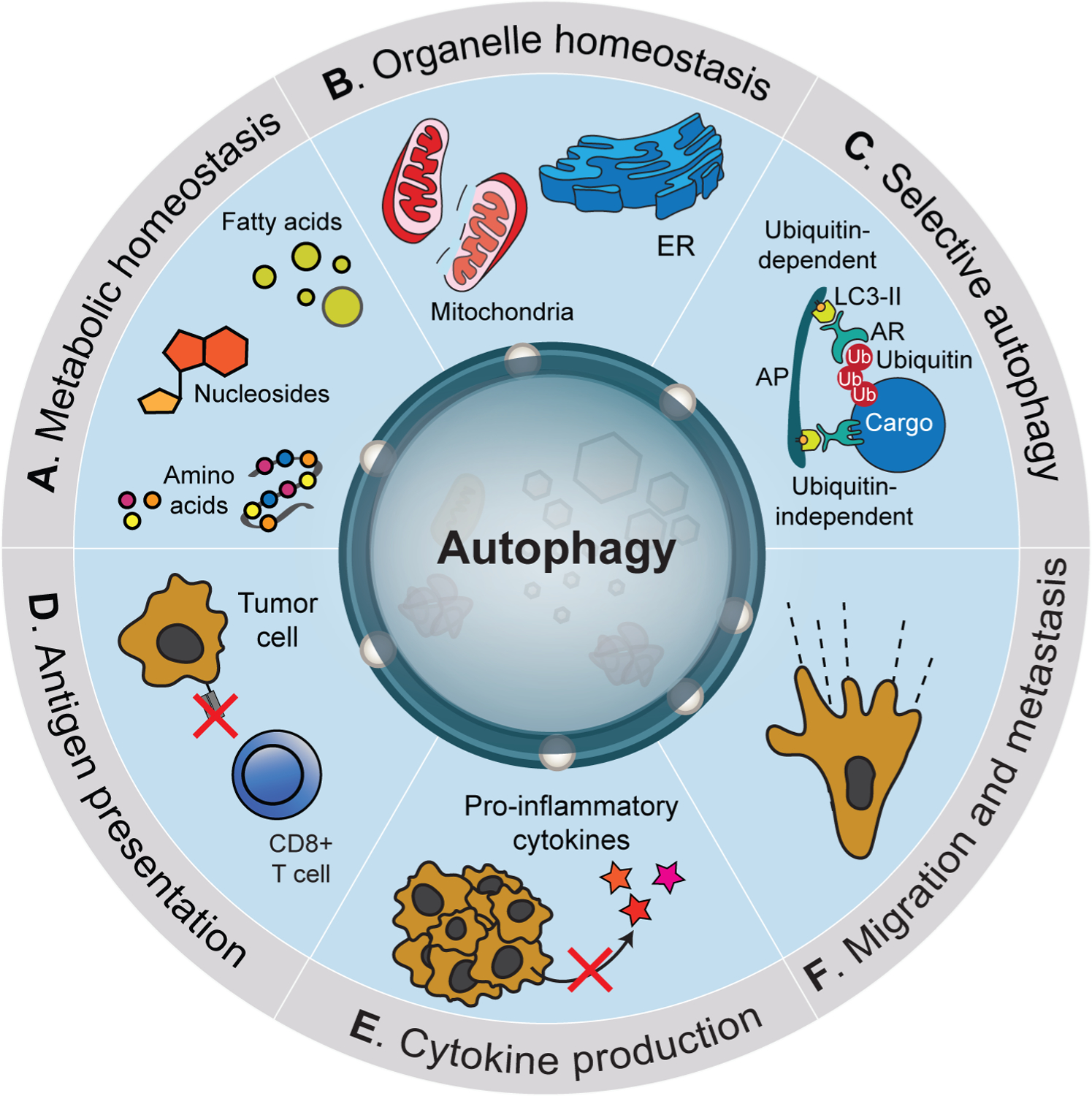
Tumor cell autonomous functions of autophagy. Cancer cells utilize autophagy to regulate diverse pro-tumorigenic functions. These include maintenance of metabolic homeostasis (A) via the capture of diverse cellular macromolecules followed by degradation and recycling in lysosomes. Autophagy is required for ensuring organelle quality control through targeted elimination of damaged components (B). Targeted elimination of cellular components is mediated through selective forms of autophagy that utilize autophagy receptors (AR) that can function in a Ub-dependent or independent manner (C). Autophagy regulates multiple components of the inflammatory/immune response including negative regulation of antigen presentation (D) and reduced production of pro-inflammatory cytokines (E). Several studies have also implicated autophagy activation during metastasis including both positive and negative roles (F).
Recent methods incorporating proximity labeling (PL) using engineered enzymes such as peroxidases [engineered ascorbate peroxidase 2 (APEX2)] (Lam et al., 2015), biotin ligases (BioID) (Kim et al., 2016; Roux et al., 2012) or TurboID (Branon et al., 2018), conjugated to LC3 (Le Guerroue et al., 2017; Leidal et al., 2020) or to autophagy receptors (Zellner et al., 2021) have also enabled high throughput autophagosome content profiling in addition to non-canonical autophagy related processes (Box 1). Leidal and colleagues (Leidal et al., 2020), utilizing LC3-conjuated to APEX, discovered a non-canonical LC3 associated pathway that regulates incorporation of RNA binding proteins and small non-coding RNAs into EVs, which are subsequently secreted (termed LDELS). Given the expanding role of EVs in inter-cellular communication, this study provides important insights into how the autophagy machinery regulates the selection of molecules destined for secretion. Zellner and colleagues, mapped the autophagic degradome under basal and stress-induced conditions by combining proximity labeling using select autophagy receptors (NBR1, OPTN, p62, NDP52, TOLLIP and TAX1BP1) with organelle enrichment techniques and quantitative proteomics (Zellner et al., 2021). The authors found that a majority of the identified interactors were shared between at least 2 of the proximity baits. A notable exception was TOLLIP, which displayed the most divergent interactome, possibly due to its function in endocytic trafficking (Jongsma et al., 2016) and endosome regulation (Katoh et al., 2004; Xiao et al., 2015), perhaps in preference to functioning as an autophagy receptor. Analysis of additional parameters, such as receptor expression levels and tissue types, potential interactions between the receptors themselves, and disease states in which a particular receptor may be more or less active, may help to clarify context dependent functions of autophagy receptors in control of cellular homeostasis in healthy and malignant cells.
Lysosomal content profiling
The lysosome is a membrane bound organelle present within virtually all eukaryotic cells and is the endpoint of autophagy as well as several additional trafficking routes in the cell (Perera and Zoncu, 2016). Profiling of lysosomal contents is a powerful and complementary approach to autophagosome profiling for detecting substrates targeted by autophagy under various cellular states. Isolation of intact lysosomes using density gradient ultracentrifugation or magnetic separation of iron-dextran loaded lysosomes, followed by mass-spectrometry based proteomic analysis, has uncovered important characteristics of lysosomes and the cargo present within their lumen (Diettrich et al., 1998; Muthukottiappan and Winter, 2021; Thelen et al., 2017) (Box 1). These studies, conducted in a variety of mammalian tissues and cell types, generated valuable proteomics datasets characterizing lysosomal composition and content (Bagshaw et al., 2005; Chapel et al., 2013; Della Valle et al., 2011; Lubke et al., 2009; Markmann et al., 2017; Muthukottiappan and Winter, 2021; Schmidtke et al., 2019; Schroder et al., 2007; Sleat et al., 2008). The repertoire of lysosomal proteins uncovered through these approaches included hydrolases, integral and peripheral membrane proteins responsible for catalysis of substrates and transport of digestion products, maintenance of lumenal pH and membrane structural integrity. Inherent drawbacks to these approaches include lack of purity due to the presence of other organelles with overlapping density profiles, particularly mitochondria. Moreover, differences in uptake rates of iron-dextran or related particles can compromise lysosome isolation efficiency making comparisons across cell types or conditions difficult. To overcome some of these challenges, immuno-isolation of lysosomes using antibodies against subunits of the V-ATPase were initially employed. Given that the V-ATPase is ubiquitous to lysosomes in all cells (Nishi and Forgac, 2002) this strategy allowed for lysosome isolation independent of their density or ability to take up external particles (Nylandsted et al., 2011). Subsequent development of lysosome membrane associated tags incorporating a lysosomal transmembrane protein conjugated to a high affinity handle (FLAG, HA, Biotin), allowed for rapid and efficient isolation of intact lysosomes from cells engineered to express the tag (Abu-Remaileh et al., 2017; Gupta et al., 2021; Ponsford et al., 2021; Wyant et al., 2018; Xiong et al., 2019; Zoncu et al., 2011) (Box 1). This approach helped demonstrate that mTORC1 localizes to the lysosome surface via interaction with the Rag GTPases (Zoncu et al., 2011), provided metabolic profiles of the lysosome lumen (Abu-Remaileh et al., 2017; Wyant et al., 2018; Xiong et al., 2019), uncovered mechanisms of cholesterol homeostasis (Davis et al., 2021) and lysosome clearance upon damage (Eapen et al., 2021) and provided a comparative analysis of non-cancer versus cancer lysosome content and composition (Gupta et al., 2021).
While the lysosome has a conserved degradative role in all cells, it is likely that heterogeneity in lysosome composition and content within a cell, across a population of cells or between different cell types, exists beyond that which occurs in response to nutrient stress. For example, comparison of non-PDA versus PDA derived lysosomes revealed a new role for Ferlin family proteins in regulation of cancer lysosome membrane integrity (Gupta et al., 2021). Analysis of lysosome proteomics across seven different cell lines (HEK293T, HeLa, HuH-7, SH-SY5Y, MEF, NIH3T3), similarly identified marked heterogeneity in both membrane associated and intraluminal proteins, including the presence of human and mouse specific lysosome associated proteins (Akter et al., 2020). Efforts to study lysosome heterogeneity have also uncovered differences in lysosome pH based on organelle position within the cell (Johnson et al., 2016) and transformation induced changes in lysosome lipid composition (Fehrenbacher et al., 2008). Efforts to assess single lysosome properties include the recent development of a single-lysosome mass spectrometry (SLMS) platform integrating lysosomal patch-clamp and mass spectrometry that enables simultaneous metabolic and electrophysiological profiling (Zhu et al., 2021).
Future studies in which profiling of lysosomes from healthy and diseased cells from in vivo tissues or following exposure to different conditions (hypoxia, nutrient starvation and microenvironmental stress) may reveal unique features and functions of the lysosome in the context of cellular transformation, cancer progression, and metastasis. Single lysosome analysis may highlight important features of lysosome evolution, degradative capacity and lifespan that could influence cell state, proliferative capacity and overall health.
ROLES OF AUTOPHAGY IN CANCER PROGRESSION
Autophagy sustains metabolic homeostasis in tumor cells
Autophagy endows cancer cells with metabolic plasticity via its ability to promote degradation of diverse cellular substrates (Kenific and Debnath, 2015). Accordingly, studies in various cancer models show that autophagy and lysosome-mediated degradation are critical sources of nucleotides (Elliott et al., 2019; Guo et al., 2016), amino acids (Bhatt et al., 2019; Perera et al., 2015; Strohecker et al., 2013), and lipids (Guo et al., 2013) that fuel cancer cell metabolism (Figure 3A–C). Ras-transformed cells and oncogenic Ras-driven tumors upregulate autophagy and rely on this pathway to help sustain metabolism in challenging microenvironments (Guo et al., 2011; Guo et al., 2013; Lock et al., 2011; Yang et al., 2011). Studies using both in vitro and in vivo Ras-driven cancer models showed that autophagy inhibition leads to defective mitochondria metabolism, including reduced respiration rates, energy charge and available metabolic intermediates (Guo et al., 2011; Karsli-Uzunbas et al., 2014; Yang et al., 2011). Similarly, pharmacological inhibition of autophagy via treatment of PDA cell lines with the lysosomal inhibitor Chloroquine led to defective mitochondrial respiration (Elliott et al., 2019; Yang et al., 2014).
The inherent function of autophagy in maintenance of organelle quality control through the selective removal, remodeling, and repair of virtually all cellular compartments, is especially important in cancer cells for maintaining metabolic fitness, high rates of protein synthesis and recycling of nutrients (Mancias and Kimmelman, 2016). Kras-mutant tumors require functional mitochondria for glucose and glutamine metabolism to support tumor growth (Guo et al., 2011; Guo et al., 2016; Humpton et al., 2019; Son et al., 2013). Maintenance of the mitochondrial network depends on a balance between de novo biogenesis, fission, and fusion of preexisting mitochondria, and mitophagy. Mitophagy is activated in response to diverse stimuli including hypoxia, nutrient deprivation, DNA damage, inflammation, and mitochondrial membrane depolarization (Drake et al., 2017). Defects in mitophagy cause accumulation of dysfunctional mitochondria, which in turn impact nutrient and energy homeostasis, cell fate and differentiation, signaling and cell death susceptibility, all of which are especially essential in cancer cells. For example, genetic inactivation of Atg7 in a GEM model of Kras-driven non-small cell lung cancer (NSCLC) led to accumulation of mitochondria with altered morphology, presumably due to defective autophagic clearance via mitophagy (Guo et al., 2013). Tumors that eventually developed in this model resembled oncocytomas – rare, benign tumors characterized by the accumulation of enlarged, defective mitochondria and defects in lipid metabolism (Guo et al., 2013). Further support for a critical role of mitophagy in supporting cancer cell viability comes from studies in which the mitophagy receptor NIX (also known as BNIP3L) was deleted in a GEM model of PDA (Humpton et al., 2019). Loss of NIX led to alterations in tumor cell metabolism, delayed tumor growth and prolonged the survival of tumor bearing mice. However, these mice eventually succumb to malignant disease, suggesting that alternative pathways for maintaining mitochondrial health may compensate for loss of NIX (Humpton et al., 2019). These studies highlight an essential role for autophagy in controlling cellular metabolism through combined regulation of metabolite levels and efficient organelle activity.
Autophagy promotes immune evasion in multiple cancers
Autophagy has a critical role in shaping both the innate and adaptive immune system and several excellent reviews have highlighted important findings in the field (Deretic and Levine, 2018; Merkley et al., 2018; Munz, 2021). Recent studies have uncovered an emerging role for autophagy in protecting cancer cells from immune-mediated elimination (Figure 3D,E).
For instance, downregulation of MHC class I (MHC-I) in PDA was recently shown to be mediated in part via an autophagy-dependent mechanism (Cheung et al., 2022; Yamamoto et al., 2020). Capture of MHC-I and targeting to the lysosome was dependent on the autophagy receptor NBR1 (Yamamoto et al., 2020) (Figure 3D). Accordingly, inhibition of autophagy, NBR1 or the lysosome was sufficient to restore total MHC-I levels, cell surface localization and elicit a potent anti-tumor immune response in human and mouse PDA models (Yamamoto et al., 2020). An independent study showed that inhibition of progranulin similarly restored MHC-I expression in PDA cells by inhibiting autophagy and lysosome activity (Cheung et al., 2022). These studies describe an important role for the autophagy-lysosome system in regulation of a critical cell surface protein necessary for immune recognition and cancer cell killing. Interestingly, autophagy dependent regulation of MHC-I did not appear to translate to other select epithelial cell surface markers or MHC-II suggesting that specificity towards MHC-I, potentially through recognition of aberrant posttranslational modifications, occurs in PDA cells (Yamamoto et al., 2020). Autophagy dependent dysregulation of MHC-I was also noted in LKB1 deficient tumors and inhibition of ULK1 was sufficient to restore antigen presentation (Deng et al., 2021). Whether autophagy more broadly reshapes the cell surface proteome remains to be fully determined and global cell surface proteomics may help to uncover the full cohort of proteins whose localization and trafficking are influenced by autophagy.
Genetic or pharmacological inhibition of autophagy across diverse cancer cell types was also shown to sensitize them to immune mediated killing (Arensman et al., 2020; Deng et al., 2021; Lawson et al., 2020; Noman et al., 2011; Noman et al., 2020; Young et al., 2020). Upregulation of inflammatory cytokines, including CCL5, CXCL5, CCL10 in autophagy deficient tumors, appears to mediate a potent anti-tumor immune response (Arensman et al., 2020; Mgrditchian et al., 2017; Noman et al., 2020; Wei et al., 2011; Yang et al., 2016) suggesting that tumor-intrinsic autophagy inhibition enhances the visibility of tumors by immune cells (Figure 3E). Unbiased genome-wide CRISPR screens in a panel of diverse cancer cell lines further identified knockout of core autophagy genes as sensitizers to TNFα dependent T cell-mediated killing (Lawson et al., 2020). Together, these studies highlighted a previously unrecognized role for tumor intrinsic autophagy in blockade of antigen presentation and immune-mediated tumor targeting.
Host autophagy supports tumor growth
In addition to studies focused on the impact of autophagy suppression in tumor cells, several recent findings also suggest that tumor cell extrinsic roles of autophagy both in cells within the local tumor microenvironment and in distant organs can have a profound effect on overall tumor growth. For instance, stellate cells utilize autophagy to generate a pool of alanine that is secreted and subsequently imported by PDA cells to fuel their mitochondrial metabolism (Sousa et al., 2016) (Figure 4A). Accordingly, autophagy suppression in stellate cells impairs tumor cell metabolism and growth. Autophagy suppression in stellate cells also promotes their quiescence and limits their ability to produce and secrete extracellular matrix in the context of PDA (Endo et al., 2017) (Figure 4B). These studies highlight a key supporting role for cells of the tumor microenvironment in promoting tumor growth in an autophagy dependent manner.
Figure 4. Functions of host autophagy in promoting tumor growth.
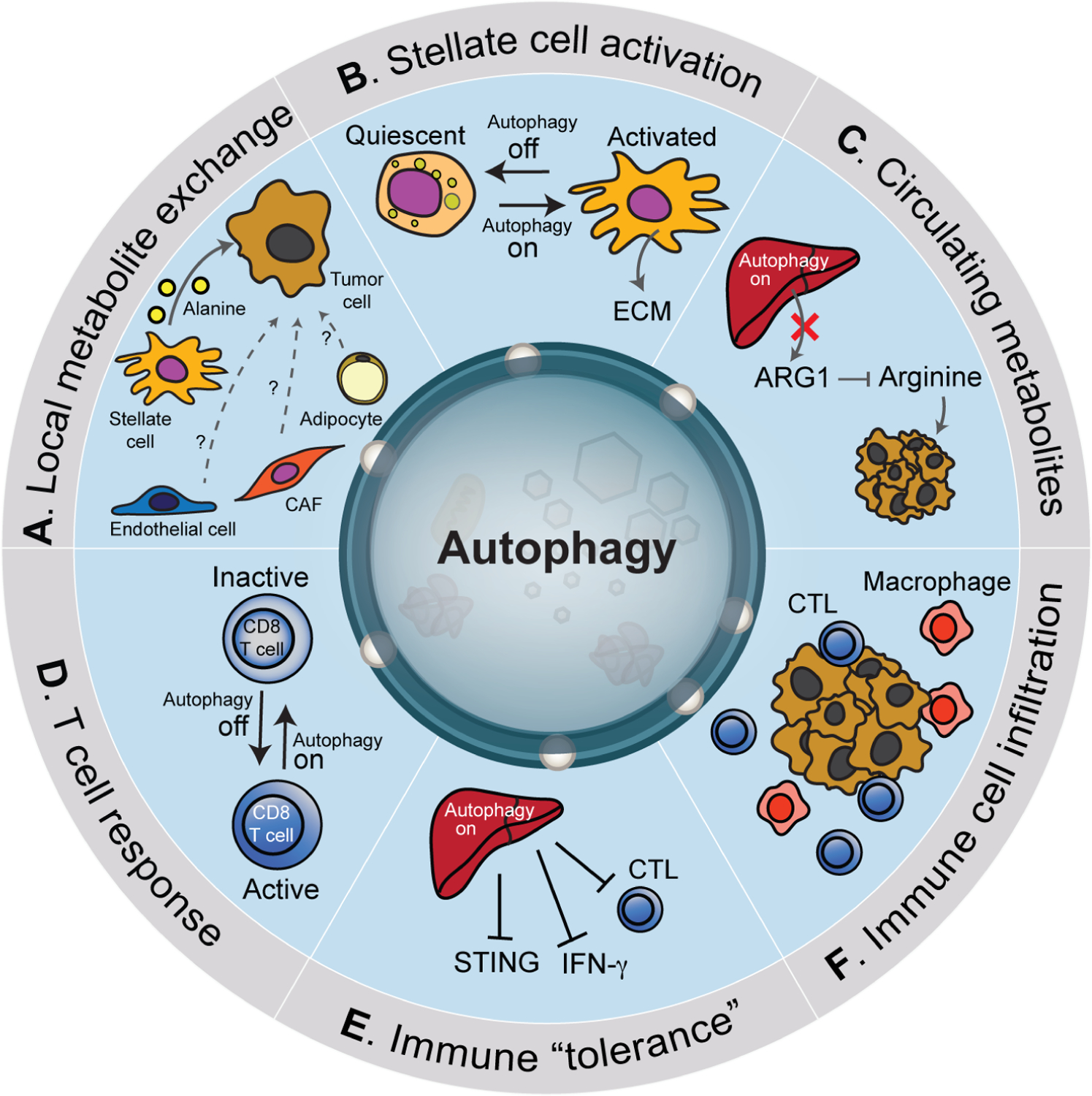
Autophagy within cells of the tumor microenvironment and distant tissues can impact tumor growth. For example, stellate cells in proximity to tumor cells utilize autophagy to generate a pool of alanine that is secreted and subsequently imported by cancer cells (A). Other cell types within the microenvironment including cancer associated fibroblasts (CAF), endothelial cells and adipocytes can also provide nutrients that fuel cancer growth however whether autophagy regulates this exchange is unclear. Autophagy suppression in stellate cells (‘off’) promotes their quiescence and reduces the deposition of extracellular matrix (ECM) (B). Likewise, autophagy in the liver helps to sustain circulating arginine necessary for supporting tumors that are arginine auxotrophs (C). Accordingly, suppression of autophagy in the liver leads to increased secretion of arginase 1 (ARG1) which reduces circulating arginine and inhibits tumor growth. Systemic host autophagy suppression (‘off’) promotes cytotoxic T lymphocytes (CTL) activation (D). A recent study showed that liver autophagy promotes tumor immune tolerance via decreased activation of CTLs, decreased interferon-γ and stimulator of interferon genes (STING) activation (E). Overall, systemic autophagy suppression promotes increased infiltration of tumors by immune cells (F).
The development of GEM models in which host autophagy could be suppressed either chronically or acutely in adult mice, combined with transplantation studies using autophagy competent tumor cells further uncovered essential functions for host tissue autophagy in supporting tumor growth. For example, inducible expression of a dominant-negative Atg4b mutant in adult mice, which effectively blocks autophagy in the host, led to delayed growth of transplanted autophagy competent PDA cells (Yang et al., 2018). Similarly, acute, systemic deletion of Atg7 in adult mice prevented the growth of allografted Kras-driven NSCLC cells to a greater extent than autophagy suppression in the tumor cells themselves (Karsli-Uzunbas et al., 2014). Importantly, tumor regression occurred prior to the onset of adverse effects resulting from whole body autophagy suppression.
Follow up studies testing the susceptibility of a wider array of cancer types (melanoma, urothelial carcinoma, and NSCLC) following transplantation into systemic Atg7 or Atg5 deleted host mice uncovered a dependency on host derived arginine for tumor growth (Poillet-Perez et al., 2018) (Figure 4C). Autophagy suppression in host tissue led to increased secretion of the arginine-degrading enzyme, arginase 1 (ARG1) leading to decreased circulating arginine. Accordingly, the tumors most susceptible to host autophagy suppression corresponded to those which were reliant on host derived arginine for growth – a phenomenon known as arginine auxotrophy – as they lacked expression of key arginine biosynthesis enzymes, including argininosuccinate lyase (ASS1) and ornithine transcarbamylase (OTC) (Poillet-Perez et al., 2018). This study indicates that host autophagy supports growth of arginine auxotrophic tumors by maintaining levels of circulating arginine. Consistent with these studies, systemic autophagy suppression via chemical or genetic approaches in Drosophila melanogaster, similarly blocked growth of RAS-driven tumors and their invasive capacity. Interestingly, transplantation of these tumors into autophagy-proficient hosts was sufficient to restore tumor growth (Katheder et al., 2017). Whether tumors less sensitive to host autophagy suppression harbor unique adaptive mechanisms that sustain metabolic homeostasis remains unclear (Venida and Perera, 2019).
Immune cells residing within the tumor microenvironment display autophagy dependency and may also contribute to tumor growth. For instance, genetic suppression of T cell autophagy via deletion of Atg5 or Atg7 led to alterations in T cell metabolism, conversion to memory effector cells and ultimately, enhanced tumor rejection in vivo (DeVorkin et al., 2019) (Figure 4D). Similarly, host suppression of Atg7 or Atg5 in tumor bearing mice promoted an anti-tumor T cell response (Poillet-Perez et al., 2020). Tumors grown on Atg7 null host mice displayed increased expression of interferon gamma (IFN-γ) pathway genes and increased expression of MHC-I and antigen presentation (Poillet-Perez et al., 2020) (Figure 4E). Collectively these studies highlight a critical function for autophagy in altering the tumor landscape to bypass detection by immune cells. Accordingly, autophagy suppression within tumor cells, immune cells or systemically appears to consistently promote immune cell infiltration and an anti-tumor immune response (Figure 4F).
Together, these findings highlight critical contributions of both tumor cell intrinsic and host autophagy in promoting tumor growth and indicate that a therapeutic window for systemic autophagy inhibition is possible. Future studies focusing on the impact of autophagy inhibition in distinct host cell types within the tumor microenvironment or distant tissues may help to define a broader role for autophagy in tumor growth and maintenance as well as during metastatic spread and seeding.
CONCLUSIONS AND FUTURE DIRECTIONS
Targeting the autophagy machinery or the lysosome remains a promising approach against a growing list of cancers that are reliant on this pathway for stress adaptation and growth. Current strategies for blocking autophagy involve targeting early stages of autophagy initiation and elongation mediated by ULK1/2, VPS34 and ATG4B. Alternatively, agents such as the FDA-approved hydroxychloroquine, target the lysosome and block late-stage cargo clearance (Amaravadi et al., 2019). These strategies have in common the ability to compromise the protective effects of autophagy in preventing accumulation of damaged or toxic macromolecules that are detrimental to the cell, compromise organelle health, cause a depletion in key metabolic substrates necessary to fuel tumor cell proliferation and expose cancer cells to the immune system. An advantage of lysosomal inhibition is the ability to simultaneously disable multiple scavenging pathways, including non-canonical autophagy pathways, which may be more effective at preventing compensatory induction of alternative routes for nutrient acquisition and recycling.
For these interventions to be maximally effective, continued investigation into the roles of autophagy in tumor progression is required. For instance, further insight into autophagy and lysosome dependence in metastatic lesions relative to the primary tumor is necessary when establishing effective therapeutic interventions that target both primary and metastatic disease. Recent studies suggest that highly metastatic bladder cancer cells are more sensitive to lysosomal inhibition relative to their less metastatic counterparts (Morgan et al., 2018) and upregulation of TFEB, lysosome biogenesis and exocytosis is associated with lung cancer metastasis (Kundu et al., 2018). These studies suggest that autophagy/lysosome suppression could be equally effective in suppressing primary and metastatic tumor growth. However, this may not be universal across cancers (Marsh et al., 2020). Therefore, in depth studies across cancer types will be necessary to fully understand autophagy-dependency and the impact of genetic versus pharmacological pathway inhibition on primary versus metastatic tumor growth.
Likewise further studies focused on the role of autophagy receptors in cancer, including how they engage cargo and the mechanisms underlying specificity for cargo selection, are important areas of investigation. While Ub remains the predominant mediator of autophagy receptor engagement, whether cooperation with additional posttranslational modifications provides additional specificity remains to be fully determined.
While the focus in cancer has been to inhibit autophagy as a therapeutic intervention, a possible alternative strategy may be to harness the intrinsic degradative power of the autophagy pathway to selectively eliminate pro-oncogenic proteins. Early efforts in this direction have generated autophagy- and lysosome-based degraders (Cassidy and Zhao, 2021) and future studies will hopefully establish their applicability to therapeutic settings.
ACKNOWLEDGEMENTS
G.A.H acknowledges National Science Foundation, Graduate Research Fellowship.
R.M.P acknowledges NIH R01CA251726.
We apologize to colleagues whose work we were unable to cite due to space limitations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, and Sabatini DM (2017). Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akter F, Ponnaiyan S, Kögler-Mohrbacher B, Bleibaum F, Damme M, Renard BY, and Winter D (2020). Multi cell line analysis of lysosomal proteomes reveals unique features and novel lysosomal proteins. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alesi N, Akl EW, Khabibullin D, Liu HJ, Nidhiry AS, Garner ER, Filippakis H, Lam HC, Shi W, Viswanathan SR, et al. (2021). TSC2 regulates lysosome biogenesis via a non-canonical RAGC and TFEB-dependent mechanism. Nat Commun 12, 4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaravadi RK, Kimmelman AC, and Debnath J (2019). Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov 9, 1167–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, and Harper JW (2018). Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat Cell Biol 20, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, Ordureau A, Korner M, Paulo JA, and Harper JW (2020). Systematic quantitative analysis of ribosome inventory during nutrient stress. Nature 583, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrejeva G, Gowan S, Lin G, Wong Te Fong AL, Shamsaei E, Parkes HG, Mui J, Raynaud FI, Asad Y, Vizcay-Barrena G, et al. (2020). De novo phosphatidylcholine synthesis is required for autophagosome membrane formation and maintenance during autophagy. Autophagy 16, 1044–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arensman MD, Yang XS, Zhong W, Bisulco S, Upeslacis E, Rosfjord EC, Deng S, Abraham RT, and Eng CH (2020). Anti-tumor immunity influences cancer cell reliance upon ATG7. Oncoimmunology 9, 1800162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagshaw RD, Mahuran DJ, and Callahan JW (2005). A proteomic analysis of lysosomal integral membrane proteins reveals the diverse composition of the organelle. Mol Cell Proteomics 4, 133–143. [DOI] [PubMed] [Google Scholar]
- Behrends C, Sowa ME, Gygi SP, and Harper JW (2010). Network organization of the human autophagy system. Nature 466, 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt V, Khayati K, Hu ZS, Lee A, Kamran W, Su X, and Guo JY (2019). Autophagy modulates lipid metabolism to maintain metabolic flexibility for Lkb1-deficient Kras-driven lung tumorigenesis. Genes Dev 33, 150–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady OA, Jeong E, Martina JA, Pirooznia M, Tunc I, and Puertollano R (2018). The transcription factors TFE3 and TFEB amplify p53 dependent transcriptional programs in response to DNA damage. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, and Ting AY (2018). Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol 36, 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy K, and Zhao H (2021). Redefining the Scope of Targeted Protein Degradation: Translational Opportunities in Hijacking the Autophagy-Lysosome Pathway. Biochemistry. [DOI] [PubMed] [Google Scholar]
- Chang C, Jensen LE, and Hurley JH (2021). Autophagosome biogenesis comes out of the black box. Nat Cell Biol 23, 450–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapel A, Kieffer-Jaquinod S, Sagne C, Verdon Q, Ivaldi C, Mellal M, Thirion J, Jadot M, Bruley C, Garin J, et al. (2013). An extended proteome map of the lysosomal membrane reveals novel potential transporters. Mol Cell Proteomics 12, 1572–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung PF, Yang J, Fang R, Borgers A, Krengel K, Stoffel A, Althoff K, Yip CW, Siu EHL, Ng LWC, et al. (2022). Progranulin mediates immune evasion of pancreatic ductal adenocarcinoma through regulation of MHCI expression. Nat Commun 13, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, Overvatn A, Stenmark H, Bjorkoy G, Simonsen A, et al. (2010). p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 6, 330–344. [DOI] [PubMed] [Google Scholar]
- Daskalaki I, Gkikas I, and Tavernarakis N (2018). Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front Cell Dev Biol 6, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis OB, Shin HR, Lim CY, Wu EY, Kukurugya M, Maher CF, Perera RM, Ordonez MP, and Zoncu R (2021). NPC1-mTORC1 Signaling Couples Cholesterol Sensing to Organelle Homeostasis and Is a Targetable Pathway in Niemann-Pick Type C. Dev Cell 56, 260–276 e267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Della Valle MC, Sleat DE, Zheng H, Moore DF, Jadot M, and Lobel P (2011). Classification of subcellular location by comparative proteomic analysis of native and density-shifted lysosomes. Mol Cell Proteomics 10, M110 006403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Thennavan A, Dolgalev I, Chen T, Li J, Marzio A, Poirier JT, Peng D, Bulatovic M, Mukhopadhyay S, et al. (2021). ULK1 inhibition overcomes compromised antigen presentation and restores antitumor immunity in LKB1 mutant lung cancer. Nat Cancer 2, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengjel J, Hoyer-Hansen M, Nielsen MO, Eisenberg T, Harder LM, Schandorff S, Farkas T, Kirkegaard T, Becker AC, Schroeder S, et al. (2012). Identification of autophagosome-associated proteins and regulators by quantitative proteomic analysis and genetic screens. Mol Cell Proteomics 11, M111 014035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V, and Levine B (2018). Autophagy balances inflammation in innate immunity. Autophagy 14, 243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, McNamara E, Huang H, Kim P, Zacharias LG, et al. (2019). Autophagy Regulation of Metabolism Is Required for CD8(+) T Cell Anti-tumor Immunity. Cell Rep 27, 502–513 e505. [DOI] [PubMed] [Google Scholar]
- Di Malta C, Siciliano D, Calcagni A, Monfregola J, Punzi S, Pastore N, Eastes AN, Davis O, De Cegli R, Zampelli A, et al. (2017). Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science 356, 1188–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diettrich O, Mills K, Johnson AW, Hasilik A, and Winchester BG (1998). Application of magnetic chromatography to the isolation of lysosomes from fibroblasts of patients with lysosomal storage disorders. FEBS Lett 441, 369–372. [DOI] [PubMed] [Google Scholar]
- Dowdle WE, Nyfeler B, Nagel J, Elling RA, Liu S, Triantafellow E, Menon S, Wang Z, Honda A, Pardee G, et al. (2014). Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat Cell Biol 16, 1069–1079. [DOI] [PubMed] [Google Scholar]
- Drake LE, Springer MZ, Poole LP, Kim CJ, and Macleod KF (2017). Expanding perspectives on the significance of mitophagy in cancer. Semin Cancer Biol 47, 110–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eapen VV, Swarup S, Hoyer MJ, Paulo JA, and Harper JW (2021). Quantitative proteomics reveals the selectivity of ubiquitin-binding autophagy receptors in the turnover of damaged lysosomes by lysophagy. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichner LJ, Brun SN, Herzig S, Young NP, Curtis SD, Shackelford DB, Shokhirev MN, Leblanc M, Vera LI, Hutchins A, et al. (2019). Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab 29, 285–302 e287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott IA, Dann AM, Xu S, Kim SS, Abt ER, Kim W, Poddar S, Moore A, Zhou L, Williams JL, et al. (2019). Lysosome inhibition sensitizes pancreatic cancer to replication stress by aspartate depletion. Proc Natl Acad Sci U S A 116, 6842–6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo S, Nakata K, Ohuchida K, Takesue S, Nakayama H, Abe T, Koikawa K, Okumura T, Sada M, Horioka K, et al. (2017). Autophagy Is Required for Activation of Pancreatic Stellate Cells, Associated With Pancreatic Cancer Progression and Promotes Growth of Pancreatic Tumors in Mice. Gastroenterology 152, 1492–1506 e1424. [DOI] [PubMed] [Google Scholar]
- Fehrenbacher N, Bastholm L, Kirkegaard-Sorensen T, Rafn B, Bottzauw T, Nielsen C, Weber E, Shirasawa S, Kallunki T, and Jaattela M (2008). Sensitization to the lysosomal cell death pathway by oncogene-induced down-regulation of lysosome-associated membrane proteins 1 and 2. Cancer Res 68, 6623–6633. [DOI] [PubMed] [Google Scholar]
- Florey O, Kim SE, Sandoval CP, Haynes CM, and Overholtzer M (2011). Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol 13, 1335–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, and Overholtzer M (2019). Macropinocytosis and autophagy crosstalk in nutrient scavenging. Philos Trans R Soc Lond B Biol Sci 374, 20180154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, Choi AM, Chu CT, Codogno P, Colombo MI, et al. (2017). Molecular definitions of autophagy and related processes. EMBO J 36, 1811–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammoh N, Fraser J, Puente C, Syred HM, Kang H, Ozawa T, Lam D, Acosta JC, Finch AJ, Holland E, et al. (2016). Suppression of autophagy impedes glioblastoma development and induces senescence. Autophagy 12, 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Kang JH, Liao Y, Ding WX, Gambotto AA, Watkins SC, Liu YJ, Stolz DB, and Yin XM (2010). Biochemical isolation and characterization of the tubulovesicular LC3-positive autophagosomal compartment. J Biol Chem 285, 1371–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge L, Zhang M, Kenny SJ, Liu D, Maeda M, Saito K, Mathur A, Xu K, and Schekman R (2017). Remodeling of ER-exit sites initiates a membrane supply pathway for autophagosome biogenesis. EMBO Rep 18, 1586–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, Kamphorst JJ, Chen G, Lemons JM, Karantza V, et al. (2011). Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev 25, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. (2013). Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 27, 1447–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JY, Teng X, Laddha SV, Ma S, Van Nostrand SC, Yang Y, Khor S, Chan CS, Rabinowitz JD, and White E (2016). Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev 30, 1704–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Yano J, Mercier V, Htwe HH, Shin HR, Rademaker G, Cakir Z, Ituarte T, Wen KW, Kim GE, et al. (2021). Lysosomal retargeting of Myoferlin mitigates membrane stress to enable pancreatic cancer growth. Nat Cell Biol 23, 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann BL, Teubner BJW, Tummers B, Boada-Romero E, Harris L, Yang M, Guy CS, Zakharenko SS, and Green DR (2019). LC3-Associated Endocytosis Facilitates beta-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer’s Disease. Cell 178, 536–551 e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. (2009). Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 20, 1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner CA, and Dikic I (2020). ER-phagy and human diseases. Cell Death Differ 27, 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humpton TJ, Alagesan B, DeNicola GM, Lu D, Yordanov GN, Leonhardt CS, Yao MA, Alagesan P, Zaatari MN, Park Y, et al. (2019). Oncogenic KRAS Induces NIX-Mediated Mitophagy to Promote Pancreatic Cancer. Cancer Discov 9, 1268–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Y, Cai H, Teplova I, Bowman-Colin C, Chen G, Price S, Barnard N, Ganesan S, Karantza V, White E, et al. (2013). Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2-associated hereditary breast cancer. Cancer Discov 3, 894–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam F, Gopalan V, Law S, Tang JC, and Lam AK (2019). FAM134B promotes esophageal squamous cell carcinoma in vitro and its correlations with clinicopathologic features. Hum Pathol 87, 1–10. [DOI] [PubMed] [Google Scholar]
- Johnson DE, Ostrowski P, Jaumouille V, and Grinstein S (2016). The position of lysosomes within the cell determines their luminal pH. J Cell Biol 212, 677–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongsma ML, Berlin I, Wijdeven RH, Janssen L, Janssen GM, Garstka MA, Janssen H, Mensink M, van Veelen PA, Spaapen RM, et al. (2016). An ER-Associated Pathway Defines Endosomal Architecture for Controlled Cargo Transport. Cell 166, 152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, et al. (2014). Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 4, 914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katheder NS, Khezri R, O’Farrell F, Schultz SW, Jain A, Rahman MM, Schink KO, Theodossiou TA, Johansen T, Juhasz G, et al. (2017). Microenvironmental autophagy promotes tumour growth. Nature 541, 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Shiba Y, Mitsuhashi H, Yanagida Y, Takatsu H, and Nakayama K (2004). Tollip and Tom1 form a complex and recruit ubiquitin-conjugated proteins onto early endosomes. J Biol Chem 279, 24435–24443. [DOI] [PubMed] [Google Scholar]
- Kenific CM, and Debnath J (2015). Cellular and metabolic functions for autophagy in cancer cells. Trends Cell Biol 25, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenific CM, Stehbens SJ, Goldsmith J, Leidal AM, Faure N, Ye J, Wittmann T, and Debnath J (2016). NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol 212, 577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Behl C, and Dikic I (2016). Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol 26, 6–16. [DOI] [PubMed] [Google Scholar]
- Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, and Roux KJ (2016). An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell 27, 1188–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, and Dikic I (2009). A role for ubiquitin in selective autophagy. Mol Cell 34, 259–269. [DOI] [PubMed] [Google Scholar]
- Kristensen AR, Schandorff S, Hoyer-Hansen M, Nielsen MO, Jaattela M, Dengjel J, and Andersen JS (2008). Ordered organelle degradation during starvation-induced autophagy. Mol Cell Proteomics 7, 2419–2428. [DOI] [PubMed] [Google Scholar]
- Kumar S, Javed R, Mudd M, Pallikkuth S, Lidke KA, Jain A, Tangavelou K, Gudmundsson SR, Ye C, Rusten TE, et al. (2021). Mammalian hybrid pre-autophagosomal structure HyPAS generates autophagosomes. Cell 184, 5950–5969 e5922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu ST, Grzeskowiak CL, Fradette JJ, Gibson LA, Rodriguez LB, Creighton CJ, Scott KL, and Gibbons DL (2018). TMEM106B drives lung cancer metastasis by inducing TFEB-dependent lysosome synthesis and secretion of cathepsins. Nat Commun 9, 2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, and Ting AY (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12, 51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T, and Johansen T (2021). Mechanisms of Selective Autophagy. Annu Rev Cell Dev Biol 37, 143–169. [DOI] [PubMed] [Google Scholar]
- Lawson KA, Sousa CM, Zhang X, Kim E, Akthar R, Caumanns JJ, Yao Y, Mikolajewicz N, Ross C, Brown KR, et al. (2020). Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586, 120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guerroue F, Eck F, Jung J, Starzetz T, Mittelbronn M, Kaulich M, and Behrends C (2017). Autophagosomal Content Profiling Reveals an LC3C-Dependent Piecemeal Mitophagy Pathway. Mol Cell 68, 786–796 e786. [DOI] [PubMed] [Google Scholar]
- Lebovitz CB, Robertson AG, Goya R, Jones SJ, Morin RD, Marra MA, and Gorski SM (2015). Cross-cancer profiling of molecular alterations within the human autophagy interaction network. Autophagy 11, 1668–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidal AM, Huang HH, Marsh T, Solvik T, Zhang D, Ye J, Kai F, Goldsmith J, Liu JY, Huang YH, et al. (2020). The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat Cell Biol 22, 187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, et al. (2012). Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14, 177–185. [DOI] [PubMed] [Google Scholar]
- Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, and Debnath J (2011). Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell 22, 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubke T, Lobel P, and Sleat DE (2009). Proteomics of the lysosome. Biochim Biophys Acta 1793, 625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, and Kimmelman AC (2016). Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J Mol Biol 428, 1659–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, and Kimmelman AC (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmann S, Krambeck S, Hughes CJ, Mirzaian M, Aerts JM, Saftig P, Schweizer M, Vissers JP, Braulke T, and Damme M (2017). Quantitative Proteome Analysis of Mouse Liver Lysosomes Provides Evidence for Mannose 6-phosphate-independent Targeting Mechanisms of Acid Hydrolases in Mucolipidosis II. Mol Cell Proteomics 16, 438–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh T, Kenific CM, Suresh D, Gonzalez H, Shamir ER, Mei W, Tankka A, Leidal AM, Kalavacherla S, Woo K, et al. (2020). Autophagic Degradation of NBR1 Restricts Metastatic Outgrowth during Mammary Tumor Progression. Dev Cell 52, 591–604 e596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Diab HI, Brady OA, and Puertollano R (2016). TFEB and TFE3 are novel components of the integrated stress response. EMBO J 35, 479–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, and Puertollano R (2017). TFEB and TFE3: The art of multi-tasking under stress conditions. Transcription 8, 48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Khor S, Hackett SR, Rabinowitz JD, Perlman DH, and White E (2014). Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol Cell 55, 916–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba K, Kotani T, Tsutsumi A, Tsuji T, Mori T, Noshiro D, Sugita Y, Nomura N, Iwata S, Ohsumi Y, et al. (2020). Atg9 is a lipid scramblase that mediates autophagosomal membrane expansion. Nat Struct Mol Biol 27, 1185–1193. [DOI] [PubMed] [Google Scholar]
- Mejlvang J, Olsvik H, Svenning S, Bruun JA, Abudu YP, Larsen KB, Brech A, Hansen TE, Brenne H, Hansen T, et al. (2018). Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J Cell Biol 217, 3640–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melia TJ, Lystad AH, and Simonsen A (2020). Autophagosome biogenesis: From membrane growth to closure. J Cell Biol 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkley SD, Chock CJ, Yang XO, Harris J, and Castillo EF (2018). Modulating T Cell Responses via Autophagy: The Intrinsic Influence Controlling the Function of Both Antigen-Presenting Cells and T Cells. Front Immunol 9, 2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mgrditchian T, Arakelian T, Paggetti J, Noman MZ, Viry E, Moussay E, Van Moer K, Kreis S, Guerin C, Buart S, et al. (2017). Targeting autophagy inhibits melanoma growth by enhancing NK cells infiltration in a CCL5-dependent manner. Proc Natl Acad Sci U S A 114, E9271–E9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N (2020). The ATG conjugation systems in autophagy. Curr Opin Cell Biol 63, 1–10. [DOI] [PubMed] [Google Scholar]
- Morgan MJ, Fitzwalter BE, Owens CR, Powers RK, Sottnik JL, Gamez G, Costello JC, Theodorescu D, and Thorburn A (2018). Metastatic cells are preferentially vulnerable to lysosomal inhibition. Proc Natl Acad Sci U S A 115, E8479–E8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munz C (2021). The Macroautophagy Machinery in MHC Restricted Antigen Presentation. Front Immunol 12, 628429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthukottiappan P, and Winter D (2021). A proteomic view on lysosomes. Mol Omics 17, 842–859. [DOI] [PubMed] [Google Scholar]
- Nakamura S, and Yoshimori T (2017). New insights into autophagosome-lysosome fusion. J Cell Sci 130, 1209–1216. [DOI] [PubMed] [Google Scholar]
- Napolitano G, Di Malta C, Esposito A, de Araujo MEG, Pece S, Bertalot G, Matarese M, Benedetti V, Zampelli A, Stasyk T, et al. (2020). A substrate-specific mTORC1 pathway underlies Birt-Hogg-Dube syndrome. Nature 585, 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nezich CL, Wang C, Fogel AI, and Youle RJ (2015). MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J Cell Biol 210, 435–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto-Torres JL, Leidal AM, Debnath J, and Hansen M (2021). Beyond Autophagy: The Expanding Roles of ATG8 Proteins. Trends Biochem Sci 46, 673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi T, and Forgac M (2002). The vacuolar (H+)-ATPases--nature’s most versatile proton pumps. Nat Rev Mol Cell Biol 3, 94–103. [DOI] [PubMed] [Google Scholar]
- Noman MZ, Janji B, Kaminska B, Van Moer K, Pierson S, Przanowski P, Buart S, Berchem G, Romero P, Mami-Chouaib F, et al. (2011). Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res 71, 5976–5986. [DOI] [PubMed] [Google Scholar]
- Noman MZ, Parpal S, Van Moer K, Xiao M, Yu Y, Viklund J, De Milito A, Hasmim M, Andersson M, Amaravadi RK, et al. (2020). Inhibition of Vps34 reprograms cold into hot inflamed tumors and improves anti-PD-1/PD-L1 immunotherapy. Sci Adv 6, eaax7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylandsted J, Becker AC, Bunkenborg J, Andersen JS, Dengjel J, and Jaattela M (2011). ErbB2-associated changes in the lysosomal proteome. Proteomics 11, 2830–2838. [DOI] [PubMed] [Google Scholar]
- Osawa T, Kotani T, Kawaoka T, Hirata E, Suzuki K, Nakatogawa H, Ohsumi Y, and Noda NN (2019). Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat Struct Mol Biol 26, 281–288. [DOI] [PubMed] [Google Scholar]
- Overbye A, Fengsrud M, and Seglen PO (2007). Proteomic analysis of membrane-associated proteins from rat liver autophagosomes. Autophagy 3, 300–322. [DOI] [PubMed] [Google Scholar]
- Perera RM, Di Malta C, and Ballabio A (2019). MiT/TFE Family of Transcription Factors, Lysosomes, and Cancer. Annu Rev Cancer Biol 3, 203–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, Stoykova S, Nicolay BN, Ross KN, Fitamant J, Boukhali M, Lengrand J, Deshpande V, Selig MK, Ferrone CR, et al. (2015). Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 524, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perera RM, and Zoncu R (2016). The Lysosome as a Regulatory Hub. Annu Rev Cell Dev Biol 32, 223–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploper D, Taelman VF, Robert L, Perez BS, Titz B, Chen HW, Graeber TG, von Euw E, Ribas A, and De Robertis EM (2015). MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. Proc Natl Acad Sci U S A 112, E420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poillet-Perez L, Sharp DW, Yang Y, Laddha SV, Ibrahim M, Bommareddy PK, Sherrie Hu Z, Vieth J, Haas M, Bosenberg MW, et al. (2020). Autophagy promotes growth of tumors with high mutational burden by inhibiting a T-cell immune response. Nat Cancer 1, 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, Su X, Maganti A, Jiang C, Lu W, et al. (2018). Autophagy maintains tumour growth through circulating arginine. Nature 563, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponsford AH, Ryan TA, Raimondi A, Cocucci E, Wycislo SA, Frohlich F, Swan LE, and Stagi M (2021). Live imaging of intra-lysosome pH in cell lines and primary neuronal culture using a novel genetically encoded biosensor. Autophagy 17, 1500–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Tortola L, Perlot T, Wirnsberger G, Novatchkova M, Nitsch R, Sykacek P, Frank L, Schramek D, Komnenovic V, et al. (2014). A dual role for autophagy in a murine model of lung cancer. Nat Commun 5, 3056. [DOI] [PubMed] [Google Scholar]
- Rigbolt KT, Zarei M, Sprenger A, Becker AC, Diedrich B, Huang X, Eiselein S, Kristensen AR, Gretzmeier C, Andersen JS, et al. (2014). Characterization of early autophagy signaling by quantitative phosphoproteomics. Autophagy 10, 356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K, Stevens C, Brunton VG, Langdon WY, Vidal M, et al. (2011). Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol 14, 51–60. [DOI] [PubMed] [Google Scholar]
- Sandilands E, Serrels B, Wilkinson S, and Frame MC (2012). Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells. EMBO Rep 13, 733–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Vasile E, White E, and Amon A (2015). Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev 29, 2010–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santanam U, Banach-Petrosky W, Abate-Shen C, Shen MM, White E, and DiPaola RS (2016). Atg7 cooperates with Pten loss to drive prostate cancer tumor growth. Genes Dev 30, 399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. (2009). A gene network regulating lysosomal biogenesis and function. Science 325, 473–477. [DOI] [PubMed] [Google Scholar]
- Schmidtke C, Tiede S, Thelen M, Kakela R, Jabs S, Makrypidi G, Sylvester M, Schweizer M, Braren I, Brocke-Ahmadinejad N, et al. (2019). Lysosomal proteome analysis reveals that CLN3-defective cells have multiple enzyme deficiencies associated with changes in intracellular trafficking. J Biol Chem 294, 9592–9604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder B, Wrocklage C, Pan C, Jager R, Kosters B, Schafer H, Elsasser HP, Mann M, and Hasilik A (2007). Integral and associated lysosomal membrane proteins. Traffic 8, 1676–1686. [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. (2011). TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. (2012). A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchors K, Massaras A, and Hanahan D (2015). Dual Targeting of the Autophagic Regulatory Circuitry in Gliomas with Repurposed Drugs Elicits Cell-Lethal Autophagy and Therapeutic Benefit. Cancer Cell 28, 456–471. [DOI] [PubMed] [Google Scholar]
- Shin HR, and Zoncu R (2020). The Lysosome at the Intersection of Cellular Growth and Destruction. Dev Cell 54, 226–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleat DE, Della Valle MC, Zheng H, Moore DF, and Lobel P (2008). The mannose 6-phosphate glycoprotein proteome. J Proteome Res 7, 3010–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, McMahon M, and White E (2013). Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov 3, 1272–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromhaug PE, Berg TO, Fengsrud M, and Seglen PO (1998). Purification and characterization of autophagosomes from rat hepatocytes. Biochem J 335 (Pt 2), 217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, and Mizushima N (2011). Autophagy-deficient mice develop multiple liver tumors. Genes Dev 25, 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thelen M, Winter D, Braulke T, and Gieselmann V (2017). SILAC-Based Comparative Proteomic Analysis of Lysosomes from Mammalian Cells Using LC-MS/MS. Methods Mol Biol 1594, 1–18. [DOI] [PubMed] [Google Scholar]
- Torti SV, and Torti FM (2020). Iron and Cancer: 2020 Vision. Cancer Res 80, 5435–5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefts E, and Shaw RJ (2021). AMPK: restoring metabolic homeostasis over space and time. Mol Cell 81, 3677–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde DP, Yu S, Boggavarapu V, Kumar N, Lees JA, Walz T, Reinisch KM, and Melia TJ (2019). ATG2 transports lipids to promote autophagosome biogenesis. J Cell Biol 218, 1787–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venida A, and Perera RM (2019). Host Control of Tumor Feeding: Autophagy Holds the Key. Cell Metab 29, 236–238. [DOI] [PubMed] [Google Scholar]
- Walczak M, and Martens S (2013). Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 9, 424–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Chen Y, Li X, Zhang W, Liu Z, Wu M, Pan Q, and Liu H (2020). Emerging role of transcription factor EB in mitochondrial quality control. Biomed Pharmacother 128, 110272. [DOI] [PubMed] [Google Scholar]
- Waters S, Marchbank K, Solomon E, Whitehouse C, and Gautel M (2009). Interactions with LC3 and polyubiquitin chains link nbr1 to autophagic protein turnover. FEBS Lett 583, 1846–1852. [DOI] [PubMed] [Google Scholar]
- Wei H, Wei S, Gan B, Peng X, Zou W, and Guan JL (2011). Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 25, 1510–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyant GA, Abu-Remaileh M, Frenkel EM, Laqtom NN, Dharamdasani V, Lewis CA, Chan SH, Heinze I, Ori A, and Sabatini DM (2018). NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 360, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Brannon MK, Zhao X, Fread KI, Ellena JF, Bushweller JH, Finkielstein CV, Armstrong GS, and Capelluto DGS (2015). Tom1 Modulates Binding of Tollip to Phosphatidylinositol 3-Phosphate via a Coupled Folding and Binding Mechanism. Structure 23, 1910–1920. [DOI] [PubMed] [Google Scholar]
- Xie X, Koh JY, Price S, White E, and Mehnert JM (2015). Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov 5, 410–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong J, He J, Xie WP, Hinojosa E, Ambati CSR, Putluri N, Kim HE, Zhu MX, and Du G (2019). Rapid affinity purification of intracellular organelles using a twin strep tag. J Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, and Ren D (2015). Lysosomal physiology. Annu Rev Physiol 77, 57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, Sohn ASW, Mukhopadhyay S, Lin EY, Parker SJ, et al. (2020). Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 581, 100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, Deng J, Hai J, Yang S, Wong KK, et al. (2018). Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discov 8, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, and Kimmelman AC (2014). Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov 4, 905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Imamura Y, Jenkins RW, Canadas I, Kitajima S, Aref A, Brannon A, Oki E, Castoreno A, Zhu Z, et al. (2016). Autophagy Inhibition Dysregulates TBK1 Signaling and Promotes Pancreatic Inflammation. Cancer Immunol Res 4, 520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, et al. (2011). Pancreatic cancers require autophagy for tumor growth. Genes Dev 25, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zheng L, Zheng X, and Ge L (2021). Autophagosomal Membrane Origin and Formation. Adv Exp Med Biol 1208, 17–42. [DOI] [PubMed] [Google Scholar]
- Young TM, Reyes C, Pasnikowski E, Castanaro C, Wong C, Decker CE, Chiu J, Song H, Wei Y, Bai Y, et al. (2020). Autophagy protects tumors from T cell-mediated cytotoxicity via inhibition of TNFalpha-induced apoptosis. Sci Immunol 5. [DOI] [PubMed] [Google Scholar]
- Zellner S, Schifferer M, and Behrends C (2021). Systematically defining selective autophagy receptor-specific cargo using autophagosome content profiling. Mol Cell 81, 1337–1354 e1338. [DOI] [PubMed] [Google Scholar]
- Zhang T, Shen S, Qu J, and Ghaemmaghami S (2016). Global Analysis of Cellular Protein Flux Quantifies the Selectivity of Basal Autophagy. Cell Rep 14, 2426–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZQ, Chen J, Huang WQ, Ning D, Liu QM, Wang C, Zhang L, Ren L, Chu L, Liang HF, et al. (2019). FAM134B induces tumorigenesis and epithelial-to-mesenchymal transition via Akt signaling in hepatocellular carcinoma. Mol Oncol 13, 792–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Li Q, Liao T, Yin X, Chen Q, Wang Z, Dai M, Yi L, Ge S, Miao C, et al. (2021). Metabolomic profiling of single enlarged lysosomes. Nat Methods 18, 788–798. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Bar-Peled L, Efeyan A, Wang S, Sancak Y, and Sabatini DM (2011). mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 334, 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]