

Abstract
The lymph node (LN) is the site of chronic lymphocytic leukemia (CLL) cell activation and proliferation. Aberrant miRNA expression has been shown to play a role in CLL pathogenesis; however a comparison of miRNA expression between CLL cells in the LN and the peripheral blood (PB) has previously not been reported. Based on analysis of 17 paired LN and PB samples from CLL patients, we identify a panel of miRNAs that are increased in LN CLL cells correlating with an activation phenotype. When evaluated in CLL cells from 38 patients pre- and post-treatment with ibrutinib, a subset of these miRNAs (miR-22, miR-34a, miR-146b, and miR-181b) was significantly decreased in response to ibrutinib. A concomitant increase in putative miRNA target transcripts (ARID1B, ARID2, ATM, CYLD, FOXP1, HDAC1, IBTK, PTEN, and SMAD4) was also observed. Functional studies confirmed targets of ibrutinib-responsive miRNAs to include mRNA transcripts of multiple tumor suppressors. Knockdown of endogenous miR-34a and miR146b resulted in increased transcription of tumor suppressors and inhibition of cell proliferation. These findings demonstrate that ibrutinib down-regulates the expression of a subset of miRNAs related to B-cell activation leading to increased expression of miRNA targets including tumor suppressors and a reduction in cell proliferation.
INTRODUCTION
Chronic lymphocytic leukemia (CLL) is the most common type of leukemia in adults in the western world. CLL is a clonal disease of mature B cells that accumulate in bone marrow (BM), lymph node (LN), spleen, and peripheral blood (PB).1 Activation of the B cell receptor (BCR) signaling pathway and dysregulated apoptosis are key features of CLL. The LN microenvironment supports CLL cell survival and proliferation and has a significant impact on cellular signaling and CLL pathogenesis.2–4 Gene expression profiling of CLL cells isolated from BM, LN, and PB revealed that the LN is the primary site of CLL proliferation; and demonstrated activation of BCR signaling in LN resident CLL cells.5 Therapeutic agents targeting BCR signaling such as the Bruton’s tyrosine kinase (BTK) inhibitor, ibrutinib, have been shown to be effective in treatment naïve6 and relapsed refractory CLL in clinical trials.7–11 Ibrutinib potently inhibits BCR signaling, downregulates NF-кB signaling, and rapidly reduces tumor burden through a combination of inhibition of tumor proliferation and increased apoptosis.9, 12, 13
MicroRNAs (miRNAs) are small (~22 nt) noncoding RNAs that play an important role in post-transcriptional regulation of their target mRNAs by binding to 3ʹ untranslated region (UTR) leading to reduced stability, degradation, and/or inhibited translation. miRNAs play key roles in regulating cellular processes including cell differentiation, proliferation, and apoptosis.14 miRNAs can function as oncogenes and/or tumor suppressors and are often disrupted by chromosomal deletions, translocations, and amplifications.15 Altered expression of miRNAs has been reported in CLL.16–23 Notably, the 13q region often deleted in CLL contains the genes encoding miR-15a and miR-16. Loss of these miRNAs promotes resistance to apoptosis by increasing Bcl-2 translation.24, 25
Aberrant miRNA expression profiles in CLL PB have been previously reported to have prognostic significance.26, 27 Calin et al. identified a panel of thirteen out of 190 miRNAs that could discriminate between aggressive CLL (ZAP-70 high expression and un-mutated IGHV) versus indolent CLL (ZAP-70 low expression and mutated IGHV).27 High levels of miR-21 are reportedly associated with a poor prognosis in CLL patients with a 17p deletion; whereaslow levels of miR-181b reportedly identify patients who may benefit from early therapeutic intervention.16 miR-155, miR-17–92, miR-181 and miR-29, have been reported to regulate an activated phenotype of CLL, impacting differential expression of these miRNAs in CLL prognostic subgroups.28 A distinct exosome miRNA signature in CLL including miR-29 family, miR-150, miR-155, and miR-223 has been reported to be associated with BCR activation induced exosome release.20
On the basis of previous studies demonstrating activation of CLL cells in the LN,5 and changes in miRNA expression in CLL cells stimulated in vitro through the BCR,29 we hypothesized that 1) miRNAs might be differentially expressed between activated CLL cells in the LN and PB; and 2) ibrutinib might alter miRNA expression in CLL, contributing to the therapeutic response by regulating the expression of miRNA target genes. We show that expression of specific miRNAs is affected by the tumor microenvironment and identify down-regulation of key miRNAs, and concomitant up-regulation of tumor suppressor targets as a previously unreported anti-tumor effect of ibrutinib.
MATERIALS AND METHODS
Patients and samples
Patient samples were obtained in accordance with the Declaration of Helsinki, with informed consent, and approval of the NHLBI institutional review board. For matched peripheral blood (PB) and lymph node (LN) samples: samples were collected from 17 untreated CLL patients (10 males and 7 females) on the same day and processed in parallel.5 Additionally, samples were collected on a phase II, open-label, single-center, investigator-initiated single agent study of ibrutinib, registered at www.clinicaltrials.gov as NCT01500733. For the pre- and post-ibrutinib samples: PB samples were obtained from 40 CLL patients (22 [55%] males and 18 [45%] females with a median age of 66 years), with 38 paired pre- and post-ibrutinib at day 28 and additional two pre-treatment CLL samples. Healthy control PB samples were obtained from 33 healthy donors (24 [73%] male and 9 [27%] female with a median age of 50 years). Additional data is in Supplementary Tables 1 and 2. The LN and PB patient and control samples evaluated were the maximum number of samples available for analysis. The sample sizes ensured adequate power to detect significant differences between designated groups using Wilcoxon’s tests. Nodal response to ibrutinib treatment was defined as a 50% reduction in lymphadenopathy determined by the percent reduction in the sum of the product of the diameters of up to four LNs on computed tomography scan after two cycles, as compared with the baseline.
CD19 + cell selection
Mononuclear cells were isolated by centrifugation using lymphocyte separation medium (Lonza, Walkersville, MD); followed by CD19+ cell selection (Miltenyi Biotec Inc., Auburn, CA) as previously described.30 Flow cytometric analysis was performed, confirming > 95% enrichment for CD19+ B-cells.
RNA isolation
RNA was isolated from CD19+B-cells and cell lines using miRNeasy Mini kit (Qiagen, Germantown, MD). RNA concentration was determined using NanoDrop 2000 instrument (Thermo Scientific, Wilmington, DE).
Quantitative real-time PCR (qRT-PCR)
For miRNA quantification, reverse transcription (RT) was performed with a polyadenylation step added before the RT step as described previously31, 32 with some modifications. Briefly, RNA (160 ng for PB CLL B-cell samples and 600 ng for cell lines) was poly-adenylated using the poly-(A) polymerase (New England Biolabs, Ipswich, MA). Reverse transcription was conducted with an anchor primer using the High Capacity cDNA Reverse Transcription kit on a GeneAmp PCR system 9700 (Life Technologies, Grand Island, NY). Real-time PCR was performed on the StepOnePlus (Life Technologies) by using a specific forward primer (mature miRNA sequence), a universal reverse primer, and a FAM-ZEN-IABKFQ-labeled TaqMan probe (Integrated DNA Technologies, Coralville, IA). Primer and probe sequences are listed in Supplementary Table 3. A Ct value of 39 was assigned to miRNAs with undetermined Ct values. To accurately determine the miRNA expression difference between PB CLL samples and healthy controls, each miRNA was synthesized (Integrated DNA Technologies) and a miRNA standard curve was generated with eight serial dilutions starting from 1000 pM and ending at 0.001 pM. The miRNA amount was interpolated from the standard curve and expressed as fmoles/ ng of total RNA as described by Kubiczkova et al.33 The R2 value for each standard curve ranged from 0.97 to 0.99. For samples from LN CLL cells and cell lines, ribosomal RNA 18S and/or small nucleolar RNAs (RNU44 or RNU48) were amplified and used as the internal control. The relative miRNA expression level was calculated as 2−delta Ct.
For mRNA quantification, RNA (1 μg for LN samples and 5 μg for cell line samples) was reverse transcribed using the High Capacity cDNA Reverse Transcription kit. Real-time PCR was performed in triplicate on the StepOnePlus by using TaqMan gene expression assays for ARID1B, ARID2, ATM, CYLD, FOXP1, HDAC1, PTEN, SMAD4, and IBTK (Life Technologies). House-keeping genes PGK1, TFRC, GUSB, and PPIA were used as internal controls. The relative gene expression level was calculated as 2−delta Ct.
NanoString nCounter assay
Gene expression of miRNA targets was quantified using the NanoString nCounter assay (nanoString Technologies, Seattle, WA) with a custom panel of 43 genes and three internal control genes (Supplementary Table 4). RNAs from 23 pairs (pre- and post-ibrutinib) of CD19+ CLL cells in the PB were assayed as previously described.30
Cell culture, drug treatment, Transfection, trypan blue staining, and water-soluble tetrazolium salt assay
Human B-cell lymphoma cell lines SP5334 and MEC-1(ATCC, Manassas, VA, USA) were cultured in RPMI1640 supplemented with 10% fetal bovine serum and 100 μg/ml penicillin/streptomycin. Cells were treated with 1 μM ibrutinib or 1 μM fludarabine (Selleckchem, Houston, TX) or approximately three million SP53 or MEC-1 cells were transfected with 150 picomoles of scrambled control, or pre-miR or anti-miR oligos (Life Technologies) using the X-treme Gene siRNA transfection reagent (Roche Applied Science, Mannheim, Germany) according to manufacturer’s recommendations . A fraction of cells were stained with trypan blue and counted to determine the dead cell percentage. The difference in cell proliferation was assessed using the water-soluble tetrazolium salts (WST) assay (Roche Applied Science). WST reagent (10 μl) was added to 100 μl of cells in culture media in a 96-well plate and incubated for 4 hours. The sample absorption was then measured against a blank control at 450 nm using the BioTek Synergy 4 (BioTek, Winooski, VT).
Western blotting
Pre-miR-34a, pre-miR-146b, and negative control oligo transfected SP53 cells were harvested at 24 h. Cell lysates were prepared in cold RIPA buffer (Sigma-Aldrich, St. Louis, MO). Protein concentration was determined using the Protein Assay Kit (Bio-Rad, Hercules, CA). Total cellular protein (20 μg) was electrophoresed in SDS-PAGE gels and transferred to PVDF membranes (Life Technologies). Protein detection was performed using the ECL kit (GE Healthcare Life Sciences, Marlborough, MA, USA). Mouse monoclonal antibody against actin (#A5316) was purchased from Sigma-Aldrich. Mouse monoclonal antibody against SMAD4 (#ab130242) and rabbit polyclonal antibody against ATM (#ab32420) were purchased from AbCam (Cambridge, MA,USA). Mouse monoclonal antibody against HDAC1 (#05–100-I) was purchased from EMD Millipore (Billerica, MA, USA). Secondary antibodies against mouse (#sc-2005) and rabbit (#sc-2004) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Protein band intensities were quantified using ImageJ.35
Statistical analysis
Statistical analyses were performed using GraphPad Prism software version 6 (GraphPad Software, La Jolla, CA, USA). Shapiro-Wilk test, t-test, Mann-Whitney and/or Wilcoxon test were used where indicated. P values of less than 0.05 were considered significant. Cluster analysis and heat map were generated using Cluster and Tree View software (Eisen Laboratory, Stanford University, Stanford, CA, USA).
RESULTS
miRNA expression is increased in LN- derived CLL cells with activated B-cell phenotype
Given the reported differences in CLL cell activation between LN and PB,5 we tested the hypothesis that miRNA in CLL cells in different sites might be differentially expressed for 22 miRNAs (Table 1), previously reported to play a role in BCR signaling and/or CLL pathogenesis.16–22 miRNAs were assayed by quantitative PCR in CD19+ selected CLL cells in paired samples from LN and PB of 17 previously untreated CLL patients and in circulating CD19+ selected B-cells from 33 healthy controls. Twenty-one of the 22 miRNAs tested were present at significantly higher levels in PB CLL cells than in the circulating B-cells from healthy controls (Supplementary Table 5). Among them, miR-15a, miR-16, miR-21, miR-22, miR-29a, miR-29c-3p, miR-34a, miR-150, miR-155, and miR-650 levels were increased by > 10-fold in CLL. Receiver operating characteristic curve analysis showed that several miRNAs had high sensitivity and specificity for discrimination between controls and CLL (data not shown).
Table 1.
Differential miRNA expression in paired CLL samples from the PB and LN of 17 untreated patients.
| miRNA | Median (IQR) in PB | Median (IQR) in LN | Fold change | P value | Change in LN |
|---|---|---|---|---|---|
| Let-7a | 565.6 (283 –1167) | 1256 (800 – 2651) | 2 | 0.02 | Up |
| Let7a-3p | 1.1 (0.3 – 2.4) | 3.1 (1.7 – 6.2) | 2.8 | 0.003 | Up |
| Let-7g | 467.2 (211–693) | 1099 (581.7–2418) | 2 | 0.002 | Up |
| miR-9 | 0.3 (0.1–5.4) | 1.9 (0.7–33) | 6 | 0.0003 | Up |
| miR-15a | 139 (42–364) | 394 (193.4–1705) | 3 | 0.0026 | Up |
| miR-16 | 10030 (2789–17081) | 17548 (12515–46571) | 1.8 | 0.015 | Up |
| miR-17 | 122.4 (19.8–176.4) | 966 (296.7–1515) | 8 | 0.0004 | Up |
| miR-20a | 131 (94–200) | 1215 (318–1652) | 9 | 0.0005 | Up |
| miR-21 | 4717 (2205–25555) | 7417 (3771–21496) | 1.6 | 0.2 | No change |
| miR-22 | 0.47 (0.2–0.9) | 1.2 (0.8–4.1) | 3 | 0.001 | Up |
| miR-29a | 2.5 (1.8–4.4) | 11.2 (4–25.2) | 4.5 | 0.003 | Up |
| miR-29c-3p | 284.5 (133.6–439.3) | 956 (353–2898) | 3 | 0.007 | Up |
| miR-29c | 2 (1.4–4.5) | 7.2 (1.8–16.5) | 4 | 0.02 | Up |
| miR-34a | 10 (5.7–18.8) | 46.6 (18–111) | 5 | 0.0005 | Up |
| miR-106b | 15.2 (8.9–24.7) | 118 (30.4–211.7) | 8 | <0.0001 | Up |
| miR-146b | 5.1 (2.3–7.7) | 18.6 (8.6–32) | 4 | 0.0067 | Up |
| miR-150 | 4593 (2348–6363) | 3748 (2258–8981) | −1.25 | 0.6 | No change |
| miR-155 | 428 (289–1236) | 867 (571–2077) | 2 | 0.04 | Up |
| miR-181b | 0. 7 (0.4–4.5) | 3.3 (1.5–22.4) | 5 | 0.0011 | Up |
| miR-185 | 4.8 (2.2–7.6) | 15.5 (7–31.7) | 3 | 0.0002 | Up |
| miR-223 | 0.9 (0.6–1.8) | 1.7 (0.9–3.6) | 2 | 0.09 | No change |
| miR-650 | 0.1 (0.09–0.2) | 0.3 (0.3–0.7) | 3 | <0.0001 | Up |
Twenty-two miRNAs were assayed by TaqMan qPCR in paired treatment naïve LN and PB CLL cells. The median and interquartile ranges of the relative miRNA expression are shown. LN, lymph node; PB, peripheral blood; IQR, interquartile range. P values calculated based on Wilcoxon paired test.
Of the 22 miRNAs assayed, 19 miRNAs were significantly increased in CLL cells from the LN when compared to matched cells from PB (Figure 1a and b and Table 1). Overall, the median fold-change between PB and LN for all 22 miRNAs was 2.8 (range 0.9 – 5.5). These findings suggest that increased expression of these miRNAs in CLL cells occurs in the setting of increased cellular activation in the LN. Further, the higher expression in circulating CLL cells compared to normal circulating B-cells likely reflects a more activated state of CLL cells compared to normal circulating naïve B cells.
Figure 1. miRNA levels are higher in CLL cells resident in the LN than in CLL cells in the PB.
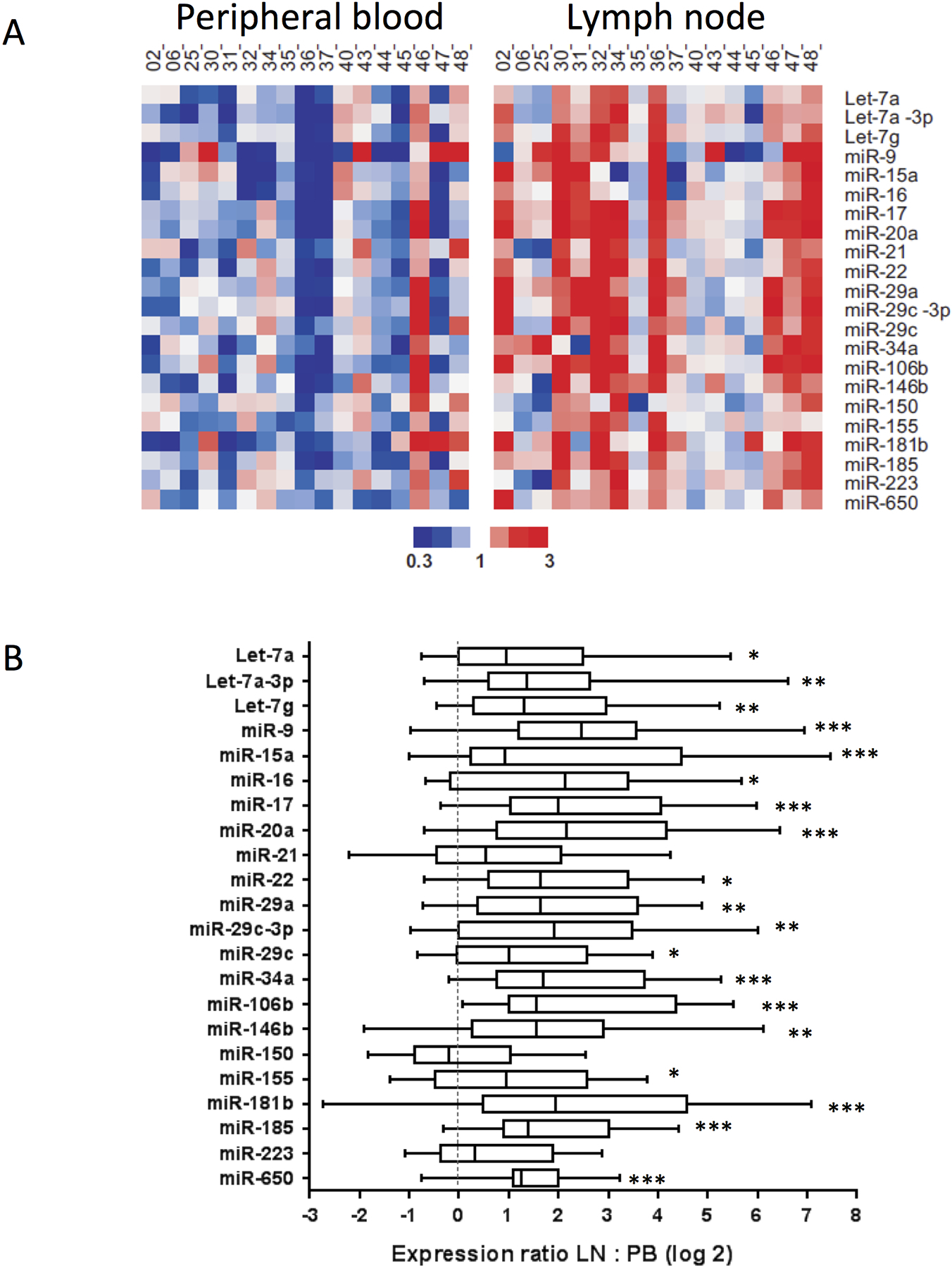
The expression levels of 22 miRNAs were assayed by TaqMan qPCR in duplicate in 17 paired, CD19+ selected CLL cells obtained concurrently from LN and PB of the same patients. (a) Heat map shows median centered expression of each miRNA across all samples graded according to the color scale shown. (b) Box plots show median and interquartile range for each miRNA. P values were calculated by the Wilcoxon paired test. *, p < 0.05, **, p < 0.01; ***, p < 0.001.
Ibrutinib treatment decreases a subset of miRNAs in CLL cells in vivo
As previously shown, BCR and NF-κB signaling are significantly inhibited by ibrutinib in vivo and the extent of target inhibition correlated with clinical response to treatment.7, 9 To test whether miRNA expression might also be altered in CLL cells in response to ibrutinib, we assayed the levels of all 22 miRNAs in paired PB CD19+ B cells from 38 CLL patients pre-treatment and at day-28 on continuous ibrutinib treatment. Expression of the miRNAs in the CLL patients was compared to expression in 33 healthy controls. Of the 22 miRNAs evaluated, four (miR-22, miR-34a, miR-146b, and miR-181b) were significantly decreased (p < 0.05) on ibrutinib treatment (Figure 2a and Supplementary Table 6). Notably, miR-22 and miR-146b levels in CLL cells on ibrutinib were not significantly different from levels in circulating B-cells from healthy controls (Figure 2B). miR-34a expression levels in CLL on ibrutinib significantly decreased and approached levels seen in B-cells from the healthy controls. miR-181b was the only miRNA in the panel that was significantly decreased in CLL cells pre-ibrutinib compared with controls and showed further decrease on ibrutinib. The remaining 18 miRNAs showed decreased expression on treatment in more than half of the patients; however across the whole group the differences were not statistically significant.
Figure 2. Expression of miR-22, miR-34a, miR-146b, and miR-181b are significantly decreased in patient CLL cells after ibrutinib treatment.
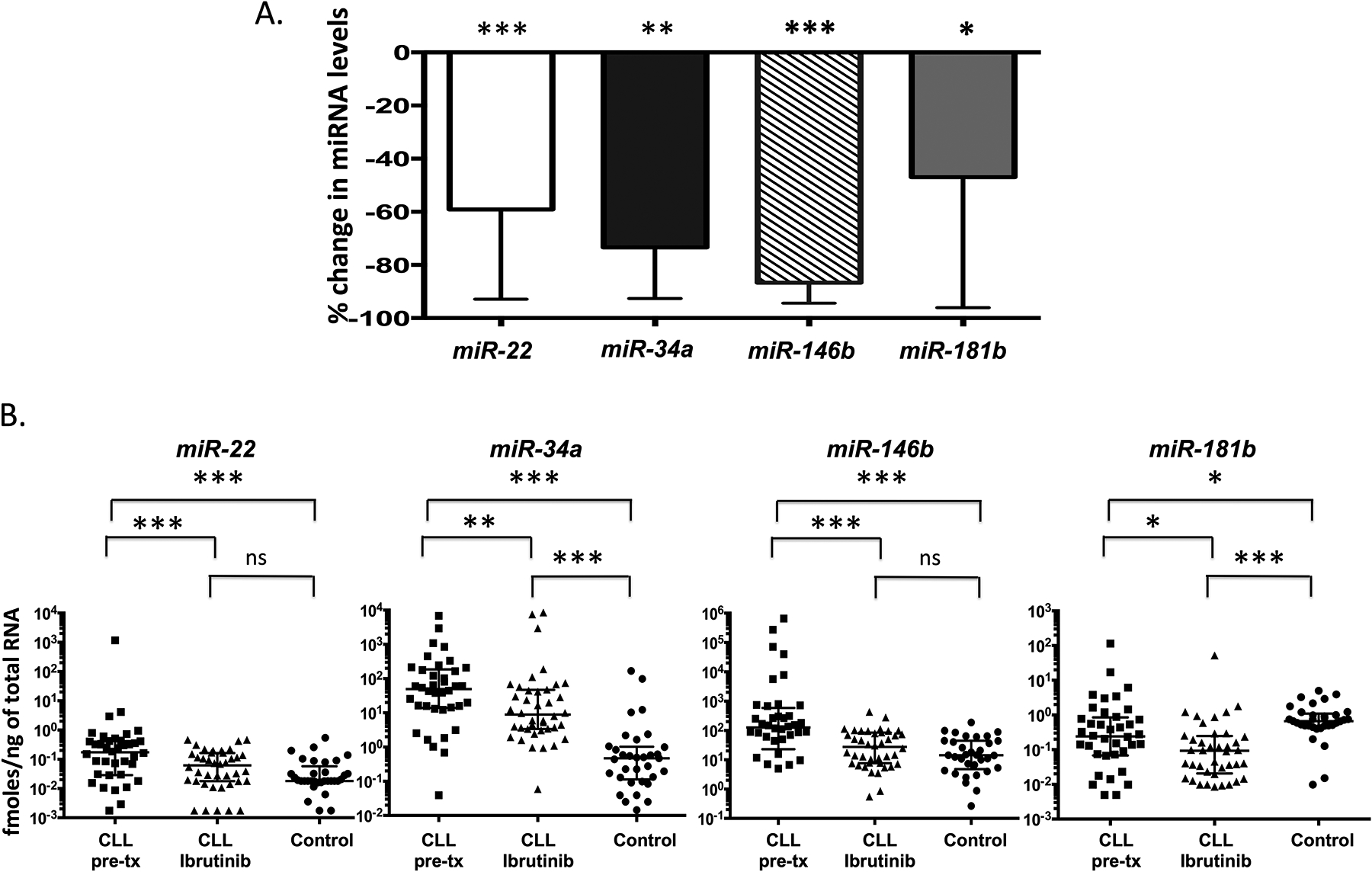
miRNA expression was determined by TaqMan qPCR in duplicate with RNA isolated from CLL cells collected from 38 patients at time points corresponding to pre- and post ibrutinib (day 28) treatment. (a.) Bar graph showing the percent changes of significant miRNAs post ibrutinib treatment. The column represents the median and the bar indicates the interquartile range. (B.) Scatter plots showing the miRNA expression in CLL patient samples pre- and post-ibrutinib treatment, in comparison to miRNA levels in circulating B-cells from 33 healthy controls. Pre-tx, pre-treatment. P values were calculated by the Wilcoxon paired test for paired samples and Wicoxon test for comparing healthy controls to pre- and post-treatment CLL. *, p < 0.05, **, p < 0.01; ***, p < 0.001; ns, not significant.
Nodal response was determined in 36 patients after two cycles of therapy. A total of 24 patientsobtained a nodal response with > 50% reduction in lymphadenopathy, and 12 were designated non-responders. miR-22 and miR-146b were significantly reduced in both responders and non-responders in response to ibrutinib (day 28) (p<0.05) (Supplementary Tables 7 and 8). In contrast miR-34a , miR-181b and miR-185 were significantly reduced only in the responder group; whereas a significant reduction in miR-155 was only obsevered in the non-responder group.
Putative targets of ibrutinib responsive miRNAs are significantly altered in CLL patient samples on ibrutinib treatment
In order to explore the potential role of the ibrutinib responsive miRNAs in the anti-tumor response, we analyzed expression of 43 mRNAs that are predicted targets of miR-22, miR-34a, miR-146b, and miR-181b (Supplementary Table 4). The target mRNAs were compiled based on TargetScan and miRWalk analyses and expression was quantified using the nanoString nCounter assay in 23 pairs of CLL samples obtained at baseline and after 4 weeks of treatment with ibrutinib. Out of the 43 evaluated mRNA targets, nine were found significantly up-regulated in the post-ibrutinib CLL cells in comparison to pre-ibrutinib levels: ARID1B, ARID2, ATM, CYLD, FOXP1, HDAC1, IBTK, PTEN, and SMAD4 (Figure 3; Supplementary Table 9). Of note, some of the genes encoding these targets have been reported to possess important tumor suppressor functions in CLL.36
Figure 3. Altered transcription of predicted miRNA targets in patient CLL cells after treatment with ibrutinib.
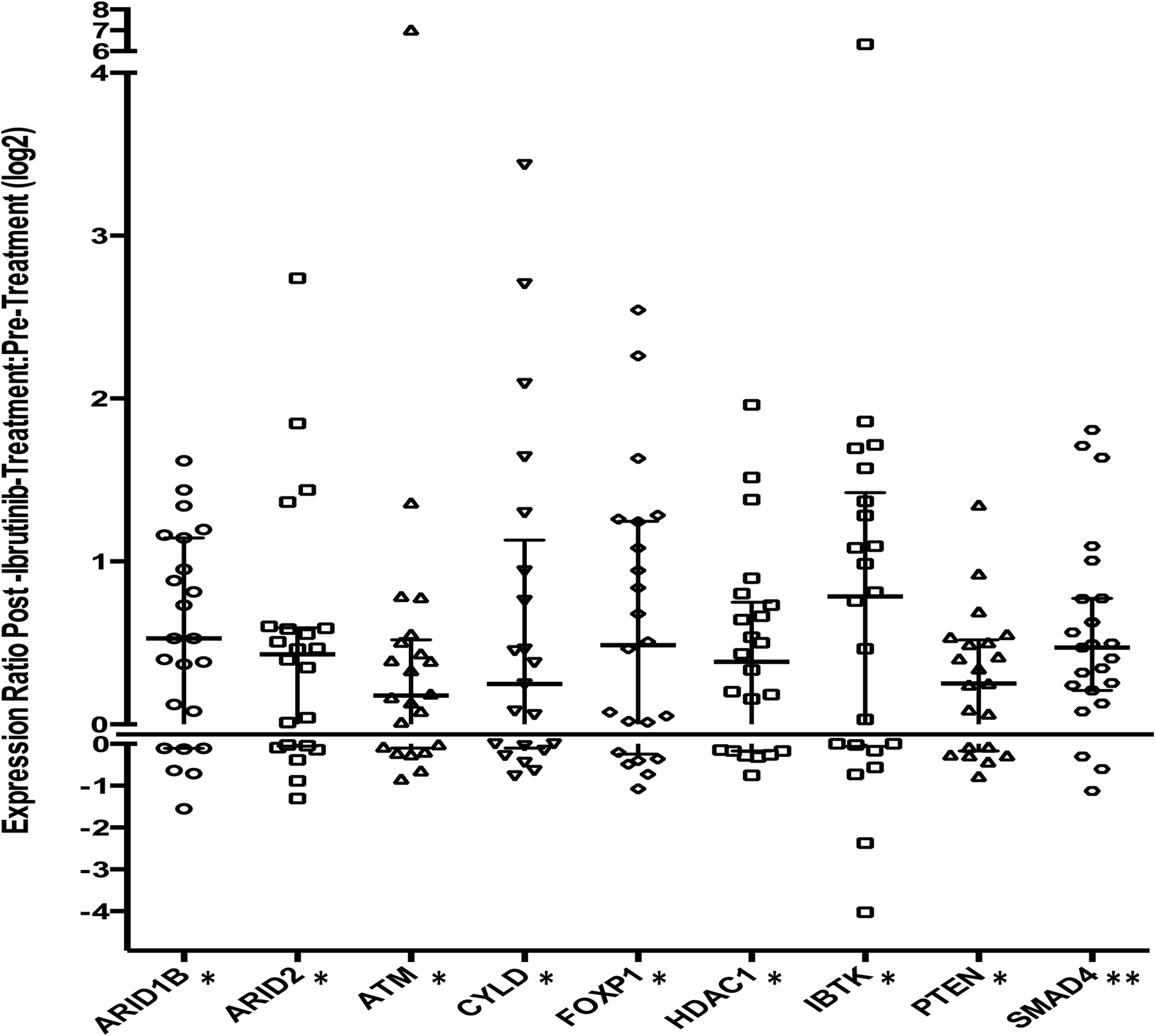
mRNA transcripts of 43 predicted targets of miR-22, miR-34a, miR-146b, and miR-181b were counted by nanoString nCounter assay with RNA isolated from 17 paired pre- and on ibrutinib treatment (day 28) CLL cells. Nine out of 43 predicted targets were significantly altered on ibrutinib (ARID1B, ARID2, ATM, CYLD, FOXP1, HDAC1, IBTK, PTEN, and SMAD4). Scatter plot graph showing the fold change post-ibrutinib:pre-ibrutinib expression ratio, for individual patient CLL cells for nine miRNA targets. A line at 0 is inserted to separate positive and negative fold changes. Median and interquartile ranges are represented. P values were calculated by Wilcoxon paired test. *, p < 0.05, **, p < 0.01.
Target mRNAs of ibrutinib responsive miRNAs are significantly down-regulated in CLL cells in the lymph node
All the ibrutinib responsive miRNAs (miR-22, miR-34a, miR-146b, and miR-181b) were also expressed at higher levels in treatment-naïve LN CLL cells in comparison to matched PB CLL cells (Figure 1; Table 1). Down-regulation of these miRNAs and concomitant upregulation of the tumor suppressor targets of these miRNAs in response to ibrutinib (Figures 2 and 3; Supplementary Tables 6 and 9) suggested that these miRNAs may play an important role in CLL tumor biology. Next we tested whether the tumor suppressor transcripts are also present at lower levels in treatment naïve LN CLL cells having increased levels of the corresponding miRNAs, than matched PB CLL cells. In 13 paired LN and PB CLL samples evaluated by qRT-PCR we found that the mRNA levels of ARID1B, CYLD, and FOXP1 were significantly down-regulated in LN CLL cells in comparison to PB CLL cells (p < 0.05) (Supplementary Table 10).
Alteration of miRNA and target mRNA expression is observed with ibrutinib and not with fludarabine
Ibrutinib is a selective BTK inhibitor37 and is relatively recently utilized as a therapeutic agent for CLL. The chemotherapeutic agent fludarabine is a purine nucleoside analog38 and has been used for CLL treatment for many years. To investigate whether miRNA and target mRNA transcript expression changes are specific to the class of kinase inhibitors represented by ibrutinib, and not a general consequence of chemotherapy, SP53 cells were treated separately with ibrutinib or fludarabine. The percentage of dead cells was counted after trypan blue staining. As shown in Figure 4A, in comparison to untreated cells, a 5.7-fold (p < 0.05) and 4.6-fold (p < 0.05) increase in dead cells was observed, respectively, 48 h after 1 μM of ibrutinib or fludarabine treatment. Assessment of cell proliferation using the WST assay showed a marked 4.6 fold (p < 0.0001) decrease in cell proliferation after ibrutinib treatment; in contrast, treatment with fludarabine resulted in only a 1.1 fold decrease in cell proliferation (p = 0.049). The findings suggest that while both ibrutinib and fludarabine cause a marked increase in cell death; ibrutinib treatment is more potent in reduction of cell proliferation.
Figure 4. Down-regulation of miRNAs (miR-22, miR-34a, miR-146b, and miR-181b), and up-regulation of putative miRNA target tumor suppressors is seen by treatment with ibrutinib and not with fludarabine.
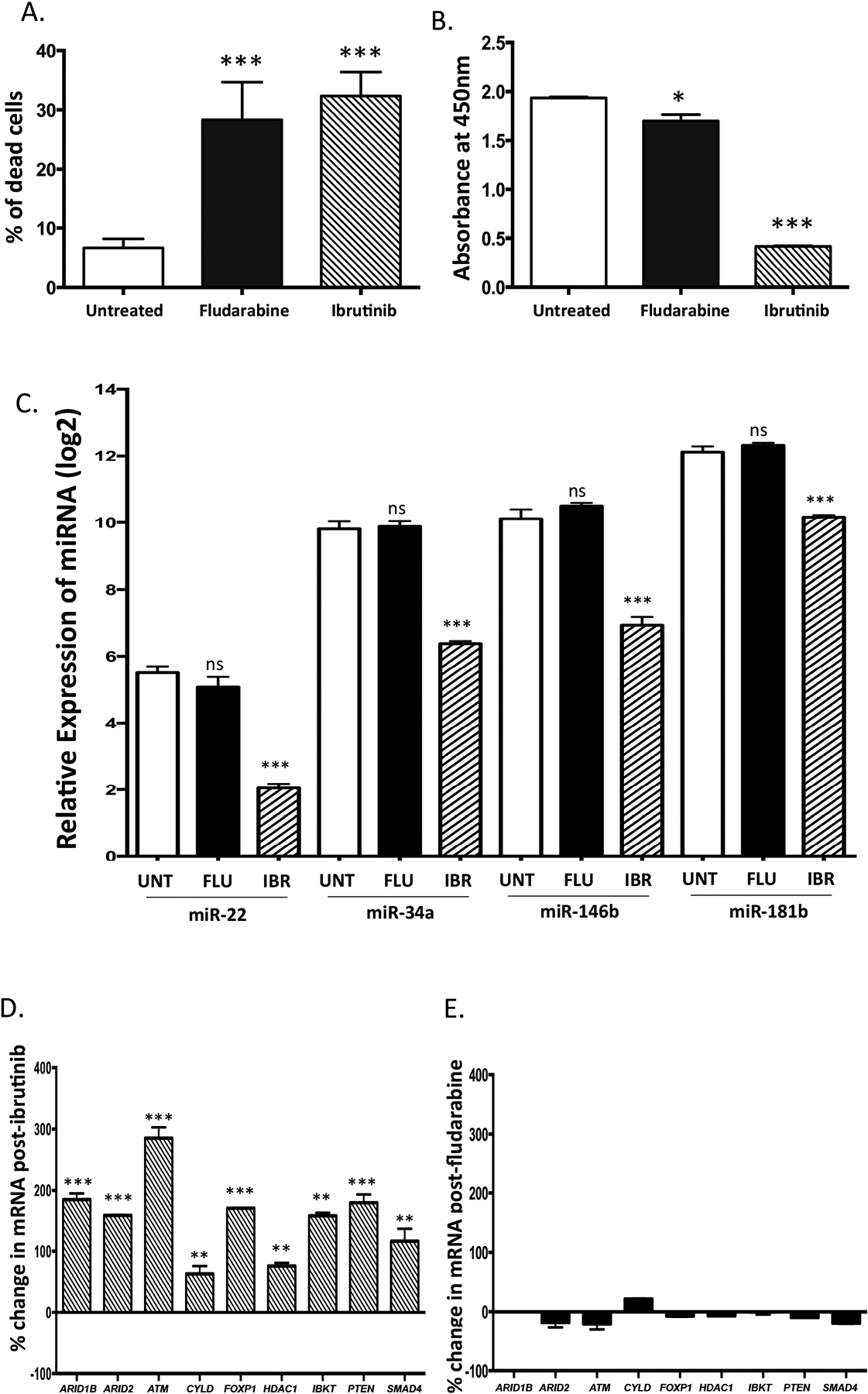
SP53 cells were treated separately with 1 μM of ibrutinib or 1 μM of fludarabine for 48 hours. Mean and standard deviation are shown. (a) The percentage of dead cells is significantly increased by both ibrutinib and fludarabine as enumerated by trypan blue staining. (b) Cell proliferation was markedly decreased with ibrutinib (p < 0.0001), and minimally decreased with fludarabine (p = 0.045) treated cells as measured by the WST assay. (c) The relative expression of miR-22, miR-34a, miR-146b, and miR-181b was significantly decreased in ibrutinib treated cells (p < 0.001) but not in fludarabine treated cells (p > 0.05) as assessed by qPCR in triplicate and compared to basal levels in untreated cells. (d) Putative miRNA targets were significantly altered in response to ibrutinib, (E) but not to fludarabine. The expression of miRNA targets was assessed by qPCR performed in triplicate. P values were calculated by the Student’s t-test. *, p < 0.05, **, p < 0.01; ***, p < 0.001. FLU, fludarabine treatment IBR, ibrutinib treatment;; ns, not significant; UNT untreated.
When miRNA expression was assayed by quantitativePCR in the ibrutinib treated cells, the levels of miR-22, miR-34a, miR146b, and miR-181b were found significantly decreased by 91%, 90%, 90%, and 80%, respectively (Figure 4C), in comparison to basal levels of the miRNA in untreated cells. In sharp contrast, no significant change in the levels of these miRNAs was observed with fludarabine treatment (Figure 4C). The transcription of the putative tumor suppressor targets (ARID1B, ARID2, ATM, CYLD, FOXP1, HDAC1, PTEN, IBTK, and SMAD4) was also assayed in these samples. The transcription of tumor suppressors was significantly increased after ibrutinib treatment, but not after fludarabine treatment (Figure 4d and e; Supplementary Table 11). Taken together, these results suggest that the changes in miRNA expression and target mRNA transcripts levels observed with ibrutinib treatment are likely specific to the mechanism of action of ibrutinib, and are not seen in the context of general antitumor effects with fludarabine.
Ibrutinib responsive miRNAs target multiple tumor suppressor transcripts
To further investigate the role of ibrutinib responsive miRNA in upregulation of tumor suppressors, we chose miR-34a and miR-146b for additional functional studies, largely based on the volume of reports implicating these two miRNAs in cancer.26, 27 miR-34a and miR-146b were both previously reported to target SMAD4;39–42 and miR-34 was reported to target HDAC143,44 in solid tumor cell lines, resulting in inhibition of both mRNA and protein expression. To confirm inhibition of mRNA transcripts, and protein expression regulated by miR-34a and miR146b in B-cell leukemia cell lines,, pre-miR oligonucleotides were transfected into SP53 and MEC1 cells. As shown in Figure 5a, miR-34a and miR-146b levels were markedly increased in both SP53 and MEC1 cells after pre-miR-34a and pre-miR-146b transfection. Expression levels of the predicted miRNA targets ARID1b, ATM, CYLD, FOXP1, HDAC1, IBKT, PTEN, and SMAD4 were assayed in both cell lines (Supplementary Table 12). Of the targets assayed after pri-miR-34a transfection, HDAC1 and SMAD4 transcripts were significantly decreased in both cell lines (Figure 5b). Of the targets assayed after miR-146b expression, ATM, HDAC1, and SMAD4 transcripts were significantly reduced in both cell lines (Figure 5b). Western blot and densitometry data analysis confirmed that protein levels of ATM, HDAC1 and SMAD4 were reduced after both miR-34a (Figure 5c) and miR-146b (Figure 5d) transfection. Transcripts of additional targets were also found to be significantly altered post pre-miR-34a or pre-miR-146b transfection (Supplementary Table 12) however, the findings were not significant across both cell lines for other targets, possibly due to differences in the genetic backgrounds of the two cell lines.
Figure 5. Ibrutinib responsive miRNAs target multiple tumor suppressors in cell lines.
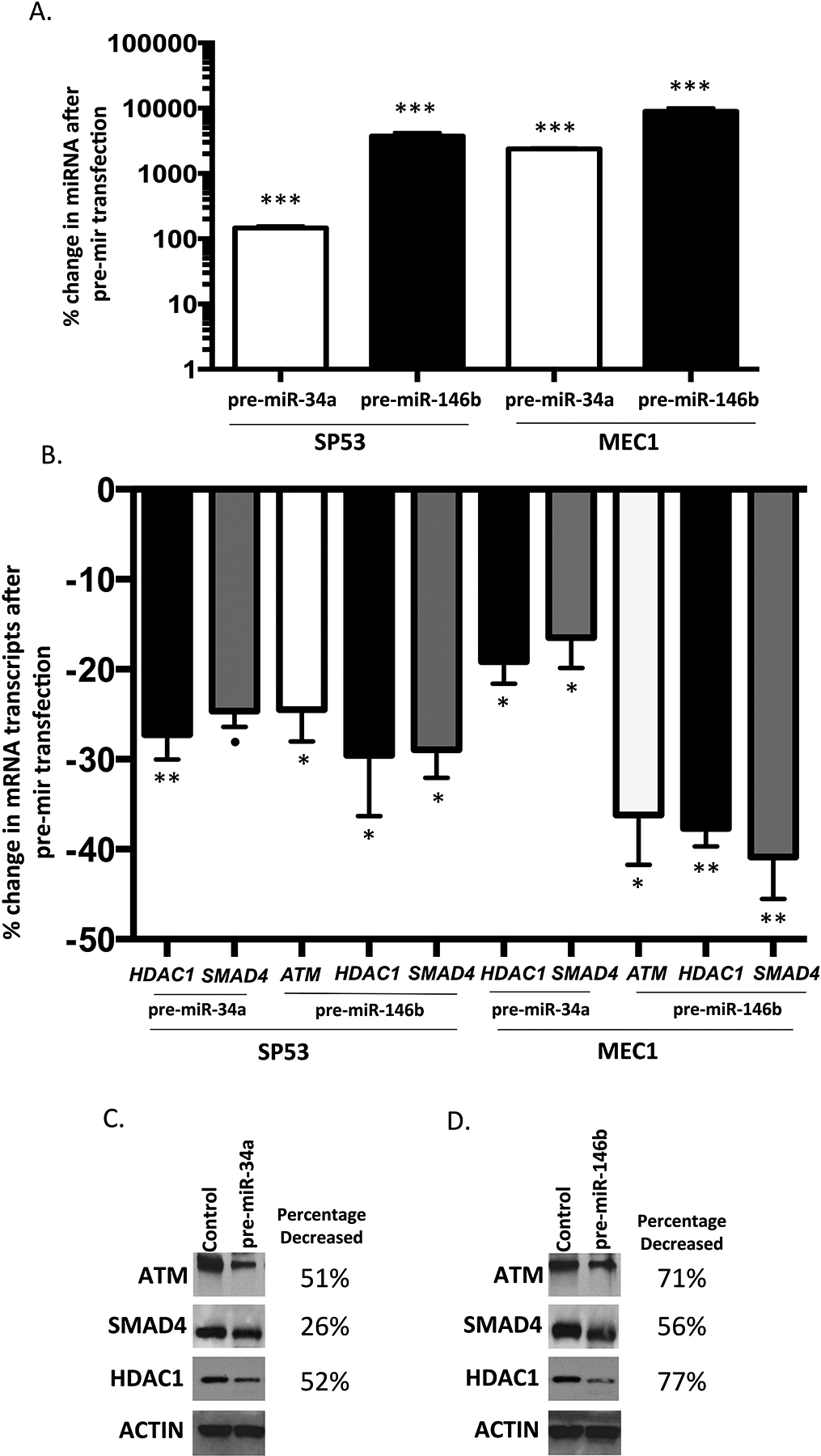
The scrambled control, pre-miR-34a and pre-miR-146b oligos were transfected into SP53 and MEC1 cells, respectively. The expression of miRNAs and putative tumor suppressor mRNAs was measured by qPCR in triplicate. Mean and standard deviation are shown. (a) The expression of miR-34a and miR-146b were markedly increased compared with the scramble control transfection in both SP53 and MEC1 cells 30 hours after pre-miR-34a and pre-miR-146b transfection. (b) The expression of the putative tumor suppressor mRNAs was significantly reduced in SP53 and MEC1 cells after pre-miR-34a and pre-miR-146b transfection. For (a) and (b), P values were calculated by the Student’s t-test. *, p < 0.05, **, p < 0.01; ***, p < 0.001; ● , p =0.06. (c) and (d) Western blot and densitometry analysis data showing decreased protein expression of ATM, SMAD4, and HDAC1 in SP53 cells transfected with pre-miR-34a (c) and pre-miR-146b (d), in comparison to scramble control.
Knock-down of endogenous miRNA results in up-regulation of tumor suppressors and inhibition of cell proliferation
In order to evaluate the effect of decreased miR-34a and miR-146b on the expression of putative tumor suppressor targets, anti-sense oligonucleotides (anti-miRs) were transfected into SP53 and MEC1 cells. Anti-miR-34a and anti-miR-146b effectively knocked down the expression of endogenous miR-34a and miR-146b in both SP53 and MEC1 cells (Figure 6a). Expression levels of predicted miRNA targets ARID1b, ATM, CYLD, FOXP1, HDAC1, IBKT, PTEN, and SMAD4 were assayed (Supplementary Table 12). Of the targets assayed after miR-34a knockdown, HDAC1 and SMAD4 were significantly increased in both cell lines post anti-miR34a transfection. Of the targets assayed under miR-146b knockdown, ATM, HDAC1, and SMAD4 were significantly increased in both cell lines (Figure 6b). Additional targets were also found to be significantly altered post anti-miR-34a or anti-miR-146b transfection (Supplementary Table 12) however, the findings were not significant across both cell lines. The pre-miR overexpression and knock-down results for miR-34a and miR-146b provide evidence that these miRs target a subset of the tumor suppressor transcripts that were upregulated in patient samples in response to ibrutinib.
Figure 6. Knock-down of endogenous miR-34a and miR-146b results in up-regulation of tumor suppressors and inhibition of cell proliferation.
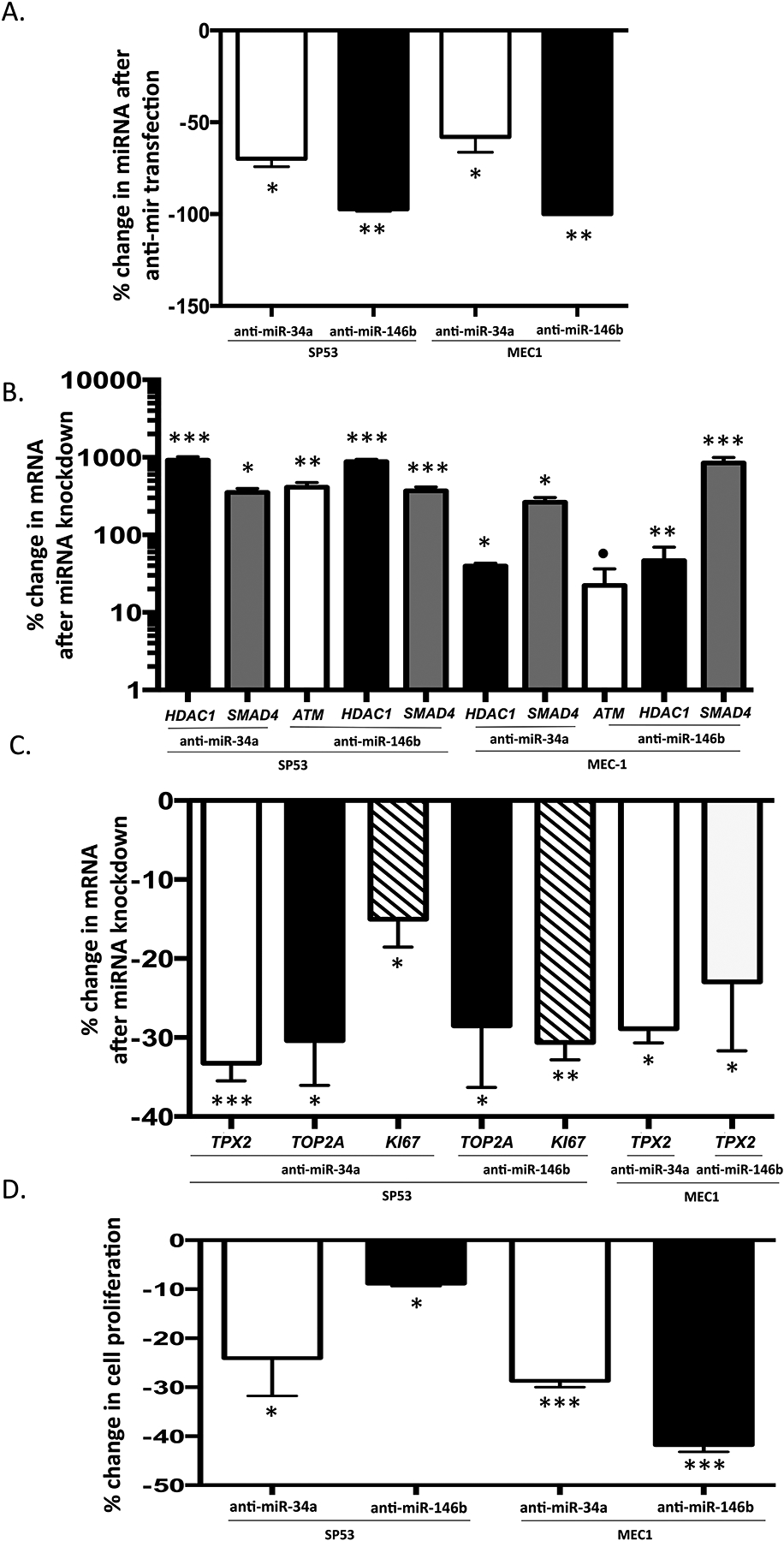
The scrambled control, anti-miR-34a and anti-miR-146b were transfected into SP53 and MEC1 cells, respectively. The expression of miR-34a and miR-146b and putative target mRNA transcripts was measured by quantitative PCR in triplicate. The change in cell proliferation rate was assessed by qPCR for cell proliferation markers and by WST assay. Mean and standard deviation are shown. (a) The expression of miR-34a and miR-146b were significantly decreased compared with the scramble control transfection in both SP53 and MEC1 cells 30 hours after the anti-miR-34a and anti-miR-146b transfection. (b) The expression of the putative target mRNA transcripts of miR-34a and miR-146b was significantly increased in SP53 and MEC1 cells after the anti-miR-34a and anti-miR-146b transfection. (c) The expression of cell proliferation markers (Ki-67, TOP2A, and TPX2) was significantly decreased in SP53 and MEC1 cells after knockdown of miR-34a and miR-146b by anti-miR-34a and anti-miR-146b transfection. (d) Change in cell proliferation is significantly decreased after knockdown of miR-34a and miR-146b as measured by the WST assay 24 h after the anti-miR-34a and anti-miR-146b transfection in SP53 and MEC1 cells. P values were calculated by the Student’s t-test. *, p < 0.05, **, p < 0.01; ***, p < 0.001; ●, p =0.08.
We postulated that decreased miR-34a and miR-146b expression and upregulation of tumor suppressor targets might have an effect on tumor cell proliferation. To evaluate the effect of reduced miR-34a and miR-146b on cell proliferation, the level of three cell proliferation markers (Ki-67, topoisomerase II alpha (TOP2A), and targeting protein for xenopus kinesin-like protein 2 (TPX2))45 were assayed by quantitative PCR from the complementary DNA of knock-down samples. In SP53 cells, knock-down of miR-34a resulted in a significant decrease in Ki-67 (−15.63%; p = 0.035), TOP2A (−30.34%; p = 0.023), and TPX2 (−34.09%; p = 0.001); and knock-down of miR-146b resulted in significant decrease in Ki-67 (−31%; p = 0.0031) and TOP2A (−27.13%; p = 0.026) (Figure 6C). In MEC1 cells TPX2 was significantly decreased after transfection of anti-miR-34a and anti-miR-146b (p<0.05) (Figure 6c and Supplementary Table 13). When the cell proliferation rates were measured from the same samples, WST assay results demonstrated that cell proliferation was significantly inhibited in both SP53 and MEC1 cells 24 hours after anti-miR-34a and anti-miR-146b transfection (Figure 6d). Knockdown of miR-34a resulted in a 24% decrease in proliferation in SP53 cells and 27% decrease in MEC-1 cells. Similarly, knock-down of miR-146b reduced cell proliferation by 9% in SP-53 cells and by 42% in MEC-1 cells (Figure 6D). Taken together, these results demonstrate that ibrutinib treatment causes down-regulation of miRNAs, including miR34a and miR-146b, which in turn results in up-regulation of tumor suppressor targets and inhibition of cell proliferation.
DISCUSSION
Herein, we report that CLL cells residing in the lymph node have a distinct increase in a subset of miRNAs associated with activated BCR and NF-kappa B signaling. This was accompanied by a corresponding decrease in miRNA targets, many of which are tumor suppressors. Among the miRNAs tested, miR-22, miR-34a, miR-146b, and miR-181b were significantly down-regulated in patient CLL cells post ibrutinib treatment. This lead to increased expression of multiple tumor suppressors including ARID1B, ARID2, ATM, CYLD, FOXP1, HDAC1, IBTK, PTEN, and SMAD4. Functional studies in cell lines confirmed that ibrutinib results in down-regulation of these four miRNAs. Knock-down of endogenous miR-34a and miR-146b leads to increased levels of ATM, HDAC1, and SMAD4 transcripts; and decreased cell proliferation as evidenced both by a significant reduction in cell proliferation markers (Ki-67, TOP2A, and TPX2) and by a significant decrease in cell proliferation rate. The findings of decreased expression of miRNAs, up-regulation of miRNA tumor suppressor targets, and inhibition of cell proliferation in response to ibrutinib provides additional insight into the mechanisms of therapeutic response of CLL to ibrutinib.
Ibrutinib exerts antitumor effects through disruption of BCR and NF-kappa B signaling,7 and induces apoptosis, decreases cell proliferation, inhibits micro-environmental survival stimuli to tumor cells, and impairs tumor-stromal adhesion.46 Yeh et al.20 recently reported that ibrutinib treatment also results in decreased exosomal secretion by CLL cells and alters exosomal miRNA profiles. Similarly, we found ibrutinib treatment results in significant decreases in the expression of miR-22, miR-34a, miR-146b, and miR-181b; among tested miRNAs. miR-22 is postulated to play an important role in CLL proliferation by targeting phosphatase and tensin homolog (PTEN), resulting in activation of the PI3K/AKT pathway.47,48 In our study, miR-22 was highly expressed in CLL cells in the LN, lower in circulating CLL cells, and even lower in circulating B-cells from healthy controls. Ibrutinib resulted in a threefold decrease in miR-22 expression in patients’ CLL cells, which was accompanied by a concomitant increase in PTEN transcripts. Similar results were also observed in experiments in cell lines suggesting that reduction in miR-22 contributes to up-regulation of PTEN in response to ibrutinib.
Higher expression of miR-146b in ZAP-70 positive and un-mutated IGHV CLL27 and in EBV positive DLBCL43 has been previously reported. Approximately 60% of the CLL cases in our study were IGHV un-mutated. We found that miR-146b was highly expressed in CLL cells in the LN and was significantly decreased (5.5-fold) in CLL cells after ibrutinib treatment. miR-146b is shown to target multiple tumor suppressors including SMAD442, which regulates the TGF-βsignaling pathway and drives increased cellular proliferation. Consistent with the reported relationship between miR-146b and SMAD4, we observed a significant increase in SMAD4 transcripts upon down-regulation of miR-146b in response to ibrutinib in patient CLL cells. These findings were confirmed in functional studies in cell lines treated with ibrutinib. Our studies also provide evidence that miR-146b targets ATM and HDAC1, which is previously unreported.
The TP53 pathway is frequently mutated in CLL. miR-34a, a target of TP53, has been reported to be altered in CLL and has been associated with disease progression and outcomes.17, 19, 50, 51 Increased miR-34a levels have been noted in several CLL studies.26, 29,52, 53 Inhibition of miR-34a by antisense RNA has been shown to sensitize lymphoma cells to apoptosis.54 We found that levels of miR-34a were 87-fold higher in circulating CLL cells in comparison to normal circulating B-cells; and miR-34a was significantly higher in CLL cells in the LN than that in the PB. miR-34a showed a 5.5-fold decrease in patient cells in response to ibrutinib. Knock-down of miR-34a in cell lines resulted in increased mRNA and protein expression of HDAC1 and SMAD4; and resulted in decreased cell proliferation. HDAC1 was previously shown to be a target of miR-34a;43, 44 and implicated as a tumor suppressor in a TP53-deleted and MYC over-expressed mouse model, whereby lymphomagenesis was accelerated by knock-down of HDAC1.44,55 miR-34a has been shown to inhibit mesenchymal transition in human cholangiocarcinoma and associate with glioblastoma survival by targeting SMAD4 of the TGF-beta signaling pathway.39,41 Several other studies have also shown that SMAD4 is a target of miR-34a in functional studies involving solid tumor cell lines.40, 56 Further studies are needed to elucidate the exact roles that miR-34a might play in CLL- in TP53-dependent and- independent settings; and in ibrutinib-induced cell growth inhibition and cell death.
Ibrutinib responsive miRNAs in our study were predicted to have multiple overlapping targets including ATM, ARID1B, ARID2, CYLD, FOXP1, SMAD4, PTEN, and IBTK. Nanostring nCounter assay results indicated that the transcription of these tumor suppressors was largely increased after ibrutinib treatment. ATM regulates cell cycle checkpoints and DNA repair process.36 ARID1B is a member of chromatin remodeling complexes that function as tumor suppressors in many cancers.57, 58 Recurrent ARID1B and ARID2 mutations have been reported in breast, pancreatic, and gastric cancers.59 CYLD encodes a deubiquitination enzyme that is reported to be a negative regulator of the NF-kappa B pathway and may play a role in CLL leukemogenesis.60 CYLD also serves as a regulator of necroptosis, an alternative cell death pathway when apoptotic pathways are suppressed or blocked.61 Absence of CYLD is reported in many cancers62 and CLL patients with high CYLD levels were reported to have improved overall survival.60 IBTK is a physiologic inhibitor of BTK.63 We found that IBTK was significantly up-regulated by ibrutinib, which may contribute to enhanced inhibition of BTK signaling and ibrutinib treated tumor response.
Ibrutinib is known to inhibit BTK and NF-kappa B signaling and to represent an effective molecularly targeted therapeutic agent in CLL. Our findings additionally suggest that the therapeutic response to ibrutinib involves the decrease of a subset of miRNAs that play a role in the up-regulation of tumor suppressors and inhibition of cell proliferation, expanding our knowledge of the mechanism of action of ibrutinib.
Supplementary Material
Acknowledgements:
This work was supported by the National Institutes of Health Division of Intramural Research, NIH Clinical Center, and National Heart, Lung and Blood Institute. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services. We thank Dr. Masanori Daibata (Kochi University Medical School, Kochi, Japan) for use of the SP53 cell line.
Footnotes
Conflict-of-interest disclosure: Adrian Wiestner has received research funding from Pharmacyclics. The remaining authors have nothing to disclose.
References
- 1.Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 Who Classification of Lymphoid Neoplasms and Beyond: Evolving Concepts and Practical Applications. Blood 2011; 117: 5019–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herishanu Y, Katz BZ, Lipsky A, Wiestner A. Biology of Chronic Lymphocytic Leukemia in Different Microenvironments: Clinical and Therapeutic Implications. Hematol Oncol Clin North Am 2013; 27: 173–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang S, Kipps TJ. The Pathogenesis of Chronic Lymphocytic Leukemia. Annu Rev Pathol 2014; 9: 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fegan C, Pepper C. Apoptosis Deregulation in Cll. Adv Exp Med Biol 2013; 792: 151–171. [DOI] [PubMed] [Google Scholar]
- 5.Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The Lymph Node Microenvironment Promotes B-Cell Receptor Signaling, Nf-Kappab Activation, and Tumor Proliferation in Chronic Lymphocytic Leukemia. Blood 2011; 117: 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as Initial Therapy for Patients with Chronic Lymphocytic Leukemia. N Engl J Med 2015; 373: 2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wiestner A The Role of B-Cell Receptor Inhibitors in the Treatment of Patients with Chronic Lymphocytic Leukemia. Haematologica 2015; 100: 1495–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting Btk with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N Engl J Med 2013; 369: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herman SE, Mustafa RZ, Gyamfi JA, Pittaluga S, Chang S, Chang B, et al. Ibrutinib Inhibits Bcr and Nf-Kappab Signaling and Reduces Tumor Proliferation in Tissue-Resident Cells of Patients with Cll. Blood 2014; 123: 3286–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farooqui MZH, Valdez J, Martyr S, Aue G, Saba N, Niemann CU, et al. Ibrutinib for Previously Untreated and Relapsed or Refractory Chronic Lymphocytic Leukaemia with Tp53 Aberrations: A Phase 2, Single-Arm Trial. The Lancet Oncology 2015; 16: 169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Claro RA, McGinn KM, Verdun N, Lee SL, Chiu HJ, Saber H, et al. Fda Approval: Ibrutinib for Patients with Previously Treated Mantle Cell Lymphoma and Previously Treated Chronic Lymphocytic Leukemia. Clin Cancer Res 2015; 21: 3586–3590. [DOI] [PubMed] [Google Scholar]
- 12.Cheng S, Ma J, Guo A, Lu P, Leonard JP, Coleman M, et al. Btk Inhibition Targets in Vivo Cll Proliferation through Its Effects on B-Cell Receptor Signaling Activity. Leukemia 2014; 28: 649–657. [DOI] [PubMed] [Google Scholar]
- 13.Herman SE, Niemann CU, Farooqui M, Jones J, Mustafa RZ, Lipsky A, et al. Ibrutinib-Induced Lymphocytosis in Patients with Chronic Lymphocytic Leukemia: Correlative Analyses from a Phase Ii Study. Leukemia 2014; 28: 2188–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bartel DP. Micrornas: Target Recognition and Regulatory Functions. Cell 2009; 136: 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Croce CM. Causes and Consequences of Microrna Dysregulation in Cancer. Nat Rev Genet 2009; 10: 704–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rossi S, Shimizu M, Barbarotto E, Nicoloso MS, Dimitri F, Sampath D, et al. Microrna Fingerprinting of Cll Patients with Chromosome 17p Deletion Identify a Mir-21 Score That Stratifies Early Survival. Blood 2010; 116: 945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dufour A, Palermo G, Zellmeier E, Mellert G, Duchateau-Nguyen G, Schneider S, et al. Inactivation of Tp53 Correlates with Disease Progression and Low Mir-34a Expression in Previously Treated Chronic Lymphocytic Leukemia Patients. Blood 2013; 121: 3650–3657. [DOI] [PubMed] [Google Scholar]
- 18.Mraz M, Chen L, Rassenti LZ, Ghia EM, Li H, Jepsen K, et al. Mir-150 Influences B-Cell Receptor Signaling in Chronic Lymphocytic Leukemia by Regulating Expression of Gab1 and Foxp1. Blood 2014; 124: 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mraz M, Malinova K, Kotaskova J, Pavlova S, Tichy B, Malcikova J, et al. Mir-34a, Mir-29c and Mir-17–5p Are Downregulated in Cll Patients with Tp53 Abnormalities. Leukemia 2009; 23: 1159–1163. [DOI] [PubMed] [Google Scholar]
- 20.Yeh YY, Ozer HG, Lehman AM, Maddocks K, Yu L, Johnson AJ, et al. Characterization of Cll Exosomes Reveals a Distinct Microrna Signature and Enhanced Secretion by Activation of Bcr Signaling. Blood 2015; 125: 3297–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu DX, Zhu W, Fang C, Fan L, Zou ZJ, Wang YH, et al. Mir-181a/B Significantly Enhances Drug Sensitivity in Chronic Lymphocytic Leukemia Cells Via Targeting Multiple Anti-Apoptosis Genes. Carcinogenesis 2012; 33: 1294–1301. [DOI] [PubMed] [Google Scholar]
- 22.Mraz M, Pospisilova S, Malinova K, Slapak I, Mayer J. Micrornas in Chronic Lymphocytic Leukemia Pathogenesis and Disease Subtypes. Leuk Lymphoma 2009; 50: 506–509. [DOI] [PubMed] [Google Scholar]
- 23.Li S, Moffett HF, Lu J, Werner L, Zhang H, Ritz J, et al. Microrna Expression Profiling Identifies Activated B Cell Status in Chronic Lymphocytic Leukemia Cells. PLoS One 2011; 6: e16956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al. Mir-15 and Mir-16 Induce Apoptosis by Targeting Bcl2. Proc Natl Acad Sci U S A 2005; 102: 13944–13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Degheidy HA, Gadalla SM, Farooqui MZ, Abbasi F, Arthur DC, Bauer SR, et al. Bcl-2 Level as a Biomarker for 13q14 Deletion in Cll. Cytometry B Clin Cytom 2013; 84: 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Negrini M, Cutrona G, Bassi C, Fabris S, Zagatti B, Colombo M, et al. Micrornaome Expression in Chronic Lymphocytic Leukemia: Comparison with Normal B-Cell Subsets and Correlations with Prognostic and Clinical Parameters. Clin Cancer Res 2014; 20: 4141–4153. [DOI] [PubMed] [Google Scholar]
- 27.Calin GA, Ferracin M, Cimmino A, Di Leva G, Shimizu M, Wojcik SE, et al. A Microrna Signature Associated with Prognosis and Progression in Chronic Lymphocytic Leukemia. N Engl J Med 2005; 353: 1793–1801. [DOI] [PubMed] [Google Scholar]
- 28.Mraz M, Kipps TJ. Micrornas and B Cell Receptor Signaling in Chronic Lymphocytic Leukemia. Leuk Lymphoma 2013; 54: 1836–1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pede V, Rombout A, Vermeire J, Naessens E, Mestdagh P, Robberecht N, et al. Cll Cells Respond to B-Cell Receptor Stimulation with a Microrna/Mrna Signature Associated with Myc Activation and Cell Cycle Progression. PLoS One 2013; 8: e60275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang W, Corrigan-Cummins M, Hudson J, Maric I, Simakova O, Neelapu SS, et al. Microrna Profiling of Follicular Lymphoma Identifies Micrornas Related to Cell Proliferation and Tumor Response. Haematologica 2012; 97: 586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luo X, Zhang J, Wang H, Du Y, Yang L, Zheng F, et al. Polya Rt-Pcr-Based Quantification of Microrna by Using Universal Taqman Probe. Biotechnol Lett 2012; 34: 627–633. [DOI] [PubMed] [Google Scholar]
- 32.Wang W, Corrigan-Cummins M, Barber EA, Saleh LM, Zingone A, Ghafoor A, et al. Aberrant Levels of Mirnas in Bone Marrow Microenvironment and Peripheral Blood of Myeloma Patients and Disease Progression. J Mol Diagn 2015; 17: 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kubiczkova L, Kryukov F, Slaby O, Dementyeva E, Jarkovsky J, Nekvindova J, et al. Circulating Serum Micrornas as Novel Diagnostic and Prognostic Biomarkers for Multiple Myeloma and Monoclonal Gammopathy of Undetermined Significance. Haematologica 2014; 99: 511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daibata M, Kubonishi I, Eguchi T, Yano S, Ohtsuki Y, Miyoshi I. The Establishment of Epstein-Barr Virus Nuclear Antigen-Positive (Sp-50b) and Epstein-Barr Virus Nuclear Antigen-Negative (Sp-53) Cell Lines with T(11;14)(Q13;Q32) Chromosome Abnormality from an Intermediate Lymphocytic Lymphoma. Cancer 1989; 64: 1248–1253. [DOI] [PubMed] [Google Scholar]
- 35.Schneider CA, Rasband WS, Eliceiri KW. Nih Image to Imagej: 25 Years of Image Analysis. Nat Methods 2012; 9: 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foa R, Del Giudice I, Guarini A, Rossi D, Gaidano G. Clinical Implications of the Molecular Genetics of Chronic Lymphocytic Leukemia. Haematologica 2013; 98: 675–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Zhang LL, Champlin RE, Wang ML. Targeting Bruton’s Tyrosine Kinase with Ibrutinib in B-Cell Malignancies. Clin Pharmacol Ther 2015; 97: 455–468. [DOI] [PubMed] [Google Scholar]
- 38.Elter T, Hallek M, Engert A. Fludarabine in Chronic Lymphocytic Leukaemia. Expert Opin Pharmacother 2006; 7: 1641–1651. [DOI] [PubMed] [Google Scholar]
- 39.Genovese G, Ergun A, Shukla SA, Campos B, Hanna J, Ghosh P et al. microRNA regulatory network inference identifies miR-34a as a novel regulator of TGF-β signaling in glioblastoma. Cancer Discov 2012; 2: 736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lal A, Thomas MP, Altschuler G, Navarro F, O’Day E, Li XL et al. Capture of microRNA-bound mRNAs identifies the tumor suppressor miR-34a as a regulator of growth factor signaling. PLoS Genet 2011; 7: e1002363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qiao P, Li G, Bi W, Yang L, Yao L, Wu D. microRNA-34a inhibits epithelial mesenchymal transition in human cholangiocarcinoma by targeting Smad4 through transforming growth factor-beta/Smad pathway. BMC Cancer 2015; 15: 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geraldo MV, Yamashita AS, Kimura ET. MicroRNA miR-146b-5p regulates signal transduction of TGF-β by repressing SMAD4 in thyroid cancer. Oncogene 2012; 31: 1910–1922. [DOI] [PubMed] [Google Scholar]
- 43.Wu MY, Fu J, Xiao X, Wu J, Wu RC. MiR-34a regulates therapy resistance by targeting HDAC1 and HDAC7 in breast cancer. Cancer Lett 2014; 354: 311–319. [DOI] [PubMed] [Google Scholar]
- 44.Zhao J, Lammers P, Torrance CJ, Bader AG. TP53-independent function of miR-34a via HDAC1 and p21(CIP1/WAF1.). Mol Ther 2013; 21: 1678–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brizova H, Kalinova M, Krskova L, Mrhalova M, Kodet R. A Novel Quantitative Pcr of Proliferation Markers (Ki-67, Topoisomerase Iialpha, and Tpx2): An Immunohistochemical Correlation, Testing, and Optimizing for Mantle Cell Lymphoma. Virchows Arch 2010; 456: 671–679. [DOI] [PubMed] [Google Scholar]
- 46.Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton Tyrosine Kinase Represents a Promising Therapeutic Target for Treatment of Chronic Lymphocytic Leukemia and Is Effectively Targeted by Pci-32765. Blood 2011; 117: 6287–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Palacios F, Prieto D, Abreu C, Ruiz S, Morande P, Fernandez-Calero T, et al. Dissecting Chronic Lymphocytic Leukemia Microenvironment Signals in Patients with Unmutated Disease: Microrna-22 Regulates Phosphatase and Tensin Homolog/Akt/Foxo1 Pathway in Proliferative Leukemic Cells. Leuk Lymphoma 2015; 56: 1560–1565. [DOI] [PubMed] [Google Scholar]
- 48.Palacios F, Abreu C, Prieto D, Morande P, Ruiz S, Fernandez-Calero T, et al. Activation of the Pi3k/Akt Pathway by Microrna-22 Results in Cll B-Cell Proliferation. Leukemia 2015; 29: 115–125. [DOI] [PubMed] [Google Scholar]
- 49.Andrade TA, Evangelista AF, Campos AH, Poles WA, Borges NM, Camillo CM, et al. A Microrna Signature Profile in Ebv+ Diffuse Large B-Cell Lymphoma of the Elderly. Oncotarget 2014; 5: 11813–11826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zenz T, Mohr J, Eldering E, Kater AP, Buhler A, Kienle D, et al. Mir-34a as Part of the Resistance Network in Chronic Lymphocytic Leukemia. Blood 2009; 113: 3801–3808. [DOI] [PubMed] [Google Scholar]
- 51.Fabbri M, Bottoni A, Shimizu M, Spizzo R, Nicoloso MS, Rossi S, et al. Association of a Microrna/Tp53 Feedback Circuitry with Pathogenesis and Outcome of B-Cell Chronic Lymphocytic Leukemia. JAMA 2011; 305: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruiz-Lafuente N, Alcaraz-Garcia MJ, Sebastian-Ruiz S, Garcia-Serna AM, Gomez-Espuch J, Moraleda JM, et al. Il-4 up-Regulates Mir-21 and the Mirnas Hosted in the Clcn5 Gene in Chronic Lymphocytic Leukemia. PLoS One 2015; 10: e0124936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Asslaber D, Pinon JD, Seyfried I, Desch P, Stocher M, Tinhofer I, et al. Microrna-34a Expression Correlates with Mdm2 Snp309 Polymorphism and Treatment-Free Survival in Chronic Lymphocytic Leukemia. Blood 2010; 115: 4191–4197. [DOI] [PubMed] [Google Scholar]
- 54.Sotillo E, Laver T, Mellert H, Schelter JM, Cleary MA, McMahon S, et al. Myc Overexpression Brings out Unexpected Antiapoptotic Effects of Mir-34a. Oncogene 2011; 30: 2587–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Santoro F, Botrugno OA, Dal Zuffo R, Pallavicini I, Matthews GM, Cluse L, et al. A Dual Role for Hdac1: Oncosuppressor in Tumorigenesis, Oncogene in Tumor Maintenance. Blood 2013; 121: 3459–3468. [DOI] [PubMed] [Google Scholar]
- 56.Huang Y, Qi Y, Du JQ, Zhang DF. MicroRNA-34a regulates cardiac fibrosis after myocardial infarction by targeting Smad4. Expert Opin Ther Targets 2014; 18: 1355–1365. [DOI] [PubMed] [Google Scholar]
- 57.Jones S, Stransky N, McCord CL, Cerami E, Lagowski J, Kelly D, et al. Genomic Analyses of Gynaecologic Carcinosarcomas Reveal Frequent Mutations in Chromatin Remodelling Genes. Nat Commun 2014; 5: 5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shao F, Guo T, Chua PJ, Tang L, Thike AA, Tan PH, et al. Clinicopathological Significance of Arid1b in Breast Invasive Ductal Carcinoma. Histopathology 2015; 67: 709–718. [DOI] [PubMed] [Google Scholar]
- 59.Shain AH, Pollack JR. The Spectrum of Swi/Snf Mutations, Ubiquitous in Human Cancers. PLoS One 2013; 8: e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu W, Zhu H, Fu Y, Shen W, Xu J, Miao K, et al. Clinical Significance of Down-Regulated Cylindromatosis Gene in Chronic Lymphocytic Leukemia. Leuk Lymphoma 2014; 55: 588–594. [DOI] [PubMed] [Google Scholar]
- 61.Liu P, Xu B, Shen W, Zhu H, Wu W, Fu Y, et al. Dysregulation of Tnfalpha-Induced Necroptotic Signaling in Chronic Lymphocytic Leukemia: Suppression of Cyld Gene by Lef1. Leukemia 2012; 26: 1293–1300. [DOI] [PubMed] [Google Scholar]
- 62.Strobel P, Zettl A, Ren Z, Starostik P, Riedmiller H, Storkel S, et al. Spiradenocylindroma of the Kidney: Clinical and Genetic Findings Suggesting a Role of Somatic Mutation of the Cyld1 Gene in the Oncogenesis of an Unusual Renal Neoplasm. Am J Surg Pathol 2002; 26: 119–124. [DOI] [PubMed] [Google Scholar]
- 63.Spatuzza C, Schiavone M, Di Salle E, Janda E, Sardiello M, Fiume G, et al. Physical and Functional Characterization of the Genetic Locus of Ibtk, an Inhibitor of Bruton’s Tyrosine Kinase: Evidence for Three Protein Isoforms of Ibtk. Nucleic Acids Res 2008; 36: 4402–4416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.