

Abstract
Genome engineering of cells using CRISPR/Cas systems has opened new avenues for pharmacological screening and investigating the molecular mechanisms of disease. A critical step in many such studies, is the intracellular delivery of the gene editing machinery and the subsequent manipulation of cells. However, these workflows often involve processes such as bulk electroporation for intracellular delivery and fluorescence activated cell sorting (FACS) for cell isolation that can be harsh to sensitive cell types such as human induced pluripotent stem cells (hiPSCs). This often leads to poor viability and low overall efficacy, requiring the use of large starting samples. In this work a fully automated version of the nanofountain probe electroporation (NFP-E) system, a nanopipette-based single-cell electroporation method is presented, that provides superior cell viability and efficiency compared to traditional methods. The automated system utilizes a deep convolutional network to identify cell locations and a cell-nanopipette contact algorithm to position the nanopipette over each cell for the application of electroporation pulses. The automated NFP-E is combined with micro-confinement arrays for cell isolation to demonstrate a workflow that can be used for CRISPR/Cas9 gene editing and cell tracking with potential applications in screening studies and isogenic cell line generation.
Keywords: Single-cell electroporation, intracellular delivery, deep learning, hiPSCs, CRISPR/Cas9
Graphical Abstract
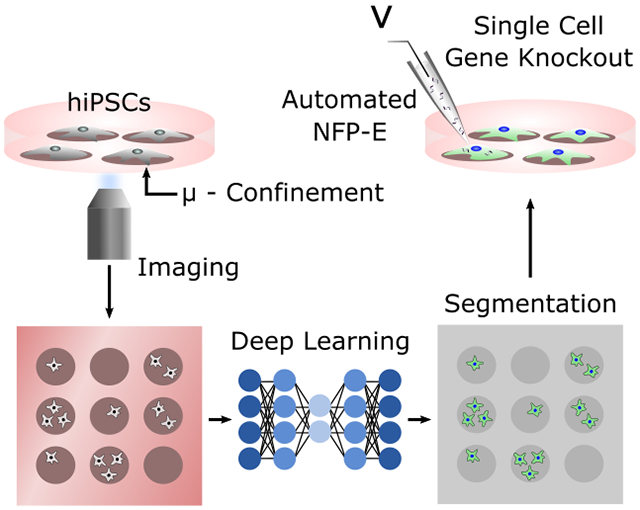
In this article, the authors present a deep learning assisted Nanofountain Probe Electroporation system in combination with micro-confinement arrays to trap and transfect single cells. Using the combined platform, the authors demonstrate automated intracellular delivery and genetic perturbation in hard-to-transfect cells followed by temporal tracking of the perturbed cell colonies.
Introduction
The development of CRISPR/Cas systems and human induced pluripotent stem cell (hiPSC) technology over the past decade has ushered in a new paradigm in disease diagnostics, therapeutic discovery, and regenerative medicine.[1, 2] The CRISPR/Cas9 technology has been frequently applied in pooled and arrayed screening formats, where cells are treated with a library of sgRNA vectors and analyzed to identify genetic targets.[3] More recently, the pooled CRISPR/Cas9 screening method has been extended to combinatorial perturbations using multiple sgRNAs and combined with single cell RNA sequencing readout to reveal genetic interactions in diseases.[4, 5] These new approaches have provided insights into the genetic background and variability of diseases previously unattainable using observational studies like Genome Wide Association Studies (GWAS) or animal models.[1]
Additionally, with the advent of the hiPSC technology, it has been possible to use the CRISPR/Cas9 system for the generation of patient-derived isogenic hiPSC lines. These hiPSC lines have been used to study the genetic basis of diseases.[6, 7] Such advancements have allowed for the design of disease modeling and drug discovery studies, where genetic targets are identified using high-throughput CRISPR based screening. This is followed by the generation of isogenic knock-out/knock-in hiPSC lines for further validation of the top targets.
A critical step in genome engineering and cell manipulation using the CRISPR/Cas9 machinery is the introduction of the functional Cas9 endonuclease and the sgRNAs into the cells of interest.[8] This is achieved either by delivering plasmid DNAs expressing the Cas9 protein and sgRNAs or via the direct intracellular delivery of Cas9/sgRNA ribonucleoprotein complexes (RNPs).[8] Commonly used methods of intracellular delivery include viral vectors, chemical carriers, and bulk electroporation (BEP).[9, 10] However, major limitations of these methods include cytotoxicity or massive cell death in the case of sensitive primary and stem cells [11, 12], low efficiency for hard-to-transfect cell types, and unintended cellular response [13, 14] which necessitates extensive optimization.[9, 10] Moreover, downstream processes such as flow sorting for enrichment, are prone to contamination and is harsh on the stem cells. This leads to poor cell survival (20-40%) [15, 16] and low overall efficacy.[17, 18] In order to overcome some of these limitations, several microfluidic approaches have been pursued by researchers. One class of methods utilizes high aspect ratio nanostructures such as nanopores,[19, 20] nanoneedles[21, 22] and nanostraws[23, 24] to either penetrate the cell membrane or electroporate the cells locally at the cell membrane-nanostructure interface for intracellular cargo delivery. These platforms have been successfully employed for cargo delivery into a wide variety of cell types with minimal perturbation including primary immune and stem cells.[25–28] However, these architectures usually involve complex fabrication protocols and do not provide selectivity over individual cells in culture. Flow-based systems represent a second class of methods that rely on perturbations in microfluidic flows for intracellular delivery. Various forms of perturbations have been used in microfluidic flows to permeabilize cells, such as constrictions or protrusions (e.g. cell squeeze),[13, 29–31] secondary inertial flows,[32, 33] and electric fields[34] to name a few. These platforms have been largely successful for engineering cells of the hematopoietic lineage with implications in immunotherapy. Yet, they have drawbacks such as cell size dependent device design and requirement to dissociate cells which may not be ideal for sensitive adherent cell types that are prone to detachment and dissociation induced apoptosis.[35, 36] Finally, probe-based methods use nanopipettes or hollow AFM tips for electroporation induced or direct intracellular delivery. Examples of this class of methods include scanning ion channel microscopy (SICM)-based nanomanipulators,[37] the FluidFM[38] and the Nanofountain Probe Electroporation (NFP-E) system,[39] a single cell electroporation method previously developed by our group. Although, probe-based methods provide high selectivity and single cell resolution delivery, they have low throughput due to their serial approach and tedious manual operation steps, thus limiting their utility in cell perturbation and manipulation workflows. To address this limitation, we present an automated NFP-E system with improved throughput, that can be used for genetic manipulation of hard-to-transfect and sensitive cells. We have previously demonstrated superior performance of the NFP-E over traditional methods in terms of cell viability, efficiency, and dosage control in a broad spectrum of cell types.[39–41] Here, we demonstrate the capability of the automated NFP-E platform to genetically engineer hard-to-transfect cells with minimal user intervention.
The automated NFP-E workflow involves different complementary functions that work in tandem. We sequentially demonstrate these functions and their specific characteristics that prove advantageous for manipulating hard-to-transfect cell types. First, we developed and optimized a deep learning framework for automated spatial localization of cells and the nanopipette. We integrated this framework with custom hardware and software for automatic cell-nanopipette contact detection[41] leading to localized electroporation of single cells without cell penetration. Moreover, we added automated calibration, positioning, and autofocus features that minimizes user input. The automated cell identification and gentle auto-contact reduces variability arising from manual operation and improves throughput (from 3-5 cells/min to 12-15 cells/min). This also improves the total number of cells addressable in a single run (~70 as compared to ~20 previously). It is worth noting that this number is limited in the current study by the total time the cultures were placed outside the incubator for the NFP-E workflow (~20 mins). This can be further improved by operating the NFP-E system in a temperature and humidity controlled environment and automating other aspects of the workflow such as cell tracking and media exchange. Once established, we used the automated NFP-E protocol to deliver plasmids and Cas9 RNPs into different hard-to-transfect cell types, for reporter expression and gene knockout, respectively. We show that this protocol efficiently delivers the exogenous cargo into cells (>90%), maintains high cell viability (~90%), and allows for working with small starting samples. Finally, we used the NFP-E to demonstrate automated transfection of hiPSCs seeded on micro-confinement arrays (micro-patterns or micro-wells) that enables Cas9 mediated gene knockout. These demonstrations highlight the potential utility of the NFP-E system in a workflow where target genes are identified using high-throughput CRISPR/Cas9 screens and then isogenic cell lines, having mutations of the target genes, are generated using the system automation features.
Results
Automated cell localization and electroporation
A critical task in automating the NFP-E (Figure 1) involves the recognition and spatial localization of cells in the field of view, to enable engagement of the nanopipette to the cells of interest and their subsequent electroporation. To this end, we explored deep learning algorithms which are increasingly being used for visual recognition over the past few years.[42] We specifically investigated fully convolutional networks (FCN), that have consistently outperformed traditional algorithms based on thresholding and morphological operations, for classification task.[43] The FCN architectures are advantageous in biomedical image segmentation where several objects must be classified as well as localized within the same image.[43] These algorithms perform semantic segmentation (pixel wise image classification) and are able to accurately identify the location and boundaries of objects. Thus, they are well suited to the task of localizing cells and their nuclei. Here, we use an FCN which is a modification of the U-Net architecture to locate the nuclei of each cell to be electroporated.[44] We previously trained our algorithm for segmentation of a wide variety of cell lines.[45] Here, we trained and optimized the algorithm to precisely identify the nuclear locations of human primary dermal fibroblasts (HDFs) and hiPSCs from phase contrast images, by using corresponding fluorescence images with nuclear stains as the ground truth. The trained network enabled localization of live cell nuclei from the phase images only, without any additional nuclear labeling. We obtained a precision and recall of 88.5% and 90.5% for the HDFs and 91.4% and 97.9% for the hiPSCs, respectively. The performance of the FCN was significantly better than an algorithm based on morphological operations[46] (Supplementary Figure 1 and Supplementary Table 1). We also applied the same FCN architecture to accurately locate the tip of the nanopipette in the XY image plane. The optimized FCN network was incorporated in the automated NFP-E electroporation software workflow (Figure 2a). This integrated software was then used for automated single cell electroporation. A single run of automated electroporation involves the autocalibration of the nanopipette tip (Figure 2b), followed by localization of the cell nuclei and nanopipette in the field of view from live phase images (Figure 2b). Intermediate autofocusing steps to switch between focal planes of the cells and the nanopipette ensures that the image segmentation is performed on images acquired at the correct focal plane (Figure 2b). The nanopipette is then automatically positioned over the cells using the XYZ piezo stages and a resistance-based contact detection algorithm[41] is used to identify cell-nanopipette contact (Supplementary Figure 2). The resistance-based contact algorithm continuously measures the system resistance as the nanopipette approaches the cell. When the nanopipette is in proximity of the cell membrane, the system resistance gradually increases, and a contact is established when this resistance rises above an empirically determined threshold. We have shown in a previous study that sufficient contact is made in this process to increase the transmembrane potential beyond the electroporation threshold.[47] We have also investigated the effect of the extent of contact on delivery efficiency and cell viability.[41] In the current study, contact is determined when the resistance change (~300 – 500 kΩ) exceed 1% of the baseline (~30 – 50 MΩ). This threshold allows for efficient intracellular delivery without any significant viability loss.[48] After establishing contact, an electroporation pulse is applied to deliver cargo into the cell. Following this, the nanopipette retracts to its original Z position and moves to the next cell after a wait time of 1 second. The entire procedure is then repeated for all cells in the field of view (see Video1). Once all the identified cells in the field of view are electroporated, the system moves to a new region of interest. Integrated software control for the microscope XY stage allows precise positioning of different regions of the substrate in the field of view, which was leveraged to electroporate cells patterned in arrays as demonstrated later. All these steps are executed sequentially by the software workflow without user intervention. The automated contact detection ensures gentle contact and reduced cell to cell variability in obtaining a good electroporation seal that may exist during manual operation. This allows for the electroporation of several cells in a single run without clogging the nanopipette as demonstrated by the delivery of fluorescent BSA in several (>60) HEK 293 cells (Figure 2c and Supplementary Figure 3).
Figure 1: NFP-E system setup and components.
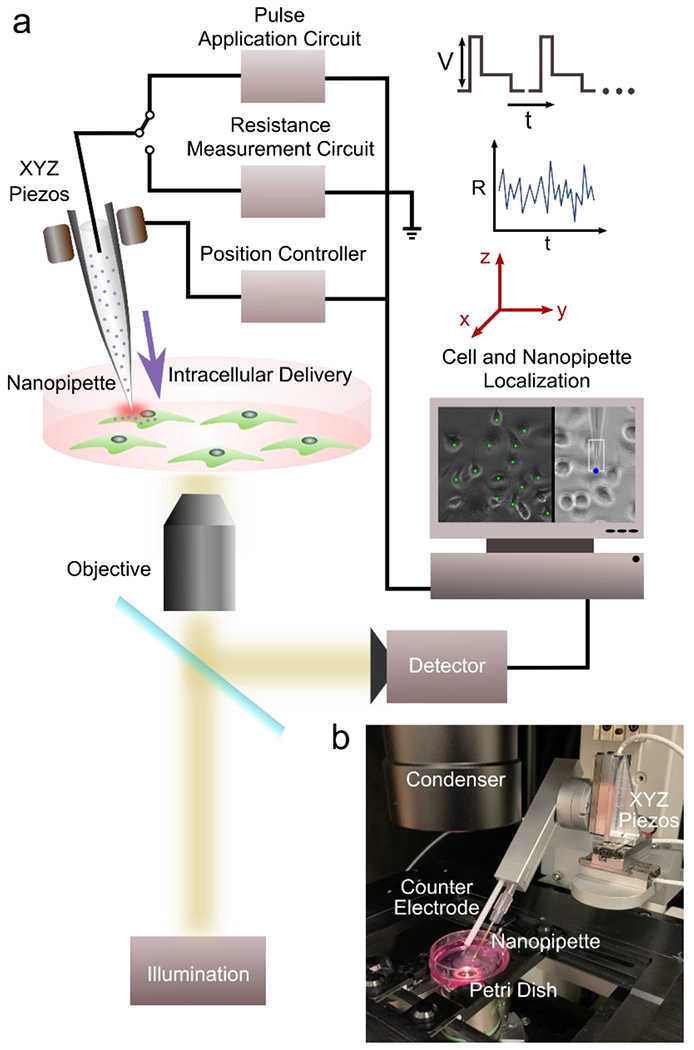
a) Schematic of major components b) Automated NFP-E setup with XYZ piezo stage and nanopipette-counter electrode holder mounted on an inverted microscope.
Figure 2: Automated NFP-E workflow.
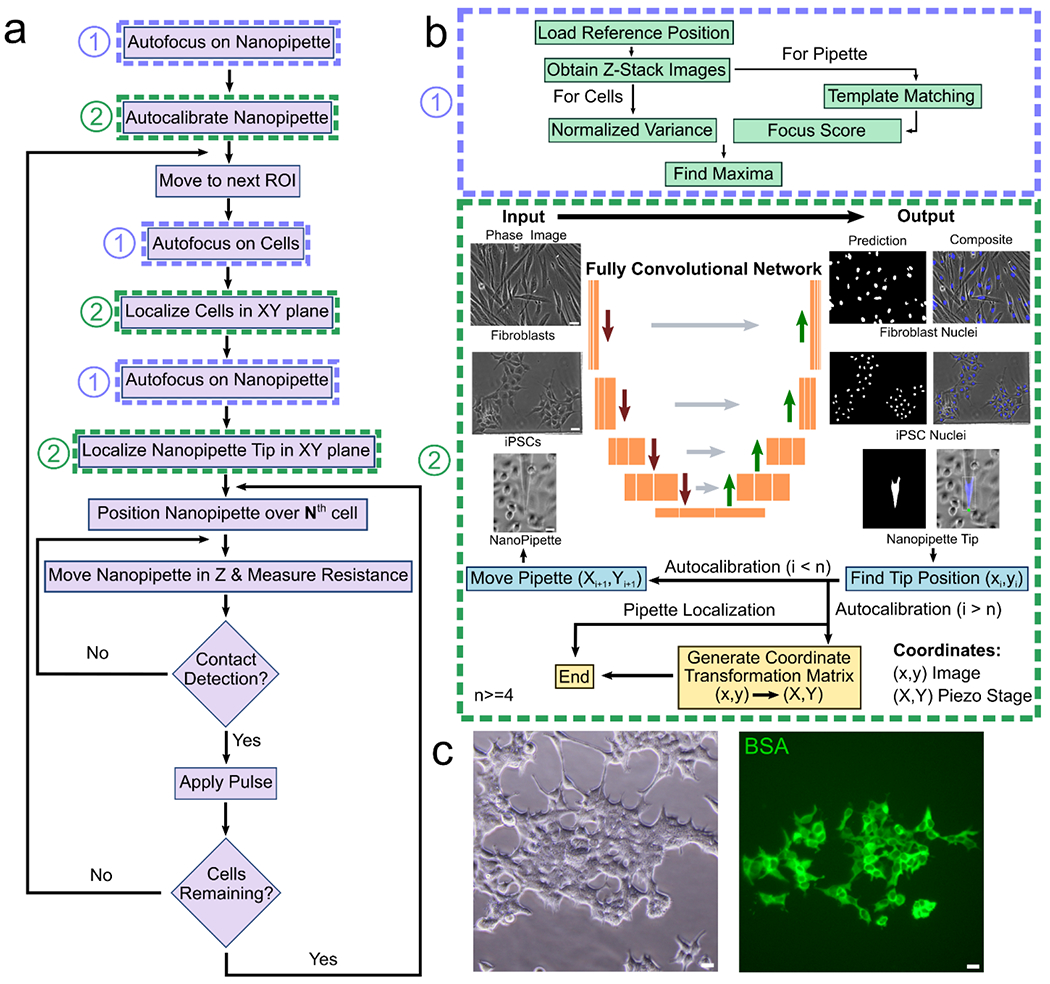
a) Flowchart showing the sequence of events for a single automated run of the NFP-E (here N goes from the 1st detected cell to the last detected cell) b) Autofocus is achieved by using a method that maximizes the normalized variance (1). The FCN in trained to detect the cell nuclei (as shown for HDFs and hiPSCs) and the nanopipette tip (2). Automated pipette detection is also used for calibrating the pipette motion c) Delivery of fluorescent BSA in 70 HEK 293 cells in a single run using the NFP-E system. Scale Bars: 20 μm.
Efficient delivery of Nucleic Acids in Primary and Stem Cells
Efficient delivery of nucleic acids such as plasmid DNA vectors, mRNA and sgRNA oligonucleotides into eukaryotic cells is an important precursor to applications such as generation of isogenic cell lines which have widespread applications in biotechnology and pharma such as high throughput drug screening assays, developing cellular therapeutics, and modeling diseases.[9] Often such applications involve the usage of primary cells from tissues or stem cells. These cells are usually hard to transfect and traditional methods such as lipid mediated delivery or bulk electroporation may lead to significant loss of cell viability or introduce toxic effects in addition to having low transfection efficiencies.[11, 12] We have previously demonstrated NFP-E based delivery of plasmid vectors in immortalized cell lines with high efficiency, leading to uniform gene expression as compared to lipid-based methods.[41] Here, we report the capability of the NFP-E to efficiently transfect primary and stem cells. First, we validated the ability of the automated NFP-E platform to deliver nucleic acids into cells and control the dosage by introducing a cy3-tagged scrambled siRNA into hiPSCs. We applied a bilevel pulse and varied the first voltage level (12V, 15V and 18V) as well as the total number of pulses applied (50 and 100 pulses) (Supplementary Figure 4). The other parameters (second voltage level, pulse width and frequency) were kept constant (see Materials and Methods). To quantify the amount of molecular cargo entering the cells, we measured the cy3 fluorescence intensity from individual cells after delivery. Our results indicate that by increasing the voltage and number of pulses, the amount of cargo delivered can be increased (Supplementary Figure 4). For subsequent experiments we fixed the pulse parameters at 15V and 100 pulses. Next, we delivered a plasmid encoding for EGFP expression into HDFs. After 24 hours we imaged the electroporated cells for the expression of the fluorescent EGFP reporter (Figure 3a). We obtained an average transfection efficiency of 76.3±5.6% which was significantly higher than that obtained using bulk electroporation (60.1±6.2%, P<0.05) (Figure 3c). We also delivered a plasmid encoding for the fluorescent protein mCherry into EGFP expressing hiPSCs using the NFP-E and observed the electroporated cells after 24 hours (Figure 3b). We found the average transfection efficiency to be 40.7±12.3% (Figure 3c). There was no significant difference between the transfection efficiency obtained using the NFP-E and that obtained using bulk electroporation (31.2±8.1%) (Figure 3c). Moreover, high viability was maintained for both HDFs (96.1±1.9%) transfected with EGFP plasmid and hiPSCs transfected with mCherry plasmid (89.97±3.9%) using the NFP-E (Figure 3d). The observed viability was significantly higher than that obtained for bulk electroporated HDFs (71.1±7.3%, P<0.05) and hiPSCs (68.8±5.5%, P<0.01) (Figure 3d). We note that the efficiency of transfection in hiPSCs using NFP-E can be further improved by using different electroporation pulse designs (e.g., exponential/bipolar) or by using higher voltage pulses at the expense of decreased viability. It is important to mention that with the NFP-E system, it is possible to address 50 to 150 cells in one treatment depending on the number of cells in the field of view whereas bulk electroporation is performed on 1 to 10 million cells. Although fewer cells can be addressed with the NFP-E, its major benefits include selective transfection of cells in a culture, low cell loss during processing steps, and much higher viability as compared to bulk electroporation. This is advantageous for handling small starting samples, temporal cell tracking based studies, and cell line generation applications.[26, 35, 41]
Figure 3: Delivery of functional biomolecules into primary and stem cells using NFP-E.
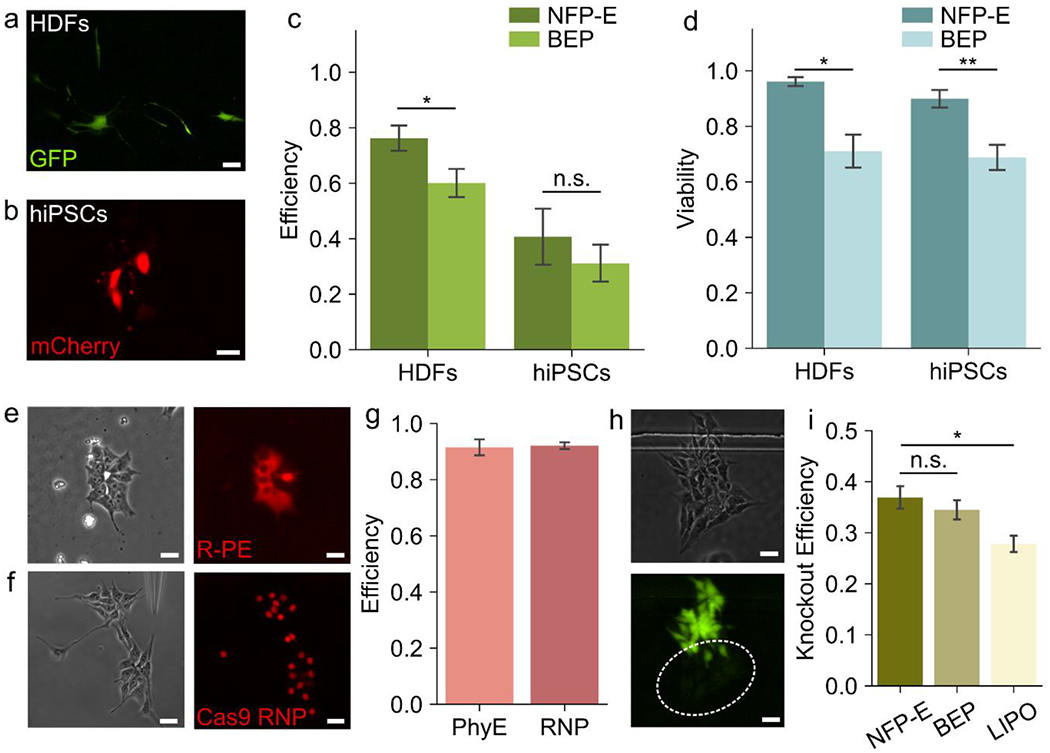
a) Fluorescence micrograph showing human primary dermal fibroblasts (HDFs) expressing GFP, after plasmid delivery using NFP-E b) Fluorescence micrograph showing Human induced pluripotent stem cells (hiPSCs) expressing mCherry, after plasmid delivery using NFP-E c) Efficiency of GFP and mCherry plasmid transfection using NFP-E and Bulk Electroporation (BEP) in HDFs and hiPSCs respectively (N=3 experiments for each, *P < 0.05, n.s. – no significance) d) Viability of HDFs transfected with EGFP plasmid and hiPSCs transfected with mCherry plasmid, 24 hours post treatment with NFF-E and BEP (N = 3 experiments of each, *P<0.05, **P<0.01) e) Phase contrast and fluorescence micrographs showing the delivery of fluorescent R-Phycoerythrin (R-PE) in hiPSCs f) Phase contrast and fluorescence micrographs showing the delivery of Atto 550 tagged Cas9/sgRNA RNP complex in hiPSCs g) Delivery efficiency of R-PE and Cas9/sgRNA RNP in hiPSCs (N=3 experiments) h) Phase contrast and fluorescence micrographs showing the knockout of EGFP in EGFP expressing hiPSCs 48 hours after Cas9/sgRNA RNP delivery using NFP-E i) Comparison of EGFP knockout efficiency between NFP-E, Lipofectamine Stem Cell Reagent (LIPO) and BEP for EGFP expressing hiPSCs (N = 3 experiments for each, *P<0.05, n.s.: no significance). Scale Bars: 20 μm, Error bars represent the S.D.
Delivery of Functional Proteins and CRISPR/Cas9 RNPs in Stem Cells
The direct delivery of Cas9 protein and sgRNA molecules into stem cells is increasingly being applied in precise genome engineering for the advancement of disease modeling and drug discovery.[8] Moreover, efficient delivery of different protein molecules into cells has potential applications in therapeutics, for example through the delivery of deficient proteins to overcome diseased cellular states.[49, 50] However, efficient delivery of proteins still presents a challenging biological problem, as proteins are largely membrane impermeable because of their size and charge. Endosomal uptake provides a means of overcoming this barrier, but endosomal escape is rare and most of the cargo is degraded.[49, 50] Proteins are also susceptible to degradation due to their poor stability unless specialized buffers are used.[49, 50] In addition, the structure and function of proteins may be compromised upon delivery. We have previously reported the delivery of fluorescently tagged BSA in immortalized cell lines as a demonstration of protein delivery into cells.[41] Here, we report the capability of the NFP-E system to directly deliver functional proteins into EGFP expressing iPSCs, bypassing the endocytic pathway. First, we delivered R-Phycoerythrin (R-PE), a 240 kDa fluorescent protein from the phycobiliprotein family into the hiPSCs using the NFP-E platform. We observed uniform fluorescence from the cytoplasm or nuclei of the cells immediately after the electroporation pulses were applied (Figure 3e). The delivery efficiency of R-PE was 91.5±3.5% (Figure 3g). This confirmed that the NFP-E technology is efficient in delivering large proteins directly and uniformly into the cytoplasm or nucleus of cells. Moreover, as the proteins retained their fluorescence post-delivery, we inferred that their structural and functional integrity is preserved. Next, we delivered a Cas9/sgRNA ribonucleoprotein (RNP) complex (>160 kDa) into the EGFP expressing iPSCs using the NFP-E system. The sgRNA was designed to knockout the EGFP gene (Supplementary Table 2) and was tagged with an Atto 550 fluorophore for visualization within the cells. Immediately after electroporation, we observed a strong and uniform fluorescence signal within the targeted cells, particularly from the nuclei, indicating successful delivery of the Cas9 RNP into the cells (Figure 3f). The delivery efficiency of the Cas9 RNP was 92.1±1.5% (Figure 3g). The high efficiency, uniform fluorescence, and the nuclear localization of the signal suggested that the NFP-E can be used for precise delivery of genome editing machinery directly into cell cytoplasm or nuclei. Here, we must note that the intracellular localization of a protein in general (cytoplasm/nucleus) is random, even though the NFP-E delivery process is performed by targeting the cell membrane over the nucleus. In the case of Cas9 RNP, the localization preferentially happens within the nuclei due to the presence of nuclear localization sequences (NLS) on the Cas9 protein. Thus, molecular design plays a key role in intracellular localization. Overall, the physical mechanisms involved in localized electroporation that determine cytoplasmic/nuclear localization of delivered cargo is poorly understood and could be an interesting future study. Next, we compared NFP-E mediated cargo delivery to that using lipofectamine. In contrast to NFP-E based delivery, the fluorescence signal of the RNP was localized in scattered intracellular vesicles after 12 hours, when the delivery was performed using lipofectamine stem cell reagent (Supplementary Figure 5). This suggests that unlike lipid mediated delivery, which follows an endocytic pathway, delivery using the NFP-E directly introduces the macromolecules into the cell and is likely less prone to endosomal degradation. Further, we observed a loss of fluorescence in several electroporated cells after 48 hours of culture, indicating a knockout of the EGFP gene (Figure 3h). Post knockout, we confirmed the presence of cells in the fluorescence depletion regions by performing a live cell staining assay (Supplementary Figure 6). The EGFP knockout efficiency in our hiPSC line using the NFP-E was found to be 36.9±2.7% which was significantly better (P <0.05) than the performance of lipofectamine stem cell reagent (27.8±1.9%) (Figure 3i and Supplementary Figure 7) and is comparable to other localized electroporation-based platforms.[20, 23] We also compared the knockout efficiency of NFP-E to that of bulk electroporation. We found that the knockout efficiency using the NFP-E is not significantly different from that of bulk electroporation (34.5±2.3%) (Figure 3i and Supplementary Figure 7). However, it must be mentioned that in bulk electroporation there is massive cell death in hiPSCs due to dissociation induced death (anoikis) and electroporation mediated stress. Anoikis in hiPSCs and poor cell recovery (>80% cell loss) has been previously reported in multiple bulk electroporation studies.[12, 36] Thus, even though the standard metrics of efficiency and viability are comparable for the NFP-E and bulk electroporation, the overall yield of the desired phenotype is much lower for bulk electroporation.
Automated Single Cell Delivery on Micro Confinement Arrays
Micro confinements such as micro-wells on polymer substrates (such as PDMS or PEG) and micro-patterns generated via micro-contact printing have been used to trap cells for applications ranging from single cell protein analysis and RNA sequencing to cell adhesion studies.[51–53] Here, we utilize the automated NFP-E in conjunction with PDMS micro-wells and, following a prior NFP-E protocol[41], arrays of micro-patterns, to demonstrate the ability of the system to perturb single cells or small cell clusters. This workflow has potential applications in cell line generation and studying dynamics of processes such as cell-cell signaling. We fabricated micro-wells by bonding a thin PDMS through-hole membrane with the polystyrene substrate using APTES (see Materials and Methods). The through holes in the PDMS membrane formed the micro-wells with the polystyrene acting as the base. Then we passivated the PDMS surface and coated the micro-well interiors with vitronectin for hiPSC attachment (Figure 4a). Alternatively, we generated the micro-patterned arrays by micro-contact printing (see Materials and Methods) a polystyrene substrate using vitronectin coated PDMS pillars and then passivating the surrounding areas (Figure 4a). Depending on the size and spacing of the micro confinements we were able to generate arrays having 400 to 1200 individual patterns or wells. We seeded EGFP expressing hiPSCs on the micro-well and micro-patterned substrates at a density of 2500 cells/cm2 and were able to capture 1 to 10 cells within each occupied patterned spot or microwell (Figure 4c). After 12 hours of culture, the cells adhered well on both the micro-wells and micro-patterns (Figure 4b) and the cells nuclei could be efficiently segmented and localized by the FCN on both substrates (Figure 4d). We note that the trained FCN used previously for cells in regular cultures was used for cell localization on the micro-confinement arrays without any degradation in localization performance. We observed the cells on these substrates for a period of 8 days and found them to proliferate, maintain a healthy morphology, and form colonies. After 8 days of culture on the micro confinements, we were able to detach the colonies with EDTA and transfer them to Matrigel coated 96 well-plates with pipettes (Supplementary Figure 8). Moreover, we were able to confine single hiPSCs in the micro substrates, which survived and produced colonies (Supplementary Figure 8). The workflow for hiPSC transfection on the micro substrates using the NFP-E system is largely the same as previously described with one additional step. After electroporating cells in one micro-compartment, the microscope stage is moved to bring the next micro-compartment of the array in the field of view, before electroporation. This process is repeated until the cells, in all micro-compartments in a selected array, have been electroporated. To investigate adverse effects on hiPSC proliferation, we electroporated cells in micro-patterned colonies using the NFP-E and tracked them for 2 days post electroporation. Although, the proliferation of NFP-E targeted colonies was lower on average as compared to non-electroporated control colonies, there was no significant difference between the two groups (Figure 4e). The slightly lower proliferation rate is likely a result of cell viability loss in some of the targeted colonies or cell stress induced due to electroporation. Previous studies have also shown proliferation rate reduction in Hematopoietic Stem Cells (HSCs) due to electroporation or Cas9 mediated DNA double-strand breaks [13, 54]. It must be noted that in this experiment we used the NFP-E pulse parameters that were most efficient for hiSPC transfection while maintaining cell viability (see Materials and Methods). In future investigations, the effects of different electroporation pulse parameters on hiPSC proliferation and function could be explored. Overall, our observations indicate that hiPSC function is not adversely affected by micro-confinement and NFP-E treatment. As a result, this protocol can be utilized in hiPSC engineering applications. For example, in future, it may be possible to use to NFP-E for delivering molecules of interest (such as Cas9 RNPs) into isolated single cells on the micro confinements, and then allowing them to proliferate before harvesting the desired clones for generating cell lines. Alternatively, a single cell within a group of cells on the micro confinements could be transfected with a plasmid vector and then a clonal colony generated by antibody selection as demonstrated by use previously using immortalized cell lines.[41] For instance, single cell electroporation with the NFP-E followed puromycin selection could be used for the generation of isogenic hiPSC lines.[17] To demonstrate the NFP-E capability to deliver functional molecules into cells cultured on the micro confinements, we delivered a fluorescently labelled BSA into the EGFP-hiPSCs cultured in micro-wells and obtained a delivery efficiency of 92.8±4.1% (Figure 4f and 4h). We also delivered Cas9/sgRNA RNP targeting the EGFP gene into EGFP-hiPSCs cultured on micro-patterns and observed EGFP knockout after 72 hours (Figure 4g). To highlight cell selectivity of the NFP-E process, a single cell was electroporated per colony in this experiment with the knockout daughter cells originating from one parent cell. We note that the algorithm was modified to stop the process after electroporating the first cell in each colony. The efficiency of colony wise knockout was 37.5±8.3% with 24 out of 64 total targeted cell colonies (N = 3 experiments) having knockout cells (Figure 4h). Such selective cell electroporation experiments within small cell populations are a unique feature of probe-based methods like the NFP-E, which cannot be performed using traditional methods like bulk electroporation or lipofection.
Figure 4: NFP-E and micro-confinement method for manipulation of small cell populations.
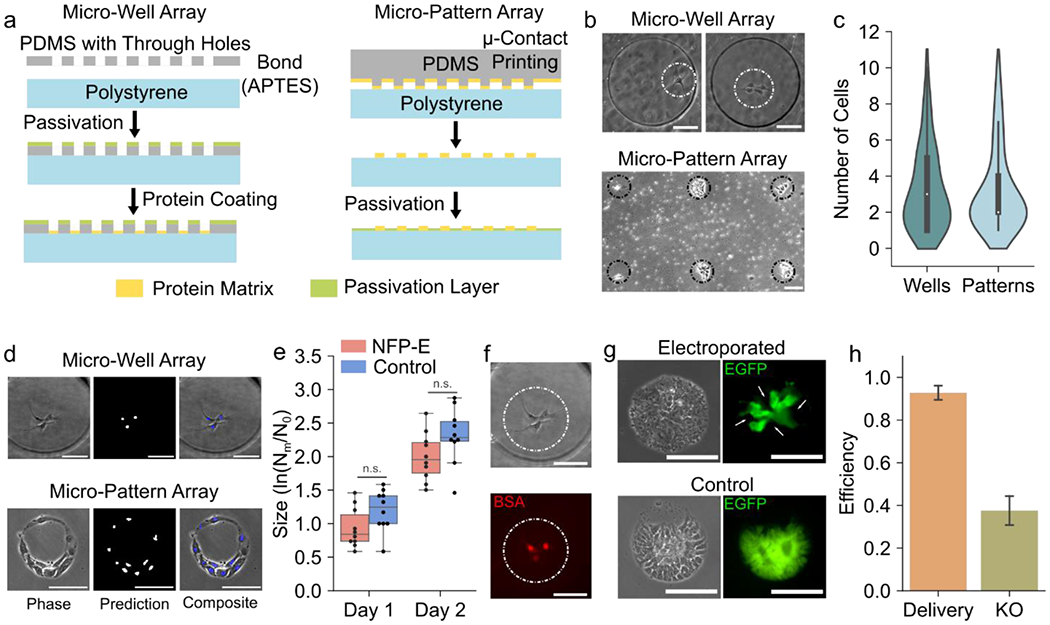
a) Schematics showing fabrication workflow for micro-well array (Left) and micro-pattern array (Right) b) Localization of a single to few cells on micro-well arrays and micro-patterned arrays c) Distribution of cells captured in micro-wells and micro-patterns. Majority of the occupied micro-confinements had 1 to 3 cells. d) Prediction of nuclear locations of hiPSCs seeded in micro-wells and micro-patterns using the FCN e) Size of control and NFP-E treated hiPSC colonies as observed over 2 days post electroporation to track the effect of electroporation on cell proliferation (N = 10 colonies for each group). The size was calculated as the logarithm of the ratio of the number of cells in a colony on a particular day (Nm) and the number of cells in the same colony on day zero (N0). (n.s.: no significant difference) f) Delivery of fluorescent BSA in hiPSCs seeded in a micro-well g) Knockout of EGFP in an hiPSC colony on a micro-pattern array observed 72 hours after Cas9/sgRNA RNP delivery. The control indicates a colony formed by non-electroporated cells. The arrows indicate knockout daughter cells originating from a single electroporated parent cell. h) Efficiency of BSA delivery (micro-well array) and EGFP knockout (micro-pattern array) in hiPSCs using the NFP-E (N = 3 experiments for both BSA delivery and EGFP knockout). Scale Bars: 100 μm, Error bars represent the S.D.
Discussions
In this work, we presented an automated version of the NFP-E single cell electroporation method, which has unique advantages over traditional intracellular delivery methods. First, the NFP-E is a generalized intracellular delivery platform capable of delivering a wide variety of molecules (plasmid DNA, molecular beacons, proteins, etc.) in several immortalized cell types as demonstrated previously.[39–41] Here, we extend the method to primary cells (HDFs) and stem cells (hiPSCs), which are sensitive and hard to transfect with traditional approaches due to high toxicity and poor cell viability. Due to the localization of the electric field applied by the NFP-E,[39] the cellular perturbation is minimal and helps preserve high cell viability even in the case of primary and stem cells. Moreover, the electroporation pulse profile, for various cell types and delivered molecules, are similar, indicating that the NFP-E method is of general applicability. As such, extensive optimization for each cell type and molecule can be avoided, which is an advantage over traditional methods.[9]
Second, the NFP-E protocol does not involve the use of specialized buffers for delivery as is the case with carrier-based methods and bulk electroporation.[9, 10] This, in combination with the low voltages applied, makes the NFP-E a suitable method for the direct delivery of recombinant proteins whose structure and function may be degraded in unsuitable buffers.[49] The efficient delivery of Cas9 nuclease and successful gene knockout results indicates that protein function is retained upon delivery via the NFP-E platform. These observations suggest that the NFP-E could potentially be used for applications such as the direct delivery of synthetic transcription factors for cellular reprogramming[55] and therapeutic proteins for studying diseases such as neurodegeneration and specific cancers.[50] It must be noted that we used cell culture media and PBS for our experiments. Additional experiments must be performed to gain insight on the range of buffers that can be used with the NFP-E platform and potential for improvements in delivery efficiency.
Third, the NFP-E allows for precise dosage control of the molecule delivered due to the electrophoretic nature of the delivery as shown by us previously.[39, 47] By modifying the voltage parameters and the threshold contact resistance, the amount of the molecule delivered can be tuned as demonstrated here and in prior studies by us,[41, 47] which may prove useful in several cases. Few pertinent examples include combinatorial delivery of several sgRNAs for multiple gene knockout that can reveal the genetic interactions[56] involved in diseases or varying the stoichiometries of different mRNAs delivered to cells to control gene expression.[23] Such control is difficult with lipid carriers or bulk electroporation where the delivery is non-uniform and stochastic.[9] Besides, due to the uniformity in delivery as demonstrated in the current study, the NFP-E method proves suitable for tracking dynamic cellular events quantitatively via imaging fluorescent markers in live cells. For instance, a technique for live imaging of genomic DNA and RNA transcripts called CRISPR LiveFISH was recently developed using Cas9, Cas13 and fluorophore tagged sgRNAs for tracking chromosomal disorders.[57] In a previous study we were able to track mRNA expression in single cells by delivering molecular beacons using the NFP-E.[40] Here, we demonstrated the delivery of a fluorophore tagged Cas9/sgRNA RNP into hiPSCs, which indicates that CRISPR LiveFISH could potentially be used in conjunction with the NFP-E to track cellular dynamics in adherent single cells at different timepoints. These studies would be challenging with traditional bulk electroporation formats, where the cells need to be resuspended for each delivery cycle making it difficult to track the same cells over time.
Additionally, the NFP-E can selectively deliver molecules into single cells within a population. With further development, this capability could be especially advantageous in studies involving multicellular systems where specific cells need to be transfected. Moreover, employing the deep learning framework for live image segmentation allows for distinguishing different cell types in a heterogeneous population as shown by others.[58, 59] By training the FCN with images of a range of cell types of interest, instances of different kinds of cells within the same field of view could be classified into distinct categories. Thus, it could be possible to automatically distinguish specific cell types in co-culture systems and deliver molecules into the desired cell type using the NFP-E system. For example, in a neuron-muscle co-culture system[60] for disease modeling, the neurons can be selectively targeted for corrective gene editing.
Finally, due to the serial nature of most probe-based single cell delivery systems and associated technical issues like probe clogging, their throughput is limited to 5-10 cells per probe.[61] Moreover, they require considerable user expertise in positioning and manipulation for operation, which proves to be a fundamental challenge while scaling up the systems to tackle biological studies of interest. In contrast, the fully automated NFP-E system uses an FCN architecture for cell localization and pipette tracking and a resistance-based cell-nanopipette contact detection algorithm, the combination of which minimizes user intervention during the automated single cell electroporation procedure. The automation partially addresses some of the issues of manual probe-based methods by eliminating tedious and repetitive manual steps and employing a gentle and tunable contact algorithm to reduce variability and debris accumulation on the nanopipette tip. These modifications allow the NFP-E to automatically electroporate 60-70 cells with a single nanopipette, which is almost an order of magnitude higher than other systems.
Leveraging the advantages of the automated NFP-E system, we combined it with micro-confinements to entrap cells and demonstrated a protocol for cell transfection and temporal tracking that can potentially be used for applications such as testing the efficacy of different sgRNAs (e.g. by transfecting groups of colonies with different Cas9/sgRNA complexes and harvesting them for downstream analysis), screening for target genes (e.g. related to a disease) and isogenic cell line generation. We must however consider the possibility of a cell detaching from its colony and adhering to an adjacent one, thus affecting clonality. To examine this scenario, cells transfected with different reporters may be tracked in time. Parameters such as distance between colonies and the degree of passivation can then be tuned to eliminate such cross-contamination.
Furthermore, the NFP-E and micro-confinement method could be augmented for cell dispensing and picking or employed in combination with commercially available ones. This would provide a fully automated cell manipulation pipeline with seamless cell dispensing in micro-confinements, followed by automated single cell electroporation, regular monitoring or imaging of the transfected cells, and picking of the desired cells for downstream analysis (DNA/mRNA sequencing) and applications. With further optimization of the protocols, the automated NFP-E method should provide high knockout efficiencies and enable combinatorial genetic manipulations (Figure 5).
Figure 5: Potential application of fully automated NFP-E system in cell line generation workflow.
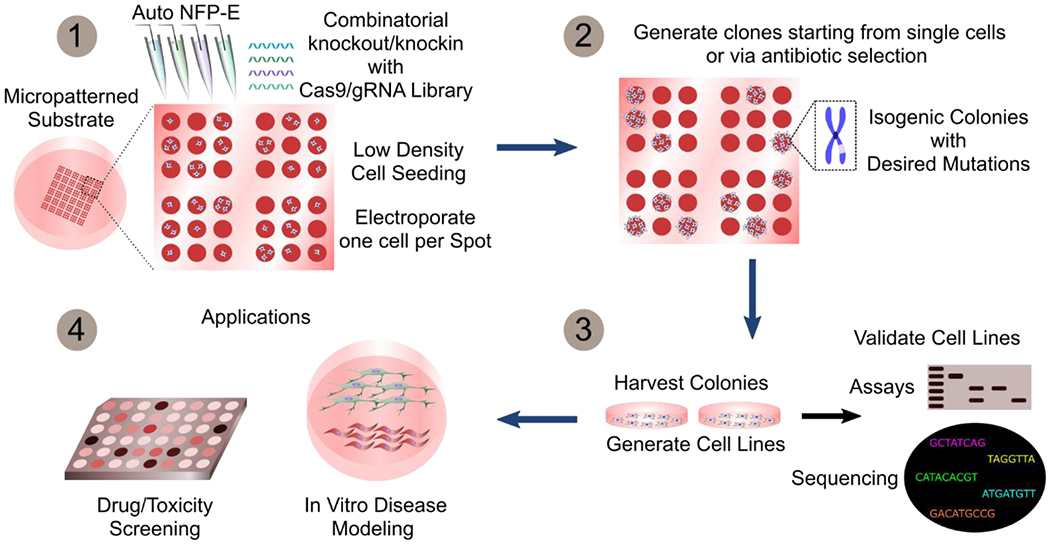
The automated NFP-E system can be used to generate isogenic cell lines with targeted mutations. Arrays of single or few cells in micro-confinements can be transfected with the Cas9/sgRNA machinery using the NFP-E (1). Single cell clones are then generated by antibiotic selection or by isolating colonies originating from single cells (2). The harvested clones may be expanded and validated to obtain isogenic cell lines (3) and then used for drug screening and disease modeling studies (4)
Conclusions
In conclusion, the automated NFP-E in combination with micro-confinement arrays provides a versatile platform for gentle single-cell manipulation for a wide variety of cell types including hiPSCs. This procedure can potentially eliminate cell manipulation steps that consume time and resources and lead to poor cell viability especially in the case of sensitive cells such as hiPSCs. Future work will involve automated transfection of specific cell types in heterogeneous co-culture populations and combinatorial gene knockouts for isogenic cell line generation that enables the study of genetic diseases.
Materials and Methods
Automated NFP-E Setup and General Electroporation Protocol
All experiments were performed using the Nano Fountain Probe Electroporation (NFP-E) system (Figure 1a and 1b). The system utilizes a glass nanopipette (Eppendorf) for targeted macromolecular delivery into single cells. The nanopipette was loaded with the desired molecular solution and fitted to a robotic arm. The robotic arm carries platinum electrodes for measurement of solution resistance and application of electroporation pulses. The XYZ motion of the robotic arm is controlled by precision linear piezo stages with 40 nm resolution. The entire system was mounted on a motorized inverted fluorescence microscope (Nikon Eclipse Ti-E). Images of cells and the nanopipette were acquired in phase contrast mode using a CMOS machine vision camera (7.1 MP, 1” format, monochromatic). The XY positions of all the cells of interest in the field of view and the nanopipette were either automatically identified (process described later) or selected by the user. A custom designed electronic PCB and software were used to measure the resistance of the system at a sampling rate of 150 Hz. The nanopipette was then automatically placed over the cell of interest and displaced in the Z direction towards the cell. Contact between the nanopipette tip and the cell was identified when the filtered baseline resistance exceeded a pre-determined threshold (typically 1% of the baseline). The desired electroporation pulses (square, bi-level or exponential) were applied and then the nanopipette engaged the next cell of interest automatically. Once all the cells of interest in the current field of view had been electroporated, the substrate was moved to the subsequent location using a motorized XY stage with 50 nm resolution, and the process was repeated. This allowed for automatic delivery of molecules into cells patterned in micro-confinement arrays. For most experiments, bilevel electroporation pulse trains (15 V, 0.5 ms; 10 V, 2.5 ms; typical) were applied at a frequency of 50 Hz. Each train consisted of 50 such bilevel pulses and typically 1-2 trains were used. For 2 train protocols the wait time between the trains was equal to the duration of 1 train. All electroporation steps were either carried out in cell culture media or PBS. After electroporation, the cells were washed with PBS and put in fresh media and returned to the incubator (37°C and 5% CO2) until further analysis.
Automated Imaging and Video Acquisition
Images were acquired in phase contrast and fluorescence modes using an inverted fluorescence microscope (Nikon Eclipse Ti-E) fitted with a CMOS machine vision camera (7.1 MP) and a scientific CMOS camera (5.5 MP). Automated image and video acquisition were performed using custom written software in C#.
Autofocus on Cells and Nanopipette
Autofocusing on the cells was performed using an algorithm based on the normalized variance method.[62] First, a z-stack of phase contrast images was obtained around a preset XY plane of the adhered cells, which served as an initial guess. Then a normalized variance metric, for each image was calculated. The image with the maximum value was chosen as the focused image. A template matching algorithm was used to focus on the nanopipette in the field of view. A z-stack of phase images were obtained around an initial guess. An ROI of a focused nanopipette was used to calculate the correlation coefficient and generate a template matching score (TMS) for each image in the z-stack. The TMS scores for all the images were normalized to the TMS of a focused image to obtain a focus score (). The pipette focal plane was determined from the highest ZTM score. Once focused, the position of the tip was located using a deep convolutional network.
Localization of cells and nanopipette
A fully convolutional network (FCN) architecture was implemented and trained to detect the nuclei of cells in phase contrast images with pixel-level resolution.[63] The FCN architecture consists of an encoder-decoder system that outputs a multiclass prediction for each pixel (interior, boundary, and exterior). An input image and its corresponding ground truth labels are passed through two convolution layers (3x3 kernel size), connected with a rectified linear unit (ReLU) nonlinear activation function, and down-sampled with a max pooling layer (stride = 2). These layers are repeated three times in the decoder region of the network to form a multilevel-architecture where the image width (W) and height (H) are halved after max-pooling. The decoder region contains one up-sampling layer, a concatenation step, and three convolution layers connected with ReLU activation. The up-sampling and concatenation steps increase the resolution of the tensor up each level of the network until the original resolution is obtained. Each pixel output contains a probability map of the three classes. A weighted soft-max cross-entropy cost function is used to measure the output error to the ground truth. The boundary class is weighted more heavily (10x) than the interior and exterior classes to separate neighboring cells.
During training, the weights of the network are updated using stochastic gradient descent (learning rate = 1 E-4, momentum = 0.9) to minimize the cost function in the backpropagation cycle. The image data was separated into training and validation sets with a 5:1 ratio, and training was stopped when the validation cost reached a minimum value to prevent overfitting. A GPU with cuDNN acceleration (NVIDIA) was used for training batches of images in parallel cores and speed up training time. We cropped an ROI (256 x 256 [px]) of the image (696 x 520 [px]) into 6 batches for training and 2 batches for validation for 100 steps/epoch (TensorFlow).
The performance of the algorithm was evaluated by measuring the overlap of the output predictions with the annotated ground-truth annotations. We computed the intersection over union (IOU) given by , where A represents the annotated image, or ground truth, and B represents the prediction. We characterized true positives (TP) as predicted labels that exceed 50% IOU. False positives (FP) correspond to predicted labels below the 50% threshold, and false negative (FN) as regions containing cells without a labeled prediction. The precision (P) and recall (R) were determined from these values with the following equations: . To improve precision and ensure that there are minimal false detects that could lead to pipette crashing on the substrate, additional watershed and area filters were applied to separate merged contours and remove outliers.
The same FCN architecture used for cell detection was utilized for the recognition of the pipette tip. To generate the ground truth labels, a custom annotation software (C#) was developed in which the user clicks on the pipette tip and a small triangular section, above the tip, is classified as the pipette for training. In this manner, hundreds of images were rapidly labeled by moving the pipette within a field of view, clicking on the tip at multiple locations, and moving to different fields of views containing backgrounds with different illumination conditions, focal positions, and cell environments. Prior to training, the images were preprocessed with a Sobel filter to emphasize the edges of the pipette. The same training, validation, and model optimization procedures used for cell detection were employed with the pipette images. Size and aspect-ratio filters were used to filter out false positive pipette objects. In the event that more than one object was recognized as a pipette in the field of view, a template matching algorithm was utilized as a secondary detection scheme to locate the position of the pipette. The FCN was used to detect the pipette tip during the tip calibration process and prior to the automated transfection procedures (see Video 1).
BSA Delivery using NFP-E
HEK 293 cells were plated on 35 mm cell culture dishes (Falcon) and allowed to adhere for 16 to 24 hours. Bovine Serum Albumin tagged to Alexa Fluor 488 (BSA-AF 488) (Thermo Fisher Scientific) was resuspended in 1X DPBS and used in the electroporation experiments at a final concentration of 2.5 mg mL−1.
siRNA Delivery using NFP-E
hiPSCs were plated on 35 mm cell culture dishes (Falcon) and allowed to adhere for 16 to 24 hours. Cy3-tagged negative control siRNA (Thermo Fisher Scientific) was resuspended in 1X DPBS and used in the electroporation experiments at a final concentration of 10 uM.
Phycoerythrin Delivery using NFP-E
Cells were plated on 35 mm cell culture dishes (Falcon) and allowed to adhere for 16 to 24 hours. R-Phycoerythrin (R-PE) (AnaSpec) was resuspended in 1X DPBS and used in the electroporation experiments at a final concentration of 1.17 mg/mL.
Cas9/sgRNA Delivery using NFP-E
The day prior delivery, the cells were plated at 2,500-5,000 cells/dish on Matrigel or vitronectin coated 35 mm gridded dishes (Ibidi) which allowed for long term tracking of the colonies. Cas9 protein (NEB) and sgRNA (IDT and Synthego) were mixed in the molar ratio 1:1 and incubated at 37°C for 10 mins to produce the active ribonucleoprotein (RNP) complex. This RNP complex was directly used for all delivery experiments at a final concentration of 1 uM.
Plasmid Delivery using NFP-E
The plasmids pEGFP and pmCherry (size ~ 4.7 kb) are composed of the pCDNA plasmid backbone of 4 kb, containing an SV40 origin of replication, with an insert of 0.7 kb that codes for a fluorescent reporter (EGFP and mCherry, respectively). Both plasmids were kept at −20°C at a concentration of 1 μg mL−1 and diluted in 1X PBS to the desired concentration (50 ng mL−1) before electroporation. Post electroporation, RevitaCell (Gibco™) was added to the cell culture media (1X final concentration) to promote viability. After 24 hours of plasmid delivery, the expression of the reporter was analyzed using fluorescence microscopy.
Plasmid Delivery using Bulk Electroporation
Bulk Electroporation was carried out using the Gene Pulser XCell System (BioRad) following the manufacturer’s protocols. Briefly, 1 × 107 cells were resuspended in 800 μL of electroporation buffer (Eppendorf) pre-chilled in ice. Prior to this step the cells were dissociated using their respective dissociation reagent (0.05% Trypsin with 0.2% EDTA for HDFs and 0.5 mM EDTA for hiPSCs). The cell suspension was mixed gently with 5-10 μg of the plasmid DNA prepared in TE buffer (1μg μL−1) and introduced into a 0.4 cm pre-chilled cuvette. The cells were electroporated using recommended electroporation parameters (e.g. 300V, 500 μF for hiPSCs). The cells were then centrifuged, washed with DPBS and transferred to well plates (USA Scientific) in their respective pre-warmed media. For hiPSCs, the well plates were coated with vitronectin for adhesion and RevitaCell (Gibco™) was added to the media (1X final concentration) to promote viability.
Cas9/sgRNA Delivery using Lipofectamine
Cells were plated in a 24 well plate and allowed to reach a confluency of 70-90% before attempting transfection with Lipofectamine CRISPRMAX (Thermo Fisher Scientific). Lipofectamine transfection was carried out using manufacturer’s protocol in opti-MEM medium (Thermo Fisher Scientific) with a 1:1 molar ratio of Cas9 (NEB) to Atto 550 tagged sgRNA (IDT). The cells were observed under a fluorescence microscope after 12 hours.
Cell Culture
Human induced pluripotent stem cells (iPSCs) expressing the enhanced green fluorescent protein (EGFP) reporter were employed. The cells were cultured in Essential 8™ Medium (basal medium and supplements, Thermo Fisher Scientific) on Matrigel (Corning) coated 6 well plates. Media was replaced every 24 hours. Cells were passaged by dissociating them in 0.5 mM EDTA (Thermo Fisher Scientific) in PBS every 4-5 days before reaching full confluency.
Human primary dermal fibroblasts (HDFs) were procured from ATCC (PCS-201-012) and cultured using fibroblast basal medium supplemented with a low-serum growth kit (ATCC PCS-201-041). The cells were passaged upon reaching confluency with 0.05% Trypsin-EDTA (Life Technologies) and trypsin inhibitor solution. Experiments were performed on cells that were passaged less than ten times (< P10).
Fabrication of Micro-Patterned Substrate
Polydimethylsiloxane (PDMS) micropillar arrays, having 150 μm pillars spaced 500 μm, were fabricated from an SU8 (Microchem) mold on a silicon wafer using standard soft lithography. A 2cm×2cm chip was cut out from the PDMS slab with the micropillar arrays and sterilized by incubating in 70% ethanol for 15 mins followed by UV treatment for 15 mins. Vitronectin solution (Sigma-Aldrich) at 0.5 μg mL−1 in PBS (Gibco) was deposited onto the PDMS chip and, after one hour at room temperature, the solution was gently removed, and the chip was dried for 5 min. The chip was then placed upside down onto a 35-mm polystyrene dish (Electron Microscopy Sciences) to allow the transfer of the vitronectin onto the substrate. After removal of the stamp, 1 mL of Pluronic F-127 (2 mg mL−1 in distilled water) (Sigma-Aldrich) was added to passivate parts of the substrate devoid of vitronectin, preventing nonspecific cellular adherence. After removal of the Pluronic F-127 solution, the surface was washed with PBS and iPSCs were added in cell culture media. The substrate was placed in an incubator (37°C and 5% CO2) overnight, allowing for the cells to adhere on the vitronectin patterns and form small colonies.
Fabrication of Micro-Well Substrate
A thin PDMS stencil (50 μm) containing an array pattern of microwells (250 μm diameter) was fabricated and bonded to a cell-culture polystyrene dish (35 mm). The stencil fabrication workflow consisted of pouring a mixture of PDMS on a SU8 mold containing the array features, placing a flexible Mylar transfer sheet on top of uncured PDMS, clamping at high force to squeeze out the PDMS from the mold features, and curing the PDMS at high temperature. The Mylar transfer-sheet, containing the PDMS stencil, was then peeled from the mold and bonded to the polystyrene dish. To sterilize the PDMS stencil dish, it was washed with ethanol and DI water followed by UV treatment for 1 hour.
Live Cell Detection Assay
NucRed Live 647 (Thermo Fisher) was added to the cell culture media (2 drops of stock solution per mL of media) and incubated for 15 minutes. The cells were then washed with PBS and fresh media was added before imaging under a fluorescence microscope.
Viability Assay
Cell viability was assessed by staining them with Hoechst 33342 (Life Technologies) and propidium iodide (PI, Life Technologies) 24 hours after intracellular delivery using manufacturer’s protocols and imaging them using florescence microscopy. The cells whose nuclei simultaneously expressed PI and Hoechst fluorescence were counted as dead while the cells whose nuclei only expressed Hoechst were counted as alive.
Image Analysis
All image analysis procedures was performed using the open source image processing package, FIJI.[64]
Statistical Analysis
Statistical comparisons were performed using two-tailed Student’s t-tests.
Supplementary Material
Acknowledgements
Research reported in this publication was supported by NIH SBIR Phase 1 award number 1R43GM128500-01 and NIH R21 award number 1R21GM132709-01. This work utilized the Argonne National Lab Center for Nanoscale Materials. Use of the Center for Nanoscale Materials, an Office of Science user facility, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Footnotes
Conflict of Interest
HDE declares majority ownership of Infinitesimal LLC, a company commercializing bio-tools for cell manipulation and analysis.
References
- [1].Fellmann C; Gowen BG; Lin P-C; Doudna JA; Corn JE, Nat. Rev. Drug Discovery 2017, 16 (2), 89–100. DOI 10.1038/nrd.2016.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Shi Y; Inoue H; Wu JC; Yamanaka S, Nat. Rev. Drug Discovery 2017, 16 (2), 115–130. DOI 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Shalem O; Sanjana NE; Zhang F, Nat. Rev. Genetics 2015, 16 (5), 299–311. DOI 10.1038/nrg3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dixit A; Parnas O; Li B; Chen J; Fulco CP; Jerby-Arnon L; Marjanovic ND; Dionne D; Burks T; Raychowdhury R; Adamson B; Norman TM; Lander ES; Weissman JS; Friedman N; Regev A, Cell 2016, 167 (7), 1853–1866.e17. DOI 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Replogle JM; Norman TM; Xu A; Hussmann JA; Chen J; Cogan JZ; Meer EJ; Terry JM; Riordan DP; Srinivas N; Fiddes IT; Arthur JG; Alvarado LJ; Pfeiffer KA; Mikkelsen TS; Weissman JS; Adamson B, Nat. Biotechnol 2020. DOI 10.1038/s41587-020-0470-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Avior Y; Sagi I; Benvenisty N, Nat. Rev. Mol. Cell Biol 2016, 17 (3), 170–182. DOI 10.1038/nrm.2015.27. [DOI] [PubMed] [Google Scholar]
- [7].Grobarczyk B; Franco B; Hanon K; Malgrange B, Stem Cell Rev. Rep 2015, 11 (5), 774–787. DOI 10.1007/s12015-015-9600-1. [DOI] [PubMed] [Google Scholar]
- [8].Yin H; Kauffman KJ; Anderson DG, Nat. Rev. Drug Discovery 2017, 16 (6), 387–399. DOI 10.1038/nrd.2016.280. [DOI] [PubMed] [Google Scholar]
- [9].Stewart MP; Sharei A; Ding X; Sahay G; Langer R; Jensen KF, Nature 2016, 538 (7624), 183–192. DOI 10.1038/nature19764. [DOI] [PubMed] [Google Scholar]
- [10].Stewart MP; Langer R; Jensen KF, Chem. Rev 2018, 118 (16), 7409–7531. DOI 10.1021/acs.chemrev.7b00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Roth TL; Puig-Saus C; Yu R; Shifrut E; Carnevale J; Li PJ; Hiatt J; Saco J; Krystofinski P; Li H; Tobin V; Nguyen DN; Lee MR; Putnam AL; Ferris AL; Chen JW; Schickel J-N; Pellerin L; Carmody D; Alkorta-Aranburu G; del Gaudio D; Matsumoto H; Morell M; Mao Y; Cho M; Quadros RM; Gurumurthy CB; Smith B; Haugwitz M; Hughes SH; Weissman JS; Schumann K; Esensten JH; May AP; Ashworth A; Kupfer GM; Greeley SAW; Bacchetta R; Meffre E; Roncarolo MG; Romberg N; Herold KC; Ribas A; Leonetti MD; Marson A, Nature 2018, 559 (7714), 405–409. DOI 10.1038/s41586-018-0326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Li X-L; Li G-H; Fu J; Fu Y-W; Zhang L; Chen W; Arakaki C; Zhang J-P; Wen W; Zhao M; Chen WV; Botimer GD; Baylink D; Aranda L; Choi H; Bechar R; Talbot P; Sun C-K; Cheng T; Zhang X-B, Nucleic Acids Res. 2018, 46 (19), 10195–10215. DOI 10.1093/nar/gky804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].DiTommaso T; Cole JM; Cassereau L; Buggé JA; Hanson JLS; Bridgen DT; Stokes BD; Loughhead SM; Beutel BA; Gilbert JB; Nussbaum K; Sorrentino A; Toggweiler J; Schmidt T; Gyuelveszi G; Bernstein H; Sharei A, Proc. Natl. Acad. Sci. U. S. A 2018, 115 (46), E10907. DOI 10.1073/pnas.1809671115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Thomas CE; Ehrhardt A; Kay MA, Nat. Rev. Genetics 2003, 4 (5), 346–358. DOI 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- [15].Yang L; Guell M; Byrne S; Yang JL; De Los Angeles A; Mali P; Aach J; Kim-Kiselak C; Briggs AW; Rios X; Huang P-Y; Daley G; Church G, Nucleic Acids Res. 2013, 41 (19), 9049–9061. DOI 10.1093/nar/gkt555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Singh AM, Front. Cell. Dev. Biol 2019, 7 (11). DOI 10.3389/fcell.2019.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Steyer B; Bu Q; Cory E; Jiang K; Duong S; Sinha D; Steltzer S; Gamm D; Chang Q; Saha K, Stem Cell Rep. 2018, 10 (2), 642–654. DOI 10.1016/j.stemcr.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ben Jehuda R; Shemer Y; Binah O, Stem Cell Rev. Rep 2018, 14 (3), 323–336. DOI 10.1007/s12015-018-9811-3. [DOI] [PubMed] [Google Scholar]
- [19].Mukherjee P; Nathamgari SSP; Kessler JA; Espinosa HD, ACS Nano 2018, 12 (12), 12118–12128. DOI 10.1021/acsnano.8b05473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cao Y; Ma E; Cestellos-Blanco S; Zhang B; Qiu R; Su Y; Doudna JA; Yang P, Proc. Natl. Acad. Sci. U. S. A 2019, 116 (16), 7899. DOI 10.1073/pnas.1818553116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chiappini C; Chen Y; Aslanoglou S; Mariano A; Mollo V; Mu H; De Rosa E; He G; Tasciotti E; Xie X; Santoro F; Zhao W; Voelcker NH; Elnathan R, Nat. Protoc 2021, 16 (10), 4539–4563. DOI 10.1038/s41596-021-00600-7. [DOI] [PubMed] [Google Scholar]
- [22].Yoh HZ; Chen Y; Aslanoglou S; Wong S; Trifunovic Z; Crawford S; Lestrell E; Priest C; Alba M; Thissen H; Voelcker NH; Elnathan R, Adv. Funct. Mater 2022, 32 (3), 2104828. DOI 10.1002/adfm.202104828. [DOI] [Google Scholar]
- [23].Cao Y; Chen H; Qiu R; Hanna M; Ma E; Hjort M; Zhang A; Lewis RS; Wu JC; Melosh NA, Sci. Adv 2018, 4 (10), eaat8131. DOI 10.1126/sciadv.aat8131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schmiderer L; Subramaniam A; Žemaitis K; Bäckström A; Yudovich D; Soboleva S; Galeev R; Prinz CN; Larsson J; Hjort M, Proc. Natl. Acad. Sci. U. S. A 2020, 117 (35), 21267. DOI 10.1073/pnas.2001367117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen Y; Wang J; Li X; Hu N; Voelcker NH; Xie X; Elnathan R, Adv.Mater 2020, 32 (40), 2001668. DOI 10.1002/adma.202001668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Brooks J; Minnick G; Mukherjee P; Jaberi A; Chang L; Espinosa HD; Yang R, Small 2020, 16 (51), 2004917. DOI 10.1002/smll.202004917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shokouhi A-R; Aslanoglou S; Nisbet D; Voelcker NH; Elnathan R, Mater. Horiz 2020, 7 (11), 2810–2831. DOI 10.1039/D0MH01016B. [DOI] [Google Scholar]
- [28].Nathamgari SSP; Mukherjee P; Kessler JA; Espinosa HD, Proc. Natl. Acad. Sci. U. S. A 2019, 116 (46), 22909. DOI 10.1073/pnas.1911718116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sharei A; Zoldan J; Adamo A; Sim WY; Cho N; Jackson E; Mao S; Schneider S; Han M-J; Lytton-Jean A; Basto PA; Jhunjhunwala S; Lee J; Heller DA; Kang JW; Hartoularos GC; Kim K-S; Anderson DG; Langer R; Jensen KF, Proc. Natl. Acad. Sci. U. S. A 2013, 110 (6), 2082. DOI 10.1073/pnas.1218705110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Deng Y; Kizer M; Rada M; Sage J; Wang X; Cheon D-J; Chung AJ, Nano Lett. 2018, 18 (4), 2705–2710. DOI 10.1021/acs.nanolett.8b00704. [DOI] [PubMed] [Google Scholar]
- [31].Liu A; Islam M; Stone N; Varadarajan V; Jeong J; Bowie S; Qiu P; Waller EK; Alexeev A; Sulchek T, Mater. Today 2018, 21 (7), 703–712. DOI 10.1016/j.mattod.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kang G; Carlson DW; Kang TH; Lee S; Haward SJ; Choi I; Shen AQ; Chung AJ, ACS Nano 2020, 14 (3), 3048–3058. DOI 10.1021/acsnano.9b07930. [DOI] [PubMed] [Google Scholar]
- [33].Hur J; Park I; Lim KM; Doh J; Cho S-G; Chung AJ, ACS Nano 2020, 14 (11), 15094–15106. DOI 10.1021/acsnano.0c05169. [DOI] [PubMed] [Google Scholar]
- [34].Ding X; Stewart MP; Sharei A; Weaver JC; Langer RS; Jensen KF, Nat. Biomed. Eng 2017, 1 (3), 0039. DOI 10.1038/s41551-017-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kang W; McNaughton RL; Espinosa HD, Trends Biotechnol. 2016, 34 (8), 665–678. DOI 10.1016/j.tibtech.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ohgushi M; Matsumura M; Eiraku M; Murakami K; Aramaki T; Nishiyama A; Muguruma K; Nakano T; Suga H; Ueno M; Ishizaki T; Suemori H; Narumiya S; Niwa H; Sasai Y, Cell Stem Cell 2010, 7 (2), 225–239. DOI 10.1016/j.stem.2010.06.018. [DOI] [PubMed] [Google Scholar]
- [37].Babakinejad B; Jönsson P; López Córdoba A; Actis P; Novak P; Takahashi Y; Shevchuk A; Anand U; Anand P; Drews A; Ferrer-Montiel A; Klenerman D; Korchev YE, Anal. Chem 2013, 85 (19), 9333–9342. DOI 10.1021/ac4021769. [DOI] [PubMed] [Google Scholar]
- [38].Guillaume-Gentil O; Potthoff E; Ossola D; Franz CM; Zambelli T; Vorholt JA, Trends Biotechnol. 2014, 32 (7), 381–388. DOI 10.1016/j.tibtech.2014.04.008. [DOI] [PubMed] [Google Scholar]
- [39].Kang W; Yavari F; Minary-Jolandan M; Giraldo-Vela JP; Safi A; McNaughton RL; Parpoil V; Espinosa HD, Nano Lett. 2013, 13 (6), 2448–2457. DOI 10.1021/nl400423c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Giraldo-Vela JP; Kang W; McNaughton RL; Zhang X; Wile BM; Tsourkas A; Bao G; Espinosa HD, Small 2015, 11 (20), 2386–2391. DOI 10.1002/smll.201401137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yang R; Lemaître V; Huang C; Haddadi A; McNaughton R; Espinosa HD, Small 2018, 14 (12), 1702495. DOI 10.1002/smll.201702495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Aloysius N; Geetha M in 2017 Int. Conf. on Commun. and Signal Process. (ICCSP), 2017, 0588–0592. DOI: 10.1109/ICCSP.2017.8286426. [DOI] [Google Scholar]
- [43].Long J; Shelhamer E; Darrell T in Proc. of the IEEE Conf. on Computer Vision and Pattern Recognit. , 2015, 3431–3440. DOI: 10.1109/CVPR.2015.7298965. [DOI] [Google Scholar]
- [44].Falk T; Mai D; Bensch R; Çiçek Ö; Abdulkadir A; Marrakchi Y; Böhm A; Deubner J; Jäckel Z; Seiwald K; Dovzhenko A; Tietz O; Dal Bosco C; Walsh S; Saltukoglu D; Tay TL; Prinz M; Palme K; Simons M; Diester I; Brox T; Ronneberger O, Nat. Methods 2019, 16 (1), 67–70. DOI 10.1038/s41592-018-0261-2. [DOI] [PubMed] [Google Scholar]
- [45].Patino CA; Mukherjee P; Lemaitre V; Pathak N; Espinosa HD, SLAS Technol. 2021, 26 (1), 26–36. DOI 10.1177/2472630320982320. [DOI] [PubMed] [Google Scholar]
- [46].Esmaeilsabzali H; Sakaki K; Dechev N; Burke RD; Park EJ, Med. Biol. Eng. Comput 2012, 50 (1), 11–21. DOI 10.1007/s11517-011-0831-2. [DOI] [PubMed] [Google Scholar]
- [47].Nathamgari SSP; Pathak N; Lemaitre V; Mukherjee P; Muldoon JJ; Peng C-Y; McGuire T; Leonard JN; Kessler JA; Espinosa HD, Small 2020, 16 (43), 2002616. DOI 10.1002/smll.202002616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].J. S. Kohoutek, IL, US), Prinz, Jonathon (Skokie, IL, US) ELECTROPORATION SYSTEM WITH MICROMANIPULATOR AND PROBE. 2019.
- [49].Scaletti F; Hardie J; Lee Y-W; Luther DC; Ray M; Rotello VM, Chem. Soc. Rev 2018, 47 (10), 3421–3432. DOI 10.1039/c8cs00008e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Lee Y-W; Luther DC; Kretzmann JA; Burden A; Jeon T; Zhai S; Rotello VM, Theranostics 2019, 9 (11), 3280–3292. DOI 10.7150/thno.34412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Prakadan SM; Shalek AK; Weitz DA, Nat. Rev. Genetics 2017, 18 (6), 345–361. DOI 10.1038/nrg.2017.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Alom Ruiz S; Chen CS, Soft Matter 2007, 3 (2), 168–177. DOI 10.1039/B613349E. [DOI] [PubMed] [Google Scholar]
- [53].Ogunniyi AO; Story CM; Papa E; Guillen E; Love JC, Nat. Protoc 2009, 4 (5), 767–782. DOI 10.1038/nprot.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Schiroli G; Conti A; Ferrari S; della Volpe L; Jacob A; Albano L; Beretta S; Calabria A; Vavassori V; Gasparini P; Salataj E; Ndiaye-Lobry D; Brombin C; Chaumeil J; Montini E; Merelli I; Genovese P; Naldini L; Di Micco R, Cell Stem Cell 2019, 24 (4), 551–565.e8. DOI 10.1016/j.stem.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kim D; Kim C-H; Moon J-I; Chung Y-G; Chang M-Y; Han B-S; Ko S; Yang E; Cha KY; Lanza R; Kim K-S, Cell Stem Cell 2009, 4 (6), 472–476. DOI 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Datlinger P; Rendeiro AF; Schmidl C; Krausgruber T; Traxler P; Klughammer J; Schuster LC; Kuchler A; Alpar D; Bock C, Nat. Methods 2017, 14 (3), 297–301. DOI 10.1038/nmeth.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang H; Nakamura M; Abbott TR; Zhao D; Luo K; Yu C; Nguyen CM; Lo A; Daley TP; La Russa M; Liu Y; Qi LS, Science 2019, 365 (6459), 1301. DOI 10.1126/science.aax7852. [DOI] [PubMed] [Google Scholar]
- [58].Chen CL; Mahjoubfar A; Tai L-C; Blaby IK; Huang A; Niazi KR; Jalali B, Sci. Rep 2016, 6 (1), 21471. DOI 10.1038/srep21471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Logan DJ; Shan J; Bhatia SN; Carpenter AE, Methods 2016, 96, 6–11. DOI 10.1016/j.ymeth.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Osaki T; Uzel SGM; Kamm RD, Sci. Adv 2018, 4 (10), eaat5847. DOI 10.1126/sciadv.aat5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Keener DG; Cheung A; Futai K, J. Neurosci. Methods 2020, 337, 108632. DOI 10.1016/j.jneumeth.2020.108632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gonzalez RC; Woods RE, Digital image processing, 4th Edition, Pearson, London, UK: 2017. [Google Scholar]
- [63].Goodfellow I; Bengio Y; Courville A, Deep Learning, The MIT Press, Cambridge, MA: 2016. [Google Scholar]
- [64].Schindelin J; Arganda-Carreras I; Frise E; Kaynig V; Longair M; Pietzsch T; Preibisch S; Rueden C; Saalfeld S; Schmid B; Tinevez J-Y; White DJ; Hartenstein V; Eliceiri K; Tomancak P; Cardona A, Nat. Methods 2012, 9 (7), 676–682. DOI 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.