

Summary:
Amyotrophic lateral sclerosis (ALS) is characterized by motor neuron degeneration accompanied by aberrant accumulation and loss-of-function of the RNA-binding protein, TDP43. Thus far it remains unresolved to what extent TDP43 loss-of-function directly contributes to motor system dysfunction. Here, we employed gene editing to ask whether the mouse ortholog of the TDP43 regulated gene, STMN2, has an important function in maintaining the motor system. Both mosaic founders and homozygous loss-of-function Stmn2 mice exhibited neuromuscular junction denervation and fragmentation, resulting in muscle atrophy and impaired motor behavior, accompanied by an imbalance in neuronal microtubule dynamics in the spinal cord. Introduction of human STMN2 through BAC transgenics was sufficient to rescue these motor phenotypes observed in Stmn2 mutant mice. Collectively, our results demonstrate that disrupting the ortholog of a single TDP43-regulated RNA is sufficient to cause substantial motor dysfunction, indicating that disruption of TDP43 function is likely a contributor to ALS.
Keywords: Stathmin 2, SCG10, Amyotrophic Lateral Sclerosis, TDP43, TARDBP, neuromuscular junction, motor neuron, CRISPR, motor neuropathy, microtubules
Graphical Abstract

eTOC Blurb
The TDP43-regulated gene STMN2, provides a potential connection between TDP43 dysfunction and motor neuropathy as observed in ALS. Guerra San Juan, Nash et al. demonstrate that mouse Stmn2 is essential for maintaining normal motor function in vivo; its deficiency results in neuromuscular junction denervation, muscle atrophy and impaired motor behavior.
Upon autopsy, up to 98% of ALS patients have been reported to exhibit a stereotypical histopathology in their spinal motor neurons in which the normally nuclear localized RNA-binding protein TDP43, becomes insoluble and excluded from the nucleus (Barmada et al., 2010; Feneberg et al., 2018; Giordana et al., 2010). The discovery that TDP43 is one of the primary components of ubiquitinated pathological inclusions in the motor neurons of ALS patients was made almost contemporaneously with the finding that mutations in the gene encoding TDP43 (TARDBP) are a cause of familial disease (Barmada et al., 2010; Ling et al., 2010; Neumann et al., 2006; Van Deerlin et al., 2008). These two findings indicate that alterations in TDP43 function are on the pathway that leads to neural degeneration in ALS (Hergesheimer et al., 2019; Igaz et al., 2011; Klim et al., 2021; Prasad et al., 2019; Vanden Broeck et al., 2014).
TDP43 is an important regulator of RNA metabolism and has been shown to influence many aspects of an RNA’s life-cycle ranging from its initial transcription, to its splicing, transport, and translation (Alami et al., 2014; Arnold et al., 2013; Briese et al., 2020; Fallini et al., 2012; Humphrey et al., 2017). Overexpression of mutant or wild-type TDP43 can act through several mechanisms to contribute to the death of various cell-types, including motor neurons (Ash et al., 2010; Fratta et al., 2018; Huang et al., 2012; Mitchell et al., 2015; Wils et al., 2010; Xu et al., 2010). However, how loss of TDP43 function through either its nuclear exclusion or compartmentalization into cytoplasmic aggregates might contribute to the selective loss or gain of mechanisms that lead to degeneration of motor neurons observed in ALS, has been less extensively explored (Klim et al., 2021).
Recently, we and others have shown that the transcript encoding the protein STMN2/SCG10 is one of the most abundant in human iPSC-derived motor neurons, and that the STMN2 transcript is tightly regulated by TDP43 (Klim et al., 2019; Melamed et al., 2019; Prudencio et al., 2020). Perturbations to cultured motor neurons that reduced TDP43 function caused premature polyadenylation of the STMN2 pre-mRNA, producing a truncated STMN2 message (Klim et al., 2021; Klim et al., 2019; Melamed et al., 2019). Strikingly, examination of STMN2 gene products within the brain and spinal cord of ALS/FTLD patients exhibiting TDP43 pathology revealed alterations identical to those identified upon TDP43 perturbation in cellular models (Klim et al., 2019; Melamed et al., 2019; Prudencio et al., 2020). STMN2 protein homologs display high levels of evolutionary conservation across mammalian species suggesting an important function (Charbaut et al., 2001; Chauvin and Sobel, 2015; Ozon et al., 1998). However, the elements in the human transcript through which TDP43 acts to control STMN2 are not conserved in either mouse or rat, providing a possible explanation for why the regulation of its expression was not previously identified (Melamed et al., 2019; Okazaki et al., 1993).
STMN2 is part of the Stathmin family of microtubule regulating proteins, which play important, but sometimes redundant, roles in regulating microtubule stability and remodeling (Charbaut et al., 2001; Chauvin and Sobel, 2015; Graf et al., 2011; Oishi et al., 2002; Okazaki et al., 1993; Ozon et al., 1998; Sugiura and Mori, 1995). More specifically, STMN2 over-expression can enhance neurite outgrowth in cultured neurons (Klim et al., 2019; Melamed et al., 2019; Morii et al., 2006; Riederer et al., 1997), while its reduction in human pluripotent stem cell (hPSC) derived motor neurons leads to deficient axonal regrowth following axotomy, which closely recapitulates the phenotype of motor neurons following a reduction in expression of TDP43 (Klim et al., 2019; Melamed et al., 2019). However, the expression pattern of the mouse ortholog within the motor system, for instance in the spinal cord, has not been examined and studies of Stmn2 mutant mice have not been conducted thus far, which would be important for assessing whether this gene and its product plays a critical role in maintaining motor axons in vivo. Thus, creating and studying mice harboring mutations in Stmn2 could provide fundamental insight into whether loss of STMN2 expression due to altered function of TDP43 is in and of itself, a credible contributor to dysfunction of motor neurons in ALS patients.
Here we report the use of CRISPR-Cas9-mediated gene editing in mouse zygotes to create animals harboring loss-of-function (LOF) mutations in Stmn2. Mice deficient in Stmn2 (Stmn2−/−) exhibited motor neuropathy as demonstrated by loss of neuromuscular junction (NMJ) innervation, accompanied by evidence for a regenerative response to muscle injury, which was further associated with a significant deficit in motor behavior. Moreover, loss of Stmn2 was shown to result in alterations to microtubule polymerization in the neuronal fraction of the mouse lumbar spinal cord and in hPSC-derived motor neurons. Interestingly, disruption to the motor system in Stmn2 mutant animals occurred without obvious signs of neuroinflammation or the onset of TDP43 pathology. Lastly, rescue with a bacterial artificial chromosome (BAC) containing the human STMN2 locus restored motor function in mutant animals. Overall, our findings are consistent with the notion that loss of STMN2 function, which occurs downstream of pathological alteration to TDP43, is a meaningful contributor to ALS.
Results
Generation of Stmn2 mutant mice
To investigate the functional importance of Stmn2 in vivo, we created LOF mutations within the Stmn2 gene using CRISPR-Cas9 genome editing in mouse zygotes. More specifically, we performed pronuclear injection with recombinant Cas9 protein complexed with two validated single-stranded guide RNAs (sgRNA) targeting exons 2 and 4 of the Stmn2 gene (Figure 1a). Our aim was to create founder animals with a variety of Stmn2 mutations in exons 2 and 4, as well as to identify animals in which the 13Kb region between exons 2 and 4 had been deleted (Figure 1a-b). To generate a set of control animals that could be phenotypically compared to any Stmn2 mutant founders, a parallel experiment involved the injection of Cas9 and guide RNA into zygotes, targeting the safe harbor locus Rosa26 (Suppl. Figure 1a-b).
Figure 1. CRISPR/Cas9 editing of Stmn2.

(A) Diagram of breeding strategy to generate Stmn2 F0, Rosa26 gRNA, and Stmn2 F2 mice. (B) Schematic representation of the STMN2 locus in which gRNAs were targeted to exons 2 and 4 to create a predicted 13 Kb deletion. Primers were designed to flank the region of the deletion region to confirm the presence or absence of mutations. (C) PCR genotyping of F2 Stmn2 mice exhibiting WT (+/+), heterozygous (+/−), and homozygous (−/−) mutations. (D) F2 Stmn2 brain mRNA levels flanking Exons 1 to 3 (left) and Exons 4 to 5 (right). (E) Spinal cord and brain Stmn2 protein levels from Western blots including the housekeeping protein GAPDH. **** p < 0.0001. In all figures: results are shown as a mean with error bars calculated as standard deviation. Detailed information (average, SD, n and detailed statistics) is shown in STAR Methods.
To identify Stmn2 mutations in putative founder Stmn2 F0 animals, two sets of primers were designed flanking each gRNA cleavage site. Primers upstream of exon 2 and downstream of exon 4 were also used to explore whether any larger deletions might have occurred between the two editing sites (Suppl. Figure 1c-e). PCR-amplification using genomic DNA from the tails of Stmn2 founders revealed 30% (5/17) of these mice showed evidence for a large deletion spanning from exon 2–4, while the remaining 70% (12/17) of the animals harbored a variety of mutations (insertions/deletions) flanking one or both of the gRNA binding sites (Suppl. Figure 1 f). Subsequent in silico predictions (Brinkman et al., 2014) from Sanger sequencing of these PCR products indicated the majority of alterations were frameshift mutations resulting in a premature stop codon (Suppl. Figure 1 j). Overall, our genotypic analysis indicated that each founder animal displayed evidence for mutagenesis at the Stmn2 locus.
The extent of mutagenesis in founder animals encouraged us to examine whether the mutations we identified were sufficient to impair Stmn2 expression. Examination of RNA isolated from the cortex of Stmn2 F0 mice demonstrated a significant decline in Stmn2 transcript abundance when compared to Rosa26 F0 controls (Suppl. Figure 1 f). Western blot analysis further showed that Stmn2 protein levels were significantly reduced in the cortex of these Stmn2 F0 animals in comparison to Rosa26 F0 (Suppl. Figure 1 g). These results indicate that while each Stmn2 mutant within the F0 cohort exhibited substantial mosaicism and allelic complexity, the levels of Stmn2 transcript and protein were greatly reduced in their nervous system, allowing us to use these animals along with Rosa26 F0 controls, to study the organismal consequence of reduced Stmn2 function.
While examination of mosaic founders provides certain advantages associated with determining whether multiple in/del alleles deliver similar phenotypes, these animals can also harbor “off-target” mutations induced during editing. To reduce the risk of improperly assigning any observed phenotypes to off-target mutations, we undertook an outcrossing strategy to isolate a 13 Kb deletion (13KbΔ) in the Stmn2 locus. Founders harboring this mutation were bred with wild-type C57BL6 mice to generate a collection of heterozygous F1 Stmn2+/− mice. We then selected F1 mice heterozygous for the 13KbΔ and continued to outcross the Stmn2+/− mutants to C57BL6 WT mice, and then intercrossed them to generate homozygous Stmn2 −/− mice (Figure 1a, Suppl. Figure 1i). A second genotyping strategy was generated that consisted of a three-primer PCR assay designed to amplify both the wild-type and deletion alleles (Figure 1c). Subsequent analysis confirmed that Stmn2−/− mice lacked detectable transcript and protein expression in their cortex when compared to +/− and +/+ littermate controls (Figure 1 d-e). Given the functional redundancy of the Stathmin protein family, transcript levels of Stmn 1, 3, and 4 were evaluated for potential compensatory expression as the result of loss of Stmn2 in the original F0 cohort. Gene expression analysis by qRT-PCR showed no significant changes in transcript levels of Stmn1, Stmn3 or Stmn4 within cortex of Stmn2 F0 mice compared to Rosa26 controls (Suppl. Figure 2 a). Western blot analysis with both N-terminal and C-terminal anti-Stmn2 antibodies readily detected a single protein of the expected size in control mice, but no detectable protein was seen with in samples from homozygous mutant animals. (Suppl. Figure 2 b). Further breeding of this LOF allele in Stmn2 allowed us to produce a colony of littermates with predictably differing Stmn2 genotypes for phenotypic studies. Notably, Stmn2 offspring were born at expected genotype frequencies both at post- and pre-natal stages (Chi-square test, p21: p=0. 0.4306; E17–18: p=0.8725), denoting that presence of the LOF allele does not result in perinatal or embryonic lethality (Suppl. Figure 2 c).
Motor impairment in Stmn2 mutants
Next, we asked whether absence of Stmn2 led to an impairment in motor function, an outward feature of ALS, by assessing motor task performance at 60 and 100 days (Figure 2 a,b). Stmn2 F0 mutant mice showed significant deficits in coordinated motor performance relative to Rosa26 F0 controls as measured by rotarod (Figure 2 c) and hanging wire test (Figure 2 d,e). Similarly, F2 Stmn2−/− mutant mice also displayed significantly reduced motor performance in both the rotarod (Figure 2 f) and hanging wire test when compared to age-matched littermate controls (Figure 2 g). Such motor dysfunction was persistent but did not significantly exacerbate over time. Collectively, these findings demonstrate that Stmn2 is necessary for maintaining proper motor behavior.
Figure 2. Stmn2 mutant mice display motor deficits.
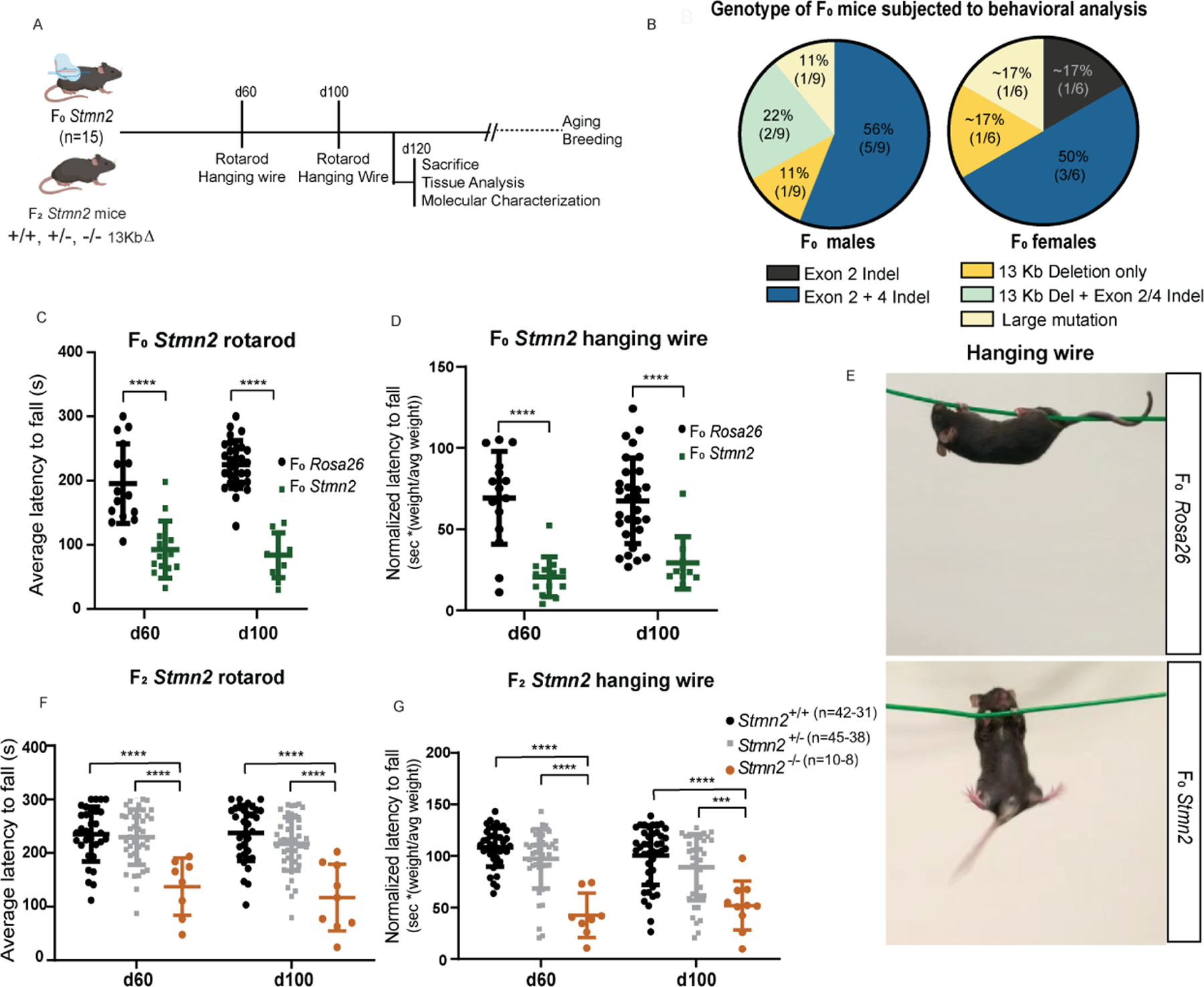
(A) Diagram of the experimental strategy to examine Stmn2 LOF on motor behavior in Stmn2 F0 mice. (B) Stmn2 F0 genotype subjected to behavioral analysis. (C) Rotarod and (D) hanging wire performance at p60 and p100 in F0 Stmn2 mutant mice and Rosa26 F0 controls, (P60: n=15 Rosa26 F0, n=13 F0 Stmn2; p100: n=29 Rosa26 F0, n=10 F0 Stmn2) (E) Graphical representation of Rosa26 F0 (top) and Stmn2 F0 (bottom) mice performing the hanging wire test. F2 Stmn2 performance on (F) rotarod and (G) hanging wire at p60 and p100. **** p < 0.0001.
Loss of Stmn2 results in an age-dependent denervation of the neuromuscular junction
Given the motor deficits observed in the Stmn2 mutant cohort, we investigated whether they were a result of alterations in both upper and lower motor neurons. We first looked for morphological changes within deeper layers of the motor cortex. Examination of the neuronal density in layer V of the primary motor cortex of p120 F2 Stmn2 mice revealed no significant changes in comparison to littermate controls (Suppl. Figure 2 d-f)
We next considered whether loss of Stmn2 function influenced the integrity of motor neurons within the spinal cord and their axon synapses at the NMJ (Figure 3, Suppl. Figure 3). To this end, we examined NMJs in whole-mount preparations of hindlimb gastrocnemius (GA) muscles from 120-day old Stmn2 F0 mutant founders and Rosa26 F0 controls. To identify NMJs, we stained with fluorescently labeled alpha-bungarotoxin (BTX), which binds directly to acetylcholine receptors (AchRs) localized at the post-synaptic apparatus on the muscle fiber. Additionally, we used antibodies specific to synaptophysin (SyPhy), which localizes to the pre-synaptic motor axon terminal. Quantification of BTX/SyPhy co-staining, indicative of an intact NMJ, demonstrated that the Stmn2 F0 mutant cohort exhibited a significant decline in the number of innervated NMJs within the GA relative to Rosa26 F0 controls (Suppl. Figure 3 a-c). Within the GA, Stmn2 F0 mice showed 45% fewer fully innervated NMJs, 12% more partially innervated NMJs, and 33% more fully denervated NMJs than Rosa26 F0 controls (Suppl. Figure 3 d-e)
Figure 3. Loss of Stmn2 leads to denervation in the hindlimb gastrocnemius.

(A-B) Diagram of the experimental strategy to examine innervation in the GA of mice. (C) Whole mount preparations from Stmn2 F2 mutant mice and littermate controls were stained with fluorescently conjugated BTX and anti-SyPhy antibodies at p21 (top panel) and p120 (bottom panel) (D) and quantified the extent of co-localization per NMJ per field of view (FOV). ns, p>0.05, **p < 0.01, **** p < 0.0001. (p21–23: n=5 Stmn2+/+and n=4 Stmn2−/− mice; p120 n=3 Stmn2+/+ and −/−). Data points for each animal are represented by different colors.
To determine the replicability of this important finding and to explore the age dependence of this observation, we examined innervation of the GA muscle at day 21 and day 120 in F2 Stmn2−/− animals harboring the 13KbΔ we identified as well as their littermate controls (Figure 3 a-c). At day 21, we did not observe a significant change in innervation of the GA muscle between mutants and controls (Figure 3 d).
In contrast, when we examined the NMJs of 13KbΔ Stmn2−/− mutant and control animals at day 120, we again observed a significant reduction in the number of intact NMJs in Stmn2 mutant muscle with approximately 33% fewer fully innervated, 10% more partially innervated, and 25% more fully denervated NMJs relative to age matched littermate controls. (Figure 3 c-d) Overall, our results suggest that in Stmn2 mutant animals, initial appropriate NMJ innervation occurs, but then NMJ denervation follows later in life.
A characteristic feature of persistent NMJ denervation is dispersion of AchR clusters at the post-synaptic junction. AchR dispersion was assessed by quantifying the number of fragmented AchR clusters within the GA of the F2 and F0 animals at p21 and p120 (Figure 4 a-c, Suppl. Figure 4 a-c). NMJs were considered fragmented when they presented with 5 or more discontinuous areas of AchR staining (Balice-Gordon et al., 1990). Skeletal muscles of both the Stmn2 F2 mutants and F0 mice displayed a significantly elevated frequency of fragmented NMJs in comparison to their respective controls at 120 days. However, this phenotype was not present at an early age in the F2 13KbΔ cohort (Figure 4 d, Suppl. Figure 4 d). Taken together, the data surrounding hindlimb muscle innervation indicate that the NMJ degeneration driven by Stmn2 loss is not a result of developmental defect but occurs after wiring of the motor system.
Figure 4. Stmn2 loss-driven denervation results in NMJ fragmentation and muscle injury.

(A) Diagram of the experimental strategy to examine level of fragmentation in the NMJs within the GA of mice. (B) Diagram exhibiting NMJ denervation over time. (C) GA isolated from Stmn2 F2 and littermate controls, stained with fluorescent conjugated BTX. (D) Quantification of fragmented NMJs per FOV in Stmn2 F2 and littermate controls. (E) Diagram of the experimental strategy to examine the level of centralized myonuclei in the GA. (F) Representative cross-sectional images of F2 Stmn2 mutants and control littermates. (G) Quantification of centralized myonuclei F2 Stmn2 and control littermates per FOV. ns, p>0.05, *** p < 0.001, **** p < 0.0001. (p21–23: n=5 Stmn2+/+ and n=4 Stmn2−/− mice; p120: n=3 Stmn2+/+ and −/− mice). Data points for each animal are represented by different colors.
Muscle atrophy and injury in Stmn2 mutants
Muscle atrophy resulting from persistent denervation is known to be associated with a regenerative response in which skeletal muscle precursor cells are activated and fuse with the muscle fibers, contributing their nuclei to the injured fiber (Folker and Baylies, 2013; Roman and Gomes, 2018). When newly fused to the myofiber, precursor nuclei are found to be centrally localized within the fiber, but as the fiber matures, nuclei become localized to the fiber’s periphery (Folker and Baylies, 2013; Roman and Gomes, 2018). Thus, an increase in centralized nuclei within skeletal myofibers is one well-established measure of an ongoing response to injury (Folker and Baylies, 2013; Roman and Gomes, 2018). To determine whether the atrophy we observed was severe enough to lead to such a response to the injury, we examined the number of centralized nuclei in both Stmn2 F0 and F2 mutant cohorts. Indeed, we found there was a significant increase in the frequency of centralized myonuclei within the GA of F0 and F2 mutant animals at 120 days, suggesting such a response to injury was occurring in adult Stmn2 deficient mice (Figure 4 e-g, Suppl. Figure 4 e-g). Conversely, % of centralized myonuclei were almost indistinguishable in the muscle fibers of younger 21-day old F2 Stmn2−/− mice from those of age-matched controls. Thus, the muscle injury we observe upon loss of Stmn2 seems to be only present in mature myofibers present in adult animals.
Stmn2 is enriched in adult spinal cord motor neurons
While studies of Stmn2 in hPSC-derived motor neurons demonstrated it to be highly expressed, we were unable to find reports of its expression in the mouse spinal cord (Klim et al., 2019; Melamed et al., 2019). To better understand Stmn2 expression in the mouse spinal cord and the extent to which we eliminated Stmn2 expression in the cord of Stmn2 F0 founder animals, we carried out immunostaining studies. In Rosa26 F0 controls, we found that within the ventral spinal cord, Stmn2 immunostaining was selective for choline acetyltransferase positive (Chat+) presumptive motor neurons (Figure 5 a-c; Suppl. Figure 5 a). A similar analysis of Stmn2 F0 mutant animals and Rosa26 F0 controls revealed that while there was not a significant difference in the number of Chat+ presumptive motor neuron cell, there was a significant decline in the number of Stmn2+/Chat+ motor neurons (Figure 5 d-f). Thus, elimination of detectable Stmn2 had occurred in the majority but not all motor neurons of Stmn2 F0 animals. We similarly examined Stmn2 expression in the spinal cords of F2 Stmn2 −/− animals relative to their littermate controls and in this case found only Chat+/Stmn2- motor neurons with no wider evidence for expression of Stmn2 protein (Figure 5). To further investigate whether loss of Stmn2 function altered motor neuron number at this time point, we carried out more extensive motor neuron counts in p120 F2 mutant animals. Again, the number of Chat+ neurons was not significantly reduced in Stmn2 −/− animals at p120, indicating that Stmn2 is not required for the development of spinal motor neuron cell bodies (Figure 5 g-j).
Figure 5. Stmn2 is selectively expressed in motor neurons within the adult ventral spinal cord.

(A) Diagram of the experimental strategy to examine Stmn2 expression in the lumbar spinal cord. (B) Stmn2 F0 genotypes in mice used for spinal cord analysis. (C) Stmn2 F0 mutant mice and Rosa26 controls cords stained with antibodies specific to Chat and Stmn2 proteins Quantification of (D) Chat positive and Stmn2 positive neurons in each hemi-section of Stmn2 F0 mutant mice and Rosa26 F0 controls. (E) Percentage of Chat positive motor neurons expressing Stmn2 protein quantified per hemi-section in Stmn2 F0 mice and Rosa26 F0 controls. (F) Ventral horn spinal cord sections from F2 Stmn2 mice and control littermates were stained with anti-Stmn2 antibodies and anti-Chat antibodies to detect motor neurons. Quantification of (G) Chat positive and Stmn2 positive neurons quantified per hemi-section in F2 Stmn2 mice and littermate controls. (H) Percent of Chat positive motor neurons containing Stmn2 protein quantified per hemi-section in F2 Stmn2 mice and littermate controls. **** p < 0.0001. Scale bars, 50 μm.
Predominant spinal cord motor neurons which innervate extrafusal muscle fiber NMJs (or α motor neurons) are composed of a mixture of fast fatigable, slow and fatigue-resistant motor pools differentiated by the contractile properties they produce in the muscle, their mode of activation, and soma size (Kanning et al., 2010). Fast-fatigable motor neurons are more abundant, have larger somas, and innervate force-generating muscles whereas slow and fatigue-resistant motor neurons are smaller in size, less abundant and generally innervate postural muscles (Kanning et al., 2010). In ALS, fast fatigable motor neurons are more vulnerable and undergo initial axonal degeneration, while slow and fatigue-resistant motor neurons degenerate at later stages in the disease (Fischer et al., 2004; Frey et al., 2000; Pun et al., 2006). As we examined its expression, we observed that Stmn2 appeared to be more highly expressed in larger and presumptive fast fatigable motor neurons. To test this idea, we measured the relationship between motor neuron soma size and Stmn2 fluorescence intensity in different lumbar regions of Rosa26 F0 control spinal cords. Interestingly, Stmn2 intensity per micron2 showed a modest but significant positive correlation to soma size (R2 = 0.089, p < 0.001), potentially suggesting selectivity for higher Stmn2 expression in motor neuron subtypes known to be most susceptible to degeneration in ALS (Suppl. Figure 5 b-d). To gain better insight into the expression of Stmn2 within specific sub-populations of α skeletal motor neurons, we co-stained wild-type p21 lumbar mouse spinal cords with a recently described adult α motor neuron marker, Spp1, which encodes for the extracellular matrix protein, Osteopontin, and Stmn2 (Blum et al., 2021; Fischer et al., 2004; Frey et al., 2000; Morisaki et al., 2016; Patel et al., 2021; Pun et al., 2006). We found Stmn2 to be expressed specifically in a subset of Spp1+ neurons, suggesting a selective expression of Stmn2 in a specific subset of presumptive α motor neurons (Suppl. Figure 5 e).
NMJ denervation occurs in absence of gliosis and TDP43 pathology.
Neuroinflammation is a hallmark of ALS and is characterized by increased activation of astrocytes and microglia (Glass et al., 2010; Philips and Robberecht, 2011). However, the factors that lead to neural inflammation in ALS and where inflammation lies on the pathway to motor neuron degeneration remain matters of intense investigation (Burberry et al., 2016; Burberry et al., 2020; Lall and Baloh, 2017; Olesen et al., 2020). To investigate whether neural inflammation might be contributing to the neuromuscular denervation observed in Stmn2 mutant animals, we performed immunostaining for antigens expressed in reactive astrocytes and microglia. Examination of the lumbar spinal cord revealed no significant microgliosis or astrogliosis as measured by % of Iba1+ and GFAP+ cells, respectively, in F0 animals at p120 (Figure 6 a-g). Similarly, the innervation defects found in the F2 cohort were not accompanied by astrogliosis at the same time point (Suppl. Figure 6 a-d). Thus, our results indicate that loss of Stmn2 function can cause neuromuscular denervation in the absence of obvious neural inflammation.
Figure 6. Absence of neural inflammation and Tdp-43 pathology in Stmn2 mutant cord.

(A) Diagram of the experimental strategy to examine astrocyte and microglia activation within the spinal cord along with Tdp-43 localization. (B) Diagram of cell types analyzed in the spinal cords. (C) Stmn2 F0 genotype corresponding to the spinal cord analysis. Hemi-sections of the lumbar spinal cord from Stmn2 F0 mice and Rosa26 F0 controls were stained for (D) GFAP, (E) Iba1, and (F) Tdp-43 and quantified in motor neurons (F). Graphs of cell counts (G), ns, p>0.05 (n=3 animals/genotype, n=5 sections/animal, for Tdp43 localization analysis: n=100 Chat+ neurons counted/animal)
While, we chose to investigate the function of Stmn2 as a downstream target of TDP43 dysfunction in motor neurons, we also noted that previous reports had shown that certain nerve injuries in rodents, which usually lead to NMJ denervation, may be sufficient to cause Tdp43 pathology (Moisse et al., 2009; Sato et al., 2009). We therefore wondered whether the NMJ denervation we observed in Stmn2 mutant mice might be in and of itself, sufficient to cause pathological localization of Tdp43. Sections of the ventral horn of spinals cords from P120 Stmn2 F0 mutant mice and Rosa26 F0 controls as well as from F2 Stmn2+/+ and Stmn2−/− animals were immunostained with Tdp43-specific antibodies to assess Tdp43 localization in motor neurons. Through these studies, we found no detectable alteration in nuclear localization of Tdp43 in the motor neurons of either Stmn2 mutants or controls (Figure 6 f-g, Suppl. Figure 6 e-f). These findings indicate that loss of Stmn2 is sufficient to cause NMJ denervation even in the absence of the broader consequences of TDP43 pathology.
Loss of Stmn2 disrupts microtubule dynamics.
As STMN2 is recognized to play important roles in microtubule regulation in neurons, (Morii et al., 2006) we investigated the impact that Stmn2 loss had on microtubule dynamics within the central nervous system. Due to Stmn2 being highly expressed within motor neurons, we chose to investigate whether microtubule alterations were present within the lumbar spinal cord of p120 Stmn2 mutants and their control littermates (Figure 7 a). Through biochemical analysis of the amount of polymerized versus free tubulin content in this tissue, we found that Stmn2−/− mice showed reduced levels of polymerized neuronal β-III-Tubulin in comparison to their wild-type littermates. Interestingly, this effect was specific to the neuronal β-III-Tubulin and did not affect total tubulin levels or ratios of free to polymerized tubulin more generally (Figure 7 c-f). These results suggest that Stmn2 might be selectively regulating microtubule polymerization in spinal cord neurons.
Figure 7. Loss of Stmn2 in the murine spinal cord and in cultured human motor neurons impairs microtubule dynamics.
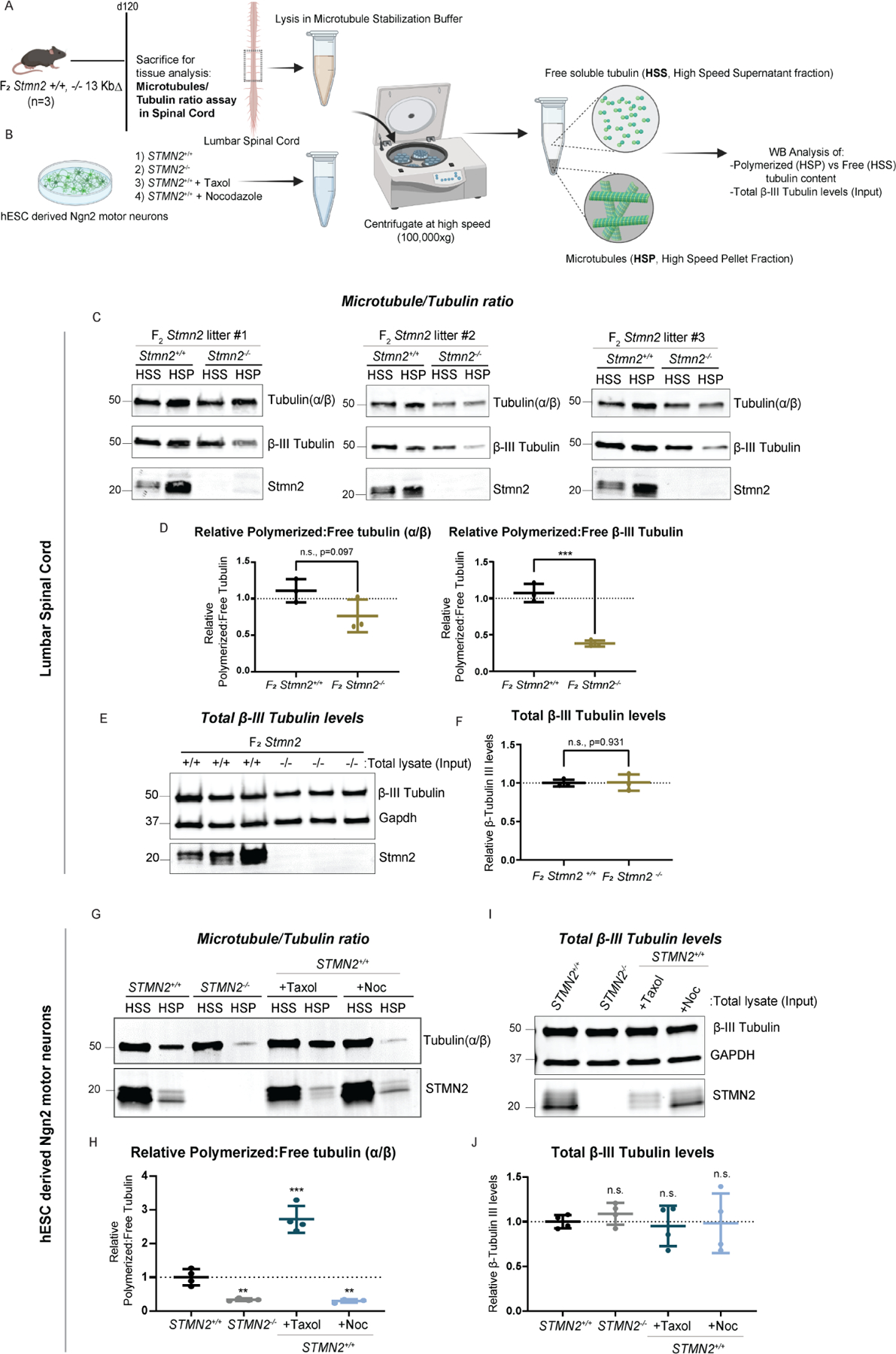
(A-B) Diagram of the experimental strategy to examine free and polymerized tubulin in Stmn2 F2 and control littermates, as well as hPSC derived Ngn2 motor neurons. (C-D) Quantified western blot analysis of the ratio of polymerized (HSP, High Speed Pellet) and free (HSS, High Speed Supernatant) tubulin (α/β and β-III) fractions isolated from the lumbar spinal cord of Stmn2−/− mice and normalized to that of Stmn2+/+ controls littermates. (E-F) Levels of overall β III tubulin in the tissue lysates (Input fraction), n=3 animals/genotype. Respective littermate controls displayed. Average of n=3 technical replicates represented. (G-H) Quantified western blot analysis of the ratio of polymerized (HSP) and free (HSS) tubulin (α/β) fractions isolated from STMN2−/− hPSC-derived Ngn2 motor neurons, normalized to corresponding controls STMN2+/+ hMNs. STMN2+/+ hMNs were also treated with microtubule regulator drugs, Taxol (1μM for 72 hours) and Nocodazole (10 μM for 45 minutes) as experimental controls. (I-J) Levels of overall β III tubulin in the cultured motor neuron lysates (Input fraction). n=2 biologically independent experiments (2 technical replicates represented for each experiment).
To explore its relevance to human neurons, the STMN2+/+ and STMN2−/− hPSCs we generated previously were differentiated into motor neurons using an Ngn2 transcriptional programming protocol, combined with activation of posteriorizing and ventralizing signaling pathways and examined for changes in their levels of free and polymerized tubulin (α/β) (Figure 7b) (Limone et al., 2022; Zhang et al., 2013). Similar to our findings in Stmn2 mutant mice, the loss of STMN2 in cultured motor neurons resulted in a significant decrease in levels of polymerized tubulin, with no observed change in the overall levels of total β III tubulin, mimicking the effects induced by Nocodazole and opposite to those induced by the microtubule polymerization enhancing drug Paclitaxel/Taxol (Figure 7 g-j). Collectively, these findings suggest that loss of Stmn2 leads to an imbalance in the dynamic process of microtubule regulation in motor neurons, which ultimately might translate to axonal defects distally at the NMJ, leading to improper synaptic stability, denervation, and muscle atrophy.
Expression of hSTMN2 is sufficient to restore motor function in mutant animals
To further confirm that loss of Stmn2 function was the lone contributor to the motor phenotypes we observed, we sought to rescue the LOF phenotypes by introduction of the human STMN2 gene through BAC transgenics. Founder BAChSTMN2 mice were generated by injecting an identified BAC containing the human STMN2 locus into C57BL/6 mouse zygotes. The resulting offspring were screened for the integration of the human gene by primers targeting the beginning, middle, and end of the gene (Intron 1, Exon 3 and Exon 5, respectively). The BAC hSTMN2 founders containing the STMN2 transgene were then crossed with WT C57BL/6 to expand the line (Figure 8 a,b). BAChSTMN2 mice were then bred into the LOF mutant heterozygous background (mStmn2+/− 13KbΔ) to generate hSTMN2xmStmn2+/−offspring and then back-crossed to produce mice completely lacking murine Stmn2, while expressing human STMN2 (hSTMN2xmStmn2−/−, Figure 8 b-d).
Figure 8. Introduction of hSTMN2 gene rescues LOF-associated motor deficits.
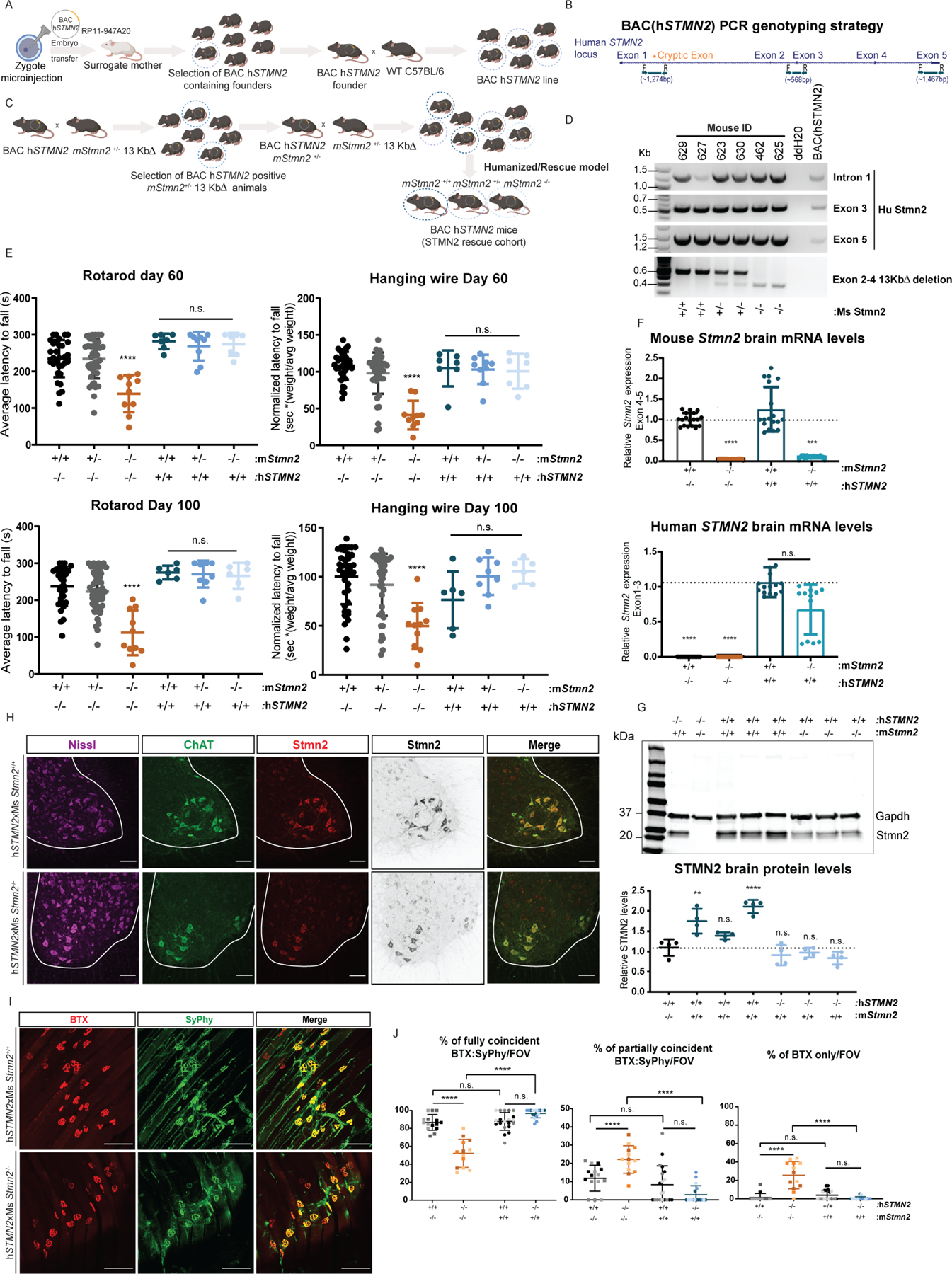
(A) Diagram of breeding strategy to generate BAChSTMN2 transgenic line and subsequent hSTMN2xmStmn2+/+, +/−, −/− lines in (B). (C) Schematic representation of the STMN2 locus in which primers were designed to flank beginning (intron 1), middle (Exon 3) and end (Exon 5). (D) PCR genotyping of hSTMN2 exhibiting presence of the hSTMN2 gene, and of WT (+/+), heterozygous (+/−), and homozygous (−/−) mutations of the mouse Stmn2 gene (E) Rotarod (left panel) and hanging wire (right panel) at p60 and p100 in hSTMN2xmStmn2+/+ and −/− compared to Stmn2 mutant mice. (p60: hSTMN2xmStmn2+/+ n=7, +/− n=9, −/− n=7; p100 hSTMN2xmStmn2+/+ n=6, +/− n=8, −/− n=6). (F) mouse Stmn2 Exons 4–5 (left) and human STMN2 flanking Exons 1–3 (right) in transgenic mice compared to Stmn2 mutant and wild-type littermate mice. (G) Brain Stmn2 protein levels from Western blots including the housekeeping protein GAPDH. (H) Representative stain of hSTMN2xmStmn2+/+ and −/− cords labeled with anti-Chat and anti-Stmn2 and counterstained for Nissl. (I) Whole mount preparations from hSTMN2xmStmn2+/+ and −/− mice were stained with fluorescently conjugated BTX and anti-SyPhy at p120 (J) and quantified the extent of co-localization per NMJ per FOV compared to Stmn2 mutant mice. * p < 0.05, **p < 0.01, *** p < 0.001, **** p < 0.0001 (n=3 animals/genotype, hSTMN2xmStmn2+/+ and −/−). Scale bars, 100 μm. Results from Stmn2 mutant mice in (E,J) are those generated in Figures 2 and 3.
Mice were subjected to the same behavioral testing described before for mStmn2−/− mutants (Figure 2) to determine the impact of hSTMN2 expression. Remarkably, expression of the hSTMN2 transgene in the mStmn2−/− background rescued the motor behavior deficits at day 60 and 100 when compared to the LOF model (Figure 8 e). Examination of RNA isolated from the cortex of hSTMN2xmStmn2+/+ and hSTMN2xmStmn2−/− transgenic mice, indicated that the human STMN2 transcript was correctly processed as determined by qPCR analysis of coding exons (Figure 8 f). Consequently, Western blot analysis detected full length STMN2 protein in the cortex of hSTMN2xmStmn2−/− animals and showed significantly higher levels in the cortex of hSTMN2xmStmn2+/+ (Figure 8 g), confirming that the anti-Stmn2 antibody recognizes the epitope in both the human and mouse protein. Examination of STMN2 expression in the spinal cord of hSTMN2xmStmn2+/+ and −/− mice by immunohistochemistry showed a similar pattern of expression to that observed in wild-type mice, with STMN2 enrichment in Chat+ ventral spinal motor neurons (Figure 8 h). Together, these data show that expression of human (wild-type) STMN2 is sufficient to restore normal motor performance in mice with an inactivate endogenous Stmn2 gene.
Finally, the NMJs of the transgenic mice were investigated to determine whether expression of the hSTMN2 locus was also sufficient to rescue the denervation identified in the mStmn2−/− 13KbΔ mice. GA NMJs were examined in whole-mount preparations from 120-day old hSTMN2xmStmn2+/+ and −/− transgenic mice. Quantification of BTX/Syphy staining demonstrated proper NMJ innervation in hSTMN2xmStmn2+/+ and −/− transgenic mice (Figure 8 i-j). Hence, expression of human STMN2 is sufficient to restore both normal motor performance and normal NMJ innervation in mice with an inactivate endogenous Stmn2 gene. This suggests that human and murine STMN2 have redundant functions and that the mouse Stmn2 mutant phenotypes we observe are relevant to the human pathology associated with STMN2 dysfunction.
Discussion
Previously, studies of TDP43 client RNAs, identified the transcript encoding STMN2 as a potential means by which altered TDP43 function could contribute to impaired motor axon function (Klim et al., 2019; Melamed et al., 2019). These same studies showed that the precise molecular alterations to STMN2 that occur upon TDP43 disruption in cultured motor neurons are widely found in postmortem spinal cord and brain tissue from ALS patients (Klim et al., 2019; Melamed et al., 2019; Prudencio et al., 2020). More recently, the length of a novel non-coding repeat in STMN2, measured in olfactory neurosphere-derived cells from sALS patients and controls, associated with earlier age of onset, disease progression and reduced survival, potentially attributed to a reduced level of STMN2 expression (Theunissen et al., 2021).
Simultaneously, the role that the closely related family of STMN paralogs play in regulating microtubule dynamics within neuronal axons has been a subject of substantial interest (Charbaut et al., 2001; Chauvin and Sobel, 2015; Graf et al., 2011; Liedtke et al., 2002; Lin and Lee, 2016). Interestingly, LOF mutations in the single fly ortholog of the STMN family, Stai, result in deficits in the stability of the NMJ (Charbaut et al., 2001; Chauvin and Sobel, 2015; Sugiura and Mori, 1995). Additionally, LOF mutations in mouse Stmn1 have been reported to cause motor neuropathy (Liedtke et al., 2002; Nguyen et al., 2019).
Prior to our study, it has remained unresolved whether STMN2, or its mouse homolog, are necessary for maintaining a functional motor system in vivo, and to what extent the loss of STMN2 protein expression that occurs in vivo (downstream of TDP43 alterations) could explain the various aspects of motor neuron disease. We attempted to address these important questions by generating mice harboring LOF mutations in the mouse ortholog of STMN2. Our evidence shows that indeed, in mice harboring Stmn2 LOF mutations, NMJ denervation occurs, which coincides with fracturing of the post-synaptic apparatus on myofibers as well as muscle damage, resulting in severe impairments in motor behavior. Overall, the selective motor neuron expression of Stmn2 we observed in the ventral horn would seem to suggest that these phenotypes arise due to motor neuron autonomous dysfunction of the motor axon. This observation would in turn be consistent with the notion that axonal dysfunction and NMJ denervation leads to a set of classical signs of degeneration of the muscle, which are also observed following other denervating injuries (Cappello and Francolini, 2017; Gonzalez-Freire et al., 2014). Thus, when we eliminated the mouse homolog of one prominent RNA target of TDP43, STMN2, we found that it was in and of itself sufficient to induce motor neuropathy, a primary contributor to the spreading paralysis that occurs in ALS patients. Consequently, expression of the human STMN2 gene was sufficient to prevent motor dysfunction.
Our work may also shine light on why motor neurons and even certain classes of motor neurons are more sensitive to degeneration in ALS than others. The gastrocnemius, the hindlimb muscle where we observe substantial NMJ denervation, contains a high proportion of fast-twitch type II fibers (Nijssen et al., 2017), and is thus heavily innervated by large, fast-fatigable motor neurons, which are reported to be most sensitive to degeneration in ALS (Pun et al., 2006). We therefore found it interesting that Stmn2 expression within the ventral horn was not only selective to Chat+ motor neurons but that it was similarly enriched in a larger subpopulation of presumptive α (Spp1+) motor neurons. These findings raise the interesting possibility that the influence of TDP43 on STMN2 expression could be one contributor to the selective degeneration of motor neuron in ALS and even the increased sensitivity of certain motor neuron sub-types. In the future, more specific, quantitative analysis of the expression pattern of STMN2 and other STMN family members in more carefully delineated motor neuron subtypes could provide deeper insight into whether their expression contributes to the phenomenon of selective vulnerability.
Given that STMN2 is only one of many client RNAs whose regulation is controlled by TDP43, we found it striking that the elimination of its ortholog could be so consequential. Machine learning algorithms based on ALS diagnoses, indicate clinical development of ALS is a multistep process requiring on average six sequential steps for the disease to manifest, whether those are environmental or genetic (Al-Chalabi et al., 2014). Here, we note that there are a variety of events that occur in ALS patients that did not occur in Stmn2 mutant animals. These included the onset of neural inflammation and TDP43 pathology at the time points analyzed (Glass et al., 2010; Philips and Robberecht, 2011). We found the absence of these phenomenon instructive in this case as they may provide some clarity on where alterations in STMN2 may reside on the pathway to motor neuron degeneration in ALS (Suppl. Figure 7)
In motor neurons of a healthy individual, TDP43 is abundant within the nucleus, and TDP43 binding sites on the STMN2 pre-mRNA are occupied as it is transcribed, allowing a full length, protein-coding message to be produced (Arnold et al., 2013; Prasad et al., 2019). As a result, there is sufficient STMN2 to regulate microtubule dynamics in the motor axons to support homeostasis and repair (Suppl. Figure 7a). In contrast, in ALS, a variety of insults (Suppl. Fig 7b) can lead to TDP43 pathology; resulting in inadequate levels of nuclear TDP43 to prevent premature truncation of STMN2, leading to accumulation of a short cryptic transcript, which cannot produce the full-length protein (Arnold et al., 2013; Highley et al., 2014; Prasad et al., 2019). Without further production of STMN2 protein, levels fall and an inability to properly regulate the motor axon arises (Suppl. Figure 7b). Loss of axonal stability in our Stmn2 mutant animals is likely explained by the observed disruption in the dynamic process of microtubule polymerization in the lumbar spinal cord. Microtubules are essential for maintaining axonal integrity, providing a dynamic tool for adaptation to environmental stress and cues (van de Willige et al., 2016). Neuronal remodeling is supported by microtubule associated proteins such as Stmn2 in which its levels are tightly correlated to development, growth and injury (Ozon et al., 1998; Shin et al., 2014). Thus, loss of Stmn2 creates an imbalance, disrupting distal remodeling at the pre-synaptic NMJ and loss of proper innervation, leading to motor neuropathy. We also take note that alterations to additional TDP43 target RNAs would be expected to further damage motor neuron function (Klim et al., 2021).
Our findings support the notion that restoration of STMN2 expression in patients who exhibit TDP43 pathology may provide a meaningful therapeutic strategy to improve motor axon function. Furthermore, the Stmn2 mutant animals we report herein will be an invaluable model for pre-clinical evaluation of such approaches. The extent of denervation and severity of motor function decline we found in these animals should readily allow the evaluation of gene transfer approaches for restoring STMN2 expression and/or therapeutics targeting axonal function, such as modulation of JNK kinases (Geisler et al., 2016; Klim et al., 2019; Shin et al., 2012; Tian et al., 2020; Turkiew et al., 2017). Perhaps more notably, the “humanized” STMN2 mouse model described here could, in the future, allow for the in vivo assessment of agents designed to restore the normal splicing of STMN2 in the presence of disease-relevant perturbations altering TDP43 function. It is noted that development of a comparable “humanized” model was instrumental in the advancement of effective gene therapies for another motor neuron disease, spinal muscular atrophy (Corey, 2017; Foust et al., 2009; Kanning et al., 2010; Meyer et al., 2015).
STAR Methods
Resource Availability
Lead Contact:
Further information and requests for resources can be directed to by the lead contact, Kevin Eggan (kevin.eggan@bmrn.com)
Materials availability statement:
Mouse lines generated in this study have been deposited to The Jackson Laboratory, Stmn2 13 Kb Δ (+/−, −/−): JAX Stock No. 036628; BAC hSTMN2xmStmn2−/−: JAX Stock No. 036775
Data and Code Availability:
The data that support the findings of this study are available from the lead contact upon request. This paper does not report original code.
Experimental Model and Subject Details
Animals.
All experimental procedures were approved by the Institutional Animal Care and Use Committee of Harvard University and were in compliance with all relevant ethical regulations.
CRISPR Guide Design, validation and generation of Stmn2 LOF mutant mice.
Stmn2 gRNAs were designed using CHOPCHOP (https://chopchop.rc.fas.harvard.edu) from the Schier Laboratory. In order to synthesize gRNAs, the Sp6 RNA polymerase promoter sequence and the transcription initiation site were added to the 5’ end of the targeting sequence. Cutting efficiency was tested of selected gRNAs and part of trans-activating CRISPR RNA (tracrRNA) sequence was added to the 3’ end of the targeting sequence. The resulting 60 bp long oligonucleotide was submitted to IDT. Oligos were first annealed with a constant oligo containing the remaining tracrRNA by denaturing at 95 °C for 5 minutes and annealing from 95 °C-85 °C at a rate of −2 °C/second, and from 85 °C-25 °C at a rate of −0.1 °C. Annealed oligos were PCR amplified to fill in 3’overhangs with AmpliTaq Gold 360 Master Mix Buffer (Life tech # 4398876) and Taq DNA polymerase (Life Tech # 10342053), by incubating at 72°C for 15 min and an additional 45 minutes after adding polymerase. PCR products were purified using Promega PCR clean-up kit (Promega #A9281). Purified dsDNA templates were in vitro transcribed using the MEGAscript kit (Life tech #AM1330) containing the Sp6 RNA polymerase and purified with the E.Z.N.A. PF Micro RNA Kit (Omega # R7036–01). Cutting efficiency was assessed of the chosen sgRNAs. Genomic DNA was extracted from Rosa26 and Stmn2 mouse tails using the DirectPCR lysis kit (Viagen Biotech Inc #102-T). The two Stmn2 target regions in Exon 2 and Exon 4 were amplified using the genotyping primers listed in the STAR Methods table using Phusion DNA Polymerase (Thermo Fisher Scientific #F549L) according to manufacturer’s protocol. Exon 2 and Exon 4 amplicons were gel purified and used as a template for the Cas9 cutting assay. In vitro transcribed sgRNA and purified gDNA were incubated with increasing concentrations of Cas9 protein (NEB #M0386T) and incubated at 37°C for 30 minutes. The cutting reaction was halted with stop reaction containing 30% Glycerol, 0.5 M Ethylenediaminetetraacetic Acid (EDTA), 2% Sodium Dodecyl Sulfate (SDS) for 10 mins at 80°C. Products were run on a 1% Agarose gel to visualize bands. Cutting efficiency was determined by measuring the relative decrease in full-length genomic DNA band intensity with increasing Cas9 concentrations compared to control. gRNAs with a cutting efficiency higher than 98% were selected for generating CRISPR mutant mice. The selected targeting sequences for submission to Synthego and to be used for zygote injections were as follows: 5’ CGCAACATCAACATCTAC 3’ (Exon 2) and 5’ AGCGAGAGGTGCTCCAGA 3’ (Exon 4). The purified guide RNAs and Cas9 mRNA were injected into C57BL/6 mouse blastocysts at the Harvard Genome Modification Facility using a standard protocol described previously (Yang et al., 2014). TIDE sequence software was utilized for deconvoluting the Sanger sequencing of the gRNA target sites and assessing the presence of the indels (Figure 1f) (Brinkman et al., 2014). Geneious software was used to determine the changes in the ORF (Figure 1e). The designed gRNAs were examined for their ability to bind to the other Stathmin-family genes using a sequence BLAST (https://blast.ncbi.nlm.nih.gov/) in which only guide 2 (targeting Exon 4) exhibited a 91% homology to Stmn4, but lacked the necessary NGG PAM site. For genotyping of the 13 Kb deletion F2 cohort, a three-primer genotyping strategy was employed, amplifying both mutant and wild-type alleles using the AmpliTaq Gold 360 Master Mix Buffer (Life tech # 4398876) and following the manufacturer’s protocol.
Generation of human STMN2 transgenic mice
Transgenic animals were produced using a BAC Clone ID# 793O4 from the RPCI – 11 human male BAC library, which contains a 174.1 Kb genomic fragment with the human STMN2 gene flanked by 66.8 Kb upstream and 70.0 Kb downstream. BAC DNA was injected into C57BL/6 zygotes at the Harvard Genome Facility and the presence of STMN2 was confirmed within the BAC by designing primers targeting the beginning, middle, and end of the gene. The regions in Intron 1, Exon 3 and Exon 5 were amplified using the genotyping primers listed in the STAR Methods table using Phusion DNA Polymerase (Thermo Fisher Scientific #F549L) according to manufacturer’s protocol. The BAC hSTMN2 founders containing the STMN2 transgene, as identified by PCR, were then crossed with WT C57BL/6 to expand the line. BAChSTMN2 mice were then bred into the LOF mutant heterozygous background (Stmn2+/− 13KbΔ) to generate hSTMN2xmStmn2+/−offspring. To generate the rescue model, the offspring were then back-crossed to produce mice completely lacking murine Stmn2, while also expressing a human STMN2 (hSTMN2xmStmn2−/−). Mice were identified by PCR amplification of the human locus to determine presence of the hSTMN2 gene and the 13 Kb deletion.
Cell culture and differentiation of hESCs into motor neurons.
Human embryonic stem cells were grown with mTeSR1 medium (Stem Cell Technologies) and maintained in 5% CO2 incubators at 37 °C. 10 μM ROCK inhibitor (Sigma, Y-27632) was added to the cultures for 24 h after dissociation to prevent cell death. hESCs were co-infected with TetO-Ngn2-Puro and reverse tetracycline-controlled transactivator (rtTA), and were plated at a density of 100,000 cells/cm2 with rock inhibitor Y27632 (Stemgent 04–0012) on Matrigel-coated culture plates. Motor neuron differentiation was achieved using a modified strategy from the previously reported NGN2-driven reprogramming protocol coupled with activation of posteriorizing and ventralizing signaling pathways for seven days (Limone et al., 2022; Zhang et al., 2013). Assays were carried out on d21 of the differentiation.
Methods Details
Behavioral Analysis.
Rotarod.
Animals were trained on rotarod at 5 RPM for 300 seconds then at 5–10 RPM for 300 seconds, one day prior to testing. The day of testing consisted of animals placed on a rotarod which accelerated from 5 to 40 RPM over 300 s. Each mouse was tested three times with each trial separated by a minimum of 20 minutes.
Hanging Wire.
Mice were placed suspended on a standard linear wire and a timer was set for a maximum of 120 seconds. The timer is stopped when the mouse falls off the wire or climbs and reaches the end of the wire. If the mouse climbs to the end of the wire it is re-positioned to the center and the timer continues. Three trials were completed per mouse with at least 20 minutes separating the trials. The average of the trials was then normalized by the ratio of the weight of the mouse relative to the average weight of the gendered cohort. Operator was blinded to genotype for all motor behavioral tests involving the F2 generation of mice.
Tissue Harvesting.
Mice were anesthetized with isoflurane and perfused using 25 mL of PBS. Tissues were harvested and fixed in 4 % PFA for immunohistochemistry, RIPA buffer (Life Technologies, 89900) for protein analysis, or RLT buffer (Qiagen 1053393) for RNA extraction. Animals were randomized and tissues were assessed for weights, protein, and RNA while blinded.
Immunocytochemistry.
Muscle.
Neuromuscular Junction. Gastrocnemius (GA) muscle was fixed with cold 4% PFA overnight at 4°C. Tissues were then removed of fat and blood, and stripped into individual fibers for TA and GA, while the diaphragm was cut in half. Muscles were placed in 0.1M glycine for 1 h. Samples were then permeabilized and blocked overnight at 4°C in 0.5% Triton-X in × 1 PBS, containing 5 % donkey serum (DS) and 3 % BSA. Muscles were then placed in primary antibody for synaptophysin 1:500 (Cell Signaling Technologies 9020 RRID: AB_2631095) at 4°C for 2–3 nights in PBS containing 3% BSA and 5 % DS. Samples are then washed 3x in PBS and incubated with secondary overnight at 1:1000 (AlexaFluor 488, Life Technologies) and α-Bungarotoxin Fluor 555 at 1:500 (Invitrogen B35451 RRID: AB_2617152). Samples are washed three times in PBS, mounted using Aqua-Poly Mount (Polysciences 18608) and visualized using the LSM 880 confocal microscope at the Harvard Center for Biological Imaging Core (RRID SCR_018673) with either a 10x or 20x objective. All comparative stains between control and mutant mice were acquired using identical laser and microscope settings, and images were processed with viewer blinded to genotype. Scoring of the NMJ features were analyzed blinded.
Cross-section Analysis.
Tissues were dissected and flash frozen by covering the muscle in OCT (StemCell Technologies) and placing it in 2-methylbutane (VWR 70000–210) cooled in liquid nitrogen. 15 μM thick muscle cross-sections were stained with H&E and approximately 200 fibers were analyzed per mouse. Scoring of the cross-sectional features were analyzed blinded.
Spinal Cord and Brain.
Spinal cords and brains were dissected and placed into cold 4% PFA for 3 nights at 4°C, washed with PBS then placed in 30 % sucrose for 2 nights at least. Lumbar spinal cords and brains are both placed in OCT and cut at 30 μM (Spinal Cord) and 35 μM (Brain) per slice. Samples are then washed with PBS 3x to remove residual OCT, blocked and permeabilized with 0.3% Triton-X in 1x PBS containing 10% Donkey Serum (DS), 0.1M glycine and Image-IT FX Signal Enhancer (Invitrogen) for 1 h at room temperature. Primary antibodies which include rabbit STMN2 (1:4,000, Novus NBP49461 RRID: AB_10011569 or 1:300 Abcam ab185956 RRID: AB_2773045), rat GFAP (1:300, Life Technologies 130300 RRID: AB_2532994), goat ChAT (1:300, Sigma Aldrich AB144P RRID: AB_2079751), guinea pig Iba1 (1:300, Synaptic Systems #234004 RRID: AB_2493179), goat Osteopontin/OPN (Spp1) (1:100, R&D Systems #AF808 RRID:AB_2194992) are added for 2 nights in PBS containing 1% DS, washed 3x using 0.05% TritonX o in PBS. Secondary antibody is added at 1:500 (AlexaFluor 488, 555, 594, and 647, Life Technologies) for 2 h at room temperature with DAPI stain for nuclei, and Nissl stain (Neurotrace 640/660 Invitrogen #N21483) for neuronal cell bodies. Samples are washed 3x, mounted using ProLong Glass Antifade Mountant (Life Technologies 36980), then visualized using the LSM 880 confocal microscope with FLIM at 10x, 20x and 40x-Oil objective, or an Axio Scan.Z1 at 20x objective at the Harvard Center for Biological Imaging Core (RRID SCR_018673). All comparative stains between control and mutant mice were acquired using identical laser and microscope settings, and images were processed with viewer blinded to genotype. Image processing was carried out on Fiji and subsequent quantifications for GFAP/Iba1% of cells and Nissl neuron counts in layer V were performed using CellProfiler. Layer V area was selected with the polygon selection tool in Fiji. The “Identify Primary Objects” module was used to select DAPI positive nuclei that were in between 10–40 pixel units in diameter in Spinal Cord (20x Objective), and 5–10 pixel units in the Brain (10x Objective). The “Measure Object Intensity” module was then used for measuring GFAP, Iba1 and Nissl signal intensity in DAPI positive cells. A threshold was applied for counting GFAP, Iba1 and Nissl cells based on their respective signal intensity distribution. Cells with ≥ twice the average intensity/section were considered GFAP or Nissl positive and those with ≥ four times the average intensity/section were considered Iba1 positive. The number of positive GFAP/Iba1 cells were divided by the total number of DAPI positive cells. For each mouse, 5–6 brain or lumbar spinal cord sections were quantified.
Microtubules/Tubulin assay:
To examine levels of free and polymerized tubulin, a microtubules/tubulin in vivo kit was used from Cytoskeleton Inc (BK038). In brief, tissue samples or cells are lysed using supplied microtubule stabilization lysis buffer (0.03 g of lumbar spinal cord tissue in 300 μL of Microtubule Stabilization buffer and 10cm dishes containing 7.5×106 Ngn2-derived motor neurons in 600 μL). Supernatant and cell pellet (tissue/cell debris) are separated via low-speed centrifugation for 5 minutes at 1,000 x g at 37°C. A small part of the supernatant (100 μL) is then centrifuged again at high speed (100,000 x g) for 60 minutes at 37°C to separate the free and polymerized tubulin while the rest of the supernatant is kept as input to quantify total tubulin levels. After the high-speed centrifugation step, the top layer of the supernatant is separated and mixed with SDS buffer (High Speed Supernatant, HSS). The pellet is then resuspended in a depolymerization buffer and incubated for 15 minutes at room temperature to help with solubilization and mixed with SDS buffer (HSP, High Speed Pellet). Tubulin quantification proceeds by SDS-PAGE and western blot analysis. Experimental controls were carried out by incubating STMN2+/+ neurons with Paclitaxel/Taxol (2mM stock diluted in DMSO provided in BK308 Kit) at 1μM for 72 hours and Nocodazole (Sigma Aldrich #M1404) at 10μM for 45 minutes. For tubulin protein quantification, tubulin α/β and β-III tubulin were probed with antibodies against Tubulin provided in BK308 Kit (1/1,1000 Cytoskeleton Inc #ATN02, RRID: AB_10708807) and β-III tubulin (1/1,000, R&D Systems MAB1195, RRID: AB 356859) respectively.
Immunoblot assays.
For examination of Stmn2 protein, brain samples were homogenized and lysed in RIPA buffer containing Halt protease and phosphatase inhibitors (Life Technologies 78441) and centrifuged at 12,000 RPM for 10 minutes at 4°C. Protein concentration was determined by a BCA assay (Thermo Scientific 23225) and 10–20 μg of total protein were separated by SDS-PAGE using a 4–20% gradient (Bio-Rad 4561094), transferred to polyvinylidene difluoride membranes (EMD Millipore IPFL00010) and probed with antibodies against GAPDH (1:2,000, EMD Millipore MAB374 RRID: AB_2107445) and STMN2 (1:2,000, Abcam EPR15286–39 RRID: AB_2773045; 1:2000 AbCam ab115513 RRID: AB_10900514). STMN2 levels were normalized to GAPDH. LiCor software (Image Studio) was used to visualize and quantitate protein signals. All immunoblots were analyzed from at least two technical replicates per mouse. Immunoblots for STMN2 levels were completed while blinded.
RNA Isolation and qRT-PCR.
RNA was isolated from brain homogenates using Qiagen RNeasy Plus Micro kit (Qiagen 74034) or Trizol (Invitrogen), in accordance with manufacturer׳s recommendations and quantified spectrophotometrically (at 260 nm). Approximately 300–500 ng of total RNA was used to synthesize cDNA using the iScript kit reverse transcriptase (Bio-Rad 1708891). The cDNA was then amplified using the SYBR Premix (iScript Advanced cDNA Synthesis Kit) or using PrimeTime™ Gene Expression Master Mix (IDT) using either CFX96 or CFX384 Touch Real-Time PCR Detection System StepOnePlus (Bio-Rad). Briefly, each 20 μl of reaction volume contained 5 μL of SYBR Green PCR Master Mix or 5 μL PrimeTime™ Gene Expression Master Mix, 0.5 μM of each primer, and UltraPure destilated water DNase and RNase free (Invitrogen). Genes were normalized to GAPDH (SYBR) or R18S (PrimeTime) expression and expressed relative to their respective control condition. All genes were tested for in technical triplicates per mouse. Analysis of mRNA levels for STMN2 were completed while blinded.
Quantification and Statistical Analysis
In figures, bars and lines represent the mean and standard deviation. Statistical calculations were performed using GraphPad prism 8.0. Equal variances were tested using Brown-Forsythe test with a p value < 0.05 as considered significant. When assumptions of normality or homogeneity of variances were met, the following parametric tests were used: Student’s t test, One-Way or Two-Way ANOVAs with a p value of < 0.05 considered as significant. Mann-Whitney U test was used for non-parametric data comparing two independent groups. For comparisons between multiple groups, normally distributed data utilized a Tukey or Dunnnet’s post-HOC test on One-Way ANOVA, while non-parametric data utilized Kruskal-Wallis’ multiple comparison test with Dunn’s correction. Tests between multiple groups over time used Two-Way ANOVA with Sidak’s multiple comparison test. Detailed information per dataset (average, SD, n and detailed statistics) is shown in table below.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Synaptophysin | Cell Signaling Technologies | Cat #9020S RRID: AB_2631095 |
| Rabbit polyclonal anti-SCG10/STMN2 | Novus Biologicals | Cat #NBP49461 RRID: AB_10011569 |
| Rabbit monoclonal anti-SCG10/STMN2 | Abcam | Cat # ab185956 RRID: AB_2773045 |
| Goat polyclonal anti-SCG10/STMN2 | Abcam | Cat# ab115513 RRID: AB_10900514 |
| Rat monoclonal anti-GFAP | Life Technologies | Cat # 130300 RRID: AB_2532994 |
| Goat polyclonal anti-ChAT | Sigma Aldrich | Cat # AB144P RRID: AB_2079751 |
| Guinea pig polyclonal anti-Iba1 | Synaptic Systems | Cat # 234004 RRID: AB_2493179 |
| Rabbit monoclonal anti-TDP-43 (G400) | Cell Signaling Technologies | Cat # #3448S RRID: AB_2271509 |
| Mouse monoclonal anti-GAPDH | EMD Millipore | Cat # MAB374 RRID: AB_2107445 |
| Mouse monoclonal anti-beta III tubulin | R & D Systems | Cat # MAB1195 RRID:AB_356859 |
| Sheep polyclonal anti-alpha/beta tubulin | Cytoskeleton Inc. | Cat # ATN02 RRID: AB_10708807 |
| Goat polyclonal anti-mouse Osteopontin | R & D Systems | Cat # AF808-SP RRID:AB_2194992 |
| Bacterial and Virus Strains | ||
| FUW-TetO-Ngn2- Puro |
Wernig Lab Zhang et al., 2013 |
|
| FUW-rtTA | Baltimore Lab Lois et al., 2002 |
|
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nocodazole ≥99% (TLC), powder | Milipore Sigma | #M1404 |
| Placitaxel 2mM | Cytoskeleton Inc. | #TXD01 |
| Critical Commercial Assays | ||
| MEGAscript SP6 Transcription Kit | Life Technologies | #AM1330 |
| E.Z.N.A. PF Micro RNA Kit | Omega | #R7036–01 |
| Promega PCR clean-up kit | Promega | #A9282 |
| DirectPCR lysis kit | Viagen Biotech | #102-T |
| RNeasy Plus Micro kit | QIAGEN | #74034 |
| Microtubules/Tubulin In vivo Assay Kit | Cytoskeleton Inc | #BK038 |
| PrimeTime Gene Expression Master Mix | IDT | #1055772 |
| Deposited Data | ||
| Raw and analyzed data | This paper | N/A |
| Experimental Models: Cell Lines | ||
| STMN2+/+,−/− WA01 hESC line |
Klim et al., 2019 Eggan Lab |
hESC cell line: WA01 |
| Experimental Models: Organisms/Strains | ||
| Mouse: F0 Stmn2 mutant C57BL/6 | This paper | N/A |
| Mouse: F0 Rosa26 mutant C57BL/6 | This paper | N/A |
| Mouse: F1 Stmn2 +/− C57BL/6 | This paper | N/A |
| Mouse: F2 Stmn2+/+, +/−, −/− 13 Kb Δ C57BL/6 | This paper |
JAX (Stock No. 036628) |
| Mouse: BAC hSTMN2 C57BL/6 | This paper | N/A |
| Mouse: hSTMN2xmStmn2+/+, +/−, −/− 13 Kb Δ C57BL/6 | This paper | JAX (Stock No. 036775) |
| Oligonucleotides | ||
| gRNA targeting sequences: 5’ CGCAACATCAACATCTAC 3’ (Exon 2) 5’ AGCGAGAGGTGCTCCAGA 3’ (Exon 4) |
This paper CHOPCHOP (Ref. 44) |
(https://chopchop.rc.fas.harvard.edu) |
| Exon 2 cut site F1: 5’ cctgatagctctgtgactatc 3’ R1: 5’ gcaagaggattgcaagttcaag 3’ Sequencing: 5’ ttaacctatgcagttcctgtcc 3’ |
IDT This paper |
N/A |
| Exon 4 cut site F2: 5’ cctgatagctctgtgactatc 3’ R2: 5’ gcaagaggattgcaagttcaag 3’ Sequencing: 5’ ttaacctatgcagttcctgtcc 3’ |
IDT This paper |
N/A |
| 5’ Mouse Stmn2 transcript qPCR primers 5’ GCAATGGCCTACAAGGAAAA 3’ (Forward) 5’ GAGCTGATCTTGAAGCCACC 3’ (Reverse) |
IDT This paper |
N/A |
| 3’ Mouse Stmn2 transcript qPCR primers 5’ AGAAGCTGATCCTGAAGATGG 3’ (Forward) 5’ TTCGCAGGAACAAGGAACT 3’ (Reverse) |
IDT This paper |
N/A |
| Mouse Gapdh transcript qPCR primers 5’ TGCGACTTCAACAGCAACTC3’ (Forward) 5’ GCCTCTCTTGCTCAGTGTCC3’ (Reverse) |
IDT This paper |
N/A |
| 13Kb Δ deletion screening primers Fcommon: 5’ cattggaaaaccaagccaag 3’ Rwt: 5’ attgacgtgctggtgaggat 3’ Rko: 5’ tttcctgcagacgttcaatg 3’ -Higher band resolution: Rwt ALT: 5’- caaacagcgatggtggtaga-3’ Rko ALT:5’- aagctctgctgcacaggaat-3’ |
IDT This paper |
N/A |
| Human genotyping primers Intron 1, forward 5’-ATTGATCTCCTTGTAGTGG-3’ reverse 5’-TGAGAGACCCTGAAATGAACTG-3’ exon 3, forward 5’-GAAGAAAGACCTGTCCCTGGAG-3’ reverse 5’-GCAGGAAAGATCTTGGAGGGA-3’. exon 5, forward 5’-AAACGTGTACTGATGCAGGTC-3’, reverse 5’-GGGGGATTTACTATTGGTGGGG-3 |
IDT This paper |
N/A |
| Primetime qPCR primers R18S Probe 5’-/ 5Cy5 /TGCTCAA TCTCGGGTGGCTGAA/31AbR QSp/−3’ forward 5’-GAGACTCTGGCATGCT AACTAG-3’ reverse 5’-GGACATCTAAGGGCATCACAG-3’ Human STMN2 1–3 Probe 5’-/5HEX/ AGCTGTCCA/ZEN/TGCTGTCACTGATCTG/31ABkFQ/−3’ 5’-CGTCTGCACATCCCT ACAATG-3’ 5’-TGCTTCACTTCCAT ATCATCGT-3’ |
IDT This paper |
N/A |
| Recombinant DNA | ||
| Software and Algorithms | ||
| ImageJ-FIJI | NIH | https://imagej.nih.gov/ij/, Fiji, RRID: SCR_002285 |
| GraphPad Prism | GraphPad software |
https://www.graphpad.com; GraphPad Prism, RRID: SCR_002798 |
| CellProfiler | CellProfiler Image Analysis Software |
http://cellprofiler.org
RRID:SCR_007358 |
| Image Studio Lite Software | Image Studio Lite Acquisition Software LI-COR Biosciences |
https://www.licor.com/bio/image-studio/ RRID:SCR_013715 |
| Geneious | Geneious Software |
http://www.geneious.com/ RRID:SCR_010519 |
| BioRender | BioRender |
http://biorender.com RRID:SCR_018361 |
KEY RESOURCES TABLE Related to quantification and statistical analysis
| Dataset | Condition | Value (Mean ± SD) |
n | p value | Statistical test |
|---|---|---|---|---|---|
| Relative Stmn2 brain transcript expression F0 cohort 5’ and 3’ transcripts (Suppl Figure 1f) |
(1) F0
Rosa26
F0 Stmn2 (2) #14 (3) #15 (4) #16 (5) #17 |
1.29 ± 0.26 (5’) 1.38 ± 0.31 (3’) 0.57 ± 0.13 (5’) 0.57 ± 0.06 (3’) 0.02 ± 0.0 (5’) 0.017 ± 0.0 (3’) 0.03 ± 0.0 (5’) 0.47 ± 0.6 (3’) 0.0002 ± 0.0 (5’) 6.675e-006 ± 0.0 (3’) |
5–4 | * p < 0.05: 1 vs 2 (5’ and 3’) ** p < 0.01: 1 vs 4 (5’ and 3’) ***p < 0.001: 1 vs 3 (5’) ****p < 0.0001:1 vs 3 (3’), 5 (5’ and 3’) |
Kruskal-Wallis with Dunn’s correction |
| Relative Stmn2 brain transcript expression F2 cohort 5’ and 3’ transcripts (Figure 1d) |
F2
Stmn2+/+ F2 Stmn2−/− (1,2,3) |
1.01 ± 0.23 (5’) 0.97 ± 0.22 (3’) 0.004 ± 0.00 (5’) (1) 0.003 ± 0.00 (3’) (1) 0.0005 ± 0.00 (5’) (2) 0.02 ± 0.00 (3’) (2) 0.0007 ± 0.00 (5’) (3) 0.02 ± 0.00 (3’) (3) |
3 | ****p < 0.0001:F2
Stmn2+/+ vs +/+ (1,2,3) (5’ and 3’) |
One-Way ANOVA (Dunnett’s post-HOC) |
| Motor performance F0 cohort Rotarod: Average latency to fall (sec) Hanging wire: Normalized latency to fall (sec*(weight/avg weight)) (Figure 2c,d) |
F0
Rosa26 F0 Stmn2 |
Rotarod 195.1 ± 62.19 (P60) 225.1 ± 37.17 (P100) Hanging wire 69.26 ± 28.58 (P60) 67.40 ± 26.27 (P100) Rotarod 92.18 ± 44.53 (P60) 83. 77 ± 34.96 (P100) Hanging wire 20.66 ± 12.31 (P60) 29.26 ± 16.15 (P100) |
P60 F0 Rosa26:15 F0 Stmn2:13 P100 F0 Rosa26:29 F0 Stmn2:10 |
****p < 0.0001:F0 Rosa26 vs F0
Stmn2: (P60 and P100, Rotarod and Hanging Wire) n.s., p > 0.05: F0 Stmn2 P60 vs P100 (Rotarod and Hanging wire) |
Two-Way ANOVA (Sidak’s multiple comparison) |
| Motor performance analysis F2 cohort Rotarod: Average latency to fall (sec) Hanging wire: Normalized latency to fall (sec*(weight/avg weight)) (Figure 2f,g) |
F2
Stmn2+/+ F2 Stmn2+/− F2 Stmn2−/− |
Rotarod P60 235 ± 50.93 (+/+) 229.1 ± 51.60 (+/−) 137.0 ± 51.60 (−/−) P100 237.1 ± 52.14(+/+) 216.7 ± 50.03 (+/−) 116.7 ± 62.36 (−/−) Hanging wire P60 108.3 ± 18.75 (+/+) 97.09 ± 28.82 (+/−) 42.33 ± 21.70 (−/−) P100 100. 4 ± 28.44 (+/+) 89.10 ± 51.82 (+/−) 51.82 ± 23.86 (−/−) |
42–31 (+/+) 45–38 (+/−) 10–8 (−/−) |
n.s., p > 0.05: F2 Stmn2−/− P60 vs P100 (Rotarod and Hanging wire) and F2 Stmn2+/+ vs +/− (P60 and P100, Rotarod and Hanging Wire) ****p < 0.0001: F2 Stmn2+/+ vs −/− (P60 and P100, Rotarod and Hanging Wire) |
Two-Way ANOVA (Sidak’s multiple comparison) |
| Relative Stmn1,3,4 brain transcript expression F0 cohort 5’ and 3’ transcripts (Suppl Figure 2a) |
(1) F0
Rosa26 F0 Stmn2 (2) #14 (3) #15 (4) #16 (5) #17 |
1.46 ± 0.31 (Stmn1), 1.41 ±0.41 (Stmn3), 1.38 ± 0.28 (Stmn4) 1.75 ± 0.06 (Stmn1) 1.84 ± 0.14 (Stmn3) 1.33 ± 0.04 (Stmn4) 1.37 ± 0.07 (Stmn1) 1.19 ± 0.04 (Stmn3) 1.47 ± 0.06 (Stmn4) 1.17 ± 0.22 (Stmn1) 1.50 ± 0.14 (Stmn3) 1.34 ± 0.05 (Stmn4) 1.31 ± 0.22 (Stmn1) 1.15 ± 0.05 (Stmn3) 1.06 ± 0.03 (Stmn4) |
5–4 | n.s., p > 0.05: 1 vs 2, 3, 4, 5 (Stmn1,3,4) |
Kruskal-Wallis with Dunn’s correction |
| Number of Nissl+ neurons/mm2 in layer V (Suppl Figure 2d) |
F2
Stmn2+/+ F2 Stmn2−/− (1,2,3) |
1275 ± 155 1294 ± 141 1209 ± 172 1327 ± 68 |
3 (5–6 sections/mouse) | n.s., p > 0.05: Stmn2+/+ vs 1, 2, 3 |
One-Way ANOVA (Dunnett’s post-HOC) |
|
Gastrocnemius NMJ innervation % of fully coincident BTX:SyPhy/FOV (% fully innervated) (%FI) % of partially coincident BTX:SyPhy/FOV (% partially innervated) (%PI) % of BTX only/FOV (% non-innervated) (%NI) (Figure 3d) |
F2
Stmn2+/+ F2 Stmn2−/− |
P21 %FI: 88.25 ± 10.95 %PI: 6.98 ± 7.59 %NI: 1.29 ± 3.108 P120 %FI: 86.64 ± 8.74 %PI: 11.83 ± 7.11 %NI: 1.52 ± 4.53 P21 %FI: 90.21 ± 8.47 %PI: 9.22 ± 6.82 %NI: 0.98 ± 2.57 P120 %FI: 52.21 ± 15.61 %PI: 22.09 ± 7.55 %NI: 25.70 ± 14.87 |
P21: 5–4 P120: 3 (5–8 FOV/mouse) |
%FI; n.s. p > 0.05: p21:Stmn2+/+ vs. p21:Stmn2−/−, p21:Stmn2+/+ vs. p120:Stmn2+/+ ****p < 0.0001: p120:Stmn2+/+ vs. p120:Stmn2−/−, p21:Stmn2−/− vs. p120:Stmn2−/− %PI; n.s. p > 0.05: p21:Stmn2+/+ vs. p21:Stmn2−/−, p21:Stmn2+/+ vs. p120:Stmn2+/+ ** p < 0.01: p120:Stmn2+/+ vs. p120:Stmn2−/− ****p < 0.0001: p21:Stmn2−/− vs. p120:Stmn2−/− %NI; n.s. p > 0.05: p21:Stmn2+/+ vs. p21:Stmn2−/−, p21:Stmn2+/+ vs. p120:Stmn2+/+ ** p < 0.01: ****p < 0.0001: p120:Stmn2+/+ vs. p120:Stmn2−/−, p21:Stmn2−/− vs. p120:Stmn2−/− |
Two-Way ANOVA (Sidak’s multiple comparison) |
|
Gastrocnemius NMJ innervation % of fully coincident BTX:SyPhy/FOV (% fully innervated) (%FI) % of partially coincident BTX:SyPhy/FOV (% partially innervated) (%PI) % of BTX only/FOV (% non-innervated) (%NI) (Suppl Figure 3e) |
F0
Rosa26 F0 Stmn2 |
%FI: 86.64 ± 8.74 %PI: 11.83 ± 7.11 %NI: 1.52 ± 4.53 %FI: 52.21 ± 15.61 %PI: 22.09 ± 7.55 %NI: 25.70 ± 14.87 |
4 (5–8 FOV/mouse) | ****p < 0.0001: F0 Rosa26 vs F0 Stmn2 (FI, PI, NI) | Mann-Whitney U test (PI,NI) Student’s t test (FI) |
|
Gastrocnemius NMJ fragmentation %fragmented NMJs/FOV (Figure 4d) |
F2
Stmn2+/+ F2 Stmn2−/− |
P21:0.08 ± 0.41 P120:0.51 ± 1.74 P21:0.14 ± 0.44 P120:6.19 ± 8.54 |
P21: 5–4 P120: 3 (5–8 FOV/mouse) |
n.s. p > 0.05: p21:Stmn2+/+ vs. p21:Stmn2−/−, p21:Stmn2+/+ vs. p120:Stmn2+/+ ***p < 0.001: p21:Stmn2−/− vs. p120:Stmn2−/− ****p < 0.0001: p120:Stmn2+/+ vs. p120:Stmn2−/− |
Two-Way ANOVA (Sidak’s multiple comparison) |
|
Gastrocnemius NMJ fragmentation %fragmented NMJs/FOV (Suppl Figure 4d) |
F0
Rosa26 F0 Stmn2 |
1.12 ± 1.75 32.62 ± 21.58 |
4 (5–8 FOV/mouse) | ****p < 0.0001: F0 Rosa26 vs F0 Stmn2 | Mann-Whitney U test |
|
Gastrocnemius muscle fiber nucleus localization %internalized nuclei/FOV (Figure 4d) |
F2
Stmn2+/+ F2 Stmn2−/− |
P21:3.75 ± 2.38 P120:1.12 ± 1.25 P21:5.500 ± 3.22 P120:6.375 ± 4.91 |
P21: 5–4 P120: 3 (5–8 FOV/mouse) |
n.s. p > 0.05: p21:Stmn2+/+ vs. p21:Stmn2−/−, ****p < 0.0001: p120:Stmn2+/+ vs. p120:Stmn2−/− |
Mann-Whitney U test (P120) Student’s t test (P21) |
|
Gastrocnemius muscle fiber nucleus localization %internalized nuclei/FOV (Suppl Figure 4d) |
F0
Rosa26 F0 Stmn2 |
0.25 ± 0.57 9.82 ± 8.03 |
4 (5–8 FOV/mouse) | ****p < 0.0001: p120: F0 Rosa26 vs F0 Stmn2 | Mann-Whitney U test |
| Number of Chat+, Stmn2+, %Chat+/Stmn2+ motor neurons in lumbar spinal cord (Figure 5d-f) |
F0
Rosa26 F0 Stmn2 |
13.36 ± 3.60 (Chat+) 11.96 ± 3.58 (Stmn2+) 92.36 ± 1.10 (%Chat+/Stmn2+) 12.81 ± 2.75 (Chat+) 3.19 ± 1.32 (Stmn2+) 25.06 ± 3.13 (%Chat+/Stmn2+) |
3 (6–9 hemisections/mouse) | n.s. p > 0.05: Chat+, F0 Rosa26 vs F0
Stmn2 ****p < 0.0001: Stmn2+, %Chat+/Stmn2+, F0 Rosa26 vs F0 Stmn2 |
Mann-Whitney U test (Stmn2+) Student’s t test (Chat+, %Chat+/Stmn2+) |
| Number of Chat+, Stmn2+, %Chat+/Stmn2+ motor neurons in lumbar spinal cord (Figure 5h-j) |
F2
Stmn2+/+ F2 Stmn2−/− |
13.36 ± 3.60 (Chat+) 11.96 ± 3.58 (Stmn2+) 97.30 ± 5.25(%Chat+/Stmn2+) 12.81 ± 2.75 (Chat+) 0.00 ± 0.00 (Stmn2+) 0.00 ± 0.00 (%Chat+/Stmn2+) |
3 (5–10 hemisections/mouse) | n.s. p > 0.05: Chat+, F2
Stmn2+/+ vs +/+ ****p < 0.0001: Stmn2+, %Chat+/Stmn2+, F2 Stmn2+/+ vs +/+ |
Mann-Whitney U test (Stmn2+) Student’s t test (Chat+, %Chat+/Stmn2+) |
| Glial cell counts, %GFAP+ and %Iba1+ cells/section, in lumbar spinal cord (Figure 6 g) | F0
Rosa26 F0 Stmn2 |
19.24 ± 1.25 (%GFAP+) 3.71 ± 0.49 (%Iba1+) 18.98 ± 0.88 (%GFAP+) 3.59 ± 0.39 (%Iba1+) |
3 (5 sections/mouse) | n.s. p > 0.05: F0 Rosa26 vs F0 Stmn2 (%GFAP+, %Iba1) | Student’s t test |
| TDP43 nuclear localization in Chat+ motor neurons of lumbar spinal cord,TDP-43/DAPI correlation coefficient (Figure 6 g) | F0
Rosa26 F0 Stmn2 |
0.89 ± 0.01 0.90 ± 0.00 |
3 (5 sections/mouse, 100 Chat+ neurons) | n.s. p > 0.05: F0 Rosa26 vs F0 Stmn2 | Student’s t test |
| Glial cell counts, %GFAP+ cells/section, in lumbar spinal cord (Suppl Figure 6 e) | F2
Stmn2+/+ F2 Stmn2−/− |
18.18± 1.53 17.69± 0.48 |
2 (5 sections/mouse) | n.s. p > 0.05: F2
Stmn2+/+ vs +/+ |
Student’s t test |
| TDP43 nuclear localization in Chat+ motor neurons of lumbar spinal cord,TDP-43/DAPI correlation coefficient (Figure 6 g) | F2
Stmn2+/+ F2 Stmn2−/− |
0.77 ± 0.12 0.78 ± 0.12 |
3 (5 sections/mouse, 100 Chat+ neurons) | n.s. p > 0.05: F2
Stmn2+/+ vs +/+ |
Student’s t test |
| Microtubules/Tubulin ratio in spinal cord Relative polymerized:free (α/β tubulin, β-III tubulin), total β-III tubulin levels (Figure 7 d,f) |
F2
Stmn2+/+ F2 Stmn2−/− |
1.10 ± 0.15 (P:F α/β) 1.07 ± 0.12 (P:F βIII) 1.00 ± 0.04 (Total βIII) 0.76 ± 0.22 (P:F α/β) 0.38 ± 0.04 (P:F βIII) 1.00 ± 0.10 (Total βIII) |
3 animals (3 technical replicates) | n.s. p > 0.05: P:F α/β and Total βIII, Stmn2+/+ vs. Stmn2−/−
***p < 0.001: P:F βIII, Stmn2+/+ vs. Stmn2−/− |
Student’s t test |
| Microtubules/Tubulin ratio in hMNs Relative polymerized:free (α/β tubulin), total β-III tubulin levels (Figure 7 h,j) |
(1) STMN2+/+ (2) STMN2−/− (3) STMN2−/− + Taxol (4) STMN2−/− + Nocodazole |
1.00 ± 0.23 (P:F α/β) 1.00 ± 0.07 (Total βIII) 0.33 ± 0.04 (P:F α/β) 1.08 ± 0.12 (Total βIII) 2.72 ± 0.39 (P:F α/β) 0.95 ± 0.22 (Total βIII) 0.29 ± 0.04 (P:F α/β) 0.98 ± 0.33 (Total βIII) |
2 independent differentiations (2 technical replicates) | n.s. p > 0.05: Total βIII, 1 vs. 2, 3, 4 **p < 0.01: P:F α/β, 1 vs. 2,4 ****p < 0.0001: P:F α/β, 1 vs. 3 |
One-Way ANOVA (Dunnett’s post-HOC) (P:F α/β) Kruskal-Wallis with Dunn’s Correction (Total βIII) |
| Motor performance analysis BAChSTMN2 line Rotarod: Average latency to fall (sec) Hanging wire: Normalized latency to fall (sec*(weight/avg weight)) (Figure 8e) |
(1) F2
Stmn2+/+ (2) F2 Stmn2+/− (3) F2 Stmn2−/− (4) hSTMN2xmStmn2+/+ (5) hSTMN2xmStmn2+/− (6) hSTMN2xmStmn2−/− |
Values for 1,2,3 in Figure 2 Rotarod P60 279.1 ± 21.39 (4) 265.9 ± 40.61 (5) 269.7 ± 32.12 (6) P100 274.9 ± 18.57 (4) 266.2 ± 37.16 (5) 260.8 ± 37.48 (6) Hanging wire P60 102.8 ± 26.38 (4) 102.9 ± 21.22 (5) 98.68 ± 25.42 (6) P100 76.54 ± 28.92 (4) 99.79 ± 20.46 (5) 104.2 ± 13.31 (6) |
Values for 1,2,3 in Figure 2 6–9 (4,5,6) |
n.s. p > 0.05: Rotarod and Hanging Wire (P60/P100), 1 vs 2, 4, 5, 6 ****p < 0.0001: Rotarod and Hanging Wire (P60/P100), 1 vs. 3 |
One-Way ANOVA (Dunnett’s post-HOC) |
| Relative mouse Stmn2 and humanSTMN2 brain transcript expression (Figure 8f) |
(1) F2
Stmn2+/+ (2) F2 Stmn2−/− (3) hSTMN2xmStmn2+/+ (4) hSTMN2xmStmn2−/− |
1.00 ± 0.15 (Ms) 6.499e-005 ± 3.368e-005 (Hu) 0.039± 0.00 (Ms) 2.311e-005 ± 5.505e-006 (Hu) 1.24 ± 0.54 (Ms) 1.066 ± 0.2126 (Hu) 0.09 ± 0.02 (Ms) 0.6768 ± 0.3558 (Hu) |
3 animals (2 technical replicates) | n.s. p > 0.05: 1 vs. 3 (Ms), 3 vs.4 (Hu) ***p < 0.001: 1 vs. 4 (Ms) ****p < 0.0001: 1 vs. 2 (Ms), 3 vs. 1, 2 (Hu) |
Kruskal-Wallis with Dunn’s correction |
| Relative STMN2 brain protein expression (Figure 8g) |
(1) F2
Stmn2+/+ (2,3,4) hSTMN2xmStmn2+/+ (5,6,7) hSTMN2xmStmn2−/− |
1.09 ± 0.20 (1) 1.75± 0.30 (2) 1.39 ± 0.08 (3) 2.11 ± 0.16 (4) 0.90± 0.25 (5) 0.97 ± 0.12 (6) 0.83 ± 0.15 (7) |
3 animals (2 technical replicates) | n.s. p > 0.05: 1 vs 2, 5, 6, 7 ***p < 0.001: 1 vs. 3 ****p < 0.0001: 1 vs. 2, 4 |
One-Way ANOVA (Dunnett’s post-HOC) |
|
Gastrocnemius NMJ innervation % of fully coincident BTX:SyPhy/FOV (% fully innervated) (%FI) % of partially coincident BTX:SyPhy/FOV (% partially innervated) (%PI) % of BTX only/FOV (% non-innervated) (%NI) (Figure 8j) |
(1) F2
Stmn2+/+
(2) F2 Stmn2−/− (3) hSTMN2xmStmn2+/+ (4) hSTMN2xmStmn2−/− |
Values for F2
Stmn2+/+ and F2
Stmn2−/− in Figure 3 %FI: 87.75 ± 9.90 %PI: 8.32 ± 10.36 %NI: 3.95 ± 5.26 %FI: 96.62 ± 4.87 %PI: 2.80 ± 4.95 %NI: 0.57 ± 1.63 |
3 (5–8 FOV/mouse) | %FI, %PI and %NI n.s. p > 0.05: 1 vs 3, 3 vs 4 ****p < 0.0001: 1 vs 2, 2 vs 4 |
Kruskal-Wallis with Dunn’s correction |
Highlights.
Loss of Stmn2 in mice leads to an age dependent NMJ denervation and motor deficits
Expression of Stmn2 is enriched in Chat+ alpha motor neurons in the spinal cord
Stmn2 deficiency disrupts microtubule dynamics in spinal cord neurons
Introduction of BAC human STMN2 in Stmn2−/− mice rescues motor function
Inclusion and Diversity Statement.
We worked to ensure sex balance in the selection of non-human subjects. One or more authors of this paper self-identifies as a member of the LGBTQ+ community. One or more authors of this paper self-identifies as living with a disability. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Acknowledgements
Support to K.E. was provided by Target ALS, NIH 5R01NS089742 and the Harvard Stem Cell Institute. I.G.S.J is supported by Target ALS. L.A.N is the recipient of the Healey Scholars Fellowship. A.B. was supported by NIH 5K99AG057808–02. J.R.K. was supported by the Tom Kirchhoff Family Postdoctoral Fellowship from Project ALS.
We thank Joanie Mok for sharing her insight and expertise on animal handling. We thank the Harvard Center for Biological Imaging, the Harvard Genome Modification Facility and the HSCRB Histology Core for infrastructure and support. Schematic diagrams in all Figures were generated with BioRender.com
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Chalabi A, Calvo A, Chio A, Colville S, Ellis CM, Hardiman O, Heverin M, Howard RS, Huisman MHB, Keren N, et al. (2014). Analysis of amyotrophic lateral sclerosis as a multistep process: a population-based modelling study. Lancet Neurol 13, 1108–1113. 10.1016/S14744422(14)70219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, et al. (2014). Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 81, 536–543. 10.1016/j.neuron.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold ES, Ling SC, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, Kordasiewicz HB, McAlonis-Downes M, Platoshyn O, Parone PA, et al. (2013). ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proceedings of the National Academy of Sciences of the United States of America 110, E736–745. 10.1073/pnas.1222809110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash PEA, Zhang Y-J, Roberts CM, Saldi T, Hutter H, Buratti E, Petrucelli L, and Link CD (2010). Neurotoxic effects of TDP-43 overexpression in C. elegans. Human molecular genetics 19, 3206–3218. 10.1093/hmg/ddq230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon RJ, and Lichtman JW (1990). In vivo visualization of the growth of pre- and postsynaptic elements of neuromuscular junctions in the mouse. J Neurosci 10(3), 894–908. 10.1523/JNEUROSCI.10-03-00894.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, and Finkbeiner S (2010). Cytoplasmic mislocalization of TDP-43 is toxic to neurons and enhanced by a mutation associated with familial amyotrophic lateral sclerosis. J Neurosci 30, 639–649. 10.1523/JNEUROSCI.4988-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum JA, Klemm S, Shadrach JL, Guttenplan KA, Nakayama L, Kathiria A, Hoang PT, Gautier O, Kaltschmidt JA, Greenleaf WJ, et al. (2021). Single-cell transcriptomic analysis of the adult mouse spinal cord reveals molecular diversity of autonomic and skeletal motor neurons. Nature Neuroscience 24, 572–583. 10.1038/s41593-020-00795-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese M, Saal-Bauernschubert L, Lüningschrör P, Moradi M, Dombert B, Surrey V, Appenzeller S, Deng C, Jablonka S, and Sendtner M (2020). Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol Commun 8, 116. 10.1186/s40478-020-00987-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168–e168. 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burberry A, Suzuki N, Wang J-Y, Moccia R, Mordes DA, Stewart MH, Suzuki-Uematsu S, Ghosh S, Singh A, Merkle FT, et al. (2016). Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med 8, 347ra393–347ra393. 10.1126/scitranslmed.aaf6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burberry A, Wells MF, Limone F, Couto A, Smith KS, Keaney J, Gillet G, van Gastel N, Wang JY, Pietilainen O, et al. (2020). C9orf72 suppresses systemic and neural inflammation induced by gut bacteria. Nature 582, 89–94. 10.1038/s41586-020-2288-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappello V, and Francolini M (2017). Neuromuscular Junction Dismantling in Amyotrophic Lateral Sclerosis. Int J Mol Sci 18, 2092. 10.3390/ijms18102092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbaut E, Curmi PA, Ozon S, Lachkar S, Redeker V, and Sobel A (2001). Stathmin family proteins display specific molecular and tubulin binding properties. The Journal of biological chemistry 276, 16146–16154. 10.1074/jbc.M010637200. [DOI] [PubMed] [Google Scholar]
- Chauvin S, and Sobel A (2015). Neuronal stathmins: a family of phosphoproteins cooperating for neuronal development, plasticity and regeneration. Progress in neurobiology 126, 1–18. 10.1016/j.pneurobio.2014.09.002. [DOI] [PubMed] [Google Scholar]
- Corey DR (2017). Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nature neuroscience 20, 497–499. 10.1038/nn.4508. [DOI] [PubMed] [Google Scholar]
- Fallini C, Bassell GJ, and Rossoll W (2012). The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Human molecular genetics 21, 3703–3718. 10.1093/hmg/dds205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feneberg E, Gray E, Ansorge O, Talbot K, and Turner MR (2018). Towards a TDP-43-Based Biomarker for ALS and FTLD. Mol Neurobiol 55, 7789–7801. 10.1007/s12035-018-0947-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, and Glass JD (2004). Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Experimental Neurology 185, 232–240. 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Folker ES, and Baylies MK (2013). Nuclear positioning in muscle development and disease. Front Physiol 4, 363-363. 10.3389/fphys.2013.00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, and Kaspar BK (2009). Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol 27, 5965. 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fratta P, Sivakumar P, Humphrey J, Lo K, Ricketts T, Oliveira H, Brito-Armas JM, Kalmar B, Ule A, Yu Y, et al. (2018). Mice with endogenous TDP-43 mutations exhibit gain of splicing function and characteristics of amyotrophic lateral sclerosis. The EMBO journal 37. 10.15252/embj.201798684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey D, Schneider C, Xu L, Borg J, Spooren W, and Caroni P (2000). Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci 20, 2534–2542. 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Doan RA, Strickland A, Huang X, Milbrandt J, and DiAntonio A (2016). Prevention of vincristine-induced peripheral neuropathy by genetic deletion of SARM1 in mice. Brain 139, 3092–3108. 10.1093/brain/aww251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordana MT, Piccinini M, Grifoni S, De Marco G, Vercellino M, Magistrello M, Pellerino A, Buccinnà B, Lupino E, and Rinaudo MT (2010). TDP-43 redistribution is an early event in sporadic amyotrophic lateral sclerosis. Brain Pathol 20, 351–360. 10.1111/j.1750-3639.2009.00284.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass CK, Saijo K, Winner B, Marchetto MC, and Gage FH (2010). Mechanisms underlying inflammation in neurodegeneration. Cell 140, 918–934. 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Freire M, de Cabo R, Studenski SA, and Ferrucci L (2014). The Neuromuscular Junction: Aging at the Crossroad between Nerves and Muscle. Front Aging Neurosci 6, 208–208. 10.3389/fnagi.2014.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Heerssen HM, Wright CM, Davis GW, and DiAntonio A (2011). Stathmin is required for stability of the Drosophila neuromuscular junction. J Neurosci 31, 15026–15034. 10.1523/JNEUROSCI.2024-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergesheimer RC, Chami AA, de Assis DR, Vourc’h P, Andres CR, Corcia P, Lanznaster D, and Blasco H (2019). The debated toxic role of aggregated TDP-43 in amyotrophic lateral sclerosis: a resolution in sight? Brain 142, 1176–1194. 10.1093/brain/awz078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highley JR, Kirby J, Jansweijer JA, Webb PS, Hewamadduma CA, Heath PR, Higginbottom A, Raman R, Ferraiuolo L, Cooper-Knock J, et al. (2014). Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathology and Applied Neurobiology 40, 670–685. 10.1111/nan.12148. [DOI] [PubMed] [Google Scholar]
- Huang C, Tong J, Bi F, Zhou H, and Xia XG (2012). Mutant TDP-43 in motor neurons promotes the onset and progression of ALS in rats. The Journal of clinical investigation 122, 107–118. 10.1172/JCI59130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey J, Emmett W, Fratta P, Isaacs AM, and Plagnol V (2017). Quantitative analysis of cryptic splicing associated with TDP-43 depletion. BMC Med Genomics 10, 38-38. 10.1186/s12920-017-0274-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T, Malunda J, Xu Y, Winton MJ, Trojanowski JQ, et al. (2011). Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. The Journal of clinical investigation 121, 726–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanning KC, Kaplan A, and Henderson CE (2010). Motor neuron diversity in development and disease. Annual review of neuroscience 33, 409–440. 10.1146/annurev.neuro.051508.135722. [DOI] [PubMed] [Google Scholar]
- Klim JR, Pintacuda G, Nash LA, Guerra San Juan I, and Eggan K (2021). Connecting TDP-43 Pathology with Neuropathy. Trends in neurosciences 44, 424–440. 10.1016/j.tins.2021.02.008. [DOI] [PubMed] [Google Scholar]
- Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, et al. (2019). ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nature neuroscience 22, 167–179. 10.1038/s41593-018-0300-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lall D, and Baloh RH (2017). Microglia and C9orf72 in neuroinflammation and ALS and frontotemporal dementia. The Journal of clinical investigation 127, 3250–3258. 10.1172/JCI90607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke W, Leman EE, Fyffe REW, Raine CS, and Schubart UK (2002). Stathmin-deficient mice develop an age-dependent axonopathy of the central and peripheral nervous systems. Am J Pathol 160, 469–480. 10.1016/S0002-9440(10)64866-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limone F, Mitchell JM, San Juan IG, Smith JLM, Raghunathan K, Couto A, Ghosh SD, Meyer D, Mello CJ, Nemesh J, et al. (2022). Efficient generation of lower induced Motor Neurons by coupling Ngn2 expression with developmental cues. bioRxiv, 2022.2001.2012.476020. 10.1101/2022.01.12.476020 [DOI] [PMC free article] [PubMed]
- Lin M-J, and Lee S-J (2016). Stathmin-like 4 is critical for the maintenance of neural progenitor cells in dorsal midbrain of zebrafish larvae. Scientific Reports 6, 36188. 10.1038/srep36188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling S-C, Albuquerque CP, Han JS, Lagier-Tourenne C, Tokunaga S, Zhou H, and Cleveland DW (2010). ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proceedings of the National Academy of Sciences 107, 13318–13323. 10.1073/pnas.1008227107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill TH, Mori N, and Cheng HW (1999). Differential regulation of the growth-associated proteins, GAP-43 and SCG-10, in response to unilateral cortical ablation in adult rats. Neuroscience 90, 1349–1360. 10.1016/s0306-4522(98)00482-5. [DOI] [PubMed] [Google Scholar]
- Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, Freyermuth F, McMahon MA, Beccari MS, Artates JW, et al. (2019). Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nature neuroscience 22, 180–190. 10.1038/s41593-018-0293-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Ferraiuolo L, Schmelzer L, Braun L, McGovern V, Likhite S, Michels O, Govoni A, Fitzgerald J, Morales P, et al. (2015). Improving single injection CSF delivery of AAV9-mediated gene therapy for SMA: a dose-response study in mice and nonhuman primates. Mol Ther 23, 477–487. 10.1038/mt.2014.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JC, Constable R, So E, Vance C, Scotter E, Glover L, Hortobagyi T, Arnold ES, Ling SC, McAlonis M, et al. (2015). Wild type human TDP-43 potentiates ALS-linked mutant TDP-43 driven progressive motor and cortical neuron degeneration with pathological features of ALS. Acta Neuropathol Commun 3, 36–36. 10.1186/s40478-015-0212-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisse K, Mepham J, Volkening K, Welch I, Hill T, and Strong MJ (2009). Cytosolic TDP-43 expression following axotomy is associated with caspase 3 activation in NFL−/− mice: support for a role for TDP-43 in the physiological response to neuronal injury. Brain research 1296, 176–186. 10.1016/j.brainres.2009.07.023 [DOI] [PubMed] [Google Scholar]
- Morii H, Shiraishi-Yamaguchi Y, and Mori N (2006). SCG10, a microtubule destabilizing factor, stimulates the neurite outgrowth by modulating microtubule dynamics in rat hippocampal primary cultured neurons. Journal of Neurobiology 66, 1101–1114. 10.1002/neu.20295. [DOI] [PubMed] [Google Scholar]
- Morisaki Y, Niikura M, Watanabe M, Onishi K, Tanabe S, Moriwaki Y, Okuda T, Ohara S, Murayama S, Takao M, et al. (2016). Selective Expression of Osteopontin in ALS-resistant Motor Neurons is a Critical Determinant of Late Phase Neurodegeneration Mediated by Matrix Metalloproteinase-9. Scientific reports 6, 27354. 10.1038/srep27354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. (2006). Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science (New York, NY) 314, 130–133. 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Nguyen TB, Prabhu VV, Piao YH, Oh YE, Zahra RF, and Chung YC (2019). Effects of Stathmin 1 Gene Knockout on Behaviors and Dopaminergic Markers in Mice Exposed to Social Defeat Stress. Brain sciences 9. 0.3390/brainsci9090215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijssen J, Comley LH, and Hedlund E (2017). Motor neuron vulnerability and resistance in amyotrophic lateral sclerosis. Acta Neuropathologica 133, 863–885. 10.1007/s00401-017-1708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi T, Higo N, Matsuda K, and Hayashi M (2002). Expression of super cervical ganglion-10 (SCG10) mRNA in the monkey cerebral cortex during postnatal development. Neurosci Lett 323, 199–202. 10.1016/s0304-3940(02)00142-8. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Yoshida BN, Avraham KB, Wang H, Wuenschell CW, Jenkins NA, Copeland NG, Anderson DJ, and Mori N (1993). Molecular diversity of the SCG10/stathmin gene family in the mouse. Genomics 18, 360–373. 10.1006/geno.1993.1477. [DOI] [PubMed] [Google Scholar]
- Olesen MN, Wuolikainen A, Nilsson AC, Wirenfeldt M, Forsberg K, Madsen JS, Lillevang ST, Brandslund I, Andersen PM, and Asgari N (2020). Inflammatory profiles relate to survival in subtypes of amyotrophic lateral sclerosis. Neurology - Neuroimmunology Neuroinflammation 7, e697. 10.1212/NXI.0000000000000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozon S, Byk T, and Sobel A (1998). SCLIP: a novel SCG10-like protein of the stathmin family expressed in the nervous system. J Neurochem 70, 2386–2396. 10.1046/j.1471-4159.1998.70062386.x. [DOI] [PubMed] [Google Scholar]
- Patel T, Hammelman J, Closser M, Gifford DK, and Wichterle H (2021). General and cell-typespecific aspects of the motor neuron maturation transcriptional program. bioRxiv, 2021.2003.2005.434185. 10.1101/2021.03.05.434185. [DOI]
- Philips T, and Robberecht W (2011). Neuroinflammation in amyotrophic lateral sclerosis: role of glial activation in motor neuron disease. The Lancet Neurology 10, 253–263. 10.1016/S1474-4422(11)70015-1. [DOI] [PubMed] [Google Scholar]
- Prasad A, Bharathi V, Sivalingam V, Girdhar A, and Patel BK (2019). Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Frontiers in molecular neuroscience 12, 25–25. 10.3389/fnmol.2019.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M, Humphrey J, Pickles S, Brown AL, Hill SE, Kachergus J, Shi J, Heckman M, Spiegel M, Cook C, et al. (2020). Truncated stathmin-2 is a marker of TDP-43 pathology in frontotemporal dementia. The Journal of clinical investigation 10.1172/JCI139741. [DOI] [PMC free article] [PubMed]
- Pun S, Santos AF, Saxena S, Xu L, and Caroni P (2006). Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nature neuroscience 9, 408–419. 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Riederer BM, Pellier V, Antonsson B, Di Paolo G, Stimpson SA, Lütjens R, Catsicas S, and Grenningloh G (1997). Regulation of microtubule dynamics by the neuronal growth-associated protein SCG10. Proceedings of the National Academy of Sciences of the United States of America 94, 741–745. 10.1073/pnas.94.2.741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman W, and Gomes ER (2018). Nuclear positioning in skeletal muscle. Seminars in Cell & Developmental Biology 82, 51–56. 10.1016/j.semcdb.2017.11.005. [DOI] [PubMed] [Google Scholar]
- Sato T, Takeuchi S, Saito A, Ding W, Bamba H, Matsuura H, Hisa Y, Tooyama I, and Urushitani M (2009). Axonal ligation induces transient redistribution of TDP-43 in brainstem motor neurons. Neuroscience 164, 1565–1578. 10.1016/j.neuroscience.2009.09.050. [DOI] [PubMed] [Google Scholar]
- Shin JE, Geisler S, and DiAntonio A (2014). Dynamic regulation of SCG10 in regenerating axons after injury. Exp Neurol 252, 1–11. 10.1016/j.expneurol.2013.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Miller BR, Babetto E, Cho Y, Sasaki Y, Qayum S, Russler EV, Cavalli V, Milbrandt J, and DiAntonio A (2012). SCG10 is a JNK target in the axonal degeneration pathway. Proceedings of the National Academy of Sciences of the United States of America 109, E3696–E3705. 10.1073/pnas.1216204109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura Y, and Mori N (1995). SCG10 expresses growth-associated manner in developing rat brain, but shows a different pattern to p19/stathmin or GAP-43. Brain Res Dev Brain Res 90, 73–91. 10.1016/0165243806(96)83488-2. [DOI] [PubMed] [Google Scholar]
- Theunissen F, Anderton RS, Mastaglia FL, Flynn LL, Winter SJ, James I, Bedlack R, Hodgetts S, Fletcher S, Wilton SD, et al. (2021). Novel STMN2 Variant Linked to Amyotrophic Lateral Sclerosis Risk and Clinical Phenotype. Frontiers in Aging Neuroscience 13. 10.3389/fnagi.2021.658226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W, Czopka T, and López-Schier H (2020). Systemic loss of Sarm1 protects Schwann cells from chemotoxicity by delaying axon degeneration. Communications biology 3, 49. 10.1038/s42003-0200776-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkiew E, Falconer D, Reed N, and Höke A (2017). Deletion of Sarm1 gene is neuroprotective in two models of peripheral neuropathy. Journal of the peripheral nervous system : JPNS 22, 162–171. 10.1111/jns.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Willige D, Hoogenraad CC, and Akhmanova A (2016). Microtubule plus-end tracking proteins in neuronal development. Cell Mol Life Sci 73, 2053–2077. 10.1007/s00018-016-2168-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB, Clay D, Wood EM, Chen-Plotkin AS, Martinez-Lage M, et al. (2008). TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol 7, 409–416. 10.1016/S1474-4422(08)70071-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Broeck L, Callaerts P, and Dermaut B (2014). TDP-43-mediated neurodegeneration: towards a loss-of-function hypothesis? Trends in molecular medicine 20, 66–71. 10.1016/j.molmed.2013.11.003. [DOI] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, and Kumar-Singh S (2010). TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proceedings of the National Academy of Sciences of the United States of America 107, 3858–3863. 10.1073/pnas.0912417107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y-F, Gendron TF, Zhang Y-J, Lin W-L, D’Alton S, Sheng H, Casey MC, Tong J, Knight J, Yu X, et al. (2010). Wild-type human TDP-43 expression causes TDP-43 phosphorylation, mitochondrial aggregation, motor deficits, and early mortality in transgenic mice. J Neurosci 30, 10851–10859. 10.1523/JNEUROSCI.1630-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, and Jaenisch R (2014). Generating genetically modified mice using CRISPR/Casmediated genome engineering. Nature Protocols 9, 1956–1968. 10.1016/j.cell.2013.08.022. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the lead contact upon request. This paper does not report original code.