

Abstract
Donor derived regulatory T lymphocytes and the JAK1/2 kinase inhibitor ruxolitinib are currently being evaluated as therapeutic options in the treatment of chronic graft versus host disease (cGvHD). In this work, we aimed to determine if the combined use of both agents can exert a synergistic effect in the treatment of GvHD. For this purpose, we studied the effect of this combination both in vitro and in a GvHD mouse model. Our results show that ruxolitinib favors the ratio of thymic regulatory T cells to conventional T cells in culture, without affecting the suppressive capacity of these Treg. The combination of ruxolitinib with Treg showed a higher efficacy as compared to each single treatment alone in our GvHD mouse model in terms of GvHD incidence, severity and survival without hampering graft versus leukemia effect. This beneficial effect correlated with the detection in the bone marrow of recipient mice of the infused donor allogeneic Treg after the adoptive transfer.
Subject terms: Bone marrow transplantation, Haematological cancer
Introduction
The allogeneic transplantation of hematopoietic stem cells (allo-HSC) represents the best therapeutic option for many patients diagnosed with hematologic malignancies. Unfortunately, a significant proportion of patients receiving an allo-HSC develop acute or chronic Graft versus Host Disease (GvHD)1–4.
The JAK kinase inhibitor ruxolitinib has been used to ameliorate the effects of different inflammatory and myeloproliferative syndromes5–13, as the JAK kinases pathway plays a key role in the transmission of the cytokine signaling in inflammatory and immune processes. With this background, different studies evaluated the efficacy of ruxolitinib in the prophylaxis and treatment of acute or chronic GvHD first in preclinical models and subsequently in prospective randomized trials14–25. With the REACH trials, ruxolitinib has become the treatment of choice for steroid refractory acute and chronic GvHD14,15,17. Interestingly, the administration of ruxolitinib in mice developing GvHD increased regulatory T cells (Treg) as compared to the non-treated mice16.
The use of donor or third party derived Treg is another promising therapy against GvHD26–36. The Treg represent a subset of CD4+ T-cells with high expression of the IL2 receptor alpha (CD25) and are also characterized by a high expression of the transcriptional factor Forkhead box p3 (Foxp3)37,38. These cells are capable to modulate the immune responses produced by the effector immune cells, having a crucial role in the development of self-tolerance, and also in the induction of tolerance of the donor cells to the recipient tissues. The absence of Treg leads to severe autoimmune complications. The Treg can regulate both innate and acquired immune responses.
Based on the hypothesis that ruxolitinib favors the ratio of Treg to conventional T cells (Tcon) in previous studies, we propose that the combined use of donor derived Treg with ruxolitinib could help to achieve therapeutic effects with lower number of Treg, allowing also the tapering of immunosuppressive treatment in GvHD patients. To address this idea, we have conducted an in vitro study and a preclinical mouse model.
Results
In vitro effect of ruxolitinib on Treg
Ruxolitinib increases the natural Treg:Tcon ratio in vitro over time
We tested the effect of the Jak1/2 inhibitor ruxolitinib in the proportion of regulatory T cells in in vitro cultures of human PBMNCs activated with anti-CD3 and anti-CD28 stimulation. As previously described16, increased concentrations of ruxolitinib reduced the activation of T cells (see decrease in CD25+ cells in Fig. 1a, Supplementary Fig. 1A). After 2 days of activation, CD4+ and CD8+ cells upregulated both CD25 and Foxp3 compared to non activated controls (Supplementary Fig. 1a). A population of CD4+ cells showed a higher expression than the CD8+ cells, and we identify them as Treg (Supplementary Fig. 1a). We detected a significant percentage of such CD4+ C25+ Foxp3high Treg cells in non-treated cultures, while cultures treated with increasing amounts of ruxolitinib showed lower percentages (Fig. 1a,b, Supplementary Fig. 1A). However, at later time points (5 and 8 days), the percentage and absolute number of Foxp3high in non-treated cultures dropped drastically, while in ruxolitinib treated cultures it increases over time. In our experiments, the optimal concentration of ruxolitinib to achieve a higher Treg:Tcon ratio was 0.3 µM (Fig. 1a,b, Supplementary Fig. 1B,C). We hypothesized that the Foxp3high cells detected in the non-treated culture after two days were induced Treg (iTreg) generated after the hyperactivation of the culture, while the Foxp3high detected in ruxolitinib treated cultures after longer periods where natural Treg (nTreg) with a stable phenotype. To test this hypothesis we stained the cultures with anti-Helios antibodies, as the Helios transcription factor is a marker of the thymic origin of the nTreg39,40. As shown in Fig. 1c,d, the ruxolitinib treated cultures were enriched in Helios+ Foxp3high cells compared with non-treated controls, and this difference is maintained along the duration of the experiment. In freshly isolated PBMNCs, the percentage of Helios positive Treg cells is around 75% (Supplementary Fig. 2A).
Figure 1.

In vitro activated huPBMNCs in the presence of ruxolitinib show increased percentages of CD4+ Foxp3 high Helios+ cells along time. (a) Representative cytometry dot plots of huPBMNCs activated with anti-CD3 and anti-CD28 at different times and ruxolitinib doses. Dot plots show CD25 and Foxp3 staining of CD4+ gated cells. (b) Quantification of Foxp3high cells of gated CD4+ huPBMNCs. Mean and standard error of the mean (S.E.M.) of a minimum of 4 independent experiments is represented for each condition. (c) Representative cytometry dot plots of huPBMNCs activated with anti-CD3 and anti-CD28 at different times and with 0 or 0.3 µM ruxolitinib. Dot plots show CD25 and Foxp3 staining of CD4+ gated cells, and density plots show Helios and PD1 staining. (d) Quantification of Helios+ cells of gated CD4+ Foxp3high huPBMNCs. n = 6. (e) Quantification of PD1+ cells of gated CD4+ Foxp3high huPBMNCs. n = 6. (f) Quantification of CTLA4+ cells of gated CD4 + Foxp3high huPBMNCs. n = 4. (g) Quantification of CD39+ cells of gated CD4+ Foxp3high huPBMNCs. n = 4. (h) Quantification of CD45RA+ cells of gated CD4+ Foxp3high huPBMNCs. n = 4. One way ANOVA test with Dunnett’s correction for multiple comparisons p values are shown. Each treatment is compared with the 0 ruxolitinib control. p values: *< 0.05, **< 0.01, ***< 0.001, ****< 0.0001.
We further characterized the Treg present in the culture. PD1 (Programmed cell Death 1) is implicated in the immune checkpoint, and in the case of Type 1 regulatory T cells (Tr1), is one of the mechanisms by which suppression is achieved41. On the other hand, it is also an exhaustion marker of activated effector T cells, and recently it has been described as a negative factor for Treg suppressive capacity in a lineage specific K.O. mouse model42. We found that PD1 expression is higher in Helios− Foxp3high cells as compared to Helios+ Foxp3high (Fig. 1c,e) showing an inverse correlation (Supplementary Fig. 2B). In freshly isolated, non-stimulated cells, most Foxp3+ cells are Helios+ and there is no correlation with PD1 expression (Supplementary Fig. 2A).
Cytotoxic T Cell Antigen 4 (CTLA4) is another immune checkpoint receptor that is required for Treg function, and for Tcon homeostasis43. While ruxolitinib reduced the expression of CTLA4 in early time points (Fig. 1f, Supplementary Fig. 2C), within higher doses, the expression was recovered over time.
CD39 is a marker that has been correlated to the suppression capacity of the Treg44,45, due to its catalytic activity producing extracellular adenosine. As shown in Fig. 1g, CD39 increases its expression along the culture in all the experimental conditions, but interestingly it was higher in ruxolitinib treated Treg as compared to non treated cells at all time points analyzed.
Finally, CD45RA expression is associated with a naïve phenotype, and in peripheral blood (PB) Treg it is used to identify a population which can be expanded in vitro maintaining the suppressive properties46–48. In our study, as shown in Fig. 1h, the CD45RA expression is lost in all populations studied along the culture, independently of the Helios expression. This indicates that all cells in the culture, including the thymic nTreg, are activated due to the anti-CD3 and CD28 stimulation, losing their naïve phenotype.
Ruxolitinib inhibits homing CCR9 and CCR5 and inflammatory CXCR3 receptors
Another aspect which could affect the efficacy of the treatment with Treg is their capacity to migrate to the GvHD target organs. Ccr9, Ccr5 and Cxcr3 have been previously correlated with the migratory capacity of T cells to the gut under pathogenic conditions49–51. Of them, Cxcr3 has been shown to be downregulated by IFN-γ signaling disruption52, either by receptor elimination or by signal transduction inhibition with ruxolitinib. We determined the effect of ruxolitinib in these three chemokine receptors expression in CD4+ Foxp3high cells compared to CD4+ Foxp3low (Fig. 2a). Ruxolitinib reduces the expression of Cxcr3 and also of Ccr9 in Tcon and Treg, and Ccr5 only in Treg. However, in all cases, the expression of the three receptors was significantly higher in Foxp3high than in Foxp3low CD4+ T cells. This suggest that homing could be less affected by ruxolitinib in Treg as compared to Tcon.
Figure 2.

Effect of ruxolitinib on homing chemokine receptors and Stat phosphorylation. (a) Percentage of Cxcr3, Ccr5 and Ccr9 positive cells in CD4+ Foxp3high and Foxp3low gated cells after 48 h of anti-CD3/CD28 stimulation of huPBMNCs. Cells were treated with the indicated amounts of ruxolitinib from the beginning of stimulation. Mean and S.E.M. of 5 independent experiments is represented. (b) Quantification of intracellular Phospho-Stat5 cytometry. huPBMNCs were cultured for 48 h in the presence of 0 or 0.3 µM ruxolitinib, with or without anti-CD3 and CD28 stimulation. Cells were gated for CD4+ Foxp3high, CD4+ Foxp3low and CD8+. Average and S.E.M. of the Median Fluorescence Intensity (M.F.I.) of two independent experiments with two technical replicates each are shown. (c) As in B, but Phospho-Stat3 staining was used instead. (d) Cytometry density plots of Phospho-Stat5 and Helios intracellular Staining of CD4+ Foxp3high gated huPBMNCs cells, cultures as in B. (e) As in B, but in this case CD4+ Foxp3high cells are also gated in Helios+ and Helios− cells. One representative of two independent experiments with two technical replicas each is shown. p values of a paired Student’s t-test are represented. p values: *< 0.05, **< 0.01, ***< 0.001, ****< 0.0001.
Phosphorylation of Stat3 and Stat5 is decreased in both Treg and Tcon after ruxolitinib treatment
In previous studies, the effect of ruxolitinib on the phosphorylation of both Stat316 and Stat553 transcription factors in CD4+ T cells has been studied. In both cases, ruxolitinib reduces the phosphorylation of these signal transducers. On the other hand, it has been described the essential role of Stat5 for the development of Treg, while Stat3 is not required54. Interestingly, it has been proposed that Stat3 phosphorylation depends mainly on Jak1 and 2, while Stat5 is targeted by Jak2 and Jak355. Thus, we decided to test whether ruxolitinib affected the phosphorylation of Stat5 and Stat3 in Treg and Tcon. After 48 h of anti-CD3 and anti-CD28 stimulation, a strong Stat5 Phosphorylation in CD8+, CD4+ Foxp3low and CD4+ Foxp3high was observed (Fig. 2b), which was completely abolished by ruxolitinib treatment in all cases. The same was true for Stat3 Phosphorylation (Fig. 2c). We checked whether or not there was any difference in Helios+ and Helios- cells, and the same result was observed for both populations (Fig. 2d,e).
Ruxolitinib does not hamper the suppressive capacity of regulatory T cells
To functionally test the suppressive capacity of the ruxolitinib-treated Treg, we first tested IL-10 production in CD4+ Foxp3high cells present in PMNCs cultures after 2 days of activation and treatment with different concentrations of ruxolitinib (Fig. 3a), compared with CD4+ Foxp3low and CD8+ cells. CD4+ Foxp3high showed a higher amount of IL-10 staining at all concentrations of ruxolitinib, with a decrease in IL-10 intracellular staining with increasing concentrations of ruxolitinib. This was true for both Helios+ and Helios− CD4 + Foxp3high cells (Fig. 3b).
Figure 3.

Quantification of intracellular IL10. (a) Quantification of intracellular IL10 staining of cells gated for CD4+ Foxp3high, CD4 + Foxp3low and CD8+. huPBMNCs were cultured for 48 h in the presence of 0, 0.1, 0.3 or 1 µM ruxolitinib, with anti-CD3 and CD28 stimulation. Average and S.E.M. of the M.F.I. of five independent experiments are shown. p values of a one-way ANOVA test with Dunnett’s correction for multiple comparisons are shown. *< 0.05, **< 0.01, ***< 0.001, ****< 0.0001. (b) As in A, but CD4 + Foxp3high cells are also gated for Helios+ and Helios− staining. A representative experiment of two biological replicates with two technical replicates is shown.
Next, we tested the suppressive capacity of the Treg in the presence of ruxolitinib in an in vitro suppression assay. Magnetic sorted purified human CD4+ CD25+ CD127low Treg were mixed with CFSE labeled huPBMNCs at different ratios, in the presence of different concentrations of ruxolitinib, and with anti-CD3 and anti-CD28 stimulation (Fig. 4). The proliferation of responder T cells was almost completely abolished in the presence of 1 µM of ruxolitinib, independently of the presence or not of Treg (Fig. 4a, lower row). However, at 0.1 µM ruxolitinib, although strongly reduced, cells were still able to proliferate in the absence of Treg, both CD4 and CD8 responders (Fig. 4a,b). The addition of Treg suppressed this proliferation in a dose-dependent manner. The quantification of the suppression capacity normalizing to the non treated controls showed the additive effect of ruxolitinib and Treg (Fig. 4c). More interestingly, when we normalize to the non Treg control of each ruxolitinib treatment, we can observe that the suppression capacity of Treg is not diminished in the presence of 0.1 µM ruxolitinib and, on the contrary, it is even higher (Fig. 4d), suggesting a synergistic effect of ruxolitinib and Treg in the suppression of the activation. We also pretreated purified Treg with ruxolitinib for 24 h, then washed and performed the suppression assay. The results show also that the suppression capacity is enhanced upon treatment of Tregs with ruxolitinib 0.1 µM (Supplementary Fig. 3).
Figure 4.

In vitro suppression assays. (a) Representative cytometry dot plots showing CD8 and CFSE staining of in vitro suppression assays. huPBMNCs were stained with CFSE, and activated with anti CD3 and anti CD8 stimulation in the presence of different ratios of huTregs and Ruxolitinib. (b) Representative histogram plots of the CFSE dilution of CD4+ and CD8+ responder Tcon cells. (c) The percentage of suppression of four independent experiments is shown, for CD4+ and CD8+ gated responder cells. The percentage of suppression was calculated using as normalization control the Tcon only ruxolitinib 0 µM samples, to measure the additive effect of Treg and ruxolitinib. Percentage of suppression is calculated as {1 − (%proliferation in the sample/%proliferation in the control)} × 100%. (d) As in (c), but in this case the values were normalized with the Tcon only Ruxolitinib 0.1 µM for the ruxolitinib treated samples, and the Tcon only Ruxolitinib 0 µM samples for the non-treated samples, to measure only the effect of Tregs. Pairwise p values of a paired Student’s t-test are shown. p values: *< 0.05, **< 0.01, ***< 0.001, ****< 0.0001.
Progressive onset GvHD mouse model
To test the therapeutic potential of a combination of ruxolitinib and donor derived Treg in the treatment of GvHD, we decided to perform a preclinical study using a progressive onset GvHD mouse model, in which a first acute GvHD phase is followed by a second phase with signs of both acute and chronic GvHD56. We used this model to test the effect of a combined treatment with Treg and ruxolitinib at day 28 post-transplant, once the first acute phase was passed and a second phase of the disease was already stablished. The scheme of treatment is depicted in Fig. 5a. Animals were treated with a single infusion of 3 × 105 GFP Treg isogenic to the donor and/or 30 mg/kg body weight-day of ruxolitinib. The survival (Fig. 5b) of the mice receiving the combined treatment was significantly higher as compared to those receiving the vehicle (p = 0.0036) or the single treatment with ruxolitinib (p = 0.0164), and although not statistically significant, it was also higher than the single treatment with Treg. Weight loss, acute and chronic GvHD clinical scores (Fig. 5c–e) also showed a significantly better behavior in the double treatment group in comparison with the control and the single treatment arms. We took blood samples of the mice at weeks 2, 4 and 6 after the onset of treatment and analyzed by cytometry and hematimetry, and at 12 weeks mice were sacrificed and blood, bone marrow (BM), spleen, thymus, Peyer’s patches, small and large intestine samples were analyzed by flow cytometry (Supplementary Fig. 4, 5). Infused GFP+ Treg (CD4+ CD25+ Foxp3high GFP+) could be detected in the BM and, to a much lower level, in the blood of Treg and Treg + ruxolitinib treated mice (Fig. 6a,b, Supplementary Fig. 4). Histological analysis of the small and large intestine and skin were performed (Supplementary Fig. 6). An improvement of skin pathological scores was observed in mice treated with ruxolitinib and ruxolitinib plus Treg, although it didn’t reach statistical significance. Selected mice were left alive and BM biopsies were performed at weeks 18, 34 and 50 (Supplementary Fig. 4D), and finally sacrificed at week 70 (Fig. 6c). Infused GFP+ Treg survived long-term and were detected in all these time points.
Figure 5.

GvHD mouse model. (a) Scheme of the mouse model. BALB/c mice were irradiated with 800–860 cGy Split in two doses with 3 h of difference. 4 h later, 2 × 106 splenocytes depleted of monocytes and 5 × 106 BM cells from C57BL/6 donors were transplanted via tail vein injection. Four weeks after transplantation, surviving mice were randomized, and divided in four treatment groups. Treg were purified from GFP mice isogenic to the transplantation donors. 3 × 105 Treg were infused in a single dose to the corresponding groups. The other groups were infused with medium. From this day, animals received ruxolitinib (30 mg/kg-day) or vehicle, via oral gavage. (b) Kaplan Meyer representation of the survival of the different treatment groups. A Log-Rank test was used to calculate the p-values of the survival differences between the double treated sample and the other groups. (c) Weight loss of the mice with the different treated mice. Statistical differences are calculated using a two way ANOVA test. (d) Acute graft versus host disease score of the different treatment groups. (e) Chronic graft versus host disease score of the different treatment groups. p values: *< 0.05, **< 0.01, ***< 0.001, ****< 0.0001.
Figure 6.

Cytometry analysis of the infused GFP+ Treg in the mice. (a) Representative cytometries of cells isolated from different organs (PB peripheral blood, BM bone marrow, Spleen and Thymus) of a GFP+ Treg infused mouse, sacrificed 10 weeks after infusion. CD3+ CD4+ Gated cells are shown. Infused Treg are detected as CD25+ GFP+ cells. (b) Foxp3 staining of the BM cells shown in (a). Cells are gated for CD4 expression. GFP+ cells show Foxp3 positive staining. (c) Cytometry of bone marrow cells from a GFP+ Treg infused mouse sacrificed 70 weeks post infusion. GFP+ CD25+ cells are still detectable.
Graft vs. leukemia effect is not hampered by the combined treatment
Previous studies have demonstrated that neither ruxolitinib alone or the infusion of Treg interfere with the Graft versus Leukemia effect after transplantation19,57. We checked the effect of the combined treatment by infusing Luciferase transduced A20 isogenic leukemic cells into BALB/c recipient mice along with C57 splenocytes (Fig. 7). While in mice not receiving splenocytes the A20 cells proliferated, they did not in mice receiving allogeneic splenocytes in all treatment groups after 2 weeks.
Figure 7.
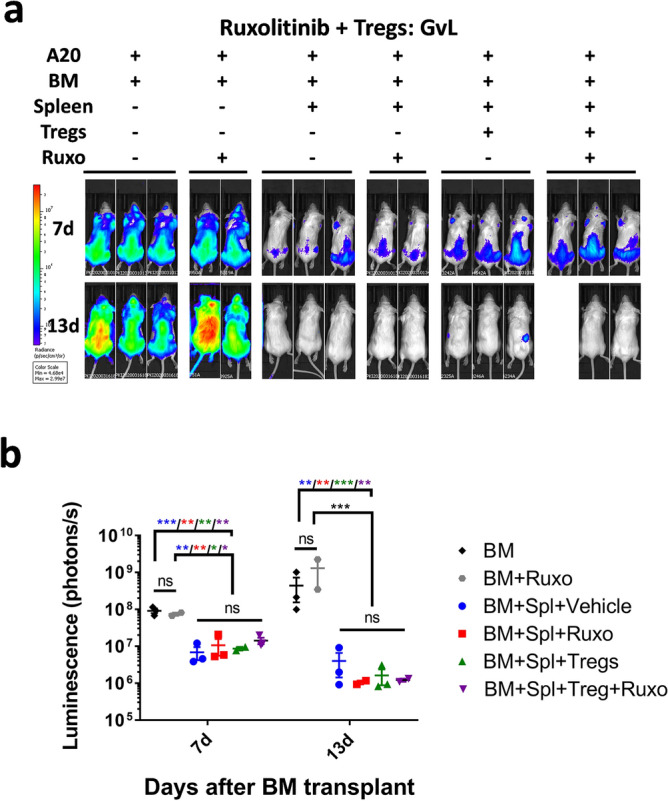
Graft vs. leukemia effect. (a) In vivo bioluminescence imaging of BALB/c mice transplanted with BM and Splenocytes from C57BL/6 donors and A20 leukemic cells expressing the Luciferase. Images were obtained at days + 7 and + 13 after transplantation. (b) Quantification of the bioluminescence shown in (a). Mean and S.E.M is represented in logarithmic scale. Statistical differences are calculated using a two way ANOVA test on the log10 bioluminescence values. p values: *< 0.05, **< 0.01, ***< 0.001, ****< 0.0001.
Discussion
Using in vitro cultures of huPBMNCs, we have shown that upon activation with anti-CD3/CD28, with no treatment, a population of CD25high Foxp3high cells arise in the early stages of the culture, and is strongly reduced along time. Addition of ruxolitinib delays the emergence of this population. Moreover, the presence of ruxolitinib increases the percentage of Helios positive cells. We reason that, in the absence of ruxolitinib, hyperactivation of the culture leads to the apparition of induced Treg from the activated Tcon. On the other hand, the presence of ruxolitinib increases the percentage of Helios+ nTreg in the culture by inhibiting Tcon proliferation without affecting nTreg. This idea is also supported by the inverse correlation of PD1 and Helios expression in these cultures, which could indicate that the Helios− fraction of CD25high FOXP3high originate from exhausted Tcon. It could also be a non suppressive CD25high FOXP3high population, as described previously58. Interestingly, in fresh non activated PBMNCs, there is no negative correlation between the expression of Helios and PD1. This result might explain the apparent discrepancies found in previous studies. Spoerl et al.16 found that the percentage of Treg is increased in mixed lymphocyte reactions with antigen presenting cells pretreated with low doses of ruxolitinib, while Parampalli Yajnanarayana et al.53 found that ruxolitinib impedes the in vitro generation of human iTreg by TGF-β and IL2 polarization. In our case, we have performed the experiments using total PBMNCs activated with anti-CD3 and anti-CD28, but we have obtained equivalent results also with purified CD4+ Tcon.
Ruxolitinib increases the expression of CD39, which is also implicated on immunomodulation via extracellular adenosine production. On the other hand, CTLA4, another important immune checkpoint molecule seems to be downregulated at earlier time points by ruxolitinib, although it is recovered along the culture with higher concentrations of ruxolitinib.
Regarding the expression of chemokine receptor related to gut migration, Ccr9, Ccr5 and Cxcr3, CD4+ Foxp3high cells seem to express higher levels than Foxp3low cells, at all concentrations of ruxolitinib, suggesting that Treg could retain better their homing capacity that Tcon upon treatment, although this effect must be confirmed with in vitro or in vivo migration assays to reach a definitive conclusion.
Stat5, but not Stat3, has been described as an essential factor for the development of Treg54. Ruxolitinib inhibits both Stat5 and Stat3 phosphorylation in Tcon16,53. However, the effect on Treg suppressive function, once they are generated might be less crucial. In our experimental conditions, ruxolitinib abolished similarly Stat5 and Stat3 phosphorylation in CD4+ Foxp3high cells than in CD4+ Foxp3low and CD8 cells. This was true for both Helios+ and Helios− Foxp3+ cells. This result however does not rule out whether or not in more physiological conditions the phosphorylation of Stat5 could be differentially affected in Treg as compared to Tcon. On the other hand, Stat5 phosphorylation might be important for the generation of Treg, through its role in Foxp3 transcriptional regulation59, but it might be dispensable once the Treg are already determined. Conditional knockout or silencing of Stat5 in already differentiated Tregs could help to address these issues.
One important question, not yet addressed, is how ruxolitinib affects the suppressive capacity of Treg. Our in vitro assays indicate that the suppressive capacity is not reduced, and might even be increased, producing more than additive effect. This result together with the fact that ruxolitinib favors the Treg to Tcon ratio both in vitro and in vivo is highly suggestive of a synergistic effect for the treatment of GvHD.
We tested this concept using a GvHD mouse model that reflects the course of the disease in the clinical setting, with an acute phase with high mortality in the early stages followed by a recovery and the subsequent development of a second phase, with characteristics of both acute GvHD, such as weight loss, and chronic GvHD, like skin damage and fibrosis60. In contrast to most preclinical studies using ruxolitinib or Treg, we have not started the treatment simultaneously to the BM and splenocytes transplantation but once the early acute phase in our model is passed and the second phase has started to show clinical signs. The fact that GvHD is already in progress instead of using it “prophylactically” might hamper response to treatment, and therefore, in our opinion, makes the results of our study more relevant. In addition to that, we have used reduced doses of both ruxolitinib (30 mg/kg-day compared to the standard dose of 60 mg/kg-day), and 1:6 Treg: splenocyte ratio, instead to the 1:1 or 1:2 ratio used in most studies. We have used these lower doses to detect the possible additive or synergistic effect of both treatment, and to demonstrate that a reduction of both treatments in the combined setting could be beneficial for the patients, reducing side effects of each treatment alone and facilitating to obtain enough Treg. With these settings, we have been able to determine that the combined treatment of GvHD with ruxolitinib and Treg, starting with the disease already established, can outperform the individual treatments, achieving significant higher survival, better clinical scores and lower weight loss, without affecting the Graft versus Leukemia effect. We have also been able to detect the infused GFP+ Treg as long as 70 weeks after infusion, in the bone marrow of Treg treated mice, demonstrating the long-term persistence of the infused Treg. These results have supported the development of a clinical trial, using donor Treg to treat GvHD patients who respond partially to ruxolitinib (NCT03683498).
Methods
Human peripheral-blood mononuclear cells (huPBMNCs) purification
Human buffy coats from healthy donors were collected from the Andalusian Health System Biobank with approval of the ethics committee of the University Hospital Virgen del Rocío (1116-n-17). All participants provided informed consent for the use of the samples and all procedures were done in accordance with the Spanish and European regulation and guidelines for research with human samples, and the Declaration of Helsinki. huPBMNCs were purified by ficoll gradient centrifugation.
Cell culture
huPBMNCs were cultured in RPMI-1640 supplemented with 10% Human AB Serum, Penycilin-Streptomycin and Glutamax. Cells were seeded at a density of 106 cell/ml in 48 well plates. Stimulation was produced with plate bound anti-humanCD3 (BD) (0.5 µg/ml), and soluble anti-humanCD28 (BD) (0.25 µg/ml). Cells were incubated at 37 °C, 5% CO2 for the indicated times. Ruxolitinib (INCB018424) was kindly provided by NOVARTIS, and stored in a DMSO stock at 10 mg/ml at – 20 °C.
Cytometry
Cells were collected, centrifuged and washed in PBS with 2% FCS. For Surface staining, cells were incubated with the corresponding fluorochrome conjugated antibodies (see Supplementary Table 1) for 15 min. at R.T. in the dark. Cells were washed with PBS with 2% FCS, and proceeded to FACs acquisition and analysis, or were processed for intracellular staining. For Intracellular Foxp3 and Helios staining, the Foxp3 staining kit (eBioscience) was used according to the manufacturer’s instructions. For IL10 staining, cells were treated with 10 µg/ml Brefeldin A (Sigma) for 4 h prior to staining. For Phospho-Stat3 and Phospho-Stat5, cells were stained using the BD Phosflow™ T Cell Activation Kit (BD), following manufacturer instructions. Data was acquired in a BD FACS Canto II cytometer, and analyzed using FlowJo v.X software. A minimum of 50,000 events were recorded for each sample. For absolute quantification, 123Count eBeads (Invitrogen) were added to the culture in a 1:50 bead:cell ratio.
In vitro suppression assays
Human Tregs were isolated from human healthy donors using the CD4+ CD25+ CD127dim/− Regulatory T Cell Isolation Kit II, human (Miltenyi) with an AUTOMACS (Miltenyi) magnetic separator. huPBMNCs responder cells were stained with Carboxyfluorescein succinimidyl ester (CFSE, Invitrogen). 5 × 104 responder were seeded in 96-well plates, and stimulated with anti-humanCD3 and anti-humanCD28. Tregs were added at decreasing ratios, in the presence or absence of ruxolitinib. Proliferation was measured after 5 days by flow cytometry as dilution of CFSE staining. The percentage of suppression was calculated as {1 − (%proliferation in the sample/%proliferation in the control)} × 100%.
Mice
BALB/c (H-2d) and C57BL/6 (H-2b) mice were purchased from Charles River Laboratories (Morrisville, NC). The green fluorescent protein (GFP) C57BL/6-Tg(ACTB-EGFP)1Osb/J) (H-2b)61 were housed in the animal facility of the IBiS. Mice between 7 and 14 weeks old were used. All procedures were approved by the Institutional Animal Care and Use Committee at Institute of Biomedicine in Seville (approval number 09/07/2019/125), and were carried out in compliance with the ARRIVE guidelines.
Mouse model of GvHD
8–12 week old BALB/c (H-2d) recipient mice were irradiated at day 0 with 800–860 cGy split it two doses separated by 3 h. Mice were transplanted with 5 × 106 BM cells and 2 × 106 splenocytes depleted of monocytes by culturing them for 2 h, from C57BL/6 (H-2b) HLA mismatched donors. After transplantation, first phase of acute GvHD is developed with a moderate percentage of mortality, and after a short recovery period, the surviving mice present a second phase of chronic GvHD56. At day 28 post-transplantation, surviving mice were randomized into four treatment groups, receiving a: 3 × 105 Tregs isolated from a GFP transgenic mice isogenic to the donor (C57Bl/6-Tg(ACTB-EGFP)1Osb/J). b: ruxolitinib (30 mg/Kg of body weight once a day) via oral gavage, 5 days a week plus two resting days, c: 3 × 105 Tregs plus ruxolitinib (30 mg/kg once a day) and d: control mice receiving the vehicle of the ruxolitinib and no Treg infusion. Ruxolitinib was prepared at 6 mg/ml in 1:3 PEG 3000:5% Dextrose and administered via oral gavage. Treg were freshly isolated from C57Bl/6-Tg(ACTB-EGFP)1Osb/J) mice spleens using the CD4+ CD25+ Regulatory T Cell Isolation Kit, mouse (Miltenyi). Acute GvHD score was assigned as previously described62. Chronic GvHD was adapted from Anderson et al.63: Skin damage with fur loss, less than 1 cm2 = 1, between 1 and 2.5 cm2 = 2, more than 2.5 cm2 = 3. Additionally, 0.2 for scaling in the tail, 0.3 points for ear damage and 0.5 points for eye lesions. At week 16 after BM transplantation, animals were sacrificed and exsanguinated. Organs of interest (spleen, liver, skin, Peyer’s patches, small intestine, colon, lung, BM, and thymus) were collected and fixed for histopathological examination. Histopathological scores were assigned by a pathologist according to published scoring system56. Cells from peripheral blood, BM, Spleen, Peyer’s patches, large intestine and small intestine were extracted for cytometry analysis as described56.
Mouse model of graft vs. leukemia
8–12 weeks old BALB/c mice were lethally irradiated with 800 cGy split in two doses separated by 3 h. 4 h after irradiation, mice were transplanted with 5 × 106 BM cells, 2 × 106 splenocytes, depleted from monocytes, and 3 × 105 freshly purified Tregs from C57BL/6 donors, and 106 A20 leukemic cells transduced with a GFP-Luciferase vector64, depending on the treatment group. Ruxolitinib (30 mg/kg day) or vehicle was administered via oral gavage from day + 1 until the end of the experiment. Luminescence was measured at days + 7 and + 13 using a IVIS Lumina III in vivo imaging system (PerkinElmer, Massachusetts, USA) as previously described64.
Statistics
Data was analyzed using GraphPad PRISM 7.03. Graphs represent Mean and Standard Error of the Mean (S.E.M). Statistical comparisons were made using Student’s t test, one or two way ANOVA test whenever appropriate. Shapiro Wilk test was used to check for normality. Survival curves were represented using the Kaplan Meyer method, and the Log-Rank test was used to determine statistical differences. p values: *< 0.05, **< 0.01, ***< 0.001, ****< 0.0001.
Supplementary Information
Acknowledgements
The authors thank Dr. João Lacerda for critical reading of the manuscript. This work was supported by grants from Novartis and the Andalusian Regional Government (P18-RT-4047, PI-0052-2018). A.R.G. and J.A.P.S. are members of CIBERONC (CB16/12/00480) and TerCel (16/0011/0035). J.V. is member of CIBERNED (CB06/05/0027). A.R.G. is funded by a Grant of the University of Seville (US-1380874) co-funded by the European Regional Development Fund (ERDF).
Author contributions
A.R.G., V.E.G. and J.A.P.S. designed the project. A.R.-G., V.E.G., M.N., F.A.S., J.A.B.G. performed the experiments. T.L.R. developed the mouse model. J.V. provided essential materials. E.G.G., C.C.C., T.C.V., C.B.G.C., P.H.D., J.L.R.O., N.R.T. and N.M.C. developed the clinical trial. A.R.G., V.E.G., J.I.R.B. and J.A.P.S. analyzed the data. A.R.G., J.I.R.B. and J.A.P.S. wrote the paper. All authors reviewed the manuscript.
Funding
This study is partially funded by Novartis.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-12407-x.
References
- 1.Blazar BR, Murphy WJ, Abedi M. Advances in graft-versus-host disease biology and therapy. Nat. Rev. Immunol. 2012;12:443–458. doi: 10.1038/nri3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pérez-Simón JA, et al. Prognostic factors of chronic graft-versus-host disease following allogeneic peripheral blood stem cell transplantation: The National Institutes Health Scale Plus the type of onset can predict survival rates and the duration of immunosuppressive therapy. Biol. Blood Marrow Transplant. 2008;14:1163–1171. doi: 10.1016/j.bbmt.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 3.Pérez-Simón JA, Sanchez-Abarca I, Diez-Campelo M, Caballero D, San Miguel J. Chronic graft-versus-host disease: Pathogenesis and clinical management. Drugs. 2006;66:1041–1057. doi: 10.2165/00003495-200666080-00002. [DOI] [PubMed] [Google Scholar]
- 4.Negrin RS. Graft-versus-host disease versus graft-versus-leukemia. Hematology. 2015;2015:225–230. doi: 10.1182/asheducation-2015.1.225. [DOI] [PubMed] [Google Scholar]
- 5.Maude SL, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125:1759–1767. doi: 10.1182/blood-2014-06-580480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Das R, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood. 2016;127:1666–1675. doi: 10.1182/blood-2015-12-684399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shide K, et al. Calreticulin mutant mice develop essential thrombocythemia that is ameliorated by the JAK inhibitor ruxolitinib. Leukemia. 2017;31:1136–1144. doi: 10.1038/leu.2016.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gallipoli P, et al. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood. 2014;124:1492–1501. doi: 10.1182/blood-2013-12-545640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karjalainen R, et al. JAK1/2 and BCL2 inhibitors synergize to counteract bone marrow stromal cell-induced protection of AML. Blood. 2017;130:789–802. doi: 10.1182/blood-2016-02-699363. [DOI] [PubMed] [Google Scholar]
- 10.Quintas-Cardama A, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: Therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Appelmann I, et al. Janus kinase inhibition by ruxolitinib extends dasatinib- and dexamethasone-induced remissions in a mouse model of Ph+ ALL. Blood. 2015;125:1444–1451. doi: 10.1182/blood-2014-09-601062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ostojic A, Vrhovac R, Verstovsek S. Ruxolitinib: A new JAK1/2 inhibitor that offers promising options for treatment of myelofibrosis. Future Oncol. 2011;7:1035–1043. doi: 10.2217/fon.11.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kong Y, et al. Ruxolitinib/nilotinib cotreatment inhibits leukemia-propagating cells in Philadelphia chromosome-positive ALL. J. Transl. Med. 2017;15:184. doi: 10.1186/s12967-017-1286-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeiser R, et al. Ruxolitinib for glucocorticoid-refractory acute graft-versus-host disease. N. Engl. J. Med. 2020;382:1800–1810. doi: 10.1056/NEJMoa1917635. [DOI] [PubMed] [Google Scholar]
- 15.Jagasia M, et al. Ruxolitinib for the treatment of steroid-refractory acute GVHD (REACH1): A multicenter, open-label phase 2 trial. Blood. 2020;135:1739–1749. doi: 10.1182/blood.2020004823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spoerl S, et al. Activity of therapeutic JAK 1/2 blockade in graft-versus-host disease. Blood. 2014;123:3832–3842. doi: 10.1182/blood-2013-12-543736. [DOI] [PubMed] [Google Scholar]
- 17.Zeiser R, et al. Ruxolitinib for glucocorticoid-refractory chronic graft-versus-host disease. N. Engl. J. Med. 2021;385:228–238. doi: 10.1056/NEJMoa2033122. [DOI] [PubMed] [Google Scholar]
- 18.Maffini E, et al. Ruxolitinib in steroid refractory graft-vs-host disease: A case report. J. Hematol. Oncol. 2016;9:67. doi: 10.1186/s13045-016-0298-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carniti C, et al. Pharmacologic inhibition of JAK1/JAK2 signaling reduces experimental murine acute GVHD while preserving GVT effects. Clin. Cancer Res. 2015;21:3740–3749. doi: 10.1158/1078-0432.CCR-14-2758. [DOI] [PubMed] [Google Scholar]
- 20.Choi J, et al. Pharmacologic blockade of JAK1/JAK2 reduces GvHD and preserves the graft-versus-leukemia effect. PLoS ONE. 2014;9:2–7. doi: 10.1371/journal.pone.0109799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi S, et al. Ruxolitinib protects skin stem cells and maintains skin homeostasis in murine graft-versus-host disease. Blood. 2018;131:2074–2085. doi: 10.1182/blood-2017-06-792614. [DOI] [PubMed] [Google Scholar]
- 22.Khoury HJ, et al. Ruxolitinib: A steroid sparing agent in chronic graft-versus-host disease. Bone Marrow Transplant. 2018;53:826–831. doi: 10.1038/s41409-017-0081-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeiser R, et al. Ruxolitinib in corticosteroid-refractory graft-versus-host disease after allogeneic stem cell transplantation: A multi-center survey. Leukemia. 2015;22:121–123. doi: 10.1038/leu.2015.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whangbo JS, et al. Dose-escalated interleukin-2 therapy for refractory chronic graft-versus-host disease in adults and children. Blood Adv. 2019;3:2550–2561. doi: 10.1182/bloodadvances.2019000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mori Y, et al. Ruxolitinib treatment for GvHD in patients with myelofibrosis. Bone Marrow Transplant. 2016;51:1584–1587. doi: 10.1038/bmt.2016.256. [DOI] [PubMed] [Google Scholar]
- 26.Di Ianni M, et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood. 2011;117:3921–3928. doi: 10.1182/blood-2010-10-311894. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, et al. Amelioration of acute graft-versus-host disease by adoptive transfer of ex vivo expanded human cord blood CD4+CD25+ forkhead box protein 3+ regulatory T cells is associated with the polarization of Treg/Th17 balance in a mouse model. Transfusion. 2012;52:1333–1347. doi: 10.1111/j.1537-2995.2011.03448.x. [DOI] [PubMed] [Google Scholar]
- 28.Ermann J, et al. Only the CD62L + subpopulation of CD4 +CD25 + regulatory T cells protects from lethal acute GVHD. Blood. 2005;105:2220–2226. doi: 10.1182/blood-2004-05-2044. [DOI] [PubMed] [Google Scholar]
- 29.June CH, Blazar BR. Clinical application of expanded CD4+25+ cells. Semin. Immunol. 2006;18:78–88. doi: 10.1016/j.smim.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Martelli MF, et al. HLA-haploidentical transplantation with regulatory and conventional T-cell adoptive immunotherapy prevents acute leukemia relapse. Blood. 2014;124:638–644. doi: 10.1182/blood-2014-03-564401. [DOI] [PubMed] [Google Scholar]
- 31.Mancusi A, Piccinelli S, Velardi A, Pierini A. CD4+FOXP3+ regulatory T cell therapies in HLA haploidentical hematopoietic transplantation. Front. Immunol. 2019;10:1–11. doi: 10.3389/fimmu.2019.02901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parmar S, et al. Third-party umbilical cord blood–derived regulatory T cells prevent xenogenic graft-versus-host disease. Cytotherapy. 2014;16:90–100. doi: 10.1016/j.jcyt.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trzonkowski P, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127− T regulatory cells. Clin. Immunol. 2009;133:22–26. doi: 10.1016/j.clim.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 34.Hannon M, et al. Infusion of clinical-grade enriched regulatory T cells delays experimental xenogeneic graft-versus-host disease. Transfusion. 2014;54:353–363. doi: 10.1111/trf.12666. [DOI] [PubMed] [Google Scholar]
- 35.Hippen KL, et al. Generation and large-scale expansion of human inducible regulatory T cells that suppress graft-versus-host disease. Am. J. Transplant. 2011;11:1148–1157. doi: 10.1111/j.1600-6143.2011.03558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramlal R, Hildebrandt GC. Advances in the use of regulatory T-cells for the prevention and therapy of graft-vs-host disease. Biomedicines. 2017;5:23. doi: 10.3390/biomedicines5020023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohkura N, Kitagawa Y, Sakaguchi S. Development and maintenance of regulatory T cells. Immunity. 2013;38:414–423. doi: 10.1016/j.immuni.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 38.Mikami N, Kawakami R, Sakaguchi S. New Treg cell-based therapies of autoimmune diseases: Towards antigen-specific immune suppression. Curr. Opin. Immunol. 2020;67:36–41. doi: 10.1016/j.coi.2020.07.004. [DOI] [PubMed] [Google Scholar]
- 39.Thornton AM, et al. Expression of helios, an ikaros transcription factor family member, differentiates thymic-derived from peripherally induced Foxp3+ T regulatory cells. J. Immunol. 2010;184:3433–3441. doi: 10.4049/jimmunol.0904028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thornton AM, et al. Helios+ and Helios− Treg subpopulations are phenotypically and functionally distinct and express dissimilar TCR repertoires. Eur. J. Immunol. 2019;49:398–412. doi: 10.1002/eji.201847935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salvany-Celades M, et al. Three types of functional regulatory T cells control T cell responses at the human maternal-fetal interface. Cell Rep. 2019;27:2537–2547. doi: 10.1016/j.celrep.2019.04.109. [DOI] [PubMed] [Google Scholar]
- 42.Tan CL, et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J. Exp. Med. 2021 doi: 10.1084/jem.20182232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jain N, Nguyen H, Chambers C, Kang J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc. Natl. Acad. Sci. 2010;107:1524–1528. doi: 10.1073/pnas.0910341107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gu J, et al. Human CD39hi regulatory T cells present stronger stability and function under inflammatory conditions. Cell. Mol. Immunol. 2017;14:521–528. doi: 10.1038/cmi.2016.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deaglio S, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Canavan JB, et al. Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut. 2016;65:584–594. doi: 10.1136/gutjnl-2014-306919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoffmann P, et al. Only the CD45RA+ subpopulation of CD4+CD25high T cells gives rise to homogeneous regulatory T-cell lines upon in vitro expansion. Blood. 2006;108:4260–4267. doi: 10.1182/blood-2006-06-027409. [DOI] [PubMed] [Google Scholar]
- 48.Booth NJ, et al. Different proliferative potential and migratory characteristics of human CD4+ regulatory T cells that express either CD45RA or CD45RO. J. Immunol. 2010;184:4317–4326. doi: 10.4049/jimmunol.0903781. [DOI] [PubMed] [Google Scholar]
- 49.Moy RH, et al. Clinical and immunologic impact of CCR5 blockade in graft-versus-host disease prophylaxis. Blood. 2017;129:906–916. doi: 10.1182/blood-2016-08-735076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schreder A, et al. Differential effects of gut-homing molecules CC chemokine receptor 9 and integrin-β7 during acute graft-versus-host disease of the liver. Biol. Blood Marrow Transplant. 2015;21:2069–2078. doi: 10.1016/j.bbmt.2015.08.038. [DOI] [PubMed] [Google Scholar]
- 51.Tan MCB, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J. Immunol. 2009;182:1746–1755. doi: 10.4049/jimmunol.182.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi J, et al. IFNγR signaling mediates alloreactive T-cell trafficking and GVHD. Blood. 2012;120:4093–4103. doi: 10.1182/blood-2012-01-403196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parampalli Yajnanarayana S, et al. JAK1/2 inhibition impairs T cell function in vitro and in patients with myeloproliferative neoplasms. Br. J. Haematol. 2015;169:824–833. doi: 10.1111/bjh.13373. [DOI] [PubMed] [Google Scholar]
- 54.Yao Z, et al. Nonredundant roles for Stat5a/b in directly regulating Foxp. Blood. 2007;109:4368–4375. doi: 10.1182/blood-2006-11-055756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Teshima T. JAK inhibitors: A home run for GVHD patients? Blood. 2014;123:3691–3693. doi: 10.1182/blood-2014-04-570325. [DOI] [PubMed] [Google Scholar]
- 56.Ramos TL, et al. Delayed administration of ixazomib modifies the immune response and prevents chronic graft-versus-host disease. Bone Marrow Transplant. 2021;56:3049–3058. doi: 10.1038/s41409-021-01452-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Edinger M, et al. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat. Med. 2003;9:1144–1150. doi: 10.1038/nm915. [DOI] [PubMed] [Google Scholar]
- 58.Miyao T, et al. Plasticity of Foxp3 + T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36:262–275. doi: 10.1016/j.immuni.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 59.Zorn E, et al. IL-2 regulates FOXP3 expression in human CD4+CD25+ regulatory T cells through a STAT-dependent mechanism and induces the expansion of these cells in vivo. Blood. 2006;108:1571–1579. doi: 10.1182/blood-2006-02-004747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramos T, et al. Pre-clinical trial to evaluate the efficacy of delayed administration of ixazomib in the prophylaxis of chronic graft-versus-host disease. Blood. 2018;132:4521–4521. doi: 10.1182/blood-2018-99-118098. [DOI] [Google Scholar]
- 61.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. ‘Green mice’ as a source of ubiquitous green cells. FEBS Lett. 1997;407:313–319. doi: 10.1016/S0014-5793(97)00313-X. [DOI] [PubMed] [Google Scholar]
- 62.Cooke KR, et al. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood. 1996;88:3230–3239. doi: 10.1182/blood.V88.8.3230.bloodjournal8883230. [DOI] [PubMed] [Google Scholar]
- 63.Anderson BE, et al. Memory CD4+ T cells do not induce graft-versus-host disease. J. Clin. Investig. 2003;112:101–108. doi: 10.1172/JCI17601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carrancio S, et al. Effects of MSC coadministration and route of delivery on cord blood hematopoietic stem cell engraftment. Cell Transplant. 2013;22:1171–1183. doi: 10.3727/096368912X657431. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.