

Abstract
Neurovascular dysfunction, as an integral part of Alzheimer’s disease, may have an important influence on the onset and progression of chronic neurodegenerative processes. The blood-brain barrier (BBB) pathway is one of the main pathways that mediates the clearance of amyloid-beta (Aβ) in the brain parenchyma. A large number of studies have shown that receptors and ATP-binding cassette transporters expressed on endothelial cells play an important role in Aβ transport across the BBB, but the specific mechanism is not clear. In this review, we summarize the possible mechanisms of Aβ production and clearance, and in particular the relationship between Aβ and brain capillary endothelial cells. Aβ is produced by abnormal cleavage of the amyloid precursor protein via amyloidogenic processing under pathological conditions. Dysregulation of Aβ clearance is considered to be the main reason for the massive accumulation of Aβ in the brain parenchyma. Several pathways mediating Aβ clearance from the brain into the periphery have been identified, including the BBB pathway, the blood-cerebrospinal fluid barrier and arachnoid granule pathway, and the lymphoid-related pathway. Brain capillary endothelial cells are the key components of Aβ clearance mediated by BBB. Receptors (such as LRP1, RAGE, and FcRn) and ATP-binding cassette transporters (such as P-gp, ABCA1, and ABCC1) expressed on endothelial cells play a critical role in Aβ transcytosis across the BBB. The toxic effects of Aβ can induce dysregulation of receptor and transporter expression on endothelial cells. Excessive Aβ exerts potent detrimental cerebrovascular effects by promoting oxidative stress, inducing chronic inflammation, and impairing endothelial structure and functions. All of these are main causes for the reduction in Aβ clearance across the BBB and the accumulation of Aβ in the brain parenchyma. Therefore, studies on the interactions between Aβ and brain capillary endothelial cells, including their receptors and transporters, studies on inhibition of the toxic effects of Aβ on endothelial cells, and studies on promoting the ability of endothelial cells to mediate Aβ clearance may provide new therapeutic strategies for Aβ clearance in Alzheimer’s disease.
Key Words: Alzheimer’s disease, amyloid beta, Aβ clearance, blood-brain barrier, cerebral amyloid angiopathy, dementia, endothelial cells, oxidative stress, review, therapeutics, transcytosis
Introduction
Alzheimer’s disease (AD) is a degenerative disease of the central nervous system (CNS) that occurs primarily in elderly individuals. It is characterized by progressive cognitive decline, memory impairment, and behavioral impairment, and accounts for 60–80% of all cases of dementia (Pflanzner et al., 2010). Given the aging of society, it is estimated that there will be more than 140 million AD patients globally by 2050, which will impose a heavy burden on both society and families of affected individuals (Tiwari et al., 2019). Familial AD (early-onset) is a rare, autosomal dominant disorder (Chai et al., 2020b; Cheng et al., 2020). Impaired amyloid beta (Aβ) clearance in sporadic (late-onset) AD, which accounts for the majority of all AD cases, is hypothesized to be the main cause of Aβ accumulation (Selkoe and Hardy, 2016).
AD is characterized pathologically by extracellular aggregation of Aβ plaques, intracellular aggregation of neurofibrillary tangles consisting of hyperphosphorylated tau protein, inflammation, microglial activation, cerebrovascular pathology, and neuronal cell death. In both familial and sporadic AD, abnormal accumulation of Aβ precedes neurodegeneration and cognitive impairment (Selkoe and Hardy, 2016; Aisen et al., 2017; Cheng et al., 2020).
The widely acknowledged amyloid hypothesis states that overproduction or impaired clearance of Aβ triggers a cascade of events that causes neuronal damage and death, manifesting as progressive clinical dementia. Aβ plays a causal role in driving the pathogenesis of AD (Selkoe and Hardy, 2016; d’Uscio et al., 2017). It is deficient Aβ clearance rather than Aβ overproduction that leads to aberrant aggregation of Aβ in the brain parenchyma (Aβ plaques) (Iturria-Medina et al., 2016). Therefore, improving Aβ clearance from the brain is a promising strategy for the treatment of AD (Cheng et al., 2020). Impaired Aβ clearance across the brain-blood barrier (BBB) plays a crucial role in the pathogenesis of AD (Yamazaki and Kanekiyo, 2017). Endothelial cells play an important role in this process. In addition, more than 80% of AD brains consistently display cerebral amyloid angiopathy (CAA), characterized by the deposition of Aβ along the cerebrovascular and pia meningeal vascular walls (Greenberg et al., 2020). Although CAA is clinically recognized as a distinct phenomenon from AD, their common cerebrovascular and neurodegenerative characteristics suggest a mechanistic link (Charidimou et al., 2017). Aβ deposition in cerebrovascular structures also causes significant damage to brain capillary endothelial cells, which triggers neurovascular inflammation, contributing to neurodegeneration (Nelson et al., 2016; Solis et al., 2020). Brain capillary endothelial cells play an important role in clearance of Aβ. The aim of this review is to address the mechanisms of Aβ production and clearance, in particular the relationship between Aβ and brain capillary endothelial cells (Figure 1), and the potential for designing novel targeting clinical therapeutics that promote Aβ clearance.
Figure 1.
The relationship between amyloid beta (Aβ) and brain capillary endothelial cells.
BBB: Brain-blood barrier.
Literature Search
PubMed was searched to retrieve studies published up to August 2021. The literature search included not only article texts, but also reference lists and supplementary files. The articles cited in this review were retrieved by replicating the search terms from the study by Behl-Tapan et al. (2021). A wide range of related search terms were used, including: Alzheimer’s disease, amyloid beta, blood-brain barrier (BBB), cerebral amyloid angiopathy, clearance, dementia, endothelial cells, therapeutics, transcytosis, and oxidative stress.
Amyloid Precursor Protein and Amyloid-Beta
AD is an important protein conformational disease (Leandro and Gomes, 2008; Adav and Sze, 2016) that is mainly caused by abnormal processing and polymerization of normal soluble proteins (Tran and Ha-Duong, 2015). Amyloid precursor protein (APP) is a transmembrane protein with extracellular domains that is highly expressed in the brain (Lopez Sanchez et al., 2019). APP and its non-amyloidogenic cleavage products (especially soluble APPα) play a neuroprotective physiological role by regulating synaptic plasticity (Steubler et al., 2021), cellular and mitochondrial metabolism (including intracellular calcium homeostasis), intraneuronal iron metabolism (Tsatsanis et al., 2020), and apoptosis signaling (Pei and Wallace, 2018; Lopez Sanchez et al., 2019). These metabolic processes are related to a variety of neurodegenerative diseases, including AD (Lionaki et al., 2015; Zorzano and Claret, 2015). Under normal conditions, APP undergoes non-amyloidogenic processing that involves constitutive and regulated cleavage by α-secretase and γ-secretase (Tiwari et al., 2019), whereas in a diseased state, APP undergoes abnormal cleavage by β-secretase, resulting in the release of a shorter ectodomain, sAPPβ (N-terminal fragment), and retention of C99 (C-terminal fragment) in the membrane. C99 is then further cleaved by γ-secretase, releasing the APP intracellular domain, and the soluble Aβ peptides further polymerize to form aggregated plaques.
Aβ is a polypeptide containing 39 to 43 amino acid residues that is produced by sequential cleavage of APP in a diseased state (Bonda et al., 2011). The two main peptide subtypes that play a direct role in inducing neurotoxicity are Aβ40, which is 40 amino acids long, and Aβ42, which is 42 amino acids long (Humpel, 2015). Aβ40 is generated in greater quantities and is less neurotoxic, while Aβ42 is less abundant and highly insoluble, causes serious neurotoxicity, and is more aggregation-prone (Ballard et al., 2011; Brothers et al., 2018). Aβ40 and Aβ42 exist in various forms, such as monomers, soluble oligomers, protofibrils, insoluble fibers, and Aβ plaques composed of mature insoluble fibers (Steiner et al., 2018). The tendency of Aβ to aggregate is concentration-dependent (Verma et al., 2015): at low nanomolar concentrations, it exists as monomers. Oligomers of Aβ, especially Aβ42, are toxic (Hladky and Barrand, 2018, McInerney et al., 2017).
Current evidence suggests that oligomers within the brain parenchyma are the main culprits leading to neurodegeneration (Nisbet et al., 2015, McIntee et al., 2016, Hladky and Barrand, 2018). The most prominent isoform of Aβ found in vascular amyloid deposits is Aβ40, while Aβ42 is the main form found in neuronal plaques in the brain parenchyma (d’Uscio et al., 2017). Aβ plaques initially form in the basal, temporal lobe and the orbitofrontal neocortex, and later spread to the diencephalon, basal ganglia, hippocampus, and amygdala (Tiwari et al., 2019). Both intracellular and extracellular Aβ accumulation have been demonstrated to blockade long-term potentiation, drive synaptic dysfunction, spread oxidative stress and neuroinflammation, facilitate tau phosphorylation, deteriorate neuronal health, and finally lead to neuronal death (Kayed and Lasagna-Reeves, 2013; Viola and Klein, 2015; Cline et al., 2018).
In vitro studies have shown that Aβ can bind to many transport proteins, such as albumin (Kim et al., 2020; Su et al., 2021), apolipoprotein J (apoJ), apolipoprotein E (apoE), transthyretin, and α2-macroglobulin (α2M) (Tokuda et al., 2000; Sousa et al., 2007). However, in human plasma, a soluble form of low density lipoprotein receptor (LDLR)-related protein 1 (SLRP1) sequesters plasma Aβ and is the main binding protein of circulating Aβ (Xu et al., 2021).
In the majority of patients with late-onset sporadic AD, the rate of Aβ production remains relatively constant, but the clearance rate decreases significantly, suggesting that that impaired Aβ clearance, rather than Aβ overproduction, leads to its accumulation (Su et al., 2021). Therefore, dysregulation of Aβ clearance is considered to be the main reason for the massive accumulation of Aβ and the key factor leading to the occurrence and development of AD.
Clearance of Amyloid-Beta
Under normal conditions, after Aβ is hydrolyzed by APP in the brain, it can be eliminated by several mechanisms, including intracellular clearance (ubiquitin-proteasome pathway and autophagy-lysosome pathway), extracellular enzymatic degradation, phagocytosis and uptake by microglial cells or astrocytes, transport to peripheral organs, and tissue clearance. Extracellular Aβ in the brain parenchyma can be degraded by proteases secreted by neurons and astrocytes, such as insulin-degrading enzyme, neprilysin, and angiotensin-converting enzyme (Su et al., 2021). It can also be recognized and phagocytized by microglia and astrocytes (Miners et al., 2008). However, it has been reported that the degree of degradation of free Aβ in the brain parenchyma is insignificant (Deane et al., 2004, 2008). sLRP1 binds 70–90% of Aβ found in the plasma, thus acting as a peripheral ‘sink’ to inhibit Aβ reentry into the brain (Tholen et al., 2016; Jin et al., 2017; Xu et al., 2021).
Several pathways mediating Aβ clearance from the brain to the periphery have been identified, the including BBB pathway (Zhao et al., 2015, Nelson et al., 2016, Storck et al., 2016), the blood-cerebrospinal fluid (CSF) barrier and arachnoid granule pathway, and the lymphoid-related pathway. Substantial evidence shows that the BBB pathway is of crucial importance for rapid removal of Aβ from the brain (Cai et al., 2018, Hladky and Barrand, 2018, Cheng and Haorah, 2019).
Blood-brain barrier pathway
Numerous studies have shown that pathological changes in the neurovascular units constituting the BBB contribute to the occurrence and development of AD, and early BBB dysfunction before neurodegeneration and/or brain atrophy has been shown to occur in AD. Therefore, there is a close relationship between BBB dysfunction and neurodegeneration (Cai et al., 2017; Sweeney et al., 2018, 2019).
Approximately 400 miles’ worth of capillaries account for more than 85% of the total length of the cerebral blood vessels in humans, providing a large microvascular surface area (approximately 20 square meters) for molecular transport between blood and the brain (Begley and Brightman, 2003) and contributing to the rapid clearance of up to 70% of excessive brain-derived Aβ (Begley and Brightman, 2003). This makes the BBB the largest surface area in the brain for exchanging and transporting substances and the main pathway for clearance of potential neurotoxic molecules (such as Aβ) produced and/or accumulated in the brain (Ramanathan et al., 2015).
The BBB is a complex cellular system composed of endothelial cells, astrocytes, pericytes, perivascular macrophages, and basement membrane (Sweeney et al., 2019). Brain capillary endothelial cells are connected by tight junction proteins and cytoplasmic accessory proteins to form a continuous endothelial monolayer (Nelson et al., 2016). The physical barrier structures in endothelial cells are essential for the regulation of many cellular functions and play a vital role in maintaining BBB permeability and brain parenchyma homeostasis (Montagne et al., 2017). At the molecular level, endothelial transporters regulate the inflow and outflow of specific molecules at the BBB (Zlokovic, 2008a; Daneman and Prat, 2015; Liebner et al., 2018).
The efficiency of Aβ clearance across the BBB is influenced by Aβ-binding transporter proteins such as apoE and clusterin (apoJ) and receptors such as LRP1 and transporters such as P-glycoprotein (P-gp), which are expressed on endothelial cells and control Aβ efflux from and influx into the brain, respectively.
Blood-CSF barrier and arachnoid granule-venous sinus pathway
The CSF-arachnoid granule-venous sinus pathway plays a lymphoid role by effectively removing metabolic waste from the brain parenchyma. Aβ in CSF passes through the blood-cerebrospinal fluid barrier (BCSFB), mediated by receptors on the choroid plexus epithelium (such as LRP1 and P-gp) (Fujiyoshi et al., 2011; Pascale et al., 2011).
CSF can be drained through arachnoid villi and granulations directly to the peripheral blood (Weller et al., 2009). Some researchers have suggested that this may occur through gaps in endothelial cells and/or by hydrostatic pressure-dependent pinocytosis (Bothwell et al., 2019). In addition, the rate of CSF production and circulation, as well as the integrity of the BCSFB, influence Aβ removal.
Lymphoid-related pathway
The lymphatic-related drainage pathways include the meningeal lymphatics, perineural outflow pathways, the basement membrane-based perivascular pathway, and the “glymphatic” pathway (Sun et al., 2018).
CSF is drained to the deep cervical lymph nodes through the meningeal lymphatics. Although interruption of the meningeal lymphatics can accelerate AD pathology in animal models, the mechanism by which the meningeal lymphatics promote Aβ removal from the brain is not clear (Da Mesquita et al., 2018). Emerging evidence suggests that CSF flows out of the cranial nerves. Perineural outflow pathways along the olfactory nerve, optic nerve, and trigeminal nerve are thought to be potential drainage pathways for Aβ removal (Ma et al., 2017, 2019).
The perivascular pathway drains interstitial fluid (ISF) from the brain parenchyma to the cervical lymph nodes along the basement membranes of capillary and cerebral artery walls (Carare et al., 2008; Weller et al., 2009). This pathway is significantly weakened in AD and cerebrovascular amyloidosis, resulting in a significant degree of Aβ deposition in the basement membrane (Albargothy et al., 2018). This defect in Aβ clearance is closely related to arterial pulsation, the degree of arteriosclerosis, the expression of apoE4 alleles, the deposition of immune complex, and the occurrence of cerebral vascular amyloidosis (Hawkes et al., 2014; Smith and Verkman, 2018; Sun et al., 2018).
The glymphatic pathway involves convective periarterial influx of CSF and efflux of ISF along venous perivascular and perineuronal spaces, clearing toxic proteinaceous metabolites, including Aβ, out of the CNS (Rasmussen et al., 2018). In addition to cerebral artery pulsation, as mentioned above, sleeping state and posture also affect the glymphatic pathway (Xie et al., 2013; Lee et al., 2015). The expression of aquaporin 4 (AQP4) on glial endfeet has been confirmed to play a key role in facilitating Aβ clearance (Xu et al., 2015). However, the idea that glymphatic function depends on astrocytic AQP4 has recently been called into question (Rasmussen et al., 2018).
In normal brain ISF, the concentration of Aβ is strictly regulated by the rate of APP production, enzymatic degradation within brain, influx into the brain, and efflux from the brain to the periphery (Storck et al., 2016; Chai et al., 2020a). In the next section, we will focus on the transport of Aβ across the BBB as mediated by capillary endothelial cells.
Brain Capillary Endothelial Cell-Mediated Amyloid-Beta Clearance
The BBB insulates the brain from blood flow and promotes the supply of nutrients and the disposal of metabolites by expressing special transporters and transcytotic receptors on polarized endothelial cells (Pflanzner et al., 2010). Clearing Aβ from the brain to blood is a two-step process. Aβ first passes through the abluminal membrane (brain side) of the brain capillary endothelial cells, and then it passes through the luminal membrane (blood side) (Hartz et al., 2010). Recently, several transporters and receptors have been identified as being involved in the transcytosis of Aβ across brain capillary endothelial cells (Figure 2).
Figure 2.
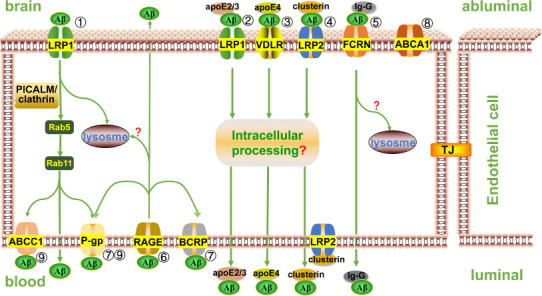
Receptors and transporters mediate Aβ transport across the blood-brain barrier.
① By binding to PICALM, the Aβ-LRP1 complex is integrated into a clathrin-coated pit and then cleared from the brain via transcytosis or directed to lysosome for degradation. ② Aβ-apoE2 or Aβ-apoE3 complex binds to LRP1 and forms endophagocytic vesicles, which are then discharged at the plasma membrane on the other side of the endothelial cell via transcytosis. ③ Aβ-apoE4 complex interacts with very low density lipoprotein receptor (VLDLR) and is slowly endocytosed, then cleared from the endothelial cells by transcytosis. ④ Aβ-clusterin complex can bind to LRP2 receptor on both sides of the endothelial cell membrane for bidirectional transport, but LRP2 is usually occupied by clusterin rather than Aβ-clusterin complex on the luminal side, so the net flow is Aβ outflow. ⑤ FcRn mediates IgG-Aβ transport from the brain into blood via transcytosis. ⑥ Luminally expressed RAGE binds to circulating Aβ and transcytoses it from blood to the brain. Both FcRn- and RAGE-mediated Aβ clearance may be involved in the endosomal/lysosomal pathway. ⑦ P-gp may assist LRP1-mediated Aβ transport and pump Aβ directly into the blood from the lumenal side of endothelial cells. P-gp and BCRP also assist in the excretion of circulating Aβ transported by RAGE into endothelial cells. ⑧ ABCA1 does not directly mediate Aβ trafficking across the BBB; it promotes proteolytic degradation of Aβ in the brain parenchyma and affects the clearance of Aβ across the BBB by regulating lipogenation of apoE. ⑨ LRP1 first mediates Aβ endocytosis, and then Aβ is transported from the abluminal side to the luminal side of the endothelial cells. Subsequently, P-gp and ABCC1 export Aβ from the luminal membrane of endothelial cells to the systemic circulation. ApoE: apolipoprotein E; Aβ: amyloid-beta; BCRP(ABCG2): breast cancer resistance protein; LRP: low density lipoprotein receptor(LDLR)-related protein; P-gp: P-glycoprotein; PICALM: phosphatidylinositol binding clathrin assembly protein; RAGE: advanced glycation end products; VLDLR: very low density lipoprotein receptor.
Receptor-mediated Aβ clearance
The transmembrane cell surface receptors expressed on brain endothelial cells are synthesized in the endoplasmic reticulum and transferred to the Golgi network for further glycosylation before being transported to the plasma membrane to participate in various processes such as cellular signal transduction and ligand internalization. After internalization, the receptor-ligand complex can enter the endosomal/lysosomal pathway that leads to intracellular degradation, and can also be transcytosed and recycled via other pathways (Pflanzner et al., 2010). Currently known receptors involved in Aβ transportation across the BBB are the low density lipoprotein receptor family members RAGE and FcRn (Table 1).
Table 1.
Studies of receptors involved in Aβ transportation across the BBB
| Authors | Experiment models /measures | Conclusion |
|---|---|---|
| Li et al., 2001 | LRP-null Chinese hamster ovary cells | Differential function of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates |
| Deane et al., 2003 | Systemic Aβ infusion and studies in genetically manipulated mice | Aβ interaction with receptor for RAGE-bearing cells in the vessel wall resulted in transport of Aβ across the BBB. |
| Ober et al., 2004 | Using single-molecule fluorescence microscopy to analyze exocytic processes in FcRn-GFP-transfected human endothelial cells | IgG may be bound to FcRn for several seconds after exocytosis. |
| Deane et al., 2008 | Male mice on a C57BL/6 background | ApoE isoforms differentially regulated Aβ clearance from the brain. |
| Storck et al., 2016 | Tamoxifen-inducible deletion of Lrp1 specifically within brain endothelial cells [Slco1c1-CreER(T2) Lrp1(fl/fl) mice] | Brain endothelial-specific Lrp1 deletion reduces plasma Aβ levels and elevates soluble brain Aβ. |
| Fang et al., 2018 | mAPP mice with genetic deletion of RAGE (mAPP/RO) | RAGE-dependent signaling pathway regulated β- and γ-secretase cleavage of APP to generate Aβ. |
| Wang et al., 2018 | db/db mice | Targeted inhibition of RAGE reduced amyloid-β influx across the BBB and improved cognitive deficits in db/db mice. |
| Kariolis et al., 2020 | Human transferrin receptor-engineered mice and cynomolgus monkeys | Fc fragment-mediated transcytosis for central nervous system delivery of biotherapeutics was performed by binding endothelial cell target. |
Aβ: Amyloid-beta; ApoE: apolipoprotein E; BBB: blood-brain barrier; LRP1: low density lipoprotein receptor (LDLR)-related protein 1; RAGE: advanced glycation end products.
The low density lipoprotein receptor family
Members of the low density lipoprotein receptor family are cell surface receptors with similar structure and function. Among them, LDLR-related protein 1 (LRP1), LRP2, and very low density lipoprotein receptor (VLDLR) are involved in endothelial cell-mediated Aβ transport. The mediation of Aβ efflux across the BBB by these receptors involves Aβ forming complexes with soluble factors including apoE and clusterin and interacting with phosphatidylinositol binding clathrin assembly protein (PICALM).
LRP1 is ubiquitously distributed in diverse cell types and is abundantly expressed on capillary endothelial cells, smooth muscle cells, pericytes, astrocytes, and neurons in the CNS (Storck et al., 2016). It is located predominantly on the abluminal membrane of capillary endothelial cells. LRP1 may be superior to other receptors in clearing Aβ, as its endocytosis rate is up to 16 times faster than that of other Aβ receptors, especially at higher concentrations (Li et al., 2001; Deane et al., 2008). Before being shuttled to the surface of the cell, LRP1 is synthesized as a precursor molecule in endoplasmic reticulum and then transferred to the Golgi network to generate a C-terminal transmembrane subunit by furin cleavage that is then noncovalently linked to the N-terminal extracellular subunit. The extracellular domain contains four ligand-binding domains that can bind up to 40 proteins, such as Aβ, apoE, and activated α2M, while in the cytoplasmic tail there are two NPxY motifs that act as dominant endocytosis signals to mediate uptake through clathrin-coated pits and an YXXL motif that promotes rapid endocytosis of LRP1 ligands (Storck et al., 2016).
LRP1 mediates endocytosis of Aβ by binding to Aβ directly or indirectly through its coreceptors or ligands. LRP1 also regulates several pathways that may also influence Aβ endocytosis (Kanekiyo and Bu, 2014). Aβ can bind directly to LRP1 to form the Aβ-LRP1 complex, which can then be integrated into a clathrin-coated pit and endocytosed. Binding to PICALM further stabilizes the complex and directs it to the lysosome for degradation (Sagare et al., 2013; Hladky and Barrand, 2018), or guides it to the luminal side of endothelial cell through a process called transcytosis, where it fuses with the plasma membrane to exocytose Aβ (Deane et al., 2008). Aβ in brain interstitial fluid can also form complexes with apoE, 2, 3, or 4 or with clusterin. Aβ-apoE2 or Aβ-apoE3 complex interacts with LRP1 to form endophagocytic vesicles and is discharged on the plasma membrane on the other side of endothelial cells through transcytosis. In contrast, Aβ-apoE4 complex inhibits LRP1-mediated endocytosis, while Aβ-apoE4 complex can bind to VLDLR and is slowly internalized into endothelial cells. This inhibition and slow transportation lead to the accumulation of Aβ in the brain, which may increase the risk for Alzheimer’s disease (Hladky and Barrand, 2018; Tachibana et al., 2019). Aβ-clusterin complex is the substrate of LRP2-mediated endocytosis, by which Aβ is transported from the brain to blood; it can also be transported in the opposite direction, but on the plasma side, LPR2 is normally saturated by clusterin rather than Aβ-clusterin, which precludes significant influx of Aβ (Li Q et al., 2018), so the net flux of this pathway is from the brain to blood (Sagare et al., 2013). Current studies suggest that endothelial LRP1 plays a key role in Aβ transcytosis across the BBB.
The receptor for advanced glycation end products
The receptor for advanced glycation end products (RAGE), a multiligand receptor that is a member of the immunoglobulin superfamily (Walker et al., 2015), contains one V-type domain responsible for extracellular ligand binding that contains Aβ, two C-type domains, a transmembrane domain, and a short cytoplasmic tail involved in RAGE-mediated cell signaling (Yan et al., 2010). RAGE is expressed on the surface of vascular endothelial cells, pericytes, glial cells, and neurons and acts as a major Aβ influx receptor (Wang et al., 2018; Lao et al., 2021). RAGE located on the luminal membrane of capillary endothelial cells not only promotes amyloid cleavage of APP by affecting the activities of β-secretase and γ-secretase, which results in an increase in Aβ production, but also binds to soluble Aβ in the nanomolar range and endocytoses and transports it from blood to the brain (Fang et al., 2018). RAGE expression is determined by the expression level of its ligand. Under normal physiological conditions, RAGE is expressed at low levels in endothelial cells (Walker et al., 2015), and soluble Aβ in the nanomolar range binds to RAGE and exerts its pathophysiologic cellular effects (Deane et al., 2003; Walker et al., 2015). RAGE expression is upregulated in inflammation-related pathological conditions such as vascular diseases and chronic neurodegenerative diseases (Zlokovic, 2008b; Cai et al., 2016; Kong et al., 2020). Dean et al. (2003) found that Aβ that is injected systemically is endocytosed by vascular endothelial cells in a RAGE-dependent manner, and that Aβ uptake and transport could be decreased by RAGE-specific immunoglobulin and soluble RAGE, which confirms the role of RAGE in Aβ transport into the brain (Smith et al., 2019). Therefore, RAGE regulates Aβ levels in the brain, and could be a useful target for novel brain homeostasis therapies for AD.
FcRn
The major histocompatibility complex class I–related protein FcRn, which is located on the abluminal membrane of capillary endothelial cells, can rapidly transport anti-Aβ antibody (IgG)/Aβ immune complexes across the BBB (Kariolis et al., 2020). FcRn is internalized with anti-Aβ antibody (IgG)/Aβ immune complexes into endosomes, where the subsequent acidification facilitates the further binding of IgG and FcRn in the vesicle. Then, the vesicle fuses with the contralateral membrane, and physiologic pH promotes the release of the IgG/Aβ immune complexes. The receptor may then be recycled for additional rounds of transcytosis (Ober et al., 2004a, b; Roopenian and Akilesh, 2007). Therefore, FcRn may play a critical role in immunotherapy for AD (Finke and Banks, 2017).
The common pathway of receptor-mediated Aβ transport across the BBB is transcytosis, that is, receptors binding Aβ internalize to form endocytic vesicles that then fuse with the plasma membrane on the contralateral side of the cells to release Aβ. In addition, there is an endosomal/lysosomal pathway that LRP1 receptor-mediated transport is known to be involved in and that RAGE- and FcRn receptor-mediated transport may also be involved in. All these receptors expressed on endothelial cells play critical roles in maintaining CNS homeostasis.
ATP-binding cassette transporter-mediated Aβ clearance
ATP-binding cassette (ABC) transporters are multi-domain integrated membrane proteins that utilize energy derived from ATP hydrolysis to transport solute across the cell membrane (Pereira et al., 2018). ABC transporters have a characteristic molecular structure that includes two nucleotide-binding domains that provide the driving force for transportation and two transmembrane domains that permit substrates to transfer across lipid membranes (Bakos and Homolya, 2007). Localized on the luminal or abluminal membrane of capillary endothelial cells, ABC transporters allow undirectional transport from the cytoplasm to the blood. Studies have shown the protective effects of P-gp (also known as ABCB1), ABCA1, breast cancer resistance protein (BCRP, otherwise called ABCG2), and ABCC1 transporters on oxidative stress, promoting brain detoxification in patients with AD by clearing neurotoxic endogenous substrates such as Aβ peptides (Abuznait and Kaddoumi, 2012) (Table 2).
Table 2.
Studies of ABC transporters involved in Aβ transportation across the BBB
| Authors | Experiment models /measures | Conclusion |
|---|---|---|
| Sugiyama et al., 2003 | Mdr1a/Mdr1b and Mrp1 knockout mice; Mrp2-deficient mutant rats | Mrp1, but not Mrp2, was involved in the excretion of E217betaG at the BBB. |
| Nies et al., 2004 | Rapidly frozen perilesional samples of several regions of adult human brain | The proteins MRP1, MRP4, and MRP5 were clearly localized on the luminal side of brain capillary endothelial cells. |
| Koldamova et al., 2005 | APP23 mice | Lack of ABCA1 considerably decreased brain ApoE level and increased amyloid deposition. |
| Tachikawa et al., 2005 | Using in situ hybridization for the mouse brain | ABCG2 showed its preferential expression on the luminal membrane of brain capillaries. |
| Wahrle et al., 2005 | PDAPP Abca1–/– mice | PDAPP Abca1–/– mice produced poorly lipidated apoE and apoE deposits co-localized with amyloid plaques. |
| Akanuma et al., 2008 | ABCA1-expressing HEK293 cells; ABCA1-deficient mice | ABCA1 did not directly transport hA beta (1–40). |
| Xiong et al., 2009 | Tg-SwDI and 3XTg mice | ABCG2 was significantly upregulated in the brains of AD/CAA mice. |
| Hartz et al., 2010 | Human amyloid precursor protein (hAPP)-overexpressing mice; Tg2576 strain | Brain capillary P-glycoprotein expression and transport activity were substantially reduced in hAPP-overexpressing mice. |
| Krohn et al., 2011 | ABCB1- and ABCC1-deficient APP/PS1mice | Deficiency of ABCC1 substantially increased cerebral Aβ levels |
| Terwel et al., 2011 | APP23 mice | Increased association of microglia and Aβ plaques was dependent on the presence of LXRα and was accompanied by increased ApoE lipidation. |
| Abuznait et al., 2012 | LS-180 cells treatment with drugs known to induce P-gp expression | The investigated drugs were able to improve the efflux of Aβ1–40 from the cells via P-gp up-regulation. |
| Do et al., 2012 | HEK293 cells transfected with human ABCG2 or mouse abcg4; Abcb1/Abcg2-deficient mice; Abcb1-deficient mice | Abcg2 was involved in the transport of Aβ at the mouse BBB and the expression of Abcg2 was increased in neurons. |
| Zhao et al., 2015 | Post-mortem paraffin embedded human frontal cortex and hippocampus samples; APPsw/0; Picalm+/– mice ; primary human brain endothelial cells | Diminished Aβ clearance and accelerated pathology were observed in APPsw/0 Picalm+/– mice; PICALM/clathrin-dependent endocytosis of Aβ-LRP1 complex was observed on endothelial cells. |
| Storck et al., 2018 | With immunoprecipitation experiments, coimmunostainings and dual inhibition of ABCB1/P-gp and LRP1 | Late-onset AD risk factor PICALM was associated with both ABCB1/P-gp and LRP1 representing a functional link and guiding both proteins through the brain endothelium. |
| Chai et al., 2020 | Brain capillaries from P-gp-knockout mice | P-gp mediated Aβ export from the BBB endothelium. |
Aβ: Amyloid-beta; ABC: ATP-binding cassette; AD: Alzheimer’s disease; ApoE: apolipoprotein E; APP: amyloid precursor protein; BBB: blood-brain barrier; CAA: cerebral amyloid angiopathy; LRP1: low density lipoprotein receptor (LDLR)-related protein 1; LXR: Liver X Receptor; Mrp1: multidrug resistance associated protein 1; P-gp: P-glycoprotein; PICALM: phosphatidylinositol binding clathrin assembly protein.
P-gp (ABCB1)
P-gp, a 170-kDa phosphorylated glycoprotein, is highly expressed on the luminal, blood-facing side of endothelial cells under normal physiological conditions. In humans, it is encoded by the ABCB1 (MDR1) gene and has more than 50 known polymorphisms at the single nucleotide level (Bartels, 2011). P-gp is expressed at lower concentrations in astrocytes, pericytes, and neuronal cells (Chai et al., 2020a), while the highest concentration of P-gp is found in the neurocapillary endothelial cells that make up the BBB (Chai et al., 2020b).
The exact mechanism by which P-gp mediates of efflux is not fully understood (Storck et al., 2018). P-gp may assist in mediating the active transfer of Aβ that is endocytosed by LRP1 or RAGE from the endothelial cells to plasma (Hladky and Barrand, 2018). Extracellular Aβ binding with LRP1 on the abluminal surface of the brain endothelial cell initiates PICALM/clathrin-dependent endocytosis, and PICALM guides trafficking of Aβ-LRP1 endocytic vesicles to Rab5-positive early endosomes and then to Rab11-positive sorting endosomes (Zhao et al., 2015), handing Aβ over to P-gp, which then pumps it out of the luminal side of endothelial cells directly into the blood (Erickson et al., 2012), or via a P-gp–independent pathway (Hartz et al., 2010). P-gp also inhibits the penetrability of circulating Aβ (transported by RAGE into the brain) from the apical to the basolateral side of neuronal capillary endothelial cells (Behl et al., 2021). It has been reported that P-gp can also export Aβ from neurons (Chai et al., 2020a). Hartz et al. (2010) suggested that Aβ efflux mediated by P-gp may be the limiting factor and a key step in Aβ clearance from the brain. Therefore, P-gp may act as a gatekeeper on the luminal side of the endothelial cells, which may have important implications for AD therapeutics.
ABCA1
ABCA1 is highly expressed in astrocytes, microglial cells, neuronal cells, and brain capillary endothelial cells in the CNS (Fujiyoshi et al., 2007). It is located on the basolateral plasma membrane of porcine brain capillary endothelial cells (Behl et al., 2021). ABCA1 promotes the lipidation of apoE by regulating the efflux of phospholipids and cholesterol from microglia cells and astrocytes (Wolf et al., 2012). ABCA1 does not directly mediate Aβ efflux transport in the brain (Akanuma et al., 2008), but influences Aβ clearance by regulating lipidation of apoE, further affecting APP processing (Puglielli et al., 2003), and facilitates proteolytic degradation of Aβ (Behl et al., 2021). ABCA1 expression induced by liver X receptor (LXR), which regulates transcription of the ABCA1 gene, resulting in a decrease in Aβ formation and secretion (Behl et al., 2021), which slows Aβ deposition (Terwel et al., 2011). Both PDAPP ABCA1–/– mice and hAPP-overexpressing ABCA1–/– mice showed decreased lipidation of apoE and low apoE expression levels, while expression levels of insoluble Aβ increased in the brain (Koldamova et al., 2005a, Ruiz et al., 2005, Wahrle et al., 2005). Therefore, it has been speculated that receptor-mediated endocytosis cannot effectively eliminate Aβ-apoE complexes (Ruiz et al., 2005). ABCA1, which acts as a cholesterol exporter and an Aβ proteolysis activator, plays a key role in Aβ elimination through the BBB.
ABCG2
The expression pattern of ABCG2, also known as breast cancer resistance protein (BCRP), overlaps with that of ABCB1 (Behl et al., 2021). It is highly expressed in multiple organs such as gastrointestinal tract, liver, kidney and the endothelium of the nervous system, and is located on the luminal plasma membrane of endothelial cells (Tachikawa et al., 2005; Fetsch et al., 2006; Robey et al., 2009). It plays a vital role in Aβ transport, blocks circulating Aβ entry into the brain, and acts as a gatekeeper for Aβ influx across the BBB (Xiong et al., 2009; Do et al., 2012). Optical imaging analysis showed that, after intravenous administration of labeled Aβ, more Aβ accumulated in ABCG2–/– mice than in wild-type mice, indicating that ABCG2 inhibits Aβ entry into the brain and safeguards the BBB (Xiong et al., 2009). In addition, another study showed that ABCG2 expression in neurons is enhanced in patients with AD, which protects against inflammatory and oxidative stress by blocking nuclear factor (NF)-κB signal transduction (Abuznait and Kaddoumi, 2012).
ABCC1
Numerous studies have shown that ABCC1 (also known as multidrug resistance protein 1) is expressed in pericytes, neurons, astrocytes, and microglia in the CNS (Wolf et al., 2012) and is present on the luminal side of the BBB (Sugiyama et al., 2003, Nies et al., 2004). ABCC1–/– APP/PS1 mice exhibit elevated Aβ expression in the brain, but the expression of Aβ-producing enzymes was unchanged, indicating that ABCC1 is involved in Aβ clearance (Krohn et al., 2011). ABCC1 may assist in trafficking Aβ from endothelial cells to the plasma via endocytosis mediated by LRP1 (Pereira et al., 2018).
In short, ABCB1, ABCG2, and ABCC1, in cooperation with LRP1, participate in Aβ outflow across the BBB. LRP1 mediates Aβ endocytosis and traffics Aβ to the luminal side of endothelial cells, then ABCB1, ABCG2, and ABCC1 export Aβ to the peripheral blood. Altered expression and/or activity of ABC transporters at the BBB may result in impaired Aβ elimination from the brain. Therefore, ABC transporters are key players in Aβ homeostasis and are potential therapeutic targets in AD.
Effects of Amyloid-Beta on Brain Capillary Endothelial Cells in Alzheimer’s disease
Although it has been largely ignored, many studies have shown that Aβ possesses neuroprotective and neurotrophic properties at physiological concentrations (Atwood et al., 2003). The average concentration of circulating Aβ physiologically produced in rodent brains is estimated to be within the picomolar range (Cirrito et al., 2003, Janelidze et al., 2016). Moreover, Aβ levels in are approximately 10-fold higher in the CSF than in plasma (Kawarabayashi et al., 2001; Janelidze et al., 2016). Low physiological concentrations of Aβ in the healthy brain are necessary for the enhancement of hippocampal long-term potentiation and memory (Puzzo et al., 2011). Cyclic guanosine monophosphate, which plays a key role in long-term potentiation signal transduction and memory (Bollen et al., 2014), positively regulates Aβ levels and function, which in turn enhances synaptic plasticity and memory (Palmeri et al., 2017).
In addition, physiological amounts of Aβ are released during neuronal activity (Cirrito et al., 2005). The metal binding and redox properties of Aβ at physiological concentrations are predicted to decrease the oxidative stress that accompanies Alzheimer’s disease. Aβ captures redox metal ions, thereby preventing them from participating in the redox cycle and activating Aβ’s antioxidant activity (Atwood et al., 2003). Aβ is released in response to injury under normal physiological conditions and provides neuroprotection against oxidative stress, after which it is cleared (Atwood et al., 2003). However, if the clearance rate is insufficient, the progressive accumulation of neuronal Aβ-Cu complex induced by oxidative stress or amyloidogenesis induced by AβPP/PS1 mutation cause H2O2 production to exceed the capacity of the antioxidant defense systems (Atwood et al., 2003). This will lead to a vicious cycle of increased Aβ and ROS production, further demonstrating the neurotoxic effects of Aβ. Therefore, Aβ acts as an antioxidant at low nanomolar (~0.1 nM) concentrations by preventing autoxidation of lipoproteins in the CSF and LDL in plasma (Kontush et al., 2001a), while at micromolar concentrations it accelerates oxidation and has toxic effects (Kontush et al., 2001b).
Brain capillary endothelial cells regulate cerebral blood flow, mediate exchanges across the BBB to form a dynamic balance between blood and the brain, participate in innate immunity, and control angiogenesis, which is very important for maintaining neurovascular homeostasis. Excessive Aβ exerts potent detrimental cerebrovascular effects that impair endothelial structure and function and alter cerebral circulation. In the following section, we will discuss the effects of Aβ on cerebral endothelial cells in AD.
Increased RAGE expression and reduced LRP1 and P-gp expression in AD
RAGE expression is increased in capillary endothelial cells in an Aβ-rich environment, such as that found in AD (Wang et al., 2018; Lao et al., 2021). RAGE mediates Aβ-induced neurotoxicity directly by activating intracelluar signal transduction, such as RAGE/NF-κB signaling, which leads to oxidative stress (Hong and An, 2018), and indirectly by amplifying Aβ-activated inflammatory responses in microglia and increasing Aβ-induced monocyte migration across human brain endothelial cell monolayers (Giri et al., 2000). RAGE mediates transcytosis of circulating Aβ across the BBB, resulting in NF-κB-dependent endothelial cell apoptosis (Deane et al., 2003), in an endothelial inflammatory response that leads to increased release of proinflammatory cytokines (tumor necrosis factor-α, interleukin-6, etc.) and adhesion molecules such as vascular cell adhesion molecule, and in dysregulation of cerebral blood flow (Deane et al., 2003). In addition, the Aβ-RAGE interaction triggers the ERK/JNK/PI3K-Egr-1 pathway, which upregulates endothelial CCR5 expression and further facilitates transendothelial migration of circulating MIP-1α-expressing T cells (Man et al., 2007). The Aβ-RAGE interaction also mediates secretion of endothelin-1, resulting in a reduction in cerebral blood flow (Zlokovic, 2008b).
LRP1 expression is reduced in brain endothelial cells in AD patients and amyloid mouse models (Hladky and Barrand, 2018). Genome-wide transcriptional profiling of human brain endothelial cells in AD demonstrated greatly reduced expression of mesenchyme homeobox gene 2 (MEOX2), which is restricted to the vascular system in adults (Wu et al., 2005). The low level MEOX2 expression in brain endothelial cells promotes proteasomal degradation of LRP1 and downregulates LRP1 expression (Wu et al., 2005). In addition, vascular-specific transcriptional cofactor myocardin (MYOCD) and serum response factor (SRF) are overexpressed in the brain vascular smooth muscle cells (VSMCs) of AD patients (Sagare et al., 2013). MYOCD and SRF overexpression in VSMCs has also been shown to lead to downregulation of LRP1 through CArG box-dependent expression of sterol regulatory element binding protein 2, which is a major LRP1 transcriptional suppressor (Llorente-Cortés et al., 2006). Because LRP1 is the major receptor trafficking Aβ from the brain to blood across the BBB, the downregulation of LRP1 in brain endothelial cells in AD patients (Deane et al., 2004, Donahue et al., 2006) will reduce the rate of Aβ clearance and promote focal accumulation of Aβ in cerebral vessels and the brain (Hladky and Barrand, 2018).
P-gp acts as a gatekeeper and plays a critical role in Aβ export from the brain. Several possible mechanisms have been proposed to explain the influence of Aβ on the reduction of P-gp expression (Chai et al., 2020b). It is possible that, in the AD brain, upregulation of DKK1 expression triggered by high Aβ concentrations results in impaired Wnt/β-catenin signaling and thus reduces P-gp expression (Wijesuriya et al., 2010; Hodges et al., 2011). Park et al. (2014) found that the interaction of RAGE with Aβ42 activates NF-κB, which in turn binds to DNA to inhibit P-gp gene transcription (Park et al., 2014). Hartz et al. found that exposure of isolated mouse brain capillaries to monomeric Aβ40 triggered ubiquitination, followed by the proteasomal degradation of P-gp and thus reduced P-gp-mediated transport activity (Hartz et al., 2016). Aβ increases the expression of pro-inflammatory cytokines, which in turn have been shown to downregulate the expression of P-gp mRNA and impair P-gp function in guinea pig brain endothelial cells in a dose-dependent manner (Iqbal et al., 2012; Alasmari et al., 2018). Therefore, the increased expression of RAGE and the decreased expression of LRP1 and P-gp further result in a reduction in Aβ clearance across the BBB and the accumulation of Aβ in the brain parenchyma.
Aβ induces endothelial cell injury by promoting oxidative stress
Aβ promotes reactive oxygen species production by inducing the activation of NADPH oxidase (NOX) in cerebral endothelial cells, which then allows reactive oxygen species to react with nitric oxide and form peroxynitrite (Niwa et al., 2000; Roher AE et al., 2009). Oxidative-nitrosation stress triggers DNA damage in endothelial cells and exerts deleterious vascular effects, including vascular inflammation, destruction of the BBB, and interruption of cerebral blood flow (Zlokovic, 2008a). Aβ induces ADP-ribose (ADPR) formation by activating poly ADP-ribose glycohydrolase (PARG), which cleaves ADPR polymers into ADPR in cerebral endothelial cells (Virág and Szabó, 2002; Putt and Hergenrother, 2004). ADPR effectively activates the opening of TRPM2 channels, leading to an increase in intracellular Ca2+, which in turn induces oxidative-nitrosation stress related to the opening of TRPM2 channels in brain endothelial cells (Buelow et al., 2008; Sumoza-Toledo and Penner, 2011). Oxidative stress also induces a significant increase in the expression of vascular endothelial growth factor (VEGF), which causes aberrant angiogenesis by binding to cognate receptors (Carmeliet and Ruiz de Almodovar, 2013; Sanchez et al., 2013). In addition, several cell culture studies have shown that Aβ inhibits VEGF-stimulated endothelial nitric oxide synthase and VEGF-induced migration of brain endothelial cells, suggesting that higher Aβ concentrations may act as a VEGF antagonist (Hayashi et al., 2009; Patel et al., 2010). Moreover, Aβ40 inhibits the proliferation of HBECs by inducing autophagy involving the intracellular regulation of class 3 phosphatidylinositol 3-kinase and Akt signaling (Hayashi et al., 2009). Overall, the evidence suggests that oxidative stress induced by Aβ interacting with endothelial cells destroys their structural integrity and functionality.
Aβ stimulates endothelial cells to release inflammatory mediators, leading to chronic neuroinflammation in AD
Brain endothelial cells connect and seal to each other through tight junction proteins. Tight junction proteins that join adjacent endothelial cells are indispensable for maintaining the function of the BBB, as well as the dynamic balance of the brain microenvironment (Cai et al., 2018). Aβ degrades tight junction proteins through inhibiting their expression and inducing the expression of matrix metalloproteinases, thereby increasing the permeability of the BBB (Porter et al., 2020). The change in BBB permeability allows circulating leukocytes to enter the CNS and cause a neuroinflammatory response. In addition, peripheral immune cells cross the damaged BBB and participate in the CNS immune response (Yamazaki et al., 2019). Immunohistochemistry analysis of hippocampal tissues showed the presence of cationic antibacterial proteins in hippocampal endothelial cells in AD. Aβ induced the expression of cationic antibacterial proteins in brain endothelial cells, which potentiated the adhesion of endothelial cells to monocytes and aggravated the endothelial cell inflammatory reaction (Stock et al., 2018). Aβ can also directly activate endothelial cells and induce the expression of endothelial selectin, intercellular cell adhesion molecule-1, and vascular cell adhesion molecule 1, which in turn promote leukocyte adhesion and transport to induce chronic neuroinflammation in AD. Therefore, activated endothelial cells that express certain adhesion molecules induce circulating immune system cells to migrate to and penetrate into the brain parenchyma, resulting in dysfunctional neurovascular units and chronic brain inflammation.
BBB impairment and diminished cerebral blood flow
Accumulation of Aβ in the brain leads to disruption of tight junction and adherens junction complexes, pericyte degeneration and loss of pericyte coverage, dysregulation of the basement membrane, loss of AQP4, and depolarization of astrocytic end-feet (Yamazaki and Kanekiyo, 2017). These structural changes lead to physical degradation of the BBB (Zlokovic, 2011). BBB breakdown causes an accumulation of plasma proteins in the brain, an event leading to perivascular edema, vascular inflammation, and suppression of cerebral blood flow (CBF) (Pacher and Szabo, 2008; Zholos et al., 2011, Zlokovic, 2011).
It is now well established that Aβ has powerful cerebrovascular effects that alter regulation of the cerebral circulation (Koizumi et al., 2016). Brain endothelial cells play an important role in the regulation of CBF by producing vasorelaxants such as nitric oxide and vasoconstrictors such as endothelin-1 (Faraci, 2011; Iadecola, 2013; Katusic and Austin, 2014). In addition, cerebral smooth muscle cells in AD patients acquire a hypercontractile phenotype, resulting in increased arteriolar contractility, which is associated with reduced resting CBF and impaired neurovascular coupling (Chow et al., 2007). Endothelial oxidative stress induced by Aβ can activate innate immunity receptors and dysregulate cerebrovascular function (Park et al., 2013; Koizumi et al., 2016). Free radical species produced by NADPH oxidase react with nitric oxide to form peroxynitrite, which has a strong regulatory effect on CBF by triggering DNA damage (Faraco and Iadecola, 2013). All of these vascular effects have been attributed to Aβ (Iadecola, 2013).
Restoration of Amyloid-beta Transport Across the Blood-Brain Barrier: Implications for Alzheimer’s Disease Therapies
The receptors and transporters expressed by brain capillary endothelial cells are very important for Aβ brain homeostasis. Changes in the expression of endothelial cell receptors and transporters may reduce Aβ deposition, suggesting that they could be useful targets for novel therapeutic agents designed to promote Aβ clearance (Figure 3).
Figure 3.

Receptors and transporters on endothelial cells may be potential targets for therapies designed to promote Aβ clearance.
Upregulation of MEOX2 expression and downregulation of SRF/MYOCD expression can restore LRP1 levels and increase Aβ removal from the brain parenchyma. Upregulation of PICALM expression can increase Aβ-LRP1 complex endothelial transcytosis. Activation of PXR can upregulate the expression of P-gp, thereby promoting Aβ efflux from the brain. Activation of LXR can mediate upregulation of ABCA1, decreasing Aβ levels in the brain. Some tertiary amides block the interaction between RAGE and Aβ, precluding Aβ from reentering the brain. Downregulation of endothelial CCR5 expression can prevent the Aβ-RAGE complex from transmitting signals to the immune system. The blue line in the figure represents mediated Aβ outflow; the brown line represents mediated Aβ inflow; the green line represents activation or inhibition; solid lines represent receptors or transporters that directly mediate Aβ transport; dashed lines represent receptors or transporters that indirectly mediate Aβ transport. Aβ: Amyloid-beta; LRP1: low density lipoprotein receptor(LDLR)-related protein 1; LXR: Liver X Receptor; MEOX2: mesenchyme homeobox gene 2; MYOCD: myocardin; P-gp: P-glycoprotein; PICALM: phosphatidylinositol binding clathrin assembly protein; PXR: pregnane X receptor; RAGE: advanced glycation end products; SRF: serum response factor.
Reducing RAGE activity
As a major Aβ influx receptor at the BBB, RAGE is a potential therapeutic target for decreasing brain Aβ burden (Lao et al., 2021). Studies have shown that some tertiary amides selected through drug screening can block the interaction between RAGE and Aβ, reduce the brain Aβ load and oxidative stress, and improve functional changes in cerebral blood flow and performance in behavioral tests in a mouse model of AD (Wang et al., 2018). By extension, these new compounds may be developed into therapies for the clinical treatment of AD. In addition, the Aβ-RAGE interaction transmits signals to the systemic immune system by upregulating endothelial CCR5 expression; therefore, determinants of CCR5 expression in brain capillary endothelial cells could also be potential new therapeutic targets for AD (Li et al., 2009).
Upregulation of LRP1 levels
LRP1 is the main Aβ efflux receptor at the BBB, and increasing its expression in brain capillary endothelial cells may improve Aβ clearance and reduce Aβ accumulation, thus precluding the progression of Aβ-dependent pathology (Nelson et al., 2016). As we have previously described, low expression of MEOX2 at the BBB and overexpression of SRF/MYOCD both induce downregulation of cell surface LRP1 levels and reduce Aβ clearance in AD. Therefore, novel drugs and/or gene therapies (e.g., those targeting MEOX2 and SRF/MYOCD) that restore LRP1 levels may be potential targets for the treatment of AD (Nelson et al., 2016). In addition, PICALM, which is abundantly expressed in brain capillary endothelial cells, regulates Aβ-LRP1 complex endothelial transcytosis and clearance from the brain; therefore, it may be another important novel target for therapeutics that promote Aβ clearance (Zhao et al., 2015).
P-gp as a potential therapeutic target in AD
Targeting ABC transporters at the BBB is another potential strategy for treating AD. However, this approach is still in its infancy, and more research is needed to address this possibility in the future (Storelli et al., 2021).
As a member of ABC transporter family, P-gp plays an important role in Aβ outflow. P-gp activity may be a novel pharmacological target in AD to limit the neurotoxic effects of excessive Aβ in the brain (Abuznait et al., 2011; Brenn et al., 2014; Hartz et al., 2018). P-gp expression is elevated in vitro by pregnane X receptor (PXR) activation (Lemmen et al., 2013). PXR is a ligand-activated nuclear receptor that can restore P-gp expression and function, thereby promoting Aβ brain efflux (Bauer et al., 2006, Lemmen et al., 2013). We predict that targeting PXR will be a valid therapeutic strategy (Wolf et al., 2012), and that upregulation of P-gp expression through PxR or other signaling pathways may reduce Aβ brain load (Hartz et al., 2010) and slow cognitive decline. The oxidative stress and neuroinflammation observed in AD can modulate P-gp expression (Sita et al., 2017; Alasmari et al., 2018). Therefore, promoting P-gp activity may have the dual advantages of reducing neuroinflammation and increasing Aβ clearance from the brain. Because of its wide range of substrate specificity, attention must be paid to minimizing the risk of toxicity (Chai et al., 2020b).
ABCA1 as a potential therapeutic target in AD
ABCA1 plays a critical role in the lipidation of apoproteins in the CNS. Reduced ABCA1 protein levels lead to lipid-poor apoE, which seems to increase Aβ accumulation in the brain (Wolf et al., 2012). Many studies have shown that LXR-mediated upregulation of ABCA1 can increase apoE levels, decrease Aβ levels in the brain, and improve cognitive function in AD model mice (Koldamova et al., 2005b; Burns et al., 2006; Lefterov et al., 2007; Riddell et al., 2007; Jiang et al., 2008; Donkin et al., 2010). Because there are many compounds that can activate LXR, this approach may be another potential therapeutic strategy for AD (Wolf et al., 2012).
Currently, approved pharmacological therapeutic strategies for AD only provide symptomatic relief (Chai et al., 2020b). Results from current clinical trials have been disappointing, as the trials lack intervention in the early stage of AD and show no effect on symptoms (Graham et al., 2017; van Dyck, 2018). Future treatment strategies should be aimed at preventing the initial accumulation of Aβ (Chai et al., 2020b). Therefore, restoring defective receptor- and/or transporter-mediated clearance to prevent the initial Aβ deposition may slow or prevent the progression of AD (Chai et al., 2020b).
Conclusions and Future Perspectives
AD is a devastating neurodegenerative disease that afflicts millions of people worldwide. Its pathogenesis is complicated, but the Aβ cascade hypothesis is the most widely accepted explanation of AD etiology. Although numerous studies suggest that Aβ transport across the BBB mediated by receptors and ABC transporters expressed by brain capillary endothelial cells plays an important role in Aβ clearance, the mechanism of impaired Aβ clearance is not completely understood at present.
In this review we summarized the current evidence for receptor- and ABC transporter–mediated Aβ elimination across the BBB and the potential interaction mechanisms between Aβ and endothelial cells.
There are many promising avenues for future research in this area. Aβ transport by receptors or transporters expressed on endothelial cells, especially intracellular processes mediated by these proteins, is worth further study. Spatial transcriptomics and other techniques could be applied to identify more receptors, transporters, and genes involved in Aβ clearance via vascular endothelial cells. In addition, the BBB should be studied in its entirety, not limited to endothelial cells. Further research on how the blood-CSF barrier and the lymphoid-related pathway mediate Aβ clearance from the brain to the periphery would also be valuable. More detailed studies should be carried out to investigate potential changes in receptor and ABC transporter expression levels and functions in brain capillary endothelial cells at specific stages of AD, in human postmortem and antemortem cohorts, as well as their roles in the treatment of many neurodegenerative diseases, including AD. All of these lines of investigation will provide an important basis for better understanding the role of neurovascular factors in AD, and hold promise for the development of novel treatments that promote Aβ clearance (Table 3).
Table 3.
A summary of future perspectives of new research strategies for amyloid-beta clearance in Alzheimer’s disease
| No. | Future perspectives |
|---|---|
| 1 | The transporting process of receptors or transporters on endothelial cells, especially the intracellular process, is worthy of further study. |
| 2 | Using space transcriptome and other techniques to find more receptors, transporters and some genes involved in amyloid-beta clearance on vascular endothelial cells. |
| 3 | The blood-brain barrier should be studied as an entirety, not limited to endothelial cells. |
| 4 | Further researches on the blood-cerebrospinal fluid barrier and lymphoid-related pathway mediating amyloid-beta clearance from the brain into the periphery. |
| 5 | Studying the changes of potential levels and functions of receptors and ABC transporters of brain capillary endothelial cells in specific stages of Alzheimer’s disease in human postmortem and antemortem cohorts. |
| 6 | The study of early reliable biological markers neuroimaging for different types of vascular cells. |
The current review has some shortcomings. For example, the discussion of Aβ clearance mediated by the BBB was limited to the role of capillary endothelial cells, while the role of other constituent cells was not fully addressed. In addition, the section on treatments is somewhat limited in that it only addresses some potential treatments related to receptors and transporters expressed on capillary endothelial cells.
Because pathophysiological changes occur decades before the onset of AD symptoms, it is important to identify reliable, early biological markers of disease. On the basis of the important role of the BBB in Aβ clearance from the brain, neuroimaging can be used to determine biomarkers for different types of vascular cells, including smooth muscle cells, pericytes, and capillary endothelial cells, which is very helpful for exploring early vascular biomarkers. In addition, the combination of new strategies with existing strategies for the treatment of AD from different perspectives may increase the chance of achieving effective treatment and providing personalized treatment that meets patient needs (Wolf et al., 2012). In this review we provided a detailed overview of Aβ clearance through the BBB, in particular the interaction between Aβ and brain capillary endothelial cells, which may provide insights into new clinical treatment strategies for promoting Aβ clearance from the brains of patients with AD.
Acknowledgments:
We thank Tian Li from School of Physiology, Shanxi Medical University, Taiyuan, Shanxi Province, China for language editing service.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Funding: This work was supported by Shanxi Provincial Special Fund for the Transformation of Scientific and Technological Achievements, No. 201704D13111584 (to JHG).
C-Editor: Zhao M; S-Editors: Wang J, Li CH; L-Editors: Crow E, Song LP; T-Editor: Jia Y
References
- 1.Abuznait AH, Cain C, Ingram D, Burk D, Kaddoumi A. Up-regulation of P-glycoprotein reduces intracellular accumulation of beta amyloid: investigation of P-glycoprotein as a novel therapeutic target for Alzheimer's disease. J Pharm Pharmacol. 2011;63:1111–1118. doi: 10.1111/j.2042-7158.2011.01309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abuznait AH, Kaddoumi A. Role of ABC transporters in the pathogenesis of Alzheimer's disease. ACS Chem Neurosci. 2012;3:820–831. doi: 10.1021/cn300077c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adav SS, Sze SK. Insight of brain degenerative protein modifications in the pathology of neurodegeneration and dementia by proteomic profiling. Mol Brain. 2016;9:92. doi: 10.1186/s13041-016-0272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aisen PS, Cummings J, Jack CR, Jr, Morris JC, Sperling R, Frölich L, Jones RW, Dowsett SA, Matthews BR, Raskin J, Scheltens P, Dubois B. On the path to 2025: understanding the Alzheimer's disease continuum. Alzheimers Res Ther. 2017;9:60. doi: 10.1186/s13195-017-0283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Akanuma S, Ohtsuki S, Doi Y, Tachikawa M, Ito S, Hori S, Asashima T, Hashimoto T, Yamada K, Ueda K, Iwatsubo T, Terasaki T. ATP-binding cassette transporter A1 (ABCA1) deficiency does not attenuate the brain-to-blood efflux transport of human amyloid-beta peptide (1-40) at the blood-brain barrier. Neurochem Int. 2008;52:956–961. doi: 10.1016/j.neuint.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Alasmari F, Ashby CR, Jr, Hall FS, Sari Y, Tiwari AK. Modulation of the ATP-binding cassette B1 transporter by neuro-inflammatory cytokines: role in the pathogenesis of Alzheimer's disease. Front Pharmacol. 2018;9:658. doi: 10.3389/fphar.2018.00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albargothy NJ, Johnston DA, MacGregor-Sharp M, Weller RO, Verma A, Hawkes CA, Carare RO. Convective influx/glymphatic system: tracers injected into the CSF enter and leave the brain along separate periarterial basement membrane pathways. Acta Neuropathol. 2018;136:139–152. doi: 10.1007/s00401-018-1862-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atwood CS, Obrenovich ME, Liu T, Chan H, Perry G, Smith MA, Martins RN. Amyloid-beta: a chameleon walking in two worlds: a review of the trophic and toxic properties of amyloid-beta. Res Brain Res Rev. 2003;43:1–16. doi: 10.1016/s0165-0173(03)00174-7. [DOI] [PubMed] [Google Scholar]
- 9.Bakos E, Homolya L. Portrait of multifaceted transporter the multidrug resistance-associated protein 1 (MRP1/ABCC1) Pflugers Arch. 2007;453:621–641. doi: 10.1007/s00424-006-0160-8. [DOI] [PubMed] [Google Scholar]
- 10.Ballard C, Gauthier S, Corbett A, Brayne C, Aarsland D, Jones E. Alzheimer's disease. Lancet. 2011;377:1019–1031. doi: 10.1016/S0140-6736(10)61349-9. [DOI] [PubMed] [Google Scholar]
- 11.Bartels AL. Blood-brain barrier P-glycoprotein function in neurodegenerative disease. Curr Pharm Des. 2011;17:2771–2777. doi: 10.2174/138161211797440122. [DOI] [PubMed] [Google Scholar]
- 12.Bauer B, Yang X, Hartz AM, Olson ER, Zhao R, Kalvass JC, Pollack GM, Miller DS. In vivo activation of human pregnane X receptor tightens the blood-brain barrier to methadone through P-glycoprotein up-regulation. Mol Pharmacol. 2006;70:1212–1219. doi: 10.1124/mol.106.023796. [DOI] [PubMed] [Google Scholar]
- 13.Begley DJ, Brightman MW. Structural and functional aspects of the blood-brain barrier. Prog Drug Res. 2003;61:39–78. doi: 10.1007/978-3-0348-8049-7_2. [DOI] [PubMed] [Google Scholar]
- 14.Behl T, Kaur I, Sehgal A, Kumar A, Uddin MS, Bungau S. The interplay of ABC transporters in Aβtranslocation and cholesterol metabolism: implicating their roles in Alzheimer's disease. Mol Neurobiol. 2021;58:1564–1582. doi: 10.1007/s12035-020-02211-x. [DOI] [PubMed] [Google Scholar]
- 15.Bollen E, Puzzo D, Rutten K, Privitera L, De Vry J, Vanmierlo T, Kenis G, Palmeri A, D'Hooge R, Balschun D, Steinbusch HM, Blokland A, Prickaerts J. Improved long-term memory via enhancing cGMP-PKG signaling requires cAMP-PKA signaling. Neuropsychopharmacology. 2014;39:2497–2505. doi: 10.1038/npp.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bonda DJ, Lee HG, Camins A, Pallàs M, Casadesus G, Smith MA, Zhu X. The sirtuin pathway in ageing and Alzheimer disease: mechanistic and therapeutic considerations. Lancet Neurol. 2011;10:275–279. doi: 10.1016/S1474-4422(11)70013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bothwell SW, Janigro D, Patabendige A. Cerebrospinal fluid dynamics and intracranial pressure elevation in neurological diseases. Fluids Barriers CNS. 2019;16:9. doi: 10.1186/s12987-019-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brenn A, Grube M, Jedlitschky G, Fischer A, Strohmeier B, Eiden M, Keller M, Groschup MH, Vogelgesang S. St. John's Wort reduces beta-amyloid accumulation in a double transgenic Alzheimer's disease mouse model-role of P-glycoprotein. Brain Pathol. 2014;24:18–24. doi: 10.1111/bpa.12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brothers HM, Gosztyla ML, Robinson SR. The physiological roles of amyloid-βpeptide hint at new ways to treat Alzheimer's disease. Front Aging Neurosci. 2018;10:118. doi: 10.3389/fnagi.2018.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buelow B, Song Y, Scharenberg AM. The poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J Biol Chem. 2008;283:24571–24583. doi: 10.1074/jbc.M802673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burns MP, Vardanian L, Pajoohesh-Ganji A, Wang L, Cooper M, Harris DC, Duff K, Rebeck GW. The effects of ABCA1 on cholesterol efflux and Abeta levels in vitro and in vivo. J Neurochem. 2006;98:792–800. doi: 10.1111/j.1471-4159.2006.03925.x. [DOI] [PubMed] [Google Scholar]
- 22.Cai W, Zhang K, Li P, Zhu L, Xu J, Yang B, Hu X, Lu Z, Chen Q. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: an aging effect. Ageing Res Rev. 2017;34:77–87. doi: 10.1016/j.arr.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai Z, Liu N, Wang C, Qin B, Zhou Y, Xiao M, Chang L, Yan LJ, Zhao B. Role of RAGE in Alzheimer's disease. Cell Mol Neurobiol. 2016;36:483–495. doi: 10.1007/s10571-015-0233-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cai Z, Qiao PF, Wan CQ, Cai M, Zhou NK, Li Q. Role of blood-brain barrier in Alzheimer's disease. J Alzheimers Dis. 2018;63:1223–1234. doi: 10.3233/JAD-180098. [DOI] [PubMed] [Google Scholar]
- 25.Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, Weller RO. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2008;34:131–144. doi: 10.1111/j.1365-2990.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 26.Carmeliet P, Ruiz de Almodovar C. VEGF ligands and receptors: implications in neurodevelopment and neurodegeneration. Cell Mol Life Sci. 2013;70:1763–1778. doi: 10.1007/s00018-013-1283-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chai AB, Hartz AMS, Gao X, Yang A, Callaghan R, Gelissen IC. New evidence for P-gp-mediated export of amyloid-βpeptides in molecular, blood-brain barrier and neuronal models. Int J Mol Sci. 2020a;22:246. doi: 10.3390/ijms22010246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chai AB, Leung GKF, Callaghan R, Gelissen IC. P-glycoprotein: a role in the export of amyloid-βin Alzheimer's disease? FEBS J. 2020b;287:612–625. doi: 10.1111/febs.15148. [DOI] [PubMed] [Google Scholar]
- 29.Charidimou A, Boulouis G, Gurol ME, Ayata C, Bacskai BJ, Frosch MP, Viswanathan A, Greenberg SM. Emerging concepts in sporadic cerebral amyloid angiopathy. Brain. 2017;140:1829–1850. doi: 10.1093/brain/awx047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng Y, Haorah J. How does the brain remove its waste metabolites from within? Int J Physiol Pathophysiol Pharmacol. 2019;11:238–249. [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng Y, Tian DY, Wang YJ. Peripheral clearance of brain-derived Aβin Alzheimer's disease: pathophysiology and therapeutic perspectives. Transl Neurodegener. 2020;9:16. doi: 10.1186/s40035-020-00195-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chow N, Bell RD, Deane R, Streb JW, Chen J, Brooks A, Van Nostrand W, Miano JM, Zlokovic BV. Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer's phenotype. Proc Natl Acad Sci U S A. 2007;104:823–828. doi: 10.1073/pnas.0608251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, DeMattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003;23:8844–8853. doi: 10.1523/JNEUROSCI.23-26-08844.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 35.Cline EN, Bicca MA, Viola KL, Klein WL. The amyloid-βoligomer hypothesis: beginning of the third decade. J Alzheimers Dis. 2018;64:S567-610. doi: 10.3233/JAD-179941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, Contarino C, Onengut-Gumuscu S, Farber E, Raper D, Viar KE, Powell RD, Baker W, Dabhi N, Bai R, Cao R, Hu S, Rich SS, Munson JM, Lopes MB, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer's disease. Nature. 2018;560:185–191. doi: 10.1038/s41586-018-0368-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7:a020412. doi: 10.1101/cshperspect.a020412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- 39.Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 41.Do TM, Noel-Hudson MS, Ribes S, Besengez C, Smirnova M, Cisternino S, Buyse M, Calon F, Chimini G, Chacun H, Scherrmann JM, Farinotti R, Bourasset F. ABCG2- and ABCG4-mediated efflux of amyloid-βpeptide 1-40 at the mouse blood-brain barrier. J Alzheimers Dis. 2012;30:155–166. doi: 10.3233/JAD-2012-112189. [DOI] [PubMed] [Google Scholar]
- 42.Donahue JE, Flaherty SL, Johanson CE, Duncan JA, 3rd, Silverberg GD, Miller MC, Tavares R, Yang W, Wu Q, Sabo E, Hovanesian V, Stopa EG. RAGE, LRP-1, and amyloid-beta protein in Alzheimer's disease. Acta Neuropathol. 2006;112:405–415. doi: 10.1007/s00401-006-0115-3. [DOI] [PubMed] [Google Scholar]
- 43.Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, Chan J, Fan J, Collins J, Wellington CL. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010;285:34144–34154. doi: 10.1074/jbc.M110.108100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.d'Uscio LV, He T, Katusic ZS. Expression and processing of amyloid precursor protein in vascular endothelium. Physiology (Bethesda) 2017;32:20–32. doi: 10.1152/physiol.00021.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA. Lipopolysaccharide impairs amyloid βefflux from brain: altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood-brain barrier. J Neuroinflammation. 2012;9:150. doi: 10.1186/1742-2094-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fang F, Yu Q, Arancio O, Chen D, Gore SS, Yan SS, Yan SF. RAGE mediates Aβaccumulation in a mouse model of Alzheimer's disease via modulation of β- and γ-secretase activity. Hum Mol Genet. 2018;27:1002–1014. doi: 10.1093/hmg/ddy017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faraci FM. Protecting against vascular disease in brain. Am J Physiol Heart Circ Physiol. 2011;300:H1566–1582. doi: 10.1152/ajpheart.01310.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Faraco G, Iadecola C. Hypertension: a harbinger of stroke and dementia. Hypertension. 2013;62:810–817. doi: 10.1161/HYPERTENSIONAHA.113.01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fetsch PA, Abati A, Litman T, Morisaki K, Honjo Y, Mittal K, Bates SE. Localization of the ABCG2 mitoxantrone resistance-associated protein in normal tissues. Cancer Lett. 2006;235:84–92. doi: 10.1016/j.canlet.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 50.Finke JM, Banks WA. Modulators of IgG penetration through the blood-brain barrier: implications for Alzheimer's disease immunotherapy. Hum Antibodies. 2017;25:131–146. doi: 10.3233/HAB-160306. [DOI] [PubMed] [Google Scholar]
- 51.Fujiyoshi M, Ohtsuki S, Hori S, Tachikawa M, Terasaki T. 24S-hydroxycholesterol induces cholesterol release from choroid plexus epithelial cells in an apical- and apoE isoform-dependent manner concomitantly with the induction of ABCA1 and ABCG1 expression. J Neurochem. 2007;100:968–978. doi: 10.1111/j.1471-4159.2006.04240.x. [DOI] [PubMed] [Google Scholar]
- 52.Fujiyoshi M, Tachikawa M, Ohtsuki S, Ito S, Uchida Y, Akanuma S, Kamiie J, Hashimoto T, Hosoya K, Iwatsubo T, Terasaki T. Amyloid-βpeptide(1-40) elimination from cerebrospinal fluid involves low-density lipoprotein receptor-related protein 1 at the blood-cerebrospinal fluid barrier. J Neurochem. 2011;118:407–415. doi: 10.1111/j.1471-4159.2011.07311.x. [DOI] [PubMed] [Google Scholar]
- 53.Giri R, Shen Y, Stins M, Du Yan S, Schmidt AM, Stern D, Kim KS, Zlokovic B, Kalra VK. beta-amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am J Physiol Cell Physiol. 2000;279:C1772–1781. doi: 10.1152/ajpcell.2000.279.6.C1772. [DOI] [PubMed] [Google Scholar]
- 54.Graham WV, Bonito-Oliva A, Sakmar TP. Update on Alzheimer's disease therapy and prevention strategies. Annu Rev Med. 2017;68:413–430. doi: 10.1146/annurev-med-042915-103753. [DOI] [PubMed] [Google Scholar]
- 55.Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, van Veluw SJ. Cerebral amyloid angiopathy and Alzheimer disease-one peptide, two pathways. Nat Rev Neurol. 2020;16:30–42. doi: 10.1038/s41582-019-0281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hartz AM, Miller DS, Bauer B. Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer's disease. Mol Pharmacol. 2010;77:715–723. doi: 10.1124/mol.109.061754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hartz AM, Zhong Y, Wolf A, LeVine H, 3rd, Miller DS, Bauer B. Aβ40reduces P-glycoprotein at the blood-brain barrier through the ubiquitin-proteasome pathway. J Neurosci. 2016;36:1930–1941. doi: 10.1523/JNEUROSCI.0350-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hartz AMS, Zhong Y, Shen AN, Abner EL, Bauer B. Preventing P-gp ubiquitination lowers Aβbrain levels in an Alzheimer's disease mouse model. Front Aging Neurosci. 2018;10:186. doi: 10.3389/fnagi.2018.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hawkes CA, Jayakody N, Johnston DA, Bechmann I, Carare RO. Failure of perivascular drainage of β-amyloid in cerebral amyloid angiopathy. Brain Pathol. 2014;24:396–403. doi: 10.1111/bpa.12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hayashi S, Sato N, Yamamoto A, Ikegame Y, Nakashima S, Ogihara T, Morishita R. Alzheimer disease-associated peptide, amyloid beta40, inhibits vascular regeneration with induction of endothelial autophagy. Arterioscler Thromb Vasc Biol. 2009;29:1909–1915. doi: 10.1161/ATVBAHA.109.188516. [DOI] [PubMed] [Google Scholar]
- 61.Hladky SB, Barrand MA. Elimination of substances from the brain parenchyma: efflux via perivascular pathways and via the blood-brain barrier. Fluids Barriers CNS. 2018;15:30. doi: 10.1186/s12987-018-0113-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hodges LM, Markova SM, Chinn LW, Gow JM, Kroetz DL, Klein TE, Altman RB. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein) Pharmacogenet Genomics. 2011;21:152–161. doi: 10.1097/FPC.0b013e3283385a1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hong Y, An Z. Hesperidin attenuates learning and memory deficits in APP/PS1 mice through activation of Akt/Nrf2 signaling and inhibition of RAGE/NF-κB signaling. Arch Pharm Res. 2018;41:655–663. doi: 10.1007/s12272-015-0662-z. [DOI] [PubMed] [Google Scholar]
- 64.Humpel C. Organotypic vibrosections from whole brain adult Alzheimer mice (overexpressing amyloid-precursor-protein with the Swedish-Dutch-Iowa mutations) as a model to study clearance of beta-amyloid plaques. Front Aging Neurosci. 2015;7:47. doi: 10.3389/fnagi.2015.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80:844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Iqbal M, Ho HL, Petropoulos S, Moisiadis VG, Gibb W, Matthews SG. Pro-inflammatory cytokine regulation of P-glycoprotein in the developing blood-brain barrier. PLoS One. 2012;7:e43022. doi: 10.1371/journal.pone.0043022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC. Early role of vascular dysregulation on late-onset Alzheimer's disease based on multifactorial data-driven analysis. Nat Commun. 2016;7:11934. doi: 10.1038/ncomms11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Janelidze S, Stomrud E, Palmqvist S, Zetterberg H, van Westen D, Jeromin A, Song L, Hanlon D, Tan Hehir CA, Baker D, Blennow K, Hansson O. Plasma β-amyloid in Alzheimer's disease and vascular disease. Sci Rep. 2016;6:26801. doi: 10.1038/srep26801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jin WS, Shen LL, Bu XL, Zhang WW, Chen SH, Huang ZL, Xiong JX, Gao CY, Dong Z, He YN, Hu ZA, Zhou HD, Song W, Zhou XF, Wang YZ, Wang YJ. Peritoneal dialysis reduces amyloid-beta plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 2017;134:207–220. doi: 10.1007/s00401-017-1721-y. [DOI] [PubMed] [Google Scholar]
- 71.Jones PM, George AM. The ABC transporter structure and mechanism: perspectives on recent research. Cell Mol Life Sci. 2004;61:682–699. doi: 10.1007/s00018-003-3336-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kanekiyo T, Bu G. The low-density lipoprotein receptor-related protein 1 and amyloid-βclearance in Alzheimer's disease. Front Aging Neurosci. 2014;6:93. doi: 10.3389/fnagi.2014.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kariolis MS, Wells RC, Getz JA, Kwan W, Mahon CS, Tong R, Kim DJ, Srivastava A, Bedard C, Henne KR, Giese T, Assimon VA, Chen X, Zhang Y, Solanoy H, Jenkins K, Sanchez PE, Kane L, Miyamoto T, Chew KS, et al. Brain delivery of therapeutic proteins using an Fc fragment blood-brain barrier transport vehicle in mice and monkeys. Sci Transl Med. 2020;12:eaay1359. doi: 10.1126/scitranslmed.aay1359. [DOI] [PubMed] [Google Scholar]
- 74.Katusic ZS, Austin SA. Endothelial nitric oxide: protector of a healthy mind. Eur Heart J. 2014;35:888–894. doi: 10.1093/eurheartj/eht544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kayed R, Lasagna-Reeves CA. Molecular mechanisms of amyloid oligomers toxicity. J Alzheimers Dis. 2013;33(Suppl 1):S67–78. doi: 10.3233/JAD-2012-129001. [DOI] [PubMed] [Google Scholar]
- 77.Kim JW, Byun MS, Lee JH, Yi D, Jeon SY, Sohn BK, Lee JY, Shin SA, Kim YK, Kang KM, Sohn CH, Lee DY. Serum albumin and beta-amyloid deposition in the human brain. Neurology. 2020;95:e815–826. doi: 10.1212/WNL.0000000000010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koizumi K, Wang G, Park L. Endothelial dysfunction and amyloid-β-induced neurovascular alterations. Cell Mol Neurobiol. 2016;36:155–165. doi: 10.1007/s10571-015-0256-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Koldamova R, Lefterov I, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG, Lazo JS. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer's disease. J Biol Chem. 2005b;280:4079–4088. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- 80.Koldamova R, Staufenbiel M, Lefterov I. Lack of ABCA1 considerably decreases brain ApoE level and increases amyloid deposition in APP23 mice. J Biol Chem. 2005a;280:43224–43235. doi: 10.1074/jbc.M504513200. [DOI] [PubMed] [Google Scholar]
- 81.Kong Y, Liu C, Zhou Y, Qi J, Zhang C, Sun B, Wang J, Guan Y. Progress of RAGE molecular imaging in Alzheimer's disease. Front Aging Neurosci. 2020;12:227. doi: 10.3389/fnagi.2020.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kontush A, Berndt C, Weber W, Akopyan V, Arlt S, Schippling S, Beisiegel U. Amyloid-beta is an antioxidant for lipoproteins in cerebrospinal fluid and plasma. Free Radic Biol Med. 2001a;30:119–128. doi: 10.1016/s0891-5849(00)00458-5. [DOI] [PubMed] [Google Scholar]
- 83.Kontush A, Donarski N, Beisiegel U. Resistance of human cerebrospinal fluid to in vitro oxidation is directly related to its amyloid-beta content. Free Radic Res. 2001b;35:507–517. doi: 10.1080/10715760100301521. [DOI] [PubMed] [Google Scholar]
- 84.Krohn M, Lange C, Hofrichter J, Scheffler K, Stenzel J, Steffen J, Schumacher T, Brüning T, Plath AS, Alfen F, Schmidt A, Winter F, Rateitschak K, Wree A, Gsponer J, Walker LC, Pahnke J. Cerebral amyloid-βproteostasis is regulated by the membrane transport protein ABCC1 in mice. J Clin Invest. 2011;121:3924–3931. doi: 10.1172/JCI57867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lao K, Zhang R, Luan J, Zhang Y, Gou X. Therapeutic strategies targeting amyloid-βreceptors and transporters in Alzheimer's disease. J Alzheimers Dis. 2021;79:1429–1442. doi: 10.3233/JAD-200851. [DOI] [PubMed] [Google Scholar]
- 86.Leandro P, Gomes CM. Protein misfolding in conformational disorders: rescue of folding defects and chemical chaperoning. Mini Rev Med Chem. 2008;8:901–911. doi: 10.2174/138955708785132783. [DOI] [PubMed] [Google Scholar]
- 87.Lee H, Xie L, Yu M, Kang H, Feng T, Deane R, Logan J, Nedergaard M, Benveniste H. The effect of body posture on brain glymphatic transport. J Neurosci. 2015;35:11034–11044. doi: 10.1523/JNEUROSCI.1625-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lefterov I, Bookout A, Wang Z, Staufenbiel M, Mangelsdorf D, Koldamova R. Expression profiling in APP23 mouse brain: inhibition of Abeta amyloidosis and inflammation in response to LXR agonist treatment. Mol Neurodegener. 2007;2:20. doi: 10.1186/1750-1326-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lemmen J, Tozakidis IE, Galla HJ. Pregnane X receptor upregulates ABC-transporter Abcg2 and Abcb1 at the blood-brain barrier. Brain Res. 2013;1491:1–13. doi: 10.1016/j.brainres.2012.10.060. [DOI] [PubMed] [Google Scholar]
- 90.Li M, Shang DS, Zhao WD, Tian L, Li B, Fang WG, Zhu L, Man SM, Chen YH. Amyloid beta interaction with receptor for advanced glycation end products up-regulates brain endothelial CCR5 expression and promotes T cells crossing the blood-brain barrier. J Immunol. 2009;182:5778–5788. doi: 10.4049/jimmunol.0803013. [DOI] [PubMed] [Google Scholar]
- 91.Li Q, Lei F, Tang Y, Pan JS, Tong Q, Sun Y, Sheikh-Hamad D. Megalin mediates plasma membrane to mitochondria cross-talk and regulates mitochondrial metabolism. Cell Mol Life Sci. 2018;75:4021–4040. doi: 10.1007/s00018-018-2847-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li Y, Lu W, Marzolo MP, Bu G. Differential functions of members of the low density lipoprotein receptor family suggested by their distinct endocytosis rates. J Biol Chem. 2001;276:18000–18006. doi: 10.1074/jbc.M101589200. [DOI] [PubMed] [Google Scholar]
- 93.Liebner S, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, Constantin G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018;135:311–336. doi: 10.1007/s00401-018-1815-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lionaki E, Markaki M, Palikaras K, Tavernarakis N. Mitochondria, autophagy and age-associated neurodegenerative diseases: new insights into a complex interplay. Biochim Biophys Acta. 2015;1847:1412–1423. doi: 10.1016/j.bbabio.2015.04.010. [DOI] [PubMed] [Google Scholar]
- 95.Llorente-Cortés V, Costales P, Bernués J, Camino-Lopez S, Badimon L. Sterol regulatory element-binding protein-2 negatively regulates low density lipoprotein receptor-related protein transcription. J Mol Biol. 2006;359:950–960. doi: 10.1016/j.jmb.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 96.Lopez Sanchez MIG, van Wijngaarden P, Trounce IA. Amyloid precursor protein-mediated mitochondrial regulation and Alzheimer's disease. Br J Pharmacol. 2019;176:3464–3474. doi: 10.1111/bph.14554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ma Q, Ineichen BV, Detmar M, Proulx ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun. 2017;8:1434. doi: 10.1038/s41467-017-01484-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ma Q, Ries M, Decker Y, Müller A, Riner C, Bücker A, Fassbender K, Detmar M, Proulx ST. Rapid lymphatic efflux limits cerebrospinal fluid flow to the brain. Acta Neuropathol. 2019;137:151–165. doi: 10.1007/s00401-018-1916-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Man SM, Ma YR, Shang DS, Zhao WD, Li B, Guo DW, Fang WG, Zhu L, Chen YH. Peripheral T cells overexpress MIP-1alpha to enhance its transendothelial migration in Alzheimer's disease. Neurobiol Aging. 2007;28:485–496. doi: 10.1016/j.neurobiolaging.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 100.McInerney MP, Short JL, Nicolazzo JA. Neurovascular alterations in Alzheimer's disease: transporter expression profiles and CNS drug access. AAPS J. 2017;19:940–956. doi: 10.1208/s12248-017-0077-5. [DOI] [PubMed] [Google Scholar]
- 101.McIntee FL, Giannoni P, Blais S, Sommer G, Neubert TA, Rostagno A, Ghiso J. In vivo differential brain clearance and catabolism of monomeric and oligomeric Alzheimer's Aβprotein. Front Aging Neurosci. 2016;8:223. doi: 10.3389/fnagi.2016.00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miners JS, Baig S, Palmer J, Palmer LE, Kehoe PG, Love S. Abeta-degrading enzymes in Alzheimer's disease. Brain Pathol. 2008;18:240–252. doi: 10.1111/j.1750-3639.2008.00132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moestrup SK, Gliemann J, Pallesen G. Distribution of the alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein in human tissues. Cell Tissue Res. 1992;269:375–382. doi: 10.1007/BF00353892. [DOI] [PubMed] [Google Scholar]
- 104.Montagne A, Zhao Z, Zlokovic BV. Alzheimer's disease: a matter of blood-brain barrier dysfunction? J Exp Med. 2017;214:3151–3169. doi: 10.1084/jem.20171406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer's disease. Biochim Biophys Acta. 2016;1862:887–900. doi: 10.1016/j.bbadis.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Nies AT, Jedlitschky G, König J, Herold-Mende C, Steiner HH, Schmitt HP, Keppler D. Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience. 2004;129:349–360. doi: 10.1016/j.neuroscience.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 107.Nisbet RM, Polanco JC, Ittner LM, Götz J. Tau aggregation and its interplay with amyloid-β. Acta Neuropathol. 2015;129:207–220. doi: 10.1007/s00401-014-1371-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Niwa K, Carlson GA, Iadecola C. Exogenous A beta1-40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J Cereb Blood Flow Metab. 2000;20:1659–1668. doi: 10.1097/00004647-200012000-00005. [DOI] [PubMed] [Google Scholar]
- 109.Ober RJ, Martinez C, Lai X, Zhou J, Ward ES. Exocytosis of IgG as mediated by the receptor, FcRn: an analysis at the single-molecule level. Proc Natl Acad Sci U S A. 2004a;101:11076–11081. doi: 10.1073/pnas.0402970101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ober RJ, Martinez C, Vaccaro C, Zhou J, Ward ES. Visualizing the site and dynamics of IgG salvage by the MHC class I-related receptor, FcRn. J Immunol. 2004b;172:2021–2029. doi: 10.4049/jimmunol.172.4.2021. [DOI] [PubMed] [Google Scholar]
- 111.Pacher P, Szabo C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am J Pathol. 2008;173:2–13. doi: 10.2353/ajpath.2008.080019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pahnke J, Walker LC, Scheffler K, Krohn M. Alzheimer's disease and blood-brain barrier function-Why have anti-beta-amyloid therapies failed to prevent dementia progression? Neurosci Biobehav Rev. 2009;33:1099–1108. doi: 10.1016/j.neubiorev.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Palmeri A, Ricciarelli R, Gulisano W, Rivera D, Rebosio C, Calcagno E, Tropea MR, Conti S, Das U, Roy S, Pronzato MA, Arancio O, Fedele E, Puzzo D. Amyloid-βpeptide is needed for cGMP-induced long-term potentiation and memory. J Neurosci. 2017;37:6926–6937. doi: 10.1523/JNEUROSCI.3607-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Park L, Zhou J, Zhou P, Pistick R, El Jamal S, Younkin L, Pierce J, Arreguin A, Anrather J, Younkin SG, Carlson GA, McEwen BS, Iadecola C. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2013;110:3089–3094. doi: 10.1073/pnas.1300021110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Park R, Kook SY, Park JC, Mook-Jung I. Aβ1-42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-κB signaling. Cell Death Dis. 2014;5:e1299. doi: 10.1038/cddis.2014.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pascale CL, Miller MC, Chiu C, Boylan M, Caralopoulos IN, Gonzalez L, Johanson CE, Silverberg GD. Amyloid-beta transporter expression at the blood-CSF barrier is age-dependent. Fluids Barriers CNS. 2011;8:21. doi: 10.1186/2045-8118-8-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Patel NS, Mathura VS, Bachmeier C, Beaulieu-Abdelahad D, Laporte V, Weeks O, Mullan M, Paris D. Alzheimer's beta-amyloid peptide blocks vascular endothelial growth factor mediated signaling via direct interaction with VEGFR-2. J Neurochem. 2010;112:66–76. doi: 10.1111/j.1471-4159.2009.06426.x. [DOI] [PubMed] [Google Scholar]
- 118.Pei L, Wallace DC. Mitochondrial etiology of neuropsychiatric disorders. JAMA Psychiatry. 2018;83:722–730. doi: 10.1016/j.biopsych.2017.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pereira CD, Martins F, Wiltfang J, da Cruz ESOAB, Rebelo S. ABC transporters are key players in Alzheimer's disease. J Alzheimers Dis. 2018;61:463–485. doi: 10.3233/JAD-170639. [DOI] [PubMed] [Google Scholar]
- 120.Pflanzner T, Kuhlmann CR, Pietrzik CU. Blood-brain-barrier models for the investigation of transporter- and receptor-mediated amyloid-βclearance in Alzheimer's disease. Curr Alzheimer Res. 2010;7:578–590. doi: 10.2174/156720510793499066. [DOI] [PubMed] [Google Scholar]
- 121.Porter KN, Sarkar SN, Dakhlallah DA, Vannoy ME, Quintana DD, Simpkins JW. Medroxyprogesterone acetate impairs amyloid beta degradation in a matrix metalloproteinase-9 dependent manner. Front Aging Neurosci. 2020;12:92. doi: 10.3389/fnagi.2020.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Puglielli L, Tanzi RE, Kovacs DM. Alzheimer's disease: the cholesterol connection. Nat Neurosci. 2003;6:345–351. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 123.Putt KS, Hergenrother PJ. A nonradiometric, high-throughput assay for poly (ADP-ribose) glycohydrolase (PARG): application to inhibitor identification and evaluation. Anal Biochem. 2004;333:256–264. doi: 10.1016/j.ab.2004.04.032. [DOI] [PubMed] [Google Scholar]
- 124.Puzzo D, Privitera L, Fa M, Staniszewski A, Hashimoto G, Aziz F, Sakurai M, Ribe EM, Troy CM, Mercken M, Jung SS, Palmeri A, Arancio O. Endogenous amyloid-βis necessary for hippocampal synaptic plasticity and memory. Ann Neurol. 2011;69:819–830. doi: 10.1002/ana.22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ramanathan A, Nelson AR, Sagare AP, Zlokovic BV. Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer's disease: the role, regulation and restoration of LRP1. Front Aging Neurosci. 2015;7:136. doi: 10.3389/fnagi.2015.00136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rasmussen MK, Mestre H, Nedergaard M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018;17:1016–1024. doi: 10.1016/S1474-4422(18)30318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Riddell DR, Zhou H, Comery TA, Kouranova E, Lo CF, Warwick HK, Ring RH, Kirksey Y, Aschmies S, Xu J, Kubek K, Hirst WD, Gonzales C, Chen Y, Murphy E, Leonard S, Vasylyev D, Oganesian A, Martone RL, Pangalos MN, et al. The LXR agonist TO901317 selectively lowers hippocampal Abeta42 and improves memory in the Tg2576 mouse model of Alzheimer's disease. Mol Cell Neurosci. 2007;34:621–628. doi: 10.1016/j.mcn.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 128.Robey RW, To KK, Polgar O, Dohse M, Fetsch P, Dean M, Bates SE. ABCG2: a perspective. Adv Drug Deliv Rev. 2009;61:3–13. doi: 10.1016/j.addr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Roher AE, Esh CL, Kokjohn TA, Castaño EM, Van Vickle GD, Kalback WM, Patton RL, Luehrs DC, Daugs ID, Kuo YM, Emmerling MR, Soares H, Quinn JF, Kaye J, Connor DJ, Silverberg NB, Adler CH, Seward JD, Beach TG, Sabbagh MN. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer's disease. Alzheimers Dement. 2009;5:18–29. doi: 10.1016/j.jalz.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 131.Ruiz J, Kouiavskaia D, Migliorini M, Robinson S, Saenko EL, Gorlatova N, Li D, Lawrence D, Hyman BT, Weisgraber KH, Strickland DK. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J Lipid Res. 2005;46:1721–1731. doi: 10.1194/jlr.M500114-JLR200. [DOI] [PubMed] [Google Scholar]
- 132.Sagare AP, Bell RD, Zlokovic BV. Neurovascular defects and faulty amyloid-βvascular clearance in Alzheimer's disease. J Alzheimers Dis 33 Suppl. 2013;1:S87–100. doi: 10.3233/JAD-2012-129037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sanchez A, Tripathy D, Luo J, Yin X, Martinez J, Grammas P. Neurovascular unit and the effects of dosage in VEGF toxicity: role for oxidative stress and thrombin. J Alzheimers Dis. 2013;34:281–291. doi: 10.3233/JAD-121636. [DOI] [PubMed] [Google Scholar]
- 134.Schmidt AM, Mora R, Cao R, Yan SD, Brett J, Ramakrishnan R, Tsang TC, Simionescu M, Stern D. The endothelial cell binding site for advanced glycation end products consists of a complex: an integral membrane protein and a lactoferrin-like polypeptide. J Biol Chem. 1994;269:9882–9888. [PubMed] [Google Scholar]
- 135.Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sita G, Hrelia P, Tarozzi A, Morroni F. P-glycoprotein (ABCB1) and oxidative stress: focus on Alzheimer's disease. Oxid Med Cell Longev 2017. 2017:7905486. doi: 10.1155/2017/7905486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Smith AJ, Verkman AS. The “glymphatic” mechanism for solute clearance in Alzheimer's disease: game changer or unproven speculation? FASEB J. 2018;32:543–551. doi: 10.1096/fj.201700999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Smith LM, Kostylev MA, Lee S, Strittmatter SM. Systematic and standardized comparison of reported amyloid-βreceptors for sufficiency, affinity, and Alzheimer's disease relevance. J Biol Chem. 2019;294:6042–6053. doi: 10.1074/jbc.RA118.006252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Solis E, Jr, Hascup KN, Hascup ER. Alzheimer's disease: the link between amyloid-βand neurovascular dysfunction. J Alzheimers Dis. 2020;76:1179–1198. doi: 10.3233/JAD-200473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sousa JC, Cardoso I, Marques F, Saraiva MJ, Palha JA. Transthyretin and Alzheimer's disease: where in the brain? Neurobiol Aging. 2007;28:713–718. doi: 10.1016/j.neurobiolaging.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 141.Steiner H, Fukumori A, Tagami S, Okochi M. Making the final cut: pathogenic amyloid-βpeptide generation by γ-secretase. Cell Stress. 2018;2:292–310. doi: 10.15698/cst2018.11.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Steubler V, Erdinger S, Back MK, Ludewig S, Fässler D, Richter M, Han K, Slomianka L, Amrein I, von Engelhardt J, Wolfer DP, Korte M, Müller UC. Loss of all three APP family members during development impairs synaptic function and plasticity, disrupts learning, and causes an autism-like phenotype. EMBO J. 2021;40:e107471. doi: 10.15252/embj.2020107471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stock AJ, Kasus-Jacobi A, Pereira HA. The role of neutrophil granule proteins in neuroinflammation and Alzheimer's disease. J Neuroinflammation. 2018;15:240. doi: 10.1186/s12974-018-1284-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Storck SE, Hartz AMS, Bernard J, Wolf A, Kachlmeier A, Mahringer A, Weggen S, Pahnke J, Pietrzik CU. The concerted amyloid-beta clearance of LRP1 and ABCB1/P-gp across the blood-brain barrier is linked by PICALM. Brain Behav Immun. 2018;73:21–33. doi: 10.1016/j.bbi.2018.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Storck SE, Meister S, Nahrath J, Meißner JN, Schubert N, Di Spiezio A, Baches S, Vandenbroucke RE, Bouter Y, Prikulis I, Korth C, Weggen S, Heimann A, Schwaninger M, Bayer TA, Pietrzik CU. Endothelial LRP1 transports amyloid-β(1-42) across the blood-brain barrier. J Clin Invest. 2016;126:123–136. doi: 10.1172/JCI81108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Storelli F, Billington S, Kumar AR, Unadkat JD. Abundance of P-glycoprotein and other drug transporters at the human blood-brain barrier in Alzheimer's disease: a quantitative targeted proteomic study. Clin Pharmacol Ther. 2021;109:667–675. doi: 10.1002/cpt.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Su Q, Li T, He PF, Lu XC, Yu Q, Gao QC, Wang ZJ, Wu MN, Yang D, Qi JS. Trichostatin A ameliorates Alzheimer's disease-related pathology and cognitive deficits by increasing albumin expression and Aβclearance in APP/PS1 mice. Alzheimers Res Ther. 2021;13:7. doi: 10.1186/s13195-020-00746-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sugiyama D, Kusuhara H, Lee YJ, Sugiyama Y. Involvement of multidrug resistance associated protein 1 (Mrp1) in the efflux transport of 17beta estradiol-D-17beta-glucuronide (E217betaG) across the blood-brain barrier. Pharm Res. 2003;20:1394–1400. doi: 10.1023/a:1025749925541. [DOI] [PubMed] [Google Scholar]
- 149.Sumoza-Toledo A, Penner R. TRPM2: a multifunctional ion channel for calcium signalling. J Physiol. 2011;589:1515–1525. doi: 10.1113/jphysiol.2010.201855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sun BL, Wang LH, Yang T, Sun JY, Mao LL, Yang MF, Yuan H, Colvin RA, Yang XY. Lymphatic drainage system of the brain: a novel target for intervention of neurological diseases. Prog Neurobiol. 2018:163–164. doi: 10.1016/j.pneurobio.2017.08.007. 118-143. [DOI] [PubMed] [Google Scholar]
- 151.Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14:133–150. doi: 10.1038/nrneurol.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: from physiology to disease and back. Physiol Rev. 2019;99:21–78. doi: 10.1152/physrev.00050.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Tachibana M, Holm ML, Liu CC, Shinohara M, Aikawa T, Oue H, Yamazaki Y, Martens YA, Murray ME, Sullivan PM, Weyer K, Glerup S, Dickson DW, Bu G, Kanekiyo T. APOE4-mediated amyloid-βpathology depends on its neuronal receptor LRP1. J Clin Invest. 2019;129:1272–1277. doi: 10.1172/JCI124853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Tachikawa M, Watanabe M, Hori S, Fukaya M, Ohtsuki S, Asashima T, Terasaki T. Distinct spatio-temporal expression of ABCA and ABCG transporters in the developing and adult mouse brain. J Neurochem. 2005;95:294–304. doi: 10.1111/j.1471-4159.2005.03369.x. [DOI] [PubMed] [Google Scholar]
- 155.Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson J, Holtzman DM, Heneka MT. Critical role of astroglial apolipoprotein E and liver X receptor-αexpression for microglial Aβphagocytosis. J Neurosci. 2011;31:7049–7059. doi: 10.1523/JNEUROSCI.6546-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tholen S, Schmaderer C, Chmielewski S, Förstl H, Heemann U, Baumann M, Steubl D, Grimmer T. Reduction of amyloid-βplasma levels by hemodialysis: an anti-amyloid treatment strategy? J Alzheimers Dis. 2016;50:791–796. doi: 10.3233/JAD-150662. [DOI] [PubMed] [Google Scholar]
- 157.Tiwari S, Atluri V, Kaushik A, Yndart A, Nair M. Alzheimer's disease: pathogenesis, diagnostics, and therapeutics. Int J Nanomedicine. 2019;14:5541–5554. doi: 10.2147/IJN.S200490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tokuda T, Calero M, Matsubara E, Vidal R, Kumar A, Permanne B, Zlokovic B, Smith JD, Ladu MJ, Rostagno A, Frangione B, Ghiso J. Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer's amyloid beta peptides. Biochem J 348 Pt. 2000;2:359–365. [PMC free article] [PubMed] [Google Scholar]
- 159.Tran L, Ha-Duong T. Exploring the Alzheimer amyloid-βpeptide conformational ensemble: A review of molecular dynamics approaches. Peptides. 2015;69:86–91. doi: 10.1016/j.peptides.2015.04.009. [DOI] [PubMed] [Google Scholar]
- 160.Tsatsanis A, Wong BX, Gunn AP, Ayton S, Bush AI, Devos D, Duce JA. Amyloidogenic processing of Alzheimer's disease β-amyloid precursor protein induces cellular iron retention. Mol Psychiatry. 2020;25:1958–1966. doi: 10.1038/s41380-020-0762-0. [DOI] [PubMed] [Google Scholar]
- 161.van Dyck CH. Anti-amyloid-βmonoclonal antibodies for Alzheimer's disease: pitfalls and promise. Biol Psychiatry. 2018;83:311–319. doi: 10.1016/j.biopsych.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Verma M, Vats A, Taneja V. Toxic species in amyloid disorders: oligomers or mature fibrils. Ann Indian Acad Neurol. 2015;18:138–145. doi: 10.4103/0972-2327.144284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Viola KL, Klein WL. Amyloid βoligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015;129:183–206. doi: 10.1007/s00401-015-1386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Virág L, Szabó C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54:375–429. doi: 10.1124/pr.54.3.375. [DOI] [PubMed] [Google Scholar]
- 165.Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, Holtzman DM. Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem. 2005;280:43236–43242. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- 166.Walker D, Lue LF, Paul G, Patel A, Sabbagh MN. Receptor for advanced glycation endproduct modulators: a new therapeutic target in Alzheimer's disease. Expert Opin Investig Drugs. 2015;24:393–399. doi: 10.1517/13543784.2015.1001490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang H, Chen F, Du YF, Long Y, Reed MN, Hu M, Suppiramaniam V, Hong H, Tang SS. Targeted inhibition of RAGE reduces amyloid-βinflux across the blood-brain barrier and improves cognitive deficits in db/db mice. Neuropharmacology. 2018;131:143–153. doi: 10.1016/j.neuropharm.2017.12.026. [DOI] [PubMed] [Google Scholar]
- 168.Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathologica. 2009;117:1–14. doi: 10.1007/s00401-008-0457-0. [DOI] [PubMed] [Google Scholar]
- 169.Wijesuriya HC, Bullock JY, Faull RL, Hladky SB, Barrand MA. ABC efflux transporters in brain vasculature of Alzheimer's subjects. Brain Res. 2010;1358:228–238. doi: 10.1016/j.brainres.2010.08.034. [DOI] [PubMed] [Google Scholar]
- 170.Wolf A, Bauer B, Hartz AM. ABC Transporters and the Alzheimer's disease enigma. Front Psychiatry. 2012;3:54. doi: 10.3389/fpsyt.2012.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wu Z, Guo H, Chow N, Sallstrom J, Bell RD, Deane R, Brooks AI, Kanagala S, Rubio A, Sagare A, Liu D, Li F, Armstrong D, Gasiewicz T, Zidovetzki R, Song X, Hofman F, Zlokovic BV. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat Med. 2005;11:959–965. doi: 10.1038/nm1287. [DOI] [PubMed] [Google Scholar]
- 172.Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, O'Donnell J, Christensen DJ, Nicholson C, Iliff JJ, Takano T, Deane R, Nedergaard M. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–377. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Xiong H, Callaghan D, Jones A, Bai J, Rasquinha I, Smith C, Pei K, Walker D, Lue LF, Stanimirovic D, Zhang W. ABCG2 is upregulated in Alzheimer's brain with cerebral amyloid angiopathy and may act as a gatekeeper at the blood-brain barrier for Abeta(1-40) peptides. J Neurosci. 2009;29:5463–5475. doi: 10.1523/JNEUROSCI.5103-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Xu L, Pan CL, Wu XH, Song JJ, Meng P, Li L, Wang L, Zhang Z, Zhang ZY. Inhibition of Smad3 in macrophages promotes Aβefflux from the brain and thereby ameliorates Alzheimer's pathology. Brain Behav Immun. 2021;95:154–167. doi: 10.1016/j.bbi.2021.03.013. [DOI] [PubMed] [Google Scholar]
- 175.Xu Z, Xiao N, Chen Y, Huang H, Marshall C, Gao J, Cai Z, Wu T, Hu G, Xiao M. Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβaccumulation and memory deficits. Mol Neurodegener. 2015;10:58. doi: 10.1186/s13024-015-0056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yamazaki Y, Kanekiyo T. Blood-brain barrier dysfunction and the pathogenesis of Alzheimer's disease. Int J Mol Sci. 2017;18:1965. doi: 10.3390/ijms18091965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yamazaki Y, Shinohara M, Shinohara M, Yamazaki A, Murray ME, Liesinger AM, Heckman MG, Lesser ER, Parisi JE, Petersen RC, Dickson DW, Kanekiyo T, Bu G. Selective loss of cortical endothelial tight junction proteins during Alzheimer's disease progression. Brain. 2019;142:1077–1092. doi: 10.1093/brain/awz011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yan SF, Ramasamy R, Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106:842–853. doi: 10.1161/CIRCRESAHA.109.212217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zhao Z, Sagare AP, Ma Q, Halliday MR, Kong P, Kisler K, Winkler EA, Ramanathan A, Kanekiyo T, Bu G, Owens NC, Rege SV, Si G, Ahuja A, Zhu D, Miller CA, Schneider JA, Maeda M, Maeda T, Sugawara T, et al. Central role for PICALM in amyloid-βblood-brain barrier transcytosis and clearance. Nat Neurosci. 2015;18:978–987. doi: 10.1038/nn.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zholos A, Johnson C, Burdyga T, Melanaphy D. TRPM channels in the vasculature. Adv Exp Med Biol. 2011;704:707–729. doi: 10.1007/978-94-007-0265-3_37. [DOI] [PubMed] [Google Scholar]
- 181.Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008a;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 182.Zlokovic BV. New therapeutic targets in the neurovascular pathway in Alzheimer's disease. Neurotherapeutics. 2008b;5:409–414. doi: 10.1016/j.nurt.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer's disease and other disorders. Nat Rev Neurosci. 2011;12:723–738. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zorzano A, Claret M. Implications of mitochondrial dynamics on neurodegeneration and on hypothalamic dysfunction. Front Aging Neurosci. 2015;7:101. doi: 10.3389/fnagi.2015.00101. [DOI] [PMC free article] [PubMed] [Google Scholar]