

Abstract
The degeneration of nerve fibres following injury was first described by Augustus Waller over 170 years ago. Initially assumed to be a passive process, it is now evident that axons respond to insult via regulated cellular signaling events resulting in their programmed degeneration. Pro-survival and pro-degenerative factors have been identified and their regulatory mechanisms are beginning to emerge. The ubiquitin system has been implicated in the pro-degenerative process and a key component is the ubiquitin E3 ligase MYCBP2 (also known as PHR1). Ubiquitin E3 ligases are tasked with the transfer of the small protein modifier ubiquitin to substrates and consist of hundreds of members. They can be classified as single subunit systems or as multi-subunit complexes. Their catalytic domains can also be assigned to three general architectures. Hints that MYCBP2 might not conform to these established formats came to light and it is now clear from biochemical and structural studies that MYCBP2 is indeed an outlier in terms of its modus operandi. Furthermore, the unconventional way in which MYCBP2 transfers ubiquitin to substrates has been linked to neurodevelopmental and pro-degenerative function. Herein, we will summarize these research developments relating to the unusual features of MYCBP2 and postulate therapeutic strategies that prevent Wallerian degeneration. These have exciting potential for providing relief from pathological neuropathies and neurodegenerative diseases.
Key Words: chemical biology, E3 ligase, MYCBP2, neurodegeneration, progammed axon death, structural biology, ubiqultin, wallerian degeneration
Introduction
First reported over 170 years ago, Wallerian degeneration (WD) is the programmed degeneration of axons following their injury and occurs in the central and peripheral nervous systems. The process is characterized by mitochondrial swelling, cytoskeletal disruption and axon fragmentation (Waller, 1851; Coleman and Hoke, 2020). As peripheral neuropathy is a significant and dose-limiting side-effect of common chemotherapeutics, pharmacological inhibition of WD might not only improve quality of life for cancer survivors, but could increase the efficacy of existing drugs in the clinic (Geisler, 2020). Furthermore, axon degeneration occurs in the early stages of amyotrophic lateral sclerosis, Parkinson’s and other age-related neurodegenerative disorders, hence WD inhibitors may confer benefits in the context of neurodegenerative diseases (Dadon-Nachum et al., 2011; Adalbert and Coleman, 2013; Tagliaferro and Burke, 2016; White et al., 2019).
Despite our understanding of the exact molecular mechanism of WD being incomplete, remarkable progress has been made in this area and pro-survival and pro-degenerative factors have been identified. Hints of the existence of a pro-survival factor came from the striking phenotype observed with a strain of mouse known as slow Wallerian degeneration (WldS), where severed axons survived for weeks after detachment from their cell bodies (Lunn et al., 1989). WldS mice were found to have undergone a chromosomal rearrangement where the gene product responsible was a fusion protein (WldS) containing the (NAD+) biosynthetic enzyme nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1). The fusion protein resulted in NMNAT1 being relocalized to axons, suggestive of NAD+ levels having a regularity role in axon integrity (Mack et al., 2001; Wang et al., 2005). It was subsequently shown in wild-type mice that levels of the neuronal NMNAT isoform, NMNAT2, are depleted post-injury and knock-down induces Wallerian-like degeneration (Gilley and Coleman, 2010). Furthermore, NMNAT2 overexpression is axon-protective so strategies that activate or stabilize NMNAT2 might have therapeutic value (Coleman and Hoke, 2020). Certain disease links to NMNAT2 levels or activity have also been established as their reduction has been associated with Alzheimer’s disease, paediatric neurological disease and fetal akinesia deformation syndrome (Ali et al., 2016; Huppke et al., 2019; Lukacs et al., 2019).
Amongst the pro-degenerative factors that have been identified in mammals is the protein Sterile Alpha and Toll/interleukin-1 receptor TIR motif containing 1 (SARM1) (Osterloh et al., 2012). Mice constitutively depleted of SARM1 appear healthy and this delays axonal degeneration by several days following axotomy. SARM1 knockout also attenuates Wallerian-like degeneration upon exposure to common chemotherapeutic drugs including vincristine and Bortezomib (Gerdts et al., 2013; Geisler et al., 2019; Geisler, 2020). It is now evident that SARM1 mediates its degenerative effect through enzymatic NAD cleavage activity (Essuman et al., 2017). This presents a tangible therapeutic target and inhibitors of SARM1 NAD cleavage activity are in active development (Krauss et al., 2020). PubMed was used to search literature and relevant work in the area of MYCBP2, in consideration of a mini review format, was used as the selection criteria for citations. Dated: October 2021.
Programmed Axon Degeneration Is Positively Regulated by the Ubiquitin System Component MYCBP2
An additional pro-degenerative factor is the protein MYCBP2. MYCBP2 is conserved from C. elegans through to mammals and the various orthologues go by a multitude of names (C. elegans Rpm-1, Drosophila Highwire, zebrafish Esrom, mouse Phr1 and human Pam). Collectively they have been coined PHR proteins (Pam/Highwire/Rpm-1) (Grill et al., 2016). MYCBP2 was first identified as a huge 510 kDa interactor with the proto-oncogene Myc (Guo et al., 1998). Although the physiological significance of this interaction has not been elaborated, genetic screens in Drosophila and C. elegans identified Highwire and Rpm-1 as important regulators of synaptogenesis (Schaefer et al., 2000; Wan et al., 2000; Zhen et al., 2000). A conserved role in synaptic development was also found in vertebrates as nerve terminal morphology is severely disrupted in zebrafish and mice constitutively lacking Esrom and MYCBP2, respectively (Burgess et al., 2004; D’Souza et al., 2005; Bloom et al., 2007). In mice the phrenic nerve does not fully innervate the diaphragm resulting in perinatal lethality due to respiratory distress. However, null and hypomorphic mutants of Drosophila Highwire were found to strongly inhibit axonal degeneration after axotomy (Xiong et al., 2012). Furthermore, conditional silencing of MYCBP2 in adult mice is tolerated for at least 6 weeks and confers axon-protective effects similar to those observed with loss of SARM1 (Babetto et al., 2013). Protection is observed in response to lesion of sciatic nerve and retinal ganglion neurons, illustrating the protective effects of MYCBP2 loss in both the peripheral and central nervous system. It has also recently been shown that loss of Highwire in Drosophila protects dopaminergic neurons and improves survival after traumatic brain injury; a major cause of human death and disability worldwide (Hill et al., 2020). Thus, despite the importance of MYCBP2 in neurodevelopment, post-developmental inhibition might be tolerated in humans. Particularly when applied to chemotherapy-induced neuropathy and neuron trauma, where acute inhibition would be sufficient.
MYCBP2 is a multi-domain E3 ubiquitin ligase (termed E3 hereon), a class of enzyme that catalyses the covalent conjugation of the small protein ubiquitin to target proteins (Zheng and Shabek, 2017). There are more than 600 ubiquitin E3s, either single polypeptides or multi-subunit complexes, that operate at the end of an enzymatic cascade involving initiating E1 activating enzymes and intermediary E2 conjugating enzymes (Hershko and Ciechanover, 1998) (Figure 1). Ubiquitination performs a wide range of cellular functions with regulation of protein substrate stability by proteasomal degradation being most notable (Pohl and Dikic, 2019). The pro-degenerative effects of MYCBP2 have been linked to its ability to destabilize NMNAT2 by neuronal proteasomal degradation (Xiong et al., 2012; Babetto et al., 2013; Desbois et al., 2018). Hence, inhibition of MYCBP2 E3 activity might be a strategy for inhibiting WD and preserving axon integrity. However, whether inhibition of MYCBP2 E3 activity specifically would have an axon-protective effect has remained unclear as neuronal phenotypes and cellular localization have been attributed to MYCBP2 regions outside the C-terminal ubiquitin ligase module (Grill et al., 2007; Abrams et al., 2008; Dorr et al., 2015).
Figure 1.
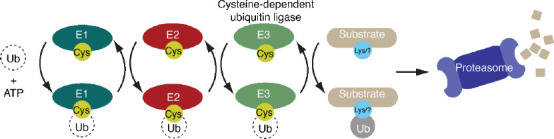
Cysteine-dependent ubiquitin ligases are key components of the ubiquitin-proteasome system.
Ubiquitin ligases operate within an enzymatic cascade consisting of E1 activating enzymes (E1s), E2 conjugating enzymes (E2s) and E3 ubiquitin ligases. Dependent on adenosine 5’-triphosphate (ATP), the E1 forms a reactive cysteine-linked intermediate with the C-terminal tail of ubiquitin. The Ub molecule is next passed onto a cysteine in an E2. Of the hundreds of ubiquitin ligase that exist, approximately 45 contain a single catalytic cysteine (transthiolation E3s) which accepts the Ub molecule from E2 via a transthiolation reaction. Protein substrates are then selected by the ubiquitin ligase and modified with Ub. This typically targets the substrate for degradation by the proteasome.
MYCBP2 Is a Novel RING-Cys-Relay Ubiquitin Ligase
In an unexpected discovery we showed that MYCBP2 is the sole member of a highly unusual class of E3 as at its extreme C-terminus resides an unprecedented 30 kDa enzymatic module termed RING-Cys-relay (RCR) (Figure 2A) (Pao et al., 2018). This was surprising because it was generally accepted that the different classes of catalytic E3 module had already been established. This discovery was enabled by chemical biological tools known as activity-based probes (Mulder et al., 2020). The activity-based probes covalently label E3s that demonstrate the transthiolation activity mediated by E3s that utilize a catalytic cysteine nucleophile (Figure 1) (Pao et al., 2016, 2018). Approximately 45 transthiolation E3s were believed to exist and MYCBP2 is a new addition to this subtype.
Figure 2.
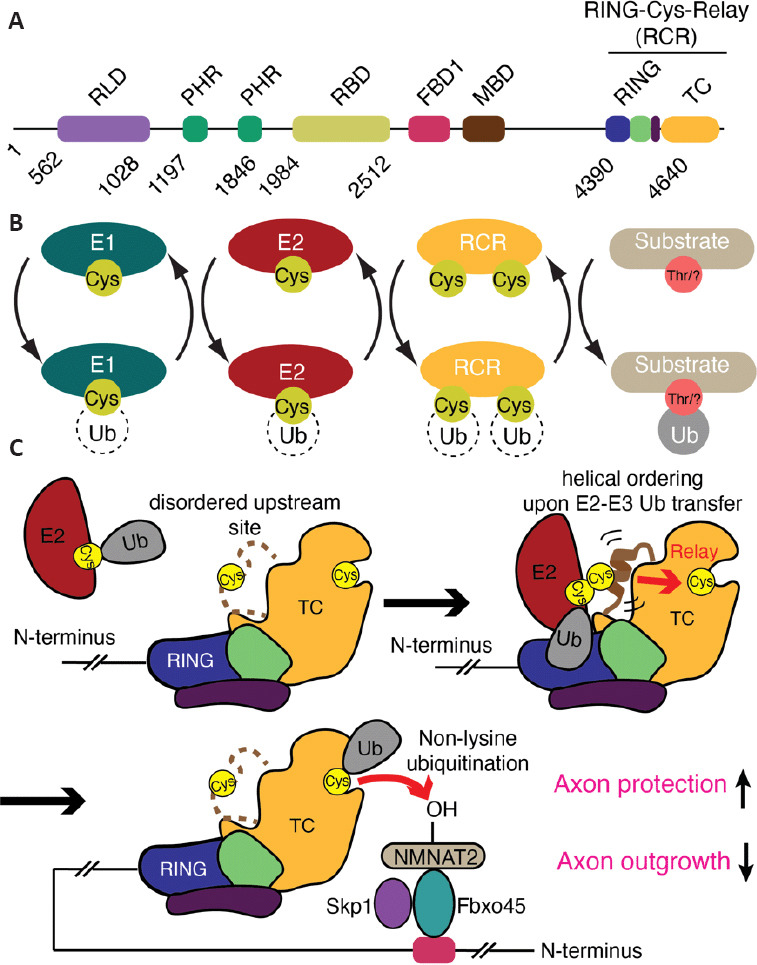
The mechanism of MYCBP2 RING-Cys-Relay activity and its role in axon protection and neural development.
(A) Domain architecture of MYCBP2 (RCC1-like GEF domain (RLD), two PHR-family-specific (PHR) domains, a RAE1 binding domain (RBD), an F-box binding domain 1 (FBD1), a Myc binding domain (MBD), C-terminal RING domain (RING) and Tandem Cysteine Domain (TC) that contains two active site cysteine residues involved in substrate ubiquitination. (B) Reaction scheme depicting how the RCR module within MYCBP2 operates within the multienzyme ubiquitin cascade. (C) The unusual ubiquitin E3 ligase MYCBP2 binds a ubiquitin-charged E2 enzyme via its RING domain. The ubiquitin molecule is transferred to an upstream catalytic cysteine within the TC domain which transiently induces local ordering of the upstream site. This might serve as an “entropic spring” allowing the ubiquitin molecule to be relayed to a downstream catalytic cysteine. Although not experimentally confirmed, the working model is that Fbxo45 and Skp1 bind to the distal N-terminal FBD1 region. They are responsible for binding substrates, including NMNAT2, and position them proximal to the downstream cysteine within the RCR module. The downstream site efficiently ubiquitinates hydroxy amino acids rather than conventional lysine residues. This unconventional mechanism of ubiquitination involving the two catalytic cysteines, termed RING-Cys-Relay (RCR), is central to neural development and its loss confers neurite protection in primary superior cervical ganglion cultures.
A highly unusual characteristic of MYCBP2 is that unlike other classes of transthiolation E3 it contains a second, downstream catalytic cysteine residue (Figure 2B). Once the initial covalent ligase-ubiquitin intermediate has been formed, the Ub molecule is intramolecularly “relayed” over a distance of ~24 Å to a downstream site. It is this downstream site that is tasked with substrate modification (Pao et al., 2018). Both of the catalytic cysteines reside within an unprecedented zinc ion-binding protein domain termed the tandem cysteine domain (Figure 2C). Further irregularities pertain to MYCBP2 because E3s studied previously invariably couple Ub to lysine amino groups via an isopeptide bond. In a striking digression from this dogma, MYCBP2 couples Ub to hydroxyl groups via an ester bond, where a strong preference is observed for threonine over serine residues in model substrates. Not only does this highlight the unappreciated substrate scope of ubiquitination, but these distinct characteristics of the RCR module might facilitate the development of specific small molecule inhibitors of MYCBP2 E3 activity.
RING-Cys-Relay Ubiquitin Ligase Activity Is Required for Normal Neural Development
Whilst MYCBP2 ligase activity had been implicated with axon degeneration by tuning NMNAT levels in Drosophila (Xiong et al., 2012), it remained unknown if MYCBP2’s newly discovered RCR mechanism was central to its pro-degenerative function. To test this, a CRISPR knock-in mouse model has been generated consisting of a constitutive mutation of the upstream cysteine to a catalytically dead alanine (Mabbitt et al., 2020). Whilst the full-length protein is preserved, ubiquitin transfer to the upstream cysteine, subsequent relay and ensuing ester-linked ubiquitination is ablated. A striking reduction in neurite outgrowth was observed in superior cervical ganglion explants from this homozygous knock-in model, reminiscent of the phenotypes observed with MYCBP2 silencing in mammals (Burgess et al., 2004; D’Souza et al., 2005; Bloom et al., 2007). Indeed, loss of RCR ubiquitin ligase activity in late-stage embryos also resulted in a marked reduction in diaphragm innervation by the phrenic nerve (Mabbitt et al., 2020). These observations indicated that the newly discovered RCR ubiquitin ligase mechanism is associated with the previously reported neurodevelopmental phenotypes.
RING-Cys-Relay Ubiquitin Ligase Activity Promotes Wallerian Degeneration
Notably, in addition to the neurodevelopmental phenotypes being recapitulated in the homozygous knock-in mouse model, so were the axon-protective effects. Despite the reduction in neurite outgrowth, loss of RCR ubiquitin ligase activity attenuated the degeneration of superior cervical ganglia explants following axotomy (Mabbitt et al., 2020). In addition, exogenous NMNAT2 was stabilized in embryonic fibroblasts from homozygous and heterozygous knock-in mice. Conditional silencing of MYCBP2 has also been shown to preserve dorsal root ganglia neurons following axotomy and vincristine treatment (Babetto et al., 2013). Whether protection is conferred after vincristine treatment in RCR-defective mice remains to be tested.
An exciting prospect is the stunted growth phenotype in the constitutive knock-in mouse model suppresses the potency of the protective effect after axotomy rather than exacerbates it. Hence, protection might be more pronounced if loss of RCR activity were to be induced pharmacologically in a post-developmental context. However, it remains unclear whether the demonstrated role of MYCBP2 RCR activity in neurodevelopment is amenable to sustained suppression in adult animals without significant side effects. Generation of a conditional knock-in mouse model, or tool inhibitors, should allow these aspects to be investigated further. Taken together, these observations suggest that the neurodevelopmental and pro-degenerative effects of MYCBP2 are largely dependent on its unusual RCR E3 activity.
Structural and Biochemical Insights into RING-Cys-Relay Ubiquitin Ligase Activity
Enabling the discovery of small molecule inhibitors of the RCR ubiquitin ligase machinery in MYCBP2 would be its structural and biochemical characterization. Blocking the binding of the ubiquitin-loaded E2 enzyme, impairing the ubiquitin relay process, or blocking the downstream site would be expected to abolish MYCBP2 ubiquitin ligase activity. Structural studies initially revealed the general architecture of the RCR module and its downstream catalytic site (Pao et al., 2018). A fortuitous packing interaction propagated throughout the crystal placed the amino acid threonine, which has a hydroxyl side chain, within the downstream active site. This provided insights into how substrate selection mediated at the catalytic site is achieved and how it might be leveraged for inhibitor design (Pao et al., 2018). However, a number of mechanistic details remained poorly understood. For example, it was not known if the RING-finger domain binds E2 enzyme as observed for other RING domain-containing E3s. The upstream site was also found to be completely disordered in the initial isolated structure. Hence, it was also unclear how the upstream cysteine accepted ubiquitin from E2 enzyme and how the relay process would deliver the ubiquitin cargo to the downstream site.
By further leveraging the activity-based probe technology a stabilized form of the otherwise labile ternary E2-E3-ubiquitin transfer intermediate was prepared and isolated (Mabbitt et al., 2020). This permitted high-resolution structure determination by X-ray crystallography. The structure revealed the molecular contacts required for E2-E3 ubiquitin transfer. In the earlier isolated structure, the region containing the upstream cysteine, termed the mediator loop, was too flexible to be observed (Pao et al., 2018). The new structure of the stabilized transfer complex revealed that the region containing the upstream cysteine forms a transiently ordered helical configuration during E2-E3 ubiquitin transfer (Figure 2C). The study also provides insights into how the intramolecular ubiquitin relay step works. Proline scanning experiments supported a model where the energy required to facilitate the striking ubiquitin relay process is generated by the transient ordering of the upstream site. This might then act like an “entropic spring” enabling the ubiquitin molecule to be catapulted to the downstream site (Figure 2C) (Mabbitt et al., 2020).
Substrate Recognition Is Achieved through a Multi-Subunit MYBCP2 Complex
The described RCR ubiquitin ligase module resides at the extreme C-terminus of the giant MYCBP2 protein but a region in the middle of the protein, known as the Fsn-1/Fbxo45 binding domain 1 (FBD1) (Figure 2A), seemingly cooperates with the RCR module. In mammals the FBD1 region in MYCBP2 binds the F-box domain containing protein Fbxo45 and the adapter protein Skp1 and this subcomplex serves as a substrate receptor module (Figure 2C) (Liao et al., 2004; Wu et al., 2007; Saiga et al., 2009). In C. elegans the same interaction is found between the orthologous proteins (Rpm-1 and Fsn-1). The formation of multi-subunit ubiquitin ligase complexes, where the substrate receptor module is a distinct polypeptide, is the hallmark of a large subset of E3s known as the Cullin E3s (Baek et al., 2020). However, unlike Cullin E3s that dynamically exchange substrate receptor modules (Wang et al., 2020), Fbxo45 is the only receptor module assigned to MYCBP2. The fact that MYCBP2, a non-Cullin family member, also utilizes a dedicated substrate receptor is a further irregularity to its E3 mechanism.
The FBD1 region in MYCBP2 is ~2000 residues N-terminal of the RCR ubiquitin ligase module (Figure 2A) (Saiga et al., 2009; Desbois et al., 2018). Knock-down of Skp1 or Fbxo45 phenocopies MYCBP2 silencing as it confers axon-protection in response to both physical and chemical injury (Yamagishi and Tessier-Lavigne, 2016). Furthermore, this has been directly linked to stabilization of NMNAT2 (Yamagishi and Tessier-Lavigne, 2016). Consistent with the role of F-box domains being direct substrate receptors, Fbxo45 interacts with NMNAT2 (Babetto et al., 2013; Desbois et al., 2018). Interestingly, disruption of the interaction between Rpm-1 and Fsn-1 in C. elegans with a transgenically expressed peptide inhibitor recapitulates the synaptic defects observed with null worms (Sharma et al., 2014). This suggests that the substrate receptor sub-complex is also a potential therapeutic target. Taken together, these findings point toward a huge multi-subunit ligase machine, where the C-terminal RCR module is the catalytic engine, being central to normal neurodevelopment and post-developmental programmed axon degeneration (Figure 2C). Hence, inhibitors that disrupt not only the RCR module, but also the formation of the multi-subunit complex, should stabilize NMNAT2 and confer axon protective effects in response to injury. Further structural characterization should enable the visualization of this complex and demonstrate how Fbxo45 binds substrates and places them proximal to the C-terminal RCR ubiquitin ligase module, and also provide insights into how this activity is regulated.
Outlook
Beyond these findings, many questions remain unanswered. Ubiquitination of NMNAT2 on an amino acid with a hydroxyl side chain is most likely to be central to destabilizing NMNAT2 and promoting programmed axon degeneration. However, the high activity of MYCBP2 towards small molecule hydroxy compounds raises the possibility that a non-proteinaceous substrate might also be involved, as has now been observed with another unusual E3 ligase that has recently come to light (Otten et al., 2021). Furthermore, we do not appreciate the limitations of MYCBP2 and SARM1 inhibition. Thus, it remains possible these are complementary targets and inhibition of one rather than the other might be preferable for certain indications or neuron types. Although there are data in support of MYCBP2 operating upstream of SARM1 in a linear pathway, Drosophila genetics indicates that MYCBP2 might operate in parallel of SARM1 (Neukomm et al., 2017). It is also important to consider the roles MYCBP2 E3 ligase activity has outside neurodevelopment and axon degeneration. Recently, Rpm-1 has been shown to suppress neuronal autophagy by destabilizing the autophagy initiator UNC51/ULK1 (Crawley et al., 2019). Whether this relates to the established neurological phenotypes and broadens or limits the therapeutic scope of MYCBP2 inhibition remains unclear. Interestingly, MYCBP2 links to cancer have also been reported as it destabilizes the tumor suppressor FBW7 thereby contributing to chemotherapy resistance (Richter et al., 2020). In addition, MYCBP2 can regulate cell growth and proliferation via the mTOR pathway (Murthy et al., 2004; Han et al., 2008, 2012).
The role of the ubiquitin system in WD does not end with MYCBP2. Forward genetic screens in Drosophila have also identified a third pro-degenerative protein (Neukomm et al., 2017). Axundead belongs to the kelch-like gene family and Axundead mutants suppress axon death across multiple neuron types and preserve axon morphology for the lifespan of the animal (Neukomm et al., 2017). Conceptually similar to Fbxo45, kelch-like proteins are the the substrate receptors for the multi-subunit Cullin E3s (Shi et al., 2019). Although conservation of this pathway in mammals remains to be confirmed, it might also afford an additional opportunity for therapeutic intervention. Furthermore, NAD+ and nicotinamide mononucleotide levels have been shown to regulate WD (Di Stefano et al., 2015; Figley et al., 2021). Strategies that modulate the axonal levels of these nucleotides might also halt or retard axonal degeneration. For example, activators of NMNAT2 would be expected to enhance NAD+ levels and inhibit WD. In summary a deeper molecular level understanding of multiple regulators of programmed axon degeneration has emerged. These might now be exploited and bring us ever closer to much needed therapeutic relief against chemotherapy-induced neuropathies, neuronal injury and neurodegenerative disease.
Acknowledgments:
The author would like to thank Dr. Marc-Andre Dery for help drafting the manuscript.
Footnotes
Conflicts of interest: SV is founder and consultant of Outrun Therapeutics, a biotechnology company working in the ubiquitin field.
Funding: This work was supported by the United Kingdom MRC (MC_UU_12016/8), the Biotechnology and Biological Sciences Research Council (BB/P003982/1), and The Michael J. Fox Foundation (to SV).
C-Editors: Zhao M, Liu WJ, Li JY; T-Editor: Jia Y.
References
- 1.Abrams B, Grill B, Huang X, Jin Y. Cellular and molecular determinants targeting the Caenorhabditis elegans PHR protein RPM-1 to perisynaptic regions. Dev Dyn. 2008;237:630–639. doi: 10.1002/dvdy.21446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adalbert R, Coleman MP. Review: Axon pathology in age-related neurodegenerative disorders. Neuropathol Appl Neurobiol. 2013;39:90–108. doi: 10.1111/j.1365-2990.2012.01308.x. [DOI] [PubMed] [Google Scholar]
- 3.Ali YO, Allen HM, Yu L, Li-Kroeger D, Bakhshizadehmahmoudi D, Hatcher A, McCabe C, Xu J, Bjorklund N, Taglialatela G, Bennett DA, De Jager PL, Shulman JM, Bellen HJ, Lu HC. NMNAT2: HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 2016;14:e1002472. doi: 10.1371/journal.pbio.1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Babetto E, Beirowski B, Russler EV, Milbrandt J, DiAntonio A. The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep. 2013;3:1422–1429. doi: 10.1016/j.celrep.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baek K, Scott DC, Schulman BA. NEDD8 and ubiquitin ligation by cullin-RING E3 ligases. Curr Opin Struct Biol. 2020;67:101–109. doi: 10.1016/j.sbi.2020.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloom AJ, Miller BR, Sanes JR, DiAntonio A. The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev. 2007;21:2593–2606. doi: 10.1101/gad.1592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgess RW, Peterson KA, Johnson MJ, Roix JJ, Welsh IC, O'Brien TP. Evidence for a conserved function in synapse formation reveals Phr1 as a candidate gene for respiratory failure in newborn mice. Mol Cell Biol. 2004;24:1096–1105. doi: 10.1128/MCB.24.3.1096-1105.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coleman MP, Hoke A. Programmed axon degeneration: from mouse to mechanism to medicine. Nat Rev Neurosci. 2020;21:183–196. doi: 10.1038/s41583-020-0269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crawley O, Opperman KJ, Desbois M, Adrados I, Borgen MA, Giles AC, Duckett DR, Grill B. Autophagy is inhibited by ubiquitin ligase activity in the nervous system. Nat Commun. 2019;10:5017. doi: 10.1038/s41467-019-12804-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D'Souza J, Hendricks M, Le Guyader S, Subburaju S, Grunewald B, Scholich K, Jesuthasan S. Formation of the retinotectal projection requires Esrom, an ortholog of PAM (protein associated with Myc) Development. 2005;132:247–256. doi: 10.1242/dev.01578. [DOI] [PubMed] [Google Scholar]
- 11.Dadon-Nachum M, Melamed E, Offen D. The “dying-back” phenomenon of motor neurons in ALS. J Mol Neurosci. 2011;43:470–477. doi: 10.1007/s12031-010-9467-1. [DOI] [PubMed] [Google Scholar]
- 12.Desbois M, Crawley O, Evans PR, Baker ST, Masuho I, Yasuda R, Grill B. PAM forms an atypical SCF ubiquitin ligase complex that ubiquitinates and degrades NMNAT2. J Biol Chem. 2018;293:13897–13909. doi: 10.1074/jbc.RA118.002176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Stefano M, Nascimento-Ferreira I, Orsomando G, Mori V, Gilley J, Brown R, Janeckova L, Vargas ME, Worrell LA, Loreto A, Tickle J, Patrick J, Webster JR, Marangoni M, Carpi FM, Pucciarelli S, Rossi F, Meng W, Sagasti A, Ribchester RR, et al. A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death Differ. 2015;22:731–742. doi: 10.1038/cdd.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dorr A, Pierre S, Zhang DD, Henke M, Holland S, Scholich K. MYCBP2 Is a guanosine exchange factor for ran protein and determines its localization in neurons of dorsal root ganglia. J Biol Chem. 2015;290:25620–25635. doi: 10.1074/jbc.M115.646901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Essuman K, Summers DW, Sasaki Y, Mao X, DiAntonio A, Milbrandt J. The SARM1 Toll/Interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron. 2017;93:1334–1343. doi: 10.1016/j.neuron.2017.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Figley MD, Gu W, Nanson JD, Shi Y, Sasaki Y, Cunnea K, Malde AK, Jia X, Luo Z, Saikot FK, Mosaiab T, Masic V, Holt S, Hartley-Tassell L, McGuinness HY, Manik MK, Bosanac T, Landsberg MJ, Kerry PS, Mobli M, et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD(+) ratio to trigger axon degeneration. Neuron. 2021;109:1118–1136. doi: 10.1016/j.neuron.2021.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geisler S. Vincristine- and bortezomib-induced neuropathies - from bedside to bench and back. Exp Neurol. 2020:113519. doi: 10.1016/j.expneurol.2020.113519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geisler S, Doan RA, Cheng GC, Cetinkaya-Fisgin A, Huang SX, Hoke A, Milbrandt J, DiAntonio A. Vincristine and bortezomib use distinct upstream mechanisms to activate a common SARM1-dependent axon degeneration program. JCI Insight. 2019;4:e129920. doi: 10.1172/jci.insight.129920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerdts J, Summers DW, Sasaki Y, DiAntonio A, Milbrandt J. Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J Neurosci. 2013;33:13569–13580. doi: 10.1523/JNEUROSCI.1197-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilley J, Coleman MP. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grill B, Murphey RK, Borgen MA. The PHR proteins: intracellular signaling hubs in neuronal development and axon degeneration. Neural Dev. 2016;11:8. doi: 10.1186/s13064-016-0063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grill B, Bienvenut WV, Brown HM, Ackley BD, Quadroni M, Jin Y. C. elegans RPM-1 regulates axon termination and synaptogenesis through the Rab GEF GLO-4 and the Rab GTPase GLO-1. Neuron. 2007;55:587–601. doi: 10.1016/j.neuron.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 23.Guo Q, Xie J, Dang CV, Liu ET, Bishop JM. Identification of a large Myc-binding protein that contains RCC1-like repeats. Proc Natl Acad Sci U S A. 1998;95:9172–9177. doi: 10.1073/pnas.95.16.9172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han S, Witt RM, Santos TM, Polizzano C, Sabatini BL, Ramesh V. Pam (Protein associated with Myc) functions as an E3 ubiquitin ligase and regulates TSC/mTOR signaling. Cell Signal. 2008;20:1084–1091. doi: 10.1016/j.cellsig.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han S, Kim S, Bahl S, Li L, Burande CF, Smith N, James M, Beauchamp RL, Bhide P, DiAntonio A, Ramesh V. The E3 ubiquitin ligase protein associated with Myc (Pam) regulates mammalian/mechanistic target of rapamycin complex 1 (mTORC1) signaling in vivo through N- and C-terminal domains. J Biol Chem. 2012;287:30063–30072. doi: 10.1074/jbc.M112.353987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 27.Hill CS, Sreedharan J, Loreto A, Menon DK, Coleman MP. Loss of highwire protects against the deleterious effects of traumatic brain injury in Drosophila melanogaster. Front Neurol. 2020;11:401. doi: 10.3389/fneur.2020.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huppke P, Wegener E, Gilley J, Angeletti C, Kurth I, Drenth JPH, Stadelmann C, Barrantes-Freer A, Bruck W, Thiele H, Nurnberg P, Gartner J, Orsomando G, Coleman MP. Homozygous NMNAT2 mutation in sisters with polyneuropathy and erythromelalgia. Exp Neurol. 2019;320:112958. doi: 10.1016/j.expneurol.2019.112958. [DOI] [PubMed] [Google Scholar]
- 29.Krauss R, Bosanac T, Devraj R, Engber T, Hughes RO. Axons matter: the promise of treating neurodegenerative disorders by targeting SARM1-mediated axonal degeneration. Trends Pharmacol Sci. 2020;41:281–293. doi: 10.1016/j.tips.2020.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Liao EH, Hung W, Abrams B, Zhen M. An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature. 2004;430:345–350. doi: 10.1038/nature02647. [DOI] [PubMed] [Google Scholar]
- 31.Lukacs M, Gilley J, Zhu Y, Orsomando G, Angeletti C, Liu J, Yang X, Park J, Hopkin RJ, Coleman MP, Zhai RG, Stottmann RW. Severe biallelic loss-of-function mutations in nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) in two fetuses with fetal akinesia deformation sequence. Exp Neurol. 2019;320:112961. doi: 10.1016/j.expneurol.2019.112961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur J Neurosci. 1989;1:27–33. doi: 10.1111/j.1460-9568.1989.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 33.Mabbitt PD, Loreto A, Dery MA, Fletcher AJ, Stanley M, Pao KC, Wood NT, Coleman MP, Virdee S. Structural basis for RING-Cys-Relay E3 ligase activity and its role in axon integrity. Nat Chem Biol. 2020;16:1227–1236. doi: 10.1038/s41589-020-0598-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, Fernando FS, Tarlton A, Andressen C, Addicks K, Magni G, Ribchester RR, Perry VH, Coleman MP. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 35.Mulder MPC, Witting KF, Ovaa H. Cracking the ubiquitin code: the ubiquitin toolbox. Curr Issues Mol Biol. 2020;37:1–20. doi: 10.21775/cimb.037.001. [DOI] [PubMed] [Google Scholar]
- 36.Murthy V, Han S, Beauchamp RL, Smith N, Haddad LA, Ito N, Ramesh V. Pam and its ortholog highwire interact with and may negatively regulate the TSC1.TSC2 complex. J Biol Chem. 2004;279:1351–1358. doi: 10.1074/jbc.M310208200. [DOI] [PubMed] [Google Scholar]
- 37.Neukomm LJ, Burdett TC, Seeds AM, Hampel S, Coutinho-Budd JC, Farley JE, Wong J, Karadeniz YB, Osterloh JM, Sheehan AE, Freeman MR. Axon death pathways converge on axundead to promote functional and structural axon disassembly. Neuron. 2017;95:78–91. doi: 10.1016/j.neuron.2017.06.031. [DOI] [PubMed] [Google Scholar]
- 38.Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Jr, Conforti L, Coleman M, Tessier-Lavigne M, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otten EG, Werner E, Crespillo-Casado A, Boyle KB, Dharamdasani V, Pathe C, Santhanam B, Randow F. Ubiquitylation of lipopolysaccharide by RNF213 during bacterial infection. Nature. 2021;594:111–116. doi: 10.1038/s41586-021-03566-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pao KC, Wood NT, Knebel A, Rafie K, Stanley M, Mabbitt PD, Sundaramoorthy R, Hofmann K, van Aalten DMF, Virdee S. Activity-based E3 ligase profiling uncovers an E3 ligase with esterification activity. Nature. 2018;556:381–385. doi: 10.1038/s41586-018-0026-1. [DOI] [PubMed] [Google Scholar]
- 41.Pao KC, Stanley M, Han C, Lai YC, Murphy P, Balk K, Wood NT, Corti O, Corvol JC, Muqit MM, Virdee S. Probes of ubiquitin E3 ligases enable systematic dissection of parkin activation. Nat Chem Biol. 2016;12:324–331. doi: 10.1038/nchembio.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pohl C, Dikic I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science. 2019;366:818–822. doi: 10.1126/science.aax3769. [DOI] [PubMed] [Google Scholar]
- 43.Richter KT, Kschonsak YT, Vodicska B, Hoffmann I. FBXO45-MYCBP2 regulates mitotic cell fate by targeting FBXW7 for degradation. Cell Death Differ. 2020;27:758–772. doi: 10.1038/s41418-019-0385-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saiga T, Fukuda T, Matsumoto M, Tada H, Okano HJ, Okano H, Nakayama KI. Fbxo45 forms a novel ubiquitin ligase complex and is required for neuronal development. Mol Cell Biol. 2009;29:3529–3543. doi: 10.1128/MCB.00364-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schaefer AM, Hadwiger GD, Nonet ML. rpm-1 a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron. 2000;26:345–356. doi: 10.1016/s0896-6273(00)81168-x. [DOI] [PubMed] [Google Scholar]
- 46.Sharma J, Baker ST, Turgeon SM, Gurney AM, Opperman KJ, Grill B. Identification of a peptide inhibitor of the RPM-1. FSN-1 ubiquitin ligase complex. J Biol Chem. 2014;289:34654–34666. doi: 10.1074/jbc.M114.614065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi X, Xiang S, Cao J, Zhu H, Yang B, He Q, Ying M. Kelch-like proteins: physiological functions and relationships with diseases. Pharmacol Res. 2019;148:104404. doi: 10.1016/j.phrs.2019.104404. [DOI] [PubMed] [Google Scholar]
- 48.Tagliaferro P, Burke RE. Retrograde axonal degeneration in Parkinson disease. J Parkinsons Dis. 2016;6:1–15. doi: 10.3233/JPD-150769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waller A. Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Edinb Med Surg J. 1851;76:369–376. [PMC free article] [PubMed] [Google Scholar]
- 50.Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, Goodman CS. Highwire regulates synaptic growth in Drosophila. Neuron. 2000;26:313–329. doi: 10.1016/s0896-6273(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Zhai Q, Chen Y, Lin E, Gu W, McBurney MW, He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang K, Deshaies RJ, Liu X. Assembly and regulation of CRL ubiquitin ligases. Adv Exp Med Biol. 2020;1217:33–46. doi: 10.1007/978-981-15-1025-0_3. [DOI] [PubMed] [Google Scholar]
- 53.White MA, Lin Z, Kim E, Henstridge CM, Pena Altamira E, Hunt CK, Burchill E, Callaghan I, Loreto A, Brown-Wright H, Mead R, Simmons C, Cash D, Coleman MP, Sreedharan J. Sarm1 deletion suppresses TDP-43-linked motor neuron degeneration and cortical spine loss. Acta Neuropathol Commun. 2019;7:166. doi: 10.1186/s40478-019-0800-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu C, Daniels RW, DiAntonio A. DFsn collaborates with Highwire to down-regulate the Wallenda/DLK kinase and restrain synaptic terminal growth. Neural Dev. 2007;2:16. doi: 10.1186/1749-8104-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiong X, Hao Y, Sun K, Li J, Li X, Mishra B, Soppina P, Wu C, Hume RI, Collins CA. The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol. 2012;10:e1001440. doi: 10.1371/journal.pbio.1001440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamagishi Y, Tessier-Lavigne M. An atypical SCF-like ubiquitin ligase complex promotes Wallerian degeneration through regulation of axonal Nmnat2. Cell Rep. 2016;17:774–782. doi: 10.1016/j.celrep.2016.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhen M, Huang X, Bamber B, Jin Y. Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron. 2000;26:331–343. doi: 10.1016/s0896-6273(00)81167-8. [DOI] [PubMed] [Google Scholar]
- 58.Zheng N, Shabek N. Ubiquitin ligases: structure, function, and regulation. Annu Rev Biochem. 2017;86:129–157. doi: 10.1146/annurev-biochem-060815-014922. [DOI] [PubMed] [Google Scholar]