

Abstract
Genomic tumour profiling informs targeted treatment options. Entrectinib is a tyrosine kinase inhibitor with efficacy in NTRK fusion‐positive (‐fp) solid tumours and ROS1‐fp non‐small cell lung cancer. FoundationOne® Liquid CDx (F1L CDx), a non‐invasive in vitro next‐generation sequencing (NGS)‐based diagnostic, detects genomic alterations in plasma circulating tumour DNA (ctDNA). We evaluated the clinical validity of F1L CDx as an aid in identifying patients with NTRK‐fp or ROS1‐fp tumours and assessed the genomic landscape pre‐ and post‐entrectinib treatment. Among evaluable pre‐treatment clinical samples (N = 85), positive percentage agreements between F1L CDx and clinical trial assays (CTAs) were 47.4% (NTRK fusions) and 64.5% (ROS1 fusions); positive predictive value was 100% for both. The objective response rate for CTA+ F1L CDx+ patients was 72.2% in both cohorts. The median duration of response significantly differed between F1L CDx+ and F1L CDx− samples in ROS1‐fp (5.6 vs. 17.3 months) but not NTRK‐fp (9.2 vs. 12.9 months) patients. Fifteen acquired resistance mutations were detected. We conclude that F1L CDx is a clinically valid complement to tissue‐based testing to identify patients who may benefit from entrectinib and those with acquired resistance mutations associated with disease progression.
Keywords: F1L CDx, genomic profiling, NTRK, resistance, ROS1
Entrectinib is a kinase inhibitor that targets both the NTRK and ROS1 oncogenes. We show that NTRK and ROS1 fusions are present in 46% and 64%, respectively, of blood samples from patients enrolled on a clinical trial, with a response rate of 72%. Profiling blood at progression showed that resistance occurs via mutations in NTRK/ROS1 or via downstream MAP‐kinase reactivation.

Abbreviations
- cfDNA
circulating cell‐free DNA
- CI
confidence interval
- CNS
central nervous system
- CR
complete response
- CRC
colorectal cancer
- CTA
clinical trial assay
- ctDNA
circulating tumour DNA
- DoR
duration of response
- ECOG PS
Eastern Cooperative Oncology Group performance status
- F1L CDx
FoundationOne® Liquid CDx
- FDA
food and drug administration
- fn
fusion‐negative
- fp
fusion‐positive
- IQR
interquartile range
- LoD
limit of detection
- MAPK
mitogen‐activated protein kinase
- MASC
mammary analogue secretory carcinoma
- N/A
not applicable
- NE
non‐evaluable
- NGS
next‐generation sequencing
- NPA
negative per cent agreement
- NPV
negative predictive value
- NSCLC
non‐small cell lung cancer
- NTRK
neurotrophic tyrosine receptor kinase
- ORR
overall response rate
- PD
progressive disease
- PPA
positive per cent agreement
- PPV
positive predictive value
- PR
partial response
- RECIST
response evaluation criteria in solid tumours
- ROS1
ROS proto‐oncogene 1
- SD
stable disease
- TKI
tyrosine kinase inhibitor
- TRK
tropomyosin receptor kinase
1. Introduction
The increasing use of targeted cancer treatments tailored to a tumour’s individual genomic profile has generated a need to develop rapid and reliable assays for comprehensive genomic profiling [1]. Next‐generation sequencing (NGS) is increasingly used to detect molecular alterations driving tumour development [2], and the genomic information provided can support physicians in making informed decisions to ensure patients have access to the most appropriate and effective treatment.
Tumour biopsies represent the most widely utilized source of tumour DNA for genomic analysis; while current tissue‐based assays show high sensitivity and specificity, several limitations are associated with solid tissue biopsies [3]. The invasiveness of biopsies makes it unsuitable for use in frail patients and repeat sampling for longitudinal tumour monitoring is undesirable. Solid tumour biopsies also cannot be taken from physically inaccessible tumours [3, 4], such as thoracic malignancies. Inherent tumour heterogeneity also means that solid tumour biopsies may fail to capture the complete genomic profile of an individual tumour [3, 5].
Technological advances have resulted in the development of liquid biopsy assays using circulating tumour DNA (ctDNA), a subset of circulating cell‐free DNA (cfDNA), extracted from blood plasma samples, as a basis for genomic analysis [6]. Liquid biopsy assays offer a minimally invasive means for genomic evaluation that is suitable for use in all patients for whom blood can be safely drawn, regardless of tumour location and patient frailty [7, 8]. Blood samples can also be taken easily, with lower burden on healthcare resources and greater patient convenience. As ctDNA is shed into blood plasma from each metastatic tumour site, a more complete picture of tumour heterogeneity is gained compared with tissue‐based sampling methods [7]. Because blood samples can be taken repeatedly for testing, liquid biopsy assays can support detection of actionable driver mutations to inform treatment selection at diagnosis, but can also monitor treatment response, detect recurrence, and identify resistance mechanisms, thereby potentially guiding therapeutic decisions [9, 10].
The FoundationOne® Liquid CDx (F1L CDx) assay (Foundation Medicine Inc., Cambridge, MA, USA) is an NGS‐based in vitro diagnostic for detecting genomic alterations in cfDNA, including gene fusions [11]. The F1L CDx assay received US Food and Drug Administration (FDA) approval as a comprehensive pan‐tumour liquid biopsy test for patients with solid tumours (August 2020) and has since received approval as a companion diagnostic in multiple indications, including non‐small cell lung cancer (NSCLC; alectinib, gefitinib, osimertinib, erlotinib), breast cancer (alpelisib), prostate cancer (olaparib, rucaparib) and ovarian cancer (rucaparib) [12].
Gene fusions of the neurotrophic tyrosine receptor kinase gene (NTRK1/2/3; coding for tropomyosin receptor kinases TRKA/B/C) and the tyrosine receptor kinase ROS proto‐oncogene 1 (ROS1) can constitutively activate kinases acting as oncogenic drivers [13, 14, 15, 16]. Entrectinib is a central nervous system (CNS)‐active potent inhibitor of TRKA/B/C and ROS1 [17, 18, 19] approved for the treatment of adult and paediatric patients aged ≥12 years with NTRK fusion‐positive (NTRK‐fp) solid tumours and adults with ROS1 fusion‐positive (ROS1‐fp) NSCLC. This was based on an integrated analysis of three Phase I/II studies (ALKA‐372‐001, EudraCT 2012–000148–88; STARTRK‐1, NCT02097810; STARTRK‐2, NCT02568267), showing strong clinical efficacy with entrectinib in patients with NTRK‐fp solid tumours [objective response rate (ORR), 57%; median duration of response (DoR), 10.4 months] and ROS1‐fp NSCLC (ORR, 77%; median DoR 24.6 months) [17, 20].
This study examined the clinical validity of F1L CDx to aid in identifying patients with NTRK‐fp solid tumours and ROS1‐fp NSCLC for entrectinib treatment and assessed the pre‐ and post‐treatment genomic landscape of patients with NTRK‐fp solid tumours and ROS1‐fp NSCLC receiving entrectinib.
2. Materials and methods
2.1. Patient population
Patients in the analysis were enrolled in the Phase II global basket study STARTRK‐2 (NCT02568267). The design of this ongoing trial has been described previously [17, 20]. Briefly, patients aged ≥18 years with locally advanced/metastatic NTRK‐fp solid tumour (96% metastatic) or ROS1‐fp NSCLC (94% metastatic) measurable by Response Evaluation Criteria in Solid Tumors (RECIST; v1.1) had received no prior tyrosine kinase inhibitor (TKI) therapy, had an Eastern Cooperative Oncology Group performance status (ECOG PS) ≤ 2, and could be enrolled with brain metastases if they were asymptomatic or had received previous treatment resulting in symptom control. Patients received entrectinib 600 mg orally once daily, until documented radiographic progression, unacceptable toxicity or consent withdrawal. The high‐level tumour types included in the NTRK‐fp cohort are presented in Table S1. Primary endpoints were ORR and median DoR, assessed by blinded independent central review using RECIST v1.1. As part of the study scheduled assessments, blood samples were collected at screening, day 1 of each treatment cycle and at end of treatment.
The presence of NTRK or ROS1 fusions was confirmed by tumour DNA‐ or RNA‐based clinical trial assays (CTAs) before enrolment in STARTRK‐2. The CTAs utilized comprised Trailblaze Pharos™ (Ignyta, F. Hoffmann‐La Roche Ltd, Basel, Switzerland) NGS central testing or local diagnostic laboratory nucleic acid‐based methodologies (e.g. NGS, Sanger sequencing, reverse transcription‐polymerase chain reaction, NanoString). If patients were enrolled via a local test and a tumour sample was available, independent central molecular NGS testing was performed post‐enrolment via the Trailblaze Pharos™ assay. For the NTRK‐fp cohort, one central and 18 local testing laboratories were used to enrol the study participants, using the following technologies: PCR (n = 1), NanoString (n = 1), RNA‐NGS (n = 29), DNA‐NGS (n = 19) and RNA‐ + DNA‐NGS (n = 4). For the ROS1‐fp cohort, one central and 11 local testing laboratories were used to enrol the study participants, using the following technologies: FISH (n = 15), RNA‐NGS (n = 27), DNA‐NGS (n = 9).
STARTRK‐2 was conducted in accordance with the principles of the Declaration of Helsinki and Good Clinical Practice guidelines. Written, informed consent was obtained from all patients. The study protocol and biosample collection were approved by all relevant institutional review boards and/or ethics committees.
2.2. Plasma samples
Patients with NTRK‐fp solid tumours or ROS1‐fp NSCLC enrolled in STARTRK‐2 (clinical cut‐off: May 2018) for whom frozen pre‐treatment plasma samples were available were eligible for inclusion in clinical bridging analyses. Those who also had available plasma samples at/after progressive disease (PD) on entrectinib treatment were eligible for resistance mutation analysis. Plasma was isolated from whole blood specimens at the time of collection and stored frozen until processing.
Additional paired plasma samples from patients with NTRK or ROS1 fusion‐negative (fn) non‐NSCLC tumour types (n = 8) were purchased from an external commercial vendor (BiolVT LCC, Westbury, NY, USA).
2.3. Evaluation of plasma samples using the F1L CDx assay
The F1L CDx assay is a qualitative assay using targeted, high‐throughput hybridization‐based capture technology and is FDA‐approved to detect substitutions, insertions, and deletions in 311 genes, rearrangements in three genes, and copy number alterations in three genes [12]. cfDNA isolated from plasma is utilized and has a median limit of detection (LoD; % variant allele frequency) for rearrangements of 0.37% in enhanced and 0.90% in standard sensitivity regions. Reproducibility for detecting gene rearrangement is 99.2% [95% confidence interval (CI): 99.1–99.3] [11].
Analysis of all pre‐treatment and PD plasma samples using the F1L CDx assay was performed in the Foundation Medicine Inc. laboratory, according to standard workflow [11, 12]. Plasma was separated from whole blood by centrifugation and cfDNA subsequently isolated using the KingFisher™ Flex Magnetic Particle Processor (Thermo Fisher, Waltham, MA, USA) and built into genomic libraries. Libraries were sequenced using the Illumina NovaSeq 6000 platform (Illumina Inc., San Diego, CA, USA). Only samples with cfDNA content >30 ng, as assessed by the Agilent 4200 TapeStation assay, were included in the analyses.
2.4. Clinical bridging analyses
2.4.1. Concordance analysis
Concordance between CTA and F1L CDx assays in detection of NTRK or ROS1 fusions was calculated as positive and negative percent agreement (PPA; NPA). Positive and negative predictive values (PPV; NPV) were derived from PPA and NPA following adjustment for fusion prevalence (NTRK, 0.32% [21]; ROS1, 1–2% [15, 16]).
Since the PPV point estimate is 1 and no statistical method is available, 18 was used as the n in the binomial distribution for a positive fusion number. The 95% CI was estimated by the Wilson score interval for PPV and the bootstrap method for NPV.
2.4.2. Clinical efficacy analysis
Clinical efficacy (ORR, DoR) was calculated for the following populations: CTA+, CTA+ F1L CDx+, CTA+ F1L CDx− and CTA+ F1L CDx unevaluable populations. Estimated clinical efficacy was then compared with entrectinib efficacy data from the primary integrated analysis of three Phase I/II studies enrolling patients with NTRK‐fp solid tumours or ROS1‐fp NSCLC [17, 20]. Two patients were removed from the original population due to prior crizotinib treatment and were therefore excluded from this analysis.
Let θ denote the estimate of overall response rate (ORR) for the FoundationOne Liquid CDx (F1L CDx)+ sub‐population. Given the concordance between CTAs and F1L CDx, θ was estimated using the following formula: [22]
where
is the estimated ORR for (F1L CDx+ & CTA+)
is the estimated ORR for (F1L CDx+ & CTA−)
P is the conditional probability; P = Pr (CTA+│F1L CDx+)
As samples obtained from patients enrolled in STARTRK‐2 were enriched for CTA+ results, and its corresponding variance var() were estimated by:
where
prevalence of NTRK or ROS1 in the intended use population
The 95% CI was calculated using bootstrap simulation results (n = 5000).
2.4.3. Sensitivity analysis
Imbalance in covariates (age, sex, baseline ECOG PS, race, smoking, baseline CNS metastases, histology) between F1L CDx‐evaluable and F1L CDx‐unevaluable groups was evaluated using non‐parametric Mann–Whitney (continuous variables) or Fisher–Freeman–Halton (Fisher’s exact) (categorical variables) tests. Association between covariates and F1L CDx result (detected vs. not detected) was evaluated via univariate logistic regression models, considering the above independent factors (excluding smoking) and clinical outcome.
To evaluate robustness of clinical efficacy estimates against missing F1L CDx results, unevaluable F1L CDx results were generated using multiple imputation with 50 imputed datasets, and PPA and NPA estimates were compared for three imputation models (Table S2). To assess sensitivity according to CTA type, PPA and NPA where then calculated and compared for the three CTA subgroups (Pharos, F1/F1Heme or other local tests; Table S3) using 50 imputed datasets and imputation model 3. Robustness of clinical endpoints was assessed using 50 imputed datasets and imputation model 1.
2.5. Resistance mutation analyses
Presence or absence of baseline primary resistance and post‐treatment acquired resistance mutations in driver proto‐oncogenes and the 10 most frequently altered genes was depicted graphically using oncoplots for each of the NTRK‐fp and ROS1‐fp populations, using matched pre‐treatment and PD plasma samples. Fisher’s exact test was performed for each gene to assess association between presence of a mutation and objective response (complete or partial response), with no multiplicity adjustments.
2.6. ctDNA fraction
To investigate whether ctDNA fraction influenced detection of fusions or secondary mutations in NTRK1/3 or ROS1, ctDNA fraction was compared using a t‐test between samples in which a target aberration was detected and samples in which the corresponding aberration was not detected. Quantification of the ctDNA fraction was measured using two complementary methods: the proprietary tumour fraction estimator (TFE) and the maximum somatic allele frequency (MSAF) method [23]. The basis of TFE is a measure of tumour aneuploidy that incorporates observed deviations in coverage across the genome for a given sample. The calibration of calculated values for this metric is done to generate an estimate of the tumour fraction against a training set based on samples with well‐defined tumour fractions. MSAF is used when the TFE's ability to return an informative estimate is limited by lack of tumour aneuploidy. In the MSAF method, the allele fraction of all known somatic, likely somatic, and variant of unknown significance coding alterations by non‐PCR duplicate read pairs is used to estimate ctDNA fraction, with the exception of germline variants and variants in genes which are associated with clonal haematopoiesis.
3. Results
3.1. Plasma samples
Ninety‐eight pre‐treatment samples were evaluable by F1L CDx, of which 85 had DNA content ≥30 ng and were included in clinical bridging analyses (NTRK‐fp, n = 38; ROS1‐fp, n = 31; NTRK/ROS1‐fn, n = 16; Fig. 1). Forty‐five patients (NTRK‐fp, n = 26; ROS1‐fp, n = 19) had evaluable plasma samples (DNA content ≥30 ng) collected pre‐treatment and at/after, PD and were included in resistance mutation analyses (Fig. 1).
Fig. 1.
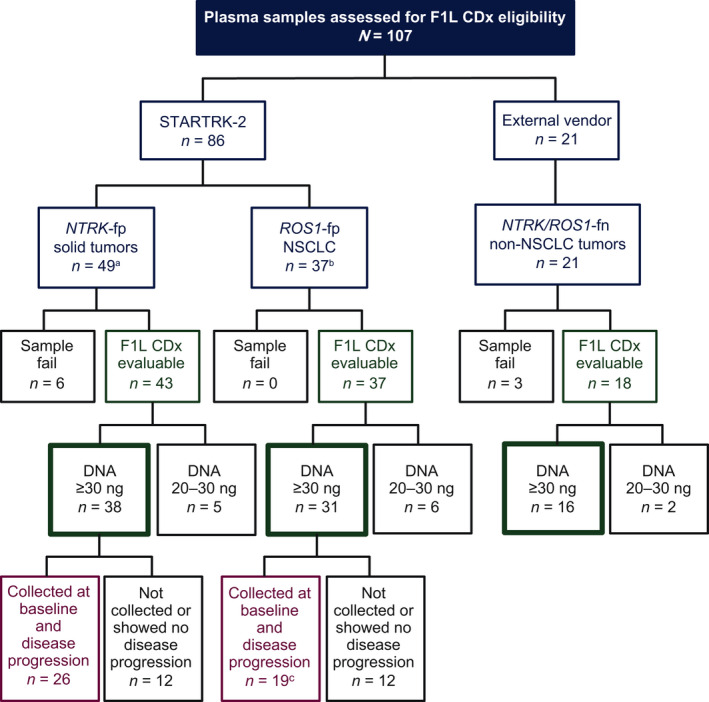
Flow chart of liquid biopsy samples used in the study. aFive samples were unavailable for testing. b16 samples were unavailable for testing. cTwo samples were excluded per FDA request.
3.2. Clinical bridging analyses
3.2.1. Concordance analysis
Concordance between the F1L CDx assay and CTAs for detection of NTRK and ROS1 gene fusions is shown in Table S4. Agreement estimates between the F1L CDx assay and CTAs are summarized (Table 1). PPA between the F1L CDx assay and CTAs was 47.4% (95% CI: 31.0–64.2) for NTRK fusions and 64.5% (95% CI: 45.4–80.8) for ROS1 fusions. PPV for the F1L CDx assay was 100% for NTRK‐fp and ROS1‐fp samples (Table 1). The assay detected NTRK and ROS1 fusions regardless of whether the patients enrolled via local or central biomarker testing (Table S5), and across a variety of fusion partners (Table S6). There was a significant difference in sum of the longest diameters between tumours where ROS1 was detected compared with those where no ROS1 fusion was detected; this was not the case for NTRK fusions (Fig. S1).
Table 1.
Performance of the F1L CDx assay for the detection of NTRK and ROS1 gene fusions.
| Parameter |
NTRK gene fusion detection N = 85 |
ROS1 gene fusion detection N = 85 |
|---|---|---|
|
PPA F1L CDx versus CTAs % (95% CI), n/N |
47.4 (31.0–64.2); 18/38 | 64.5 (45.4–80.8); 20/31 |
|
NPA F1L CDx versus CTAs % (95% CI), n/N |
100 (92.5–100.0); 47/47 | 100 (93.4–100); 54/54 |
|
PPV F1L CDx a % (95% CI) |
100 (82.4–100.0) | 100 (83.9–100) |
|
NPV F1L CDx b % (95% CI) |
99.8 (99.8–99.9) | 99.6 (99.4–99.8) |
The 95% CI of PPV was estimated by the Wilson score interval.
The 95% CI for NPV was estimated using the bootstrap method.
3.2.2. Clinical efficacy analysis
Overall response rate (ORR) and DoR estimates according to CTA and F1L CDx results are presented for NTRK‐fp and ROS1‐fp cohorts (Table 2). Among patients with NTRK‐fp solid tumours, those classified as CTA+ F1L CDx− or CTA+ F1L CDx unevaluable had lower ORR (ORR 55.0% and 43.8%, respectively) than those classified as CTA+ F1L CDx+ (ORR 72.2%). However, this was not statistically significant and was based on a small sample size therefore insufficient to conclude any differences between groups. As shown in Table 2 and Fig. S2, among patients with NTRK‐fp solid tumours and ROS1‐fp NSCLC, those classified as CTA+ F1L CDx− or CTA+ F1L CDx unevaluable had longer median DoR (NTRK‐fp, 12.9 and 14.1 months, respectively; ROS1‐fp, 17.3 and 13.3 months, respectively) than those classified as CTA+ F1L CDx+ (NTRK‐fp, 9.2 months; ROS1‐fp, 5.6 months); however, this was only significant for patients with ROS1‐fp NSCLC (CTA+ F1L CDx+ vs. CTA+ F1L CDx−: ROS1‐fp, P = 0.009; NTRK‐fp, P = 0.434). F1L CDx clinical efficacy estimate, accounting for concordance between F1L CDx and CTA, gave an ORR of 72.2% (95% CI: 50.0–88.9) in the CTA+ F1L CDx+ NTRK‐fp solid tumour and ROS1‐fp NSCLC cohorts. As PPV was 100%, ORR for the overall F1L CDx+ population was equal to the ORR in the CTA+ F1L CDx+ cohort (72.2%).
Table 2.
Clinical efficacy according to F1L CDx assay and CTA results. IQR, interquartile range.
| CTA+ a | CTA+ F1L CDx evaluable | CTA+ F1L CDx unevaluable | P‐values d | ||||
|---|---|---|---|---|---|---|---|
| CTA+ F1L CDx+ | CTA+ F1L CDx− | P‐values c | Total | ||||
| Patients with an NTRK‐fp solid tumour | |||||||
| ORR (95% CI) |
57.4 (43.2–70.8) n = 54 |
72.2 (46.5–90.3) n = 18 |
55.0 (31.5–76.9) n = 20 |
0.446 |
63.2 (47.3–76.6) n = 38 |
43.8 (19.8–70.1) n = 16 |
0.24 |
| Median DoR, months (IQR) |
10.4 (5.7–15.1) n = 54 |
9.2 (5.8–12.8) n = 18 |
12.9 (6.7–14.0) n = 20 |
0.434 |
9.3 (5.7–12.9) n = 38 |
14.1 (8.9–19.2) n = 16 |
0.40 |
| Patients with ROS1‐fp NSCLC | |||||||
| ORR (95% CI) |
78.4 (64.8–88.7) n = 51 b |
72.2 (46.5–90.3) n = 18 |
72.7 (39.0–94.0) n = 11 |
1.00 |
72.4 (54.3–85.3) n = 29 |
86.4 (65.1–97.1) n = 22 |
0.31 |
| Median DoR, months (IQR) |
12.0 (5.6–17.2) n = 51 |
5.6 (3.5–11.4) n = 18 |
17.3 (13.9–18.8) n = 11 |
0.009 |
10.4 (3.7–17.2) n = 29 |
13.3 (8.5–16.4) n = 22) |
0.31 |
ORR values in the CTA+ groups were derived in the ALKA‐372‐001/STARTRK‐1/STARTRK‐2 integrated analysis (May 2018 cut‐off) [17, 20].
Two ROS1‐fp patients were removed per FDA request.
P‐values derived with Wilcoxon rank‐sum test, for comparison between F1L CDx+ and F1L CDx− groups.
P‐values derived from Fisher exact test for categorical factors between the F1L CDx evaluable set and the F1L CDx unevaluable set.
3.2.3. Sensitivity analysis
For NTRK‐fp samples, covariates were balanced across evaluable (n = 38) and unevaluable (n = 16) F1L CDx groups (Table S7). For ROS1‐fp samples, covariate analysis indicated an unbalanced distribution in histology (P < 0.0001) and sex (P = 0.04) between evaluable (n = 29) and unevaluable (n = 22) F1L CDx groups (Table S8). F1L CDx outcome was significantly associated with histology (P = 0.03) using the NTRK‐fp evaluable set and with CNS lesions (P = 0.02) using the ROS1‐fp evaluable set (Table S9).
Estimations of PPA and NPA using imputation for F1L CDx unevaluable samples (NTRK‐fp 11/54; ROS1‐fp 22/51) were consistent across the three models, indicating robustness of the results (Table S10). PPA and NPA estimates according to CTA used were consistent across all three CTA subsets for NTRK‐fp samples. Variability was observed between Pharos and Others versus F1/F1 Haem for ROS1‐fp samples, but the latter population was too small for meaningful interpretation (Table S11). ORR estimations using imputation for NTRK‐fp (Table S12) and ROS1‐fp (Table S13) samples unevaluable by F1L CDx were similar to observed data.
3.3. Resistance mutation analyses
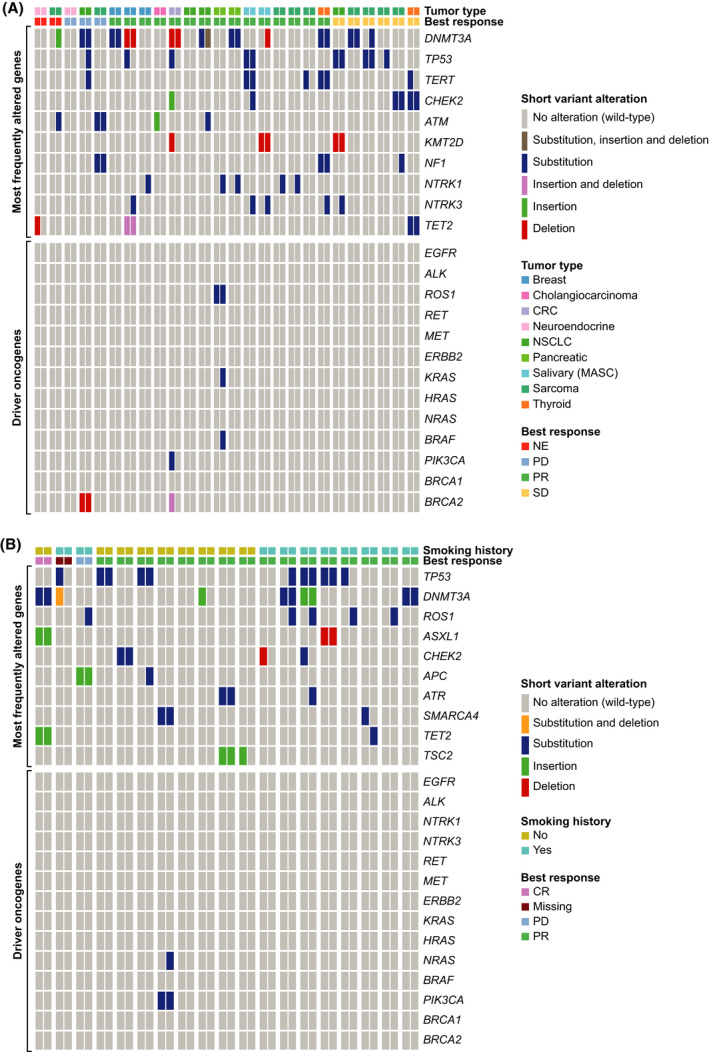
Baseline co‐mutations present in NTRK‐fp (n = 46) and ROS1‐fp (n = 37) samples are shown in Fig. 2. In the NTRK‐fp cohort, the most frequently co‐mutated genes were DNMT3A, TP53, TET2 and TERT, and no gene mutations were significantly associated with clinical response following entrectinib treatment. The most frequently co‐mutated genes in ROS1‐fp samples were DNMT3A, TP53, CHEK2 and SETD2. For ROS1‐fp samples, a significant association was observed between presence of APC mutations and clinical non‐response following entrectinib treatment (P ≤ 0.05). Consistent with previous results [24, 25], neither of the NTRK‐fp or ROS1‐fp samples showed co‐mutation in other driver proto‐oncogenes (e.g. EGFR, BRAF, KRAS). We note that the detection of DNMT3A, TET2 and ASXL1 variants could also be explained by the common biological phenomenon of clonal haematopoiesis; however, as peripheral blood mononuclear cells were not collected and analysed as part of this study, we cannot definitely make this conclusion [26, 27].
Fig. 2.

Primary resistance detected by F1L CDx, in patient ctDNA. Each column represents a single patient, with the genomic alterations indicated with non‐grey lines. Grey lines represent no alteration detected. Each sample was assayed a single time. Statistical analysis was carried out using Fisher’s exact test. (A) Patients with NTRK‐fp solid tumours; (B) Patients with ROS1‐fp NSCLC. CR, complete response; CRC, colorectal cancer; MASC, mammary analogue secretory carcinoma; N/A, not applicable; NE, non‐evaluable; PR, partial response; SD, stable disease.
In the NTRK‐fp solid tumour cohort, acquired resistance mutations were observed across a range of tumour types (Fig. 3A). Ten patients (38%) had a detectable NTRK solvent front mutation at PD (NTRK1G595R , n = 5; NTRK3G623E/K/R , n = 5), which were not detected pre‐treatment. BRAFV600E and KRASG12D mutations were detected in the PD sample from a patient with pancreatic cancer who had an initial partial response to entrectinib (DoR, 12.9 months). Notably, allele fractions of the BRAFV600E and KRASG12D mutations were 0.51% and 5.15%, respectively, suggesting that these are independent resistance mechanisms arising from different metastatic lesions.
Fig. 3.
Acquired resistance mutations detected by F1L CDx, in patient ctDNA. Each paired column represents a single patient at two timepoints (pre‐treatment and end of study), with the genomic alterations indicated with a non‐grey line. Grey lines represent no alteration detected. Each sample was assayed a single time. Statistical analysis was carried out using Fisher’s exact test. (A) Patients with NTRK‐fp solid tumours; (B) Patients with ROS1‐fp NSCLC. CR, complete response; CRC, colorectal cancer; MASC, mammary analogue secretory carcinoma; NE, non‐evaluable; PR, partial response; SD, stable disease.
Fig. 3B presents acquired resistance mutations in pre‐treatment and post‐PD samples from the ROS1‐fp NSCLC cohort. Five patients (26%), four with tumours harbouring CD74‐ROS1 fusions and one with a SLC34A2‐ROS1 fusion‐positive tumour showed emergence of acquired ROS1 resistance mutations (ROS1G2032R ; ROS1F2004C/I ) at PD, which were not present pre‐treatment.
An NRASQ61K mutation was detected in the end of treatment sample from one patient with a partial response to entrectinib. Three patients with NTRK‐fp solid tumours and one with ROS1‐fp NSCLC had detectable mutations in TP53 at/after PD that were not detected pre‐treatment.
3.4. ctDNA fraction
As detection of genomic alterations by ctDNA is highly dependent on the amount of tumour DNA present in blood, we calculated ctDNA fraction at baseline and end of study (Fig. S3). No significant association was observed between baseline NTRK1/3 fusion detection or non‐detection and ctDNA fraction level (P = 0.152; Fig. S3A). Conversely, higher ctDNA fraction levels were associated with higher likelihood of ROS1 fusion detection (P = 0.027; Fig. S3B). When comparing ctDNA fraction and detection levels of secondary NTRK or ROS1 resistance mutations at end of study, no significant association was found between ctDNA fraction and detection of secondary point mutations in NTRK1/3 (P = 0.479; Fig. S3C). However, higher ctDNA fraction was associated with higher likelihood of secondary ROS1 fusion detection (P = 0.140; Fig. S3D).
4. Discussion
Availability of non‐invasive genomic profiling assays, such as F1L CDx, enables testing for clinically actionable oncogenic biomarkers. Repeated F1L CDx testing opens up the possibility of informed clinical decisions at all stages of a patient’s cancer management. Our analysis of pre‐treatment samples with F1L CDx yielded a 100% PPV, reflecting high confidence in detected ROS1 and NTRK fusions. The clinical efficacy (ORR) estimate of 72.2% in CTA+ F1L CDx+ NTRK‐fp and ROS1‐fp cohorts trended to be higher than observed in the overall CTA+ NTRK‐fp cohort (57%) [20] and similar to the overall CTA+ ROS1‐fp cohort (77%) [17], supporting F1L CDx assay use in patients for whom tissue samples are unavailable or inadequate for NGS testing. Among patients with comparable NGS results, 38% with NTRK‐fp solid tumours and 26% with ROS1‐fp NSCLC acquired possible resistance mutations detected by F1L CDx with potential therapeutic implications, such as second‐generation tyrosine kinase inhibitor therapy.
Compared with predominantly tissue‐based CTAs (n = 83/85; 97.6%), the F1L CDx assay showed moderate PPAs of 47.4% and 64.5% for detection of NTRK and ROS1 fusions, respectively, likely reflecting methodological differences (i.e. plasma vs. solid tissue and DNA‐ vs. RNA‐based CTAs, differences in diagnostic methodology between laboratories, tumour shedding variability among patients [28]). These values are consistent with plasma versus tissue PPA estimates for FDA‐approved plasma CDx assays, such as Therascreen® PIK3CA RGQ PCR Kit [54.6%; breast cancer (alpelisib)] and cobas® EGFR Mutation Test v2 [≥58.7%; NSCLC (erlotinib, osimertinib, gefitinib)] [29, 30]. Additionally, given the aforementioned influence of differing methodology, our results are consistent with F1L CDx performance in detection of PIK3CA mutations in breast cancer (PPA, 71.7%) [31]. The high prevalence‐adjusted results are similar to those for F1L CDx detection of ALK rearrangements in NSCLC for alectinib treatment [12]. A 100.0% PPV for both NTRK‐fp and ROS1‐fp samples shows that, while F1L CDx may not detect all fusions present, positive results are very reliable and can rapidly inform clinical decisions. If fusions are not detected by F1L CDx because of patient variability in tumour type, stage, size, shedding, etc., further tissue‐based testing should be explored. The F1L CDx assay was able to detect gene fusions across a wide number of fusion partners, indicating no bias according to fusion partner.
A recent study also supports the detection of NTRK fusions across nine cancer types [32]. In this study, the authors report a PPV of 88% (CI 51–98%), which is similar to that observed here. However, direct comparison of these results should be taken with caution as the authors identified the NTRK‐fp samples using plasma and retrospectively tested for NTRK fusions with tissue. In addition, the NPV or ORR was not reported in the study.
High response rates were observed with entrectinib in patients classified as CTA+ F1L CDx+. This is consistent with a number of studies correlating liquid biopsy results and patient response to therapy [33, 34]. In the Phase III ALEX study of alectinib versus crizotinib in patients with advanced ALK‐fp NSCLC, although patients with high baseline cfDNA showed high response rates to targeted therapy, duration of response was much lower than observed for those with low baseline cfDNA [35]. This result was explained by baseline characteristics associated with higher tumour shedding (e.g. high tumour burden) and the authors concluded that baseline cfDNA acted as a prognostic factor for patient outcome. In our study, we observed a shorter median DoR in patients who were CTA+ F1L CDx+ versus CTA+ F1LCDx−, supporting that detection by F1L CDx could act as a prognostic factor for poorer patient outcome. However, this difference was only statistically significant in the ROS1‐fp cohort, likely reflecting the diverse cancer population of the NTRK‐fp cohort. CNS lesions are associated with more advanced disease [36] and, in our study, were also significantly associated with F1L CDx results in the ROS1‐fp cohort. Although the small sample size makes it difficult to draw definitive conclusions, these findings support the hypothesis that the likelihood of detecting gene fusions by F1L CDx may be higher in samples from patients with higher tumour burden and tumour shedding [37]. Similarly, within the ROS1‐fp cohort, ctDNA fraction, which may be a surrogate for tumour size and/or shedding, was higher in patients with a detected baseline fusion.
As is common in targeted therapy, emergence of mutations associated with re‐activation of the target gene [e.g. ROS1 or NTRK (Fig. 4)] or the downstream mitogen‐activated protein kinase (MAPK) pathway have been described at progression, including entrectinib and larotrectinib [38, 39]. In vitro resistance studies have also predicted KRAS/NRAS mutations to drive resistance to ROS1 inhibitors [40]. In the current study, acquired MAPK mutations were observed in one patient in each of the NTRK‐fp and ROS1‐fp cohorts, providing further evidence to support MAPK reactivation as a key mechanism of entrectinib resistance. One patient with pancreatic cancer showed mutations in both the KRAS and BRAF genes, despite previous evidence that these mutations are mutually exclusive due to overlapping functionality [41]. We hypothesize that KRAS and BRAF mutations are unlikely to develop within the same lesion given differences in allele frequencies and may reflect tumour heterogeneity detected by the F1L CDx assay. A relatively high frequency of TP53 mutations was observed in NTRK‐fp and ROS1‐fp samples, which are a hallmark of a hypermutation phenotype [42, 43]. Their presence may therefore suggest a hypermutation phenotype rather than site‐specific mutation in these fusion‐positive tumours.
Fig. 4.
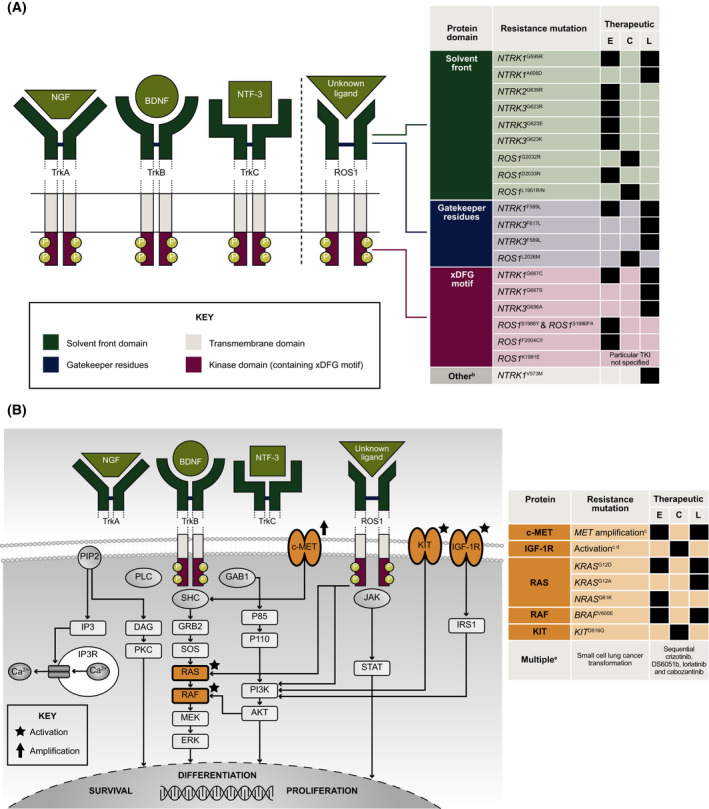
Mechanisms of acquired resistance to TKIs in NTRK‐fp solid tumours and ROS1‐fp NSCLC: (A) On‐target resistance; (B) Off‐target resistance. Black square indicates clinical resistance observed following treatment with therapeutic. aDual mutation. bFunctional domain/structure not reported/unknown. cSpecific mutation unknown/not reported. dReported in patients with ALK fusion‐positive lung cancer only. eComplete genomic profile of transformed SCLC not fully elucidated. ALK, anaplastic lymphoma kinase; BDNF, brain‐derived neurotrophic factor; C, crizotinib; DFG, Asp‐Phe‐Gly motif; E, entrectinib; KIT, KIT proto‐oncogene, receptor tyrosine kinase; KRAS, Kirsten rat sarcoma virus; L, larotrectinib; MET, mesenchymal epithelial transition; NGF, nerve growth factor beta; NRAS, neuroblastoma RAS viral (v‐ras) oncogene homolog; NTF‐3, Neurotrophin‐3; TKI, tyrosine kinase inhibitor; Trk, tropomyosin receptor kinase.
Within the NTRK‐fp cohort, we did not find co‐mutations present at baseline that were significantly associated with clinical benefit following entrectinib treatment. We hypothesize that this may be partly due to the heterogeneity in tumour type in our sample for this cohort, as co‐mutations may vary between tumour types and may therefore have confounded this result. In the ROS1 cohort, the APC gene was associated with poor clinical response to entrectinib; however, this was based on a small number of cases and requires further study in a larger population.
This study was retrospective and has a relatively small sample size, reflecting the rarity of NTRK and ROS1 fusions. However, samples and comprehensive clinical data were collected as part of a prospective, global clinical trial leading to the approval of entrectinib. The availability of matched samples before treatment initiation and following progression also facilitated detailed examination of resistance mutations arising following entrectinib treatment. The current analyses thus comprise a unique dataset, due to the rarity of the tumour types studied and the scarcity of prospective data on resistance mechanisms.
5. Conclusion
Patients with NTRK‐fp solid tumours or ROS1‐fp NSCLC detected by CTAs and F1L CDx assay demonstrate high response rates to entrectinib, which can complement tissue‐based testing for the identification of patients who may benefit from entrectinib treatment. Additionally, the F1L CDx assay can identify acquired resistance mechanisms in patients who experience progression following entrectinib. The F1L CDx assay can therefore be used to inform clinical decisions at all stages of the patient journey from diagnosis to disease progression, to ensure rapid access to targeted treatment in fusion‐positive patients, including patients for whom tissue biopsies are contraindicated or where tumour samples are limited. Assessment at progression will support initiation of second‐generation TRK or ROS1 inhibitors, or potential combination regimens, if resistance mutations are detected.
Conflict of interest
The authors declare the following competing financial interests: RD has received consultancy/advisory fees from F. Hoffmann‐La Roche Ltd, Foundation Medicine Inc., Pfizer, AstraZeneca, Celon Pharma, Bristol‐Myers Squibb, Merck, MSD, Regeneron, Takeda, Seattle Genetics, Novartis; and has received travel or accommodation expenses from F. Hoffmann‐La Roche Ltd and AstraZeneca. TH, VC, CW, CWC, BS, and TR are employees of Genentech, Inc. AD reports honoraria/advisory fees from Ignyta/Genentech, Inc./F. Hoffmann‐La Roche Ltd, Loxo/Bayer/Lilly, Takeda/Ariad/Millenium, TP Therapeutics, AstraZeneca, Pfizer, Blueprint Medicines, Helsinn, Beigene, BergenBio, Hengrui Therapeutics, Exelixis, Tyra Biosciences, Verastem, MORE Health, Abbvie, 14ner/Elevation Oncology, Remedica Ltd, ArcherDX, Monopteros, Novartis, EMD Serono, Melendi, Liberum, Repare RX, Nuvalent, Merus; research grant/funding (institution) from Pfizer, Exelixis, GlaxoSmithKlein, Teva, Taiho, PharmaMar; research grant (self) from Foundation Medicine; royalties from Wolters Kluwer; expenses from Merck, Puma, Merus, Boehringer Ingelheim; CME honoraria from Medscape, OncLive, PeerVoice, Physicians Education Resources, Targeted Oncology, Research to Practice, Axis, Peerview Institute, Paradigm Medical Communications, WebMD, MJH Life Sciences, Med Learning, Imedex, Answers in CME, Clinical Care Options. RCD declares consulting fees from Ignyta, Genentech, Inc./F. Hoffmann‐La Roche Ltd, AstraZeneca, Anchiano, and Rain Therapeutics; royalties or licensing fees for intellectual property from Ignyta, Abbott Molecular, Genentech, Inc./F. Hoffmann‐La Roche Ltd, Foundation Medicine, Black Diamond, Pearl River, Voronoi, Takeda, Scorpion, and Rain Therapeutics; stock ownership in Rain Therapeutics; is an employee of Rain Therapeutics. FB reports consulting/advisory role for F. Hoffmann‐La Roche Ltd/Genentech, Inc., Pfizer, Novartis, Pierre Fabre, Bristol‐Myers Squibb, AstraZeneca/MedImmune, Boehringer Ingelheim, Lilly, Merck Serono, MSD Oncology, Takeda, Bayer; travel/accommodation/expenses from F. Hoffmann‐La Roche Ltd/Genentech, Inc., Bristol‐Myers Squibb, AstraZeneca/MedImmune, MSD Oncology; honoraria from F. Hoffmann‐La Roche Ltd/Genentech, Inc., Pfizer, Pierre Fabre, AstraZeneca, Bristol‐Myers Squibb, Boehringer Ingelheim, Lilly, Novartis, Pierre Fabre, Merck Serono, MSD Oncology, Takeda, Bayer; research funding (institution) from F. Hoffmann‐La Roche Ltd/Genentech, Inc., AstraZeneca/MedImmune, Bristol‐Myers Squibb, Pierre Fabre, Abbvie, Amgen, Bayer, Boehringer Ingelheim, Eisai, Lilly, Ipsen, Innate Pharma, Novartis, Merck Serono, MSD Oncology, Pfizer, Sanofi/Aventis, Takeda. TRW is an employee of Genentech, Inc., and has stock and ownership interest in F. Hoffmann‐La Roche Ltd. KW was an employee of Foundation Medicine, Inc. at the time of the study but is now an employee of the US Food and Drug Administration. JS, LD, RW, and ML are employees of Foundation Medicine, Inc.
Author contributions
TH, RW, CW and TRW were involved in conception and design of the analysis. TH, CW and TRW were involved in conduct of the analysis. All authors were involved in data interpretation/analysis. All authors were involved in manuscript drafting, writing and editing. All authors approved the manuscript for submission.
Supporting information
Fig. S1. Differences in sum of the longest diameters between tumours where ROS1 or NTRK fusions were detected by F1L CDx versus those with no detected fusion.
Fig. S2. Duration of response in patients with NTRK fusion‐positive solid tumours and ROS1 fusion‐positive NSCLC by ctDNA status.
Fig. S3. F1L CDx detection of primary and secondary mutations in NTRK1/2/3 and ROS1 fusion‐positive samples. (A) Primary NTRK1/2/3 fusion. (B) Primary ROS1 fusion. (C) Secondary NTRK1/2/3 mutation. (D) Secondary ROS1 mutation.
Table S1. Number of patients by NTRK‐fp solid tumour type within each CTA+ subgroup.
Table S2. Imputation models used in sensitivity analyses.
Table S3. CTAs used to confirm the presence of NTRK or ROS1 gene fusions in tumour samples from patients enrolled in STARTRK‐2 with valid F1L CDx results.
Table S4. Concordance between the F1L CDx assay and CTAs for the detection of NTRK and ROS1 gene fusions.
Table S5. F1L CDx NTRK and ROS1 fusion detection and method of enrolment (central vs local).
Table S6. List and prevalence of NTRK and ROS1 fusion partners detected by the F1L CDx assay.
Table S7. Demographic and clinical characteristics for patients with NTRK‐fp solid tumours.
Table S8. Demographic and clinical characteristics for patients with ROS1‐fp NSCLC.
Table S9. Univariate logistic regression model evaluating relationships between covariates and F1L CDx test results (Positive vs Negative).
Table S10. Sensitivity of PPA and NPA according to baseline characteristics, Pharos result and response.
Table S11. Sensitivity of PPA and NPA according to CTA used.
Table S12. Robustness of ORR using imputation Model 1 for NTRK‐fp samples.
Table S13. Robustness of ORR using imputation Model 1 for ROS1‐fp samples.
Acknowledgements
This work was supported by F. Hoffmann‐La Roche Ltd. The authors thank the patients, their families, and the participating study centre. Medical writing support for the development of this manuscript, under the direction of the authors, was provided by Cassidy Bayley, PhD, Emily Morton, BSc, and Lucy Carrier, PhD of Ashfield MedComms, an Ashfield Health company, and funded by F. Hoffmann‐La Roche Ltd.
Rafal Dziadziuszko and Tiffany Hung contributed equally to this article
Todd Riehl is deceased. This paper is dedicated to his memory
Data accessibility
For eligible studies, qualified researchers may request access to individual patient level clinical data through a data request platform. At the time of writing, this request platform is Vivli: https://vivli.org/ourmember/roche/. For up‐to‐date details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here: https://go.roche.com/data_sharing. Anonymized records for individual patients across more than one data source external to Roche cannot, and should not, be linked due to a potential increase in risk of patient re‐identification.
References
- 1. Ribeiro TB, Ribeiro A, Rodrigues LO, Harada G, Nobre MRC. U.S. Food and Drug Administration anticancer drug approval trends from 2016 to 2018 for lung, colorectal, breast, and prostate cancer. Int J Technol Assess Health Care. 2020;36:20–8. 10.1017/S0266462319000813 [DOI] [PubMed] [Google Scholar]
- 2. Liu Z, Zhu L, Roberts R, Tong W. Toward clinical implementation of next‐generation sequencing‐based genetic testing in rare diseases: where are we? Trends Genet. 2019;35:852–67. 10.1016/j.tig.2019.08.006 [DOI] [PubMed] [Google Scholar]
- 3. Ilié M, Hofman P. Pros: can tissue biopsy be replaced by liquid biopsy? Transl Lung Cancer Res. 2016;5:420–3. 10.21037/tlcr.2016.08.06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lim C, Tsao MS, Le LW, Shepherd FA, Feld R, Burkes RL, et al. Biomarker testing and time to treatment decision in patients with advanced nonsmall‐cell lung cancer. Ann Oncol. 2015;26:1415–21. 10.1093/annonc/mdv208 [DOI] [PubMed] [Google Scholar]
- 5. Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. 10.1056/NEJMoa1113205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corcoran RB, Chabner BA. Application of cell‐free DNA analysis to cancer treatment. N Engl J Med. 2018;379:1754–65. 10.1056/NEJMra1706174 [DOI] [PubMed] [Google Scholar]
- 7. Rolfo C, Mack PC, Scagliotti GV, Baas P, Barlesi F, Bivona TG, et al. Liquid biopsy for advanced non‐small cell lung cancer (NSCLC): a statement paper from the IASLC. J Thorac Oncol. 2018;13:1248–68. 10.1016/j.jtho.2018.05.030 [DOI] [PubMed] [Google Scholar]
- 8. De Rubis G, Rajeev Krishnan S, Bebawy M. Liquid biopsies in cancer diagnosis, monitoring, and prognosis. Trends Pharmacol Sci. 2019;40:172–86. 10.1016/j.tips.2019.01.006 [DOI] [PubMed] [Google Scholar]
- 9. Ou SI, Nagasaka M, Zhu VW. Liquid biopsy to identify actionable genomic alterations. Am Soc Clin Oncol Educ Book. 2018;38:978–97. 10.1200/EDBK_199765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Mattos‐Arruda L, Weigelt B, Cortes J, Won HH, Ng CKY, Nuciforo P, et al. Capturing intra‐tumor genetic heterogeneity by de novo mutation profiling of circulating cell‐free tumor DNA: a proof‐of‐principle. Ann Oncol. 2018;29:2268. 10.1093/annonc/mdx804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Woodhouse R, Li M, Hughes J, Delfosse D, Skoletsky J, Ma P, et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324‐Gene cfDNA‐based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS One. 2020;15:e0237802. 10.1371/journal.pone.0237802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. F1L CDx summary of safety and effectiveness. 2020. Available from: https://www.accessdata.fda.gov/cdrh_docs/pdf20/P200006B.pdf
- 13. Amatu A, Sartore‐Bianchi A, Bencardino K, Pizzutilo EG, Tosi F, Siena S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol. 2019;30:viii5–15. 10.1093/annonc/mdz383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov. 2015;5:25–34. 10.1158/2159-8290.CD-14-0765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Drilon A, Jenkins C, Iyer S, Schoenfeld A, Keddy C, Davare MA. ROS1‐dependent cancers—biology, diag‐nostics and therapeutics. Nat Rev Clin Oncol. 2020;18:35–55. 10.1038/s41571-020-0408-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davies KD, Doebele RC. Molecular pathways: ROS1 fusion proteins in cancer. Clin Cancer Res. 2013;19:4040–5. 10.1158/1078-0432.CCR-12-2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Drilon A, Siena S, Dziadziuszko R, Barlesi F, Krebs MG, Shaw AT, et al. Entrectinib in ROS1 fusion‐positive non–small‐cell lung cancer: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21:261–70. 10.1016/s1470-2045(19)30690-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Menichincheri M, Ardini E, Magnaghi P, Avanzi N, Banfi P, Bossi R, et al. Discovery of entrectinib: a new 3‐aminoindazole as a potent anaplastic lymphoma kinase (ALK), c‐ros oncogene 1 kinase (ROS1), and pan‐tropomyosin receptor kinases (Pan‐TRKs) inhibitor. J Med Chem. 2016;59:3392–408. 10.1021/acs.jmedchem.6b00064 [DOI] [PubMed] [Google Scholar]
- 19. Fischer H, Ullah M, de la Cruz CC, Hunsaker T, Senn C, Wirz T, et al. Entrectinib, a TRK/ROS1 inhibitor with anti‐CNS tumor activity: differentiation from other inhibitors in its class due to weak interaction with P‐glycoprotein. Neuro Oncol. 2020;22:819–29. 10.1093/neuonc/noaa052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Doebele RC, Drilon A, Paz‐Ares L, Siena S, Shaw AT, Farago AF, et al. Entrectinib in patients with advanced or metastatic NTRK fusion‐positive solid tumours: integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020;21:271–82. 10.1016/s1470-2045(19)30691-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Okamura R, Boichard A, Kato S, Sicklick JK, Bazhenova L, Kurzrock R. Analysis of NTRK alterations in pan‐cancer adult and pediatric malignancies: implications for NTRK‐targeted therapeutics. JCO Precis Oncol. 2018;2018:PO.18.00183. 10.1200/PO.18.00183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li M. Statistical consideration and challenges in bridging study of personalized medicine. J Biopharm Stat. 2015;25:397–407. 10.1080/10543406.2014.920340 [DOI] [PubMed] [Google Scholar]
- 23. Tukachinsky H, Madison RW, Chung JH, Gjoerup OV, Severson E, Dennis L, et al. Genomic analysis of circulating tumor DNA in 3,334 patients with advanced prostate cancer identifies targetable BRCA alterations and AR resistance mechanisms. Clin Cancer Res. 2021;27(11):3094–105. 10.1158/1078-0432.CCR-20-4805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rosen EY, Goldman DA, Hechtman JF, Benayed R, Schram AM, Cocco E, et al. TRK fusions are enriched in cancers with uncommon histologies and the absence of canonical driver mutations. Clin Cancer Res. 2020;26:1624–32. 10.1158/1078-0432.CCR-19-3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wilson TR, Sokol ES, Trabucco SE, Newberg JY, Simmons B, Riehl T, et al. Genomic characteristics and predicted ancestry of NTRK1/2/3 and ROS1 fusion‐positive tumours from >165,000 pan‐solid tumours. Ann Oncol. 2019;30:v161–2. 10.1093/annonc/mdz244.005 [DOI] [Google Scholar]
- 26. Silver AJ, Bick AG, Savona MR. Germline risk of clonal haematopoiesis. Nat Rev Genet. 2021;22:603–17. 10.1038/s41576-021-00356-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jaiswal S, Ebert BL. Clonal hematopoiesis in human aging and disease. Science. 2019;366:eaan4673. 10.1126/science.aan4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng ML, Pectasides E, Hanna GJ, Parsons HA, Choudhury AD, Oxnard GR. Circulating tumor DNA in advanced solid tumors: clinical relevance and future directions. CA Cancer J Clin. 2021;71:176–90. 10.3322/caac.21650 [DOI] [PubMed] [Google Scholar]
- 29. Ohashi R, Watanabe R, Esaki T, Taniguchi T, Torimoto‐Katori N, Watanabe T, et al. Development of simplified in vitro P‐glycoprotein substrate assay and in silico prediction models to evaluate transport potential of P‐glycoprotein. Mol Pharm. 2019;16:1851–63. 10.1021/acs.molpharmaceut.8b01143 [DOI] [PubMed] [Google Scholar]
- 30. therascreen® PIK3CA RGQ PCR Kit instructions for use (handbook). 2019. Available from: https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190001C.pdf
- 31. cobas® EGFR mutation test v2. 2015. Available from: https://www.accessdata.fda.gov/cdrh_docs/pdf12/P120019S007c.pdf
- 32. Rolfo C, Drilon A, Hong D, McCoach C, Dowlati A, Lin JJ, et al. NTRK1 fusions identified by non‐invasive plasma next‐generation sequencing (NGS) across 9 cancer types. Br J Cancer. 2022;126(3):514–20. 10.1038/s41416-021-01536-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Russano M, Napolitano A, Ribelli G, Iuliani M, Simonetti S, Citarella F, et al. Liquid biopsy and tumor heterogeneity in metastatic solid tumors: the potentiality of blood samples. J Exp Clin Cancer Res. 2020;39:95. 10.1186/s13046-020-01601-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Taus Á, Camacho L, Rocha P, Hardy‐Werbin M, Pijuan L, Piquer G, et al. Dynamics of EGFR mutation load in plasma for prediction of treatment response and disease progression in patients with EGFR‐mutant lung adenocarcinoma. Clin Lung Cancer. 2018;19:387–94.e382. 10.1016/j.cllc.2018.03.015 [DOI] [PubMed] [Google Scholar]
- 35. Dziadziuszko R, Peters S, Mok TSK, Camidge DR, Noé J, Nowicka M, et al. Circulating free DNA as a prognostic biomarker in patients with advanced ALK+ NSCLC treated with alectinib from the global phase III ALEX trial. J Clin Oncol. 2019;37:9053. 10.1200/JCO.2019.37.15_suppl.9053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Remon J, Besse B. Brain metastases in oncogene‐addicted non‐small cell lung cancer patients: incidence and treatment. Front Oncol. 2018;8:1–15. 10.3389/fonc.2018.00088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cho MS, Park CH, Lee S, Park HS. Clinicopathological parameters for circulating tumor DNA shedding in surgically resected non‐small cell lung cancer with EGFR or KRAS mutation. PLoS One. 2020;15:e0230622. 10.1371/journal.pone.0230622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Russo M, Misale S, Wei G, Siravegna G, Crisafulli G, Lazzari L, et al. Acquired resistance to the TRK inhibitor entrectinib in colorectal cancer. Cancer Discov. 2016;6:36–44. 10.1158/2159-8290.CD-15-0940 [DOI] [PubMed] [Google Scholar]
- 39. Cocco E, Schram AM, Kulick A, Misale S, Won HH, Yaeger R, et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat Med. 2019;25:1422–7. 10.1038/s41591-019-0542-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cargnelutti M, Corso S, Pergolizzi M, Mevellec L, Aisner DL, Dziadziuszko R, et al. Activation of RAS family members confers resistance to ROS1 targeting drugs. Oncotarget. 2015;6:5182–94. 10.18632/oncotarget.3311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Calhoun ES, Jones JB, Ashfaq R, Adsay V, Baker SJ, Valentine V, et al. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol. 2003;163:1255–60. 10.1016/S0002-9440(10)63485-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Loeb LA. A mutator phenotype in cancer. Cancer Res. 2001;61:3230–9. 10.1007/3-540-30683-8_1140 [DOI] [PubMed] [Google Scholar]
- 43. Kron A, Alidousty C, Scheffler M, Merkelbach‐Bruse S, Seidel D, Riedel R, et al. Impact of TP53 mutation status on systemic treatment outcome in ALK‐rearranged non‐small‐cell lung cancer. Ann Oncol. 2018;29:2068–75. 10.1093/annonc/mdy333 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Differences in sum of the longest diameters between tumours where ROS1 or NTRK fusions were detected by F1L CDx versus those with no detected fusion.
Fig. S2. Duration of response in patients with NTRK fusion‐positive solid tumours and ROS1 fusion‐positive NSCLC by ctDNA status.
Fig. S3. F1L CDx detection of primary and secondary mutations in NTRK1/2/3 and ROS1 fusion‐positive samples. (A) Primary NTRK1/2/3 fusion. (B) Primary ROS1 fusion. (C) Secondary NTRK1/2/3 mutation. (D) Secondary ROS1 mutation.
Table S1. Number of patients by NTRK‐fp solid tumour type within each CTA+ subgroup.
Table S2. Imputation models used in sensitivity analyses.
Table S3. CTAs used to confirm the presence of NTRK or ROS1 gene fusions in tumour samples from patients enrolled in STARTRK‐2 with valid F1L CDx results.
Table S4. Concordance between the F1L CDx assay and CTAs for the detection of NTRK and ROS1 gene fusions.
Table S5. F1L CDx NTRK and ROS1 fusion detection and method of enrolment (central vs local).
Table S6. List and prevalence of NTRK and ROS1 fusion partners detected by the F1L CDx assay.
Table S7. Demographic and clinical characteristics for patients with NTRK‐fp solid tumours.
Table S8. Demographic and clinical characteristics for patients with ROS1‐fp NSCLC.
Table S9. Univariate logistic regression model evaluating relationships between covariates and F1L CDx test results (Positive vs Negative).
Table S10. Sensitivity of PPA and NPA according to baseline characteristics, Pharos result and response.
Table S11. Sensitivity of PPA and NPA according to CTA used.
Table S12. Robustness of ORR using imputation Model 1 for NTRK‐fp samples.
Table S13. Robustness of ORR using imputation Model 1 for ROS1‐fp samples.
Data Availability Statement
For eligible studies, qualified researchers may request access to individual patient level clinical data through a data request platform. At the time of writing, this request platform is Vivli: https://vivli.org/ourmember/roche/. For up‐to‐date details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here: https://go.roche.com/data_sharing. Anonymized records for individual patients across more than one data source external to Roche cannot, and should not, be linked due to a potential increase in risk of patient re‐identification.