

Abstract
Enterococci have emerged among the leading causes of nosocomial infection. With the goal of analyzing enterococcal genes differentially expressed in environments related to commensal or environmental colonization and infection sites, we adapted and optimized a method more commonly used in the study of eukaryotic gene expression, random arbitrarily primed PCR (RAP-PCR). The RAP-PCR method was systematically optimized, allowing the technique to be used in a highly reproducible manner with gram-positive bacterial RNA. In the present study, aerobiosis was chosen as a variable for the induction of changes in gene expression by Enterococcus faecalis. Aerobically and anaerobically induced genes were detected and identified to the sequence level, and differential gene expression was confirmed by quantitative, specifically primed RT-PCR. Differentially expressed genes included several sharing identity with those of other organisms related to oxygen metabolism, as well as hypothetical genes lacking identity to known genes.
Enterococci have emerged as leading causes of nosocomial infection, including urinary tract infections, wound infections, and bacteremia (10, 12, 25, 33, 37, 39, 41, 44). Among these and other types of enterococcal infection, approximately 80% are associated with Enterococcus faecalis (21) and the majority are nosocomial (34). Enterococcal infections are especially troublesome because of the high level of intrinsic antibiotic resistance and because they have acquired resistance determinants capable of rendering the organism completely resistant to all currently approved antibiotics (21).
Because of the emergence of enterococci as leading nosocomial pathogens and the development of broad antimicrobial resistance, particularly to vancomycin, an understanding of host-parasite interactions is a key to the development of new prophylactic and therapeutic strategies. Only a few traits are known to contribute to the pathogenesis of enterococcal infection. The enterococcal cytolysin contributes to toxicity, loss of organ function, and lethality during enterococcal infection (6, 24, 26), and cytolytic strains of E. faecalis have been observed to be enriched among clinical isolates, especially those from the bloodstream (21–23). Enterococcal aggregation substance has been demonstrated to contribute to increased cardiac vegetation size (6) and has been associated with enhanced virulence in a rabbit model of endocarditis (45). Other putative virulence factors include surface carbohydrates (18, 19) and enterococcal lipoteichoic acid (2), but their specific roles in enterococcal infection have yet to be determined. Since enterococci evolved as commensal organisms, the contribution of specific factors to pathogenesis is typically subtle. Thus, sensitive techniques are required for the identification of such factors.
Differential-display PCR (DD-PCR) is a relatively new method for observing the differential expression of genes in comparative studies within a large number of experimental systems (31, 35, 51). The technique was originally developed for use in the study of eukaryotic gene expression, and this continues to be its most common application (31, 35). A derivative of DD-PCR, random arbitrarily primed PCR (RAP-PCR), utilizes an arbitrary primer at a low annealing temperature for both the first- and second-strand cDNA synthesis reactions (51). At such low-stringency temperatures the arbitrary primer is able to bind at random sites within the template that show limited, but not complete, complementarity. Because the same arbitrary primer is used as both the 5′ and the 3′ primers, the primer sequence is incorporated at both ends of the resulting double-stranded products. The products are amplified by standard PCR by using the arbitrary primer, resolved by polyacrylamide electrophoresis, and visualized by autoradiography. Differences in gene expression can be inferred from the resulting “fingerprint” by observing the presence or absence of specific products between different populations of cells. By eliminating the use of an oligo(dT) primer, the 3′ bias toward the polyadenylate tail in eukaryotic mRNA is eliminated. Thus, priming of first-strand synthesis potentially can occur at any point within the RNA. For this reason, RAP-PCR may be used for amplification of RNAs that are not polyadenylated, such as bacterial RNA.
This technology has been applied to only a few prokaryotic systems (1, 52) because of high sample-to-sample variability, as was encountered in initial attempts to study E. faecalis gene expression by using available methods (51, 52). We therefore systematically optimized the RAP-PCR method and identified several key refinements which ultimately allowed the technique to be used in a highly reproducible manner with gram-positive bacterial RNA. In the present study, aerobiosis was chosen as a variable for the induction of changes in gene expression by E. faecalis. Aerobically and anaerobically induced genes were detected and identified to the sequence level, and differential gene expression was confirmed by quantitative, specific reverse transcriptase PCR (RT-PCR).
MATERIALS AND METHODS
Bacterial strains and growth conditions.
A strain of E. faecalis, MMH594 (23), that caused multiple infections in a hospital ward outbreak was used to study differential gene expression under either aerobic or anaerobic conditions. Escherichia coli XL-1 Blue (Stratagene, La Jolla, Calif.) was used for cloning differentially expressed RAP-PCR products. For aerobically cultured E. faecalis, a 10-ml overnight culture was grown in a 50-ml centrifuge tube for 18 h in brain heart infusion (BHI) broth (Becton Dickinson, Cockeysville, Md.) at 37°C with shaking at 300 rpm. The overnight culture was diluted 1:100 in 10 ml of fresh BHI broth and subcultured in a 50-ml centrifuge tube to mid-log phase (optical density at 560 nm [OD560] of 1.0) at 37°C with shaking at 300 rpm for 4 h prior to RNA isolation. For anaerobically cultured E. faecalis, a 10-ml overnight culture was grown in a 50-ml centrifuge tube in BHI broth at 37°C without shaking for 18 h in an anaerobic hood. The overnight culture was diluted 1:100 in 10 ml of fresh BHI broth and subcultured anaerobically in a 50-ml centrifuge tube to mid-log phase (OD560 = 1.0) at 37°C without shaking for 3.5 h prior to RNA isolation. E. coli was cultured on plates containing Luria-Bertani (LB) media and 1.5% Bacto-Agar (Difco, Detroit, Mich.), and appropriate transformants were subsequently cultured in LB broth (43). Antibiotics used for selection of transformed E. coli included ampicillin (100 μg/ml) and tetracycline (12.5 μg/ml) (Sigma, St. Louis, Mo.). For detection of insertional inactivation of the lacZα gene contained in the cloning vectors, 50 μM isopropyl-β-d-thiogalactopyranoside (Sigma) and 0.01% 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (Sigma) were added to the media.
RNA extraction and preparation.
Total RNA was isolated as previously described (5) with minor modifications. Briefly, each 10-ml subculture of E. faecalis was centrifuged (2,500 × g for 2 min at 4°C), and the bacterial pellet was resuspended in 1.5 ml of Tri Reagent (Sigma). The cell suspension was immediately transferred to a 2-ml microcentrifuge tube (BioSpec Products, Bartlesville, Okla.) containing 0.5 ml of 100-μm-diameter zirconia-silica beads (BioSpec Products). The tube was immediately placed in a high-speed reciprocating shaker (BioSpec Products) and horizontally shaken at 5,000 rpm for 1 min to lyse the bacterial cells. After lysis, the tube was placed on ice and the beads were allowed to settle out of the lysis mixture. The lysate was clarified by centrifugation (12,000 × g for 10 min at 4°C). The supernatant was recovered and extracted with 300 μl of chloroform. After a manual shaking for 15 s, the mixture was placed on ice for 15 min prior to centrifugation (12,000 × g for 10 min at 4°C). After centrifugation the aqueous phase was recovered, and the RNA was precipitated by adding 750 μl of isopropyl alcohol. After incubation on ice for 10 min, the RNA was pelleted by centrifugation (12,000 × g for 10 min at 4°C). The RNA pellet was washed with 1.7 ml of 75% ethanol, air dried for 5 to 10 min, and resuspended in 200 μl of diethylpyrocarbonate (DEPC)-treated water.
Residual contaminating genomic DNA was removed in the following manner. After resuspension of the total RNA, a 0.25 volume of transcription-optimized buffer (Promega, Madison, Wis.) containing 200 mM Tris-HCl (pH 7.9), 30 mM MgCl2, 10 mM spermidine, and 50 mM NaCl was added. Five units of RQ1 RNase-free DNase (Promega) was added, and the mixture was incubated at 37°C for 15 min. After incubation, 250 μl of phenol-water (3.75:1 [vol/vol]; Life Technologies, Grand Island, N.Y.) was added. Additionally, 250 μl of chloroform was added, and the solution was centrifuged (12,000 × g for 10 min at 4°C). RNA was recovered in the aqueous phase, and the phenolic phase was extracted with 250 μl of TE buffer and centrifuged to ensure quantitative recovery. After centrifugation, the second aqueous phase was recovered and mixed with the first. RNA was precipitated by the addition of 1 ml of 100% ethanol and incubation at −70°C for at least 30 min. RNA was pelleted by centrifugation (12,000 × g for 20 min at 4°C), washed with 75% ethanol, and air dried for 5 to 10 min. DNase-treated total RNA was resuspended in 50 μl of DEPC-treated water containing 0.1 mM EDTA and stored at −70°C.
The integrity of the RNA was assessed by electrophoresis of 2 μl of each sample through a 1.2% agarose–0.66 M formaldehyde gel in MOPS running buffer (20 mM MOPS [morpholinepropanesulfonic acid; pH 7.0], 8 mM sodium acetate, 1 mM EDTA [pH 8.0]) at a power of 3 to 4 V/cm (43). RNA concentration was determined spectrophotometrically by measuring the A260/A280 ratio of a 1:50 dilution in DEPC-treated water.
RAP-PCR.
To initiate each experimental reaction, 14.5 μl containing 1 μg of total E. faecalis RNA, diluted in DEPC-treated water as needed, was heated in a 0.65-ml thin-wall PCR tube to 70°C for 10 min and immediately placed on ice. After 1 min of incubation on ice and brief centrifugation pulse to collect contents, 5 μl of buffer containing 50 mM Tris-HCl (pH 8.3), 75 mM KCl, 3 mM MgCl2, a 1.25 μM concentration of an arbitrarily chosen primer (Stratagene), 1.25 mM concentrations of each deoxynucleoside triphosphate (dNTP), and 40 U of RNase Block RNase inhibitor (Stratagene) was added. Contents were mixed, collected by brief centrifugation, and allowed to equilibrate to 37°C for 5 min. After equilibration, 25 U of Moloney murine leukemia virus (MMLV) RT (Stratagene) was added for a final volume of 20 μl. The reaction was incubated at 37°C for 1 h. Upon completion of first-strand cDNA synthesis, the reaction was heated to 90°C for 5 min to inactivate the RT and then immediately placed on ice for 10 min. The reaction was diluted 1:10 in sterile water and either used immediately in a second-strand synthesis reaction or stored at −20°C.
For second-strand synthesis, 10 μl of the cDNA preparation was mixed with 39.8 μl of a standard PCR mixture in a 0.65-ml thin-wall PCR tube for a final reaction volume of 50 μl containing 10 mM Tris-HCl (pH 9.0), 50 mM KCl, 3 mM MgCl2, 50 μM concentrations of each dNTP, 10 μCi of [α-33P]dCTP (DuPont NEN, Boston, Mass.), and a 1 μM concentration of the same arbitrary primer used in first-strand cDNA synthesis. After overlay of 50 μl of light mineral oil, the reaction was incubated at 96°C for 10 min, followed by incubation at 36°C for 15 min. After the extended equilibration at 36°C, 1 U of Taq polymerase was added with gentle mixing of contents. The reaction was allowed to equilibrate at 36°C for an additional 15 min, followed by a 5-min incubation at 72°C. The following parameters were used for an additional 39 cycles of PCR: 94°C (1 min), 50°C (1 min), 72°C (2 min), and a final extension at 72°C for 10 min. Upon completion, the reaction was stored at 0 to 4°C. The sequences of the primers used in separate RAP-PCR experiments were AATCTAGAGCTCTCCAGC (primer 1) and AATCTAGAGCTCCCTCCA (primer 2).
To visualize RAP-PCR products, 5 μl from each reaction was mixed with 10 μl of stop buffer containing 80% formamide, 50 mM Tris-HCl (pH 8.3), 1 mM EDTA, 0.1% (wt/vol) xylene cyanol, and 0.1% (wt/vol) bromophenol blue. The samples were heated to 96°C for 2 min, and the contents were collected by brief centrifugation. Five microliters of each reaction was loaded on a 6% polyacrylamide sequencing gel prepared in TBE buffer (Life Technologies). Electrophoresis was performed at 1,500 V and continued until the xylene cyanol dye had migrated to 2.5 cm above the bottom of the gel. The gel was transferred to 3MW paper (Midwest Scientific, Valley Park, Mo.) and dried under vacuum at 80°C for 40 min. Autoradiography was performed by exposing the gel to Kodak BioMax MR film for 18 h at an ambient temperature.
Isolation, cloning, and sequencing of RAP-PCR products.
The RAP-PCR gel and the autoradiogram were aligned by using radioactive ink marks placed on the gel prior to autoradiography. By using the autoradiogram as a template, individual bands representing differentially expressed products were cut and removed from the gel with a sterile scalpel (30, 52). Each isolated piece of acrylamide gel with filter paper was cut into smaller pieces with a sterile scalpel, and the pieces were collected in a microcentrifuge tube. Fifty microliters of elution buffer (0.5 M ammonium acetate, 10 mM magnesium acetate, 1 mM EDTA [pH 8.0], 0.1% sodium dodecyl sulfate) (1, 43) was added to the tube, and the mixture was heated to 100°C for 30 min. The eluate from the reaction was collected, and the DNA was precipitated by the addition of 0.1 volume of 3 M sodium acetate (pH 5.17) and 2.5 volumes of 100% ethanol followed by incubation at −70°C for 2 h. DNA was pelleted by centrifugation at 12,000 × g for 30 min at 4°C. The DNA pellet was washed with 2.5 volumes of 75% ethanol, air dried for 5 min, and resuspended in 10 μl of sterile water.
The individual products of interest were reamplified with the same primer incorporated in both first- and second-strand synthesis reactions of RAP-PCR. The entire volume of recovered DNA was amplified in a standard 50-μl PCR mixture containing 10 mM Tris-HCl (pH 9.0), 50 mM KCl, 3 mM MgCl2, a 50 μM concentration of each dNTP, a 1 μM concentration of the arbitrary primer, and 1 U of Taq polymerase (Promega). The following parameters were used in the PCR: 94°C (1 min), 50°C (1 min), 72°C (2 min), and a final extension at 72°C for 10 min. The reamplified RAP-PCR products were analyzed on a 1% low-melting-point agarose gel and purified using a Geneclean kit (Bio 101, Vista, Calif.) according to the manufacturer’s recommendations.
Each purified RAP-PCR product was ligated into either the EcoRV site of the pBluescript cloning vector (Stratagene) or the XcmI site of the pKRX cloning vector (46) by standard techniques for blunt-end ligation (43). Ligation products were used to transform E. coli XL-1 Blue (Stratagene) according to established protocols (43). Transformed E. coli were identified by using inactivation of the lacZα gene as a phenotypic marker (43), and plasmids containing the RAP-PCR product of interest were isolated by using the Wizard Plus Miniprep kit (Promega) according to the manufacturer’s recommendations.
DNA sequence information was obtained by using standard chain termination reactions employing fluorescein-labeled M13 forward and reverse primers (Pharmacia Biotech, Piscataway, N.J.) and a T7 DNA polymerase-based AutoRead Sequencing Kit (Pharmacia Biotech), with a Pharmacia LKB A.L.F. DNA Sequencer. GCG utilities on a VAX mainframe were used in BLAST searches for sequence homologies (49).
Confirmation of differential gene expression.
Sequence data from each differentially expressed RAP-PCR product was used to develop specific primers for use in specifically primed, quantitative RT-PCR to confirm differential gene expression. In each reaction, 1 μg of total RNA, diluted in DEPC-treated water as needed, was incubated at 85°C for 3 min and immediately placed on ice. After a 1-min incubation on ice and brief centrifugation to collect contents, 8 μl of a standard RT-PCR reaction mixture was added in a final reaction volume of 20 μl containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 500 μM concentrations of each dNTP, 50 pmol of the specific reverse primer, 10 U of RNase inhibitor (Ambion, Austin, Tex.), and 100 U of MMLV RT (Ambion). After gentle mixing and brief centrifugation, each reaction was incubated at 42°C for 60 min followed by a 10-min incubation at 92°C. The cDNA from each reaction was either used immediately in PCR or stored at −20°C.
For amplification, 5 μl of the cDNA preparation was mixed with 45 μl of a standard PCR mixture in a final reaction volume of 50 μl containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 125 μM concentrations of each dNTP, 50 pmol each of both the specific forward and reverse primers, and 5 U of Taq polymerase (Promega). Each reaction was gently mixed and overlaid with 50 μl of light mineral oil. After a 5-min incubation at 95°C, amplification was performed for 20 cycles by using the following parameters: 94°C (20 s), 55°C (30 s), and 72°C (40 s), with a final extension at 72°C for 5 min. Five microliters of each PCR product was analyzed by electrophoresis in a 1% agarose gel stained with ethidium bromide following normalization to an internal control product amplified in parallel. Relative quantities of each RT-PCR product amplified from both aerobic and anaerobic RNA were analyzed by using SigmaGel image analysis software (Jandel Corp., San Rafael, Calif.). Paired reactions lacking RT were used as negative controls to detect contamination of RNA by residual genomic DNA.
RESULTS
RNA isolation.
Cells of E. faecalis MMH594 were cultured either aerobically or anaerobically to mid-log phase in BHI broth, isolated, and resuspended in a chaotropic RNA extraction reagent. Cell lysis was achieved by horizontally shaking the cells mixed with zirconia-silica beads on a reciprocating high-speed shaking device, and RNA was isolated as described. RNA was isolated from six independent aerobic cultures and six independent anaerobic E. faecalis cultures. After electrophoresis, bands indicating the 23S, 16S, and 5S rRNA species were clearly identifiable (data not shown). Each band was well defined with no smearing, indicating that no overt degradation of the RNA occurred during isolation. The A260/A280 ratios ranged from 1.88 to 2.00 for the 12 RNA preparations.
RAP-PCR.
To assess the effect of RNA concentration in RAP-PCR, various amounts of total RNA (0.125, 0.25, 0.5, 1, and 2 μg) were used in the first-strand synthesis reactions in a series of amplifications. For standardization, the same template RNA was used in each independent amplification. The results of the titration experiments are shown in Fig. 1. Although broadly similar from lane to lane, there were RNA-dependent differences in the number of RAP-PCR products detected. As the amount of RNA in the first-strand reaction increased from 0.125 to 2 μg, the aggregate number of RAP-PCR products increased, with a plateau in the number of well resolved and easily detected products beginning at ca. 1 μg of RNA in the first-strand reaction. Therefore, 1 μg of RNA was selected for use in subsequent first-strand reactions.
FIG. 1.

Effects of RNA concentration on RAP-PCR. Different amounts of the same RNA were amplified by RAP-PCR. In each lane the following amounts of total RNA were used during first-strand synthesis: lane 1, 125 ng; lane 2, 250 ng; lane 3, 500 ng; lane 4, 1 μg; and lane 5, 2 μg. A paired control reaction without RT showed no products derived from genomic DNA contamination.
Because RAP-PCR depends on the random priming of an arbitrary primer at low annealing temperatures, the effect of different annealing temperatures in the second-strand synthesis reaction was investigated. A series of annealing reactions was performed with the same template RNA and a range of temperatures to characterize the impact on the quantity and quality of the RAP-PCR products generated. The reactions were performed as described by using 1 μg of template RNA with the following range of annealing temperatures in the second-strand synthesis reaction: 20, 25, 30, 36, and 40°C. As the second-strand annealing temperature increased from 20 to 36°C, the total number of RAP-PCR products decreased, and the resolution of the larger products increased (Fig. 2). With interest in maximizing both the number and the resolution of RAP-PCR products, 36°C was chosen as the second-strand annealing temperature in subsequent reactions.
FIG. 2.
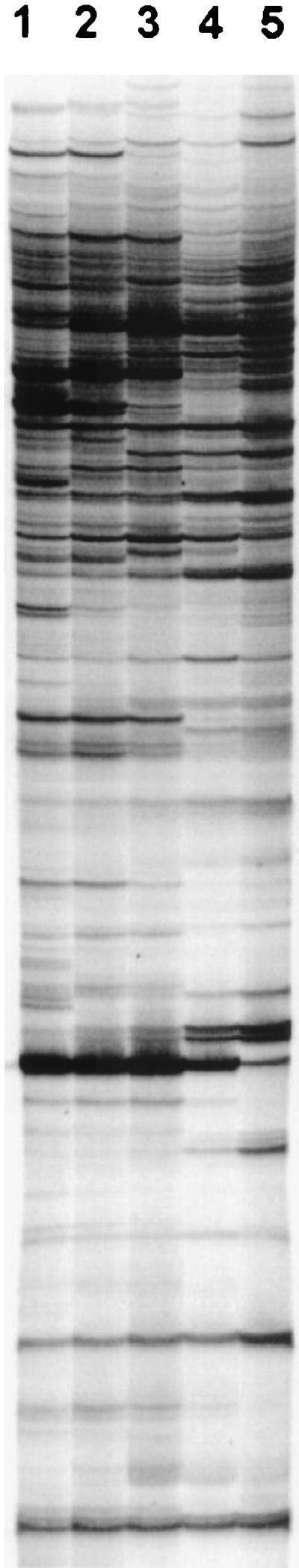
Effects of annealing temperatures during second-strand synthesis on RAP-PCR. For each reaction, 1 μg of the same total RNA was amplified by RAP-PCR. In each lane the following annealing temperatures were used during second-strand synthesis: lane 1, 20°C; lane 2, 25°C; lane 3, 30°C; lane 4, 36°C; and lane 5, 40°C. A paired control reaction without RT showed no products derived from genomic DNA contamination.
Extended equilibration of the samples during first- and second-strand synthesis proved to be one of the most important parameters for achieving reproducibility. Amplification products that resulted from RAP-PCR amplification of aerobic RNA, either with or without extended temperature equilibration during first- and second-strand synthesis (5 min at 37°C for the former and 15 min at 36°C for the latter), are shown in Fig. 3. In lanes 1 to 3, three aerobic RNA samples were amplified by RAP-PCR without allowing for extended equilibration of the samples prior to the addition of appropriate enzymes for DNA synthesis. Thus, RT and Taq polymerase were added immediately after denaturation. In lanes 4 to 6, the same three aerobic RNA samples were amplified by RAP-PCR as detailed in the Materials and Methods section. Many of the products were amplified equivalently regardless of the inclusion or exclusion of extended equilibration, reflective of favorable priming at certain sites that did not depend on the stringency of control during first- and second-strand synthesis. However, a more consistent and reproducible product profile was generated when extended equilibration was included during amplification. Additionally, there appeared to be a subtle yet distinct increase in the number of products amplified when extended equilibration is included.
FIG. 3.

Effects of temperature equilibration during first- and second-strand synthesis on reproducibility of RAP-PCR amplification. Total RNA from three independent aerobic cultures of E. faecalis was amplified by RAP-PCR either without (lanes 1 to 3) or with (lanes 4 to 6) extended equilibration during first- and second-strand synthesis prior to the addition of the appropriate enzymes for DNA synthesis. Arrows indicate areas of irreproducibility in the reactions performed without extended equilibration.
To assess the reproducibility of the RAP-PCR technique described in the present work and to determine whether it could be used to identify differences in enterococcal gene expression, RAP-PCR was performed on total RNA isolated from aerobically and anaerobically cultured E. faecalis MMH594 by using the optimized reaction parameters. Aerobiosis was used as a variable to induce potential changes in enterococcal gene expression. Aerobiosis was chosen as a test variable because it is significantly different between sites of enterococcal colonization (e.g., the gastrointestinal tract) and disease (e.g., the bloodstream). Thus, it may provide an important environmental cue for regulation of the expression of enterococcal genes. The amplification products that resulted from RAP-PCR amplification of aerobic and anaerobic RNA are shown in Fig. 4. To facilitate discrimination of differentially expressed products from spurious artifacts and to assess the reproducibility of the optimized protocol, RAP-PCR was performed with RNA purified from six independent aerobic and six independent anaerobic E. faecalis cultures.
FIG. 4.

RAP-PCR products derived from samples of total RNA from aerobically and anaerobically cultured E. faecalis. Lanes: 1 to 6, aerobic samples 1 to 6; 7 to 12, anaerobic samples 1 to 6. Arrows indicate differentially amplified products. Primer 2 was used for amplification. For each experimental reaction, a paired negative control reaction lacking RT during first-strand synthesis was performed. These controls were used to qualitatively assess the levels of genomic DNA contamination of each RNA sample. No products derived from residual genomic DNA contamination were identified in any of the negative control reactions.
As expected, a majority of the RAP-PCR products were common to amplifications of RNA from aerobically and anaerobically cultured E. faecalis (Fig. 4). However, RAP-PCR products that were unique to either the aerobic or the anaerobic RNA samples could be identified. Importantly, products unique to RNA derived from either environment occurred reproducibly. Some RAP-PCR products were amplified at qualitatively different levels between RNA derived from both environments, based on variations in the intensities of the bands in the autoradiograph. The relative amount of a specific product amplified by RAP-PCR closely parallels the relative amount of the specific corresponding RNA in a given sample of total RNA (51). Thus, the intensity of the bands in the autoradiograph can serve as a qualitative index of the relative abundance of a particular transcript. Therefore, RAP-PCR appears to provide a sensitive technique for detecting both gross and subtle differences in gene expression between populations of E. faecalis.
Sequence identification of differentially expressed RAP-PCR products.
To identify genes within the emerging E. faecalis genome database that are differentially expressed in response to changes in oxygen tension, the differentially amplified RAP-PCR products were recovered and reamplified by standard PCR with the same arbitrary primer used in RAP-PCR. After reamplification, the products were gel purified and ligated into a TA cloning vector for transformation of E. coli. Plasmids were isolated, and the inserts were subjected to automated sequence analysis. Specific primer pairs for each RAP-PCR product were designed by using the sequence data and then used in RT-PCR to confirm differential gene expression (Fig. 5).
FIG. 5.

RT-PCR analysis to confirm differential gene expression. RT-PCR was performed with both aerobic (A) and anaerobic (An) RNA with primers specific for each product. Product numbers correspond to those given in Table 1. Relative quantities of each RT-PCR product amplified from both aerobic and anaerobic RNA were analyzed after normalization to the internal control. Paired control reactions lacking RT showed no products derived from genomic DNA.
Of 13 total RAP-PCR products amplified with the first application of the optimized protocol, 4 were identified as false positives for differential gene expression upon visual examination after RT-PCR with specific primers (data not shown) and were eliminated from further analysis. DNA sequence information from the nine RAP-PCR products confirmed by RT-PCR to be derived from differentially expressed genes was compared to that in the GenBank database by using the BLAST search algorithm (49) to identify similarities to known sequences (Table 1). Cloned inserts of the RAP-PCR products ranged in size from 181 to 425 bp. Four of the RAP-PCR products were amplified by using random primer 1, and five were amplified by using random primer 2. Sequence analysis showed that each random primer amplified different products, with no duplications either within or between the two groups of products. Additionally, a similar number of either aerobically or anaerobically induced products were identified. Sequencing revealed a previously undetected enterococcal gene that was aerobically induced and exhibited significant similarity (smallest sum probability score of <10−6) to a Bacillus subtilis gene coding for catalase (32). Two aerobically induced products with different sequences had significant similarity to a B. subtilis gene coding for an oxidoreductase. Anaerobically induced genes included one with significant similarity to an ABC transporter from B. subtilis, as well as one with significant similarity to a B. subtilis gene coding for seryl-tRNA synthetase (40). Two of the anaerobically induced products, both of different sequences, showed limited similarity (smallest sum probability score of >10−6) to a mammalian gene coding for NADH dehydrogenase. Of the RAP-PCR products cloned and sequenced and whose differential expression was confirmed by RT-PCR, 22% demonstrated no significant similarity to known sequences.
TABLE 1.
Sequence data for cloned RAP-PCR products from aerobically and anaerobically cultured E. faecalis
| Product | Primer | Environment | Size (bp) | Similarity (accession no.) | Fold induction | Sequence similaritya |
|---|---|---|---|---|---|---|
| 1 | 2 | Anaerobic | 181 | Ycdl ABC transporter (AB000617) | 2.05 | 2.8−24 |
| 2 | 1 | Anaerobic | 339 | Seryl-tRNA synthetase (P37464)48 | 581 | 2.8−41 |
| 3 | 1 | Anaerobic | 350 | NADH dehydrogenase (496166) | 3.11 | 0.0020 |
| 4 | 1 | Anaerobic | 350 | NADH dehydrogenase (496166) | 6.72 | 0.00082 |
| 5 | 2 | Aerobic | 253 | Oxidoreductase (P25887)11 | 1029 | 3.0−27 |
| 6 | 2 | Aerobic | 260 | No known similarity | 8.28 | |
| 7 | 2 | Aerobic | 350 | No known similarity | 1.56 | |
| 8 | 2 | Aerobic | 355 | Oxidoreductase (P25887) | 50.3 | 3.5−54 |
| 9 | 1 | Aerobic | 425 | Catalase (Z82044)8 | 12.19 | 5.8−81 |
Values represent the lowest sum probability score.
Data generated by RT-PCR (Fig. 5) was used to quantitate the level to which genes coding for each RAP-PCR product were specifically induced (Table 1). RT-PCR products were normalized to an internal control product identified by RAP-PCR, the expression of which was not affected by aerobiosis or anaerobiosis. The control product showed significant similarity (smallest sum probability of 2.2 × 10−56) to a locus coding for 23S rRNA in Staphylococcus aureus. After normalization to the non-differentially expressed internal control, RT-PCR products were visualized by electrophoresis and analyzed by image analysis to determine relative quantities. For purposes of calculating the variability of lane-to-lane comparisons, the four products identified as false positives upon visual examination after RT-PCR were used to determine the standard error of the analysis. Each of the four products demonstrated relative induction levels of <1.20 between aerobic and anaerobic RNA samples (data not shown). For purposes of defining differential gene expression, comparisons demonstrating differential expression greater than 2 standard deviations from the mean of these measures (>26% differential expression) were considered significant. The specific level of induction for each of the RAP-PCR products is indicated in Table 1. RAP-PCR products derived from genes that showed a confirmed, significant difference in expression between an aerobic versus an anaerobic environment exhibited a broad range of differential expression of 1.56- to 1,029-fold.
DISCUSSION
In the present study we optimized a technique that has been applied broadly to analyze patterns of eukaryotic gene expression in order to permit its reproducible use in studies of gene expression by a gram-positive bacterium. Initial efforts to use existing protocols for RAP-PCR in a prokaryotic genetic background (1, 52) highlighted several extant pitfalls, most notably a lack of experimental reproducibility. More specifically, we were unable to generate similar RAP-PCR product patterns upon repeated amplification from either the same RNA sample or the same cDNA sample. Moreover, RAP-PCR amplification of RNA from bacteria cultured independently under identical conditions yielded widely different product patterns. Therefore, several modifications, which are detailed in the preceding sections, were made to the RAP-PCR protocol that ultimately allowed the technique to be used in a reproducible manner in the study of differential enterococcal gene expression. Although the optimized RAP-PCR protocol was optimized while studying differential gene expression in E. faecalis, the same protocol has been used by others in our laboratory to reproducibly identify products from differentially expressed genes by Streptococcus gordonii and Actinomyces naeslundii. Additionally, the modifications included in the optimized RAP-PCR protocol may serve to guide others who are interested in applying this technology to other bacteria but who are unable to apply the precise parameters we used.
Of the changes made to previously defined protocols for use of RAP-PCR to study bacterial genetics, the most significant modifications were made to the first-strand and second-strand synthesis reactions. Both steps involve random priming by an arbitrary primer at a low annealing temperature. We found that extended equilibration of each reaction to the low annealing temperature prior to the addition of the appropriate enzyme was essential for achieving reproducibility among similar RNA samples and upon repeated amplifications of the same sample. Thus, during first-strand synthesis, each reaction is allowed to equilibrate to 37°C for 5 min prior to the addition of RT. Similarly, during second-strand synthesis, each reaction is allowed to equilibrate to 36°C for 15 min prior to the addition of Taq polymerase. It was not until these subtle yet significant changes were made that reproducibility in both reactions was achieved. Upon RAP-PCR amplification with the optimized protocol, each of the six aerobic RNA preparations produced similar product patterns, as did each of the six anaerobic RNA preparations (Fig. 4). Thus, there is reproducibility in RAP-PCR amplification of independent samples of RNA from E. faecalis cultured in a similar manner. Perhaps most importantly, three independent but identical amplifications of each RNA preparation yielded identical product patterns (for example, compare Fig. 4, lane 6, with Fig. 1, lane 4, and Fig. 2, lane 4). Thus, there is reproducibility upon repeated RAP-PCR amplification of a single RNA preparation.
Equilibration to the low annealing temperatures in both first- and second-strand synthesis prior to the addition of the appropriate enzymes is essential for eliminating the variability inherent to random priming. For instance, during second-strand synthesis, the RNA-cDNA duplex is initially denatured. As the reaction is cooled from the denaturation temperature of 96°C to the low-stringency annealing temperature of 36°C, it passes through a range of temperatures at which the random primer can bind. If Taq polymerase were present in the reaction during this period, polymerization would begin at various points within the temperature ramp. This introduces experimental variability, because the time course of cycling to the low annealing temperature may be different between independent amplifications of the same sample or between simultaneous amplifications of independent samples. In standard PCR this variability is eliminated by using a specific primer at an annealing temperature at which it must recognize its complement for priming to occur. However, in RAP-PCR such subtle experimental variability is potentiated by using a random primer at low stringency. Equilibration of each reaction to 36°C prior to the addition of Taq polymerase eliminates this variability. By “clamping” each reaction at the low annealing temperature before synthesis, time is allowed for the primer to recognize all potential binding sites. Thus, each sample is randomly primed essentially to completeness prior to the initiation of polymerization, and polymerization is initiated under identical conditions. The same rationale applies for equilibration of the first-strand synthesis reaction to 37°C prior to the addition of RT.
Using the optimized protocol detailed in the Materials and Methods section, RAP-PCR was used to identify products from genes differentially expressed when E. faecalis was cultured in an aerobic versus an anaerobic environment. Additionally, sequence data generated after RAP-PCR was used to both confirm and quantitate, by specific RT-PCR, differences in expression for genes coding for the RAP-PCR products. Several of the RAP-PCR products that were cloned and sequenced demonstrated significant levels of similarity to known sequences in current databases, including several involved in the respiration of B. subtilis. The role of these genes in enterococcal biology in either an aerobic or anaerobic environment is unexplored. However, the data show that the RAP-PCR protocol may be utilized to reproducibly identify products that derive from differentially expressed genes and that the RAP-PCR products may ultimately be used to identify these specific differentially expressed genes.
In the present work, 22% of the RAP-PCR products demonstrated no similarity to sequences in the databases. Recent sequencing of other bacterial genomes has demonstrated that many of the inferred proteins show no homologies to known proteins. For example, 38% of the E. coli K-12 genome (3) and 42% of the Haemophilus influenzae Rd genome (14) code for proteins with no known homologies. Similarly, sensitive techniques for the identification of bacterial genes whose expression is induced by or within host cells have produced similar data (4, 20, 36, 48). The detection of unique enterococcal genes most likely reflects gaps in the understanding of E. faecalis genetics and physiology.
Our current research is directed toward the application of RAP-PCR to studies of enterococcal gene expression in in vitro and in vivo models of enterococcal disease. Additionally, we are employing RAP-PCR in initial studies of gene expression patterns in and among streptococci and actinomyces and are achieving the same level of reproducibility and quality. Thus, the refinements detailed in the present work allow the RAP-PCR protocol to serve as a general and highly reproducible technique for the study of prokaryotic gene expression.
ACKNOWLEDGMENTS
This work was supported by NIH grants AI 41108 and EY08289 to Michael S. Gilmore.
REFERENCES
- 1.Abu Kwaik Y, Pederson L L. The use of differential display-PCR to isolate and characterize a Legionella pneumophila locus induced during the intracellular infection of macrophages. Mol Microbiol. 1996;21:543–556. doi: 10.1111/j.1365-2958.1996.tb02563.x. [DOI] [PubMed] [Google Scholar]
- 2.Bhakdi S, Klonisch T, Nuber P, Fischer W. Stimulation of monokine production by lipoteichoic acids. Infect Immun. 1991;12:4614–4620. doi: 10.1128/iai.59.12.4614-4620.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blattner, F. R., et al. The complete genome sequence of Escherichia coli K-12. Science 277:1452–1462. [DOI] [PubMed]
- 4.Camilli A, Mekalanos J J. Use of recombinase gene fusions to identify Vibrio cholerae genes induced during infection. Mol Microbiol. 1995;18:671–683. doi: 10.1111/j.1365-2958.1995.mmi_18040671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheung A L, Eberhardt K J, Fischetti V A. A method to isolate RNA from gram-positive bacteria and mycobacteria. Anal Biochem. 1994;222:511–514. doi: 10.1006/abio.1994.1528. [DOI] [PubMed] [Google Scholar]
- 6.Chow J W, Thal L A, Perri M B, Vasquez J A, Donabedian S M, Clewell D B, Zervos M J. Plasmid-encoded hemolysin and aggregation substance production contribute to virulence in experimental enterococcal endocarditis. Antimicrob Agents Chemother. 1993;37:2474–2477. doi: 10.1128/aac.37.11.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cummings N J, Connerton I F. The Bacillus subtilis 168 chromosome from sspE to katA. Microbiology. 1997;143:1855–1859. doi: 10.1099/00221287-143-6-1855. [DOI] [PubMed] [Google Scholar]
- 8.Dunny G M. Genetic functions and cell-cell interactions in the pheromone-inducible plasmid transfer system of Enterococcus faecalis. Plasmid. 1990;4:689–696. doi: 10.1111/j.1365-2958.1990.tb00639.x. [DOI] [PubMed] [Google Scholar]
- 9.Eick-Helmerich K, Braun V. Import of biopolymers into Escherichia coli: nucleotide sequences of the exbB and exbD genes are homologous to those of the tolQ and tolR genes, respectively. J Bacteriol. 1989;171:5117–5126. doi: 10.1128/jb.171.9.5117-5126.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Emori T G, Gaynes R P. An overview of nosocomial infections, including the role of the microbiology laboratory. Clin Microbiol Rev. 1993;6:428–442. doi: 10.1128/cmr.6.4.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ena J, Dick R W, Jones R N, Wenzel R P. The epidemiology of intravenous vancomycin usage in a university hospital: a 10-year study. JAMA. 1993;269:598–602. [PubMed] [Google Scholar]
- 12.Evans A C, Chinn A L. The enterococci: with special reference to their association with human disease. J Bacteriol. 1947;54:495–512. doi: 10.1128/jb.54.4.495-512.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fagon J Y, Chastre J, Vuagnat A, Trouillet J L, Novara A, Gibert C. Nosocomial pneumonia and mortality among patients in intensive care units. JAMA. 1996;275:866–869. [PubMed] [Google Scholar]
- 14.Fleischmann, R. D., et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269:496–512. [DOI] [PubMed]
- 15.Galli D, Lottspeich F F, Wirth R. Sequence analysis of Enterococcus faecalis aggregation substance encoded by the sex pheromone plasmid pAD1. Mol Microbiol. 1990;4:895–904. doi: 10.1111/j.1365-2958.1990.tb00662.x. [DOI] [PubMed] [Google Scholar]
- 16.Girou E, Brun-Buisson C. Morbidity, mortality, and the cost of nosocomial infections in critical care. Curr Opin Crit Care. 1996;2:347–351. [Google Scholar]
- 17.Gold H S, Moellering R C., Jr Antimicrobial-drug resistance. N Engl J Med. 1996;335:1445–1453. doi: 10.1056/NEJM199611073351907. [DOI] [PubMed] [Google Scholar]
- 18.Guzmàn C A, Pruzzo C, LiPira G, Calegari L. Role of adherence in pathogenesis of Enterococcus faecalis urinary tract infection and endocarditis. Infect Immun. 1989;57:1834–1838. doi: 10.1128/iai.57.6.1834-1838.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guzmàn C A, Pruzzo C, Platè M, Guardati M C, Calegari L. Serum dependent expression of Enterococcus faecalis adhesins involved in the colonization of heart cells. Microb Pathog. 1991;11:399–409. doi: 10.1016/0882-4010(91)90036-a. [DOI] [PubMed] [Google Scholar]
- 20.Hensel M, Shea J E, Gleeson C, Jones M D, Dalton E, Holden D W. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269:400–403. doi: 10.1126/science.7618105. [DOI] [PubMed] [Google Scholar]
- 21.Huycke M M, Sahm D F, Gilmore M S. Multiply resistant enterococci: the nature of the problem and an agenda for the future. Emerg Infect Dis. 1998;4:239–249. doi: 10.3201/eid0402.980211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huycke M M, Gilmore M S. Frequency of aggregation substance and cytolysin genes among enterococcal endocarditis isolates. Plasmid. 1995;34:152–156. doi: 10.1006/plas.1995.9992. [DOI] [PubMed] [Google Scholar]
- 23.Huycke M M, Spiegel C A, Gilmore M S. Bacteremia caused by hemolytic, high-level gentamicin resistant Enterococcus faecalis. Antimicrob Agents Chemother. 1991;35:1626–1634. doi: 10.1128/aac.35.8.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ike Y, Hashimoto H, Clewell D B. Hemolysin of Streptococcus faecalis subspecies zymogenes contributes to virulence in mice. Infect Immun. 1984;45:528–530. doi: 10.1128/iai.45.2.528-530.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jett B D, Huycke M M, Gilmore M S. Virulence of enterococci. Clin Microbiol Rev. 1994;7:462–478. doi: 10.1128/cmr.7.4.462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jett B D, Jensen H G, Nordquist R E, Gilmore M S. Contribution of the pAD1-encoded cytolysin to the severity of experimental Enterococcus faecalis endophthalmitis. Infect Immun. 1992;60:2445–2452. doi: 10.1128/iai.60.6.2445-2452.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaye D. Enterococci: biologic and epidemiologic characteristics and in vitro susceptibility. Arch Intern Med. 1982;142:2006–2009. doi: 10.1001/archinte.142.11.2006. [DOI] [PubMed] [Google Scholar]
- 28.Kreft B, Marre R, Schramm U, Wirth R. Aggregation substance of Enterococcus faecalis mediates adhesion to cultured renal tubular cells. Infect Immun. 1992;60:25–30. doi: 10.1128/iai.60.1.25-30.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leclercq R, Derlot E, Duval J, Courvalin P. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med. 1988;319:157–161. doi: 10.1056/NEJM198807213190307. [DOI] [PubMed] [Google Scholar]
- 30.Liang P, Averboukh L, Pardee A B. Distribution and cloning of eukaryotic mRNAs by means of differential display: refinements and optimization. Nucleic Acids Res. 1993;21:3269–3275. doi: 10.1093/nar/21.14.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liang P, Pardee A B. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- 32.Loewen P C, Switala J. Multiple catalases in Bacillus subtilis. J Bacteriol. 1987;169:3601–3607. doi: 10.1128/jb.169.8.3601-3607.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.MacCallum W G, Hastings T W. A case of acute endocarditis caused by Micrococcus zymogenes (nov. spec.), with a description of the organism. J Exp Med. 1899;4:521–534. doi: 10.1084/jem.4.5-6.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maki D G, Agger W A. Enterococcal bacteremia: clinical features, the risk of endocarditis, and management. Medicine (Baltimore) 1988;67:248–269. [PubMed] [Google Scholar]
- 35.McClelland M, Mathieu-Daude F, Welsh J. RNA fingerprinting and differential display using arbitrarily primed PCR. Trends Genet. 1995;11:242–246. doi: 10.1016/s0168-9525(00)89058-7. [DOI] [PubMed] [Google Scholar]
- 36.Mei, J.-M., F. Nourbakhsh, C. W. Ford, and D. W. Holden. Identification of Staphylococcus aureus virulence genes in a murine model of bacteraemia using signature-tagged mutagenesis. Mol. Microbiol. 26:399–407. [DOI] [PubMed]
- 37.Moellering R C., Jr Emergence of enterococcus as a significant pathogen. Clin Infect Dis. 1992;14:1173–1178. doi: 10.1093/clinids/14.6.1173. [DOI] [PubMed] [Google Scholar]
- 38.National Nosocomial Infections Surveillance System. National nosocomial infections surveillance (NNIS) report, data summary from October 1986–April 1997, issued May 1997. Am J Infect Control. 1997;25:477–487. [PubMed] [Google Scholar]
- 39.Nichols R L, Muzik A C. Enterococcal infections in surgical patients: the mystery continues. Clin Infect Dis. 1992;15:72–76. doi: 10.1093/clinids/15.1.72. [DOI] [PubMed] [Google Scholar]
- 40.Ogasawara N, Nakai S, Yoshikawa H. Systematic sequencing of the 180 kilobase region of the Bacillus subtilis chromosome containing the replication origin. DNA Res. 1994;1:1–14. doi: 10.1093/dnares/1.1.1. [DOI] [PubMed] [Google Scholar]
- 41.Rantz L A, Kirby W M K. Enterococcic infections: an evaluation of the importance of fecal streptococci and related organisms in the causation of human disease. Arch Intern Med. 1943;71:516–528. [Google Scholar]
- 42.Ruoff K L, De La Maza L, Murtagh M J, Spargo J D, Ferraro M J. Species identities of enterococi isolated from clinical specimens. J Clin Microbiol. 1990;28:435–437. doi: 10.1128/jcm.28.3.435-437.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 44.Schaberg D R, Culver D H, Gaynes R P. Major trends in the microbial etiology of nosocomial infection. Am J Med. 1991;91(Suppl. 3B):72S–75S. doi: 10.1016/0002-9343(91)90346-y. [DOI] [PubMed] [Google Scholar]
- 45.Schlievert P M, Gahr P J, Assimacopoulos A P, Dinges M M, Stoehr J A, Harmala J W, Hirt H, Dunny G M. Aggregation and binding substances enhance pathogenicity in rabbit models of Enterococcus faecalis endocarditis. Infect Immun. 1998;66:218–223. doi: 10.1128/iai.66.1.218-223.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schutte B C, Ranade K, Pruessner J, Dracopoli N. Optimized conditions for cloning PCR products into an XcmI T-vector. BioTechniques. 1997;22:40–44. doi: 10.2144/97221bm06. [DOI] [PubMed] [Google Scholar]
- 47.Uttley A H, Collins C H, Naidoo J, George R C. Vancomycin-resistant enterococci. Lancet. 1988;1:57–58. doi: 10.1016/s0140-6736(88)91037-9. . (Letter.) [DOI] [PubMed] [Google Scholar]
- 48.Valdivia, R. H., and S. Falkow. Fluorescence-based isolation of bacterial genes expressed within host cells. Science 277:2007–2011. [DOI] [PubMed]
- 49.Warren G, States D J. Identification of protein coding regions by database similarity search. Nat Genet. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- 50.Wells C L, Erlandsen S L. Localization of translocating Escherichia coli, Proteus mirabilis, and Enterococcus faecalis within cecal and colonic tissues of monoassociated mice. Infect Immun. 1991;59:4693–4697. doi: 10.1128/iai.59.12.4693-4697.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Welsh J, Chada K, Dalal S S, Cheng R, Ralph D, McClelland M. Arbitrarily primed PCR fingerprinting of RNA. Nucleic Acids Res. 1992;20:4965–4970. doi: 10.1093/nar/20.19.4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong K K, McClelland M. Stress-inducible gene of Salmonella typhimurium identified by arbitrarily primed PCR of RNA. Proc Natl Acad Sci USA. 1994;91:639–643. doi: 10.1073/pnas.91.2.639. [DOI] [PMC free article] [PubMed] [Google Scholar]