

Abstract
Staphylococcus aureus (SA) bloodstream infections cause high morbidity and mortality (20–30%) despite modern supportive care. In a human bacteremia cohort, development of thrombocytopenia was correlated to increased mortality and increased α-toxin expression by the pathogen. Platelet-derived antibacterial peptides are important in bloodstream defense against SA, but α-toxin decreased platelet viability, induced platelet sialidase to cause desialylation of platelet glycoproteins, and accelerated platelet clearance by the hepatic Ashwell-Morell receptor (AMR). Ticagrelor (Brilinta), a commonly prescribed P2Y12 receptor inhibitor used post-myocardial infarction, blocked α-toxin-mediated platelet injury and resulting thrombocytopenia, thus providing protection from lethal SA infection in a murine intravenous challenge model. Genetic deletion or pharmacological inhibition of AMR stabilized platelet counts and enhanced resistance to SA infection, and the anti-influenza sialidase inhibitor oseltamivir (Tamiflu) provided similar therapeutic benefit. Thus a “toxin-platelet-AMR” regulatory pathway plays a critical role in the pathogenesis of SA bloodstream infection, and its elucidation provides proof-of-concept for repurposing two commonly prescribed drugs as adjunctive therapies to improve patient outcomes.
One-sentence summary:
The clinically approved drugs ticagrelor and oseltamivir fortify a regulatory pathway of platelet-mediated immunity to clear staphylococcal bacteremia
INTRODUCTION
Staphylococcus aureus (SA) is one of the most important human bacterial pathogens, as the second leading cause of bloodstream infections (bacteremia) and the leading cause of infective endocarditis (1). Despite modern supportive measures, overall mortality in SA bacteremia has not declined in decades and remains unacceptably high (20–30%), with substantial risk of complications including sepsis syndrome, endocarditis, and metastatic foci of infection (for example osteomyelitis) (2). High risk populations include the elderly, diabetics, surgical and hemodialysis patients (3). Multidrug resistance (such as with methicillin-resistant SA, MRSA) is prevalent and associated with adverse outcome and increased medical costs (4).
The high incidence of SA bacteremia signifies a remarkable capacity of the organism to resist host innate defense mechanisms that function to prevent pathogen bloodstream dissemination (5). Extensive research has focused on SA virulence factors that counteract opsonization by serum complement (6), surface-anchored protein A that impairs the Fc function of antibodies (7), and the pathogen’s numerous resistance mechanisms to avoid phagocytosis and oxidative burst killing by neutrophils (8).
Comparatively less is understood about how SA interacts with circulating platelets. These abundant, small anucleate cells are best known for their central role in hemostasis but are increasingly appreciated to possess bioactivities relevant to immune defense (9). Platelets can act as mechano-scavengers to bundle bacteria (10) and enhance the function of professional phagocytic cell types such as neutrophils (11), macrophages (12), and hepatic Kuppfer cells (13, 14). Platelets express several Toll-like receptors that recognize pathogen-associated molecular patterns (15) to activate their release of pro-inflammatory cytokines (for example interleukin-1β) (16) and antimicrobial peptides including platelet microbicidal protein (tPMP) and human beta-defensin-1 (hBD-1) with direct antibacterial actions (12, 17, 18). SA activates platelets via integrin GP11b/IIIa, FcγRIIa receptor, and ADAM-10-dependent pathways (19–21), and the pathogen induces platelet aggregation via its “clumping factors” ClfA and ClfB (22, 23).
In a cohort of 49 patients with SA bacteremia, we report a strong association of mortality with lowered platelet count (thrombocytopenia) rather than changes in leukocyte count. Of note, platelet depletion in mice was recently shown to impair SA clearance (12, 24). Our mechanistic analysis with human platelets ex vivo and murine platelets in vivo revealed a critical activity of platelets in direct killing of SA. The pathogen attempts to counteract this defense by deploying a pore-forming toxin (α-toxin) to both disrupt platelet antimicrobial activity and accelerate sialidase-dependent platelet clearance through the hepatic Ashwell-Morell receptor (AMR). Elucidation of this “toxin-platelet-AMR” regulatory pathway guided us to therapeutic repurposing of two U.S. Federal Drug Administration (FDA)-approved drugs to preserve platelet homeostasis, thereby providing significant host protection in experimental SA bloodstream infection.
RESULTS
Platelets are essential for blood immunity against SA bacteremia
The normal human platelet count ranges from 150,000–450,000/mm3 of blood. In 49 consecutive patients with SA bacteremia (blood culture growing MRSA or MSSA) identified prospectively at an academic medical center in Madison, WI (25), we observed a strong association of patient mortality with thrombocytopenia (platelet count <100,000/mm3) on the initial blood sample and without abnormally elevated or reduced leukocyte count (Fig. 1, A and B). Indeed, two patients with thrombocytopenia failed to clear their bacteremia despite antibiotic therapy for more than 60 days before succumbing. No significant correlation was observed between platelet count and serum concentrations of pro-inflammatory cytokine IL-1β nor APACHE score, a clinical metric of disease severity (Fig. 1A). These data are consistent with a prior single center study in Israel showing thrombocytopenia (and not leukocyte count) to be a significant risk factor for 30-day all-cause mortality in SA bacteremia (unadjusted OR, 2.41; 95% CI, 1.76–3.32; P < 0.001), although overall mortality rates were much higher than other published series (56.2% in thrombocytopenic group vs. 34% with normal platelet counts) (26). These clinical studies suggest that circulating platelets, and not white blood cells, could play the dominant role in resolution of SA during bloodstream infection. Indeed, when directly compared, the second most common Gram-positive bacterial pathogen associated with human bacteremia, Streptococcus pneumoniae (1), SA was 49.3% more resistant to killing by purified human neutrophils, but 63.7% more susceptible to killing by human platelets (Fig. 1C).
Fig. 1. Platelets are essential for blood immunity against Staphylococcus aureus (SA) bacteremia and SA α-toxin induces thrombocytopenia to evade platelet-mediated microbicidal activity.
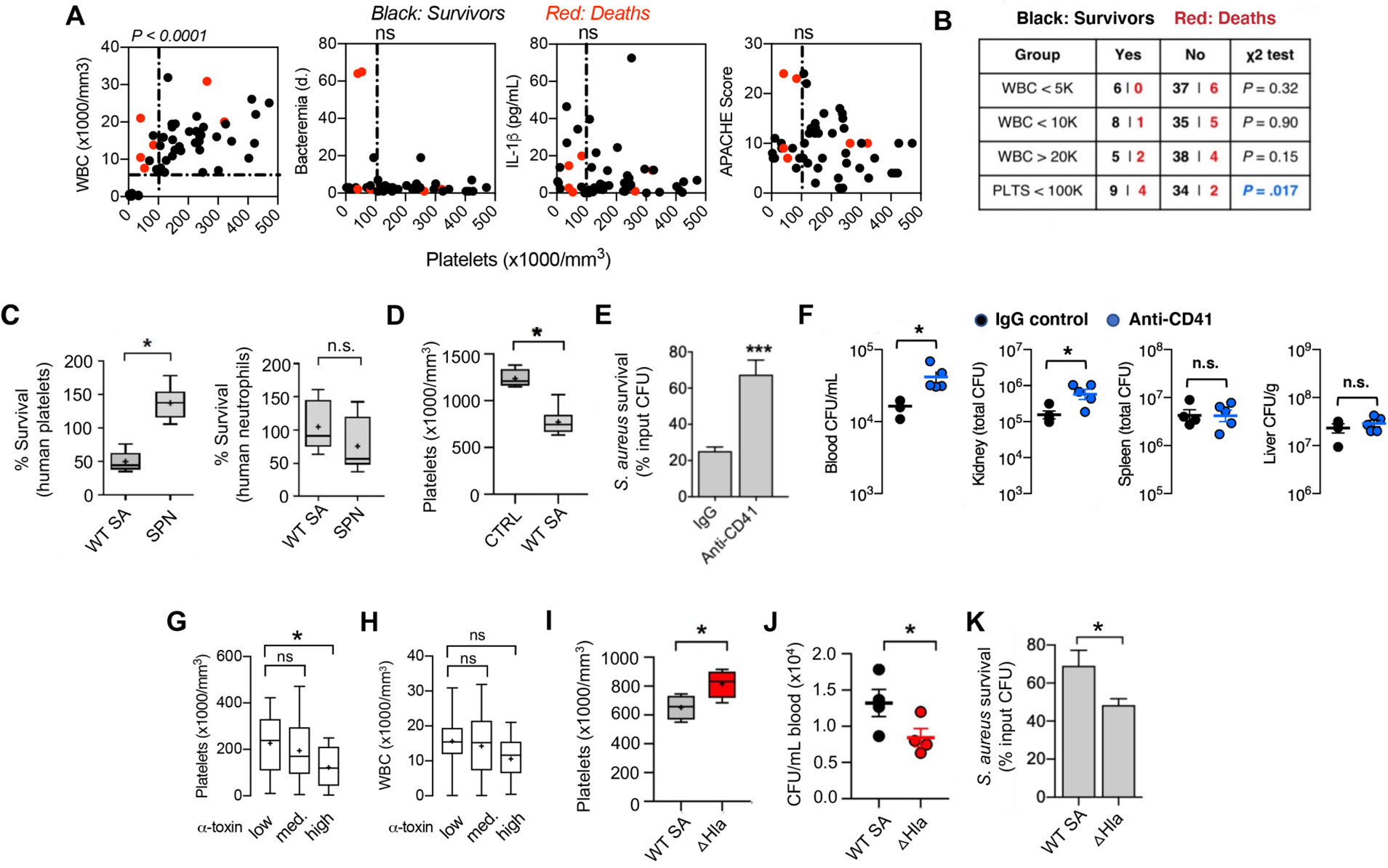
(A) Correlation of circulating platelet counts with leukocyte counts and duration of bacteremia in 49 consecutive patients with SA bacteremia from a tertiary medical center. Spearman’s rank correlation coefficient compared variables. (B) Mortality in patient cohort associated with different white blood cell (WBC) and platelet (PLTS) count cutoffs; Chi-square without Yates correction. (C) Washed isolated human platelets and neutrophils exposed to SA or Streptococcus pneumoniae (SPN) at MOI = 0.01 for 2 h. Samples were sonicated, serial diluted, and plated on THA plates for enumeration of bacterial colony forming units (CFU). (D) Reduction in platelet count 2 h after intravenous infection of mice with SA (n = 8) vs. non-infected littermate controls (n = 4). Two biological replicates were performed and data pooled; data represented as mean ± SEM. (E) Ex vivo killing of SA upon 2 h co-incubation with blood collected from mice 16 h after treatment with anti-CD41 antibody (n= 9) or IgG control (n= 12). (F) Mice treated with platelet depleting anti-CD41 antibody (n = 5) or IgG control (n = 4) for 16 h prior to intravenous SA infection. Organs harvested and CFU enumerated 2 h post-infection in triplicate for each sample. (G) Assessment of α-toxin production by the infecting SA isolate in 49 consecutive bacteremia cases and its association with patient platelet counts and (H) WBC counts. (I-J) Platelet counts (I) and CFUs (J) in outbred CD-1 mice intravenously challenged with wild-type SA (n = 4) or isogenic ΔHla mutant (n = 4). Blood harvested by cardiac puncture, complete blood count performed and colony forming units (CFUs) enumerated 4 h post-infection. (K) Ex vivo killing of SA by freshly isolated human platelets (2 h co-incubation) vs. isogenic ΔHla mutant. All data represented as mean ± SEM and representative of at least 3 independent experiments. Statistical significance was determined by unpaired two-tailed Student’s t-test (C-F, I, J, K) or one way analysis of variance (ANOVA) with Bonferroni’s multiple comparisons test (G,H). *P < 0.05. For floating bar graphs, + denotes the mean, whiskers represent min. to max, and floating box represents 25th to 75th percentile.
We pursued this association further using an in vivo model of SA bacteremia (MRSA strain USA300 TCH1516) established by intravenous (i.v.) tail vein injection in mice, where normal platelet count ranges from 900,000 to 1.4 million cells/mm3 blood (27). Within 2 h of SA challenge, the circulating platelet count of infected mice was reduced by 37.6% from baseline (1237 ± 49.82 vs. 772 ± 49.91) (Fig. 1D). To verify that reduced platelet count was indicative of a functional immune deficiency, we used an anti-CD41 antibody to deplete mice of platelets to 17% of baseline abundance (fig. S1). The drawn blood of the thrombocytopenic animals was impaired in ex vivo killing of SA (67.5 ± 7.9% surviving CFU vs. 25 ± 2.1% surviving CFU in normal blood) (Fig. 1E), and bacterial burdens in blood and kidneys of thrombocytopenic mice were significantly (P < 0.05) increased vs. untreated mice within 2 h following i.v. SA challenge (Fig. 1F).
SA α-toxin induces thrombocytopenia to evade platelet-mediated microbicidal activity
A major SA secreted virulence factor, the pore-forming α-toxin (Hla), induces platelet cytotoxicity and aberrant aggregation after binding its protein receptor A-disintegrin metalloprotease-10 (ADAM-10) on the platelet surface (14, 20, 28). Using ImageJ densitometric analysis of anti-Hla western immunoblots, we grouped the SA bacteremia isolates from our clinical cohort into low- (n= 18), medium- (n=15), and high- (n=16) α-toxin producers (fig. S2). A significant association (P < 0.05) was seen between thrombocytopenia and high-level α-toxin production (Fig. 1G) but not the low- or medium-level α-toxin production. There was no significant association between α-toxin production and leukocyte counts (Fig. 1H). For comparison to the wild-type (WT) parent SA strain in analyses of platelet interactions, we constructed a Δhla knockout strain by precise allelic replacement (fig. S3). In the mouse i.v. challenge model, the SA Δhla mutant induced less thrombocytopenia (Fig. 1I) and was more rapidly cleared from the blood circulation (Fig. 1J) than the WT parent strain. Ex vivo, the SA Δhla mutant was more susceptible to killing by purified human platelets (Fig. 1K). Together these studies indicate that the virulence effects of SA α-toxin extend to evasion of direct platelet-mediated antibacterial killing.
FDA-approved P2Y12 inhibitor ticagrelor blocks SA α-toxin-mediated platelet cytotoxicity
Inhibition of platelet activation is the target of antithrombotic drug therapy designed to reduce the risk of cardiovascular death, myocardial infarction (MI), and stroke in patients with acute coronary syndrome or a history of MI, beginning with classical studies of aspirin (acetylsalicylic acid, ASA) in the 1970s, then extending to newer selective inhibitors of adenosine signaling through the platelet P2Y12 receptor (clopidogrel, prasugrel, ticagrelor) (29). However, the effect of “antiplatelet” drugs on the direct antibacterial properties of platelets has not been reported. Of potential relevance, a clinical study of 224 consecutive patients with community-acquired pneumonia found that those receiving antiplatelet therapy (ASA and/or clopidogrel) for secondary prevention of cardiovascular disease reduced need for intensive care unit treatment (odds ratio 0.19, 95% confidence interval 0.04−0.87) and shorter hospital stays (13.9 ± 6.2 vs. 18.2 ± 10.2 days) compared to their age-matched cohort (30). Additional human retrospective or matched cohort studies of endocarditis, bacteremia, or sepsis (not restricted to SA) have provided similar hints of improved clinical outcome among patients receiving antiplatelet drugs (31–34).
To discriminate the effect of the two antiplatelet drug classes on SA killing, we pretreated freshly isolated human platelets for 15 min with ASA or ticagrelor (chosen because clopidogrel is a prodrug requiring hepatic conversion in vivo) and co-incubated them with the bacteria. Within 2 h, ticagrelor-treated platelets showed a 2.2-fold enhancement of SA killing vs. untreated controls (Fig. 2A), whereas ASA did not significantly alter platelet antibacterial activity. In contrast, ticagrelor did not promote macrophage or neutrophil killing of SA, did not alter neutrophil extracellular trap production, and did not directly inhibit SA growth (fig. S4, A–D). Upon direct co-incubation of SA with human platelets in a tissue culture well, severe platelet damage was evident by transmission electron microscopy; however, ticagrelor treatment preserved platelet structural integrity against SA-induced injury (Fig. 2B). As α-toxin is the principal driver of SA platelet toxicity, we measured α-toxin-induced lactate dehydrogenase (LDH) release from platelets treated with ticagrelor, ASA (COX-1 inhibitor), or specific small molecule inhibitors of other known platelet activation receptors (CD41, PAR-1, PAR4). Among these agents, only ticagrelor significantly inhibited SA α-toxin-induced platelet LDH release (Fig. 2C), doing so in a dose-dependent manner (Fig. 2D). The deleterious effect of α-toxin on platelets involves activation of its receptor protease ADAM-10 leading to intracellular Ca2+ mobilization (20), and these biological effects were both inhibited by ticagrelor as measured in specific assays (Fig. 2, E and F). Whereas ticagrelor did not alter the amount of ADAM-10 expressed on the platelet surface (Fig. 2G), the P2Y12 inhibitor drug blocked SA-induced ADAM-10-dependent shedding of platelet glycoprotein-6 (GP6) (Fig. 2H). SA exposure also induced P-selectin, a transmembrane protein specific to alpha granules that is translocated to the platelet surface upon activation (Fig. 2I), and increased surface expression of CD63, a marker of dense granule mobilization (fig. S5A). The SA-induced upregulation of P-selectin and CD63 were both blocked upon ticagrelor treatment (Fig. 2I, fig. S5A). Neither SA nor ticagrelor produced a statistically significant change in platelet beta-galactosidase activity, another lysosomal marker (P value above 0.05 for both, fig. S5B).
Fig. 2. FDA-approved P2Y12 inhibitor ticagrelor blocks SA α-toxin-mediated platelet cytotoxicity.

(A) Effect of 10 μM aspirin (ASA) or 10 μM ticagrelor (TICA) (15 min pretreatment ex vivo) on human platelet killing of MRSA for 2 h (n = 9). Experiments were performed in triplicate and repeated three times. (B) Representative transmission electron microscopy image of platelets pre-treated with or without 10 μM TICA and exposed to MRSA at multiplicity of infection (MOI) 0.1 for 2 h. Scale bar = 5 μM. (C) P2Y12 inhibitor (TICA) pretreatment blocks human platelet cytotoxicity by 5 μg/ml purified α-toxin as measured by LDH release (n = 3) in a (D) dose-dependent manner. Inhibitors: P2Y12 (ticagrelor), GPIIb/IIIa (eptifibatide), COX-1 (SC560), PAR-1 (vorapaxar), and PAR-4 (ML-354). (E) TICA (blue line) treatment of human platelets reduces proteolytic cleavage of an ADAM10-specific fluorogenic substrate compared to PBS control (black line). Data representative of three independent experiments and statistical significance determined by least squares ordinary fit, *P < 0.5. (F) TICA (blue line) reduces intracellular calcium levels in human platelets loaded with 2 μM Fluo-3 dye and stimulated with 5 μg/mL recombinant α-toxin compared to PBS control (black line); calcium influx was measured every 30 sec by fluorescence. For both (E) and (F), α-toxin stimulated platelets (whether TICA or PBS treated) were normalized to their respective non-stimulated platelet controls. (G) TICA treatment of human platelets did not alter surface ADAM-10 expression as determined by flow cytometry. (H) Human platelets with or without TICA treatment were infected with MRSA at MOI = 0.1 for 90 min. Surface glycoprotein-6 (GP6) was measured by flow cytometry and perecent decrease in expression (GP6 shedding) calculated. (I) Human platelet P-selectin expression indicating platelet activation measured by flow cytometry with or without MRSA challenge (MOI = 0.1) and with or without TICA for 90 min. All data represented as mean ± SEM and are representative of at least 3 independent experiments. Statistical significance was determined by One way ANOVA with Bonferroni’s multiple comparisons test (A,C,G), unpaired two-tailed Student’s t-test (H) and two-way analysis of variance (ANOVA) with Bonferroni’s multiple comparisons posttest (I). *P< 0.05, **P < 0.005. PBS, phosphate buffered saline; ns, not significant.
FDA-approved P2Y12 inhibitor ticagrelor protects against SA bacteremia
Inhibition of α-toxin mediated platelet cytotoxicity suggested that P2Y12 inhibition using ticagrelor could mitigate the toxin’s virulence role in driving SA-induced thrombocytopenia to promote bloodstream survival of the pathogen. IV SA challenge in mice drove down platelet counts beginning as early as 4 h (35% decrease, P < 0.0005) and continuing through 24 h (63% decrease, P < 0.0005), with partial recovery by 72 h (35% decrease, P < 0.005) (fig. S6A); bone marrow analysis at 72 h revealed increased thrombopoiesis as evidenced by greater megakaryocyte number and by ploidy distribution (fig. S6B and C). The rapid SA-induced thrombocytopenia was associated with platelet GP6 shedding (fig. S6D and E) and platelet microparticulation (fig. S6F), the latter determined by in vitro studies to be α-toxin-dependent (fig. S6G). Indeed, mice treated with ticagrelor maintained higher numbers of circulating platelets compared to control animals following SA i.v. infection (Fig. 3A), significantly reducing the bacterial burden in the blood (P < 0.05, Fig. 3B) and in systemic organs (kidney P < 0.005, liver P < 0.0005, spleen P < 0.0005, Fig. 3C), ultimately improving survival in a 10-day mortality study (Fig. 3D). Blinded histological examination of tissues by a veterinary pathologist revealed treatment-associated differences in the kidneys (Fig. 3E) and the heart (fig. S7). A 4- to 10-fold reduction in bacterial micro-abscesses was identified within the renal glomeruli, tubules, and blood vessels of ticagrelor-treated mice vs. PBS control animals, corroborating the CFU quantification data. Renal microabscesses in the control group were generally larger and more densely packed with bacteria, whereas those in ticagrelor-treated mice were frequently disrupted by immune cell infiltrates. Together these data suggest that platelet P2Y12 inhibition blocks α-toxin and SA-mediated platelet cytotoxicity and consequent thrombocytopenia, thus enhancing the platelet-mediated clearance of the pathogen in vitro and in vivo.
Fig. 3. FDA-approved P2Y12 inhibitor ticagrelor protects against SA bacteremia.
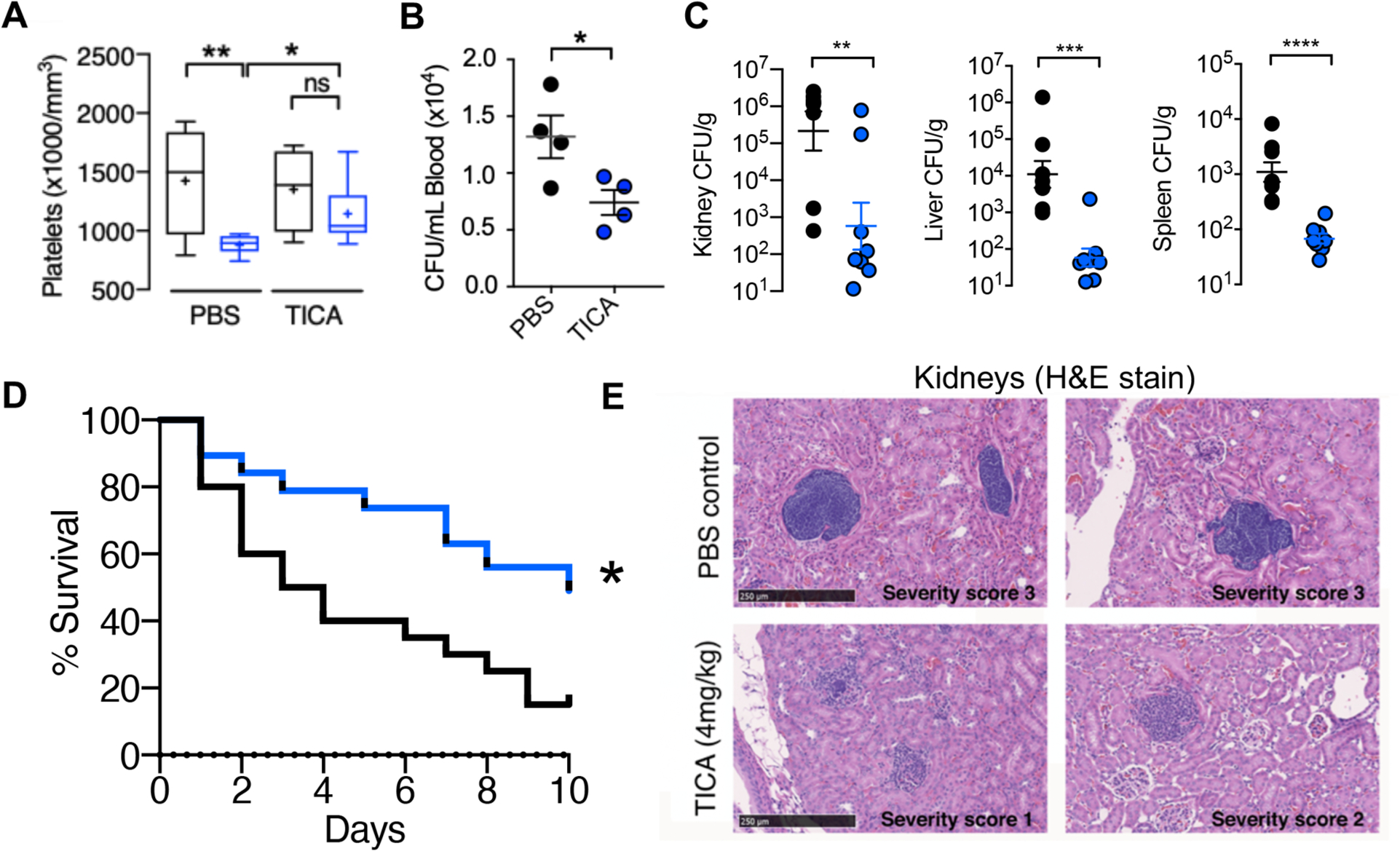
(A and B) Outbred CD-1 mice treated with 4 mg/ml ticagrelor (TICA; n = 9) or PBS (n = 9) control every 12 h for 3 days, then challenged intravenously with 1 × 108 colony forming units (CFUs) of SA; blood platelets and bacterial CFU burden enumerated 4 h post-infection. (C) Enumeration of bacterial CFU burden at 72 h in organs of mice pretreated with vehicle (PBS) or 4 mg/kg ticagrelor 12 h prior to intravenous SA and q 12 h thereafter; (n= 8). (D) Mortality curves of outbred CD-1 mice pretreated with vehicle (PBS) or 4 mg/kg ticagrelor beginning 24 h prior to intravenous SA infection then q 12 h over a 10-day observation period (n = 20). Independent experiments were repeated twice and data pooled. (E) Hematoxylin and eosin stain (H&E) of representative histological kidney sections from mice pre-treated with PBS vehicle or 4 mg/kg ticagrelor 12 h prior to SA infection and q 12 h thereafter for 72 h; (n = 8). Yellow stars denote formation of dense bacterial colonies and black arrows represent immune infiltrate. All histological sections are representative photos of at least 6 samples per two independent experiments. Where applicable, results are represented as mean ± SEM and statistical significance was determined by unpaired two-tailed Student’s t-test (B,C), and two-way ANOVA with Bonferroni’s multiple comparisons posttest (A). For survival curves, statistical significance determined by Log-rank Mantel-Cox test (D); *P < 0.05. For floating bar graphs, + denotes the mean, whiskers represent min. to max, and floating box represents 25th to 75th percentile. *P < 0.05, **P < 0.005, ***P < 0.0005.
SA α-toxin activates endogenous platelet sialidase activity
Our clinical data and those of others (26), coupled with our experimental work and prior platelet depletion studies (12, 24), suggest platelet count per se is important in determining SA clinical outcome. Since α-toxin production correlated to thrombocytopenia in patients and in experimental mouse infection, we hypothesized that SA deploys the toxin as a means to deplete the host of an effective circulating innate immune cell. Yet, platelet senescence and clearance are tightly regulated by multiple mechanisms, in particular the highly conserved hepatic transmembrane heterodimeric Ashwell-Morell receptor (AMR) (35). The AMR clears “aging” platelets with reduced terminal α2,3-linked sialic acids on their surface glycoproteins and glycolipids by engaging the exposed underlying galactose. We asked if the observed therapeutic effect of ticagrelor in SA sepsis was solely based on inhibiting platelet cytotoxicity or further intersected with this important mechanism of platelet homeostasis.
To assess platelet sialylation state during SA bacteremia, we obtained frozen plasma from 10 randomly selected adult patients with SA bacteremia, 10 patients with Escherichia coli bacteremia, and 5 healthy subjects. We found an increase in exposed galactose (indicative of desiaylation) on the platelets of SA-infected patients compared to the two other groups (Fig. 4A). However, SA lacks a bacterial sialidase (neuraminidase) present in other pathogens including S. pneumoniae (Fig. 4B). Rather, we found that WT SA induced sialidase activity (P < 0.005) on purified human platelets, whereas its isogenic Δhla mutant derivative did not (Fig. 4C). An increase in platelet sialidase activity in response to WT SA or purified α-toxin, present within the platelet pellet but not released into the media, was detected in independent assays using lectin affinity and a fluorescent substrate (fig. S8A, Fig. 4D). Although the precise mechanism of its transfer is not established, Neu1 is the main endogenous sialidase that can translocate from lysosomal stores to the platelet surface to target glycoproteins and expose AMR ligands (galactose) (36, 37), and we confirmed its upregulation in response to SA challenge by flow cytometry (fig. S8B). Probing the observed therapeutic effect of P2Y12 inhibition in this context, we found ticagrelor strongly inhibited SA-induced platelet sialidase activity (Fig. 4E). These results suggest that SA α-toxin-induced thrombocytopenia may not depend on wholescale platelet injury, but instead involve accelerated hepatic AMR-dependent clearance of desialylated platelets upon surface mobilization of Neu1. Binding of ADP to P2Y12 elevates cytosolic calcium (Ca2+) concentrations by stimulating phospholipase C-mediated production of inositol-1,4,5-trisphosphate (IP3), which in turn releases Ca2+ from the intracellular stores through IP3 receptor channels. Because Ca2+ is a major signaling molecule that allows for ADP-induced lysosomal secretion, P2Y12 inhibition in theory would block this process. However, a singular correlation between intracellular Ca2+ concentrations and granular secretion remains ambiguous, as our data, as well as older literature, suggest that there are both Ca2+-dependent and Ca2+-independent granular secretory pathways (38, 39). That said, by linking sialidase activity to the ticagrelor therapeutic effect, additional target points for pharmacological support of platelet defense against SA came into view.
Fig. 4. SA α-toxin activates endogenous platelet sialidase activity.
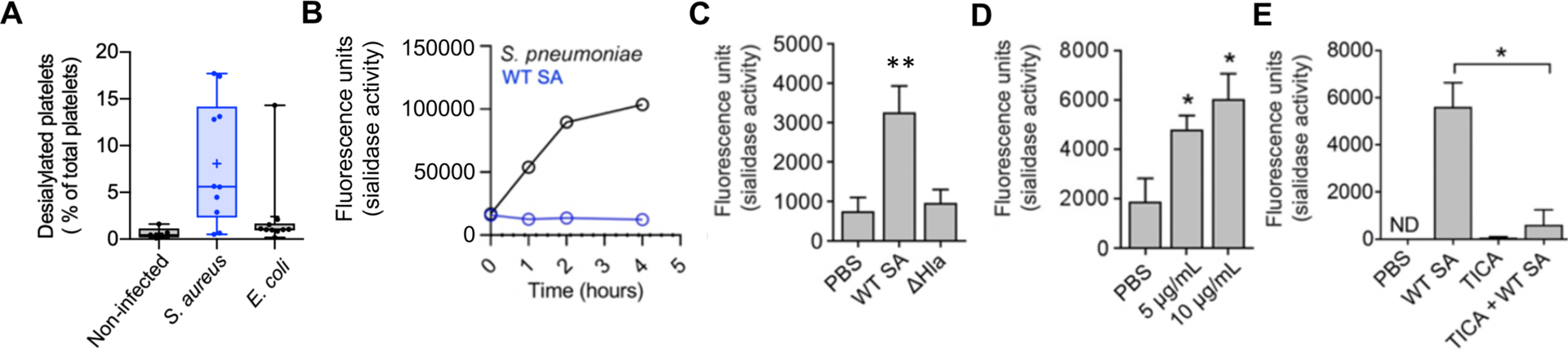
(A) Percent desialylated platelets in platelet rich plasma from non-infected subjects or patients with SA or E. coli bacteremia measured by flow cytometry. (B) SA and S. pneumoniae sialidase activity assessed for over 4 h. (C) Sialidase activity examined on washed human platelets exposed to WT SA or isogenic ΔHla for 1 h or (D) sialidase activity examined on washed human platelets exposed to 5 μg/mL and 10 μg/mL recombinant α-toxin for 30 min. (E) Sialidase assay performed on washed human platelets treated with or without 10 μM ticagrelor and exposed to WT SA for 1 h. Where applicable, all data represented as mean ± SEM and are representative of at least 3 independent experiments. Statistical significance determined by one way ANOVA with Bonferroni’s multiple comparisons test (A, C, D, E). *P < 0.05. PBS, phosphate buffered saline; ns, not significant. ND, not detectable.
Inhibition of the hepatic Ashwell-Morell receptor (AMR) supports platelet-mediated defense against SA bacteremia
In previous work, we showed that moderate thrombocytopenia mediated by AMR-dependent clearance of desialylated platelets was protective in experimental sepsis caused by S. pneumoniae, a sialidase-expressing pathogen (40, 41). However, as shown earlier in this study, S. pneumoniae is resistant to human platelet killing, and therefore removal of desialylated and hypercoagulable platelets does not deplete the bloodstream of an effective antimicrobial effector cell type. We asked whether the innate immune calculus could prove different for platelet-sensitive SA by challenging WT and AMR-deficient (Asgr2−/−) mice in the C57bl/6 background. Whereas WT mice remained highly sensitive to SA α-toxin-induced thrombocytopenia, platelet counts in Asgr2−/− mice did not drop following bacterial challenge (Fig. 5A), indicating that recruitment of AMR clearance was the main pathogenic driver of platelet clearance during infection. And in contrast to findings in S. pneumoniae infection (40), Asgr2−/− mice, which are resistant to pathogen-induced thrombocytopenia, exhibited a strong survival advantage against lethal SA challenge (Fig. 5B). This genetic association could be reproduced pharmacologically in WT mice, where asialofetuin, a competitive glycoprotein inhibitor of the hepatic AMR (Fig. 5C), improved mouse survival in lethal SA challenge by maintaining platelet count during infection (Fig. 5D) and by reducing bacterial burden in the kidney, liver, and spleen (Fig. 5E). Corroborating that α-toxin-dependent desialylation drove the accelerated platelet clearance, no SA-induced reduction in platelet count was seen in mice lacking the hepatic AMR (Fig. 5A, Fig. 5F).
Fig. 5. Inhibition of the hepatic Ashwell-Morell receptor (AMR) supports platelet-mediated defense against SA bacteremia.
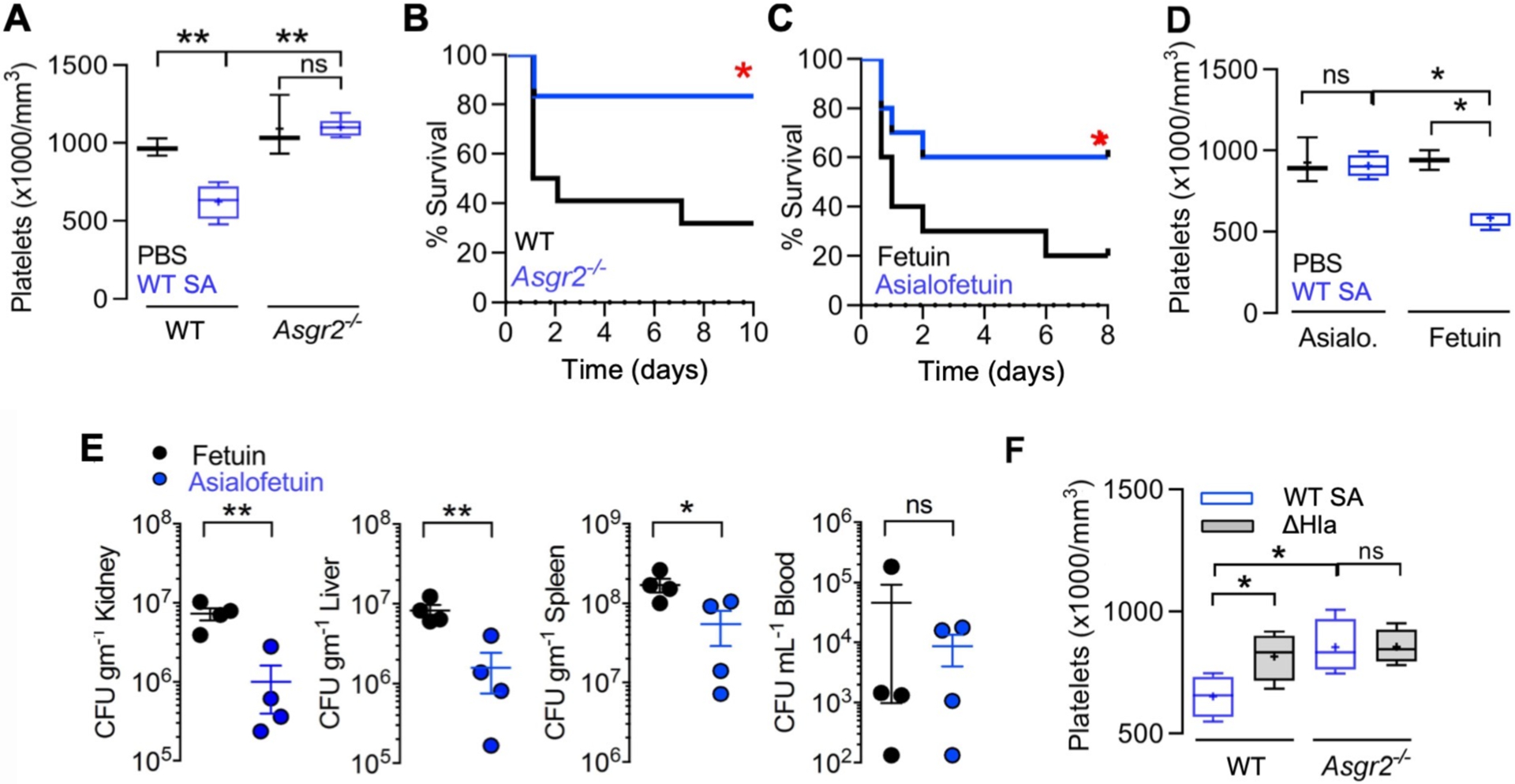
(A) C57/Bl6 (n = 4) and Asgr2−/− (n = 6) mice were challenged by intraperitoneal injection with SA, blood harvested by cardiac puncture, and platelet count enumerated. (B) 10-day mortality study with C57/Bl6 (n = 22) and Asgr2−/− mice (n = 16) challenged by intraperitoneal injection with SA. Study performed two independent times and data pooled. (C) 8-day mortality study with C57/Bl6 treated with fetuin (n = 10) or asialofetuin (n = 10) and challenged by intraperitoneal injection with SA. (D) C57/Bl6 mice treated with asialofetuin (n = 4) or fetuin (n = 4) and challenged by intraperitoneal injection with SA, platelet count enumerated, and (E) kidneys, liver, spleen, and blood harvested 24 h post-infection for bacterial CFU enumeration. (F) C57/Bl6 and Asgr2−/− mice challenged with wild-type MRSA or the isogenic ΔHla mutant. 4 h post-infection, blood was harvested by cardiac puncture for enumeration of platelet count. Statistical significance was determined by unpaired two-tailed Student’s t-test (E), two-way ANOVA with Bonferroni’s multiple comparisons posttest (A,D,F) or log-rank (Mantel-Cox) Test (B,C) for the survival curves. For floating bar graphs, + denotes the mean, whiskers represent min. to max, and floating box represents 25th to 75th percentile. *P < 0.05, **P < 0.005. PBS, phosphate buffered saline; ns, not significant.
FDA-approved sialidase inhibitor oseltamivir blocks AMR-mediated platelet clearance and protects against SA bacteremia
The above results showed that mice were protected against SA infection by ticagrelor, which inhibits α-toxin-induced platelet desialylation, or by genetic or pharmacological inactivation of the AMR, which blocks hepatic clearance of desialylated platelets. Further corroboration of the importance of platelet sialylation for maintaining bloodstream defense against SA bacteremia was obtained using mice lacking the St3gal4 sialyltransferase gene, which show diminished platelet sialylation and baseline thrombocytopenia (42). Compared to WT C57bl6 mice, the St3gal4−/− mice had accelerated mortality upon IV SA infection (Fig. 6A), but no further SA-induced reduction in their already low platelet counts (~25% of normal mice, Fig. 6B). Because sialidase (Neu1) activity appears central to the “toxin-platelet-AMR” pathway driving deleterious thrombocytopenia in SA bloodstream infection, we considered the possibility that pharmacological sialidase inhibition could be of therapeutic benefit. Oseltamivir (Tamiflu) is a commonly prescribed FDA-approved drug designed to target influenza sialidase (neuraminidase) and lessen the severity of flu symptoms. However, oseltamivir has a degree of non-selectivity in its sialidase inhibition, as the drug was recently recognized to raise platelet counts in mice with anti-GPIbα-mediated thrombocytopenia (43). Using Asgr2−/− mice to prevent immediate clearance of desialylated platelets, we confirmed that oseltamivir inhibited platelet desialylation in vivo during SA infection (P < 0.005, Fig. 6C). Then using WT mice, we showed that oseltamivir significantly reduced = the degree of α-toxin-induced thrombocytopenia during WT SA infection (P < 0.005, Fig. 6D). Both oseltamivir and established human Neu1-selective sialidase inhibitor C9-butyl-amide-2-deoxy-2,3-dehydro-N-acetylneuraminic acid (DANA) significantly improved survival outcomes in lethal SA bacteremia (P < 0.05, Fig. 6E).
Fig. 6. FDA-approved sialidase inhibitor oseltamivir blocks AMR-mediated platelet clearance and protects against SA bacteremia.
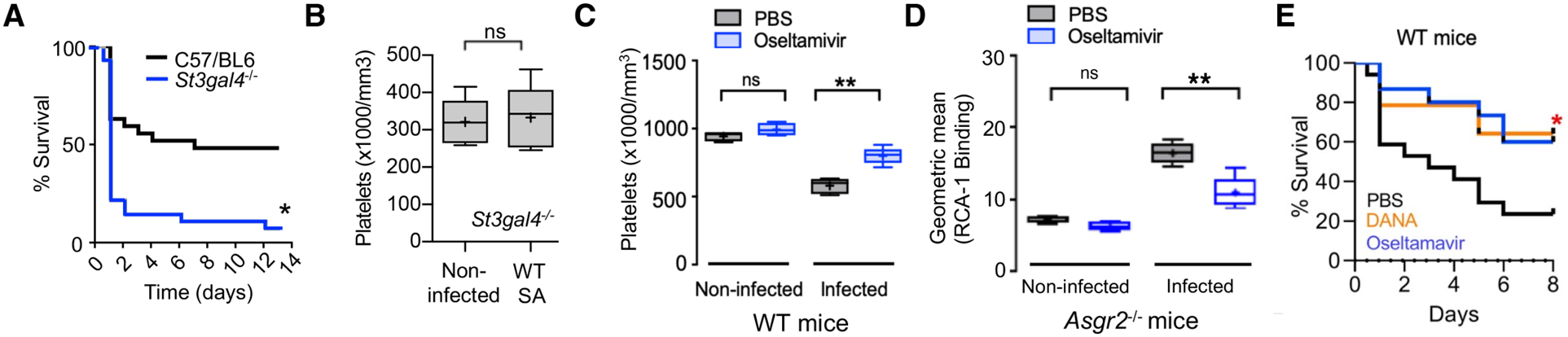
(A) St3gal4−/− mice that have decreased platelet sialylation and thrombocytopenia show accelerated mortality upon SA bloodstream infection (n = 10 per group). (B) circulating platelet count 4 h after IV SA challenge in WT vs. St3gal4−/− mice (n = 10 per group). (C) Platelets isolated from Asgr2−/− mice treated with or without oseltamavir and infected with MRSA were assessed for RCA-1 lectin binding. (D) C57/Bl6 mice were treated with oseltamavir (n = 6) or PBS control (n = 5) and infected with WT SA by intraperitoneal injection. Blood was harvested 24 h after infection and platelet counts collected. (E) 8-day mortality study conducted on C57/Bl6 mice treated with DANA (n = 16), oseltamavir (n = 16), or PBS control (n = 16). Statistical significance was determined by unpaired two-tailed Student’s t-test (B), two-way ANOVA with Bonferroni’s multiple comparisons posttest (C,D), or Log-rank (Mantel-Cox) Test (A,E). For floating bar graphs, + denotes the mean, whiskers represent min. to max, and floating box represents 25th to 75th percentile. Unless otherwise stated, *P < 0.05, **P < 0.005. PBS, phosphate buffered saline; ns, not significant.
Last, to support all elements of the elucidated “toxin-platelet-AMR” pathway, we repeated several key in vivo experiments (the ticagrelor, oseltamivir, and asialofetuin treatment studies as well as challenge of Asgr2−/− mice) with the SA ΔHla knockout mutant. Given the attenuated virulence of this mutant, establishment of bacteremia in the murine IV model required an 8-fold higher challenge inoculum. Blood harvested 4 h post-infection in all the experiments showed that the platelet count drop associated with WT infection was not seen in ΔHla-infected mice in any of the models, even at the 8-fold higher bacterial challenge inoculum (fig. S9A). Likewise, equal CFU counts of the ΔHla knockout mutant SA were recovered in the kidney, liver, spleen, and blood of control mice vs. mice treated with either TICA, asialofetuin, or oseltamivir, as well as WT mice when compared to Asgr2−/− animals (fig. S9B). As predicted by the model, deletion of α-toxin “phenocopies” the therapeutic benefits of the drug and genetic interventions to support platelet defense against SA bloodstream infection.
DISCUSSION
SA is a leading agent of human bloodstream infection, with higher morbidity and mortality rates than other common bacterial pathogens. Successful treatment of SA bacteremia remains vexing, as the pathogen deploys diverse mechanisms for resistance to immune and antibiotic clearance. Life-threatening complications of SA bacteremia such as metastatic infections, infective endocarditis, and disseminated intravascular coagulation drive worsened patient outcomes. Thrombocytopenia (platelet count <150,000/mm3 blood) is a common phenotype observed during bacteremia and is the most predictive independent risk factor for bacteremia-associated mortality, especially in cases of neonatal sepsis and critically ill septic patients in the intensive care unit (44, 45). Although the underlying cause of thrombocytopenia is multifactorial, our mechanistic analysis of platelet-mediated defense establishes a central pathophysiologic framework aggravating platelet depletion during SA bacteremia. We show that for SA to evade platelet microbicidal activity, the pathogen deploys the cytotoxic α-toxin, which injures platelets and stimulates the release of endogenous sialidase, thereby dysregulating the platelet clearance mechanism involving the hepatic AMR. Pharmacological targeting of multiple levels of this “toxin-platelet-AMR” pathway revealed new strategies to mitigate the progression of this immunocompromised state and protect against lethal SA bacteremia (fig. S10).
Sialidase transfer from lysosomal compartments to the platelet cell surface may potentially be elicited by multiple surface-bound receptors including but not limited to P2Y12, PAR-1, and PAR-4 (46). These receptors have converging intracellular signaling pathways, and studies indicate that P2Y12 functions in crosstalk with PAR receptors (47). We find here that ticagrelor, a commonly prescribed FDA-approved P2Y12 inhibitor, hastened clearance of SA bacteremia in vivo and enhanced human platelet killing of SA ex vivo. Here we describe effects of the drug in reducing SA α-toxin-mediated platelet cytotoxicity, inhibiting activation of endogenous platelet sialidase activity, and preventing AMR-dependent platelet clearance. As thrombocytopenia was not observed in SA-challenged mice lacking the AMR, ticagrelor’s ability to counteract wholescale SA-induced platelet damage may be unique to the high bacterial concentrations and close platelet contact present in our in vitro assays, and perhaps only relevant in vivo within an infected thrombus. Conceivably, ticagrelor’s primary therapeutic indication for acute coronary syndrome, reduction of platelet aggregation, may further provide protective benefit in SA bacteremia as the pathogen produces two clotting factors, coagulase (Coa) and von Willebrand factor binding protein (vWbp), that contribute to abscess formation and systemic virulence (48). Of note, one prior report linked in vitro P2Y12 activation to the release of platelet antimicrobial peptides active against SA (49). This paradoxical result may in part reflect the particular SA strain (ISP479C) used in the study, which harbors a chromosomal Tn551 insertion with a pleiotropic effect on several extracellular and cell wall proteins, including elimination of measurable α-toxin activity (50). Platelet release of antimicrobial peptides active against SA is also activated by additional pathways, including thrombin-mediated enzymatic activation of cell surface protease-activated receptor-1 (PAR-1) still operative during P2Y12 blockade (12). The feasibility of ticagrelor as an adjunctive therapy for SA bacteremia in complex ICU patients is likely enhanced by its reversible binding to the P2Y12 receptor binding providing a very rapid onset and offset of action (51).
Oseltamivir, a commonly used FDA-approved influenza sialidase inhibitor, maintained platelet sialylation to delay AMR-dependent platelet clearance and thus provided protection against mortality in SA bloodstream infection. Although one enzymatic study suggested that oseltamivir had only limited inhibitory activity against human sialidases (52), humans prescribed oseltamivir show higher platelet counts than matched controls (independent of proven influenza, (53)), and two independent case studies report successful use of the drug to restore platelet counts in a patient with immune thrombocytopenia (54, 55). Bacterial co-infection is estimated to have contributed to nearly all influenza deaths in the 1918 influenza pandemic and up to one-third of 2009 pandemic influenza A (H1N1) infections managed in ICUs worldwide (56). In particular, the potential for lethal synergism between SA and influenza virus has recently been documented in U.S. clinical epidemiologic studies of adult and pediatric patients (57, 58). In laboratory-confirmed influenza, an inverse relationship between virus load and platelet count is seen, and viral-induced thrombocytopenia can be recapitulated in the ferret model (59). We speculate that the “two-hit” scenario of influenza neuraminidase on top of α-toxin-induced endogenous sialidase activation may accelerate platelet clearance, depleting the host of a critical frontline defense against SA bloodstream dissemination, thus increasing the odds of fatal outcome.
An important limitation of pharmacological targeting AMR-dependent clearance of desialylated platelets to treat bacteremia is its dependency on sensitivity of the offending pathogen to platelet antimicrobial activity. A definitive or strong presumptive microbiologic diagnosis of SA would be required, precluding its use as empiric therapy wherein other pathogens such as platelet-resistant S. pneumoniae could yield adverse results (41). Multiple pathogenic mechanisms contribute to sepsis and intrinsic host factors can have differing roles depending on the pathogen involved (60). In this case, AMR function may serve protective and disadvantageous roles depending upon the pathogen and the balance of platelet action in thrombosis vs. antimicrobial activity. Prior research indicates that loss of AMR can increase platelet count and regulate thrombopoietin production (61). Mechanisms of physiologic platelet turnover remain to be fully established and are likely to contribute to therapeutic modulation in the future. However, it is unlikely that either ticagrelor or oseltamivir administered late in the course of severe SA-induced thrombocytopenia could quickly restore platelet counts. There, perhaps platelet transfusion could augment anti-SA killing capacity in blood, wherein the pharmacological agents could mitigate against further α-toxin driven accelerated desialylation and AMR clearance of the donor platelets. The pathological process can also be targeted upstream at the level of the inciting SA α-toxin, where important research on neutralizing antibodies (for example Medimmune 4893) and receptor antagonists (for example GI254023X) have shown promising results (14, 62).
Therapeutic drug repurposing is an important avenue of exploration to improve clinical outcomes in serious infections where high rates of treatment failure and antibiotic resistance jeopardize patients. Elucidation of sialidase-dependent platelet homeostasis as a key battleground in host defense against SA bloodstream infection revealed the potential utility of P2Y12 and sialidase inhibition as adjunctive agents to antibiotic treatment and ICU supportive care for the critically ill. The most effective physiological concentrations to inhibit platelet cytotoxicity and sialidase activity and protect against SA bacteremia in humans are currently unknown. As FDA-approved drugs with excellent safety profiles in each class are readily at hand, we hope that carefully designed clinical investigation to validate or refute our experimental observations may follow.
MATERIALS AND METHODS
Study Design
The objective of this study was to understand the mechanistic basis of platelet homeostasis and function during SA bacteremia to guide future therapeutic approaches. We analyzed patient data and SA isolates from a published 2009–10 IRB-approved study (25) of SA bacteremia at the University of Wisconsin Hospital (a 493-bed academic medical center in Madison, WI) to link thrombocytopenia to patient mortality and elevated α-toxin production. We corroborated both associations in a UC San Diego IACUC-approved murine model of SA bacteremia. Additional UC San Diego IRB-approved ex vivo studies with freshly isolated human platelets found that the FDA-approved P2Y12 antagonist ticagrelor blocked α-toxin-induced platelet injury and sialidase activation, improving microbial killing. Infection studies in WT and isogenic AMR-deficient mice were used to link α-toxin-mediated platelet sialidase activation to accelerated thrombocytopenia and impaired SA clearance, which was counteracted by ticagrelor or the FDA-approved sialidase inhibitor oseltamivir.
Ethics statement
Animal studies were conducted in accord with protocols approved by the UC San Diego Institutional Animal Care and Use Committee; all efforts were made to minimize animal numbers and suffering. Blood for platelet isolation was obtained via venipuncture from healthy volunteers under written informed consent approved by the UC San Diego Human Research Protection Program.
S. aureus patient isolates
Consecutive patients from the above previously published, IRB-approved study (25) and its ongoing continuation (IRB #2018-0098) with blood cultures of SA from April 2009 through March 2010 at the University of Wisconsin Hospital were analyzed for α-toxin expression by western immunoblot and densitometry band analysis by Image J. Levels of α-toxin expression were grouped in the following order: low: >10,000; medium: 10,000 – 20,000; high: >20,000. Patient demographics, blood work (including platelet and leukocyte counts), and infection source were collected at time of administration. Bacterial isolates obtained at the onset of presentation and stored at −80°C until analysis. All laboratory tests were performed by investigators blinded to patient information.
Asgr2−/−, St3gal4−/−, and AMR inhibitor mouse infection studies
Eight- to 12-week-old Asgr2−/− mice (68) or 10 to 14-week-old St3gal4−/− mice on a C57/Bl6 (Jackson Laboratories) genetic background (69) and WT mice bred and raised in the same room were used. WT SA was grown overnight shaking at 37°C in THB, washed once in 1x PBS, and 1 × 108 CFU injected intraperitoneally (i.p.) unless otherwise specified in the Figure Legend, and mortality observed over the course of 10 days. For AMR inhibitor studies, C57/Bl6 mice were treated with 25mg/mL asialofetuin or fetuin prior to i.p. challenge with 1 × 108 CFU SA; mortality was observed over the course of 8 days. For both studies, mice that appeared moribund were euthanized by CO2 asphyxiation. Platelet count enumerations were performed 4 h post infection. For AMR inhibitor CFU enumeration, mice were euthanized 24 h post-infection, organs harvested, and dilution plated onto THA.
Sialidase inhibitor mouse infection studies
Eight- to 10-week-old wild-type C57/Bl6 mice were treated with oseltamivir (5 mg/kg) in 100 μL PBS or Neu1-selective inhibitor C9-butyl-amide-2-deoxy-2,3-dehydro-N-acetylneuraminic acid (DANA) (2 mg/kg) at the time of- and 3 h-post intraperitoneal infection with 1 × 108 CFU SA. Platelets were enumerated and sialidase activity assessed using 2′-(4-methylumbelliferyl)-α-D-N-acetylneuraminic acid (4MU; Sigma) from blood collected 4 h post infection per using a previously described protocol (4). For ex vivo sialidase analysis, murine platelet rich plasma (PRP) was isolated by cardiac puncture with a 25G needle attached to a syringe containing 100 μL ACD and centrifuged at 100 × g for 10 min without braking. Following isolation, 25 μL of PRP was added to wells of white 96-well plate (Costar) with 25 μL RPM I+ 125 μM 4MU. The plate was incubated at 37°C + 5% CO2 for 30 min, followed by an addition of 1M Na2CO3, and fluorescence determined at excitation 530 nm and emission 585nm.
Ticagrelor treatment mouse infection studies
SA was grown shaking overnight at 37°C in THB, washed once in PBS, and 1 × 108 CFU injected intravenously (i.v.) into outbred 8- to 10-week-old CD1 (Charles River Laboratories) mice. Where indicated, ticagrelor (4 mg/kg) or vehicle (water) was delivered by oral gavage 24 h prior- and every 24 h post-infection over a course of 10 days. Mice that appeared moribund were euthanized by CO2 asphyxiation. For quantification of CFU burden and histological preparation, mice treated with ticagrelor (4 mg/kg) or vehicle (water) 12 h prior- and every 24 h post-intravenous injection of 1 × 108 CFU SA. At 12 h post-infection, two mice from each group were euthanized by CO2 asphyxiation and the kidneys, spleen, heart, and liver harvested and fixed 10% neutral-buffered formalin for 24 h, then routinely processed and paraffin-embedded for histological analysis. Five-micron thick hematoxylin and eosin-stained sections of each tissue were examined by a veterinary pathologist blinded to the treatment group. Distinct bacterial colonies visible at 4x magnification were counted in three longitudinal sections of heart and six longitudinal sections of kidney. Lesions related to bacterial infection were described and graded (minimal-1, mild-2, moderate-3, or severe-4) based on degree of tissue damage. At 72 h post infection, remaining surviving mice were euthanized by CO2 asphyxiation, blood collected by cardiac puncture, and organs excised. Blood and organ homogenate (MagNA Lyser instrument (Roche Diagnostics Corporation) were serially diluted in molecular grade H2O and plated onto THA for bacterial CFU enumeration. The study was performed three independent times and data from a representative experiment shown. For platelet quantification, mice were treated with ticagrelor (4 mg/kg) or vehicle (water) every 12 h for 72 h prior to i.v. injection of 1 × 108 CFU SA. Four h post-infection, blood was collected by cardiac puncture with a 25G needle attached to a syringe containing 100 mL ACD buffer, transferred into EDTA tubes and a complete blood count (CBC) was obtained.
Statistical Analysis
All in vitro, ex vivo, and in vivo data were collected from three or more (≥3) independent experiments with ≥3 biological replicates and are represented as mean ± standard error mean (SEM), unless otherwise stated. For descriptive data (transmission electron microscopy and histopathologic staining), experiments were performed at least twice independently with ≥3 biological replicates and illustrated as best representative images. The alpha level used for all tests was 0.05; the data were normalized (single outliers removed via Grubbs’ test if applicable) and unpaired Student’s t-test, one-way ANOVA with Bonferroni’s multiple comparisons test, or two-way ANOVA with Bonferroni’s multiple comparisons test performed as explained in figure legends to determine statistical significance. For comparison of survival curves, a log-rank (Mantel-Cox) test was performed. Statistical analyses were done using GraphPad Prism, version 8.42 (GraphPad Software Inc.). P values < 0.05 were considered statistically significant.
Supplementary Material
Materials and Methods
Fig. S1. Thrombocytopenia established by anti-CD41 antibody treatment of mice.
Fig. S2. Grouping of α-toxin expression of SA isolates from bacteremia patients.
Fig. S3. Generation of an isogenic SA ΔHla mutant.
Fig. S4. Exclusion of ticagrelor off-target effects on SA immune cell interactions and growth.
Fig. S5. Additional effects of SA or TICA on human platelet phenotypes in vitro.
Fig. S6. Effects of SA challenge on platelets and thrombopoiesis in vivo.
Fig. S7. Organ pathology in murine SA infection with or without ticagrelor treatment.
Fig. S8. Neu1 is predominantly detected on the platelet cell surface and is induced by SA exposure.
Fig. S9. SA α-toxin deletion mutant phenocopies protective effects of TICA, oseltamivir, and AMR loss or inhibition.
Fig. S10. Schematic illustration of proposed “toxin-platelet-AMR” pathway exploited by SA in the pathogenesis of bloodstream infection.
ACKNOWLEDGEMENTS
The authors thank Ajit Varki for his critical insights on human and murine sialobiology.
Funding:
This work was supported by NIH grants HL125352 (J.D.M., V.N.), HL107150 (V.N.), HL131474 (J.D.M.), AI124326 (G.S., V.N.), HD090259 (G.S., V.N.) and AI13262 (W.E.R.). J.S. was supported the UCSD PharmD/PhD Program and I.C. by the UCSD Research Training Program for Veterinarians (NIH OD017863).
Footnotes
SUPPLEMENTARY MATERIALS
data file s1: Excel spreadsheet of primary values for main and supplemental figures
Competing interests: W.E.R. has received speaking honoraria from Melinta unrelated to the current study. G.S. has consulted for Allergan, Paratek, and Octapharma unrelated to the current study. V.N. has consulted for Cellics Therapeutics, Vaxcyte, Clarametyx Biosciences, SNIPR Biome, Boehringer Ingelheim, and Iogen unrelated to the current study. The authors do not hold patents related to the current study.
Data and materials availability:
All data associated with the current study are present in the paper or supplementary materials.
REFERENCES AND NOTES
- 1.Laupland KB, Incidence of bloodstream infection: a review of population-based studies, Clin. Microbiol. Infect 19, 492–500 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Kaasch AJ, Barlow G, Edgeworth JD, Fowler VG Jr, Hellmich M, Hopkins S, Kern WV, Llewelyn MJ, Rieg S, Rodriguez-Baño J, Scarborough M, Seifert H, Soriano A, Tilley R, Tőrők ME, Weiß V, Wilson APR, Thwaites GE, ISAC, INSTINCT, SABG, UKCIRG, and Colleagues, Staphylococcus aureus bloodstream infection: a pooled analysis of five prospective, observational studies, J. Infect 68, 242–251 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Del Rio A, Cervera C, Moreno A, Patients at risk of complications of Staphylococcus aureus bloodstream infection, Clin. Infect. Dis 48 Suppl 4:S246–53 (2009) [DOI] [PubMed] [Google Scholar]
- 4.Joo E-J, Park DA, Kang C-I, Chung DR, Song J-H, Lee SM, Peck KR, Reevaluation of the impact of methicillin-resistance on outcomes in patients with Staphylococcus aureus bacteremia and endocarditis, Korean J. Intern. Med 34, 1347–1362 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thomer L, Schneewind O, Missiakas D, Pathogenesis of Staphylococcus aureus bloodstream infections, Ann. Rev. Pathol 11, 343–364 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laarman A, Milder F, van Strijp J, Rooijakkers S, Complement inhibition by Gram-positive pathogens: molecular mechanisms and therapeutic implications, J. Mol. Med 88, 115–120 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim HK, Thammavongsa V, Schneewind O, Missiakas D, Recurrent infections and immune evasion strategies of Staphylococcus aureus, Curr. Opin. Microbiol 15, 92–99 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGuinness W, Kobayashi S, DeLeo F, Evasion of neutrophil killing by Staphylococcus aureus, Pathogens 5, 32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeaman MR, Platelets: at the nexus of antimicrobial defence, Nat. Rev. Microbiol 12, 426–437 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Gaertner F, Ahmad Z, Rosenberger G, Fan S, Nicolai L, Busch B, Yavuz G, Luckner M, Ishikawa-Ankerhold H, Hennel R, Benechet A, Lorenz M, Chandraratne S, Schubert I, Helmer S, Striednig B, Stark K, Janko M, Böttcher RT, Verschoor A, Leon C, Gachet C, Gudermann T, Schnitzler M. M. y., Pincus Z, Iannacone M, Haas R, Wanner G, Lauber K, Sixt M, Massberg S, Migrating platelets are mechano-scavengers that collect and bundle bacteria, Cell 171, 1368–1382.e23 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Kim S-J, Jenne CN, Role of platelets in neutrophil extracellular trap (NET) production and tissue injury, Semin. Immunol 28, 546–554 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Ali RA, Wuescher LM, Dona KR, Worth RG, Platelets Mediate Host Defense against Staphylococcus aureus through direct bactericidal activity and by enhancing macrophage activities, J. Immunol 198, 344–351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wong CHY, Jenne CN, Petri B, Chrobok NL, Kubes P, Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance, Nat. Immunol 14, 785–792 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Surewaard BGJ, Thanabalasuriar A, Zeng Z, Tkaczyk C, Cohen TS, Bardoel BW, Jorch SK, Deppermann C, Wardenburg JB, Davis RP, Jenne CN, Stover KC, Sellman BR, Kubes P, α-Toxin induces platelet aggregation and liver injury during Staphylococcus aureus sepsis, Cell Host Microbe 24, 271–284.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cognasse F, Nguyen KA, Damien P, McNicol A, Pozzetto B, Hamzeh-Cognasse H, Garraud O, The inflammatory role of platelets via their TLRs and Siglec receptors, Front. Immunol 6, 83 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anabel A-S, Eduardo P-C, Antonio H-CP, Carlos S-M, Juana N-M, Honorio T-A, Nicolás V-S, Roberto A-RS, Human platelets express Toll-like receptor 3 and respond to poly I:C, Hum. Immunol, 75, 1244–1251 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, Kahr WHA, Lindemann S, Seizer P, Yost CC, Zimmerman GA, Weyrich AS, Novel anti-bacterial activities of β-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation, PLoS Pathog. 7, e1002355 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeaman MR, Norman DC, Bayer AS, Staphylococcus aureus susceptibility to thrombin-induced platelet microbicidal protein is independent of platelet adherence and aggregation in vitro, Infect. Immun 60, 2368–2374 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolff M, Handtke S, Palankar R, Wesche J, Kohler TP, Kohler C, Gruel Y, Hammerschmidt S, Greinacher A, Activated platelets kill Staphylococcus aureus, but not Streptococcus pneumoniae—The role of FcγRIIa and platelet factor 4/heparin antibodies, J. Thromb. Haemost 18, 1459–1468 (2020). [DOI] [PubMed] [Google Scholar]
- 20.Powers ME, Becker REN, Sailer A, Turner JR, Wardenburg JB, Synergistic action of Staphylococcus aureus α-toxin on platelets and myeloid lineage cells contributes to lethal sepsis, Cell Host Microbe 17, 775–787 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzgerald JR, Loughman A, Keane F, Brennan M, Knobel M, Higgins J, Visai L, Speziale P, Cox D, Foster TJ, Fibronectin-binding proteins of Staphylococcus aureus mediate activation of human platelets via fibrinogen and fibronectin bridges to integrin GPIIb/IIIa and IgG binding to the FcgammaRIIa receptor, Mol. Microbiol 59, 212–230 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Loughman A, Fitzgerald JR, Brennan MP, Higgins J, Downer R, Cox D, Foster TJ, Roles for fibrinogen, immunoglobulin and complement in platelet activation promoted by Staphylococcus aureus clumping factor A, Mol. Microbiol 57, 804–818 (2005). [DOI] [PubMed] [Google Scholar]
- 23.ÓBrien L, Kerrigan SW, Kaw G, Hogan M, Penadés J, Litt D, Fitzgerald DJ, Foster TJ, Cox D, Multiple mechanisms for the activation of human platelet aggregation by Staphylococcus aureus: roles for the clumping factors ClfA and ClfB, the serine-aspartate repeat protein SdrE and protein A. Mol. Microbiol 44, 1033–1044 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Wuescher LM, Takashima A, Worth RG, A novel conditional platelet depletion mouse model reveals the importance of platelets in protection against Staphylococcus aureus bacteremia J. Thrombos. Haemostas 13, 303–313 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rose WE, Eickhoff JC, Shukla SK, Pantrangi M, Rooijakkers S, Cosgrove SE, Nizet V, Sakoulas G, Elevated serum interleukin-10 at time of hospital admission is predictive of mortality in patients with Staphylococcus aureus bacteremia, J. Infect. Dis 206, 1604–1611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gafter-Gvili A, Mansur N, Bivas A, Zemer-Wassercug N, Bishara J, Leibovici L, Paul M, Thrombocytopenia in Staphylococcus aureus bacteremia: risk factors and prognostic importance, Mayo Clinic Proc., 86, 389–396 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peters LL, Cheever EM, Ellis HR, Magnani PA, Svenson KL, Von Smith R, Bogue MA, Large-scale, high-throughput screening for coagulation and hematologic phenotypes in mice Physiol. Genom 11, 185–193 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Bhakdi S, Muhly M, Mannhardt U, Hugo F, Klapettek K, Mueller-Eckhardt C, Roka L, Staphylococcal α toxin promotes blood coagulation via attack on human platelets, J. Exp. Med, 168, 527–542 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Damman P, Woudstra P, Kuijt WJ, de Winter RJ, James SK, P2Y12 platelet inhibition in clinical practice, J. Thrombos. Thrombolys 33, 143–153 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winning J, Reichel J, Eisenhut Y, Hamacher J, Kohl M, Deigner HP, Claus RA, Bauer M, Lösche W, Anti-platelet drugs and outcome in severe infection: Clinical impact and underlying mechanisms, Platelets 20, 50–57 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Pepin J, Tremblay V, Bechard D, Rodier F, Walker C, Dufresne D, Lafontaine A, Li N, Lacroix C, Lanthier L, Chronic antiplatelet therapy and mortality among patients with infective endocarditis, Clin. Microbiol. Infect 15, 193–199 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Osthoff M, Sidler JA, Lakatos B, Frei R, Dangel M, Weisser M, Battegay M, Widmer AF, Low-dose acetylsalicylic acid treatment and impact on short-term mortality in Staphylococcus aureus bloodstream infection: a propensity score–matched cohort study, Crit. Care Med 44, 773 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Trauer J, Muhi S, McBryde ES, Al Harbi SA, Arabi YM, Boyle AJ, Cartin-Ceba R, Chen W, Chen Y-T, Falcone M, Gajic O, Godsell J, Gong MN, Kor D, Lösche W, McAuley DF, O’Neal HR Jr, Osthoff M, Otto GP, Sossdorf M, Tsai M-J, Valerio-Rojas JC, van der Poll T, Violi F, Ware L, Widmer AF, Wiewel MA, Winning J, Eisen DP, Quantifying the effects of prior acetyl-salicylic acid on sepsis-related deaths: an individual patient data meta-analysis using propensity matching, Crit. Care Med 45, 1871–1879 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winning J, Neumann J, Kohl M, Claus RA, Reinhart K, Bauer M, Lösche W, Antiplatelet drugs and outcome in mixed admissions to an intensive care unit, Crit. Care Med 38, 32–37 (2010). [DOI] [PubMed] [Google Scholar]
- 35.Hoffmeister KM, Falet H, Platelet clearance by the hepatic Ashwell-Morrell receptor: mechanisms and biological significance, Thrombos. Res, 141, S68–S72 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jansen AJG, Gerard Jansen AJ, Josefsson EC, Rumjantseva V, Liu QP, Falet H, Bergmeier W, Cifuni SM, Sackstein R, von Andrian UH, Wagner DD, Hartwig JH, Hoffmeister KM, Desialylation accelerates platelet clearance after refrigeration and initiates GPIbα metalloproteinase-mediated cleavage in mice, Blood 119, 1263–1273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang X-H, Wang Q-M, Zhang J-M, Feng F-E, Wang F-R, Chen H, Zhang Y-Y, Chen Y-H, Han W, Xu L-P, Liu K-Y, Huang X-J, Desialylation is associated with apoptosis and phagocytosis of platelets in patients with prolonged isolated thrombocytopenia after allo-HSCT, J. Hematol. Oncol, 8, 116 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rink TJ, Sanchez A, Hallam TJ, Diacylglycerol and phorbol ester stimulate secretion without raising cytoplasmic free calcium in human platelets, Nature 305, 317–319 (1983). [DOI] [PubMed] [Google Scholar]
- 39.Knight DE, von Grafenstein H, Athayde CM, Calcium-dependent and calcium-independent exocytosis, Trends Neurosci. 12, 451–458 (1989). [DOI] [PubMed] [Google Scholar]
- 40.Grewal PK, Uchiyama S, Ditto D, Varki N, Le DT, Nizet V, Marth JD, The Ashwell receptor mitigates the lethal coagulopathy of sepsis, Nat. Med 14, 648–655 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grewal PK, Aziz PV, Uchiyama S, Rubio GR, Lardone RD, Le D, Varki NM, Nizet V, Marth JD, Inducing host protection in pneumococcal sepsis by preactivation of the Ashwell-Morell receptor, Proc. Natl. Acad. Sci. U. S. A 110, 20218–20223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ellies LG, Ditto D, Levy GG, Wahrenbrock M, Ginsburg D, Varki A, Le DT, Marth JD, Sialyltransferase ST3Gal-IV operates as a dominant modifier of hemostasis by concealing asialoglycoprotein receptor ligands, Proc. Natl. Acad. Sci. U. S. A 99, 10042–10047 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li J, van der Wal DE, Zhu G, Xu M, Yougbare I, Ma L, Vadasz B, Carrim N, Grozovsky R, Ruan M, Zhu L, Zeng Q, Tao L, Zhai Z-M, Peng J, Hou M, Leytin V, Freedman J, Hoffmeister KM, Ni H, Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia, Nat. Commun 6 (2015), 6:7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ree IMC, Fustolo-Gunnink SF, Bekker V, Fijnvandraat KJ, Steggerda SJ, Lopriore E, Thrombocytopenia in neonatal sepsis: Incidence, severity and risk factors, PLoS One 12, e0185581 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Claushuis TAM, van Vught LA, Scicluna BP, Wiewel MA, Klein PM, Hoogendijk AJ, Ong DSY, Cremer OL, Horn J, Franitza M, Toliat MR, Nürnberg P, Zwinderman AH, Bonten MJ, Schultz MJ, van der Poll T, Thrombocytopenia is associated with a dysregulated host response in critically ill sepsis patients, Blood 127, 3062–3072 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Södergren AL, Svensson Holm A-CB, Ramström S, Lindström EG, Grenegård M, Öllinger K, Thrombin-induced lysosomal exocytosis in human platelets is dependent on secondary activation by ADP and regulated by endothelial-derived substances, Platelets 27, 86–92 (2016). [DOI] [PubMed] [Google Scholar]
- 47.Smith TH, Li JG, Dores MR, Trejo J, Protease-activated receptor-4 and purinergic receptor P2Y12 dimerize, co-internalize, and activate Akt signaling via endosomal recruitment of β-arrestin. J. Biol. CHem 292, 13867–13878 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng AG, McAdow M, Kim HK, Bae T, Missiakas DM, Schneewind O, Contribution of coagulases towards Staphylococcus aureus disease and protective immunity, PLoS Pathog. 6, e1001036 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trier DA, Gank KD, Kupferwasser D, Yount NY, French WJ, Michelson AD, Kupferwasser LI, Xiong YQ, Bayer AS, Yeaman MR, Platelet antistaphylococcal responses occur through P2X1 and P2Y12 receptor-induced activation and kinocidin release Infect. Immun 76, 5706–5713 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheung AL, Wolz C, Yeaman MR, Bayer AS, Insertional inactivation of a chromosomal locus that modulates expression of potential virulence determinants in Staphylococcus aureus,,\ J. Bacteriol 177, 3220–3226 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goel D, Ticagrelor: The first approved reversible oral antiplatelet agent Int. J. Appl. Basic Med. Res 3, 19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hata K, Koseki K, Yamaguchi K, Moriya S, Suzuki Y, Yingsakmongkon S, Hirai G, Sodeoka M, von Itzstein M, Miyagi T, Limited inhibitory effects of oseltamivir and zanamivir on human sialidases Antimicrob. Agents Chemother 52, 3484–3491 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jansen AJG, Peng J, Zhao H-G, Hou M, Ni H, Sialidase inhibition to increase platelet counts: A new treatment option for thrombocytopenia Am. J. Hematol 90, E94–E95 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Bigot P, Auffret M, Gautier S, Weinborn M, Ettahar N-K, Coupé P, Unexpected platelets elevation in a patient with idiopathic thrombocytopenia treated with oseltamivir for influenza infection Fundamen. & Clin. Pharmacol 30, 483–485 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Alioglu B, Tasar A, Ozen C, Selver B, Dallar Y, An experience of oseltamivir phosphate (Tamiflutm) in a pediatric patient with chronic idiopathic thrombocytopenic purpura: a case report Pathophys. Haemostas. Thrombos 37, 55–58 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Chertow DS, Memoli MJ, Bacterial coinfection in influenza, JAMA 309, 275 (2013). [DOI] [PubMed] [Google Scholar]
- 57.McDanel JS, Perencevich EN, Storm J, Diekema DJ, Herwaldt L, Kristie Johnson J, Winokur PL, Schweizer ML, Increased mortality rates associated with Staphylococcus aureus and influenza co-infection, Maryland and Iowa, USA, Emerg. Infect. Dis 22, 1253–1256 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Finelli L, Fiore A, Dhara R, Brammer L, Shay DK, Kamimoto L, Fry A, Hageman J, Gorwitz R, Bresee J, Uyeki T, Influenza-associated pediatric mortality in the United States: increase of Staphylococcus aureus coinfection, Pediatrics 122, 805–811 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Jansen AJG, Gerard Jansen AJ, Low HZ, van den Brand J, van Riel D, Osterhaus A, van der Vries E, Platelets can phagocytose influenza virus which may contribute to the occurrence of thrombocytopenia during influenza infection, Blood 128, 1358–1358 (2016). [Google Scholar]
- 60.Yang WH, Heithoff DM, Aziz PV, Haslund-Gourley B, Westman JS, Narisawa S, Pinkerton AB, Millán JL, Nizet V, Mahan MJ, Marth JD, Accelerated aging and clearance of host anti-inflammatory enzymes by discrete pathogens fuels sepsis, Cell Host Microbe 24, 500–513.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grozovsky R, Begonja AJ, Liu K, Visner G, Hartwig JH, Falet H, Hoffmeister KM, The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling, Nat. Med 21, 47–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sampedro GR, DeDent AC, Becker REN, Berube BJ, Gebhardt MJ, Cao H, Wardenburg JB, Targeting Staphylococcus aureus α-toxin as a novel approach to reduce severity of recurrent skin and soft-tissue infections, J. Infect. Dis 210, 1012–1018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bae T, Schneewind O, Allelic replacement in Staphylococcus aureus with inducible counter-selection, Plasmid 55, 58–63 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Potier M, Mameli L, Bélisle M, Dallaire L, Melançon SB, Fluorometric assay of neuraminidase with a sodium (4-methylumbelliferyl-α-d-N-acetylneuraminate) substrate, Anal. Biochem 94, 287–296 (1979). [DOI] [PubMed] [Google Scholar]
- 65.Jansen AJG, Gerard Jansen AJ, Josefsson EC, Rumjantseva V, Liu QP, Falet H, Bergmeier W, Cifuni SM, Sackstein R, von Andrian UH, Wagner DD, Hartwig JH, Hoffmeister KM, Desialylation accelerates platelet clearance after refrigeration and initiates GPIbα metalloproteinase-mediated cleavage in mice Blood 119, 1263–1273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanaji T, Vo M-N, Kanaji S, Zarpellon A, Shapiro R, Morodomi Y, Yuzuriha A, Eto K, Belani R, Do M-H, Yang X-L, Ruggeri ZM, Schimmel P, Tyrosyl-tRNA synthetase stimulates thrombopoietin-independent hematopoiesis accelerating recovery from thrombocytopenia, Proc. Natl. Acad. Sci. U. S. A 115, E8228–E8235 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Park EI, Mi Y, Unverzagt C, Gabius H-J, Baenziger JU, The asialoglycoprotein receptor clears glycoconjugates terminating with sialic acidα2,6GalNAc, Proc. Natl. Acad. Sci. U. S. A 102, 17125–17129 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ellies LG, Ditto D, Levy GG, Wahrenbrock M, Ginsburg D, Varki A, Le DT, Marth JD, Sialyltransferase ST3Gal-IV operates as a dominant modifier of hemostasis by concealing asialoglycoprotein receptor ligands, Proc. Natl. Acad. Sci. U. S. A 99, 10042–10047 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Materials and Methods
Fig. S1. Thrombocytopenia established by anti-CD41 antibody treatment of mice.
Fig. S2. Grouping of α-toxin expression of SA isolates from bacteremia patients.
Fig. S3. Generation of an isogenic SA ΔHla mutant.
Fig. S4. Exclusion of ticagrelor off-target effects on SA immune cell interactions and growth.
Fig. S5. Additional effects of SA or TICA on human platelet phenotypes in vitro.
Fig. S6. Effects of SA challenge on platelets and thrombopoiesis in vivo.
Fig. S7. Organ pathology in murine SA infection with or without ticagrelor treatment.
Fig. S8. Neu1 is predominantly detected on the platelet cell surface and is induced by SA exposure.
Fig. S9. SA α-toxin deletion mutant phenocopies protective effects of TICA, oseltamivir, and AMR loss or inhibition.
Fig. S10. Schematic illustration of proposed “toxin-platelet-AMR” pathway exploited by SA in the pathogenesis of bloodstream infection.
Data Availability Statement
All data associated with the current study are present in the paper or supplementary materials.