

Abstract
During metastasis cancer cells must adapt to survive loss of anchorage and evade anoikis. An important pro-survival adaptation is the ability of metastatic tumor cells to increase their antioxidant capacity and restore cellular redox balance. Although much is known about the transcriptional regulation of antioxidant enzymes in response to stress, how cells acutely adapt to alter antioxidant enzyme levels is less well understood. Using ovarian cancer cells as a model, we demonstrate that an increase in mitochondrial superoxide dismutase SOD2 protein expression is a very early event initiated in response to detachment, an important step during metastasis that has been associated with increased oxidative stress. SOD2 protein synthesis is rapidly induced within 0.5–2 h of matrix detachment, and polyribosome profiling demonstrates an increase in the number of ribosomes bound to SOD2 mRNA, indicating an increase in SOD2 mRNA translation in response to anchorage-independence. Mechanistically, we find that anchorage-independence induces cytosolic accumulation of the RNA binding protein HuR/ELAVL1 and promotes HuR binding to SOD2 mRNA. Using HuR siRNA-mediated knockdown, we show that the presence of HuR is necessary for the increase in SOD2 mRNA association with the heavy polyribosome fraction and consequent nascent SOD2 protein synthesis in anchorage-independence. Cellular detachment also activates the stress-response mitogen-activated kinase p38, which is necessary for HuR-SOD2 mRNA interactions and induction of SOD2 protein output. These findings illustrate a novel translational regulatory mechanism of SOD2 by which ovarian cancer cells rapidly increase their mitochondrial antioxidant capacity as an acute stress response to anchorage-independence.
Graphical abstract
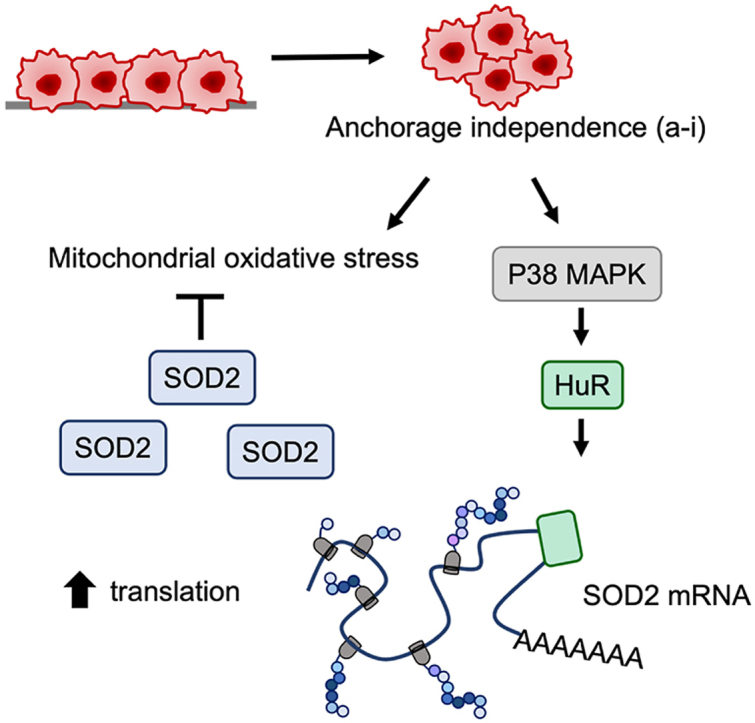
1. Introduction
In vivo studies have demonstrated that increased antioxidant enzyme expression and small molecule antioxidant treatments promote the metastatic spread of melanoma and breast cancer cells [1,2], suggesting that the maintenance of redox homeostasis is a key adaptation during metastasis. Manganese superoxide dismutase (SOD2) is an important mitochondrial antioxidant enzyme that resides in the mitochondrial matrix and is responsible for scavenging the majority of superoxide produced as a biproduct of respiration. SOD2 is often upregulated during tumor progression and its expression is important for successful metastasis of cancer cells [[3], [4], [5], [6], [7], [8]]. A key step during metastasis is a tumor cell's ability to survive in non-adherent conditions and to evade anchorage-independent cell death, known as anoikis. This process has been associated with an increased capacity of tumor cells to scavenge reactive oxygen species that are elevated in response to detachment [9,10]. We previously demonstrated that epithelial ovarian cancer cells increase their mitochondrial antioxidant capacity after matrix detachment, by upregulating the transcription and activity of the deacetylase sirtuin 3 (SIRT3), and its target protein SOD2 [6]. Both proteins conferred anoikis resistance and promoted transcoelomic spread of ovarian cancer cells in vivo [6].
Given the pivotal function of SOD2 in oxidant scavenging, SOD2 is susceptible to extensive stress-specific transcriptional regulation [7]. Nrf2 (encoded by NFE2L2), a major transcription factor responsive to oxidants, has been implicated in regulating increased SOD2 expression in tumor cells including breast and ovarian clear cell carcinomas [4,11]. SOD2 transcription can also be induced by the sirtuin-regulated transcription factor Foxo3A [12], and by NF-κB following matrix detachment of breast cancer cells [13]. Although much emphasis has been placed on the transcriptional mechanisms of SOD2 expression in different contexts, its regulation at the mRNA level remains less well established in tumor cells.
Posttranscriptional and translational regulatory mechanisms are crucial for fine-tuning of gene expression and enabling cells to rapidly adjust for protein abundance in response to specific cues. In particular, the interplay among mRNAs, miRNAs, and RNA-binding proteins has been implicated in the regulation of protein expression during cancer development and metastasis [[14], [15], [16]]. HuR (encoded by ELAVL1) is a well-known cancer-associated RNA-binding protein that has been implicated in the regulation of mRNAs that encode proteins involved in oncogenic signaling [[17], [18], [19]], anti-apoptotic mechanisms [20], cell cycle regulation [21,22], and chemoresistance [23]. By binding to the AU- and U-rich elements (AREs) in the 3’ UTR of target mRNAs, HuR exerts multiple functions including RNA splicing, regulation of mRNA stability and translation [24]. Importantly, HuR cytoplasmic translocation and mRNA binding is induced upon genotoxic or extracellular stress stimuli [21,25], which suggests that HuR-dependent alterations in expression may be a critical stress adaptation utilized by cancer cells. Analyses across different malignancies, including ovarian cancer, show that HuR expression and cytoplasmic accumulation correlate with advanced tumor stage and poor patient prognosis [[26], [27], [28], [29]].
A transcriptome-wide RNA-binding analysis identified multiple HuR binding sites in the 3’ UTR of SOD2 mRNA [30]. However, the functional consequences of these sites and potential regulatory role of HuR in SOD2 mRNA translation have not been investigated in cancer. In the present work, we show that SOD2 mRNA is a target of HuR binding and that the interaction of HuR with SOD2 mRNA is enhanced and required for rapid de novo SOD2 protein synthesis after matrix detachment in ovarian cancer cells with lower basal SOD2 expression. Our study provides evidence for a novel mechanism of rapid SOD2 regulation in response to acute stress associated with anchorage-independence, a key step during metastasis.
2. Results
2.1. SOD2 protein expression increases rapidly in response to anchorage-independence
Ovarian cancer cells adaptat to stress associated with matrix detachment and anchorage-independence during transcoelomic metastasis in the peritoneal cavity. Similar to previous reports we observed an increase in mitochondrial oxidative stress when cancer cells were placed in anchorage-independent conditions (Suppl Fig. 1A–C) [9,10]. However, we found that MitoSox oxidation was rapidly abrogated in OVCA433 and OVCAR10 cells within hours of culture in ultra-low attachment conditions, suggesting that ovarian cancer cells are able to acutely enhance their mitochondrial oxidant scavenging, likely prior to any transcriptional reprogramming (Suppl Fig. 1). The degree and level of recovery from mitochondrial oxidative stress in response to detachment was cell line dependent. OVCA433 cells experienced a more robust increase in MitoSox oxidation in response to detachment compared to OVCAR10 cells, suggesting that OVCAR10 cells are inherently more resistant to mitochondrial oxidant bursts in response to anchorage-independence. Even though a higher initial MitoSox oxidation was observed in OVCA433 cells, this rapidly declined within 4 h of detachment, suggesting that OVCA433 cells rely strongly on mechanisms that acutely increase mitochondrial oxidant scavenging in response to anchorage-independent stress.
Assessment of SOD2 expression demonstrated a rapid 2.7- and 2.5-fold increase in SOD2 protein levels within 0.5 and 2 h following detachment of OVCA433 and OVCAR10 cells, respectively (Fig. 1A). Treatment with the protein synthesis inhibitor cycloheximide demonstrated that these increases likely represent nascent SOD2 protein pools when cells are cultured in ultra-low attachment conditions (Fig. 1A). 35S-Met/Cys incorporation assays and immunoprecipitation of SOD2 demonstrated a 1.8-fold increase in 35S-Met/Cys incorporation into the SOD2 protein compared to attached conditions in OVCA433 (Fig. 1B, Suppl Fig. 2A–C). These changes were again abrogated by cycloheximide treatment, suggesting increased de novo SOD2 protein synthesis in short-term anchorage-independent conditions. A similar trend was observed in OVCAR10 cells, yet this did not reach significance (Fig. 1B). Although 35S-Met/Cys incorporation assays suggested a global increase in Methionine/Cysteine containing proteins following detachment (Suppl Fig. 2C), this was not observed using the methionine analog l-azidohomoalanine (AHA) as an alternate method to label newly synthesized proteins (Suppl Fig. 2D). Due to the limited number of methionine residues in SOD2, we were unable to use AHA to label nascent SOD2 proteins.
Fig. 1.

SOD2 protein expression increases rapidly in response to anchorage-independence. A. Total SOD2 protein levels were assessed by immunoblotting in response to culture in anchorage-independent conditions and protein synthesis inhibited by cycloheximide (CHX, 20 μg/mL; n = 4, one-way ANOVA, P < 0.0001, Tukey's multiple comparison test *P < 0.05; **P < 0.01). B. 35S-Met/Cys incorporation assay followed by SOD2 IP (Suppl Fig. 2A&B), demonstrates increased 35S-Met/Cys incorporation into SOD2 under anchorage-independence compared to attached cells, which is abrogated in the presence of cycloheximide (n = 4, one-way ANOVA, OVCA433 P < 0.0001, OVCAR10 P = 0.0057, Tukey's multiple comparison test **P < 0.01; ****P < 0.0001). C. The cytosolic SOD2 protein pool increases rapidly in response to anchorage-independence (a–i), compared to attached culture conditions (A). Cells were maintained for indicated times in ULA plates and SOD2 protein expression assessed following cellular fractionation and immunoblotting. Fold change in SOD2 cytosolic protein expression in response to anchorage-independent (a–i) culture was quantified using densitometry, normalized to β-tubulin loading control and expressed relative to attached (A) culture conditions (n = 4, one-way ANOVA, OVCA433 P = 0.0015, OVCAR10 P = 0.0744, Dunnett's multiple comparison test *P < 0.05; **P < 0.01). D. Fold change in SOD2 mRNA in response to short term anchorage-independent culture was assessed using semi-quantitative real time RT-PCR (n = 3–4, one-way ANOVA, OVCA433 P = 0.0069, OVCAR10 P = 0.2946, Dunnett's multiple comparison test *P < 0.05; **P < 0.01).
Thus, to further elucidate whether SOD2 protein expression is increased in response to anchorage-independence, we assessed changes in SOD2 levels within the cytosolic fraction , representing the newly synthesized protein pool of SOD2. Subcellular fractionation demonstrated an average 4.7-fold increase in OVCA433 cytosolic SOD2 expression after 0.5 h of detachment compared to attached cells, while a more nuanced maximal 1.5-fold increase was observed after 2 h in anchorage-independent conditions in OVCAR10 cells (Fig. 1C). Concomitant with the observations that OVCAR10 cells experience less MitoSox oxidation in anchorage-independent conditions, these cells also demonstrated higher basal cytosolic SOD2 protein levels and higher levels of mitochondrial SOD2 compared to OVCA433 in attached conditions (Fig. 1C, Suppl Fig 2D), suggesting that due to these high basal expression levels an increase in SOD2 protein synthesis in response to detachment may not be as critical to this cell line as compared to OVCA433 [5]. Increases in SOD2 mRNA levels trailed the surges in SOD2 protein expression in OVCA433 cells, suggesting that the rapid rise in SOD2 protein levels following detachment is likely independent of increases in transcription in this cell line (Fig. 1D).
To further confirm that the increase in SOD2 expression is due to de novo protein synthesis in OVCA433 cells, ribosome-mediated mRNA translation was assessed using polyribosome profiling (Fig. 2). Following centrifugation, sucrose gradients were separated into four fractions and RNA was isolated from each fraction. Fraction 1 contains mRNAs not associated with ribosomes, fraction 2 contains mRNAs associated with one or two ribosomes, fraction 3 contains mRNAs associated with 3–6 ribosomes (referred to hereafter as ‘light polysomes’), and fraction 4 contains mRNAs associated with >6 ribosomes (referred to as ‘heavy polysomes’; Fig. 2A). In attached conditions, SOD2 mRNA was primarily found in fraction 3 (Fig. 2B&C), suggesting that SOD2 is translated at a constitutive level in this basal condition, which is evident by ready detection of SOD2 protein by western blotting. In anchorage-independent conditions, the relative proportion of SOD2 mRNA shifted to fractions 3 and 4. In particular, anchorage-independent cells showed a significant shift towards an enrichment of SOD2 mRNA in the heavy polyribosome fraction 4 (Fig. 2B&C), demonstrating a larger number of ribosomal units associated with SOD2 mRNA and increased translation efficiency of SOD2 mRNA in anchorage-independent conditions. As a point of comparison, mRNA encoding the key nutrient stress response protein ATF4 was primarily found in the mono/subpolysomal fraction 2 in attached conditions and a larger proportion of ATF4 could be detected in fraction 4 in response to anchorage-independence (Fig. 2B&C), as did PTGS2/COX2 mRNA, a known target of the stress-response RNA binding protein HuR (ELAVL1; Fig. 5B & Suppl Fig 4D), as discussed further below [31].
Fig. 2.

Polysome profiling demonstrates increased SOD2 mRNA translation efficiency in anchorage-independence. A. Polyribosome profiling was carried out after OVCA433 cells were cultured in attached (A) and anchorage-independent (a–i) conditions (0.5 h) and analyzed following sucrose density gradient centrifugation. Four fractions were collected as indicated, RNA extracted and equal quantities of RNA reverse transcribed to assessed for presence of mRNAs in panel B. B. Representative images of SOD2 and ATF4 RT-PCR from RNA isolated from each polysomal fraction demonstrates an increase in the percentage of mRNA in the heavy polysomal fraction 4 in response to anchorage-independence. C. Quantification of relative SOD2 and ATF4 mRNA levels in each fraction demonstrates increased proportion of SOD2 in fraction 4 following culture in anchorage-independent conditions (n = 3; t-test, *P < 0.05; ***P < 0.001).
Fig. 5.

HuR enhancesSOD2mRNA translation in response to anchorage-independence. A. HuR/ELAVL1 knock-down abrogates increases in cytosolic SOD2 expression in short term anchorage-independence (a-i, OVCA433 0.5 h; OVCAR10 2 h) compared to attached cultures (A; n = 3–4, one-way ANOVA, OVCA433 P = 0.012, OVCAR10 P = 0.0001; Tukey's multiple comparison test *P < 0.05, **P < 0.01, ****P < 0.0001; quantification of HuR knockdown Suppl Fig. 4A). B. Polysome profiling of OVCA433 cells cultured in attached (A) and anchorage-independent (a-i, 0.5 h) conditions following siRNA-mediated HuR/ELAVL1 knockdown demonstrates that HuR loss abrogates a shift of SOD2 and PTGS2 mRNA into fraction 4 in response to anchorage-independence (a–i). Representative images of RT-PCR from polyribosome fractions shown (polysome plots Suppl Fig. 4C). C. Quantification of relative SOD2 mRNA levels in each fraction demonstrates HuR/ELAVL1 knock-down prevents increase of SOD2 in fraction 4 following culture in anchorage-independent conditions (n = 3; t-test, *P < 0.05; quantification of PTGS2 and GAPDH in Suppl Fig. 4D). D. SOD2-3′UTR-pmirGLO-driven firefly luciferase (FLuc) activity corrected for renilla luciferase standard (RLuc) was assessed in OVCA433 and OVCAR10 cells for SOD2-3′UTR Fragment 1 and Fragment 3 respectively. Fragment 1, which contains an HuR site closest to the CDS of SOD2 significantly increase luciferase reporter activity of pmirGLO in anchorage-independent conditions, and this was abrogated following HuR knock-down, n = 3, one way ANOVA P = 0.0021, Tukey's post test: *P < 0.05, **P < 0.01).
2.2. HuR accumulates in the cytosol and binds SOD2 mRNA in response to anchorage-independence
Regulation of gene expression at the translational level is mediated by the interplay between mRNAs and RNA binding proteins. HuR (encoded by the gene ELAVL1) is a major RNA binding protein that has been implicated with alternative splicing, mRNA stability, and translation during stress conditions [21,25,32]. HuR recognizes and binds to AU-/U-rich elements in target mRNA transcripts. Analysis of HuR RNA binding by screening of publicly available RNA immunoprecipitation sequencing (RIP-seq; ENCODE: ENCSR000CWW, ENCSR000CWZ) [33,34] and photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP; GSE29943) [30] transcriptome-wide data sets revealed that the SOD2 mRNA contains multiple binding sites for HuR within 3.5 kb downstream of the STOP codon in the 3′ UTR (Fig. 3A, Suppl Fig. 3A). While the 5′ UTR of SOD2 is less than 75 bp in length, the complete SOD2 3′ UTR spans 13,424 bp (Fig. 3A, Variant 1: NM_000636). SOD2 transcripts with variable 3′ UTR lengths have previously been reported (Suppl Fig 3A) [53,54]. Using RT-PCR we confirmed that OVCA433 and OVCAR10 cells express the longer 4.2 kb 3’ UTR containing the majority of HuR sites identified (Suppl Fig. 3B).
Fig. 3.

HuR accumulates in the cytosol and bindsSOD2mRNA in response to anchorage-independence. A. HuR/ELAVL1 binding profiles on the SOD2 mRNA was assessed using ENCODE RIP-seq data sets ENCSR000CWW and ENCSR000CWZ, and PAR-CLIP data set GSE29943. B. HuR accumulates in the cytosol in response to anchorage-independence (n = 4, one-way ANOVA, OVCA433 P < 0.0001, OVCAR10 P = 0.0248, Dunnett's multiple comparison test **P < 0.01; ***P < 0.001). C. Anchorage-independence induces HuR binding to SOD2 and PTGS2 mRNA, as assessed by Ribonucleoprotein Immunoprecipitation and SOD2/PTGS2 RT-PCR following OVCA433 culture in attached or anchorage-independent conditions (a-i, OVCA433: 0.5 h; OVCAR10: 2 h).
To examine if HuR regulates SOD2 protein expression in response to anchorage-independence, cytosolic translocation of HuR in response to culture in ULA plates was first determined. Concurrent with the increases in SOD2 protein expression, HuR cytosolic protein levels significantly increased 4-fold in OVCA433 within 0.5 h of anchorage-independence and 3-fold within 2 h in OVCAR10 cells (Fig. 3B). We next investigated if HuR binds to SOD2 mRNA in anchorage-independent conditions using formaldehyde-mediated crosslinking and ribonucleoprotein immunoprecipitation to capture the HuR-bound mRNAs (Fig. 3C, Supp Fig 3C). SOD2 mRNA was more readily detected by RT-PCR in HuR immunoprecipitates from OVCA433 and OVCAR10 cells cultured in anchorage-independence than attached conditions, indicating that matrix detachment may induce the binding of HuR to SOD2 mRNA (Fig. 3C). The mRNA of PTGS2, also known as COX2, is a previously described HuR target [31]. Using PTGS2 mRNA as a positive control, HuR was shown to associate with PTGS2 mRNA in ribonucleoprotein immunoprecipitation assays in OVCA433 cells (Fig. 3C). PTGS2 de novo protein synthesis was also increased in anchorage-independent conditions in OVCA433 cells (Suppl Fig 2D). OVCAR10 cells did not display HuR-PTGS2 mRNA interaction, and it should be noted that this cell line expressed relatively little PTGS2 mRNA (see total RNA input, Fig. 3C).
Since HuR binds to SOD2 mRNA shortly after matrix detachment, we investigated the functional consequences of the HuR-SOD2 mRNA interaction using siRNA mediated knockdown of HuR/ELAVL1. An established function of HuR as a stress response RNA binding protein is its role in mRNA stabilization within the cytosol [20,35]. To determine if HuR has an effect on SOD2 mRNA stability, we treated ovarian cancer cells with the transcription inhibitor actinomycin D. Compared to attached conditions, anchorage-independence did not significantly alter SOD2 mRNA stability in OVCA433 cells (Fig. 4A), while decreased SOD2 mRNA stability in anchorage-independence was observed in OVCAR10 cells compared to attached conditions (Fig. 4B, two-way ANOVA, P = 0.0104), indicating that these cells differ in mechanisms regulating SOD2 mRNA stability. However, HuR knockdown did not significantly alter SOD2 mRNA levels in response to actinomycin D treatment in anchorage-independent or attached culture conditions (Fig. 4), suggesting that HuR binding has a minimal impact on SOD2 mRNA stability.
Fig. 4.

HuR knock-down does not affectSOD2mRNA stability in attached or anchorage-independent conditions. mRNA stability was determined by actinomycin D treatment (n = 4; two-way ANOVA: ns). HuR knock-down was assessed by semi quantitative real time RT-PCR (t-test, ****P < 0.0001). A: OVCA433 B: OVCAR10.
2.3. HuR enhances SOD2 mRNA translation under anchorage-independence
We next tested if HuR is necessary for enhanced SOD2 mRNA translation in anchorage-independence. The matrix detachment-induced increases in SOD2 cytosolic protein levels were significantly abrogated when HuR expression was downregulated using siRNA-mediated HuR (ELAVL1) knockdown (Fig. 5A, Suppl Fig 4A), demonstrating a potential involvement of HuR in SOD2 translation. Similarly, a loss of HuR expression inhibited the increase in PTGS2 protein levels in response to OVCA433 culture in anchorage-independence (Suppl Fig. 4B-), while PTSG2 expression was not detected to appreciable levels in OVCAR10 cells, correlating with the lack of observed PTSG2 mRNA binding to HuR in these cells (Fig. 3C).
To further demonstrate that increased SOD2 protein synthesis is HuR-dependent in anchorage-independent cells, polyribosome profiling following siRNA mediated HuR knock-down was carried out (Fig. 5B). As expected from Fig. 2, OVCA433 cells transfected with a scramble control siRNA showed a shift of SOD2 mRNA towards the heavy polyribosome fraction (fraction 4) in anchorage-independence (Fig. 5B&C, Suppl Fig 4C). This shift of SOD2 mRNA into fraction 4 could no longer be observed following HuR knockdown (Fig. 5B). An HuR-dependent shift into the heavy polysome was similarly observed for PTGS2 (Fig. 5B, Suppl Fig 4D). There was no difference in SOD2 mRNA abundance in the subpolysome fractions (fractions 1 & 2) following HuR knockdown, but rather a shift of relative SOD2 and PTSG2 mRNA levels from fraction 4 to fraction 3. This potentially indicates that a loss of HuR does not lead to a complete ablation of SOD2 protein synthesis, but rather that HuR primarily functions to boost SOD2 mRNA translation in anchorage-independent conditions.
In addition, we tested if predicted HuR binding sites on the SOD2 transcript influence 3′UTR-driven translation in anchorage-independence using a translation reporter assay. To first determine which regions of the SOD2 3′UTR contribute to SOD2 translation in anchorage-independence, 6 fragments of the 3′UTR were cloned into the 3′ end of the pmirGLO Dual-Luciferase Expression vector (Suppl Fig 5A). Following normalization to Renilla luciferase, we observed that the first HuR binding site following the SOD2 coding sequence (CDS), represented as SOD2 3′UTR pmirGLO fragment 1 (F1), had the highest luciferase activity in anchorage-independent conditions in OVCA433 cells (Suppl Fig 5A). OVCAR10 exhibited the highest expression at the third ENCODE RIP-seq/PAR-CLIP overlapping HuR binding region, denoted as SOD2 3′UTR pmirGLO F3 (Suppl Fig. 5A). To determine the direct effects of HuR on the activity of the SOD2-3′UTR pmirGLO constructs, we assessed luciferase activity of the SOD2-3′UTR pmirGLO constructs with highest activity in their respective cell lines following HuR knockdown (Fig. 5D, Suppl Fig 5B). OVCA433 cells exhibited HuR-dependent increases in luciferase activity driven by SOD2 3′UTR Fragment 1 in anchorage-independent conditions. Fragment 3, which contains a predicted HuR binding site located ∼2500bp downstream of the STOP codon of the SOD2 CDS, displayed highest luciferase activity in OVCAR10 cells (Suppl Fig. 5A and 5D). Although there was a slight decrease in reporter activity in both attached and anchorage-independent conditions, HuR/ELAVL1 knock-down did not significantly affect the activity of fragment 3 in OVCAR10 cells (Fig. 5D, Suppl Fig 5B).
2.4. Inhibition of p38 MAPK activation in response to anchorage-independence abrogates increases in SOD2 protein expression and HuR-SOD2 mRNA binding
HuR can be activated in response to cellular stress via the p38 MAPK kinase signaling pathway [36,37]. p38 MAP kinase signaling is frequently activated and uncoupled from pro-apoptotic pathways in cancer cells to ensure cell survival under stress conditions and during metastatic progression [[38], [39], [40]]. An increase in p38 MAPK phosphorylation was previously reported in ovarian cancer cell lines cultured in long-term anchorage-independence (24–48 h) [41]. We were able to show that short-term anchorage-independence (0.5–2 h) was sufficient to also increase p38 MAPK phosphorylation in OVCA433 and OVCAR10 cell lines (Fig. 6A). To determine if the p38 MAPK pathway is involved in the observed increases in cytosolic SOD2 protein expression during this time, cells were treated with the p38 MAPK inhibitor, SB203580. SB203580 inhibited the phosphorylation of the p38 target MAPKAPK2 and abrogated the increases in SOD2 protein expression observed in OVCA433 and partially reversed SOD2 increases in OVCAR10 cells under anchorage-independent conditions (Fig. 6B). In addition, the formation of the HuR-SOD2 mRNA complex was monitored in the presence of p38 MAPK inhibition. Similar to Fig. 3, anchorage-independent conditions increased SOD2 and control PTGS2 mRNA binding to HuR, while SB203580 disrupted these interactions (Fig. 6C). The above demonstrates a link between p38 MAPK signaling, HuR binding to the SOD2 mRNA and SOD2 expression in response to cellular detachment. p38 MAPK has previously been shown to phosphorylate Thr118 of HuR [25,42]. In the absence of a commercially available phosphoThr118 HuR specific antibody, we were unable to successfully demonstrate that anchorage-independence or p38 MAPK inhibition influences phosphorylation of HuR using HuR IP and a pan phosphorylated-Thr antibody (data not shown). In OVCAR10 cells, p38 MAPK inhibition resulted in slight decreases in cytosolic HuR accumulation in response to anchorage-independence, while this could not be consistently observed in OVCA433 cells (Fig. 6D). Thus, the above data suggest that p38 signaling primarily regulates HuR SOD2 mRNA binding rather than HuR cellular localization in OVCA433 cells.
Fig. 6.

Inhibition of p38 MAPK abrogates increases in SOD2 protein expression and HuR-SOD2mRNA binding in response to anchorage-independence. A. p38 MAPK (Thr180/Tyr182) phosphorylation is induced in response to culture in anchorage-independent culture conditions (a-i OVCA433 0.5 h, OVCAR10 2 h; n = 4, T-test, **P < 0.01, ****P < 0.0001). B. p38 MAPK inhibition abrogates a-i induced increases in SOD2 expression (n = 3, one-way ANOVA P < 0.0001, Tukey's multiple comparison test **P < 0.01, ****P < 0.0001). C. p38 MAPK inhibition abrogates HuR binding to SOD2 and PTGS2 mRNA in anchorage-independence, as assessed by RNA immunoprecipitation. D. Effects of p38 MAPK inhibition on cytosolic HuR levels (n = 4–5, one-way ANOVA, OVCA433 P = 0.0053, OVCAR10 P = 0.0221, Tukey's multiple comparison test **P < 0.01, ****P < 0.0001).
3. Discussion
Recent studies have highlighted that tumor cells need an adequate antioxidant system to deal with intrinsic and extrinsic increases in ROS associated with metastatic progression [1,2,6]. Tumor cells must therefore readily adapt to increase their antioxidant capacity at the transcriptional and post-transcriptional levels. In line with these findings, in previous work we showed that SIRT3-mediated deacetylation of SOD2 drives transcoelomic metastasis by increasing mitochondrial antioxidant capacity in anchorage-independent ovarian cancer cells [6]. We previously demonstrated that certain ovarian cancer subtypes and cell lines, including clear cell carcinomas basally express high levels of SOD2 transcript and protein, while some high grade serous tumor cell lines are able to further upregulate their SOD2 activity in response to anchorage-independence via the deacetylase SIRT3 [5,6]. Thus, even under lower basal SOD2 expression cells are able to rapidly increase SOD2 in response to stress associated with anchorage-independence. In the present work we demonstrate that SOD2 translation represents an additional acute adaptation to elevate SOD2 levels in ovarian cancer cell lines in response to anchorage-independence (Fig. 1, Fig. 2). The present work demonstrates that translational control of SOD2 occurs during early-stage anchorage-independence as a consequence of the p38 MAPK-HuR axis, which promotes SOD2 mRNA translation for rapid protein synthesis (Fig. 3, Fig. 5, Fig. 6). Our work also demonstrates anchorage-independence as a novel stress cue which triggers HuR activity as a regulator of mRNA translation. It remains to be determined if cellular oxidants or changes in signaling related to cellular detachment are the major activators of the p38 MAPK-HuR axis [[38], [39], [40]]. OVCA433 cells which initially experience a larger surge in mitoSox oxidation in response to anchorage-independence (Suppl Fig 1) are able to rapidly recover from this and display a more robust and rapid HuR-dependent increase in SOD2 translation under these conditions than OVCAR10 cells. With higher basal SOD2 levels OVCAR10 cells also display less mitochondrial oxidative stress in response to detachment and therefore additional SOD2 protein synthesis may not be a critical requirement for this cell line. These data demonstrate that some cancer cells may be more dependent on translational regulation of antioxidant enzymes. While we have previously demonstrated that SOD2 knock-down decreases transcoelomic spread in mouse IP xenografts [6], the need for acute SOD2 translational regulation needs to be further tested in vivo and in patient-ascites derived cells. It needs to be determined if clonal cell populations that display low basal SOD2 expression are more sensitive to anoikis during metastatic spread when SOD2 translational regulation is inhibited. Moreover, these models will better represent stress encountered by cells during metastasis, such as hypoxia and exogenous oxidants in the tumor environment that have the potential to further drive activation of the p38-HuR-SOD2 axis.
Aberrant HuR expression has been reported in several malignancies including ovarian cancer [[26], [27], [28]]. HuR's pro-tumorigenic function involves selective mRNA binding, mRNA stabilization and/or increased translation of target mRNAs. Previously identified HuR targets include mRNAs that promote carcinogenesis, like PTGS2, encode pro-survival and anti-apoptotic proteins, such as Bcl-2, and proteins that support invasion and metastasis, and angiogenic factors, such as VEGF [20,22,36,[43], [44], [45], [46]]. HuR promotes glioma cell growth in anchorage-independence and decreases apoptosis by increasing Bcl-2 mRNA stability and protein output [20]. Moreover, HuR regulation can interplay with miRNAs to further fine tune gene expression in cancer, as has been demonstrated in ovarian cancer with miR-200c [47]. The growing repertoire of cancer-related mRNAs regulated by HuR highlights a critical role of this RNA binding protein in cancer etiology. Our data identify SOD2, an important antioxidant enzyme for the maintenance of mitochondrial redox homeostasis, as a novel HuR target during early-stages of anchorage-independence.
HuR is a predominantly nuclear protein which translocates to the cytoplasm upon extrinsic or intrinsic stimuli and stress signals. Depending on the location of target HuR amino acid residues, posttranslational modifications of HuR by different signaling pathways have been shown to affect its RNA binding affinity, nucleo-cytoplasmic shuttling, and HuR protein stability [24]. Among different kinases activated during stress, p38 MAPK-dependent phosphorylation on Thr118 induces cytoplasmic accumulation of HuR and increased p21 mRNA binding after exposure to ionizing radiation [25] and enhanced mRNA binding upon IL-1β treatment [42]. Consistent with these previous findings, we found that stress associated with matrix detachment activated p38 MAPK (Fig. 6). Importantly, activation of the p38 MAPK pathway increased SOD2 cytosolic protein expression under anchorage-independence and we found that the association of HuR with SOD2 mRNA was also p38 MAPK-dependent (Fig. 6). It remains to be determined whether HuR is phosphorylated on Thr118 in anchorage-independent cells, or if p38 MAPK indirectly activates HuR to bind SOD2 mRNA. Although p38 has previously been implicated in cytosolic shuttling of HuR in response to stress [36,37,48], cytosolic HuR accumulation was not greatly affected by the p38 MAPK inhibition in anchorage-independence, unlike SOD2 mRNA binding (Fig. 6). This raises the possibility that additional stress signaling pathways could contribute to the HuR nucleo-cytoplasmic shuttling observed following matrix detachment, and points to the previously reported multifaceted and context-dependent regulation of HuR. For example, post-translational modifications of residues within HuR's RNA recognition motifs lead primarily to changes in HuR RNA binding, while phosphorylation of the hinge region affects nuclear to cytoplasmic shuttling [49,50]. Threonine 118, the target of p38 signaling, is located in one of the RNA recognition motifs [25], which may explain why the activation of p38 signaling in anchorage-independence primary affects HuR Sod2 mRNA binding. The exploration of additional HuR mRNA targets following matrix detachment and mechanisms linking the p38 MAPK pathway to HuR activation require further investigation to unveil novel stress response translational pathways under conditions of anchorage-independence.
While the transcriptional regulation of antioxidant enzymes has been studied widely in the context of antioxidant response elements and stress response transcription factors, such as Nrf2, fewer studies have focused on their translational regulation. In earlier work, the presence of an un-identified redox-sensitive SOD2 mRNA binding protein was reported in rat lung extracts [51]. Further analysis identified that RNA binding occurred at a cis-regulatory region located 111 bp downstream of the stop codon in the rat SOD2 mRNA [52]. The 3′ UTR of human SOD2 mRNA shares ∼75% homology with the rat 3′ UTR. Based on sequence comparison, the previously identified rat RNA protein binding region partially overlaps with the first HuR binding sites from PAR-CLIP analysis (Fig. 3A) [30,52], suggesting that this region could be an important RNA regulatory domain of SOD2 mRNA. This also represents the SOD2 3′UTR fragment with highest, translation reporter activity in anchorage independent conditions in OVCA433 cells, activity of which was HuR dependent (Fig. 5D; Supp Fig 5).
Among the different SOD2 mRNA splice variants, different 3′ UTRs have been reported (Supp Fig 3A). Variant 2 (NM_001024465) has a short 3′ UTR composed of a spliced region that excludes the majority of the HuR sites identified. Variant 1 (NM_000636) has been annotated to contain a 13.4 kb 3′ UTR. However, past studies have shown that the two most common SOD2 transcripts contain either a short 240 bp or a 3,439 bp segment of this 3′ UTR, which arise from use of a proximal and distal polyadenylation site, respectively (Supp Fig 3A) [53,54]. Interestingly, Chaudhuri et al. reported that the expression of these two SOD2 transcripts is altered between quiescent and proliferating cells, with the shorter transcript being associated with quiescence and increased protein expression [53]. Moreover, radiation increased levels of the shorter SOD2 transcript levels of the 1.5 kb MnSOD transcript, with expression of the longer form remaining unaltered [53]. The mechanisms for this radiation-induced increase in the short 3′ UTR transcript remain unclear. We verified that ovarian cancer cells used in the present work express the transcript containing the longer 3′ UTR (Suppl Fig 3B), and that HuR can induce translation from the HuR site immediately adjacent to the SOD2 CDS in anchorage-independent conditions (Fig. 5D). Further studies are needed to determine if and how these alternate 3′ UTR SOD2 transcripts are processed in response to different sources of stress, and how their transcription co-operates with translational regulation through the activation of cell-specific RNA binding proteins, as well as the interplay with non-coding RNAs, such as miRNAs. A screen for miRNA binding reveals that the SOD2 mRNA contains potential binding sites for miRNAs throughout the length of the 3′ UTR. While most are located toward the far upstream region, several overlap with identified HuR binding sites. Several studies have investigated the role of miRNAs in regulating SOD2 expression and miRNAs identified that either positively or negatively regulate SOD2 levels in cancer (reviewed in Ref. [7]). Given that we see reduced pmirGLO activity with some 3′UTR fragments, suggests that miRNAs also play a role in fine tuning SOD2 expression in ovarian cancer cells (Suppl 5A, F5 & F6). Although we did not specifically observe differences between attached and anchorage-independent conditions in this context, it remains to be investigated if changes in miRNA binding further influence the regulation of SOD2 at the posttranscriptional level in anchorage-independence, and if this interplays with the regulation by HuR.
In conclusion, we show for the first time that SOD2 mRNA is an HuR target in anchorage-independent ovarian cancer cells. The present findings uncover a novel stress response mechanism at the level of translation that enables tumor cells to rapidly increase the expression of SOD2 in response to anchorage-independence.
4. Materials and methods
4.1. Cell culture and reagents
OVCA433 and OVCAR10 cells were provided by Dr. Susan K. Murphy (Duke University) and Dr. Katherine Aird (Penn State University & University of Pittsburgh), respectively. OVCA433 and OVCAR10 were grown in RPMI1640 supplemented with 10% FBS at 37 °C with 5% CO2. STR profiling is carried out routinely to validate cell identity, which revealed at the commencement of this work that OVCAR10 cells share the same STR profile as NIH-OVCAR3 cells. It is unclear if the OVCAR10 cell line was initially derived from the same patient as OVCAR3, or if OVCAR10 cells represent a sub-line derived from OVCAR3 cells. The protein synthesis inhibitor cycloheximide (Sigma) was added at a concentration of 20 μg/mL in fully supplemented growth media. For mRNA stability assays, actinomycin D (Sigma) was added at 10 μg/mL. The p38 MAPK inhibitor SB203580 was used at a final concentration of 20 μM.
4.2. Cell culture in adherent and ultra-low attachment (ULA) conditions
For attached conditions, cells were plated in 150-mm dishes and grown to ∼80% confluency. For anchorage-independent cell culture, cells were trypsinized and seeded at a density (300,000 cells/2 mL media/well) in 6-well ULA (ultra-low attachment) plates (Corning: 3471) and collected at different time points for downstream analyses.
4.3. siRNA-mediated HuR/ELAVL1 knock-down
Cells were transfected with scramble non-targeting SMARTpool control (Dharmacon: D-001810-10-05) or HuR (ELAVL1)-specific SMARTpool siRNA oligonucleotides (Dharmacon: L-003773-00-0005) using Lipofectamine RNAiMAX (Invitrogen), and knock-down confirmed by western blotting.
4.4. Subcellular fractionation
Cells in adherent and ULA plates were collected and the cell pellets washed with ice-cold PBS. The cell pellets were processed as described in Sugiura et al. [55]. Briefly, cells were centrifuged and resuspended in 200–500 μl of ice-cold homogenization buffer (10 mM HEPES pH 7.4, 220 mM mannitol, 70 mM sucrose, Roche protease and phosphatase inhibitor cocktails). The lysates were homogenized by several passages through 27-G needles. Lysates were centrifuged at 800 g for 10 min, followed by centrifugation of the supernatants at 2,500 g for 15 min at 4 °C. The mitochondrial pellets were resuspended in homogenization buffer and the supernatants were centrifuged at 100,000g for 1 h at 4 °C using a Beckman Coulter Optima MAX Ultracentrifuge. Post-centrifugation supernatants containing cytosolic fractions were transferred to new tubes and used for immunoblotting.
4.5. Immunoblotting
Protein concentrations were measured using the Pierce BCA protein assay kit. An equal amount of protein lysates was loaded onto 4–20% SDS-PAGE gels. Following electrophoresis, proteins were transferred to PVDF membranes. For detection of proteins, the membranes were incubated with the following antibodies overnight at 4 °C: SOD2 (A-2, Santa Cruz: sc-133134, 1:500 dilution); β-tubulin (9F3, Cell Signaling Technology: 2128, 1:1,000 dilution), ATP5A (Abcam: ab14748, 1:1000 dilution), β-actin (Thermo: AM4302, 1:10,000 dilution), HuR/ELAVL1 (3A2, Santa Cruz: sc-5261, 1:500 dilution), Phospho-p38 MAPK (Thr180/Tyr182, Cell Signaling Technology: 9211, 1:1000 dilution), p38 MAPK (A-12, Santa Cruz Biotechnology: sc-7972, 1:1000 dilution), MAPKAPK-2 (Cell signaling technology: 3042, 1:1000 dilution), Cox2/PTGS2 (D5H5, Cell Signaling: 12282, 1:1000 dilution). The blots were developed using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo: 34096) after incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies (Amersham Biosciences) for 1 h at RT.
4.6. Immunoprecipitation (IP)
1–1.5 mg of cell lysates were pre-cleared by incubating with 2 μg normal rabbit IgG (Cell Signaling Technology: 2729S) or normal mouse IgG (Millipore: 12–371) on a rotator for 1 h at 4 °C followed by an additional 1 h incubation with protein A- (Thermo: 20333) or protein G- agarose beads (50 μL; Thermo: 20399) at 4 °C. Following centrifugation at 3000g for 10 min supernatants were transferred to clean tubes and incubated with either IgG or primary antibodies overnight at 4 °C. 50 μL of agarose beads were added to the lysates for 1–2 h at 4 °C and the antibody-bead complexes were washed three times in IP lysis buffer and further processed for downstream assays.
4.7. 35S protein radiolabeling
Cells in adherent and ULA plates were treated with EasyTag Express35S Protein Labeling Mix (PerkinElmer: NEG772), using 40 μl 35S (440 μCi) per 20 mL media in 150-mm dish, 4 μl 35S (44 μCi)/2 mL media/well in ULA plates, according to a protocol adapted from Gallagher et al. (Gallagher et al., 2008). Following 2 h incubation in the presence of 35S-l-methionine and 35S-l-cysteine (35S-Met/Cys), cells were collected, washed with ice-cold PBS, and harvested using RIPA buffer supplemented with protease and phosphatase inhibitors. The cell lysates were rotated for 30 min at 4 °C, centrifuged at 12,000 rpm for 30 min at 4 °C and supernatants transferred to new tubes. After pre-clearing, the lysates were incubated overnight with 2 μg of normal rabbit IgG or SOD2 antibody (Abcam: Ab13533). Following SOD2 IP, the lysates were resolved in SDS-PAGE gels. The SOD2 band in each lane was cut with a razor blade and weighed. The bands were dissolved in 1 mL of 1X TGS running buffer overnight on a rocker at 4 °C. Next day, dissolved gel pieces were further heated for 20 min at 60 °C. The dissolved radioactive sample solutions were transferred to glass vials containing 10 mL of Opti-Fluor (PerkinElmer) in duplicate (500 μl per vial). Liquid scintillation counting was performed using a Beckman Coulter Scintillation Counter. The readouts were normalized against the values from untreated samples.
4.8. Ribonucleoprotein immunoprecipitation & RT-PCR
Cells were cultured in attached and anchorage-independent conditions as described above. Before harvesting cells, 0.3% formaldehyde was added for 10 min at 37 °C for crosslinking followed by addition of glycine (final concentration 0.25 M) for 5 min for quenching. RNP-IP was performed as described in Refs. [23,56] with modifications. Briefly, crosslinked cells were lysed in 500–1,000 μl NT1 buffer (100 mM KCl, 5 mM MgCl2, 10 mM HEPES, [pH 7.0], 0.5% Nonidet P40 [NP40], 1 mM dithiothreitol [DTT], 100 units/mL SUPERase·In RNase Inhibitor [Invitrogen: AM2694], protease inhibitors [Thermo: 78429], 0.2% vanadyl ribonucleoside complexes [New England Biolabs: S1402S]). After centrifugation of lysates at 16,000 g for 15 min, the supernatants were used for IP with normal mouse IgG or HuR antibody. The antibody-bead mixtures were washed several times with NT2 buffer (50 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM MgCl2, 0.05% NP40, RNAse inhibitor, protease inhibitor). IP samples for RNA elution were incubated with proteinase K (30 μg/100 μl NT2 buffer with 0.1% SDS) for 30 min at 60 °C. RNA was extracted using TRIzol, followed by cDNA synthesis (Quantabio: 95047) and SOD2 RT-PCR using the PrimeSTAR polymerase (Takara: R010A) with the following cycles: 98 °C for 10 s, 98 °C for 10 s + 60 °C for 10 s + 72 °C for 20 sec X 35–38 cycles, followed by a final extension step at 72 °C for 2 min. PCR products were analyzed by 2% agarose gel electrophoresis or 10% PAGE.
4.9. Polysome profiling by sucrose density gradient centrifugation
Cells in adherent and ULA plates were incubated with cycloheximide (100 μg/mL) for 10 min at 37 °C before harvesting and were washed twice with ice cold 1X PBS containing cycloheximide. The cells were homogenized in 500 μl lysis buffer (50 mM HEPES, 75 mM KCl, 5 mM MgCl2, 250 mM sucrose, 100 μg/mL cycloheximide, 2 mM DTT, 20 U/μl SUPERase·In RNase Inhibitor [Invitrogen: AM2694], 10% Triton X-100, 13% NaDOC) and polysome profiling carried out as previously described [57]. Lysates were placed on ice for 10 min and centrifuged at 3000 g for 15 min at 4 °C. 500 μl supernatants were loaded on linear sucrose gradients ranging from 20% to 47% (10 mM HEPES, KCl 75 mM, 5 mM MgCl2, 0.5 mM EDTA) and were separated by ultracentrifugation in a SW41 rotor at 34,000 rpm for 4 h 15 min at 4 °C (Beckman Coulter). Subsequently, four sucrose fractions were collected using a UV/VIS absorbance detector. TRIzol reagent (Invitrogen) was added to each fraction for RNA isolation. Briefly, post-centrifugation at 3,200g for 20 min after addition of 1/5 volume of chloroform, the aqueous layer was transferred, and 1/2 volume of isopropanol was added for overnight precipitation at −20 °C. RNA was pelleted by centrifugation at 4,640 rpm for 55 min at 4 °C. RNA pellets were washed with 70% ethanol twice and dissolved in RNAse-free water. RNA quantities were determined from each fraction and an equal amount of RNA from each fraction was collected and converted into cDNA. After cDNA synthesis and qPCR reactions, final PCR products were analyzed on 2% agarose gels.
4.10. Semi-quantitative real-time PCR
Total RNA was isolated by RNA isolation kit (Zymo Research: R2052) and used for cDNA synthesis (Quantabio: 95047) according to the manufacturer's instruction. cDNA was mixed with iTaq™ Universal SYBR® Green Supermix (BioRad) and the primers listed in Table 1. Semi-quantitative real time RT-PCR was carried out using a BioRad qRT-PCR machine (BioRad), data normalized to the geometric mean of four housekeeping genes (Table 1), and expressed as fold-change in expression using the 2−ΔΔCT formula.
Table 1.
Primers used for RT-PCR and semi-quantitative real time PCR.
| Primer | Sense | Antisense |
|---|---|---|
| SOD2 CDS | 5′-TCCACTGCAAGGAACAACAG-3′ | 5′-CGTGGTTTACTTTTTGCAAGC-3′ |
| SOD2 3′UTR-A | 5′-ATAATGCTGGGGTGAGCAAC-3′ | 5′-GCTGAGGTGGGACAATCACT-3′ |
| SOD2 3′UTR-B | 5′-TGTGTATGCATGCTTGTGGA-3′ | 5′CCACCTTGCCCGTCTATTTA-3′ |
| ATF4 | 5′- TGTCCTCCACTCCAGATCAT | 5′-GGCTCATACAGATGCCACTATC-3′ |
| ELAVL1 | 5′-CGCAGAGATTCAGGTTCTCC-3′ | 5′-CCAAACCCTTTGCACTTGTT-3′ |
| PTGS2 CDS | 5′- CTGGCGCTCAGCCATACAG-3′ | 5′-CCGGGTACAATCGCACTTAT-3′ |
| GAPDH | 5′-TGCACCACCAACTGCTTAGC-3′ | 5′-GGCATGGACTGTGGTCATGAG-3′ |
| Housekeeping genes for semiquantitative real-time RT-PCR: | ||
| GAPDH | 5′-GAGTCAACGGATTTGGTCGT-3′ | 5′-TTGATTTTGGAGGGATCTCG-3′ |
| 18S | 5′-AGAAACGGCTACCACATCCA-3′ | 5′- CACCAGACTTGCCCTCCA-3′ |
| HPRT1 | 5′-TGACCTTGATTTATTTTGCATACC-3′ | 5′-CGAGCAAGACGTTCAGTCCT-3′ |
| TBP | 5′-TTGGGTTTTCCAGCTAAGTTCT-3′ | 5′-CCAGGAAATAACTCTGGCTCA-3′ |
4.11. Luciferase reporter assay
Based on ENCODE RIP-seq data sets ENCSR000CWW and ENCSR000CWZ, and PAR-CLIP data set GSE29943, overlapping predicted HuR binding sites within the 3′UTR of SOD2 were generated by PCR using genomic DNA derived from OVCA433 cells and the primers listed in Table 2, followed by cloning into the pmirGLO Dual-Luciferase Expression vector (Promega E1330) via NEBuilder® HiFi DNA Assembly. Cells were seeded into 6-well plates at ∼70% confluency and transfected with their respective dual luciferase reporter vectors via Lipofectamine 3000 (Invitrogen L3000001). Luciferase activity in anchorage-independence and attached conditions was measured 48-h post-transfection with the Dual-Glo® Luciferase Assay System (Promega). 3′UTR-modulated expression was reported through Firefly luciferase and normalized to internal control Renilla luciferase expression. For HuR knockdown experiments, cells were first transfected with their respective siRNA, then transfected again with the dual luciferase reporter vectors. Luciferase expression was measured 24-h later.
Table 2.
Primers used for generating SOD2 3′UTR luciferase reporter plasmids.
| Plasmid | 3′UTR fragment position (following stop codon) | Forward Primer | Reverse Primer |
|---|---|---|---|
| Fragment 1 | 1–191 | 5′-GTTTAAACGAGCTCG CTAGCACCACGATCGTTATGC-3′ |
5′-CCTGCAGGTCGACTCTAG ATTTTCAATCACTTGCCC-3′ |
| Fragment 2 | 1–441 | 5′-TAAACGAGCTCGCTAGCA CCACGATCGTTATGCTG-3′ |
5′-GCCTGCAGGTCGACTCTA GACGACAAAAAGTGCAATTTTC-3′ |
| Fragment 3 | 2067–2499 | 5′-TTAAACGAGCTCGCTAGC AAGCGGTGTGTGTATGTG-3′ |
5′-GCCTGCAGGTCGACTCTAGA CTCTTAAAATCAATCTATC-3′ |
| Fragment 4 | 2939–3349 | 5′- TTAAACGAGCTCGCTAGC TGTTGAGAAATCACAAC-3′ |
5′- GCCTGCAGGTCGACTCTAGA CAGTCAAAAACTTCACAC-3′ |
| Fragment 5 | 1–1662 | 5′-GTTTAAACGAGCTCGCTA GCACCACGATCGTTATGC-3′ |
5′-TGCAGGTCGACTCTAGA GGCTGAGGTGGGACAATCACTT-3′ |
| Fragment 6 | 1660–3401 | 5′-GTTTAAACGAGCTCG CTAGCTCCTGAATAGCTGGG-3′ |
5′- CCTGCAGGTCGACTCTAG AACATGCAAGCATAATGCAG-3′ |
4.12. Click-&-go™ protein capture of Azide-modified proteins
Adherent cells at ∼70% confluency were placed in methionine-free medium for 30 min at 37 °C. Following, 20 μM SB203580 or 20 μg/mL cycloheximide was added to respective conditions for 30 min at 37 °C. Cells in adherent or ULA plates were left to incubate with 50 μM l-azidohomoalanine (AHA, Invitrogen, C10102) for 2 h at 37 °C. Cells were washed once with ice-cold 1X PBS and harvested in 50 mM Tris-HCl pH 8.0, 1% SDS, Roche protease and phosphatase inhibitor cocktails. Lysates were prepared by cold bath sonication (50–60% amplitude, 10 s pulses, ∼10 times) followed by centrifugation at max speed for 15 min at 4 °C. Protein concentrations were measured by Pierce BCA protein assay kit and 1.5 mg of protein was biotinylated by click chemistry (Click Chemistry Tools, 1440) for 2 h with end-over rotation. Proteins were acetone-precipitated, followed by resuspension in 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% SDS by cold bath sonication (70–80% amplitude, 30 s pulses, ∼10 times) and end-over incubated with streptavidin agarose resin for 2 h minimum. Beads were washed 2 times in 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% SDS, and 1 time in 1X PBS, 1% SDS with a 27G needle and proteins eluted in 0.4 vol RIPA, 0.4 vol 10% SDS, 25 mM biotin and 1X loading dye + βME. 40 μg of input protein and eluted samples were then immunoblotted. Total protein images were acquired by stain-free imaging via the BioRad Chemi-Doc platform.
4.13. Statistical analysis
All data are representatives of at least three independent experiments. Data are presented as mean ± SEM with individual replicate values superimposed. Statistical analysis was performed using GraphPad Prism Software v9, with statistical tests chosen based on experimental design, as described in figure legends.
Author contributions
Y.S.K. and P.W.T. designed the conceptual framework and experiments of the study, carried out the majority of the experiments and data analysis, prepared the figures and wrote the manuscript. W.P., J.E.W., Z.J. and A.T.E. assisted with experimental execution, and manuscript editing. K.M. and S.R.K. contributed to experimental design, data interpretation and manuscript editing. N.H. supervised and conceived the study, contributed to experimental design, assisted in data analysis, and assisted in writing and editing of the manuscript.
Declaration of competing interest
None.
Acknowledgements
The authors would like to thank Sara Shimko and Lydia Kutzler for technical assistance. This work was supported by the U.S. National Institutes of Health grants R01CA242021 (N.H.) and R01CA230628 (N.H. & K.M.), and R01DK015658 (S.R.K.).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102329.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Davison C.A., et al. Antioxidant enzymes mediate survival of breast cancer cells deprived of extracellular matrix. Cancer Res. 2013;73(12):3704–3715. doi: 10.1158/0008-5472.CAN-12-2482. [DOI] [PubMed] [Google Scholar]
- 2.Piskounova E., et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527(7577):186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Connor K.M., et al. Manganese superoxide dismutase enhances the invasive and migratory activity of tumor cells. Cancer Res. 2007;67(21):10260–10267. doi: 10.1158/0008-5472.CAN-07-1204. [DOI] [PubMed] [Google Scholar]
- 4.Hart P.C., et al. Caveolin-1 regulates cancer cell metabolism via scavenging Nrf2 and suppressing MnSOD-driven glycolysis. Oncotarget. 2016;7(1):308–322. doi: 10.18632/oncotarget.5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hemachandra L.P., et al. Mitochondrial superoxide dismutase has a protumorigenic role in ovarian clear cell carcinoma. Cancer Res. 2015;75(22):4973–4984. doi: 10.1158/0008-5472.CAN-14-3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim Y.S., et al. Context-dependent activation of SIRT3 is necessary for anchorage-independent survival and metastasis of ovarian cancer cells. Oncogene. 2020;39(8):1619–1633. doi: 10.1038/s41388-019-1097-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim Y.S., et al. Insights into the dichotomous regulation of SOD2 in cancer. Antioxidants. 2017;6(4) doi: 10.3390/antiox6040086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hempel N., Carrico P.M., Melendez J.A. Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anti Cancer Agents Med. Chem. 2011;11(2):191–201. doi: 10.2174/187152011795255911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jiang L., et al. Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature. 2016;532(7598):255–258. doi: 10.1038/nature17393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schafer Z.T., et al. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461(7260):109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konstantinopoulos P.A., et al. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res. 2011;71(15):5081–5089. doi: 10.1158/0008-5472.CAN-10-4668. [DOI] [PubMed] [Google Scholar]
- 12.Kenny T.C., et al. Selected mitochondrial DNA landscapes activate the SIRT3 axis of the UPR(mt) to promote metastasis. Oncogene. 2017;36(31):4393–4404. doi: 10.1038/onc.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamarajugadda S., et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. 2013;4:e504. doi: 10.1038/cddis.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Audic Y., Hartley R.S. Post-transcriptional regulation in cancer. Biol. Cell. 2004;96(7):479–498. doi: 10.1016/j.biolcel.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 15.van Kouwenhove M., Kedde M., Agami R. MicroRNA regulation by RNA-binding proteins and its implications for cancer. Nat. Rev. Cancer. 2011;11(9):644–656. doi: 10.1038/nrc3107. [DOI] [PubMed] [Google Scholar]
- 16.Wurth L., Gebauer F. RNA-binding proteins, multifaceted translational regulators in cancer. Biochim. Biophys. Acta. 2015;1849(7):881–886. doi: 10.1016/j.bbagrm.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 17.Epis M.R., et al. The RNA-binding protein HuR opposes the repression of ERBB-2 gene expression by microRNA miR-331-3p in prostate cancer cells. J. Biol. Chem. 2011;286(48):41442–41454. doi: 10.1074/jbc.M111.301481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazan-Mamczarz K., et al. Post-transcriptional gene regulation by HuR promotes a more tumorigenic phenotype. Oncogene. 2008;27(47):6151–6163. doi: 10.1038/onc.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J., et al. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int. J. Mol. Sci. 2013;14(5):10015–10041. doi: 10.3390/ijms140510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filippova N., et al. The RNA-binding protein HuR promotes glioma growth and treatment resistance. Mol. Cancer Res. 2011;9(5):648–659. doi: 10.1158/1541-7786.MCR-10-0325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lal S., et al. HuR posttranscriptionally regulates WEE1: implications for the DNA damage response in pancreatic cancer cells. Cancer Res. 2014;74(4):1128–1140. doi: 10.1158/0008-5472.CAN-13-1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang W., et al. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000;19(10):2340–2350. doi: 10.1093/emboj/19.10.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raspaglio G., et al. HuR regulates beta-tubulin isotype expression in ovarian cancer. Cancer Res. 2010;70(14):5891–5900. doi: 10.1158/0008-5472.CAN-09-4656. [DOI] [PubMed] [Google Scholar]
- 24.Abdelmohsen K., Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA. 2010;1(2):214–229. doi: 10.1002/wrna.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lafarga V., et al. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol. Cell Biol. 2009;29(16):4341–4351. doi: 10.1128/MCB.00210-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Denkert C., et al. Overexpression of the embryonic-lethal abnormal vision-like protein HuR in ovarian carcinoma is a prognostic factor and is associated with increased cyclooxygenase 2 expression. Cancer Res. 2004;64(1):189–195. doi: 10.1158/0008-5472.can-03-1987. [DOI] [PubMed] [Google Scholar]
- 27.Denkert C., et al. Expression of the ELAV-like protein HuR is associated with higher tumor grade and increased cyclooxygenase-2 expression in human breast carcinoma. Clin. Cancer Res. 2004;10(16):5580–5586. doi: 10.1158/1078-0432.CCR-04-0070. [DOI] [PubMed] [Google Scholar]
- 28.Miyata Y., et al. High expression of HuR in cytoplasm, but not nuclei, is associated with malignant aggressiveness and prognosis in bladder cancer. PLoS One. 2013;8(3) doi: 10.1371/journal.pone.0059095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mrena J., et al. Cyclooxygenase-2 is an independent prognostic factor in gastric cancer and its expression is regulated by the messenger RNA stability factor HuR. Clin. Cancer Res. 2005;11(20):7362–7368. doi: 10.1158/1078-0432.CCR-05-0764. [DOI] [PubMed] [Google Scholar]
- 30.Lebedeva S., et al. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol. Cell. 2011;43(3):340–352. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 31.Young L.E., et al. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol. Cancer Res. 2012;10(1):167–180. doi: 10.1158/1541-7786.MCR-11-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Akaike Y., et al. HuR regulates alternative splicing of the TRA2beta gene in human colon cancer cells under oxidative stress. Mol. Cell Biol. 2014;34(15):2857–2873. doi: 10.1128/MCB.00333-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis C.A., et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46(D1):D794–D801. doi: 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.EncodeProjectConsortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jakstaite A., et al. HuR mediated post-transcriptional regulation as a new potential adjuvant therapeutic target in chemotherapy for pancreatic cancer. World J. Gastroenterol. 2015;21(46):13004–13019. doi: 10.3748/wjg.v21.i46.13004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tran H., Maurer F., Nagamine Y. Stabilization of urokinase and urokinase receptor mRNAs by HuR is linked to its cytoplasmic accumulation induced by activated mitogen-activated protein kinase-activated protein kinase 2. Mol. Cell Biol. 2003;23(20):7177–7188. doi: 10.1128/MCB.23.20.7177-7188.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang W., et al. HuR regulates p21 mRNA stabilization by UV light. Mol. Cell Biol. 2000;20(3):760–769. doi: 10.1128/mcb.20.3.760-769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dolado I., et al. p38alpha MAP kinase as a sensor of reactive oxygen species in tumorigenesis. Cancer Cell. 2007;11(2):191–205. doi: 10.1016/j.ccr.2006.12.013. [DOI] [PubMed] [Google Scholar]
- 39.Emerling B.M., et al. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol. Cell Biol. 2005;25(12):4853–4862. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang S., et al. Urokinase plasminogen activator/urokinase-specific surface receptor expression and matrix invasion by breast cancer cells requires constitutive p38alpha mitogen-activated protein kinase activity. J. Biol. Chem. 2000;275(16):12266–12272. doi: 10.1074/jbc.275.16.12266. [DOI] [PubMed] [Google Scholar]
- 41.Carduner L., et al. Cell cycle arrest or survival signaling through alphav integrins, activation of PKC and ERK1/2 lead to anoikis resistance of ovarian cancer spheroids. Exp. Cell Res. 2014;320(2):329–342. doi: 10.1016/j.yexcr.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 42.Liao W.L., et al. The RNA-binding protein HuR stabilizes cytosolic phospholipase A2α mRNA under interleukin-1β treatment in non-small cell lung cancer A549 Cells. J. Biol. Chem. 2011;286(41):35499–35508. doi: 10.1074/jbc.M111.263582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ishimaru D., et al. Regulation of Bcl-2 expression by HuR in HL60 leukemia cells and A431 carcinoma cells. Mol. Cancer Res. 2009;7(8):1354–1366. doi: 10.1158/1541-7786.MCR-08-0476. [DOI] [PubMed] [Google Scholar]
- 44.Levy N.S., et al. Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J. Biol. Chem. 1998;273(11):6417–6423. doi: 10.1074/jbc.273.11.6417. [DOI] [PubMed] [Google Scholar]
- 45.Kim G.Y., Lim S.J., Kim Y.W. Expression of HuR, COX-2, and survivin in lung cancers; cytoplasmic HuR stabilizes cyclooxygenase-2 in squamous cell carcinomas. Mod. Pathol. 2011;24(10):1336–1347. doi: 10.1038/modpathol.2011.90. [DOI] [PubMed] [Google Scholar]
- 46.Dixon D.A., et al. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J. Clin. Invest. 2001;108(11):1657–1665. doi: 10.1172/JCI12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prislei S., et al. MiR-200c and HuR in ovarian cancer. BMC Cancer. 2013;13:72. doi: 10.1186/1471-2407-13-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Slone S., et al. Activation of HuR downstream of p38 MAPK promotes cardiomyocyte hypertrophy. Cell. Signal. 2016;28(11):1735–1741. doi: 10.1016/j.cellsig.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doller A., et al. Tandem phosphorylation of serines 221 and 318 by protein kinase Cdelta coordinates mRNA binding and nucleocytoplasmic shuttling of HuR. Mol. Cell Biol. 2010;30(6):1397–1410. doi: 10.1128/MCB.01373-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grammatikakis I., Abdelmohsen K., Gorospe M. Posttranslational control of HuR function. Wiley Interdiscip Rev RNA. 2017;8(1) doi: 10.1002/wrna.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fazzone H., Wangner A., Clerch L.B. Rat lung contains a developmentally regulated manganese superoxide dismutase mRNA-binding protein. J. Clin. Invest. 1993;92(3):1278–1281. doi: 10.1172/JCI116700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chung D.J., Wright A.E., Clerch L.B. The 3' untranslated region of manganese superoxide dismutase RNA contains a translational enhancer element. Biochemistry. 1998;37(46):16298–16306. doi: 10.1021/bi980935g. [DOI] [PubMed] [Google Scholar]
- 53.Chaudhuri L., et al. Preferential selection of MnSOD transcripts in proliferating normal and cancer cells. Oncogene. 2012;31(10):1207–1216. doi: 10.1038/onc.2011.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Church S.L. Manganese superoxide dismutase: nucleotide and deduced amino acid sequence of a cDNA encoding a new human transcript. Biochim. Biophys. Acta. 1990;1087(2):250–252. doi: 10.1016/0167-4781(90)90213-l. [DOI] [PubMed] [Google Scholar]
- 55.Sugiura A., et al. Newly born peroxisomes are a hybrid of mitochondrial and ER-derived pre-peroxisomes. Nature. 2017;542(7640):251–254. doi: 10.1038/nature21375. [DOI] [PubMed] [Google Scholar]
- 56.Tenenbaum S.A., et al. Ribonomics: identifying mRNA subsets in mRNP complexes using antibodies to RNA-binding proteins and genomic arrays. Methods. 2002;26(2):191–198. doi: 10.1016/S1046-2023(02)00022-1. [DOI] [PubMed] [Google Scholar]
- 57.Dang Do A.N., et al. eIF2alpha kinases GCN2 and PERK modulate transcription and translation of distinct sets of mRNAs in mouse liver. Physiol. Genom. 2009;38(3):328–341. doi: 10.1152/physiolgenomics.90396.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.