

Abstract

The tumor suppressor protein fragile histidine triad (Fhit) is known to be associated with genomic instability and apoptosis. The tumor-suppressive function of Fhit depends on the interaction with the alarmone diadenosine triphosphate (Ap3A), a noncanonical nucleotide whose concentration increases upon cellular stress. How the Fhit–Ap3A complex exerts its signaling function is unknown. Here, guided by a chemical proteomics approach employing a synthetic stable Fhit–Ap3A complex, we found that the Fhit–Ap3A complex, but not Fhit or Ap3A alone, impedes translation. Our findings provide a mechanistic model in which Fhit translocates from the nucleolus into the cytosol upon stress to form an Fhit–Ap3A complex. The Fhit–Ap3A complex impedes translation both in vitro and in vivo, resulting in reduced cell viability. Overall, our findings provide a mechanistic model by which the tumor suppressor Fhit collaborates with the alarmone Ap3A to regulate cellular proliferation.
Introduction
At present, the database Catalogue Of Somatic Mutations In Cancer (COSMIC) lists that from 4437 tested tumor samples, 2890 (65%) were found to be mutated in the tumor suppressor protein fragile histidine triad (Fhit).1,2 These include a variety of cancers including lung, cervical, and esophageal and squamous cell carcinoma,3−7 suggesting that in many different types of cancer, Fhit plays an important role in suppressing the formation of cancer development. Fhit is believed to exert its tumor suppressor function when in complex with the nucleotide diadenosine triphosphate (Ap3A). Ap3A (Figure 1a) is an “alarmone” as its intracellular concentration is rising upon cellular stress.8,9 Fhit significantly influences intracellular Ap3A concentrations10 as Fhit is also the main hydrolase for Ap3A.11 However, it was found that Fhit acts as a tumor suppressor by binding to Ap3A but independently of its capabilities to hydrolyze Ap3A.12 This emphasizes the importance of the Fhit-Ap3A complex formation for signaling. Several important cancer-related genes and pathways have been linked to Fhit. For example, Ras/Rho GTPases are known to interact with Fhit.13 Additionally, Fhit serves as a physiological target of the Src tyrosine kinase14 and the apoptotic pathway induced by Fhit was shown to be mediated by FADD and by the activation of caspase-8-, −9-, and −3-associated cell death pathways.15−17 Nevertheless, evidence based on Fhit variants supports the notion that the formation of the Fhit–Ap3A complex is crucial in growth inhibition.18,19 However, the molecular mechanism by which the Fhit–Ap3A complex exerts its tumor suppressor function remains elusive.
Figure 1.

Synthesis of the covalent Fhit–Ap3A complex Fhit*. (a) Structure of Ap3A. (b) Schematic representation of the generation of Fhit variant bound to Ap3A*. The complex formation is achieved by nucleophilic attack of the thiol group in the binding pocket of Fhit to the chloroacetamide group of Ap3A*. (c) LC/ESI-MS chromatogram of the Fhit variant (18675 Da) bound to Ap3A* showing mass corresponding to the Fhit variant covalent bound to the Ap3A analogue (+896 Da). (d) Crystal structure of Fhit bound to Ap3A*. Two chains are shown (A and C) bound to AMP* moiety. The binding pocket of Ap3A is visualized in light purple (chain C) and light blue (chain A). (e) Simulated annealing omit map for AMP* moiety countered at 2σ is shown as gray mesh.
We speculated that the elucidation of the interactome of the Fhit–Ap3A complex, in contrast to Fhit and Ap3A alone, respectively, will provide indications into the tumor suppression mechanism of the complex. Leveraging chemical biology and chemical proteomic approaches employing a synthetic stable Fhit–Ap3A complex, we succeeded in elucidating its interactome in a human non-small-cell lung carcinoma cell line H1299.
Guided by these results, we found that the Fhit–Ap3A complex interacts with the ribosome and thereby impedes translation, leading to an increased propensity of cell death and in consequence tumor suppression.
Results and Discussion
Generation and Structure of a Covalent Fhit–Ap3A Complex
Previous studies indicate that the binding of Ap3A is crucial for the tumor suppressor function of Fhit.19 To uncover the Fhit-Ap3A interactome and to gain insights into its molecular mechanisms and tumor suppressor function, we generated a covalent Fhit-Ap3A complex to be subsequently used in affinity purification mass spectrometry (AP-MS). Therefore, Ap3A was equipped with an electrophilic chloroacetamide function resulting in Ap3A* (Figure 1b). The chloroacetamide was introduced via a linker at the N6-position of one adenosine moiety since it has been shown that Fhit accepts Ap3A analogues with a modification at this position.20,21 The synthesis of Ap3A* is described in detail in the Supporting Information (Figure S1). Ap3A* was reacted with an Fhit variant carrying a nucleophilic cysteine instead of a histidine in position 8 of the binding pocket. Further, histidine 96 was mutated to an asparagine to obtain a hydrolytic inactive Fhit. Of note, it was reported that this inactivating mutation does not change the tumor suppressor function of Fhit.12 The synthesized complex of the mutated Fhit and Ap3A* is henceforth termed Fhit* (Figure 1b). The integrity of Fhit* was proven by LC/ESI-MS analysis revealing exclusive species with masses for the Fhit variant plus the Ap3A analogue (+896 Da) (Figure 1c). In contrast, when wild-type Fhit was incubated with Ap3A*, no new signal that would indicate covalent binding of the nucleotide to Fhit was detected and only the mass corresponding to wild-type Fhit was detected (Figure S2). To investigate the impact of Ap3A* binding on the global Fhit structure, we solved the crystal structure of Fhit* at a resolution of 2.34 Å (PDB: 7P8P).
Electron density of Fhit and the linkage of Ap3A* revealed that Ap3A* is correctly positioned and covalently attached to the introduced Cys8 residue of Fhit (Figures 1d and S3a). The attached nucleobase fits perfectly in the primary binding pocket and analysis of the surface indicated the location of Ap3A* inside the protein (Figures 1e and S3b). Then, we compared the structures by superimposing to the published crystal structure of Fhit bound to nonhydrolyzable Ap3A (nhAp3A, PDB: 1FHI).22 Overlay of the structures demonstrated an overall alignment of 0.3 ± 0.1 Å root-mean-square deviation (rmsd), showing a high similarity of both structures (Figure S3c,d). These results demonstrate that Ap3A* binding does not alter the structural characteristics of Fhit and proves the regioselectivity of the reaction of Ap3A* with the mutated Fhit. Thus, Fhit* provides an excellent mimic of the native Fhit-Ap3A complex.
Elucidation of the Ap3A-Dependent Fhit Interactome
To identify the stress-specific cellular interactome of Fhit, we deployed an affinity purification approach using Fhit and Fhit* as baits and H1299 whole cell lysates (Figure 2a). Of note, the human non-small-cell lung carcinoma cell line H1299 is deficient of endogenous Fhit.23 To monitor the stability of Fhit* in the presence of cell lysate, we incubated Fhit* with H1299 cell lysates and resolved the samples on SDS-PAGE demonstrating the stability of Fhit* under the chosen conditions (Figure S4a). Next, Fhit and Fhit* were used as baits and immobilized on streptavidin-conjugated agarose and subsequently incubated with the respective cell lysates (Figure S4b), using H2O2 as cellular stress signal. Bound proteins were eluted with biotin and resolved on SDS-PAGE before tryptic digestion (Figure 2b). For LC-MS/MS analysis of the bound proteins, triplicates of the experiments were performed. We conducted several control experiments including wild-type Fhit as bait and the matrix without bait protein incubated with lysates from H1299 cells either grown under physiological conditions or subjected to H2O2 treatment prior to harvesting. We observed a stress-specific enrichment of proteins for Fhit* (Figure 2b). No significant enrichment of proteins was observed when lysates of H2O2-treated and unstressed cells were applied to wild-type Fhit as bait and also when the Fhit* bait was subjected to lysates of unstressed cells. Next, the eluted proteins resolved on SDS-PAGE were digested with trypsin for subsequent LC-MS/MS analysis.
Figure 2.

Identification of the Ap3A-dependent cellular Fhit interactome. (a) Schematic representation of the affinity purification MS (AP-MS) workflow using Fhit* as bait protein in H1299 cell lysates. (b) SDS-PAGE of elution fractions before LC-MS/MS analysis; (**) Fhit dimer, (*) Fhit monomer. (c) Hierarchical clustering (Euclidean distance) of statistically significantly enriched interactors following ANOVA analysis (S0 = 5, FDR = 0.005). Columns represent bait proteins, rows show interacting proteins, and ribosomal proteins and elongation factors are highlighted in blue on the left side. H1299 cells were exposed to oxidative stress with 1 mM H2O2 10 min before harvesting. Empty beads with unstressed cells (lane 1) and stressed cells (lane 2), Fhit with (lane 3) and without (lane 4) stressed cells and Fhit* with lysate from unstressed cells (lane 5) were added as controls. Lane 6 shows Fhit* with stressed cells. Clusters of proteins with similar enrichment patterns are depicted on the right. AP-MS experiments were carried out in biological triplicates and measured as technical duplicates. (d) Venn diagram of main steps in data analysis of AP-MS. Total number of identified proteins after LC-MS/MS analysis, number of proteins after filtering, and number of statistically enriched proteins after ANOVA analysis, enriched proteins specifically for Fhit* with cells after H2O2 treatment. (e) GO-term analysis for biological processes of enriched proteins interacting with Fhit* under stressed conditions based on DAVID classification. (f) KEGG-pathway analysis of enriched proteins with Fhit* under stressed conditions based on DAVID classification. Proteomic data are provided as a Supporting Data File.
The three replicates were analyzed as technical duplicates by LC-MS/MS to give a total of six measurements per applied condition. Enriched proteins were identified by MaxQuant with its integrated search engine Andromeda followed by label-free quantification (LFQ).24−27 The data were further analyzed by Perseus,26 resulting in 1559 confidently identified proteins in total. A total of 1032 proteins were obtained (66% of the uploaded proteins) after filtering and validation that they were present in at least five of the six replicate experiments. For these proteins, missing values imputation (downshift 1.8, width 0.3) in the total matrix and multiple-sample test (FDR = 0.005, S0 = 5) was performed. Under these conditions, 183 constantly and significantly enriched proteins by ANOVA statistics were obtained (18% of 1032 proteins, Supporting Data 1). All significantly enriched proteins were visualized by correlation-based hierarchical clustering and depicted in a heatmap, resulting in a total of two protein clusters that show similar binding behavior (Figure 2c). The color scale represents the median Z-score normalized LFQ value as calculated by Perseus.
Interestingly, we found that the largest cluster (cluster 2) harbors proteins that were identified with the Fhit* bait and lysates from stressed cells. Proteins represented in the ANOVA statistics with a minimum Z-score of ≥0.5 were considered for further analysis. After filtering against the control experiments, we identified 132 proteins that were significantly and consistently enriched using Fhit* incubated with lysates of H2O2-treated H1299 cells (Figure 2d). Statistical analysis (Figure 2c and Supporting Data 1) and bioinformatic clustering showed enrichment for translation, in particular for translation initiation (Figure 2e and Supporting Data 2) and a specific enrichment for ribosomal proteins of both 40S and 60S subunits (Figure 2f and Supporting Data 2). We found that at least 40% of the 60S and 45% of the 40S subunit proteins were enriched.
Action of Fhit on Translation
Guided by our finding that translational proteins are enriched in the Fhit*-based AP-MS study, we evaluated the action of Fhit and Ap3A on translation and developed an in vitro translation assay based on transcription/translation-coupled rabbit reticulocyte lysate. Translation of the target protein was detected by the incorporation of 35S-methionine followed by SDS-PAGE analysis and autoradiography. First, we investigated the effect of Fhit on the translation efficiency in the presence of increasing amounts of Ap3A (Figure 3a). We found no effect on translation efficiency when Fhit or Ap3A, respectively, was applied (Figure 3a, lanes 1–3).
Figure 3.

Functional characterization of Fhit within the translation. (a) Radioactive readout of in vitro translation with a transcript/translation-coupled reticulocyte lysate of a DNA vector for Mdm2. Where indicated, 40 μM Fhit and Ap3A in increasing concentrations (0–180 μM) were added. The experiment was performed in biological triplicates, the value of the control experiment was set to unity, and all other values were calculated accordingly. Shown is the mean ± s.e.m. (b) In vitro translation assay with a DNA vector for Mdm2, the addition of Fhit ATP, GTP, nonhydrolyzable ATP analogues (AppCH2p, ApCH2pp), and dinucleotide polyphosphates (Ap5A, Ap4A, Gp3G, nhAp3A, Ap3A). The experiment was performed in biological triplicates, the value of the control experiment was set to unity, and all other values were calculated accordingly. Shown is the mean ± s.e.m. (c) H1299 cells were transfected with an expression vector for Fhit and an empty vector (empty). The cells were starved for 2 h in methionine-free medium, extracellular stress was induced with 0.5 mM H2O2 for 20 min at 37 °C. The methionine-free medium was supplemented with 0.1% 35S-methionine, and the incorporation of 35S-Met was analyzed 2 h after stress induction at 37 °C via SDS-PAGE. Shown are two experiments out of three with similar outcomes. (d) Analysis of protein synthesis in H1299 cells. The radioactive intensity of H1299 cells transfected with empty vector (−) was set to unity, and all other values were calculated accordingly. Samples were measured in biological triplicates. Shown is the mean ± s.e.m. (e) Cell viability of H1299 cells. H1299 cells were transfected with an expression vector for Fhit, eGFP, and H96N. The value of H1299 cells transfected with empty vector was set to unity, and all other values were calculated accordingly. Extracellular stress was induced with 0.5 mM H2O2 for 20 min at 37 °C. Samples were measured in biological triplicates. Shown is the mean ± s.e.m.
In contrast, when both Fhit and Ap3A were present in the reaction mix, a stark Ap3A concentration-dependent decline of target protein expression was observed (Figure 3a, lanes 4–12). We found that upon the addition of 40 μM Fhit and 80 μM Ap3A (Fhit/Ap3A 1:2), a 2-fold decrease in translation was observed. When increasing the Ap3A concentration even further to 180 μM (Fhit/Ap3A 1:4.5), a decrease in translation efficiency by 89% was observed. Since control experiments employing either Fhit or Ap3A alone revealed no effect on translation efficiencies, we concluded that only Fhit in complex with Ap3A affects translation.
To ensure that the decrease in translation is not due to inhibition of transcription, we performed additional experiments using a pretranscribed mRNA template encoding for Firefly luciferase and subsequently measured the resulting luminescence. Again, a decrease in protein translation was correlated with increasing Ap3A concentrations and was only observed in the presence of Fhit (Figure S5a), which further corroborates our notion that Fhit only in complex with Ap3A affects protein synthesis at the translation step.
Next, we investigated the nucleotide specificity of the effect of Fhit on translation and employed various nucleotides (ATP, GTP, nonhydrolyzable ATP analogues (AppCH2p and ApCH2pp), Ap4A, Ap5A, Gp3G, nhAp3A, and Ap3A (structures are shown in Figure S6)) in our translation assay (Figures 3b and S5b). Among the multiple nucleotides evaluated, only Ap3A and its nonhydrolyzable analogue nhAp3A caused a significant decrease in translation (Figure 3b, lanes 10, 11), stressing the importance of Fhit-Ap3A complex formation for the inhibitory effect. Furthermore, the observation that hydrolysis of Ap3A is not required for this function is along the lines with the observation that the tumor suppressor activity is independent of Ap3A hydrolysis.12
To investigate the potential effect of Fhit on translation, we monitored the protein synthesis after cells were subjected to H2O2 treatment. Therefore, H1299 cells were transfected with an expression vector encoding for Fhit. As a control, we also transfected the cells with an empty vector, to investigate the effect of transfection. The cells were subjected to H2O2 treatment, and methionine-free medium was supplemented with radioactive 35S-methionine to monitor the protein synthesis after extracellular oxidative stress by the incorporation of 35S-methionine followed by SDS-PAGE analysis and autoradiography. The determined values of H1299 cells transfected with the empty vector were set to unity, and sample values were calculated accordingly. No Fhit-dependent alterations of the cellular protein level were observed when the cells were grown under physiological conditions (Figures 3c,d and S5c). However, when the cells were subjected to H2O2 treatment, we observed a decrease in protein synthesis of cells expressing Fhit (Figures 3c,d and S5c). Interestingly, the Fhit variant H96N shows a similar decrease in the total cellular protein concertation (Figure S5d), demonstrating that the activity of Fhit is independent of its hydrolytic activity of Ap3A.
Conclusively, these results demonstrated that the presence of Fhit in H1299 cells results in a decrease in total protein synthesis in this cell line. To further examine the potential influence of Fhit on cell viability, we performed an ATP-dependent luminescent cell viability assay in H1299 cells (Figure 3e). We observed a decrease in cell viability of 22% of cells overexpressing eGFP when cells were stressed. Interestingly, the Fhit variant H96N (66%) shows a similar decrease in cell viability compared to the wild-type enzyme (68%) (Figure 3e). These findings demonstrate that the effect of Fhit is independent of its hydrolytic activity of Ap3A. These results suggest that the expression of Fhit could also enhance oxidative stress-mediated apoptosis.
Fhit Binding to the Ribosome and Polysomes in Human Cells
As we had identified the ribosome as the main interactor in our Fhit*-based AP-MS study, we wondered if the inhibitory effect that we have observed for Fhit on translation might be related to an interaction of Fhit with the ribosome. We therefore further analyzed the interaction of Fhit with the 80S ribosomal fraction. Previous reports had suggested that the interaction of Fhit with binding partners elongates the lifetime of the Fhit-Ap3A complex by modulating the hydrolase activity of Fhit.28
Given that the hydrolysis of Ap3A is not crucial for the inhibitory effect of Fhit, we investigated if a potential interaction of Fhit with the ribosome would influence the hydrolysis of Ap3A by Fhit. To address this, we established an analytical RP-HPLC-based assay to monitor the turnover of Ap3A by Fhit within fractions containing 80S ribosomes that we isolated from H1299 cells. We found that the incubation of Ap3A with Fhit resulted in more than 98% of Ap3A being hydrolyzed to AMP and ADP (Figures 4a and S7). Interestingly, in the presence of 80S ribosomes, Fhit exhibits a decrease in the turnover of Ap3A (Figure 4a). To investigate whether intact 80S ribosomes indeed influence the turnover of Ap3A, we incubated the ribosomes with RNaseA before the addition of Fhit and Ap3A. By incubating the ribosomes with RNaseA, the ribosomal RNA is degraded and the ribosomal proteins can dissociate from the ribosomal complex.29,30 We found that Fhit exhibits some decrease in hydrolysis activity (<5%), indicating that the intact 80S ribosome is relevant for the interaction with Fhit. Conclusively, these results show that ribosome binding of the Fhit-Ap3A complex modulates Ap3A hydrolysis by Fhit.
Figure 4.

Fhit binding to the ribosome and polysomes in human cells. (a) HPLC-based assay to quantify Ap3A levels. Incubation of Fhit with 400-fold Ap3A excess for 30 min at 37 °C. Samples are Fhit without ribosome, Fhit with purified 80S ribosome from H1299 cells, Ap3A 400-fold excess, and pre-inactivation of ribosomes by incubation with RNaseA. Samples were measured in biological triplicates. Shown is the mean ± s.e.m. (b) Polysome profile of HEK 293T cells. HEK 293T cells were grown under physiological conditions (gray) or subjected to 0.5 mM H2O2 for 20 min at 37 °C (black). The translation was stopped with cycloheximide. Cleared cell lysate was separated in 5–50% sucrose gradient by ultracentrifugation and analyzed by OD 254 nm. Fractions were collected after 40 s each. (c) Western blot analysis of fractions from polysome profile. Antibody against uL4 for 60S ribosomal subunit, eS6 for 40S ribosomal subunit and Fhit. Cells without H2O2 treatment (top), and cells with H2O2 treatment (bottom). The experiments were performed twice with matching results.
To further investigate the influence of Fhit on translational processes in cells, the overall change in the polysome formation was analyzed. HEK 293T cells that express Fhit endogenously were grown under physiological conditions or subjected to H2O2 treatment before harvesting. We analyzed the polysome formation in actively proliferating cells. The polysome profile of HEK 293T cells grown under physiological conditions shows a significantly higher distribution of polysomes compared to cells treated with H2O2 (Figure 4b). The polysome fractions are more abundant than unbound 80S ribosomes and free ribosomal subunits under physiological conditions.
Remarkably, cells subjected to oxidative stress show a decrease in polysomes and a significant shift from the polysomes toward the unbound 80S ribosomes and separated ribosome subunits 40S and 60S. Consistently, our immunoblot analysis of the separated fractions showed a significant shift of the 60S subunit protein uL4 and the 40S subunit protein eS6 in the polysome fractions from stressed cells compared to unstressed cells (Figure 4c). A decrease in uL4 intensity was observed in the later fractions 5–10 after cells were subjected to H2O2 treatment. In addition, a decrease in eS6 intensity was observed, too. Interestingly, immunoblot analysis revealed the presence of Fhit within the polysome profile, but mainly when cells were subjected to H2O2 treatment (Figures 4c and S10d,e). Fhit accumulated especially in the early fractions 3–5 where also uL4 and eS6 are enhanced. The shift toward unbound ribosomal subunits, the decrease in polysome formation, and the presence of Fhit in the polysome fraction only upon stress imposition strongly indicate that during stress response, Fhit interacts with the free ribosomal subunits and the 80S ribosomes, which might prevent the formation of polysomes.
Structural Analysis of Fhit and the Ribosome
To investigate site-specific interactions of Fhit with ribosomes, we performed chemical crosslinking coupled to mass spectrometry (XL-MS).32,33 Fhit was incubated with purified 80S ribosomal fraction isolated from H1299 cells in the presence and absence of Ap3A, respectively. Crosslinking of Fhit to proximal proteins was performed by the addition of the homobifunctional active ester bis(sulfosuccinimidyl)suberate (BS3).34 The crosslinked proteins were tryptically digested, and crosslinked peptides were enriched by size exclusion chromatography. Crosslinked peptides were detected by LC-MS/MS and used to indicate interacting regions and domains33 (Supporting Data 3).
The data analysis revealed a large number of high-confidence crosslinks both within different ribosomal subunits and between Fhit and ribosomal subunits, confirming the interaction of Fhit with the ribosome (Figure 5). While some ribosomal interactors stay consistent and thus independent of Ap3A binding (Figures 5a,b, S8, and S9), some appear to exist in particular only in the Ap3A-bound state (Figure 5b).
Figure 5.

Crosslinking of Fhit and the 80S ribosome. High-confidence crosslink network for Fhit in the absence of Ap3A (a) or the presence of Ap3A (b) with purified 80S ribosomal fraction from H1299 cells. (c) Crosslinked proteins of the ribosome with Fhit in the presence of Ap3A were highlighted in the corresponding structure of 80S ribosome (PDB 4UG0),31 containing the landmark of the 40S and 60S ribosomal subunits in a pretranslocation state in which the 40S subunit is nonrotated.
For example, for the large 60S subunit, uL2, uL4, and eL6 were only identified in the presence of Ap3A, while uL16 appears to be specific for the Ap3A independent state. In total, nine direct interaction sites with Fhit in the presence of Ap3A were identified. To generate a model of the interaction sites of Fhit within the ribosome, we mapped our crosslinks to the structures of Fhit, 40S subunit, and 60S subunit.31,35,36 The mapping of our crosslinking data onto the published cryo-EM reconstitution of the 80S ribosome points to the head of the small 40S ribosomal subunit and the bottom of the large 60S subunit as potential interaction sites (Figures 5c and S9). Besides showing that Fhit-Ap3A is indeed binding the ribosome, our XL-MS study provides insights into the interaction mode of Fhit with the ribosome in the presence or absence of Ap3A. In presence of Ap3A, Fhit seems to interact with the proteins uL4 and uL6 of the larger 60S subunit. Recent studies showed that both proteins are necessary for the assembly of the 60S subunit.37−39 Interestingly, the interactions of Fhit in the presence of Ap3A with the protein eS25 located near the mRNA binding tunnel of the small 40S subunit and the protein uL2 near the E-site tRNA of the large 60S subunit were observed. The protein uL2 is located near the ribosomal peptidyl transferase center (PTC) and is essential for the catalytic activity of the ribosome.40 The eIF3 protein identified as an interaction partner by crosslinking as well as the eIF4 and eIF2 proteins identified in the affinity enrichment for Fhit with Ap3A are essential for mRNA binding to the 40S subunit and the correct formation of 40S pre-initiation complex.41,42
Taken together, our crosslinking data show an interaction of Fhit with the ribosome, which appears to intensify in the presence of Ap3A. Interestingly, the interactions of Fhit with the small 40S subunit were observed, suggesting that the binding of Fhit at this position would alter the binding of mRNA and the assembly of the pre-initiation complex, resulting in the inhibition of translation. Additionally, the crosslinking data are in good agreement with ribosomal proteins determined by our AP-MS study of Fhit* under stressed conditions.
Cellular Level and Location of Fhit
To determine the cellular level of Fhit in cells, we grew HEK 293T cells that express Fhit endogenously, both under physiological conditions and after H2O2 treatment, and monitored Fhit protein levels by immunoblot analysis.
We found that Fhit levels are observed in a similar range for cells grown under physiological conditions and cells subjected to H2O2 treatment (Figure 6a), demonstrating that the protein concentration of Fhit is not altered by extracellular stress. To investigate mRNA levels, we performed RT-qPCR for Fhit. As control, we used the housekeeping enzyme RNaseP. The mRNA levels were in a similar range for cells grown under physiological conditions and cells subjected to extracellular stress (97.3 ± 2.6% when stressed) (Figures 6b and S10a).
Figure 6.

Localization of Fhit in HEK 293T cells. Fhit levels were analyzed in HEK 293T cells grown under physiological conditions or after subjection to extracellular stress (H2O2 treatment). (a) Immunoblot (IB) analysis of Fhit with (+) and without (−) H2O2 treatment. Immunoblot against tubulin was used as a control. Shown is one experiment out of three with similar outcomes. (b) mRNA level of Fhit in HEK 293T cells. The mRNA levels were normalized to the level of RNaseP. The value of untreated HEK 293T cells was set to unity, and the stressed value was calculated accordingly. Data are given in percentage, representing the mean of n = 3 independent experiments. Shown is the mean ± s.e.m. (c) Immunofluorescence microscopy of Fhit in cellulo. Fluorescence microscopy of HEK 293T cells treated with (+) or without (−) H2O2. Antibodies Fhit (green) and Calnexin (red); Hoechst 33342 staining (blue) and overlay. Scale bar: 20 μm.
Taken together, these observations demonstrate that the Fhit level, at least in HEK 293T cells, is neither affected on the protein level nor the mRNA level when cells are subjected to extracellular oxidative stress. This could indicate that Fhit is stored in a reservoir within the cells until its use in the stress response. To test this hypothesis, we analyzed HEK 293T cells by fluorescence microscopy. After fixation, we permeabilized the cells and incubated them with an antibody against Fhit and calnexin, a protein present in the endoplasmic reticulum (ER) and thus a marker for ribosomes.43 Fluorescence microscopy revealed that endogenously expressed Fhit is located mainly in the nucleolus (Figure 6c). In contrast, when cells were subjected to oxidative extracellular stress, Fhit was mainly located in the cytosol. Additionally, by immunoblot analysis of cytosolic and nuclear fractions from HEK 293T cells, we demonstrated that Fhit translocates from the nuclear fraction to the cytosol under oxidative extracellular stress (Figure S10b).
Conclusions
Even though the Ap3A-dependent hydrolase activity of Fhit was discovered in the 1990s12 the cellular role and function of the complex as a tumor suppressor remain enigmatic. Here, we report on a new mechanism by which the Fhit-Ap3A complex signals stress and introduces stress-dependent cell death. First, we investigated the proteome-wide interaction map of the Fhit-Ap3A complex in the cellular stress response by employing a synthetic covalent complex of Fhit-Ap3A. Structural studies reveal that this complex well mimics the natural complex. This complex was subsequently used in AP-MS studies. Most of the identified proteins appear to be involved in translation processes (Figure 2e,f); more than 26% of these proteins are ribosomal proteins, indicating a role of Fhit in the cellular process of translation upon extracellular stress. In comparison, earlier studies of interaction partners of Fhit44−47 focused on interaction partners specifically for Fhit without Ap3A employing co-immunoprecipitation experiments or in cell crosslinking experiments in unstressed cells and identified one to five proteins only.
Guided by our findings from the AP-MS study, we investigated the action of Fhit on translation. We found that Fhit only impedes translation in an Ap3A concentration-dependent manner. Moreover, this effect is specific to Ap3A and independent of Ap3A hydrolysis since an nhAp3A analogue shows the same effect in the presence of Fhit.
Encouraged by these findings, we investigated if this effect holds also true in living cells. Therefore, we ectopically expressed Fhit in H1299 cells that do not express Fhit endogenously.23 We speculated that in stressed cells, Ap3A levels increase as reported9 and that inhibition of translation by the then higher levels of Fhit-Ap3A complex in these cells manifests itself in decreasing total protein levels compared with nonstressed cells or cells that do not express Fhit. Indeed, that is what we observed (Figure 3c) providing evidence that inhibition of translation also happens in living cells. Next, we investigated cell viability and found that indeed Fhit overexpression leads to an increase in cell death when the cells are stressed with H2O2 in comparison to unstressed cells (Figure 3d). Cellular stress like oxidative stress imposes a threat on the genome integrity and results in damaged nucleobases.48 If not repaired efficiently, remaining damaged nucleobases lead to increased levels of mutations that might be drivers for carcinogenesis.49 Thus, translation inhibition by Fhit in complex with the stress-sensitive alarmone Ap3A might prevent cells that are damaged by cellular stress from proliferation, ultimately leading to cell death and thereby preventing cells from becoming degenerated.
Since we found many ribosomal proteins enriched in our AP-MS studies, we speculated that the ribosome might be the target of the Fhit–Ap3A complex. Therefore, we investigated if an interaction with purified 80S ribosomes affects the turnover of Ap3A by Fhit. Indeed, we found that Ap3A hydrolysis is impeded by the ribosome leading to an extension of the Fhit-Ap3A complex lifetime and points to the ribosome as being the binding target of the Fhit-Ap3A complex.
These results are in accordance with previous reports that implicated that the interaction of Fhit with binding partners elongates the lifetime of the Fhit-Ap3A complex and inhibits the hydrolase activity of Fhit.28 Th investigation of the polysome formation of HEK 293T cells revealed an increase of free 80S ribosomes and a decrease of polysomes in the presence of Fhit when HEK 293T cells were subjected to H2O2 treatment. This, in combination with results obtained from the XL-MS studies (Figure 5), indicates that the Fhit-Ap3A complex binds to the ribosomal subunits.
To gain insights into the origin of Fhit in the stress response, we investigated the Fhit protein and mRNA levels in HEK 293T cells and found that both levels are unchanged independent of the growth conditions (Figure 6a,b). This finding suggests that Fhit synthesis is not stimulated by cellular stress but rather that localization of the protein is modulated upon stress. The localization of Fhit in HEK 293T cells was investigated by fluorescence microscopy. Under physiological conditions, we observed the location of Fhit in the nucleolus (Figure 6c). However, when cells are subjected to H2O2 treatment, Fhit is translocated from the nucleolus to the cytosol (Figure 6c). These results are in line with the report that Fhit is localized in the nucleus fraction under physiological conditions shown by immunoblotting of subcellular fractions.50,51
Based on our findings, we propose a mechanistic model for the tumor suppressor function of Fhit (Figure 7). Extracellular stress leads to an increased concentration of the alarmone Ap3A.9 This recruits the tumor suppressor Fhit, which is stored under normal growth conditions in the nucleolus, from the nucleolus to the cytosol and especially to the ER. Fhit and Ap3A form a complex that binds to the ribosome, which in turn leads to perturbation of translation as long as the complex is intact, which eventually may lead to cell death. In this way, Fhit-Ap3A protects severely damaged cells from further and possibly uncontrolled proliferation. The stress signal is maintained as long as Ap3A is not hydrolyzed or continuously generated and will probably fade upon Ap3A hydrolysis by Fhit. This could allow the system to adapt its response to different degrees of cellular damage. Hence, the discovery of ribosome inhibition as a mechanism for tumor suppression of Fhit will inspire the development of new therapeutic approaches in the future.
Figure 7.
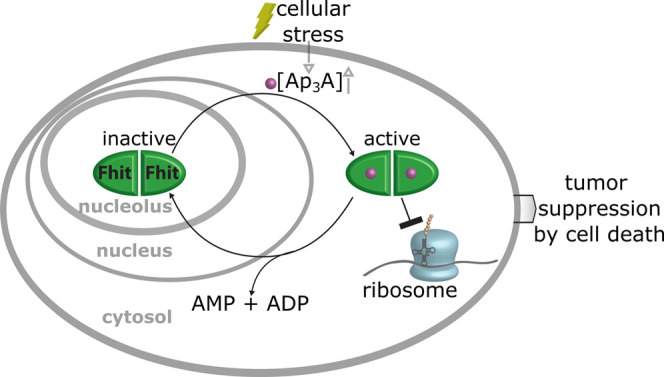
Model for tumor suppression as a stress response of Fhit–Ap3A complex. Fhit is stored in the nucleolus; however, upon stress stimuli, the Ap3A concentration increases and the cellular localization of Fhit shifts from the nucleolus to the cytosol. Thereby, Fhit binds Ap3A and interacts with the ribosome and impedes translation.
Acknowledgments
The authors thank A. Julier (Department of Biology, University of Konstanz) for providing pcDNA Mdm2 construct and S. Ludmann (Department of Chemistry, University of Konstanz) for helping with figure design and illustration of the crosslinking data. They also thank A. Marquardt and A. Sladewska-Marquardt (Proteomics Center, University of Konstanz) for performed measurements. The authors thank the team of the Bioimaging Center, University of Konstanz, and especially M.T. Stöckl for help with microscopy experiments. D.H., J.J., and M.M. thank the Konstanz Research School Chemical Biology for support.
Glossary
Abbreviations
- Fhit
fragile histidine triad
- Ap3A
diadenosine triphosphate
- nhAp3A
nonhydrolyzable diadenosine triphosphate
- Ap4A
diadenosine tetraphosphate
- Ap5A
diadenosine pentaphosphate
- Gp3G
diguanosine triphosphate
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.2c00815.
General chemical synthesis of Ap3A*, biological methods and procedures, tables of primer, and additional crystallography data (PDF)
Annotation information of each of the LC-MS/MS identified peptides (XLSX)
Annotation information of each DAVID analysis (XLSX)
Annotation Information of XL-MS analysis (XLSX)
This work was supported by the European Research Council (ERC-AdG 101019280) and the Deutsche Forschungsgemeinschaft (MA 2288/19-1) to A.M. and within the Emmy Noether Program (STE 2517/1-1) to F.S. that is gratefully acknowledged.
The authors declare no competing financial interest.
Supplementary Material
References
- Sanger Institute. COSMIC, The Catalogue of Somatic Mutations in Cancer. https://cancer.sanger.ac.uk/cosmic (accessed March 18, 2022).
- Tate J. G.; Bamford S.; Jubb H. C.; Sondka Z.; Beare D. M.; Bindal N.; Boutselakis H.; Cole C. G.; Creatore C.; Dawson E.; Fish P.; Harsha B.; Hathaway C.; Jupe S. C.; Kok C. Y.; Noble K.; Ponting L.; Ramshaw C. C.; Rye C. E.; Speedy H. E.; Stefancsik R.; Thompson S. L.; Wang S.; Ward S.; Campbell P. J.; Forbes S. A. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sard L.; Accornero P.; Tornielli S.; Delia D.; Bunone G.; Campiglio M.; Colombo M. P.; Gramegna M.; Croce C. M.; Pierotti M. A.; Sozzi G. The tumor-suppressor gene FHIT is involved in the regulation of apoptosis and in cell cycle control. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 8489–8492. 10.1073/pnas.96.15.8489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujan O.; Oliver R.; Roz L.; Sozzi G.; Ribeiro N.; Woodwards R.; Thakker N.; Sloan P. Fragile histidine triad expression in oral squamous cell carcinoma and precursor lesions. Clin. Cancer Res. 2006, 12, 6723–6729. 10.1158/1078-0432.CCR-06-1475. [DOI] [PubMed] [Google Scholar]
- Mori M.; Mimori K.; Shiraishi T.; Alder H.; Inoue H.; Tanaka Y.; Sugimachi K.; Huebner K.; Croce C. M. Altered expression of Fhit in carcinoma and precarcinomatous lesions of the esophagus. Cancer Res. 2000, 60, 1177–1182. [PubMed] [Google Scholar]
- Connolly D. C.; Greenspan D. L.; Wu R.; Ren X.; Dunn R. L.; Shah K. V.; Jones R. W.; Bosch F. X.; Muñoz N.; Cho K. R. Loss of fhit expression in invasive cervical carcinomas and intraepithelial lesions associated with invasive disease. Clin. Cancer Res. 2000, 6, 3505–3510. [PubMed] [Google Scholar]
- Zavalhia L. S.; Weber Medeiros A.; Oliveira Silva A.; Vial Roehe A. Do FHIT gene alterations play a role in human solid tumors?. Asia Pac. J. Clin. Oncol. 2018, 14, e214–e223. 10.1111/ajco.12868. [DOI] [PubMed] [Google Scholar]
- Baltzinger M.; Ebel J.-P.; Remy P. Accumulation of dinucleoside polyphosphates in Saccharomyces cerevisiae under stress conditions. High levels are associated with cell death. Biochimie 1986, 68, 1231–1236. 10.1016/S0300-9084(86)80069-4. [DOI] [PubMed] [Google Scholar]
- Krüger L.; Albrecht C. J.; Schammann H. K.; Stumpf F. M.; Niedermeier M. L.; Yuan Y.; Stuber K.; Wimmer J.; Stengel F.; Scheffner M.; Marx A. Chemical proteomic profiling reveals protein interactors of the alarmones diadenosine triphosphate and tetraphosphate. Nat. Commun. 2021, 12, 5808 10.1038/s41467-021-26075-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy G. A.; Halliday D.; McLennan A. G. The Fhit Tumor Suppressor Protein Regulates the Intracellular Concentration of Diadenosine Triphosphate but not Diadeonsine Tetraphsophate. Cancer Res. 2000, 60, 2342–2344. [PubMed] [Google Scholar]
- Campiglio M.; Bianchi F.; Andriani F.; Sozzi G.; Tagliabue E.; Ménard S.; Roz L. Diadenosines as FHIT-ness instructors. J. Cell. Physiol. 2006, 208, 274–281. 10.1002/jcp.20633. [DOI] [PubMed] [Google Scholar]
- Barnes L. D.; Garrison P. N.; Siprashvili Z.; Guranowski A.; Robinson A. K.; Ingram S.; Croce C. M.; Ohta M.; Huebner K. Fhit, a Putative Tumor Suppressor in Humans, Is a Dinucleoside 5′,5‴-P1,P3-Triphosphate Hydrolase. Biochemistry 1996, 35, 11529–11535. 10.1021/bi961415t. [DOI] [PubMed] [Google Scholar]
- Jayachandran G.; Sazaki J.; Nishizaki M.; Xu K.; Girard L.; Minna J. D.; Roth J. A.; Ji L. Fragile histidine triad-mediated tumor suppression of lung cancer by targeting multiple components of the Ras/Rho GTPase molecular switch. Cancer Res. 2007, 67, 10379–10388. 10.1158/0008-5472.CAN-07-0677. [DOI] [PubMed] [Google Scholar]
- Pekarsky Y.; Garrison P. N.; Palamarchuk A.; Zanesi N.; Aqeilan R. I.; Huebner K.; Barnes L. D.; Croce C. M. Fhit is a physiological target of the protein kinase Src. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 3775–3780. 10.1073/pnas.0400481101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roz L.; Andriani F.; Ferreira C. G.; Giaccone G.; Sozzi G. The apoptotic pathway triggered by the Fhit protein in lung cancer cell lines is not affected by Bcl-2 or Bcl-x(L) overexpression. Oncogene 2004, 23, 9102–9110. 10.1038/sj.onc.1208142. [DOI] [PubMed] [Google Scholar]
- Dumon K. R.; Ishii H.; Fong L. Y.; Zanesi N.; Fidanza V.; Mancini R.; Vecchione A.; Baffa R.; Trapasso F.; During M. J.; Huebner K.; Croce C. M. FHIT gene therapy prevents tumor development in Fhit-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 3346–3351. 10.1073/pnas.061020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H.; Dumon K. R.; Vecchione A.; Trapasso F.; Mimori K.; Alder H.; Mori M.; Sozzi G.; Huebner K.; Croce C. M.; et al. Effect of Adenoviral Transduction of the Fragile Histidine Triad Gene into Esophageal Cancer Cells. Cancer Res. 2001, 61, 1578–1584. [PubMed] [Google Scholar]
- Siprashvili Z.; Sozzi G.; Barnes L. D.; McCue P.; Robinson A. K.; Eryomin V.; Sard L.; Taglibue E.; Greco A.; Fusetti L.; Schwartz G.; Pierotti M. A.; Croce C. M.; Huebner K. Replacement of Fhit in cancer cells suppresses tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 13771–13776. 10.1073/pnas.94.25.13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapasso F.; Krakowiak A.; Cesari R.; Arkles J.; Yendamuri S.; Ishii H.; Vecchione A.; Kuroki T.; Bieganowski P.; Pace H. C.; Huebner K.; Croce C. M.; Brenner C. Designed FHIT alleles establish that Fhit-induced apoptosis in cancer cells is limited by substrate binding. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 1592–1597. 10.1073/pnas.0437915100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker S. M.; Mortensen F.; Scheffner M.; Marx A. Selective monitoring of the enzymatic activity of the tumor suppressor Fhit. Angew. Chem., Int. Ed. 2014, 53, 10247–10250. 10.1002/anie.201405259. [DOI] [PubMed] [Google Scholar]
- Lange S.; Hacker S. M.; Schmid P.; Scheffner M.; Marx A. Small-Molecule Inhibitors of the Tumor Suppressor Fhit. ChemBioChem 2017, 18, 1707–1711. 10.1002/cbic.201700226. [DOI] [PubMed] [Google Scholar]
- Pace H. C.; Garrison P. N.; Robinson A. K.; Barnes L. D.; Dragenescu A.; Roesler A.; Blackburn M. G.; Siprashvili Z.; Croce C. M.; Huebner K.; Brenner C. Genetic, biochemical, and crystallographic characterization of Fhit-substrate complex as the active signaling form of Fhit. Proc. Natl. Acad. Sci. U.S.A. 1998, 5484–5489. 10.1073/pnas.95.10.5484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T.; Otsuka A.; Girard L.; Sato M.; Iwakawa R.; Ogiwara H.; Sanchez-Cespedes M.; Minna J. D.; Yokota J. A catalog of genes homozygously deleted in human lung cancer and the candidacy of PTPRD as a tumor suppressor gene. Genes Chromosomes Cancer 2010, 49, 342–352. 10.1002/gcc.20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Hein M. Y.; Luber C. A.; Paron I.; Nagaraj N.; Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics 2014, 13, 2513–2526. 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Tyanova S.; Temu T.; Sinitcyn P.; Carlson A.; Hein M. Y.; Geiger T.; Mann M.; Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- Cox J.; Neuhauser N.; Michalski A.; Scheltema R. A.; Olsen J. V.; Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Golebiowski F.; Szulc A.; Szutowicz A.; Pawelczyk T. Ubc9-induced inhibition of diadenosine triphosphate hydrolase activity of the putative tumor suppressor protein Fhit. Arch. Biochem. Biophys. 2004, 428, 160–164. 10.1016/j.abb.2004.05.020. [DOI] [PubMed] [Google Scholar]
- Moore S.; Stein W. H. Chemical Structures of Pancreatic Ribonuclease and Deoxyribonuclease. Science 1973, 180, 458–464. 10.1126/science.180.4085.458. [DOI] [PubMed] [Google Scholar]
- Raines R. T. Ribonuclease A. Chem. Rev. 1998, 98, 1045–1066. 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- Khatter H.; Myasnikov A. G.; Natchiar S. K.; Klaholz B. P. Structure of the human 80S ribosome. Nature 2015, 520, 640–645. 10.1038/nature14427. [DOI] [PubMed] [Google Scholar]
- Leitner A.; Walzthoeni T.; Aebersold R. Lysine-specific chemical cross-linking of protein complexes and identification of cross-linking sites using LC-MS/MS and the xQuest/xProphet software pipeline. Nat. Protoc. 2014, 9, 120–137. 10.1038/nprot.2013.168. [DOI] [PubMed] [Google Scholar]
- Sailer C.; Offensperger F.; Julier A.; Kammer K.-M.; Walker-Gray R.; Gold M. G.; Scheffner M.; Stengel F. Structural dynamics of the E6AP/UBE3A-E6-p53 enzyme-substrate complex. Nat. Commun. 2018, 9, 4441 10.1038/s41467-018-06953-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A.; Faini M.; Stengel F.; Aebersold R. Crosslinking and Mass Spectrometry: An Integrated Technology to Understand the Structure and Function of Molecular Machines. Trends Biochem. Sci. 2016, 41, 20–32. 10.1016/j.tibs.2015.10.008. [DOI] [PubMed] [Google Scholar]
- Erzberger J. P.; Stengel F.; Pellarin R.; Zhang S.; Schaefer T.; Aylett C. H. S.; Cimermančič P.; Boehringer D.; Sali A.; Aebersold R.; Ban N. Molecular architecture of the 40S·eIF1·eIF3 translation initiation complex. Cell 2014, 158, 1123–1135. 10.1016/j.cell.2014.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban N.; Beckmann R.; Cate J. H. D.; Dinman J. D.; Dragon F.; Ellis S. R.; Lafontaine D. L. J.; Lindahl L.; Liljas A.; Lipton J. M.; McAlear M. A.; Moore P. B.; Noller H. F.; Ortega J.; Panse V. G.; Ramakrishnan V.; Spahn C. M. T.; Steitz T. A.; Tchorzewski M.; Tollervey D.; Warren A. J.; Williamson J. R.; Wilson D.; Yonath A.; Yusupov M. A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol. 2014, 24, 165–169. 10.1016/j.sbi.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelter P.; Huber F. M.; Kunze R.; Flemming D.; Hoelz A.; Hurt E. Coordinated Ribosomal L4 Protein Assembly into the Pre-Ribosome Is Regulated by Its Eukaryote-Specific Extension. Mol. Cell 2015, 58, 854–862. 10.1016/j.molcel.2015.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson D. M.; Li Y.; LaPeruta A.; Gamalinda M.; Gao N.; Woolford J. L. Structural insights into assembly of the ribosomal nascent polypeptide exit tunnel. Nat. Commun. 2020, 11, 5111 10.1038/s41467-020-18878-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borkiewicz L.; Mołoń M.; Molestak E.; Grela P.; Horbowicz-Drożdżal P.; Wawiórka L.; Tchórzewski M. Functional Analysis of the Ribosomal uL6 Protein of Saccharomyces cerevisiae. Cells 2019, 8, 718 10.3390/cells8070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanshina D. D.; Bulygin K. N.; Malygin A. A.; Karpova G. G. Hydroxylated histidine of human ribosomal protein uL2 is involved in maintaining the local structure of 28S rRNA in the ribosomal peptidyl transferase center. FEBS J. 2015, 282, 1554–1566. 10.1111/febs.13241. [DOI] [PubMed] [Google Scholar]
- Trachsel H.; Erni B.; Schreier M. H.; Staehelin T. Initiation of Mammalian Protein Synthesis:The Assembly of the Initiation Complex with Purified Initiation Factors. J. Mol. Biol. 1977, 116, 755–767. 10.1016/0022-2836(77)90269-8. [DOI] [PubMed] [Google Scholar]
- Lee A. S. Y.; Kranzusch P. J.; Doudna J. A.; Cate J. H. D. eIF3d is an mRNA cap-binding protein that is required for specialized translation initiation. Nature 2016, 536, 96–99. 10.1038/nature18954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan C. H.; Williams C. C.; Weissman J. S. Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science 2014, 346, 1257521 10.1126/science.1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizaki M.; Sasaki J.-I.; Fang B.; Atkinson E. N.; Minna J. D.; Roth J. A.; Ji L. Synergistic tumor suppression by coexpression of FHIT and p53 coincides with FHIT-mediated MDM2 inactivation and p53 stabilization in human non-small cell lung cancer cells. Cancer Res. 2004, 64, 5745–5752. 10.1158/0008-5472.CAN-04-0195. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Zou M.; Farid N. R.; Paterson M. C. Association of FHIT (fragile histidine triad), a candidate tumour suppressor gene, with the ubiquitin-conjugating enzyme hUBC9. Biochem. J. 2000, 352, 443–448. 10.1042/bj3520443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri A. R.; Khan I. A.; Prasad V.; Robinson A. K.; Ludueña R. F.; Barnes L. D. The tumor suppressor protein Fhit. A novel interaction with tubulin. J. Biol. Chem. 1999, 274, 24378–24382. 10.1074/jbc.274.34.24378. [DOI] [PubMed] [Google Scholar]
- Druck T.; Cheung D. G.; Park D.; Trapasso F.; Pichiorri F.; Gaspari M.; Palumbo T.; Aqeilan R. I.; Gaudio E.; Okumura H.; Iuliano R.; Raso C.; Green K.; Huebner K.; Croce C. M. Fhit-Fdxr interaction in the mitochondria: modulation of reactive oxygen species generation and apoptosis in cancer cells. Cell Death Dis. 2019, 10, 147 10.1038/s41419-019-1414-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanvah S.; Joseph J.; Schuster G. B.; Barnett R. N.; Cleveland C. L.; Landman U. Oxidation of DNA: damage to nucleobases. Acc. Chem. Res. 2010, 43, 280–287. 10.1021/ar900175a. [DOI] [PubMed] [Google Scholar]
- Hu J. J.; Dubin N.; Kurland D.; Ma B.-L.; Roush G. C. The effects of hydrogen peroxide on DNA repair activities. Mutat. Res. 1995, 336, 193–201. 10.1016/0921-8777(94)00054-A. [DOI] [PubMed] [Google Scholar]
- Golebiowski F.; Kowara R.; Pawelczyk T. Distribution of Fhit protein in rat tissues and its intracellular localization. Mol. Cell. Biochem. 2001, 226, 49–55. 10.1023/A:1012729601270. [DOI] [PubMed] [Google Scholar]
- Bianchi F.; Sasso M.; Turdo F.; Beretta G. L.; Casalini P.; Ghirelli C.; Sfondrini L.; Ménard S.; Tagliabue E.; Campiglio M. Fhit Nuclear Import Floowing EGF Stimulation Sustains Proliferation of Bereast Cancer Cells. J. Cell. Physiol. 2015, 230, 2661–2670. 10.1002/jcp.24968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.