

Abstract

Urolithins (dibenzo-pyran-[b,d]-6 one derivatives) are human gut microbiota metabolites produced from the natural food antioxidant ellagic acid. Urolithins are better absorbed than ellagic acid and demonstrate biological activities that suggest that they are responsible for the health effects observed after consuming ellagitannin- and ellagic acid-containing foods. Urolithins occur in the systemic circulation as glucuronide conjugates following phase II metabolism. These phase II conjugates are essential for testing the urolithin mechanisms of action in human cell line bioassays. Urolithin glucuronides are not commercially available, and their biosynthesis leads to mixtures of regional isomers. This study describes a novel and regioselective synthesis of urolithin A (3,8-dihydroxy urolithin) 3- and 8-glucuronides and isourolithin A (3,9-dihydroxy urolithin) 3- and 9-glucuronides. The metabolites were characterized using 1H and 13C NMR spectroscopy and UV spectrophotometry. The presence of these metabolites in human subjects belonging to different urolithin metabotypes was also investigated.
Keywords: ellagic acid, gut microbiota metabolites, urolithins, glucuronides, synthesis
Introduction
Urolithins (hydroxy-6H-dibenzo[b,d]pyran-6-one derivatives) are gut microbiota metabolites produced from the natural polyphenolic antioxidants ellagic acid and ellagitannins.1 These antioxidants are present in significant quantities in foods, including pomegranates, muscadine grapes, berries (strawberries, raspberries, and blackberries), nuts (walnuts and pecans), tropical fruits (camu-camu and jaboticaba), tea, oak-aged wines, spirits, and many herbal medicinal products.2,3 Compared to ellagic acid, urolithins are better absorbed in humans and display biological activities that suggest that they are responsible for the health effects observed after consuming ellagitannin-containing foods.3,4 These include cardiovascular effects, anticancer activities, antiaging effects, and gut and systemic anti-inflammatory effects that also have impact in neurocognitive disorders, as shown in recent literature reviews.5−7 In the systemic circulation, urolithins occur mainly as glucuronide conjugates following phase II metabolism, enhancing their solubility and, therefore, their urinary excretion.1,8,9 Thus, these phase II conjugates are essential metabolites for testing the biological effects of urolithins on in vitro human cell line bioassays.9,10
The synthesis of glucuronide conjugates of phenolic compounds (4-hydroxycinnamic, urolithin B, hydroxytyrosol, resveratrol, citrus flavanones) has been attempted using different approaches based on the use of methyl-2,3,4-tri-O-acetyl-1-O-(trichloroacetimidoyl)-α-d-glucuronide, although in these studies, regioisomeric mixtures were obtained.11−13
The different regioisomeric urolithin glucuronides have not been available through chemical synthesis so far. In an early attempt, the preparation of both urolithin A 8- and urolithin A 3-glucuronide (18 and 22, respectively) was described but not in a regioselective manner. In turn, these urolithins were obtained as a mixture of both regioisomers.14 Urolithin aglycones have been chemically synthesized using different methods.15−19 However, syntheses of the main circulating glucuronide metabolites, as single individual regioisomers, have not been reported. In fact, only the synthesis of urolithin B-glucuronide (urolithin 3-glucuronide) (23) was previously reported13 because urolithin B has only one hydroxyl group for glucuronidation, and this makes the synthesis straightforward. However, syntheses of the main circulating glucuronide metabolites have not been reported, probably because of difficulties in producing and isolating the two isomers of urolithin A and those of isourolithin A (Figure 1).
Figure 1.

Structures of the urolithin glucuronide metabolites. *(Nomenclature following Kay et al., 2020).23
These different glucuronides may exert dissimilar biological effects or be produced in diverse quantities in different individuals due to enzyme polymorphisms as was shown for hesperetin glucuronide conjugates.20 However, this has not been demonstrated yet for urolithins due to the lack of authentic standards for these metabolites.
This study describes a novel and regioselective synthesis of urolithin A 3- and 8-glucuronides and isourolithin A 3- and 9-glucuronides and their characterization using 1H 13C NMR spectroscopy, mass spectrometry, and UV spectrophotometry.
Materials and Methods
Reagents and Chemicals
All solvents and reagents were purchased from commercial sources and were analytically pure and used as purchased. NaOH, CuSO4, 2-bromo-4-methoxybenzoic acid, 2-bromo-5-methoxybenzoic acid, resorcinol, triisopropylsilyl chloride (TIPS-Cl), imidazole, dimethylformamide (DMF), BBr3, OEt2, dichloromethane (DCM), K2CO3, KF, MeOH, H2O, pivaloyl chloride, pyridine, and BF3 were bought from Sigma-Aldrich (St. Louis). Methyl-(2,3,4-tri-O-acetyl-α-d-glucopyranosyl trichloroacetimidate) uronate. CAS Number: 92420-89-8. From Combi-Blocks (San Diego, CA).
General Synthetic Procedure for Compound 3 (3-Hydroxy-9-methoxy-6H-dibenzo[b,d]pyran-6-one)
The mixture of 2-bromo-4-methoxybenzoic acid 1 (2.0 g, 8.65 mmol), resorcinol 2 (2. g, 18.17 mmol), and NaOH (0.72 g, 18.17 mmol) in H2O (9.1 mL) was heated at 80 °C for 30 min. Then, aqueous 5% CuSO4 (3.6 mL) was added to the mixture and heated for additional 10 min. HCl (37% 1.0 mL) was added, and the resulting precipitate was filtered and washed with H2O (5.00 mL) and MeOH (3.0 mL). The filter cake was transferred to a flask, and MeOH (10 mL) was added. The suspension in MeOH was stirred at 50 °C for 10 min, newly filtered and washed with Et2O, and dried under a high vacuum to yield compound 3 as a colorless powder (1.50 g., 72%). ESI-MS (m/z): [M + H]+ calcd, 242.06; found, 242.90
General Synthetic Procedure for Compound 4 (9-Methoxy-3-((triisopropylsilyl)oxy)-6H-benzo[c]chromen-6-one)
To an ice–water-cooled solution of 3 (0.365 g; 1.507 mmol) in dry DMF (8.0 mL) were sequentially added triisopropyl silyl chloride (0.969 mg, 4.521 mmol) and imidazole (0.205 g, 3.014 mmol) and stirred until reaching room temperature (rt) overnight. Additional triisopropyl silyl chloride (0.969 mg, 4.521 mmol) and imidazole (0.205 g, 3.014 mmol) were added at rt, and the reaction mixture was stirred for further 3 h. The solvent was removed under reduced pressure, and the residue was partitioned between CH2Cl2 and H2O. The organic layer was evaporated under reduced pressure, and the residue was newly partitioned between EtOAc and H2O, and the layers were separated. The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The resulting crude material was purified by MPLC (SiO2, EtOAc/heptane 0:100 until 15:85) to afford 4 as a colorless solid (0.597 g, 94% yield). ESI-MS (m/z): [M + H]+ calcd, 398.19; found, 399.10.
General Synthetic Procedure for Compound 5 (9-Hydroxy-3-((triisopropylsilyl)oxy)-6H-benzo[c]chromen-6-one)
To a cooled (−80 °C) solution of 4 (1.300 g, 3.262 mmol) in dry CH2Cl2 (90.0 mL), BBr3 (1.886 mL, 19.572 mmol) was added dropwise. The reaction mixture was gradually warmed to rt and stirred for 12 h. The reaction mixture was poured into ice/water (50 mL) and extracted with EtOAc (3 × 30 mL). The combined organic layers were dried over Mg2SO4, filtered, and then concentrated under reduced pressure. The resulting crude material was purified by MPLC (SiO2, EtOAc/heptane 0:100 until 15:85) to afford 5 as a colorless solid (0.711 g, 70% yield). ESI-MS (m/z): [M + H]+ calcd, 384.55; found, 385.30.
General Synthetic Procedure for Compound 7 (2S,3S,4S,5R,6R)-2-(Methoxycarbonyl)-6-(((6-oxo-3-(triisopropylsilyl)oxy)-6H-benzo[c]chromen-9-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyltriacetate
A freshly prepared 0.1 M solution in CH2Cl2 of BF3. Et2O (1.43 mL) was added to a CH2Cl2 (2 mL) solution of 6 (0.274 g, 0.572 mmol) and 5 (0.220 g, 0.572 mmol) in dry CH2Cl2 (2.0 mL) at rt. The mixture was stirred for 1 h at rt. The reaction mixture was concentrated under reduced pressure, and the resulting crude material was purified by MPLC (SiO2, EtOAc/heptane 0:100 until 30:70) to afford 7 as colorless foam (0.289 g, 72% yield). ESI-MS (m/z): [M + H]+ calcd, 700.82; found, 543.00 [M-TiPS]+.
General Synthetic Procedure for Compound 8 (2S,3S,4S,5R,6R)-3,4,5-Trihydroxy-6-((3-hydroxy-6-oxo-6H-benzo[c]chromen-9-yl)oxy)tetrahydro-2H-pyran-2-carboxylic Acid
To a mixture of 7 (0.289 g, 0.412 mmol), KF (0.048 g, 0.824 mmol), and K2CO3 (0.114 g, 0.824 mmol), MeOH–H2O (10 mL, 5:1) was added. The resulting mixture was stirred at rt for 16 h. The solvent was removed under vacuum, and the mixture was dissolved in H2O and purified by RP-HPLC to afford 8 (0.080 g, 48% yield). ESI-MS (m/z): [M + H]+ calcd, 404.33; found, 403.00 [M – H]+.
General Synthetic Procedure for Compound 9 (6-Oxo-3-((triisopropylsilyl)oxy)-6H-benzo[c]chromen-9-yl pivalate)
To a solution of 5 (0.146 g, 0.38 mmol) in pyridine (10 mL), pivaloyl chloride (0.935 mL, 7.60 mmol) was added dropwise at rt. The resulting mixture was stirred at rt for 18 h. The solvent was removed under reduced pressure, and the resulting crude material was purified by MPLC (SiO2, EtOAc/heptane 0:100 until 90:10) to afford 9 as a colorless solid (0.157 g, 88% yield). ESI-MS (m/z): [M + H]+ calcd, 468.23; found, 469.30.
General Synthetic Procedure for Compound 10 (3-Hydroxy-6-oxo-6H-benzo[c]chromen-9-yl pivalate)
To a solution of 9 (0.157 g, 0.335 mmol) in methanol (3.5 mL), KF (0.021 g, 0.368 mmol) was added. The resulting mixture was stirred at rt for 1 h. The solvent was removed under reduced pressure, and the resulting crude material was purified by MPLC (SiO2, EtOAc/heptane 0:100 until 20:80) to afford 9 as a colorless solid (0.062 g, 59% yield). ESI-MS (m/z): [M + H]+ calcd, 312.23; found, 311.00 [M – H]+.
General Synthetic Procedure for Compound 11 ((2S,3S,4S,5R,6R)-2-(Methoxycarbonyl)-6-((6-oxo-9-(pivaloyloxy)-6H-benzo[c]chromen-3-yl)oxy) tetrahydro-2H-pyran-3,4,5-triyltriacetate)
A freshly prepared 0.1 M solution in CH2Cl2 of BF3. Et2O (0.5 mL) was added to a CH2Cl2 (2 mL) solution of 6 (0.095 g, 0.199 mmol) and 10 (0.062 g, 0.199 mmol) in dry CH2Cl2 (2.0 mL) at room temperature. The mixture was stirred for 1 h at rt. The reaction mixture was concentrated under reduced pressure, and the resulting crude material was purified by MPLC (SiO2, EtOAc/heptane 0:100 until 30:70) to afford 7 as colorless foam (0.089 g, 72% yield). ESI-MS (m/z): [M + H]+ calcd, 628.18; found, 543.00 [M-t-BuCO]+.
General Synthetic Procedure for Compound 12 ((2S,3S,4S,5R,6R)-3,4,5-Trihydroxy-6-((9-hydroxy-6-oxo-6H-benzo[c]chromen-3yl)oxy) tetrahydro-2H-pyran-2-carboxylic Acid)
To a mixture of 11 (0.084 g, 0.134 mmol), KF (0.016 g, 0.268 mmol), and K2CO3 (0.037g, 0.268 mmol), MeOH–H2O (2,8 mL, 5:1) was added. The resulting mixture was stirred at rt for 16 h. The solvent was removed under vacuum, and the mixture was dissolved in H2O and purified by RP-HPLC to afford 12 (0.032 g., 59% yield). ESI-MS (m/z): [M + H]+ calcd, 404.33; found, 403.00 [M – H]+.
General Synthetic Procedure for Compound 14 (3-Hydroxy-8-methoxy-6H-dibenzo[b,d]pyran-6-one)
The mixture of 2-bromo-5-methoxybenzoic acid 13 (0.500 g., 2.164. mmol), resorcinol 2 (1.430. g, 12.98 mmol), and NaOH (0.672 g., 16.80 mmol) in H2O (25 mL) was refluxed for 1 h. Then, aqueous 28% CuSO4 (25 mL) was added to the mixture and heated for an additional 10 min. On cooling to rt, the precipitate formed was filtered and washed several times with ice–H2O and finally with Et2O and dried under high vacuum to yield compound 14 as a colorless powder (0.476 g., 91%). ESI-MS (m/z): [M + H]+ calcd, 242.06; found, 243.00
General Synthetic Procedure for Compound 15 (8-Methoxy-3-((triisopropylsilyl)oxy)-6H-benzo[c]chromen-6-one)
To an ice–water-cooled solution of 3-hydroxy-8-methoxy-6H-dibenzo[b,d]pyran-6-one, 3, (0.050 g; 0.206 mmol) in dry DMF (8.0 mL) were sequentially added triisopropyl silyl chloride (0.132 mg, 0.618 mmol) and imidazole (0.028 g, 0.412 mmol) and stirred until rt overnight. The solvent was removed under reduced pressure, and the residue was partitioned between CH2Cl2 and H2O. The organic layer was evaporated under reduced pressure, and the residue was newly partitioned between EtOAc and H2O, and the layers were separated. The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The resulting crude material was purified by MPLC (SiO2, EtOAc/heptane 0:100 until 15:85) to afford 15 as a colorless solid (0.069 g, 84% yield). ESI-MS (m/z): [M + H]+ calcd, 398.19; found, 399.10.
General Synthetic Procedure for Compound 16 (8-Hydroxy-3-((triisopropylsilyl)oxy)-6H-benzo[c]chromen-6-one)
To a cooled (−80 °C) solution of 9-methoxy-3-((triisopropylsilyl)oxy)-6H-benzo[c]chromen-6-one 4 (1.0 g, 2.509 mmol) in dry CH2Cl2 (89 mL), BBr3 (1.45 mL, 15.05 mmol) was added dropwise. The reaction mixture was gradually warmed to rt and stirred for 12 h. The reaction mixture was poured into ice/water and extracted with CH2Cl2 (2×) and EtOAc (1×). The combined organic layers were dried over Mg2SO4, filtered, and then concentrated under reduced pressure. The resulting crude material was purified by MPLC (SiO2, EtOAc/heptane 0:100 until 10:90) to afford 16 as a colorless solid (1.0 g, 73% yield). ESI-MS (m/z): [M + H]+ calcd, 384.55; found, 385.40.
General Synthetic Procedure for Compound 17 ((2S,3S,4S,5R,6R)-2-(Methoxycarbonyl)-6-((6-oxo-3-((triisopropylsilyl)oxy)-6H-benzo[c]chromen-8-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate)
A freshly prepared 0.1 M solution in CH2Cl2 of BF3.Et2O (0.940 mL) was added to a CH2Cl2 (3 mL) solution of 6 (0.224 g, 0.468 mmol) and 16 (0.180 g, 0.468 mmol) in dry CH2Cl2 (2.0 mL) at room temperature. The mixture was stirred for 30 min at rt. The reaction mixture was concentrated under reduced pressure, and the resulting crude material was purified by MPLC (SiO2, EtOAc/heptane 0:100 until 30:70) to afford 17 as a colorless solid (0.248 g, 76% yield). ESI-MS (m/z): [M + H]+ calcd, 700.26; found, 542.90 [M-TiPS]+.
General Synthetic Procedure for Compound 18 ((2S,3S,4S,5R,6R)-3,4,5-Trihydroxy-6-((3-hydroxy-6-oxo-6H-benzo[c]chromen-8-yl)oxy) tetrahydro-2H-pyran-2-carboxylic Acid)
To a mixture of 17 (0.294 g, 0.419 mmol), KF (0.048 g, 0.838 mmol), and K2CO3 (0.115g, 0.838 mmol), MeOH–H2O (20 mL, 5:1) was added. The resulting mixture was stirred at rt for 16 h. The solvent was removed under vacuum, and the mixture was dissolved in H2O and purified by RP-HPLC to afford 18 as a colorless solid (0.078 g, 46% yield). ESI-MS (m/z): [M + H]+ calcd, 404.33; found, 403.00 [M – H]+.
General Synthetic Procedure for Compound 19 (3-Hydroxy-6-oxo-6H-benzo[c]chromen-8-yl pivalate)
To a solution of 16 (0.050 g, 0.107 mmol) in pyridine (3 mL), pivaloyl chloride (0.320 mL, 2.60 mmol) was added dropwise at rt. The resulting mixture was stirred at rt for 16 h. The solvent was removed under reduced pressure to afford a solid that was filtered and washed with cold MeOH to afford 19 as a colorless solid (0.043 g, 71% yield). ESI-MS (m/z): [M + H]+ calcd, 468.23; found, 469.20.
General Synthetic Procedure for Compound 20 (3-Hydroxy-6-oxo-6H-benzo[c]chromen-8-yl pivalate)
To a solution of 19 (0.050 g; 0.107 mmol) in MeOH (1 mL) was added KF (0.006 mg, 0.107 mmol). The reaction mixture was stirred at rt for 2 h. The solvent was evaporated under reduced pressure, and the resulting crude material was purified by MPLC (SiO2, EtOAc/heptane 0:100 until 50:50) to afford 20 as a colorless solid (0.025 g, 75% yield). ESI-MS (m/z): [M + H]+ calcd, 310.32, found, 313.00.
General Synthetic Procedure for Compound 21 ((2S,3S,4S,5R)-2-(Methoxycarbonyl)-6-((6-oxo-8-(pivaloyloxy)-6H-benzo[c]chromen-3-yl)oxy)tetrahydro-2H-pyran-3,4,5-triyl Triacetate)
A freshly prepared 0.1 M solution in CH2Cl2 of BF3·Et2O (2.1 mL) was added to a CH2Cl2 (2 mL) solution of 6 (0.398 g, 0.832 mmol) and 20 (0.260 g, 0.832 mmol) in dry CH2Cl2 (3.0 mL) at 0 °C. The mixture was stirred for 1 h at rt. The reaction mixture was concentrated under reduced pressure, and the resulting crude material was purified by MPLC (SiO2, EtOA/heptane 0:100 until 50:50) to afford 21 as a colorless solid (0.179 g, 99% yield). ESI-MS (m/z): [M + H]+ calcd, 628.18; found, 646.40 [M + H2O]+.
General Synthetic Procedure for Compound 22 ((2S,3S,4S,5R,6R)-3,4,5-Trihydroxy-6-((8-hydroxy-6-oxo-6H-benzo[c]chromen-3-yl)oxy) tetrahydro-2H-pyran-2-carboxylic Acid)
To a mixture of 21 (0.318 g, 0.506 mmol), KF (0.059 g, 1.012 mmol), and K2CO3 (0.140 g, 1.012 mmol), MeOH–H2O (10 mL, 5:1) was added. The resulting mixture was stirred at rt for 16 h. The solvent was removed under vacuum, and the mixture was dissolved in H2O and purified by RP-HPLC to afford 12 (0.094 g., 46% yield). ESI-MS (m/z): [M + H]+ calcd, 404.07; found, 403,10 [M – H]+.
HPLC–DAD-MS Analysis
Analysis of the synthesized standards and urine samples was developed using a 1200 HPLC chromatograph coupled in series with a photodiode array detector and a 6120 single-quadrupole mass spectrometer [HPLC–DAD-ESI-Q (MS)] (Agilent Technologies, Santa Clara CA). A method previously optimized for analyzing urolithins in biological samples was applied.9
NMR Analyses
The NMR spectra were recorded on a Brüker 500 MHZ Advance with a Cryofit (Bruker, Bremen, Germany) in dimethyl sulfoxide-d6 (DMSO-d6) with tetramethylsilane (TMS) as an internal standard. 1H NMR, 13C NMR, and 2D-HSQC experiments were completed.
Urine Collection from Volunteers Belonging to Urolithin Metabotypes A and B
Two healthy volunteers characterized as belonging to urolithin metabotypes A and B19 consumed 30 g of walnuts/day for 3 days. Walnuts were purchased at a local supermarket. After the last intake, a urine sample was collected and immediately stored at −20 °C, until analysis.21 Institutional ethical approvals were unnecessary as the experiments were carried out with freely available foodstuff, and only urine samples were collected, as advised by the Ethical Committee for previous studies.22 The volunteers gave written informed consent.
Results and Discussion
Synthesis of Isourolithin A (3,9-Dihydroxy Urolithin) Glucuronides
For isourolithin A 9-glucuronide (8), the synthetic sequence was initiated through the preparation of 9-O-methyl isourolithin A (3), in 72% yield, according to a known methodology based on the condensation between resorcinol (2) and a benzoic acid derivative (1).13,15,16 Next, protection of the phenolic group using TiPS-Cl in the presence of imidazole afforded the corresponding derivative 4 in 94% isolated yield after chromatographic purification. Very important in this approach is that the triisopropylsilyl protecting group in 4 showed to be stable enough toward the demethylation reaction with BBr3 in DCM at 0 °C, thus leading to pure phenol derivative 5 in 70% isolated yield after chromatographic purification.
Finally, glycosylation of the acceptor isourolithin derivative 5 with commercially available glucuronosyl donor 6 in using BF3·OEt2 as a promoter13 afforded 7 in 72% isolated yield. One-pot, simultaneous desilylation/saponification reaction of 7 with KF/K2CO3 in MeOH–H2O afforded the desired isourolithin A 9-glucuronide (8) in 48% after RP-HPLC purification and with >99% purity (Scheme 1).
Scheme 1. Synthesis of Isourolithin A 9-Glucuronide (8).
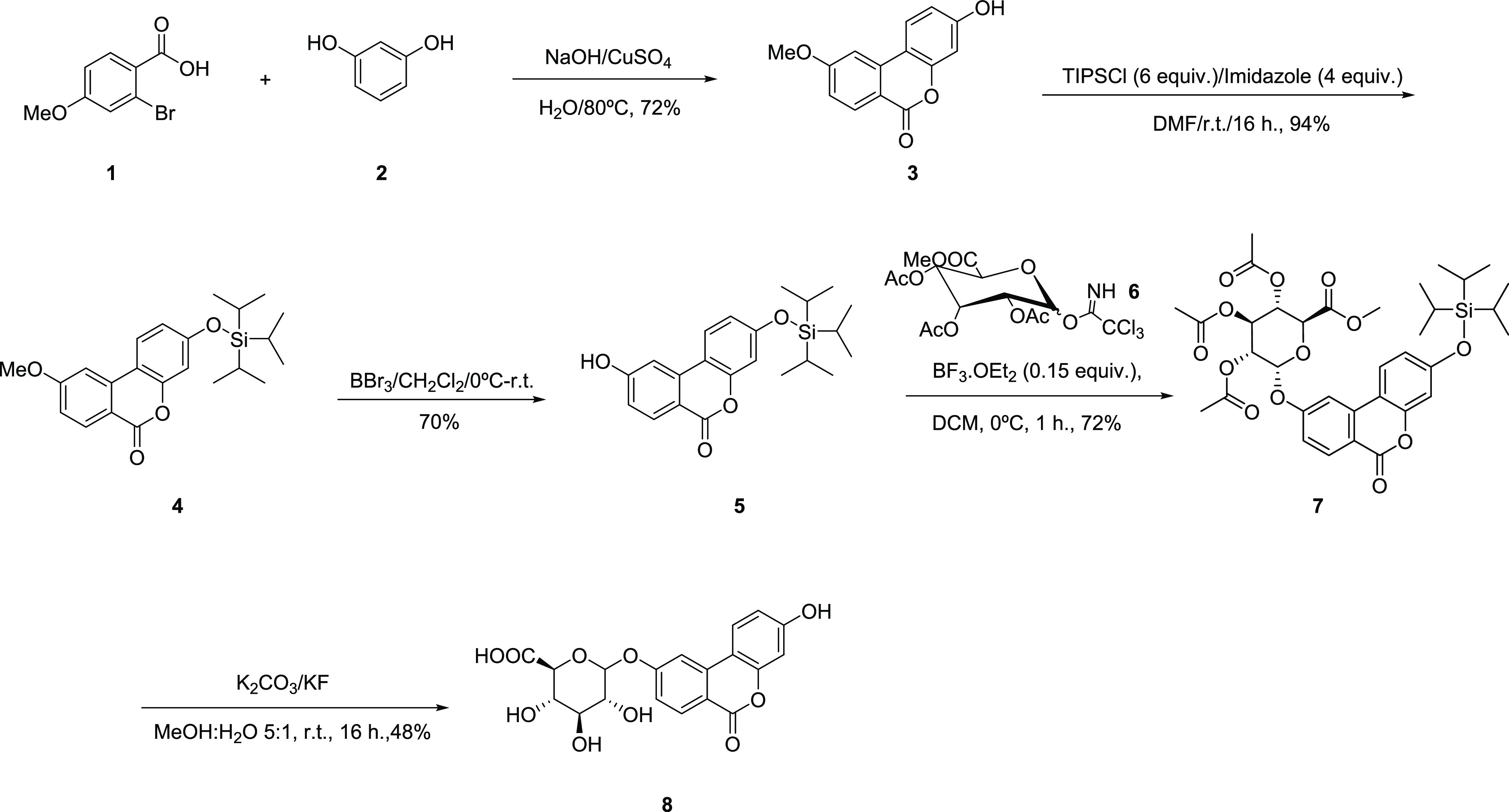
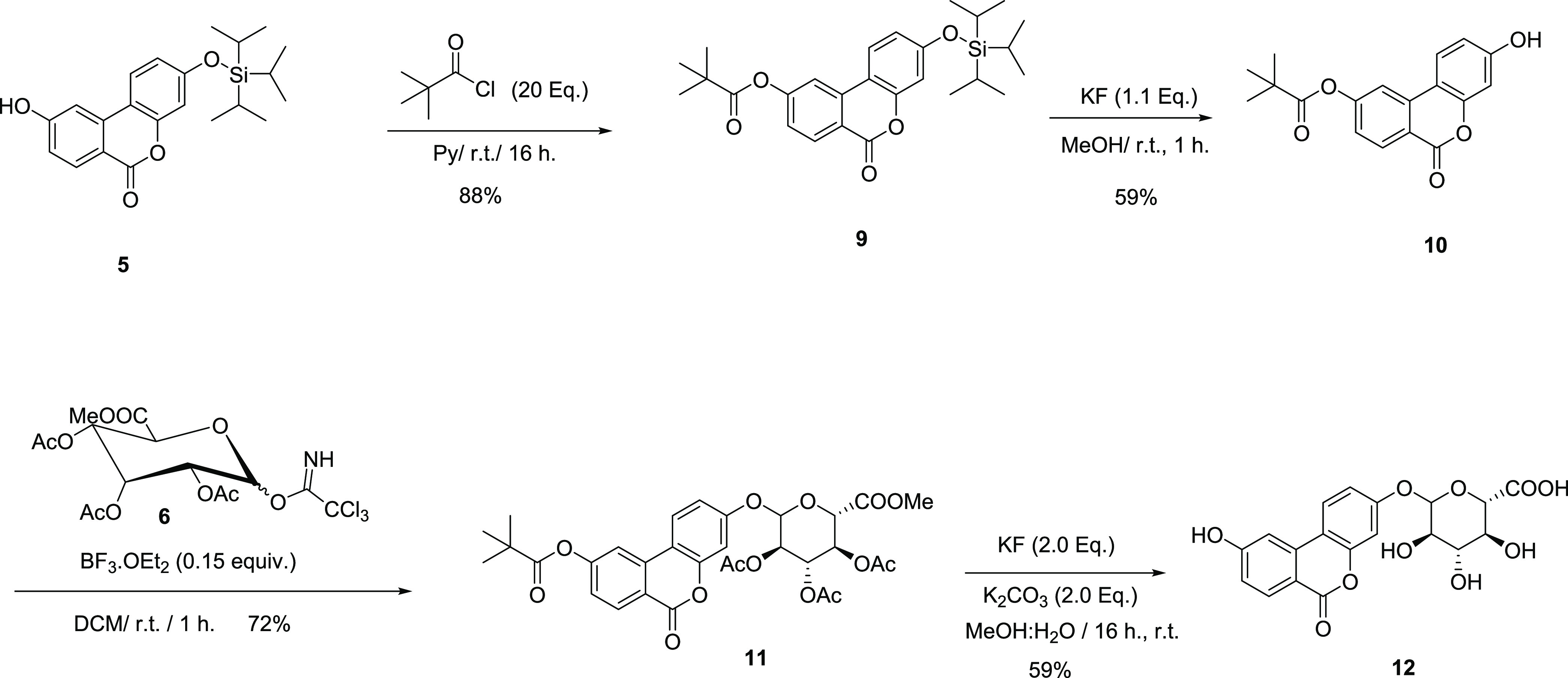
Alternatively, also from intermediate 5, protection of the free 9-OH with pivaloyl chloride under standard conditions afforded the fully protected intermediate 9 in 88% yield. Selective deprotection of the silyl group with KF in MeOH afforded the desired regioisomeric phenol 10 in 59% yield with a free −OH group now at the 3-position. Glycosidation of 10 promoted by BF3·Et2O under analogous conditions as for isourolithin A 9-glucuronide 8 afforded the fully protected glucuronide 11 (72%), which was completely saponified in a one-pot reaction with KF/K2CO3 in MeOH/H2O to afford the desired isourolithin A 3-glucuronide (12) in 49% isolated yield after RP-HPLC purification and with >99% purity (Scheme 2).
Scheme 2. Synthesis of Isourolithin A 3-Glucuronide (12).
Synthesis of Urolithin A (3,8-Dihydroxy Urolithin) Glucuronides
With a robust methodology in hands toward the regioselective preparation of isourolithin A 9- and 3-glucuronides in the pure form and satisfactory yields, 8 and 12, respectively, we next addressed the synthesis of urolithin A 3- and 8-glucuronides using essentially an analogous approach.
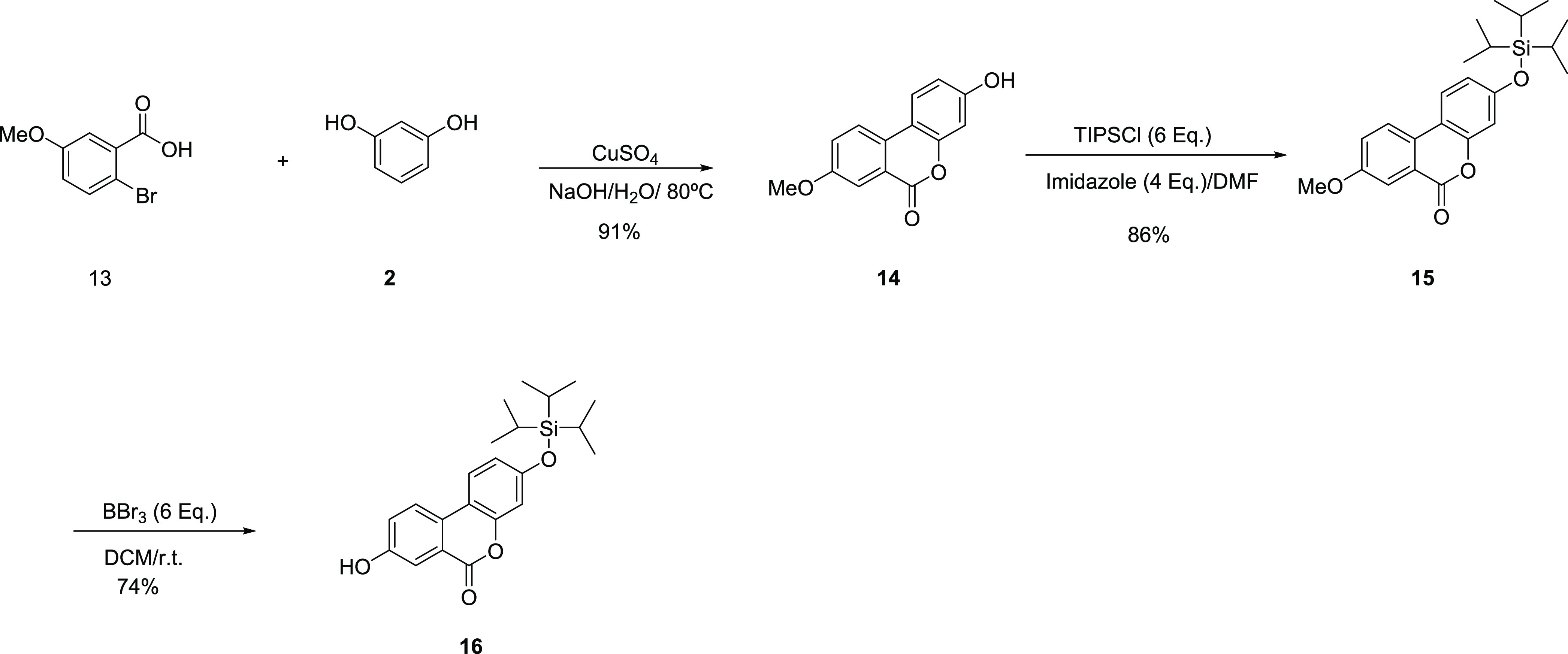
Thus, from commercially available bromobenzoic acid (13) and resorcinol (2), 8-methoxy urolithin A (14) was obtained under standard conditions of CuSO4 in basic media, in 91% isolated yield.13,15,16 Protection with TiPS-chloride led to 15 that underwent selective demethylation with BBr3 in DCM, producing the desired key intermediate 16 (Scheme 3).
Scheme 3. Synthesis of Urolithin A Intermediate 16.
Next, following the same strategy and analogous reaction conditions as those employed for isourolithin-A derivatives 8 and 12, glycosylation of 16 under standard conditions afforded intermediate 17 in 76% yield, which after subsequent one-pot, full deprotection, led to urolithin A 8-glucuronide (18) as a single regioisomer in 46% yield, after RP-HPLC purification and with >99% purity (Scheme 4).
Scheme 4. Synthesis of Urolithin A 8-Glucuronide (18) and 3-Glucuronide (22).
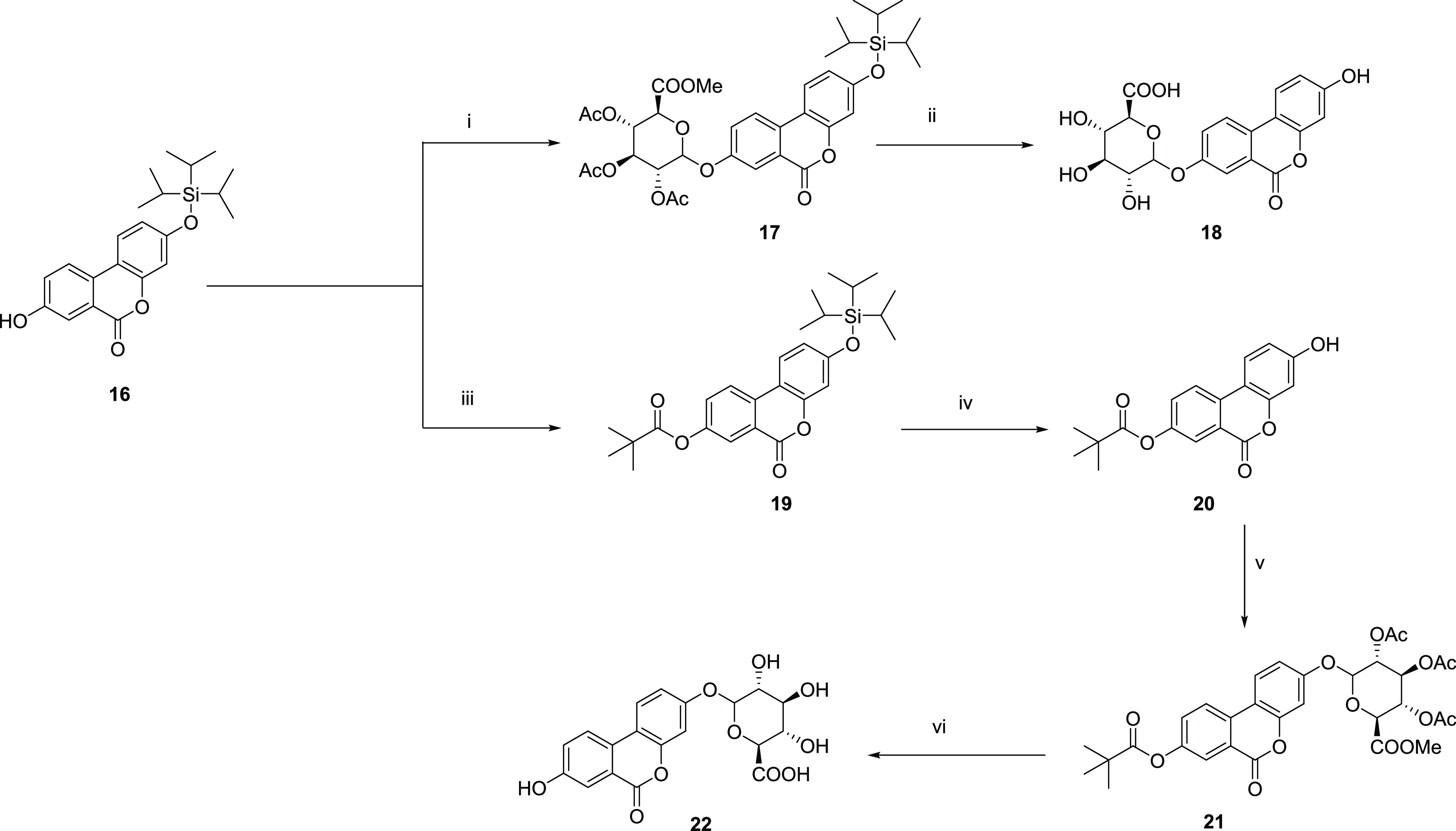
Reagents and conditions: (i) 6, BF3.Et2O (0.15 equiv), CH2Cl2, rt, 1 h., 76% yield; (ii) KF (2 equiv), K2CO3 (2 equiv), MeOH/H2O, rt, 16 h., 46% yield; (iii) pivaloyl chloride (20 equiv), Py, rt, 16 h; 71% yield; (iv) KF (1.1 equiv), MeOH, rt, 1 h., 75% yield; (v) 6, BF3.Et2O (0.15 equiv), CH2Cl2, rt, 1 h, 99% yield; and (vi) KF (2 equiv), K2CO3 (2 equiv), MeOH/H2O (5:1), rt, 16 h, 46%.
Alternatively, in turn, introduction of the pivaloyl group in 16 afforded 19 in 71% yield. Selective removal of the silyl protecting group using KF in MeOH led to the phenol 20 in 75% yield, which was subjected to glycosylation under analogous conditions to afford 21 and hence to the desired urolithin A 3-glucuronide (22) upon one-pot removal of the corresponding protecting groups in satisfactory overall yields and with >99% purity (Scheme 4).
Characterization of the Synthesized Urolithin Glucuronide Conjugates
The structures of the synthesized metabolites (Figure 1) were confirmed by 1H NMR and 13C NMR (500 MHz). The NMR results of the glucuronides dissolved in DMSO-d6 are summarized in Tables 1 and 2. The chemical shifts were consistent with those previously published for the available urolithin glucuronides isolated from human urine.24 However, in this previous study, some of the metabolites were not isolated due to difficulties in the chromatographic separation of urolithin A 3-glucuronide (22), urolithin A 8-glucuronide (18), and isourolithin A 9-glucuronide (12), which coeluted as a single peak, and the 1H NMR of the mixture was reported.24 All the synthesized urolithin conjugates were β-glucuronides as the H-1 anomeric signal (GlucU 1 in Table 1) appeared as a doublet between 5.13 and 5.51 ppm. Coupling constants of about 7 Hz were consistent with those values previously reported for urolithin glucuronides.24 The downfield shifts for H-2 and H-4 signals of the 3-glucuronides (at 6.82–7.03 and 6.74–7.07 ppm, respectively) confirmed the position of glucuronidation, which was consistent with previous results.24,25 A similar behavior was observed for H-7 and H-9 of the 8-glucuronide (shifts at 7.54–7.72 and 7.35–7.55 ppm, respectively) and also for the H-8 and H-10 of the 9-glucuronide (shifts at 6.48–7.18 ppm and 6.65–7.76 ppm, respectively) when compared with the results for the corresponding metabolites with free hydroxyls at the 3-, 8-, or 9-positions (Table 1).
Table 1. 1H NMR Spectroscopy Data (500 MHz) DMSO-d6a.

| metabolite | urolithin A 3-glucuronide | urolithin A 8-glucuronide | isourolithin A 3-glucuronide | isourolithin A 9-glucuronide | urolithin B 3-glucuronide |
|---|---|---|---|---|---|
| 8 | 12 | 18 | 22 | 23 | |
| protons | |||||
| 1 | 8.15 (d, J = 8.7) | 8.09 (d, J = 8.8) | 7.99 (d, J = 8.7) | 8.16 (d, J = 8.8) | 8.31 (d, J = 8.9) |
| 2 | 7.03 (dd, J = 8.7, 2.4) | 6.82 (dd, J = ′8.8, 2.3) | 6.98 (dd, J = 8.7, 2.1) | 6.83 (dd, J = 8.8, 2.3) | 7.08 (dd, J = 8.9, 2.4) |
| 3 | 10.27 | ||||
| 4 | 7.07 (d, J = 2.4) | 6.74 (d, J = 2.3) | 6.96 (d, J = 2.1) | 6.72 (d, J = 2.3) | 7.12 (d, J = 2.4) |
| 7 | 7.54 (d, J = 2.7) | 7.72 (d, J = 2.7) | 7.97 (d, J = 8.8) | 8.12 (d, J = 8.8) | 8.36 (d, J = 8.3) |
| 8 | 6.98 (dd, J = 8.8, 2.2) | 7.18 (dd, J = 8.8, 2.2) | 7.63 (t, J = 7.7) | ||
| 9 | 7.35 (dd, J = 8.8, 2.7) | 7.55 (dd, J = 8.9, 2.7) | 7.93 (t, J = 7.7) | ||
| 10 | 8.19 (d, J = 8.8) | 8.22 (d, J = 8.9) | 7.37 (d, J = 2.0) | 7.76 (d, J = 2.2) | 8.23 (d, J = 7.7) |
| GlucU 1 | 5.17 (d, J = 7.1) | 5.17 (d, J = 7.1) | 5.13 (d, J = 7.06) | 5.42 (d, J = 7.0) | 5.51 (d, J = 4.7) |
| GlucU 2 | 3.23–3.45 | 3.23–3.45 | 3.56–3.72 | 3.24–3.49 | 5.19–5.31 |
| GlucU 3 | 3.23–3.45 | 3.23–3.45 | 3.56–3.72 | 3.24–3.49 | 3.34–3.40 |
| GlucU 4 | 3.23–3.45 | 3.23–3.45 | 3.56–3.72 | 3.24–3.49 | 3.34–3.40 |
| GlucU 5 | 3.94 (d, J = 9.1) | 3.91 (d, J = 7.8) | 3.95 (d, J = 9.4) | 3.95 (d, J = 8.9) | 4.01 (d, J = 9.3) |
d: Doublet; dd: double doublet.
Table 2. 13C NMR Spectroscopy Data (500 MHz) DMSO-d6.

| metabolite | urolithin A 3-glucuronide | urolithin A 8-glucuronide | isourolithin A 3-glucuronide | isourolithin A 9-glucuronide | urolithin B 3-glucuronide |
|---|---|---|---|---|---|
| 8-hydroxy-urolithin 3-glucuronide | 3-hydroxy-urolithin 8-glucuronide | 9-hydroxy-urolithin 3-glucuronide | 3-hydroxy urolithin 9-glucuronide | Urolithin 3-glucuronidea | |
| carbonsb | |||||
| 1 | 124.19 | 124.82 | 124.79 | 125.61 | 125.29 |
| 2 | 114.09 | 113.62 | 113.85 | 113.49 | 114.04 |
| 3 | 157.94 | 159.69 | 159.24 | 162.83 | 159.06 |
| 4 | 104.33 | 103.34 | 104.15 | 103.33 | 104.34 |
| 4a | 151.00 | 151.83 | 152.47 | 152.93 | 152.29 |
| 6 | 160.87 | 160.82 | 160.61 | 160.65 | 160.91 |
| 6a | 121.21 | 120.53 | 111.04 | 113.46 | 119.99 |
| 7 | 113.98 | 115.38 | 132.67 | 132.59 | 130.19 |
| 8 | 158.03 | 156.67 | 117.52 | 117.18 | 128.89 |
| 9 | 124.59 | 125.34 | 164.63 | 160.58 | 135.86 |
| 10 | 124.55 | 124.09 | 106.98 | 107.36 | 122.63 |
| 10a | 112.86 | 109.81 | 112.13 | 109.77 | 112.45 |
| 10b | 126.72 | 130.18 | 137.15 | 137.74 | 135.00 |
| GlucU 1 | 100.09 | 100.72 | 99.99 | 99.40 | 99.90 |
| GlucU 2 | 73.35 | 73.45 | 73.41 | 73.37 | 73.31 |
| GlucU 3 | 76.31 | 76.11 | 76.70 | 76.49 | 76.20 |
| GlucU 4 | 71.86 | 71,87 | 72.29 | 71.92 | 71.76 |
| GlucU 5 | 75.58 | 75.73 | 74.66 | 75.40 | 75.78 |
| GlucU 6 | 170.82 | 170.75 | 172.22 | 171.00 | 170.59 |
The 13C NMR analysis (Table 2), DEP, and the HSQC analyses (Supporting Information Figure 1) also confirmed the structure of the synthesized urolithin glucuronide conjugates.
The HPLC analysis of the synthesized urolithin glucuronides also demonstrated that urolithin A 3-glucuronide (22) (8-hydroxy-urolithin 3-glucuronide) and the isomeric urolithin A 8-glucuronide (18) (3-hydroxy-urolithin-8-glucuronide) could not be resolved on reversed-phase columns. However, urolithin A 8-glucuronide eluted slightly earlier than the 3-glucuronide (Figure 2A). Isourolithin A 9-glucuronide (8) (3-hydroxy-urolithin 9-glucuronide) also eluted at a similar retention time, complicating the analysis. Only isourolithin A 3-glucuronide (12) (9-hydroxy-urolithin 3-glucuronide) and urolithin B glucuronide (23) (urolithin 3-glucuronide) were sufficiently resolved.
Figure 2.

HPLC–DAD chromatogram (305 nm) (A) and UV spectra (B) of the synthesized urolithin glucuronides. Urolithin A 8-glucuronide (18); urolithin A 3-glucuronide (22); isourolithin A 9-glucuronide (8); and isourolithin A 3-glucuronide (12).
The UV spectra of the different metabolites recorded by HPLC–DAD were similar to those previously published,9 with the most remarkable differences observed between the spectra of the urolithin A (3,8-dihydroxy-urolithin) and isourolithin A (3,9-dihydroxy-urolithin) conjugates (Figure 2B). The MSMS spectra revealed similar fragmentation patterns for all the compounds with the main fragments at m/z 227 and 113, as previously reported.9
Urine samples from individuals belonging to the main urolithin metabotypes A and B21 were collected after walnut ellagitannin intake (30 g walnuts for 3 days) and analyzed by HPLC (Figure 3). The results showed that isourolithin A 3- (12) and 9 -glucuronides (8) separated neatly. However, isourolithin A 9-glucuronide was not visible in the chromatogram from metabotype B urine as coeluted with the urolithin A glucuronides (18 and 22) (Figure 3B). The analysis of urine from the metabotype A individual revealed that the two urolithin A conjugates coeluted in a broad peak. Both metabolites were visible although not resolved in the chromatographic peak (Figure 3A). Urolithin A 8-glucuronide eluted first (18), and 3-glucuronide (22) eluted as a shoulder of the 8-glucuronide chromatographic peak.
Figure 3.

HPLC–DAD chromatograms at 305 nm of urine samples from volunteers belonging to urolithin metabotype A (A) and urolithin metabotype B (B). Urolithin A 8-glucuronide (18); urolithin A 3-glucuronide (22); isourolithin A 3-glucuronide (12); and urolithin B 3-glucuronide (23).
These results indicate that further studies are required to optimize the separation of all the possible urolithin conjugates in biological samples, to improve the urolithin metabotype assignment of individuals and to explore the glucuronyl transferase polymorphisms20 that can also affect inter-individual variations in ellagitannin metabolism and their effects in human health.26,27
Acknowledgments
The authors are grateful to María Dolores Frutos Lisón for her help with the HPLC analyses and to José Enrique Yuste for NMR assistance.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jafc.2c00170.
Full 2D-HSQC experiments of urolithin B 3-glucuronide and urolithin A 8-glucuronide (PDF)
The present study was funded by the project TURSP-HC2021/3 from Taif University, Saudi Arabia. “Polyphenols from fruits and vegetables relevant to Saudi Arabia. Their metabolism and bioactivation to enhance activity.”
The authors declare no competing financial interest.
Supplementary Material
References
- Cerdá B.; Espín J. C.; Parra S.; Martínez P.; Tomás-Barberán F. A. The potent in vitro antioxidant ellagitannins from pomegranate juice are metabolised into bioavailable but poor antioxidant hydroxy-6H-dibenzopyran-6-one derivatives by the colonic microflora in healthy humans. Eur. J. Nutr. 2004, 43, 205–220. 10.1007/s00394-004-0461-7. [DOI] [PubMed] [Google Scholar]
- Larrosa M.; García-Conesa M. T.; Espín J. C.; Tomás-Barberán F. A. ellagitannins, ellagic acid and vascular health. Mol. Aspects Med. 2010, 31, 513–539. 10.1016/j.mam.2010.09.005. [DOI] [PubMed] [Google Scholar]
- Espín J. C.; Larrosa M.; García-Conesa M. T.; Tomás-Barberán F. A. Biological significance of urolithins, the gut microbial ellagic acid-derived metabolites: The evidence so far. Evidence-Based Complementary Altern. Med. 2013, 2013, 270418 10.1155/2013/270418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomás-Barberán F. A.; González-Sarrías A.; García-Villalba R.; Nuñez-Sanchez M. A.; Selma M. V.; García-Conesa M. T.; Espin J. C. Urolithins, the rescue of ‘old’ metabolites to understand a ‘new’ concept; Metabotypes as a nexus between phenolic metabolism, microbiota dysbiosis and host health status. Mol. Nutr. Food Res. 2017, 61, 1500901 10.1002/mnfr.201500901. [DOI] [PubMed] [Google Scholar]
- Al-Harbi S. A.; Abdulrahman A. O.; Zamzami M. A.; Khan M. I. Urolithins: The gut based polyphenol metabolites of ellagitannins in cancer prevention. A review. Front. Nutr. 2021, 8, 647582 10.3389/fnut.2021.647582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Amico D.; Andreux A. A.; Valdés P.; Singh A.; Rinsch C.; Auwerx D. Impact of the natural compound urolithin A on health, disease and aging. Trends Mol. Med. 2021, 27, 687–699. 10.1016/j.molmed.2021.04.009. [DOI] [PubMed] [Google Scholar]
- García-Villalba R.; Gimenez-Bastida J. A.; Cortes-Martin A.; Avila-Galvez M. A.; Tomas-Barberan F. A.; Selma M. V.; Espin J. C.; Gonzalez-Sarrias A. Urolothins: a comprehensice update on their metabolism, bioactivity, and associated gut microbiota. Mol. Nutr. Food Res. 2022, e2101019 10.1002/mnfr.202101019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerdá B.; Periago P.; Espín J. C.; Tomás-Barberán F. A. Identification of urolithin A as a metabolite produced by human colon microflora from ellagic acid and related compounds. J. Agric. Food Chem. 2005, 53, 5571–5576. 10.1021/jf050384i. [DOI] [PubMed] [Google Scholar]
- García-Villalba R.; Espín J. C.; Tomás-Barberán F. A. Chromatographic and spectroscopic characterization of urolithins for their determination in biological samples after the intake of foods containing ellagitannins and ellagic acid. J. Chromatogr. A 2016, 1428, 162–175. 10.1016/j.chroma.2015.08.044. [DOI] [PubMed] [Google Scholar]
- Giménez-Bastida J. A.; González-Sarrías A.; Larrosa M.; Tomás-Barberán F. A.; Espín J. C.; García-Conesa M. T. Ellagitannin metabolites, urolithin A glucuronide and its aglycone urolithin A, ameliorate TNF-α induced inflammation and associated molecular markers in human aortic endothelial cells. Mol. Nutr. Food Res. 2012, 56, 784–796. 10.1002/mnfr.201100677. [DOI] [PubMed] [Google Scholar]
- Pearson A. G.; Kiefel M. J.; Feno V.; von Itzstein M. Towards the synthesis of aryl glucuronides as potential heparanase probes. An interesting outcome in the glycosidation of glucuronic acid with 4-hydroxycinnamic acid. Carbohydr. Res. 2005, 340, 2077–2085. 10.1016/j.carres.2005.06.029. [DOI] [PubMed] [Google Scholar]
- Khan M. K.; Rakotomanomana N.; Loonis M.; Dangles O. Chemical synthesis of citrus flavanone glucuronides. J. Agric. Food Chem. 2010, 58, 8437–8443. 10.1021/jf1010403. [DOI] [PubMed] [Google Scholar]
- Lucas R.; Alcántara D.; Morales J. C. A Concise Synthesis of Glucuronide metabolites of urolithin-B, resveratrol, and hydroxytyrosol. Carbohydr. Res. 2009, 344, 1340–1346. 10.1016/j.carres.2009.05.016. [DOI] [PubMed] [Google Scholar]
- González-Sarrías A.; Miguel V.; Merino G.; Lucas R.; Morales J. C.; Tomas-Barberan F. A.; Alvarez A. I.; Espín J. C. The Gut Microbiota Ellagic Acid-Derived Metabolite Urolithin A and Its Sulfate Conjugate Are Substrates for the Drug Efflux Transporter Breast Cancer Resistance Protein (ABCG2/BCRP). J. Agric. Food Chem. 2013, 61, 4352–4359. 10.1021/jf4007505. [DOI] [PubMed] [Google Scholar]
- Pandey J.; Jha A. K.; Hajela K. Synthesis and biological activities of some new dibenzopyranones and dibenzopyrans: search for potential oestrogen receptor agonists and antagonists. Bioorg. Med. Chem. 2004, 12, 2239–2249. 10.1016/j.bmc.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Orellana-Moraleda G.; Moreno-Bondi M. C.; Descalzo-Lopez A. B.; Urraca-Ruiz J. L.; Ahmed G. A.; Hany R.. Materials for Selective Recognition of Alternaria Mycotoxins (Alternariol, and Alternariol Monomethyl Ether). WO Patent WO2013144394A12013.
- Bialonska D.; Kasimsetty S. G.; Khan S. I.; Ferreira D. Urolithins, intestinal microbial metabolites of pomegranate ellagitannins, exhibit potent antioxidant activity in a cell-based assay. J. Agric. Food Chem. 2009, 57, 10181–10186. 10.1021/jf9025794. [DOI] [PubMed] [Google Scholar]
- Rinsch C. L.; Müller R.; Skrane W.. Process-Scale Synthesis of Urolithins. US Patent US9,394B22016.
- Noshadi B.; Ercetin T.; Luise C.; Yuksel M. Y.; Sippl W.; Sahin M. F.; Gazi M.; Gulcan H. O. Synthesis, characterization, molecular docking, and biological activities of some natural and synthetic urolithin analogs. Chem. Biodiversity 2020, 17, e2000197 10.1002/cbdv.202000197. [DOI] [PubMed] [Google Scholar]
- Nishioka A.; Tobaruela E. C.; Fraga L. N.; Tomás-Barberán F. A.; Lajolo F. M.; Hassimotto N. M. A. Stratification of volunteers according to flavanone metabolite excretion and phase II metabolism profile after single doses of ‘Pera’ orange and ‘Moro’ blood orange juices. Nutrients 2021, 13, 473. 10.3390/nu13020473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomás-Barberán F. A.; García-Villalba R.; González-Sarrías A.; Selma M. V.; Espín J. C. Ellagic acid metabolism by human gut microbiota: Consistent observation of three urolithin phenotypes in intervention trials, independent of food source, age, and health status. J. Agric. Food Chem. 2014, 62, 6535–6538. 10.1021/jf5024615. [DOI] [PubMed] [Google Scholar]
- Tomás-Navarro M.; Navarro J. L.; Vallejo F.; Tomás-Barberán F. A. Novel urinary biomarkers of orange juice consumption, inter-individual variability, and differences with processing methods. J. Agric. Food Chem. 2021, 69, 4006–4017. 10.1021/acs.jafc.0c08144. [DOI] [PubMed] [Google Scholar]
- Kay C. D.; Clifford M. N.; Mena P.; McDougall G. J.; Andres-Lacueva C.; Cassidy A.; Del Rio D.; Kuhnert N.; Manach C.; Pereira-Caro G.; Rodriguez-Mateos A. M.; Scalbert A.; Tomás-Barberán F. A.; Williamson G.; Wishart D. S.; Crozier A. Recommendations for standardizing nomenclature for dietary (poly)phenol catabolites. Am. J. Clin. Nutr. 2020, 112, 1051–1068. 10.1093/ajcn/nqaa204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piwowarski J. P.; Stanisławska I.; Granica S.; Stefańska J.; Kiss A. K. Phase II conjugates of urolithins isolated from human urine and potential role of β-glucuronidases in their disposition. Drug Metab. Dispos. 2017, 45, 657–665. 10.1124/dmd.117.075200. [DOI] [PubMed] [Google Scholar]
- Piwowarski J. P.; Granica S.; Stefańska J.; Kiss A. K. Differences in metabolism of ellagitannins by human gut microbiota ex vivo cultures. J. Nat. Prod. 2016, 79, 3022–3030. 10.1021/acs.jnatprod.6b00602. [DOI] [PubMed] [Google Scholar]
- Milenkovic D.; Morand C.; Cassidy A.; Konic-Ristic A.; Tomás-Barberán F. A.; Ordovas J. M.; Kroon P.; De Caterina R.; Rodriguez-Mateos A. Interindividual variability in biomarkers of cardiometabolic health after consumption of major plant-food bioactive compounds and the determinants involved. Adv. Nutr. 2017, 8, 558–570. 10.3945/an.116.013623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney E. R.; Milenkovic D.; Combet E.; Ruskovska T.; Greyling A.; González-Sarrías A.; de Roos B.; Tomás-Barberán F.; Morand C.; Rodriguez-Mateos A. Factors influencing the cardiometabolic response to (poly)phenols and phytosterols: A review of the COST action POSITIVe activities. Eur. J. Nutr. 2019, 58, 37–47. 10.1007/s00394-019-02066-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.