

Graphical Abstarct
We report innovative scalable, vertical, ultra-sharp nanowire arrays that are individually addressable to enable long-term, native recordings of intracellular potentials. Stable amplitudes of intracellular potentials from 3D tissue-like networks of neurons and cardiomyocytes are obtained. Individual electrical addressability is necessary for high-fidelity intracellular electrophysiological recordings. This study paves the way toward predictive, high-throughput, and low-cost electrophysiological drug screening platforms.
Keywords: nanowire, intracellular, neuron, cardiomyocyte, tissue, culture
Intracellular access with high spatiotemporal resolution can enhance our understanding of how neurons or cardiomyocytes regulate and orchestrate network activity, and how this activity can be affected with pharmacology or other interventional modalities. Nanoscale devices often employ electroporation to transiently permeate the cell membrane and record intracellular potentials, which tend to decrease rapidly with time. Here, we report innovative scalable, vertical, ultra-sharp nanowire arrays that are individually addressable to enable long-term, native recordings of intracellular potentials. We report electrophysiological recordings that are indicative of intracellular access from 3D tissue-like networks of neurons and cardiomyocytes across recording days and that do not decrease to extracellular amplitudes for the duration of the recording of several minutes. Our findings are validated with cross-sectional microscopy, pharmacology, and electrical interventions. Our experiments and simulations demonstrate that individual electrical addressability of nanowires is necessary for high-fidelity intracellular electrophysiological recordings. This study advances our understanding of and control over high-quality multi-channel intracellular recordings, and paves the way toward predictive, high-throughput, and low-cost electrophysiological drug screening platforms.
1. Introduction
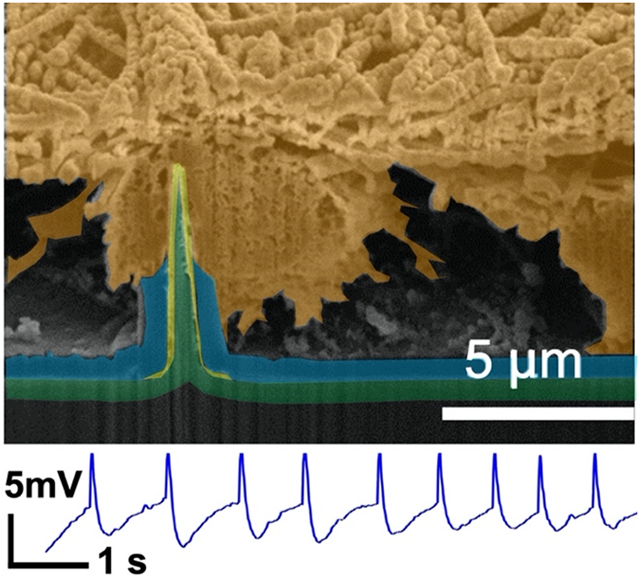
Reliable intracellular electrophysiological access of neurons is essential to properly measure and interrogate the ionic conductances that underscore neuronal activity. The gold standard technique for intracellular recordings is whole cell patch clamp electrophysiology, which has revolutionized our understanding of the neuronal dynamics of individual excitable cells. A typical patch clamp recording trace[1] and simplified circuit model of the patch pipette-neuron interface shown in Figure 1a, where high coupling coefficient and large dynamic range is observed through leak-tight, Giga-ohm seal interface between the pipette and the cell membrane. In patch clamp electrophysiology, access to the intracellular currents and potentials are obtained at the cost of the health of the cell; most intracellular recordings obtained with patch clamp are extremely labor intensive and provide less than 30-60 minutes of data.[2-4] Instead, evolving technological advances include vertical, nanoscale interface electrodes which are widely utilized to allow intracellular access and recording, with varied structures and interface coupling strategies such as membrane penetration, engulfment, electroporation, and optoporation.[5-17] These techniques can scale intracellular electrophysiological recordings to a large number of cells in extended networks with high spatial resolution. Additionally, due to their nanoscale size, they are minimally destructive and are envisioned to extend the recording duration for days to weeks, all while maintaining the high sensitivity comparable to that of patch clamp recordings. However, most state-of-the-art vertical nanoelectrodes are required to actively deliver electroporation currents to temporarily and destructively permeate neuronal membranes to have intracellular access. Unfortunately, that process is also accompanied with large, irreversible micrometer-scale gas bubble formations[5-7] due to water electrolysis at the surface of the high-impedance nanoelectrodes.
Figure 1.

a) Simplified circuit model and typical recording segment from patch clamp electrode-neuron interface from in vivo cat neocortical neurons. Reprinted with permission from Steriade et al. J. Neurophys., 2001, 85, 1969. b) Simplified circuit model and an example recording segment observed from mouse hippocampal neurons at 13 DIV for NW electrode-neuron interface with TDT system.
One successful example of the vertical electrode type is the μm-scale gold mushroom-shaped micro-electrode pillar arrays. The gold mushroom arrays demonstrated intracellular “in-cell” recordings with proper engulfment of the electrodes by the target cell membrane with a short electroporation pulse.[7] Xie et al.[8] similarly utilized electroporation with their sub-100 nm, 4x4 platinum nanopillar pad arrays to achieve intracellular action potential (AP) recordings that yielded maximum potentials of 11.8 mV post-electroporation, but which eventually attenuated to 30% of the initial amplitude after 2 minutes and to 1% after 10 minutes reaching an extracellular potential of 200 μV. Abbott et al.[10] attained intracellular recordings by electroporation from arrays composed of 4096 sites built atop complementary metal-oxide semiconductor (CMOS) acquisition electronics. Each electrode contact was composed of nine nanowires coated with low-impedance platinum-black which reduced bubble formation during electroporation. To eliminate the often-uncontrolled destructive process of electroporation, others have used nanowires to mechanically puncture the cell membrane to achieve intracellular recordings.[9, 16-17] Zhao et al.[17] gained intracellular access with their nanowire field-effect transistor into both neuron and cardiomyocytes in culture by probing the cells with an U-shaped 15-nm-diameter Si nanowire. Other nanostructures than 1D nanowires have also been investigated for intracellular recordings. Desbiolles et al.[11] achieved passive intracellular recording with their novel 3D nanovolcano structure comprising of sub-20 nm nanoring. The aforementioned advances have demonstrated intracellular recordings with either cardiomyocyte cells or neurons. Neuronal recording is more challenging because unlike cardiomyocytes, most neurons do not have regular, periodic firing, and also have interspersed axons that result in different waveform and potential dynamics based on the electrode and cell interface locations.[13] Furthermore, neurons possess relatively softer elastic characteristics and lower stiffness that require more contact forces to penetrate their membranes.[18-19] Of the vertical nanoelectrodes, ultra-sharp nanowires (USNWs) hold the most promise for recording intracellular potentials[20-25] and subthreshold synaptic potentials.[26] Generally, decreasing the NW tip diameter and increasing the NW height allows easier intracellular access via penetration.[19, 27-28] Using Si NWs with tip diameters as small as 60 nm, we recorded in vitro from mouse hippocampal neurons APs with amplitudes up to 99 mV in AP, a similar magnitude to that recorded with patch clamp, and with subthreshold sensitivity.[26] These potentials were recorded with a Tucker-Davis Technologies electrophysiological recording system using their MZ60 microelectrode array (MEA) interface with vertical NW array platform.[26] Using the same recording system with our USNWs subject of this work and the same mouse hippocampal neurons, we have also recorded APs with peak-to-peak amplitudes of 106 mV (Figure 1b). As illustrated in Figure 1b and further discussed in this work, APs recorded with NWs exhibit temporal broadening compared to APs recorded with patch-clamp due to increased time constants that arise from parasitic capacitances and large electrochemical interfacial impedance between the NW electrode and the intracellular medium, consistent with prior results.[6, 17, 26] To obtain reliable recordings from USNW arrays, we used a recording setup we have successfully implemented in recording electrophysiological activity from intact brains in birds,[29-30] rodents,[30-32], non-human primates,[30] and humans.[33-37] The maximum potentials that can be recorded with this setup using amplifiers from Intan Technologies LLC is ±6.4 mV. Therefore, the recorded action potentials for most of the measurements reported in this work are truncated at 6.4 mV. Despite these instrumentational shortcomings, we demonstrate that USNWs yield higher fidelity in intracellular recordings from neurons and cardiomyocytes than any prior nanoscale devices and illustrate with experiments and modeling that individual electrical addressability is an essential precursor for obtaining a large dynamic range in these recordings, a design flaw in prior work that used nanowires to interface with neurons and cardiomyocytes.
2. Results and Discussion
We demonstrate a scalable, silicon-based USNW array interface system that natively permeates cell membranes and records intracellular APs from cultured rat cortical neurons and from induced pluripotent stem cell (iPSC)-derived cardiovascular progenitor cells (iPSC-CVPCs). Critically, these intracellular recordings are achievable without electroporation. Each contact is composed of a small footprint metallic pad with a diameter of 2 μm, addressing an individual USNW electrode. We observed significantly higher peak-to-peak signal amplitude from recordings with a single USNW compared to those from multiple USNWs, demonstrating the importance of independent electrical addressability for recording high amplitude intracellular potentials.[26] Using sequential focused ion beam (FIB) sectioning to reveal the NW-neuron interface, we assessed the relative position of our metal-coated USNW tips with diameters in the range of 30 – 70 nm with respect to the neuronal cells. Significantly, we detected graded membrane potentials prior to the recorded APs from three-dimensional (3D) multi-layered ‘tissue’ like neuronal networks, justifying the innate ability of our USNWs to record subthreshold potentials, and pharmacologically modulated network activities. Our recordings were not affected by the maturation of the primary culture and glial proliferation, as we achieved high quality recordings from cultured primary rat cortical neurons up to 19 days in vitro (DIV). We also observed clear AP propagation in cardiac networks that can be interrogated by electrical stimulation. Our USNW platform development and their remaining limitations are discussed in detail, uncovering the promise of USNW-neuron interfaces and the challenges set forth to fulfill their full potential.
2.1. Fabrication of USNW Arrays
To achieve sub-10 nm vertical USNW arrays, we employed successive and selective oxidation and etching of top-down etched Si USNWs on a Si substrate, invoking standard integrated circuit fabrication technologies in innovative combinations. Figure 2 and Figure S1- S6 exhibit details of the fabrication process. Our process involves the dry etching of Si USNW arrays followed by selective oxidation and oxide etching to thin down the NW tips, and the electron-beam lithography and photolithography to attain individually addressable USNW arrays as illustrated in the process schematics (Figure S1). Achieving sub-10 nm diameter USNW tips involves multiple selective oxidations and oxide stripping steps (Figure 2b-f), which yield a fully oxidized Si NW with a smooth surface morphology (Figure 2g). Typical cycles of oxidation and stripping include 10 min – 2 hours of thermal oxidation at 1100 °C and 10 sec – 2 min of buffered oxide etch. To achieve electrical insulation in between individual USNWs, a final oxidation step is used to fully react with the Si NW and the surface of the substrate. The resulting SiO2 USNW is then selectively coated with a Pt metal layer (Figure S3) and a blanket plasma enhanced chemical vapor deposition (PECVD) step is used to deposit 500 nm thick SiO2 passivation layer above the metal leads which is then selectively etched to expose the NW tip (Figure 2b and Figure S4).
Figure 2.

Si USNW array fabrication and characterization of structure and the electrode-neural interface. Overview of the fabrication process. a) Top-view optical microscope image of patterned Ni dot arrays for Si nanowire etching. The center dot diameter was 800 nm. The peripheral dot diameter was 300 nm. b-d) Example SEM images that shows sequential oxidation and oxide stripping leading to reduced diameter of the NWs in the array, smoothening of the NW surface and the tapered structure. e,f) Example SEM images that shows dry etching, sequential oxidation and oxide stripping leading to smoothening of the surface of the USNWs in the array and the reduction of their diameter to sub-10 nm. g) SEM image of a sub-10 nm Si USNW tip prior to Pt coating. h) SEM image of a single Si USNW showing the tapered structure and exposed Pt tip, and i, overall view of Si USNW array. j-o) Colorized SEM images after rat cortical neuron cell fixation showing j) morphology of the cultured rat cortical neurons exhibiting high-density and neurite growth evidencing healthy cell culture and successful network formation, k) cross-section of the cultured neurons exhibiting a multi-layer structure, l) ‘satellite’-like interconnected multi-layer structures on the substrate surface, m) sequential FIB sectioning revealing first the tip of the USNW inside the soma and n) the whole USNW/neuron cross-section, o) wide-view SEM image showing the relative position of multiple USNW electrode with respect to the neuron’s somas.
Our first set of devices with 300 nm PECVD deposited SiO2 layer was not sufficient to passivate and prevent the delamination of the 10 nm Cr/100 nm Au metal interconnects in in vitro cell culture after 14 days (Figure S5a-c). We added a 10 nm thin Ti layer atop the Cr/Au metal leads to promote the adhesion between thinly formed TiO2 layer and the PECVD deposited SiO2 layer on top of the 10 nm Ti layer, and increased the thickness of the SiO2 layer to 500 nm. Accelerated aging experiments by submerging the device into saline solution at 60 °C for three days, equivalent of 15 days at 37 °C, demonstrated robustness of the platform against delamination (Figure S5d, e).
Deposited PECVD oxide layers that had a root-mean-square (rms) surface roughness of less than one nm were too smooth to promote neuronal cell culture adhesion even under the presence of adhesion promoters such as PEI or Matrigel.[38-39] We calibrated the surface roughness of SiO2 (thermally grown and PECVD deposited, Figure S6), and found that the rough surface with rms roughness of 2 - 5 nm promotes the cell adhesion according to our initial cell culture experiments. In addition to the SiO2 passivation, we added a 500 nm thick parylene C layer and roughened its surface with O2 plasma treatment (Figure S7; rms roughness of 7nm), resulting in better cell viability in comparison to the bare SiO2 surface. The mechanism for this increased cell viability is thought to arise from the inherent nanoscale surface roughness of parylene C and its stable hydrophilic surface for cell adherence after plasma treatment.[40]
Devices passivated with additional parylene C coating were used in our experiments. A picture of the overall packaged device is shown in Figure S8. The optimized surface materials and roughness were found to help neurite growth (Figure S12) and neuronal and cardiac network formation on the USNW array platform (Figure S13-17).
Figure 2j shows representative top-view scanning electron microscope (SEM) images of the cultured rat cortical neurons on the surface of our devices. We observed the formation of continuous layers (Figure S14a,b) and of satellite neuronal clusters (Figure S14c,d). FIB sectioning revealed that the cultured neurons exhibit multi-layer structure (Figure 2k and Figure S15: non-colorized SEM image) with extensive neurite connection that suggest excellent cell viability on parylene C-coated vertical USNW arrays. FIB-sectioning in the USNW array region illustrated that the USNW tip resides inside the neuron soma as shown in the time-sequenced SEM images (Figures 2m-o and Figure S16: non-colorized SEM images). While the resolution of these SEM images does not conclusively rule out that the USNW is indeed inside the soma, these images coupled with the recorded and relatively significant intracellular potentials support our hypothesis of intracellular access.
As fabricated, the USNWs exhibit impedances exceeding 10 MΩ at 1 kHz (Figure S9) that limit a stimulation current to several nanoamperes to avoid electrolyzing water (Figure S11a,c). However, in silent or moderately active recording, we don’t know a priori which USNWs are capable of intracellular stimulation. To enable extracellular current stimulation at the periphery of the USNW array, we selectively coated poly(3, 4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) on selected USNWs (Figure S10) which reduced the electrode impedance by ~ 50X and enabled microampere levels of current to stimulate without causing water electrolysis (Figure S11b,d). However, for all recordings in this manuscript, we did not use the PEDOT:PSS for stimulation. We stimulated the USNWs for 20 to 40 cycles of 10 nA, 500 μs current pulses injected by a 128-channel Intan RHS2000 stimulation controller and subsequently recorded the electrophysiological activity with the USNWs for several minutes post stimulation (see Supplementary Materials Section 5). Each tested device consisted of four arrays, 32 channels each, with a USNW pitch of 5, 10, 30, and 70 μm (Groups A, B, C, and D, respectively, Figure S4a-d).
2.2. In vitro Culture Recordings
2.2.1. Recordings from Rat Cortical Neurons
Multiple in vitro cultures from dissociated rat cortical neurons were recorded starting at 7 DIV. Recordings performed from 11 to 19 DIV exhibited diverse characteristics of biphasic and positive monophasic APs (Figure 3a-h). The raw signals show peak-to-peak amplitudes ranging from approximately 500 μV to 1 mV and upwards towards 10 - 12 mV. The maximum recorded potentials were truncated by the Intan amplifier recording limit of ±6.4 mV. Using USNW arrays with a different type of recording system, we have previously recorded isolated APs with 99 mV amplitude;[26] it is likely that the maximum amplitudes of the AP reported in this work exceed the ±6.4 mV limits of our recording system. The diverse waveform characteristics on adjacent channels at 11 DIV (Figure 3a) illustrate minimal cross-coupling in between channels in our USNW arrays. We demonstrate representative recordings from all 32 channels in group B (single array with 32 USNWs at 10 μm pitch) from 11 to 19 DIV (Figure 3 a-h). At earlier than 11 DIV (Figure 3a), we only observed a limited number of USNWs with intracellular activity (15/32 channels, determined by the amplitude and waveforms being recorded by each channel over 240 s). Starting from 13 DIV and onwards towards 19 DIV, almost all channels exhibited similar intracellular waveforms (31/32, 32/32, and 31/32 channels respectively for 13, 15, and 19 DIV), indicating synchronous AP firing and intimate, intracellular USNW-neuron interfaces. Development of multi-layered neural networks is observed directly by gradual increase in spike frequency with the neurons getting mature (Figure 3d,f,h and Figure S25b-d) and the evolution of cross-correlation between the channels from low to high synchrony (Figure S25e-h). The synchronized neuronal activity is a common phenomenon in neuronal cultures with multi-electrode arrays[41] and is related to the stage of neuron development and the neuron-glia interactions,[41-42] and plays a crucial role in complex brain function and neurological diseases.[43-45] The observed spike synchronicity likely originated from our multi-layered, largely neuronal cultures (Figure S12) with reduced isolation between neurons by glia and without the in vivo molecular heterogeneity composed of extracellular matrices that usually result in heterogeneity in firing in the brain. Thus, 13 and 15 DIV traces show two or multiple continuous spikes. Clear variance in spike counts across 32 channels in Figure 3g at 19 DIV and the evident variations in graded subthreshold potentials before AP spikes in Figure 3i confirm non-shorted, strong intracellular USNW-neuron interfaces across the recorded channels. This is further corroborated with evidence presented in this work that shorted nanowires must exhibit small AP amplitudes (Figure 5). By taking the difference in the spike counts (across the recording duration of 240 s) measured from each channel at 13, 15, and 19 DIV to the spike counts of the same channel at 11 DIV, a contour map across the USNW placement is obtained (Figure 3k and Figure S19): for longer culture duration, we observed expansion of contour regions from 13 to 19 DIV, signifying an increase in regions of spike activities from few local areas towards the majority of the USNW array. Between different recording days, channels observed shift in USNW-neuron interface conditions (visibly seen for channels 12 and 4 respectively in Figure 3c,g), corresponding to either neuron movement during culture or medium changes between days, which thereby may have exposed the particular USNWs to the culture medium. A representative channel of 7 s recording segment exhibiting significant intracellular-like APs is shown in Figure 3b,d,f,h across 11 - 19 DIV.
Figure 3.


Example electrophysiological recording from one array (pitch=10 μm) and pharmacological interrogation. a, c, e, g) Overall 5 s electrophysiological recording segments of multiple USNWs from group B (10 μm electrode pitch) at 11, 13, 15, and 19 DIV respectively. b, d, f, h) Close-up image of a 7 s recording segment of selected channel 31 from group B exhibiting intracellular AP waveforms at 11, 13, 15, and 19 DIV respectively. a) Overall 5 s recording segments at 11 DIV (different time segments are plotted for each channel to illustrate the overall electrophysiological activities at 11 DIV). b) Close-up image of 7 s recording segment, 11 DIV. c) Overall 5 s recording segments at 13 DIV. d) Close-up image of 7 s recording segment, 13 DIV. Potential shift in USNW-neuron interface is illustrated with green and purple borders (channels 12 and 4 respectively) between 13 and 19 DIV. e) Overall 5 s recording segments at 15 DIV. f) Close-up image of 7 s recording segment, 15 DIV. g) Overall 5 s recording segments at 19 DIV, exhibiting wide variance in spike counts across channels. h) Close-up image of 7 s recording segment, 19 DIV. i) Waterfall plot of APs and varying subthreshold potentials arranged in a descending order across 32 channels from group B at 19 DIV. j) Continuous, 6 minutes recording segment from channel 31 USNW from group B (10 μm electrode pitch) at 19 DIV, showing consistent high amplitude without attenuation. k) Contour map of normalized spike activity differences of 32 channels from 13, 15, and 19 DIV with spike activities from corresponding channels at 11 DIV, group B. The black, blue, and red contours represent regions exhibiting spike activity increases from 11 to 13 DIV, 11 to 15 DIV, and 11 to 19 DIV respectively. Contour colormap from 19 DIV is depicted (normalized to the maximum spike activity difference across 32 channels). l, m, n) Sequential recording segment of sequential pharmacological drug test performed at 8 DIV with 7nM CNQX, 33 nM DAP-5, 33 nM PTX, and 1 nM TTX. o, p, q) Representative spike plots from 8 DIV from baseline recording, after CNQX, DAP-5, and PTX application, and final washout. r, s, t) Sequential recording segment of sequential pharmacological drug test performed at 7 DIV with 33 nM PTX and 1 nM TTX. The arrow pointing at the initial section mark the exact moment the drug was applied to the cultured neurons. PTX application results in heightened spike frequencies and TTX application following PTX application and recording results in reduction in spiking activities. u, v, w) Plots of spike rate over time for baseline, PTX, and TTX recording segments. Spike rate change according to pharmacological drug applications.
Figure 5.

Sample recording traces and amplitude contrasts between individually addressable USNW and multiple USNWs per recording sites and small signal circuit model simulation results. a-f) SEM images of individually addressable, single USNW, 16 USNWs, and 625 USNWs and sample 10 s recording trace from recordings performed at 11 DIV. Single USNW - channel 31 from group B (10 μm electrode pitch), 16 USNWs – channel 31 from group B (10 μm electrode pitch), 625 USNWs – channel 21 from group C (30 μm electrode pitch). Vertical scale bars for zoomed-in recording segments for 16 USNWs and 625 USNWs are: 500 μV and 200 μV respectively. The time scale bar for both segments is 250 ms. g) Histogram of peak-to-peak signal amplitude between single USNW and multi-USNWs per site. There are clear, distinguishable differences in the distribution of amplitudes. Two distributions most likely correspond to the intracellular and extracellular recording interface setup the USNW formed with the neurons. h) Circuit model of single USNW penetrating the cultured neuron. i) Circuit model of multi-USNW setup with only one USNW penetrating the cultured neuron. j) Simulated intracellular signal amplitude based on the number of extracellular USNWs connected to the intracellular USNW. k) Simulated plot of coupling coefficient and temporal spreading coefficient versus the number of extracellular USNWs. Signal attenuation decays exponentially with increasing the number of extracellular USNWs.
In addition, the electrophysiological activity can be strongly modulated by pharmacological intervention. On 7 DIV, the addition of the GABAA receptor antagonist picrotoxin (PTX; 33 nM) gradually increased the frequency and the amplitude of APs, consistent with the loss of GABAergic inhibition (Figure 3s). In contrast, we found that the addition of the sodium channel blocker tetrodotoxin (TTX; 1 nM) suppressed firing (Figure 3t). To quantitatively evaluate the effect of pharmacological intervention, we implemented an auto-thresholding algorithm[46-48] on the high-pass filtered recorded waveforms from 32 channels, with a criterion of positive/negative threshold of amplitudes above standard deviation values of the baseline oscillation by 4 – 6.5 σ. As expected, the addition of PTX increased the spike frequency against the baseline recording (Figure 3v) and the application of TTX decreased the spike firing rates (Figure 3w). The change of electrophysiological activity after the drug applications lagged for both PTX and TTX, most likely due to the inherent delay arising from diffusion time of the applied drug molecules through overlaying neuronal layers (Figure 2k) to reach the bottom-most layer of recorded neurons. Additionally, on 8 DIV, another pharmacology modulation study was performed to validate that the pre-spike activities observed at Figure S18c-e were excitatory postsynaptic potentials (EPSPs). Starting from baseline recording with clear AP activities (Figure 3l, o), solutions of cyanquixaline (CNQX; 7 nM), D-(−)-2-Amino-5-phosphonopenatanoic acid (D-AP5; 33 nM), and picrotoxin (PTX; 33 nM) were added to respectively block AMPA, NMDA, and GABAA receptors. Following their applications, there were clear decrease in AP and pre-potential activities, as shown in Figure 3m, p and with noticeable reduction in spike frequency as shown in Figure S21g-i. After washout, the AP and pre-spike activities recover (Figure 3n, q), signifying that the pre-spike activities observed previously were most likely EPSPs.
Overall, we observed more than 46,000 intracellular AP spikes from the rat cortical neurons across 11 – 19 DIV on the 32 channels of group B. The longest recording segments with continuous, intracellular activities were approximately 6 minutes (Figure 3j and Figure S25). Our measured APs retain their amplitudes and shapes over the duration of the recording (Figure 3j and Figure S18). The histogram of the interspike interval (Figure S18c) reveals variability in spike bursting and an exponentially decaying tail, corresponding to AP refractory periods and spike triggering from random processes, alternating between resting and spiking phases. The mean and mode of the interspike intervals (Figure S18g) range from 500 to 700 ms. For a long recording segment at 11 DIV (Figure S18a), in the first 2 s time snippet (Figure S18b), we observed multiple lower-amplitude spikes within the dampened and broadened temporal response of the USNW array. Such spikes likely correspond to the superposition of high frequency AP spikes that are faster than the temporal response of the USNW array and in some recordings appeared at a small time offset from the peak of the AP (e.g., Figure 3h). For this channel, the spike rate was then modulated and was terminated at ~ 210 s. Smaller amplitude potentials that were recorded prior and within the spike trains appear after the 210 s (Figure S18f).
Given that our recording setup is limited to measuring amplitudes below 6.4 mV, we assessed the temporal spread of the waveforms, because it is well established in USNW recordings that longer AP durations are associated with larger AP amplitudes. We compared AP spikes above and below 5 mV, considering that the best amplifier linearity for the Intan amplifier is found was between −5 and 5 mV. Below 5 mV, the temporal spread has a distribution centered around 50 ms whereas those above 5 mV exhibited a temporal spread centered around 75 ms (Figure S20a), further suggesting the high sensitivity of our intracellular USNWs. There is a noticeable distribution difference in the spike width, providing evidence the signals were indeed clipped and with recording system with greater limits, the amplitude would have been larger than what was recorded.
2.2.2. Recordings from iPSC-derived Cardiovascular Progenitor Cells
We further investigated the capabilities of our USNW array platform in recording intracellular and network-level activity in vitro in iPSC-derived cardiovascular progenitor cells (iPSC-CVPCs)[49] with the culture procedures detailed in the Supplementary Materials Section 3.2. Figure 4a shows voltage traces for 52 channels of two separate arrays (out of 64 total channels, 32 channels per array) recorded from the iPSC-CVPCs at day 34 of differentiation (5 DIV on USNW platform). The zoom-in plot of single spikes across the array (Figure 4b) shows a clear peak time delay revealing tissue-wide propagation of APs, afforded by the high temporal resolution of the recording (0.033 ms for sampling rates at 30 kS s−1). Similar to the electrophysiological recordings from neurons, significant intracellular amplitudes from the iPSC-CVPCs were also recorded for the overall duration of the experiment. The maximum voltage amplitudes of the APs are clipped at 6.4 mV in our Intan setup (Figure S22); however, with the TDT system with its PZ5 Neurodigitizer with ±500 mV amplifier input range, we were able to record AP with a maximum peak-to-peak voltage of 60 mV (Figure 4e). The maximum voltage amplitudes of the representative channel at Figure 4c shows consistent firing with no amplitude decay during a 372 s recording time (beating interval of 2.05 s; Figure 4c,d). The iPSC-CVPCs’ recorded APs exhibit atrial-like intracellular attributes with minimal plateau phase, shorter durations, and higher spontaneous beating rates compared to ventricular-like APs.[50] We measured the time difference between the AP duration at 30% to 40% repolarization (APD30-40) and the time difference between APD70 to APD80 (APD70-80), and then the calculated ratio of APD30-40/APD70-80 is close to unity (Figure 4d), evidencing that the recorded shape is that of atrial-like quality. Furthermore, our iPSC-CVPCs cell line show atrial-like cell phenotype verified by immunofluorescence staining images from our past works.[49, 51] At the early stage of the iPSC differentiation, not all the ion channels are expressed; thus, the CMs cannot show signals as conveyed from mature CMs, such as ventricular-like signals with longer duration; rather, CMs at the early stage of the iPSC differentiation have demonstrated to have shorter APD.[52] In recent research by Zhang et al.[53] regarding iPSC-CMs differentiation with nearly identical cell type, cell age (D35), and protocol, atrial-like CM signals were recorded via patch clamp, with similar atrial-like AP shape and duration as our recorded iPSC-CVPC AP. Importantly, cells even at D60 derived in the same protocol have shown similar, atrial-like AP properties.[53]
Figure 4. Network-level intracellular recording of in vitro iPSC-derived cardiovascular progenitor cells (iPSC-CVPCs) and active spatiotemporal modulation of APs.

a) 52-Channel voltage traces of two arrays recorded from the iPSC-CVPCs at day 34 of differentiation (5 DIV). b) Zoom-in view of the second column of traces in a) shows a single-spiked AP recording. c) Intracellular recordings of cardiac activity from a representative channel (No. 48) show consistent spiked APs with no amplitude decay during the 372 s recording time. d) Zoom-in view of 47 randomly selected raw waveforms of a representative spike-sorting APs and spike averaged waveform, as shown in black. e) 60 mV AP measured via TDT system. f) Selected time segment of 52-channel voltage traces. g-j) Mapping of AP propagation patterns across the two arrays at different time points before (at 0.57 s in g) and at 75.06 s in h)) and after (at 238.76 s in i) and at 350.15 s in j)) electrical stimulation. Two intracellular recordings before electrical stimulation (at 0.57 s and at 75.06 s) show AP propagation from left to right, whereas intracellular recordings after electrical stimulation show an evolution from homogeneous propagation at 238.76 s to reversed direction that originates from right to left, where the AP propagation direction start from the simulating electrode. The original pacemaker foci location are labeled with arrows. The biphasic-pulse stimulation peak width, amplitude and frequency were 0.5 ms, 10 nA and 1 Hz, respectively.
Our USNW platform enabled us to natively record significant intracellular potentials, demonstrating network-level intracellular recording from the tightly connected in vitro cultured iPSC-CVPCs 2D tissues (Figure S17) covering two arrays of USNWs. We viewed the difference of the activation time across channels at the same time point (Figure 4f), and we mapped the AP propagation patterns across the two arrays at different time points before (at 0.57 s and at 75.06 s) and after (at 238.76 s and at 350.15 s) electrical stimulation (Figure 4g-j, and Figure S23,24). Two intracellular recordings before electrical stimulation (at 0.57 s and at 75.06 s) show AP propagation from left to right. We used active electrical stimulation and mapping capabilities to illustrate spatial modulation of the AP propagation direction within cardiac tissues. Specifically, a USNW–cardiac tissue sample with the original pacemaker foci located at channel No. 32 (Figure 4g) was sequentially paced by stimulator electrodes located at channel No. 52 (see Figure S24c). After electrical stimulation, the activity was synchronized across all channels (Figure 4i, 238.76 s) and then reversed propagation direction from right to left (Figure 4j, 350.15 s). The new AP propagation direction starts from the simulated electrode of channel No. 52 (see Figure S24c). The biphasic-pulse stimulation peak width, amplitude and frequency were 0.5 ms, 10 nA and 1 Hz, respectively, applied for 10 s. The results presented here show high-spatiotemporal-resolution electrophysiological mapping and simultaneous interrogation in cardiac tissues for control of cardiac activity, and offer the potential to affect several areas of cardiac research including in vitro models for drug-screening, and patient specific models related to cardiac differentiation from progenitor cells or stem cells into damaged tissues with integrated self-mapping and self-modulation functionality.
2.3. Performance from Individual Addressability of USNWs
2.3.1. Amplitude Comparison between Numbers of USNWs per Channel
Finally, we explored the benefit of conducting recordings with individually electrically addressable USNWs. The same fabrication process was implemented for USNW electrodes with 16 and 625 USNWs shorted on a single pad/channel (Figure 5a,c,e). Rat cortical neurons were simultaneously cultured on the three types of samples and they were all recorded from 11 DIV to 19 DIV. Sample recording traces for 10 s are shown for single USNW (Figure 5b), 16 USNWs (Figure 5d), and 625 USNWs (Figure 5f), where we observed the highest recorded APs with graded subthreshold potentials from the sample with an individual USNW per channel. The amplitude of the recorded potentials decreased with the increase of the number of USNWs per channel. To quantitatively assess the amplitudes of the recorded APs with respect to the number of USNWs per channel, we plotted the histogram of the peak-to-peak amplitudes in a semi-log scale (Figure 5g). We selected channels exhibiting high amplitude signals (15 to 32 out of 32 channels for single USNW, 27 out of 32 channels for 16 USNWs, 30 out of 64 channels for 625 USNWs from 11 to 19 DIV), and compared amplitude distribution with a similar number of detected spikes (2133 spikes, randomly truncated without repetitions from a total of around 6000 detected spikes, from one channel for single USNW, 2133 spikes for 16 USNWs, and 1773 spikes for 625 USNWs). As with pharmacological drug-modulated data analysis, spikes were sorted through an auto-threshold algorithm with bipolar thresholds (Supplementary Materials Section 6). The resulting amplitude distributions for different number of USNWs per site were disparate from one another: as the number of USNWs per site decreased, the distribution of peak-to-peak amplitude increased (Figure 5g). The amplitude distributions were centered around approximately 4 mV and 9 mV, 500 μV and 2 mV, and 60 μV and 300 μV for 1, 16, and 625 NWs per channel, respectively. The different amplitude distributions indicate variability in the capability of each type of USNWs for recording intracellular potentials. However, the plots together with the exhibited potential waveforms clearly demonstrate that individual USNWs per channel yield the highest amplitude APs and can record subthreshold potentials. Neither of these attributes can be clearly discerned from the recordings made with 16 and 625 USNWs per channel.
2.3.2. Small Signal Circuit Modeling
The circuit models presented in Figure 5h-i demonstrate a critical design flaw with placing multiple USNWs on a single recording channel, wherein the intracellular potential can be electrically shorted to the grounded extracellular potential. This model builds off the simulations done by Hai et al. [54] by investigating the effects of having a portion of the USNWs penetrate a cell body, while the remaining USNWs remain extracellular. The details of this model are discussed in the Supplementary Materials Section 7. Using measurements of transmembrane current during an AP to ground our models, we simulated the relevant signal paths for several situations. We first modeled the ideal case where a single USNW penetrates the cell membrane and forms a tight seal. We then modeled the effects of adding up to five additional extracellular USNWs on the same recording channel. The resulting potential waveforms shown in Figure 5i demonstrated a maximum signal amplitude for the single USNW case and a decrease in amplitude as we increased the number of extracellular USNWs. Furthermore, when we plotted the ratio of the peak intracellular potential over the peak potential seen by the amplifier (a proxy for signal gain), denoted as the coupling coefficient,[54] we saw that this coefficient decayed exponentially with increasing number of extracellular USNWs (Figure 5k). To develop intuition on the frequency-dependent signal distortion, we computed the ratio of pulse widths of the simulated signal at the input to the amplifier over that of the potential inside of the cell, which we denoted as a temporal spreading coefficient (Figure 5k). These modeling results agree well with the experimental results (Figure 5a-g).
3. Limitations
There are three main experimental limitations in this study: The first being that our experimental setup requires recording outside the cell culture incubator, where the duration of the recording is limited to several minutes before returning the devices to the incubator in order to maintain constant temperature CO2 concentration and pH levels and to avoid contamination,[55-56] thereby excluding our ability to assess the longitudinal intracellular recording capacity. This could be tested using cell culture methods compatible with an ex-incubator environment, e.g., HEPES-buffered media and a heated recording platform. Further, sample movements inside and out of the incubator could also alter the USNW-neuron interface to positively or negatively affect the recordings (provide intimate, intracellular interface or expose USNW to culture medium without neuron interface respectively). This limitation can be addressed by performing recordings inside the incubator, which has been demonstrated by Abbot et al.[10] The second limitation is that our recording amplifiers are designed for extracellular multi-electrode recordings, which do not perform parasitic capacitance cancellation, and as a result distorts the temporal fidelity of the recordings, broadens and attenuates the recorded waveforms. This could be addressed using amplifiers with a wide dynamic range for intracellular recordings from thousands of channels, which are available in research labs,[57] where specialized circuits can be custom-designed to restore the temporal resolution of the USNWs for known USNW and parasitic impedances. Lastly, our fabrication method involved multiple dry oxidation cycles to sharpen the USNW tips at 1100 °C, which is not CMOS compatible. We envision acquisition integrated circuits to be integrated on the periphery of our USNW arrays rather than below them to maintain the capabilities and the advantages that this platform brings over the state of the art.
4. Conclusion
The present study unequivocally demonstrates high yield fabrication process for long (> 6 μm) vertical USNW arrays with sub-10 nm tips that are individually electrically addressable for electrophysiological recordings of intracellular potentials. This work demonstrates that vertical USNW arrays and individual electrical addressability can record subthreshold and APs with significant amplitudes by the natural internalization. We mapped the neuronal/cardiac intracellular activity at the single cell resolution, observed clear synaptic network activity between neurons and cardiac activity propagation in the extended networks, manipulated the neuronal activities by pharmacological test, and manipulated the cardiac signal propagation direction by electrical stimulation. Using both experiments and simulations, we validated that multiple USNWs per single channel reduce the amplitude and sensitivity of the recordings compared to single USNW per channel recording. We believe that these results underscore a significant advancement in our understanding and control over the USNW-neural interface. Novel integration of the USNW with depth probes or 2D soft substrate may be applied in vivo for the potential intracellular recordings from intact brains. With the demonstrated sensitivity, this platform paves the way for novel scientific and technological undertakings that aim to establish large-scale bidirectional biotic-abiotic interfaces with intracellular access for drug screening, disease modeling, and beyond.
5. Experimental Section/Methods
5.1. USNW Electrode Design, Fabrication, Packaging, and Characterization:
Our Si USNW platform consists of total of 128 channels divided into 4 subgroups with different USNW pitch of 5, 10, 30, 70 μm (Figure S4) to provide a judicious range of USNW density to maximize the probability of membrane permeability by changing the electrode-neuron interface tension. Generally, narrower pitch requires taller nanoelectrodes for effective membrane penetration, which is accounted for with our USNW’s height, standing between 6 to 7 μm.[58]
The array fabrication begins with e-beam lithography (EBL) patterning of center dots with 800 nm diameter and peripheral dots with 300 nm diameter in the resist on the prime-grade p-type doped Si substrate (Figure 2a and Figure S1,2). 200 nm of Ni is then evaporated and lifted off, forming dot-like arrays that form the etching masks for Si. A SF6/C4F8 based inductively coupled plasma (ICP)/reactive ion etching (RIE) process is then utilized to selectively etch the uncovered Si substrate by 6–7 μm to form vertically standing Si nanowires (Figure S1,2). These nanowires initially have flat tips following the planar Ni discs. The resulting nanowires are then processed through multiple thermal oxidations and buffered oxide etch (BOE) cycles to provide smooth surface, achieve desired tapering for robust support, and thin USNW tips as shown in Figure S1,2. Following the USNW formation, the whole device undergoes a long, unmasked, thermal oxidation to fully oxidize the vertically standing USNWs and the surface of the substrate to electrically isolate all USNWs. Subsequently, patterns of USNW electrode and center interconnects are aligned and defined by EBL, and 10 nm/100 nm of Cr/Pt are deposited by conformal electron-beam evaporation through sample rotation to provide independent electrical addressability for each USNW (Figure S3). To avoid electrochemical coupling and corrosion in the culture medium, the metal interconnects were passivated by using PECVD SiO2. Control experiments demonstrated that a 500 nm thick SiO2 passivation layer is needed for stable passivation against delamination in a wet environment (Figure S5). Additionally, we employed a 500 nm thick parylene C layer for both passivation and improvement of adhesion of cells and substrate. Both the parylene C and the SiO2 layer were selectively etched from the USNW tips by recessing a resist layer and employing an O2 plasma etch for parylene C and BOE etch for SiO2, exposing the Pt coated USNW tips while the remained of the USNW remains unaltered (Figure S6,7). Alongside SiO2, parylene C exhibits excellent mammalian cell adhesion properties after proper surface preparation via O2 plasma treatment.[40] For in vitro cell culture, the final device is bonded with a culture chamber via polydimethylsiloxane (PDMS) application to delineate the region of cell culture interest with the rest of the device. Finally, the USNW electrodes are electrically connected with flexible flat cable (FFC) via anisotropic conductive film (ACF) bonding (Figure S8). The resulting USNW structure is composed of an exposed metal tip and a passivated bottom layer for good electrical isolation. The ideal USNW tips are sharpened to sub-10 nm range before conformal metal coating with proper masking and optimized etching process (Figure 2i, Figure S1f). Overall, USNWs formed consistently throughout the array with uniform electrochemical impedance. A representative characterization result of the 128 channel devices showed 100% yield and average impedance of 14.95 MΩ at 1 kHz in Figure S9.
5.2. Rat Cortical Neuron Culture on USNW Array Electrodes:
The integration of USNW array electrodes with cells followed immediately from fabrication and packaging of the devices, which involved the following steps (steps 1 to 4 were also applied before iPSC-CVPCs cultures):
(1) The USNW array device was sterilized by 1) DI water rinse for three times, and 2) 70% ethanol for at least 30 min.
(2) The device was washed with phosphate buffered saline (PBS) for three times followed by placing 20 μl drop of (0.1% w/v) Polyethylenimine (PEI, Sigma-Aldrich) solution on the USNW array and incubating them at 37 °C incubator for 1 hour.
(3) The PEI was aspirated and washed with 500 μl double distilled water (ddH2O) for 4 times. The device was dried in the incubator for 5 hours/overnight.
(4) A spot of 20 μl laminin (20 μg ml−1, Sigma-Aldrich) was added to the USNW array and incubated at 37 °C for 1 hour.
(5) Neuronal cell culture medium (Neurobasal (Thermo Fisher Scientific) +2% B27 (Thermo Fisher Scientific) +1% P/S (Corning) + 10% FBS) was prepared for plating rat cortical neurons on the USNW arrays.
(6) The cryopreserved rat neurons cryovial (from Thermo Fisher Scientific) was removed from the liquid nitrogen storage container, warmed in a 37 °C water bath for exactly 2.5 min, sprayed the outside with 70% ethanol, wiped dry, and placed in a tissue culture hood.
(7) The contents of the cryovial were carefully transferred to a 15 mL centrifuge tube using a 1 mL pipettor. The inside of the cryovial was carefully washed with 1 mL of room temperature neuronal cell culture medium (~1 drop s−1). 3 mL of room temperature neuronal cell culture medium was slowly added to the tube (~1-2 drops s−1). The contents were carefully mixed by inverting the tube 2-3 times. The total number of cells in suspension was determined via hemocytometer count.
(8) The cells were concentrated by centrifuging 1100 rpm for 3 min. Laminin was aspirated and 20 μl cell suspension was plated directly on the USNW arrays with the cell density at 200k cells/array. The USNW devices with seeded neurons were incubated in a cell culture incubator at 37 °C, 5% CO2 for 40-50 min. Next, 500 μL of warm Neurobasal medium was carefully added to chamber from the side of the device. To avoid adding the medium too fast to cause the detachment of the adhered neurons on the USNWs, the USNW devices with seeded neurons were put back to the cell culture incubator at 37 °C, 5% CO2 for another 35-40 min. Then, another 500 μL of warm Neurobasal medium was carefully added to chamber from the side of the device to reach a volume of 1.5 ml per device. Finally, the USNW devices with seeded neurons were put back to the cell culture incubator at 37 °C, 5% CO2.
(9) On the next day, the medium was changed to Brainphys Complete (Brainphys basal medium (Stemcell technologies)+1% N2 supplement (Thermofisher) +2% B27 supplement (Thermofisher) +1% Pen/Strep (Gibco/Life Technologies)) with the addition of the following supplements: Brainderived Neurotrophic Factor (BDNF, 20 ng ml−1; Peprotech,), Glia-derived Neurotrophic Factors (GDNF, 20 ng ml−1; Peprotech), ascorbic acid (AA, 200 nM; Sigma), dibutyryl cyclic AMP (cAMP, 1mM Sigma) and laminin (1 μg ml−1; Invitrogen). Then, half the medium was replaced with fresh medium every other day with Brainphys Complete and supplements till 7 DIV, then switched back to Brainphys Complete with no added supplements.
5.3. FIB-SEM Characterization of USNW-Neuron Interface:
To precisely investigate the biological interface formed between the USNWs and the cultured neurons after the completion of electrophysiological recording sessions, a sequential FIB cut was performed to reveal the cross-section of USNW regions after appropriate Pt plating for protection from ion-milling damages. The successive FIB cuts show both proper engulfment and clear intracellular penetration of the USNW into the cultured rat neurons’ soma, which align well to the positive-phase potentials measured in our electrophysiological recordings, shown in followed analysis. The intracellular nanoelectrode-neuron interface observed here is based on native penetration deriving from the ultra-sharp USNW tips, without any additional surface chemical treatments such as peptide-modification[59] and widespread electroporation for increased cell membrane permeability. The SiO2 USNWs were rigid and free-standing for stable interaction with the neurons without any USNW breakage.
5.4. Neuronal Recording and Pharmacological Drug Application:
Three minutes of baseline activity was recorded, followed by 33 nM application of the GABAA-R blocker picrotoxin (PTX, Tocris) to the device chamber. The activity was then recorded for 3 to 5 minutes to observe the effect of blocking inhibition on the electrical activity of the neuronal network. Finally, 1 nM tetrodotoxin (TTX, Abcam) was added to the solution to block voltage-gated sodium channels (Nav) and prevent generation of APs.
Supplementary Material
Acknowledgements
This work was performed with the gracious support of National Science Foundation Award No. 1728497 under the stewardship of Dr. Khershed Cooper and of the National Institutes of Health Award No. NBIB DP2-EB029757 under the stewardship of Dr. Michael Wolfson. S.A.D., J.S.M. and R.L. also acknowledge the gracious support of the UC-National Laboratory in Residence Graduate Fellowships (UC-NLGF), Award No. 477131. This work was performed, in part, at the Center for Integrated Nanotechnologies, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science. Los Alamos National Laboratory, an affirmative action equal opportunity employer, is managed by Triad National Security, LLC for the U.S. Department of Energy’s NNSA, under contract 89233218CNA000001 and Sandia National Laboratories (Contract No. DE-AC04-94AL85000) through a CINT user proposal. This work was performed in part at the San Diego Nanotechnology Infrastructure (SDNI) of UCSD, a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (Grant ECCS-1542148). The authors thank technical support from the Integration Laboratory at CINT and from the nano3 clean room facilities at UC San Diego’s Qualcomm Institute. R.L. and S.A.D. would like to acknowledge inspiring technical discussions with Dr. Renjie Chen, and Dr. Atsunori Tanaka, as well as discussions and technical support from Dr. Katherine L. Jungjohann, Anthony R. James, Douglas V. Pete, and Denise B. Webb of Sandia National Laboratories.
Footnotes
Supporting Information
Supporting information is available in the online version of the paper from the Wiley Online Library.
Contributor Information
Ren Liu, Integrated Electronics and Biointerfaces Laboratory, Department of Electrical and Computer Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA.
Jihwan Lee, Integrated Electronics and Biointerfaces Laboratory, Department of Electrical and Computer Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA.
Youngbin Tchoe, Integrated Electronics and Biointerfaces Laboratory, Department of Electrical and Computer Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA.
Deborah Pre, Conrad Prebys Center for Chemical Genomics, Sanford Burnham Prebys Medical Discovery Institute, 10901 North Torrey Pines Road, La Jolla, CA 92037, USA.
Andrew M. Bourhis, Integrated Electronics and Biointerfaces Laboratory, Department of Electrical and Computer Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
Agnieszka D'Antonio-Chronowska, Department of Pediatrics, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA.
Gaelle Robin, Conrad Prebys Center for Chemical Genomics, Sanford Burnham Prebys Medical Discovery Institute, 10901 North Torrey Pines Road, La Jolla, CA 92037, USA.
Sang Heon Lee, Integrated Electronics and Biointerfaces Laboratory, Department of Electrical and Computer Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA.
Yun Goo Ro, Integrated Electronics and Biointerfaces Laboratory, Department of Electrical and Computer Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA.
Ritwik Vatsyayan, Integrated Electronics and Biointerfaces Laboratory, Department of Electrical and Computer Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA.
Karen J. Tonsfeldt, Integrated Electronics and Biointerfaces Laboratory, Department of Electrical and Computer Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA; Center for Reproductive Science and Medicine, Department of Obstetrics, Gynecology, and Reproductive Sciences, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
Lorraine A. Hossain, Integrated Electronics and Biointerfaces Laboratory, Department of Electrical and Computer Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
M. Lisa Phipps, Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, NM 87545, USA.
Jinkyoung Yoo, Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, NM 87545, USA.
John Nogan, Center for Integrated Nanotechnologies, Sandia National Laboratories, Albuquerque, NM 87185, USA.
Jennifer S. Martinez, Center for Materials Interfaces in Research and Applications and Department of Applied Physics and Materials Science, Northern Arizona University, 624 S. Knoles Dr. Flagstaff, AZ 86011
Kelly A. Frazer, Department of Pediatrics, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
Anne G. Bang, Conrad Prebys Center for Chemical Genomics, Sanford Burnham Prebys Medical Discovery Institute, 10901 North Torrey Pines Road, La Jolla, CA 92037, USA
Shadi A. Dayeh, Integrated Electronics and Biointerfaces Laboratory, Department of Electrical and Computer Engineering, University of California San Diego, 9500 Gilman Drive, La Jolla, CA 92093, USA
Uncategorized References
- [1].Steriade M, Timofeev I, Grenier F, Journal of neurophysiology 2001, 85, 1969. [DOI] [PubMed] [Google Scholar]
- [2].Neher E, Sakmann B, Steinbach JH, Pflügers Archiv 1978, 375, 219. [DOI] [PubMed] [Google Scholar]
- [3].Martina M, Vida I, Jonas P, Science 2000, 287, 295. [DOI] [PubMed] [Google Scholar]
- [4].Henze D, Buzsaki G, Neuroscience 2001, 105, 121. [DOI] [PubMed] [Google Scholar]
- [5].Hai A, Shappir J, Spira ME, Nature methods 2010, 7, 200. [DOI] [PubMed] [Google Scholar]
- [6].Robinson JT, Jorgolli M, Shalek AK, Yoon M-H, Gertner RS, Park H, Nature nanotechnology 2012, 7, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Spira ME, Shmoel N, Huang S-HM, Erez H, Frontiers in neuroscience 2018, 12, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xie C, Lin Z, Hanson L, Cui Y, Cui B, Nature nanotechnology 2012, 7, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Duan X, Gao R, Xie P, Cohen-Karni T, Qing Q, Choe HS, Tian B, Jiang X, Lieber CM, Nature nanotechnology 2012, 7, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Abbott J, Ye T, Krenek K, Gertner RS, Ban S, Kim Y, Qin L, Wu W, Park H, Ham D, Nature biomedical engineering 2020, 4, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Desbiolles B, de Coulon E, Bertsch A, Rohr S, Renaud P, Nano letters 2019, 19, 6173. [DOI] [PubMed] [Google Scholar]
- [12].Desbiolles BX, de Coulon E, Maïno N, Bertsch A, Rohr S, Renaud P, Microsystems & nanoengineering 2020, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dipalo M, Amin H, Lovato L, Moia F, Caprettini V, Messina GC, Tantussi F, Berdondini L, De Angelis F, Nano letters 2017, 17, 3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dipalo M, Rastogi SK, Matino L, Garg R, Bliley J, Iachetta G, Melle G, Shrestha R, Shen S, Santoro F, Science Advances 2021, 7, eabd5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lin ZC, McGuire AF, Burridge PW, Matsa E, Lou H-Y, Wu JC, Cui B, Microsystems & nanoengineering 2017, 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Qing Q, Jiang Z, Xu L, Gao R, Mai L, Lieber CM, Nature nanotechnology 2014, 9, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhao Y, You SS, Zhang A, Lee J-H, Huang J, Lieber CM, Nature nanotechnology 2019, 14, 783. [DOI] [PubMed] [Google Scholar]
- [18].Tyler WJ, Nature Reviews Neuroscience 2012, 13, 867. [DOI] [PubMed] [Google Scholar]
- [19].Xie X, Xu AM, Angle MR, Tayebi N, Verma P, Melosh NA, Nano letters 2013, 13, 6002. [DOI] [PubMed] [Google Scholar]
- [20].Kim W, Ng JK, Kunitake ME, Conklin BR, Yang P, Journal of the American Chemical Society 2007, 129, 7228. [DOI] [PubMed] [Google Scholar]
- [21].Piret G, Perez M-T, Prinz CN, Biomaterials 2013, 34, 875. [DOI] [PubMed] [Google Scholar]
- [22].Hanson L, Lin ZC, Xie C, Cui Y, Cui B, Nano letters 2012, 12, 5815. [DOI] [PubMed] [Google Scholar]
- [23].Gällentoft L, Pettersson LM, Danielsen N, Schouenborg J, Prinz CN, Linsmeier CE, Biomaterials 2015, 42, 172. [DOI] [PubMed] [Google Scholar]
- [24].Patolsky F, Timko BP, Yu G, Fang Y, Greytak AB, Zheng G, Lieber CM, Science 2006, 313, 1100. [DOI] [PubMed] [Google Scholar]
- [25].Cohen-Karni T, Qing Q, Li Q, Fang Y, Lieber CM, Nano letters 2010, 10, 1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Liu R, Chen R, Elthakeb AT, Lee SH, Hinckley S, Khraiche ML, Scott J, Pre D, Hwang Y, Tanaka A, Nano letters 2017, 17, 2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lou H-Y, Zhao W, Zeng Y, Cui B, Accounts of chemical research 2018, 51, 1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Capozza R, Caprettini V, Gonano CA, Bosca A, Moia F, Santoro F, De Angelis F, ACS applied materials & interfaces 2018, 10, 29107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hermiz J, Hossain L, Arneodo EM, Ganji M, Rogers N, Vahidi N, Halgren E, Gentner TQ, Dayeh SA, Gilja V, Frontiers in neuroscience 2020, 14, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ganji M, Paulk AC, Yang JC, Vahidi NW, Lee SH, Liu R, Hossain L, Arneodo EM, Thunemann M, Shigyo M, Nano letters 2019, 19, 6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hossain L, Thunemann M, Lee K, Dayeh S, Devor A, presented at Optical Techniques in Neurosurgery, Neurophotonics, and Optogenetics 2021. [Google Scholar]
- [32].Ryu SB, Paulk AC, Yang JC, Ganji M, Dayeh SA, Cash SS, Fried SI, Lee SW, Journal of Neural Engineering 2020, 17, 056036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rogers N, Hermiz J, Ganji M, Kaestner E, Kılıç K, Hossain L, Thunemann M, Cleary DR, Carter BS, Barba D, PLoS computational biology 2019, 15, e1006769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Paulk AC, Yang JC, Cleary DR, Soper DJ, Halgren M, O’Donnell AR, Lee SH, Ganji M, Ro YG, Oh H, Cerebral Cortex 2021, 31, 3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yang JC, Paulk AC, Salami P, Lee SH, Ganji M, Soper DJ, Cleary D, Simon M, Maus D, Lee JW, Clinical Neurophysiology 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hermiz J, Rogers N, Kaestner E, Ganji M, Cleary DR, Carter BS, Barba D, Dayeh SA, Halgren E, Gilja V, NeuroImage 2018, 176, 454. [DOI] [PubMed] [Google Scholar]
- [37].Ganji M, Kaestner E, Hermiz J, Rogers N, Tanaka A, Cleary D, Lee SH, Snider J, Halgren M, Cosgrove GR, Advanced Functional Materials 2018, 28, 1700232. [Google Scholar]
- [38].Khan SP, Auner GG, Newaz GM, Nanomedicine: Nanotechnology, Biology and Medicine 2005, 1, 125. [DOI] [PubMed] [Google Scholar]
- [39].Bonde S, Berthing T, Madsen MH, Andersen TK, Buch-Månson N, Guo L, Li X, Badique F, Anselme K, Nygård J, ACS applied materials & interfaces 2013, 5, 10510. [DOI] [PubMed] [Google Scholar]
- [40].Chang TY, Yadav VG, De Leo S, Mohedas A, Rajalingam B, Chen C-L, Selvarasah S, Dokmeci MR, Khademhosseini A, Langmuir 2007, 23, 11718. [DOI] [PubMed] [Google Scholar]
- [41].Huang Y-T, Chang Y-L, Chen C-C, Lai P-Y, Chan C, PloS one 2017, 12, e0187276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lam D, Enright HA, Cadena J, Peters SK, Sales AP, Osburn JJ, Soscia DA, Kulp KS, Wheeler EK, Fischer NO, Scientific reports 2019, 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tang-Schomer MD, Jackvony T, Santaniello S, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wachowiak M, Wesson D, Pírez N, Verhagen J, Carey R, Annals of the New York Academy of Sciences 2009, 1170, 286. [DOI] [PubMed] [Google Scholar]
- [45].Bonnefond M, Kastner S, Jensen O, eneuro 2017, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tanskanen JM, Kapucu FE, Vornanen I, Hyttinen JA, presented at 2016 24th European Signal Processing Conference (EUSIPCO) 2016. [Google Scholar]
- [47].Tanskanen JM, Kapucu FE, Hyttinen JA, presented at 2015 7th International IEEE/EMBS Conference on Neural Engineering (NER) 2015. [Google Scholar]
- [48].Tanskanen JM, Kapucu FE, Vornanen I, Lenk K, Hyttinen J. [Google Scholar]
- [49].D'Antonio-Chronowska A, Donovan MK, Greenwald WWY, Nguyen JP, Fujita K, Hashem S, Matsui H, Soncin F, Parast M, Ward MC, Stem cell reports 2019, 13, 924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Doss MX, Di Diego JM, Goodrow RJ, Wu Y, Cordeiro JM, Nesterenko VV, Barajas-Martinez H, Hu D, Urrutia J, Desai M, PloS one 2012, 7, e40288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].D’Antonio-Chronowska A, D’Antonio M, Frazer KA, Bio-protocol 2020, 10, e3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ma J, Guo L, Fiene S, Anson B, Thomson J, Kamp T, Kolaja K, Swanson B, January C, American journal of physiology Heart and circulatory 598 physiology 2011, 301, 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Zhang JZ, Termglinchan V, Shao N-Y, Itzhaki I, Liu C, Ma N, Tian L, Wang VY, Chang AC, Guo H, Cell Stem Cell 2019, 24, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hai A, Dormann A, Shappir J, Yitzchaik S, Bartic C, Borghs G, Langedijk J, Spira ME, Journal of The Royal Society Interface 2009, 6, 1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rajan DK, Verho J, Kreutzer J, Välimäki H, Ihalainen H, Lekkala J, Patrikoski M, Miettinen S, presented at 2017 IEEE International Symposium on Medical Measurements and Applications (MeMeA) 2017. [Google Scholar]
- [56].Michl J, Park KC, Swietach P, Communications biology 2019, 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang J, Paul A, Zhang D, Wu J, Xu Y, Zou Y, Kim C, Cauwenberghs G, presented at 2020 IEEE Symposium on VLSI Circuits 2020. [Google Scholar]
- [58].Khraiche ML, El Hassan RH, Journal of Science: Advanced Materials and Devices 2020. [Google Scholar]
- [59].Lee J-H, Zhang A, You SS, Lieber CM, Nano letters 2016, 16, 1509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.