

Abstract
Background
Listeria monocytogenes (Lm) present in farming soil and food‐processing facilities threatens food safety, but little is known about the carriage of Lm by wildlife.
Objectives
We estimated the prevalence of faecal Lm shedding among wildlife admitted to a veterinary medical teaching hospital in central New York and characterized a subset of the Lm isolates.
Methods
Wildlife samples were collected between May 2018 and December 2019. We characterized the Lm isolates by assessing the growth at three temperatures approximating the body temperatures of reptiles (25°C), mammals (37°C), and birds (42°C) and identifying genotypic characteristics related to transmission and virulence.
Results
The apparent prevalence of faecal Lm shedding was 5.6% [18/324; 95% confidence interval (CI), 3.3%–8.6%]. Among 13 isolates that represented two lineages and 11 clonal complexes, three and five isolates were grouped into the same SNP clusters with human clinical isolates and environmental isolates, respectively. However, specific SNP difference data showed that Lm from wildlife was generally not closely related (>22 SNP differences) to Lm from human clinical sources and the food‐processing environment. While the stress response locus SSI‐2 was absent, SSI‐1 was found in four isolates. Virulence genes prfA, plcA, hly, mpl, actA, plcB, inlA, inlB, inlC, inlE, inlH, inlJ, and inlK were present, without any premature stop codons, in all isolates. Virulence loci Listeria pathogenicity island 3 (LIPI‐3) and LIPI‐4, which have been linked to hypervirulence, and inlG were found in four, three, and seven isolates, respectively.
Conclusions
Wildlife represents a potential reservoir for genetically diverse and putatively hypervirulent Lm strains. No statistically significant association between growth parameters and hosts was observed. However, compared to lineage I isolates, lineage II isolates showed significantly (p < 0.05) faster growth at 25°C and significantly slower growth at 42°C, suggesting that wildlife Lm isolates that belong to lineages I and II differ in their ability to grow at 25°C and 42°C.
Keywords: epidemiology, Listeria, wildlife, zoonoses
We estimated the prevalence of fecal Listeria monocytogenes (Lm) shedding among wildlife admitted to a veterinary medical teaching hospital and characterized the isolates. The apparent prevalence of fecal Lm shedding among central New York wildlife was approximately 6%. The Lm isolates had no mutations associated with virulence attenuation and harbored genes associated with hypervirulence; thus, wildlife may be reservoirs or carriers of potentially hypervirulent Lm.

1. INTRODUCTION
Listeria monocytogenes (Lm) is a Gram‐positive, rod‐shaped, intracellular foodborne pathogen that causes listeriosis. The consequences of listeriosis in people include septicaemia, encephalitis, meningoencephalitis, abortion, and even death (Vazquez‐Boland, Dominguez‐Bernal, et al., 2001). Among the major foodborne pathogens, Lm accounts for an estimated 19% of all deaths due to domestically acquired foodborne illness caused by known pathogens in the United States (Scallan et al., 2011). Based on genetic characterization, Lm is divided into four evolutionary lineages (I, II, III, and IV) (Orsi et al., 2010). Most Lm isolated from natural environments and farm animals belong to lineages I and II (Sauders et al., 2006). Lineage I is typically over‐represented among human listeriosis cases, while lineage II is typically over‐represented among environmental and food isolates; however, lineage II isolates also cause a number of sporadic human cases of listeriosis (Gray et al., 2004). Even though lineage II is widespread, most human listeriosis outbreaks are associated with lineage I (Vazquez‐Boland, Dominguez‐Bernal, et al., 2001). Lineage III is more frequently found in animal cases of listeriosis than in human listeriosis (Jeffers et al., 2001; Roberts et al., 2006). Lineage IV was first reported as a separate lineage in 2008; prior to this, lineage IV isolates were classified as lineage III (Ward et al., 2008). While specific data on lineage IV frequency are thus difficult to ascertain from studies published before 2008, both lineages III and IV appear to be rarely isolated from various sources, specifically food and environmental samples.
Lm virulence depends on several virulence factors, most of which are present in the Listeria pathogenicity island 1 (LIPI‐1). LIPI‐1 contains prfA, which encodes the major positive transcriptional regulator of other virulence genes, as well as hly, plcA, plcB, actA, and mpl, which are involved in the Lm intracellular life cycle and help Lm escape from the phagosomes inside animal host cells (Radoshevich & Cossart, 2018; Vazquez‐Boland, Kuhn, et al., 2001). Other Lm virulence factors include more than 25 internalin proteins, which are encoded in a number of distinct loci throughout the Lm genome. Some of these internalin proteins have been shown to play an important role in host–cell invasion; among them, internalin A (encoded by inlA) is particularly crucial for the virulence of Lm (Lecuit et al., 2001). Internalin A is a surface molecule that interacts with the host cell surface protein E‐cadherin and is critical for Lm invasion into the host intestinal epithelial cells. Sequences of inlA with premature stop codons (PMSC) leading to a truncated internalin A protein are associated with compromised invasion capability and hypovirulence (Nightingale et al., 2008, 2005; Orsi et al., 2007). Compared to lineage I, lineage II isolates more commonly show hypovirulence due to premature stop codon mutations in inlA as well as other virulence genes (Nightingale et al., 2005; Orsi et al., 2007; Roche et al., 2005). While some Lm subtypes are hypovirulent, other Lm subtypes have been hypothesized to be hypervirulent based on their increased infectivity and severity of symptoms in infected individuals (Maury et al., 2016). For example, subtypes associated with infection of the human central nervous system and maternal‐neonatal infections have been considered hypervirulent (Maury et al., 2016). Lm hypervirulence is also associated with presence of Listeria pathogenicity island 3 (LIPI‐3) and Listeria pathogenicity island 4 (LIPI‐4) (Cotter et al., 2008; Maury et al., 2016).
Lm can survive and grow under different stress conditions including low pH, high osmolarity, low temperature, and oxidative stress, making it a difficult organism to control in food‐processing settings. In addition, the ability to survive in certain stress conditions allows Lm to infect a wide range of animal hosts. The general alternative sigma factor, Sigma B, is responsible for regulating the expression of several genes involved in stress survival, including the genes present in the stress survival islet 1 (SSI‐1). SSI‐1 and SSI‐2 are islets containing genes involved in survival and growth under high osmolarity and low pH (Ryan et al., 2010) and survival under high pH and oxidative stress (Harter et al., 2017), respectively.
The ability to survive and grow under different conditions likely facilitates the wide distribution and frequent presence of Lm across different environments, leading to Lm often being characterized as ‘ubiquitous.’ For example, Lm has frequently been found in soil and water in natural and urban environments, and it can contaminate food‐processing plants where it may subsequently contaminate food products. Likewise, Lm has been isolated from different animals, including reptiles, birds, and mammals. The reported Lm prevalence in wildlife varies across multiple studies depending on habitats and hosts. Studies in China and the Mediterranean found relatively low Lm prevalence in wildlife, whereas studies in Finland, Japan, and Germany reported high Lm prevalence (Cao et al., 2018; Hellstrom et al., 2008; Najdenski et al., 2018; Weis & Seeliger, 1975; Yoshida et al., 2000). In the study reported here, all the animal hosts could be classified into three categories: birds, mammals, and reptiles (plus one amphibian). Birds’ body temperature can range from approximately 39.2 to 41.2˚C (Marshall, 1961). Mammals’ body temperatures vary among species based on the scaling of body temperature with body mass by species‐level phylogenetic analysis (Clarke et al., 2010). The body temperature of turtles is stable at around 22˚C (Cabanac & Bernieri, 2000).
Whole‐genome sequencing (WGS) allows for the identification of virulence genes and stress response genes, as well as for the assessment of genome‐wide diversity. Genome data can also be used for in silico multilocus sequencing typing (MLST), which has previously been used to develop a clonal framework for Lm (Ragon et al., 2008). Hence, in this study, WGS data of Lm isolates obtained from wildlife in central New York were used to (i) assess the diversity of the isolates using in silico MLST, (ii) identify virulence genes associated with hypervirulence, (iii) identify mutations in virulence genes associated with hypovirulence, and (iv) identify stress survival islets associated with stress survival and growth. In addition, the Lm wildlife isolates were phenotypically assessed for their ability to grow under three different temperatures approximating the body temperatures of the original hosts from which the isolates were obtained.
2. MATERIALS AND METHODS
2.1. Isolation of L. monocytogenes from wildlife
Wildlife patients admitted to the Janet L. Swanson Wildlife Hospital at Cornell University were sampled for this study between May 2018 and December 2019. Isolates confirmed to be Lm by the end of September 2019 (n = 13) were characterized by WGS and phenotypic analysis. Voided faecal samples (birds and mammals) and cloacal swab samples (reptiles and amphibians) were obtained from patients at the Wildlife Hospital, with all admitted patients being eligible for sample collection. Samples were collected upon admission or within 24 h of admission, then placed in Cary–Blair medium and held at room temperature until Listeria isolation was performed at the Cornell Food Safety Laboratory. This protocol was approved by the Cornell University Institutional Animal Care and Use Committee. A Buffered Listeria Enrichment Broth Base (BLEB) enrichment protocol (U.S. FDA, 2018) was used to isolate Listeria spp. from the faecal samples. Briefly, for 5 ml of faecal sample, 45 ml of BLEB (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) was added (1:10 enrichment). Then, enrichments were incubated for a total of 48 h at 30°C with BLEB Selective Supplement (Thermo Fisher Scientific, Waltham, MA, USA) added after 4 h of incubation. Each enriched sample was streaked onto one Modified Oxford Agar (MOX) plate (Becton, Dickinson and Company; Thermo Fisher Scientific) and one L. monocytogenes plating medium (LMPM) plate (R & F Products, Downers Grove, IL), with 50 μl aliquots of the enrichment media on each plate, after 24 and 48 h of incubation (for a total of two MOX and two LMPM plates per enriched sample). MOX and LMPM plates were incubated for 48 h at 30°C (Curtis et al., 1989) and 35°C, respectively. Putative positive colonies were subjected to sigB allelic typing (AT) (Liao et al., 2017) to confirm their Listeria classification and preserved in glycerol stock at –80˚C.
2.2. Whole‐genome sequencing and analyses
Lm isolates were further characterized by WGS, conducted by the New York State Department of Health using the Illumina NextSeq500 system. WGS data were submitted to NCBI where they were assigned to SNP clusters in the Pathogen Detection database if they matched to at least one other isolate with <50 SNP differences (https://www.ncbi.nlm.nih.gov/pathogens). Sequence type, clonal complex, and lineage assignments were obtained in silico using the Lm MLST scheme reported by Ragon et al. (2008) and available from the Institut Pasteur MLST and Whole Genome MLST Databases (https://bigsdb.pasteur.fr/listeria/). The nucleotide sequences of all available alleles of (i) 15 virulence genes (prfA, plcA, hly, mpl, actA, plcB, inlA, inlB, inlC, inlE, inlF, inlG, inlH, inlJ, and inlK), (ii) the genes located in LIPI‐3 and LIPI‐4, and (iii) the genes located in two stress response operons (SSI‐1 and SSI‐2) were downloaded from Institut Pasteur MLST and Whole Genome MLST Databases (https://bigsdb.pasteur.fr/listeria/). These sequences were used for identification of the corresponding virulence and stress response genes, as well as mutations leading to premature stop codons in the Lm isolates using Basic Local Alignment Search Tool (BLAST) (Altschul et al., 1990). For identification of premature stop codons, nucleotide sequences were translated into amino acid sequences and searched for the ‘*’ symbol, representing a stop codon, inside the sequence using MEGA7 (Kumar et al., 2016).
2.3. Growth curves
Isolates were assessed for their ability to grow in Brain Heart Infusion (BHI) media at 25, 37, and 42˚C using the Synergy H1 Microplate Reader instrument. Each of the Lm isolates was retrieved from frozen glycerol stock and streaked on a separate BHI plate, which was incubated for 24 h at 37˚C. Five colonies were selected from each of the plates and inoculated into five separate tubes with 5 ml BHI broth. The 65 culture tubes (representing the 13 test isolates and five colonies from each test isolate) plus a blank tube with 5 ml of uninoculated BHI broth were incubated overnight at 37°C with 200 RPM agitation. For each overnight culture tube, 500 μl of the broth was transferred to one corresponding well in a 0.8 ml deep 96‐well plate. The location of each Lm isolate/replicate was randomly assigned within the 96‐well plate for each temperature separately using the ‘sample’ function implemented in R. Serial 10‐fold dilutions were made to reach 10−4 of the original concentration, which was used to start the growth curve experiments. Bacterial growth at 25˚C, 37˚C, and 42˚C was measured in 96‐well plates using the absorbance capabilities at a wavelength of 600 nm, with measurements conducted every 10 min for 18, 26, and 30 h for cells grown at 42, 37, and 25°C, respectively.
2.4. Statistical analysis
All data analyses were performed in R Statistical Programming Environment (Pinheiro et al., 2018). The threshold of statistical significance for all analyses was p = 0.05.
The ability of the Lm isolates to grow at 25, 37, and 42°C was assessed using the growthcurver (v 0.3.0) package (Sprouffske & Wagner, 2016). Growth curve data were fitted with a standard form logistic model using the non‐linear least‐squares Levenberg–Marquardt algorithm. Two growth parameters were extracted from each of the growth curves, including the area under the curve (auc_l), which was obtained by taking the integral of the logistic equation, and the maximum population size (k). The area under the curve can summarize a growth curve, as it incorporates the initial population size, growth rate, and maximum population size into a single value and shows a strong correlation with the growth rate (Sprouffske & Wagner, 2016).
For each temperature (25, 37, and 42°C), the auc_l data were fitted with a simple linear regression model, using the stats (v 4.0.2) package (Team, 2020), with isolate as the independent variable; temperature was not included as an independent variable because growth duration differed between temperatures preventing auc_l comparisons across temperatures. Additionally, the auc_l data were fitted with a mixed effects linear model using the lme4 (v 1.1.17) package (Bates et al., 2015), with lineage as the fixed effect and the BHI media plate used for streaking the stock culture as the random effect. A one‐way analysis of variance (ANOVA) was performed on the simple linear model and the mixed effects linear model to explore the difference in auc_l at the isolate and the lineage levels, respectively. Upon the identification of a significant difference by ANOVA, a Tukey's HSD test was performed for the pairwise comparison of auc_l across isolates or lineages using the emmeans (v 1.4.4) package (Lenth et al., 2020).
The data for k were fitted with a simple linear model with isolate and temperature as the independent variables, and with a mixed effects linear model with lineage and temperature as fixed effects and BHI media plate as a random effect. The interaction between temperature and lineage was included in both models. The effect of isolate, temperature, and their interaction on k was explored by performing a two‐way ANOVA followed by a Tukey's HSD test. Similarly, the effect of lineage, temperature, and their interaction on k was explored by performing the same statistical tests on the mixed effects linear model.
3. RESULTS
3.1. Prevalence of L. monocytogenes
Individual samples were collected from 324 wildlife patients between May 2018 and December 2019. The prevalence of faecal Lm shedding was 5.6% [18/324; 95% confidence interval (CI), 3.3%–8.6%]. Reptiles had the highest prevalence (12%, 2/17), followed by mammals (8%, 5/64) and birds (4.5%, 11/242); the one amphibian was negative.
3.2. Genetic diversity of L. monocytogenes
Among the 13 wildlife Lm isolates that were characterized (confirmed as Lm by the end of September 2019; Table 1), six and seven isolates were classified as lineage I and lineage II, respectively, based on their sigB AT classification. In addition, the 13 isolates could be classified into 11 sequence types (STs) and clonal complexes (CC), with three isolates representing ST 388 and CC 388 and all other isolates representing one ST and CC each (Table 2). Upon submission of the WGS data to NCBI´s genome databases, five isolates were placed into four different NCBI Pathogen Detection SNP clusters, and eight did not match (i.e. >50 SNP differences) with any other isolate in the NCBI Pathogen Detection database and hence were not placed in a cluster. Thus, the 13 wildlife isolates are genetically diverse. Five isolates (FSL‐R9‐8346, FSL‐R9‐8380, FSL‐R9‐8822, FSL‐R9‐9447, and FSL‐R9‐9449) were placed within four SNP clusters (PDS000024423.3, PDS000003170.45, PDS000045213.3, and PDS000043859.2) that also contained animal clinical isolates, food isolates, and food‐environment isolates (Table 2). Detailed information about these five isolates can be found in the NCBI database (https://www.ncbi.nlm.nih.gov/pathogens/isolates/#FSL‐R9‐8346%20FSL‐R9‐8380%20FSL‐R9‐8822%20FSL‐R9‐9447%20FSL‐R9‐9449). Three of these five isolates (FSL‐R9‐8346, FSL‐R9‐8380, and FSL‐R9‐9447) were placed in three SNP clusters (PDS000024423, PDS000003170, and PDS000043859) that also included human clinical isolates. FSL‐R9‐8346, FSL‐R9‐8380, and FSL‐R9‐9447 showed 28, 27, and 23 SNP differences (i.e. SNP differences shown on NCBI Pathogen Detection) to the closest human isolates in their SNP clusters, respectively. Two of the three ST 388 isolates (i.e. FSL R9‐8822 and FSL R9‐9449) showed only one SNP difference (as shown on NCBI Pathogen Detection) and were placed in the same SNP cluster (PDS000045213). These two isolates were obtained from a mouse and a snapping turtle, suggesting a potential common source of these isolates.
TABLE 1.
Listeria monocytogenes isolates used in this study and their respective hosts
| Isolate ID | Host | Host type | Date of sample collection (YYYYMMDD) |
|---|---|---|---|
| FSL‐R9‐8346 | Beaver | Mammal | 20180607 |
| FSL‐R9‐8348 | Gray squirrel | Mammal | 20180525 |
| FSL‐R9‐8350 | Snapping turtle | Reptile | 20180529 |
| FSL‐R9‐8352 | European Starling | Bird | 20180531 |
| FSL‐R9‐8380 | Red‐winged Blackbird | Bird | 20180623 |
| FSL‐R9‐8502 | American Robin | Bird | 20180718 |
| FSL‐R9‐8704 | Eastern chipmunk | Mammal | 20180901 |
| FSL‐R9‐8818 | Scarlet Tanager | Bird | 20190514 |
| FSL‐R9‐8822 | Mouse | Mammal | 20190517 |
| FSL‐R9‐9355 | Rock Pigeon | Bird | 20190606 |
| FSL‐R9‐9447 | Canada Goose | Bird | 20190612 |
| FSL‐R9‐9449 | Snapping turtle | Reptile | 20190620 |
| FSL‐R9‐9581 | Gray Catbird | Bird | 20190924 |
TABLE 2.
Listeria monocytogenes diversity
| Isolate ID | sigB AT | SNP cluster ‡ | ST | CC | Lineage |
|---|---|---|---|---|---|
| FSL R9‐8346 | 61 | PDS000024423.3 | 389 | 389 | I |
| FSL R9‐8348 | 57 | – | 795 | 795 | II |
| FSL R9‐8350 | 57 | – | 37 | 37 | II |
| FSL R9‐8352 | 86 | – | 391 | 89 | II |
| FSL R9‐8380 | 61 | PDS000003170.45 | 554 | 554 | I |
| FSL R9‐8502 | 57 | – | 365 | 14 | II |
| FSL R9‐8704 | 57 | – | 368 | 368 | II |
| FSL R9‐8818 | 57 | – | 2469 | 2469 | II |
| FSL R9‐8822 | 58 | PDS000045213.3 | 388 | 388 | I |
| FSL R9‐9355 | 58 | – | 388 | 388 | I |
| FSL R9‐9447 | 58 | PDS000043859.2 | 1 | 1 | I |
| FSL R9‐9449 | 58 | PDS000045213.3 | 388 | 388 | I |
| FSL R9‐9581 | 59 | – | 2428 | 1790 | II |
Note: A ‘–’ indicates that the isolate was not placed into a SNP cluster (i.e. the isolate did not match any other isolate in the NCBI PD database by <50 SNPs).
Abbreviations: CC, clonal complex; ST, sequence type; sigB AT, sigB allelic type; SNP, single nucleotide polymorphism.
SNP cluster: single nucleotide polymorphism cluster as assigned by NCBI Pathogen Detection database. A set of isolates different by less than 50 SNP difference in the whole genome is grouped within one SNP cluster.
3.3. Stress‐response loci, virulence attenuation, and hypervirulence
While the stress response islet SSI‐2 was not present in any isolate, SSI‐1 was found in four lineage II isolates (FSL‐R9‐8350, FSL‐R9‐8704, FSL‐R9‐8818, and FSL‐R9‐9581). The virulence genes prfA, plcA, hly, mpl, actA, and plcB (representing LIPI‐1) as well as inlA, inlB, inlC, inlE, inlH, inlJ, and inlK were present in all isolates, and no premature stop codon was found in any of these genes. The virulence loci LIPI‐3 and LIPI‐4, as well as inlF and inlG, were found in four, three, ten, and seven isolates, respectively (Table 3).
TABLE 3.
Identification of virulence genes a and stress genes in Listeria monocytogenes isolates
| Isolate ID | Lineage | LIPI‐3 | LIPI‐4 | inlF | inlG | SSI‐1 | SSI‐2 |
|---|---|---|---|---|---|---|---|
| FSL R9‐8346 | I | + | – | + | – | – | – |
| FSL R9‐8380 | I | + | – | + | – | – | – |
| FSL R9‐8822 | I | – | + | – | – | – | – |
| FSL R9‐9355 | I | – | + | – | – | – | – |
| FSL R9‐9447 | I | + | – | + | – | – | – |
| FSL R9‐9449 | I | – | + | – | – | – | – |
| FSL R9‐8348 | II | – | – | + | + | – | – |
| FSL R9‐8350 | II | – | – | + | + | + | – |
| FSL R9‐8352 | II | – | – | + | + | – | – |
| FSL R9‐8502 | II | – | – | + | + | – | – |
| FSL R9‐8704 | II | – | – | + | + | + | – |
| FSL R9‐8818 | II | + | – | + | + | + | – |
| FSL R9‐9581 | II | – | – | + | + | + | – |
Note: ‘+’ and ‘–’ indicate presence and absence of the loci, respectively.
The following virulence genes were identified in all isolates: prfA, plcA, hly, mpl, actA, plcB, inlA, inlB, inlC, inlE, inlF, inlH, inlJ, and inlK.
3.4. Growth performance
A significant difference (p < 0.05) in auc_l (area under curve) was identified across isolates at 25 and 42°C, but not 37°C (Figure 1). At 42˚C, FSL‐R9‐8352 showed significantly impaired growth performance (based on auc_l values) compared with other isolates, but it had auc_l values at 25 and 37˚C that did not significantly differ from other isolates (Figure 1). Compared to lineage I, the auc_l of lineage II isolates was significantly higher (p < 0.05) at 25°C, not different at 37°C, and significantly lower (p < 0.05) at 42°C (Figure 2).
FIGURE 1.
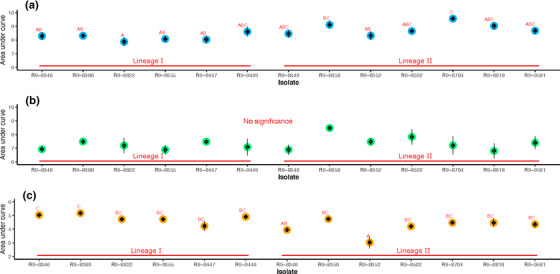
Comparison of area under curve of Listeria monocytogenes (Lm) growth across isolates at (a) 25°C, (b) 37°C, and (c) 42°C. The parameter auc_l represents overall fitness of isolates. The big circle refers to the model estimates, while the diamonds and the error bar present the mean and standard error of the raw data, respectively. Isolates with overlapping letters (on top of data points) are not significantly different from each other. Isolates with distinct letters are significantly different from each other (Tukey´s p < 0.05)
FIGURE 2.
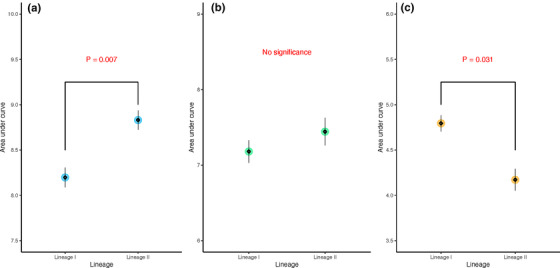
Comparison of area under curve of Listeria monocytogenes (Lm) growth between lineages at (a) 25°C, (b) 37°C, and (c) 42°C. The big circle refers to the model estimates, while the diamonds and the error bar present the mean and standard error of the raw data, respectively
A significant difference (p < 0.05) in maximum population density was identified across isolates at 37°C, but not at 25 and 42°C (Figure 3). The k‐value also showed no significant difference between lineages at 25˚C. Compared to lineage I isolates, lineage II isolates had significantly higher k‐value at 37˚C and a significantly lower k‐value at 42°C (Figure 4).
FIGURE 3.
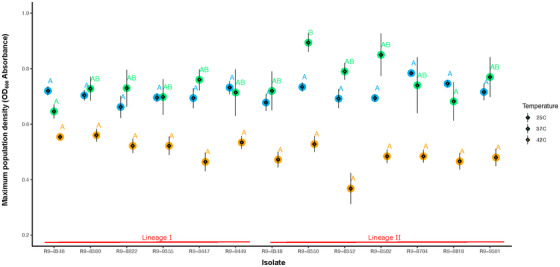
Comparison of maximum population density of Listeria monocytogenes (Lm) growth across isolates at three temperatures. The big circle refers to the model estimates, while the diamonds and the error bar present the mean and standard error of the raw data, respectively. Isolates with overlapping letters (on top of data points) are not significantly different from each other. Isolates with distinct letters are significantly different from each other (Tukey´s p < 0.05)
FIGURE 4.
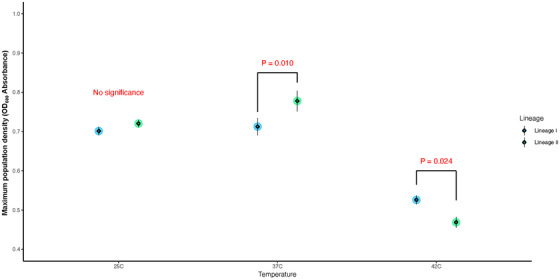
Comparison of maximum population density of Listeria monocytogenes (Lm) growth between lineages at three temperatures. The big circle refers to the model estimates, while the diamonds and the error bar present the mean and standard error of the raw data, respectively
4. DISCUSSION
4.1. Prevalence of faecal L. monocytogenes shedding among wildlife was 6%
Although Lm has previously been reported to be more commonly associated with urban environments than with natural environments (Sauders et al., 2006, 2012), wildlife such as black bears and deer have been reported as potential reservoirs of Lm (Parsons et al., 2020; Weis & Seeliger, 1975). For example, previous studies reported an Lm prevalence of 45% among 231 black bears captured around Asheville, NC (Parsons et al., 2020) and 10% among 102 deer faecal samples collected in Germany (Weis & Seeliger, 1975). Among wildlife in Japan, six Lm‐positive samples were obtained from a total of 623 mammalian samples (1.0% prevalence), while five Lm‐positive samples were obtained from 996 avian samples (0.5% prevalence) (Yoshida et al., 2000). In other studies, the Lm prevalence in wild birds varied widely. A previous study in China found an Lm prevalence of 1.0% among 895 Black‐headed Gull faecal samples (Cao et al., 2018), whereas studies in Finland (Hellstrom et al., 2008) and Germany (Weis & Seeliger, 1975) reported an Lm prevalence as high as 36.0% and 17.3%, respectively. One study isolated Lm from two out of 34 cloacal samples from adult European pond turtles (6% prevalence) (Nowakiewicz et al., 2015), which was lower than the Lm prevalence in reptiles in our study. Although the methods used for isolation of Lm from wildlife samples may have differed among the aforementioned studies, the Lm prevalence observed here among mammals, reptiles, and birds was within the range of the Lm prevalence estimates obtained in these previous studies around the world.
4.2. L. monocytogenes isolates obtained from wildlife faecal samples were genetically diverse but included a substantial number of possibly hypervirulent strains
The 13 Lm isolates characterized here by WGS represented considerable diversity, including two lineages and 11 STs. The isolates classified into lineage II, which in a previous study was reported to be more prevalent in natural environments than lineage I (Sauders et al., 2006), included one isolate (FSL‐R9‐8350) that was classified into ST 37; this ST was previously reported to be predominant in Austrian soil samples (Linke et al., 2014). Classification of the WGS data for the 13 isolates showed that some of the wildlife isolates collected here are closely related to isolates contaminating foods and food‐processing environments (26–36 SNP differences), while other isolates were found to group with isolates from human cases (23–28 SNP differences). It is important to note, however, that previous studies have indicated mutation rates of 0.18–0.38 SNPs per year (Harrand et al., 2020; Knudsen et al., 2017; Moura et al., 2016); hence, 23–28 SNP differences indicate that at least a few decades elapsed since the most recent common ancestor between the wildlife isolates and the human and food‐associated isolates. In comparison to our findings, a study in Finland found that 75 out of 212 avian Lm isolates had the same pulsotypes as Lm isolates from food‐processing plants based on pulsed‐field gel electrophoresis (PFGE) subtyping (Hellstrom et al., 2008).
WGS data supported that wildlife Lm isolates obtained here have the potential to cause human disease, as initially supported by the fact that all 13 isolates contained genes that encoded a full length complement of key virulence factors, including inlA, inlB, inlC, inlE, inlH, inlJ, plcA, and prfA. While inlA and inlB gene are critical for cell invasion (Vazquez‐Boland, Kuhn, et al., 2001), hly, mpl, actA, and plcB are crucial for Lm intracellular survival (Vazquez‐Boland, Kuhn, et al., 2001). In addition to the virulence genes found in all 13 isolates, inlG and inlF were found in all seven lineage II isolates, while inlF was found in three out of six lineage I and inlG was not found in any lineage I isolates. Our results are similar to those obtained by previous studies in which inlG was found to be present exclusively in lineage II isolates, while inlF was found to be more predominant in lineage II, although also found among lineage I isolates (Jia et al., 2007; Toledo et al., 2018).
WGS‐based characterization of the wildlife isolates obtained here also provided evidence that wildlife isolates include clonal groups that may be hypervirulent or at least be likely to cause human disease given exposure to a sufficiently high dose. In addition to 6/13 Lm isolates being classified into lineage I, which has been reported to be over‐represented among human clinical cases, one of the 13 Lm wildlife isolates (FSL‐R9‐9447) was classified into a hypervirulent CC (i.e. CC1) (Maury et al., 2016); most clinical Lm isolates in France belong to seven distinct clones, and CC1 is considered the most diverse and genetically stable (Ragon et al., 2008). In addition, LIPI‐3 and LIPI‐4, which have been associated with hypervirulence (Maury et al., 2016), were found in four and three wildlife isolates, respectively. Furthermore, none of the wildlife Lm isolates in this study showed premature stop codons (PMSC) in inlA or prfA; this is important, as previous studies have demonstrated that PMSC mutations in inlA and prfA in lineage II isolates may cause virulence attenuation (Maury et al., 2017; Nightingale et al., 2008, 2005). Although PMSC mutations in inlA are relatively common in lineage II Lm isolates, such mutations are rarely observed in linage I isolates (Jacquet et al., 2004; Nightingale et al., 2008; Van Stelten et al., 2010). This may partially explain the lower occurrence of listeriosis outbreaks caused by lineage II isolates as compared to lineage I isolates (Chen et al., 2006).
In addition to evidence that the Lm isolated here from wildlife are likely to be fully virulent, including some possibly hypervirulent isolates, we also found that four Lm isolates (FSL‐R9‐8350, FSL‐R9‐8704, FSL‐R9‐8818, and FSL‐R9‐9581) carried SSI‐1 and hence may show enhanced resistance to some stress conditions. The SSI‐1 operon has previously been associated with enhanced capability of survival and growth under suboptimal conditions of low pH and high osmolarity. These conditions can be encountered by Lm in foods and in the host gastrointestinal tract (Ryan et al., 2010).
4.3. No association between original host body temperature and ability to grow at different temperatures was observed, suggesting no genetic adaptation to the original hosts
We tested the growth ability of Lm under temperatures (25, 37, and 42˚C) that resemble the body temperature of different animal hosts (reptiles, mammals, and birds, respectively). We hypothesized that the growth parameters of Lm isolates would show better fitness under temperatures approximating their hosts’ body temperatures. However, we did not find associations between hosts’ body temperatures and the growth fitness of isolates, suggesting that these isolates are not pre‐adapted to the body temperatures of their original hosts. Although these isolates are not genetically adapted to the hosts’ body temperature, they show adaptation to certain growth temperatures (25, 37, and 42˚C) depending on lineages. Compared to lineage II, isolates from lineage I demonstrated better growth at 42˚C, but reduced growth at 25˚C. Interestingly, in a previous study in Switzerland, lineage I isolates (serotype 4b) were more prevalent in water samples in the summer, while isolates from lineage II (serotype 1/2a) were more prevalent in the fall (Schaffter & Parriaux, 2002), which suggests that future studies on the survival of different Lm lineages in different environments and under different temperature conditions may be valuable.
5. CONCLUSIONS
Animals may contaminate pre‐harvest environments (i.e. farms) and post‐harvest environments (i.e. food‐processing facilities) via Lm shedding. The Lm transmission cycle between animals, environments, humans, sewage, and food‐processing plants potentially increases the load of Lm in hosts and habitats (Ivanek et al., 2006; Sanaa, et al., 1993; Schlech et al., 1983). Thus, research aimed at understanding the Lm transmission pathways between wildlife and humans is needed. This project showed that wildlife may be reservoirs or carriers of potentially hypervirulent Lm, but further studies will be needed to assess the virulence of the isolates used in this study. As various wildlife species have been reported as potential sources of zoonotic disease transmission (Lempp et al., 2017), our findings emphasize the value of zoonotic pathogen surveillance among wildlife that are geographically close to human habitats and the food supply chain to support ‘One Health.’
CONFLICT OF INTEREST
The authors declare no conflict of interest. The viewpoints expressed in this study do not represent the official positions of the New York State Department of Health.
AUTHOR CONTRIBUTIONS
Formal analysis, investigation, methodology, and writing—original draft: Tong Chen. Conceptualization, methodology, supervision, writing—original draft, writing—review, and editing: Renato H. Orsi. Formal analysis and writing—original draft: Ruixi Chen. Data curation, investigation, methodology, and supervision: Maureen Gunderson. Data curation and methodology: Sherry Roof. Conceptualization, funding acquisition, project administration, resources, supervision, writing—review, and editing: Martin Wiedmann. Resources, supervision, writing—review, and editing: Sara E. Childs‐Sanford. Conceptualization, funding acquisition, project administration, supervision, writing—review, and editing: Kevin J. Cummings.
ACKNOWLEDGEMENT
We thank the Wadsworth Center of the New York State Department of Health for conducting the whole‐genome sequencing.
Chen, T. , Orsi, R. H. , Chen, R. , Gunderson, M. , Roof, S. , Wiedmann, M. , Childs‐Sanford, S. E. , & Cummings, K. J. (2022). Characterization of Listeria monocytogenes isolated from wildlife in central New York. Veterinary Medicine and Science, 8, 1319–1329. 10.1002/vms3.758
DATA AVAILABILITY STATEMENT
The WGS data that support the findings of this study are openly available in NCBI's short read archive (SRA) database at https://www.ncbi.nlm.nih.gov/sra, accession numbers SRR10356384 to SRR10356396.
REFERENCES
- Altschul, S. F. , Gish, W. , Miller, W. , Myers, E. W. , & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Bates, D. , Mächler, M. , Bolker, B. , & Walker, S. (2015). Fitting linear mixed‐effects models using lme4. Journal of Statistical Software, 67, 48. [Google Scholar]
- Cabanac, M. , & Bernieri, C. (2000). Behavioral rise in body temperature and tachycardia by handling of a turtle (Clemmys insculpta). Behavioural Processes, 49, 61–68. [DOI] [PubMed] [Google Scholar]
- Cao, X. , Wang, Y. , Wang, Y. , & Ye, C. (2018). Isolation and characterization of Listeria monocytogenes from the black‐headed gull feces in Kunming, China. Journal of Infection and Public Health, 11, 59–63. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Ross, W. H. , Gray, M. J. , Wiedmann, M. , Whiting, R. C. , & Scott, V. N. (2006). Attributing risk to Listeria monocytogenes subgroups: Dose response in relation to genetic lineages. Journal of Food Protection, 69, 335–344. [DOI] [PubMed] [Google Scholar]
- Clarke, A. , Rothery, P. , & Isaac, N. J. (2010). Scaling of basal metabolic rate with body mass and temperature in mammals. Journal of Animal Ecology, 79, 610–619. [DOI] [PubMed] [Google Scholar]
- Cotter, P. D. , Draper, L. A. , Lawton, E. M. , Daly, K. M. , Groeger, D. S. , Casey, P. G. , Ross, R. P. , & Hill, C. (2008). Listeriolysin S, a novel peptide haemolysin associated with a subset of lineage I Listeria monocytogenes . PLoS Pathogens, 4, e1000144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, G. D. W. , Nichols, W. W. , & Falla, T. J. (1989). Selective agents for Listeria can inhibit their growth. Letters in Applied Microbiology, 8, 169–172. [Google Scholar]
- Gray, M. J. , Zadoks, R. N. , Fortes, E. D. , Dogan, B. , Cai, S. , Chen, Y. , Scott, V. N. , Gombas, D. E. , Boor, K. J. , & Wiedmann, M. (2004). Listeria monocytogenes isolates from foods and humans form distinct but overlapping populations. Applied and Environmental Microbiology, 70, 5833–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrand, A. S. , Jagadeesan, B. , Baert, L. , Wiedmann, M. , & Orsi, R. H. (2020). Evolution of Listeria monocytogenes in a food processing plant involves limited single‐nucleotide substitutions but considerable diversification by gain and loss of prophages. Applied and Environmental Microbiology, 86, e02493–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harter, E. , Wagner, E. M. , Zaiser, A. , Halecker, S. , Wagner, M. , & Rychli, K. (2017). Stress survival islet 2, predominantly present in Listeria monocytogenes strains of sequence type 121, is involved in the alkaline and oxidative stress responses. Applied and Environmental Microbiology, 83, e00827–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom, S. , Kiviniemi, K. , Autio, T. , & Korkeala, H. (2008). Listeria monocytogenes is common in wild birds in Helsinki region and genotypes are frequently similar with those found along the food chain. Journal of Applied Microbiology, 104, 883–888. [DOI] [PubMed] [Google Scholar]
- Ivanek, R. , Grohn, Y. T. , & Wiedmann, M. (2006). Listeria monocytogenes in multiple habitats and host populations: Review of available data for mathematical modeling. Foodborne Pathogens and Disease, 3, 319–336. [DOI] [PubMed] [Google Scholar]
- Jacquet, C. , Doumith, M. , Gordon, J. I. , Martin, P. M. , Cossart, P. , & Lecuit, M. (2004). A molecular marker for evaluating the pathogenic potential of foodborne Listeria monocytogenes . Journal of Infectious Diseases, 189, 2094–2100. [DOI] [PubMed] [Google Scholar]
- Jeffers, G. T. , Bruce, J. L. , McDonough, P. L. , Scarlett, J. , Boor, K. J. , & Wiedmann, M. (2001). Comparative genetic characterization of Listeria monocytogenes isolates from human and animal listeriosis cases. Microbiology, 147(Pt 5), 1095–1104. [DOI] [PubMed] [Google Scholar]
- Jia, Y. , Nightingale, K. K. , Boor, K. J. , Ho, A. , Wiedmann, M. , & McGann, P. (2007). Distribution of internalin gene profiles of Listeria monocytogenes isolates from different sources associated with phylogenetic lineages. Foodborne Pathogens and Disease, 4, 222–232. [DOI] [PubMed] [Google Scholar]
- Knudsen, G. M. , Nielsen, J. B. , Marvig, R. L. , Ng, Y. , Worning, P. , Westh, H. , & Gram, L. (2017). Genome‐wide‐analyses of Listeria monocytogenes from food‐processing plants reveal clonal diversity and date the emergence of persisting sequence types. Environmental Microbiology Reports, 9, 428–440. [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , & Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Molecular Biology and Evolution, 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit, M. , Vandormael‐Pournin, S. , Lefort, J. , Huerre, M. , Gounon, P. , Dupuy, C. , Babinet, C. , & Cossart, P. (2001). A transgenic model for listeriosis: Role of internalin in crossing the intestinal barrier. Science, 292, 1722–1725. [DOI] [PubMed] [Google Scholar]
- Lempp, C. , Jungwirth, N. , Grilo, M. L. , Reckendorf, A. , Ulrich, A. , van Neer, A. , Bodewes, R. , Pfankuche, V. M. , Bauer, C. , Osterhaus, A. D. , Baumgärtner, W. , & Siebert, U. (2017). Pathological findings in the red fox (Vulpes vulpes), stone marten (Martes foina) and raccoon dog (Nyctereutes procyonoides), with special emphasis on infectious and zoonotic agents in Northern Germany. PLoS One, 12, e0175469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenth, R. , Singmann, H. , Love, J. , Buerkner, P. , & Herve, M. (2020). R package emmeans: Estimated marginal means. R package version 1.4.4. https://github.com/rvlenth/emmeans
- Liao, J. , Wiedmann, M. , & Kovac, J. (2017). Genetic stability and evolution of the sigB allele, used for Listeria sensu stricto subtyping and phylogenetic inference. Applied and Environmental Microbiology, 83, e00306–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke, K. , Ruckerl, I. , Brugger, K. , Karpiskova, R. , Walland, J. , Muri‐Klinger, S. , Tichy, A. , Wagner, M. , & Stessl, B. (2014). Reservoirs of Listeria species in three environmental ecosystems. Applied and Environmental Microbiology, 80, 5583–5592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, A. J. (1961). Biology and Comparative Physiology of Birds (Vol. 2): Academic Press. [Google Scholar]
- Maury, M. M. , Chenal‐Francisque, V. , Bracq‐Dieye, H. , Han, L. , Leclercq, A. , Vales, G. , Moura, A. , Gouin, E. , Scortti, M. , Disson, O. , Vázquez‐Boland, J. A. , & Lecuit, M. (2017). Spontaneous loss of virulence in natural populations of Listeria monocytogenes . Infection and Immunity, 85, e00541–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury, M. M. , Tsai, Y. H. , Charlier, C. , Touchon, M. , Chenal‐Francisque, V. , Leclercq, A. , Criscuolo, A. , Gaultier, C. , Roussel, S. , Brisabois, A. , Disson, O. , Rocha, E. P. C. , Brisse, S. , & Lecuit, M. (2016). Uncovering Listeria monocytogenes hypervirulence by harnessing its biodiversity. Nature Genetics, 48, 308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moura, A. , Criscuolo, A. , Pouseele, H. , Maury, M. M. , Leclercq, A. , Tarr, C. , Björkman, J. T. , Dallman, T. , Reimer, A. , Enouf, V. , Larsonneur, E. , Carleton, H. , Bracq‐Dieye, H. , Katz, L. S. , Jones, L. , Touchon, M. , Tourdjman, M. , Walker, M. , … Brisse, S. (2016). Whole genome‐based population biology and epidemiological surveillance of Listeria monocytogenes . Nature Microbiology, 2, 16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najdenski, H. , Dimova, T. , Zaharieva, M. M. , Nikolov, B. , Petrova‐Dinkova, G. , Dalakchieva, S. , Popov, K. , Hristova‐Nikolova, I. , Zehtindjiev, P. , Peev, S. , Trifonova‐Hristova, A. , Carniel, E. , Panferova, Y. A. , & Tokarevich, N. K. (2018). Migratory birds along the Mediterranean—Black Sea Flyway as carriers of zoonotic pathogens. Canadian Journal of Microbiology, 64(12), 915–924. [DOI] [PubMed] [Google Scholar]
- Nightingale, K. K. , Ivy, R. A. , Ho, A. J. , Fortes, E. D. , Njaa, B. L. , Peters, R. M. , & Wiedmann, M. (2008). inlA premature stop codons are common among Listeria monocytogenes isolates from foods and yield virulence‐attenuated strains that confer protection against fully virulent strains. Applied and Environmental Microbiology, 74, 6570–6583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale, K. K. , Windham, K. , Martin, K. E. , Yeung, M. , & Wiedmann, M. (2005). Select Listeria monocytogenes subtypes commonly found in foods carry distinct nonsense mutations in inlA, leading to expression of truncated and secreted internalin A, and are associated with a reduced invasion phenotype for human intestinal epithelial cells. Applied and Environmental Microbiology, 71, 8764–8772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowakiewicz, A. , Ziolkowska, G. , Zieba, P. , Dziedzic, B. M. , Gnat, S. , Wojcik, M. , Dziedzic, R. , & Kostruba, A. (2015). Aerobic bacterial microbiota isolated from the cloaca of the European pond turtle (Emys orbicularis) in Poland. Journal of Wildlife Diseases, 51, 255–259. [DOI] [PubMed] [Google Scholar]
- Orsi, R. H. , den Bakker, H. C. , & Wiedmann, M. (2010). Listeria monocytogenes lineages: Genomics, evolution, ecology, and phenotypic characteristics. International Journal of Medical Microbiology, 301, 79–96. [DOI] [PubMed] [Google Scholar]
- Orsi, R. H. , Ripoll, D. R. , Yeung, M. , Nightingale, K. K. , & Wiedmann, M. (2007). Recombination and positive selection contribute to evolution of Listeria monocytogenes inlA . Microbiology, 153(Pt 8), 2666–2678. [DOI] [PubMed] [Google Scholar]
- Parsons, C. , Niedermeyer, J. , Gould, N. , Brown, P. , Strules, J. , Parsons, A. W. , Bernardo Mesa‐Cruz, J. , Kelly, M. J. , Hooker, M. J. , Chamberlain, M. J. , Olfenbuttel, C. , DePerno, C. , & Kathariou, S. (2020). Listeria monocytogenes at the human‐wildlife interface: Black bears (Ursus americanus) as potential vehicles for Listeria . Microbial Biotechnology, 13, 706–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro, J. , Bates, D. , DebRoy, S. , Sarkar, D. , & Team, R. C. (2018). nlme: Linear and nonlinear mixed effects models. https://CRAN.R‐project.org/package=nlme
- Radoshevich, L. , & Cossart, P. (2018). Listeria monocytogenes: Towards a complete picture of its physiology and pathogenesis. Nature Reviews Microbiology, 16, 32–46. [DOI] [PubMed] [Google Scholar]
- Ragon, M. , Wirth, T. , Hollandt, F. , Lavenir, R. , Lecuit, M. , Le Monnier, A. , & Brisse, S. (2008). A new perspective on Listeria monocytogenes evolution. PLoS Pathogens, 4, e1000146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, A. , Nightingale, K. , Jeffers, G. , Fortes, E. , Kongo, J. M. , & Wiedmann, M. (2006). Genetic and phenotypic characterization of Listeria monocytogenes lineage III. Microbiology, 152(Pt 3), 685–693. [DOI] [PubMed] [Google Scholar]
- Roche, S. M. , Gracieux, P. , Milohanic, E. , Albert, I. , Virlogeux‐Payant, I. , Temoin, S. , Grépinet, O. , Kerouanton, A. , Jacquet, C. , Cossart, P. , & Velge, P. (2005). Investigation of specific substitutions in virulence genes characterizing phenotypic groups of low‐virulence field strains of Listeria monocytogenes . Applied and Environmental Microbiology, 71, 6039–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan, S. , Begley, M. , Hill, C. , & Gahan, C. G. (2010). A five‐gene stress survival islet (SSI‐1) that contributes to the growth of Listeria monocytogenes in suboptimal conditions. Journal of Applied Microbiology, 109, 984–995. [DOI] [PubMed] [Google Scholar]
- Sanaa, M. , Poutrel, B. , Menard, J. L. , & Serieys, F. (1993). Risk factors associated with contamination of raw milk by Listeria monocytogenes in dairy farms. Journal of Dairy Science, 76, 2891–2898. [DOI] [PubMed] [Google Scholar]
- Sauders, B. D. , Durak, M. Z. , Fortes, E. , Windham, K. , Schukken, Y. , Lembo, A. J. Jr. , Akey, B. , Nightingale, K. K. , & Wiedmann, M. (2006). Molecular characterization of Listeria monocytogenes from natural and urban environments. Journal of Food Protection, 69, 93–105. [DOI] [PubMed] [Google Scholar]
- Sauders, B. D. , Overdevest, J. , Fortes, E. , Windham, K. , Schukken, Y. , Lembo, A. , & Wiedmann, M. (2012). Diversity of Listeria species in urban and natural environments. Applied and Environmental Microbiology, 78, 4420–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scallan, E. , Hoekstra, R. M. , Angulo, F. J. , Tauxe, R. V. , Widdowson, M. A. , Roy, S. L. , Jones, J. L. , Griffin, P. M. (2011). Foodborne illness acquired in the United States—Major pathogens. Emerging Infectious Diseases, 17, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffter, N. , & Parriaux, A. (2002). Pathogenic‐bacterial water contamination in mountainous catchments. Water Research, 36, 131–139. [DOI] [PubMed] [Google Scholar]
- Schlech, W. F. 3rd , Lavigne, P. M. , Bortolussi, R. A. , Allen, A. C. , Haldane, E. V. , Wort, A. J. , Hightower, A. W. , Johnson, S. E. , King, S. H. , Nicholls, E. S. , & Broome, C. V. (1983). Epidemic listeriosis—Evidence for transmission by food. New England Journal of Medicine, 308, 203–206. [DOI] [PubMed] [Google Scholar]
- Sprouffske, K. , & Wagner, A. (2016). Growthcurver: An R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinformatics, 17, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team, R. C. (2020). R: A language and environment for statistical computing. https://www.R‐project.org/
- Toledo, V. , den Bakker, H. C. , Hormazabal, J. C. , Gonzalez‐Rocha, G. , Bello‐Toledo, H. , Toro, M. , & Moreno‐Switt, A. I. (2018). Genomic diversity of Listeria monocytogenes isolated from clinical and non‐clinical samples in Chile. Genes, 9, 396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. FDA. (2018). Bacteriological Analytical Manual (BAM). https://www.fda.gov/food/laboratory‐methods‐food/bacteriological‐analytical‐manual‐bam [Google Scholar]
- Van Stelten, A. , Simpson, J. M. , Ward, T. J. , & Nightingale, K. K. (2010). Revelation by single‐nucleotide polymorphism genotyping that mutations leading to a premature stop codon in inlA are common among Listeria monocytogenes isolates from ready‐to‐eat foods but not human listeriosis cases. Applied and Environmental Microbiology, 76, 2783–2790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez‐Boland, J. A. , Dominguez‐Bernal, G. , Gonzalez‐Zorn, B. , Kreft, J. , & Goebel, W. (2001). Pathogenicity islands and virulence evolution in Listeria. Microbes and Infection, 3, 571–584. [DOI] [PubMed] [Google Scholar]
- Vazquez‐Boland, J. A. , Kuhn, M. , Berche, P. , Chakraborty, T. , Dominguez‐Bernal, G. , Goebel, W. , González‐Zorn, B. , Wehland, J. , & Kreft, J. (2001). Listeria pathogenesis and molecular virulence determinants. Clinical Microbiology Reviews, 14, 584–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, T. J. , Ducey, T. F. , Usgaard, T. , Dunn, K. A. , & Bielawski, J. P. (2008). Multilocus genotyping assays for single nucleotide polymorphism‐based subtyping of Listeria monocytogenes isolates. Applied and Environmental Microbiology, 74, 7629–7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis, J. , & Seeliger, H. P. (1975). Incidence of Listeria monocytogenes in nature. Applied Microbiology, 30, 29–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, T. , Sugimoto, T. , Sato, M. , & Hirai, K. (2000). Incidence of Listeria monocytogenes in wild animals in Japan. Journal of Veterinary Medical Science, 62, 673–675. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The WGS data that support the findings of this study are openly available in NCBI's short read archive (SRA) database at https://www.ncbi.nlm.nih.gov/sra, accession numbers SRR10356384 to SRR10356396.