

Abstract
A sensitive, fast and robust liquid chromatography--tandem mass spectrometry (LC–MS-MS) method was developed and validated for the determination of usaramine (URM) and usaramine N-oxide (UNO) in rat plasma. The separation was conducted on an ACQUITY UPLC BEH C18 Column (50 × 2.1 mm, 1.7 μm) and gradient eluted with mobile phase A (0.1% formic acid with 5 mM ammonium acetate in water) and B (0.1% formic acid in acetonitrile/methanol, 9/1, v/v). The method was linear over the range of 1–2,000 ng/mL for both analytes. The validated method was applied to investigate the pharmacokinetic behaviors and sex differences of URM and its N-oxide metabolite in rats. After intravenous administration of URM at 1 mg/kg, the AUC0-t values for URM and UNO were 363 ± 65 and 172 ± 32 ng/mL*h in male rats, while 744 ± 122 and 30.7 ± 7.4 ng/mL*h in females, respectively. The clearance of URM was significantly higher in male rats than in females (2.77 ± 0.50 vs 1.35 ± 0.19 L/h/kg, P < 0.05). After oral administration of URM at 10 mg/kg, the AUC0-t values of URM and UNO were 1,960 ± 208 and 1,637 ± 246 ng/mL*h in male rats, while 6,073 ± 488 and 300 ± 62 ng/mL*h in females, respectively. The oral bioavailability of URM in female rats (81.7%) was much higher than in males (54.0%). In conclusion, sex-based differences were observed in the pharmacokinetics, N-oxide metabolism and oral bioavailability of URM.
Introduction
Pyrrolizidine alkaloids (PAs, Figure 1A) are a class of toxic secondary metabolites that are produced by over 6,000 plants in the world, representing about 3–5% of flowering plants (1, 2). To date, more than 660 PAs and their N-oxides have been identified from more than 13 plant families (3, 4). PAs with 1,2-unsaturated necine bases exhibit hepatotoxicity, pneumotoxicity, cytotoxicity, genotoxicity and neurotoxicity (5–9). The main PA-induce acute toxicity is hepatic sinusoidal obstruction syndrome that is a distinctive and potentially fatal form of hepatic injury that occurs predominantly (10, 11). Recently, several studies demonstrated that PA N-oxides exert toxicity via intestinal and hepatic biotransformation to the corresponding PAs (10, 12–14). Currently, the major sources of PAs to humans include herbal medicines, (herbal) teas, food supplements, honey and milk (15–20). A report on occurrence of PAs in food in six European countries published by Mulder et al. (16) revealed that PAs were found in 6% of milk samples at low concentration levels (0.05–0.17 µg/L), in 91% of the (herbal) teas with an average PA content of 454 μg/kg and in 60% of the food supplements (16, 19). The occurrence of PAs in food supplements was highly diverse, ranging from the highest content up to 2.4 g/kg in the supplements (16), based on known PA-containing plants, to the relatively low content at 1846 μg/kg in bee pollen (20). A representative report on PA in honey samples demonstrated that 94% of the retail honeys originated in various countries were contaminated with PAs, with the average PA content of 26 μg/kg (20).
Figure 1.
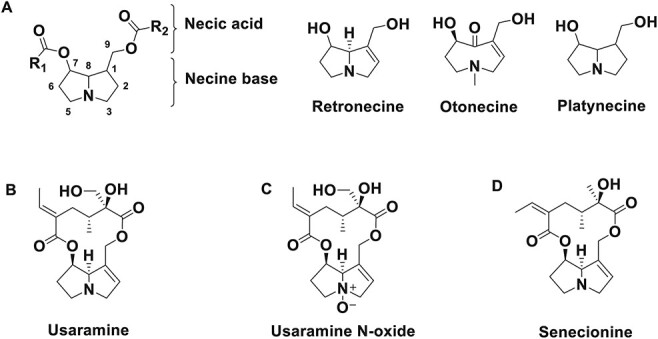
Structures of PAs. (A) Common structures of pas; (B) URM; (C) UNO and (D) senecionine (IS).
Usaramine (URM), a retronecine-type PA, is mainly found in Gynura divaricata (Compositae family) (21, 22). Gynura divaricata, also named as “Bai Zi Cai” or “Bai Bei San Qi”, is a new source of food approved by National Health Commission (China) in 2010 (21). It is also a traditional Chinese medicine, which used for the treatment of diabetes, hypertension and hyperlipidemia (21, 23–25). Despite the toxicity of this retronecine-type PA, few metabolic studies of URM have been reported. Moreover, clear sex differences in toxicity or metabolism of other PAs such as riddelliine (26, 27), senecionine (SCN) (28–30), seneciphylline (29) and clivorine (31, 32) were reported. Consequently, the following studies were conducted:(i) a sensitive, fast and robust LC–MS-MS method was developed to quantify URM and usaramine N-oxide (UNO) in Sprague–Dawley (SD) rat plasma; (ii) pharmacokinetic behaviors of URM and its N-oxide metabolite in rats were investigated and (iii) the sex differences in oral bioavailability of URM in rats were compared.
Materials and Methods
Chemicals and reagents
URM (Figure 1B, purity >98%) was purchased from Absin Bioscience Inc. (Shanghai, China). UNO (Figure 1C, purity >98%) and SCN (Figure 1D, purity >98%) were purchased from Chengdu Biopurify Phytochemicals Ltd. (Chengdu, China). High-performance liquid chromatography (HPLC) grade acetonitrile, HPLC grade methanol and liquid chromatography coupled with mass spectrometry (LC–MS) grade ammonium acetate were purchased from Sigma-Aldrich (St. Louis, MO, USA). LC–MS grade formic acid (purity >98%) was obtained from J&K Technology Co., Limited (Beijing, China). Deionized water was prepared by a Milli-Q ultra-pure water purification system (Millipore, Molsheim, France).
Preparation of stock solutions, calibration and quality control samples
The stock solutions of URM, UNO and SCN were prepared in methanol at 2 mg/mL, respectively. A series of calibration standards of URM and UNO were prepared in rat plasma as follows: 1, 2, 10, 30, 100, 300, 1,000 and 2,000 ng/mL. Quality control (QC) samples were diluted in rat plasma at concentration of 1, 3, 150, 750 and 1,500 ng/mL using a separate stock solution. SCN (internal standard, IS) working solution at 100 ng/mL was diluted in methanol/water (1/1, v/v).
Sample preparation
A 10-μL aliquot of plasma sample was mixed with 10 μL of IS working solution (100 ng/mL of SCN) in a 96-well microwell plate (Nunc, Copenhagen, Denmark) and mixed for 1 min. Then, 90 μL of acetonitrile/methanol (1/1, v/v) was added for protein precipitation. The mixtures were vortexed for 5 min, and then, centrifuged for 5 min at 4,000 rpm. A 40-μL aliquot of supernatant was transferred to a 384-well conical-bottom plate (Greiner Bio-One, Frickenhausen, Germany), and then, 1 μL was injected to LC–MS-MS for analysis.
Instrumentation and LC–MS-MS conditions
Plasma levels of URM and UNO in rat plasma were determined on a SCIEX Triple QuadTM 5500 mass spectrometer with Turbo VTM source (AB SCIEX, Concord, Ontario, Canada) coupled with Waters AcquityTM I-class system (Waters Corp, Milford, MA, USA). The chromatographic separation was performed on a Waters UPLC BEH C18 column (50 × 2.1 mm, 1.7 μm; Waters Corp), and the column was maintained at 45°C. The mobile phase consisting of (A) 0.1% formic acid with 5 mM ammonium acetate in water and (B) 0.1% formic acid in acetonitrile/methanol (9/1, v/v). Analysis was performed using the following gradient elution at a flow rate of 0.5 mL/min: 0–0.2 min, 10% B; 0.2–1.0 min, 10% B to 60% B; 1.0–1.1 min, 60% to 95% B; 1.1–1.5 min, maintained at 95% B; 1.5–2.0 min, re-equilibration with initial condition. The injection volume was 1 μL.
The MS was operated in positive ionization mode with electrospray voltage 5,500 V, source temperature of 550°C and collision activation dissociation of 7 psi. The detection was performed in the multiple reaction monitoring (MRM) mode at m/z 352.1→120.0 (collision energy (CE) 37 eV) for URM, 368.1→120.0 (CE 42 eV) for UNO and 336.1→120.1 (CE 36 eV) for SCN (IS), respectively, and declustering potential was 150 V for the analytes and IS. Data were acquired and processed in Analyst 1.6.3 software (AB SCIEX, Concord, Ontario, Canada).
Method validation
The method validation conducted in accordance with the US Food and Drug Administration (FDA) Guidance for Method Validation (33). The items for method validation included: selectivity, matrix effect, linearity, sensitivity, carryover, accuracy, precision, recovery and stability of plasma samples.
Selectivity was evaluated by analyzing six different sources of blank plasma to confirm whether there was endogenous interference at the retention times of URM, UNO and IS. The peak areas of the analytes in blank samples should be no more than 20% of the corresponding mean peak area of the lowest limit of quantification (LLOQ) samples, and the peak area of IS in blank samples should be less than 5%.
The matrix effect was evaluated by the comparison of the peak area ratio in the presence of matrix from six different sources to the peak area ratio in the absence of matrix at low and high QC concentrations. The inter-source variability of matrix effect per concentration level should be no more than 15%.
The linearity was evaluated over the concentration range of 1–2,000 ng/mL for URM and UNO, respectively. Each concentration level samples of calibration curve were analyzed in duplicate. Calibrators should have back-calculated concentration within ±15.0% of nominal except at the LLOQ where ±20.0% of the nominal is acceptable.
Carryover was evaluated by the blank sample immediately injected after ULOQ samples.
Within-run and between-run accuracy and precision were assessed six replicate QCs at LLOQ, low, low-mid, mid and high QCs for three independent runs conducted over 2 days. The overall accuracy bias should be within ±15.0% of nominal concentration, except ±20.0% at LLOQ. The precision should be no more than 15%, except 20.0% at LLOQ.
The recovery was assessed at low, low-mid, mid and high QC concentrations. The recovery values of URM, UNO and IS were calculated by dividing the mean peak area of extracted spiked samples by the mean peak area of blanks with the analytes and IS post-extraction.
The stability experiments of URM and UNO were performed in three replicates at low and high QC concentrations for bench-top, freeze-thaw and long-term stability. The acceptable stable results were the accuracy bias of nominal concentration at each level within ±15.0%.
Pharmacokinetic study
Ten male (180–220 g, 6–9 weeks) and 10 female (160–200 g, 6–9 weeks) SD rats were obtained from HFK Bio-technology Co. Ltd (Beijing, China). After being kept in fast but with free access to water for 12 h, rats were assigned to two groups (five animals/sex/group) randomly. A group of rats received a single intravenous administration of 1 mg/kg URM via tail vein, while another group of rats received a single oral administration of 10 mg/kg URM. For both intravenous and oral administrations, URM was dissolved in saline. Blood samples (∼ 50 μL) were collected via saphenous vein into heparinized tubes at the time points of 0.05, 0.167, 0.5, 1, 2, 4, 8 and 24 h after intravenous treatment, while at 0.167, 0.333, 0.667, 1, 2, 4, 8 and 24 h after oral treatment, and centrifuged for 10 min at 10,000 rpm to harvest plasma samples. The plasma samples were stored at < −60°C before analysis.
The animal study was conducted in compliance with the Guide for the Care and Use of Laboratory Animals (34) and was approved by Institutional Animal Care and Use Committee (IACUC) of Shanghai Institute of Materia Medica, Chinese Academy of Sciences (IACUC number 2020-02-YY-10).
A non-compartmental pharmacokinetic analysis of URM and UNO was performed using Phoenix WinNonlin 6.4 (Pharsight Corporation, Mountain View, CA, USA). The maximum concentration (Cmax) and time to reach Cmax (Tmax) were determined from the plasma concentration versus time data. The area under the plasma concentration–time curve from time 0 to the time of the last measurable concentration (AUC0–t) was determined using the linear trapezoidal method. The area under the plasma concentration–time curve from time 0 to infinity concentration (AUC0–∞) was determined using the linear trapezoidal method. The elimination half-life (T1/2) was calculated using ln (2)/ke. The clearance (CL) was the ratio of the dose to the AUC0–t. The oral bioavailability (F) was the ratio of the dose-corrected AUC for oral to intravenous administration.
Statistical analysis of pharmacokinetic parameters between male and female rats was performed by Student’s t-test, and P-values < 0.05 were considered statistically significant.
Results and Discussion
Method development
The chromatographic conditions were optimized to obtain reproducible symmetric peak shapes with a short run time. During method development, four-type columns from Waters including ACQUITY BEH C18, HSS T3, CORTEST C18+ and CSH C18 were tested with various solvent and buffers as mobile phases. Finally, a mobile phase consisted of 0.1% formic acid with 5 mM ammonium acetate and 0.1% formic acid with acetonitrile/methanol (9/1, v/v) using a Waters ACQUITY BEH C18 column (50 × 2.1 mm, 1.7 μm) provided satisfactory peak shapes, resolution and a short run time (2 min). Further, the gradient program was optimized to eliminate the carryover. For 0–0.2 min, 10% of mobile phase B was adopted to improve the solubility of analytes in initial mobile phase and eliminate the carryover in the injection system.
Method validation
Selectivity and matrix effect
No endogenous interferences were observed at the retention times of URM (0.77 min), UNO (0.79 min) and IS (0.89 min). The representative chromatograms are shown in Figure 2.
Figure 2.
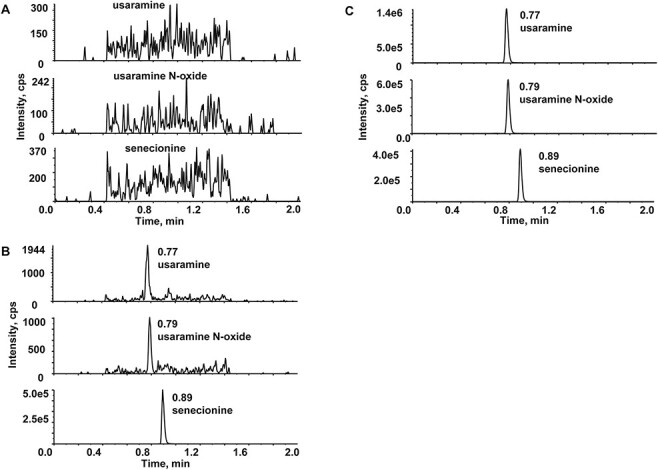
Representative chromatograms in rat plasma (A) blank plasma, (B) plasma spiked with analytes (1 ng/mL, LLOQ) and IS (100 ng/mL) and (C) 1 h post-dose plasma sample after oral administration of 10 mg/kg URM.
The matrix effects ranged from 96.1 to 98.7% for URM and 95.2–98.1% for UNO, while the coefficient of variations (CVs) for the analytes were less than 8.0%. The results indicated the impact of the matrix was considered negligible.
Linearity, sensitivity and carry-over
The calibration curves were linearity in rat plasma over the concentration ranges of 1–2,000 ng/mL for URM and UNO with correlation coefficient (r2) greater than 0.990, respectively. The accuracy bias of the nominal concentrations at all concentration levels in three validation runs ranged from −5.3% to 3.1% and the CVs were less than 7.5%. The LLOQ was 1.0 ng/mL for both URM and UNO. The typical equations of URM and UNO in plasma were y = 0.00365x + 0.00053 (r2 = 0.9974) and y = 0.0026x + 0.00004 (r2 = 0.9950), respectively.
The acceptable carryover of analytes and IS in the blank samples immediately after ULOQ samples were observed in all test runs.
Accuracy and precision
The within-run and between run accuracy and precision were within acceptable ranges, and the results are summarized in Table I. It was demonstrated the developed method was accuracy and reliability.
Table I.
Accuracy and Precision for URM and UNO in Rat Plasma
| Spiked (ng/mL) |
URM | UNO | ||
|---|---|---|---|---|
| Accuracy (%) | Precision (%) | Accuracy (%) | Precision (%) | |
| Within run (n = 6) | ||||
| 1 | 105.8 | 6.7 | 96.8 | 6.5 |
| 3 | 102.6 | 5.1 | 98.2 | 8.7 |
| 150 | 99.9 | 2.5 | 101.7 | 2.5 |
| 750 | 99.6 | 1.5 | 100.2 | 3.0 |
| 1,500 | 101.0 | 2.3 | 97.0 | 2.7 |
| Between run (n = 18) | ||||
| 1 | 105.9 | 10.6 | 96.2 | 11.6 |
| 3 | 101.0 | 5.2 | 99.4 | 6.8 |
| 150 | 100.4 | 2.4 | 102.0 | 2.9 |
| 750 | 100.7 | 2.3 | 99.8 | 3.1 |
| 1,500 | 98.7 | 3.9 | 96.7 | 3.4 |
Recovery
The mean recovery of URM and UNO from plasma ranged from 87.2 to 90.2% and 87.9 to 94.4%, and the mean recovery of IS was 91.7%. The CVs for the analytes at all concentration levels and IS were less than 6.6%.
Stability
The stabilities of URM and UNO in rat plasma samples were analyzed under various conditions. The results are shown in Table II, which demonstrated the analytes were stable at room temperature for 8 h, after three freeze-thaw cycles and at < −60°C for 2 weeks.
Table II.
Stability of URM and UNO in Rat Plasma
| Conc. (ng/mL) |
URM | UNO | ||
|---|---|---|---|---|
| Mean accuracy (%) |
CV (%) | Mean accuracy (%) |
CV (%) | |
| Room temperature for 8 h | ||||
| 3 | 101.0 | 6.9 | 101.4 | 2.7 |
| 1,500 | 99.7 | 1.0 | 97.3 | 4.6 |
| Three freeze-thaw cycles | ||||
| 3 | 101.7 | 2.1 | 104.8 | 4.5 |
| 1,500 | 102.2 | 3.1 | 99.2 | 2.6 |
| < − 60°C for 2 weeks | ||||
| 3 | 100.8 | 6.8 | 98.4 | 4.8 |
| 1,500 | 101.8 | 2.0 | 97.8 | 3.5 |
Pharmacokinetics
The validated analytical method was successfully applied to study the pharmacokinetic behavior and sex differences of URM in rats after intravenous and oral administrations.
The plasma concentration profiles of URM and UNO after intravenous or oral administration in male and female rats are plotted in Figure 3. The PK parameters of URM and UNO are listed in Table III.
Figure 3.
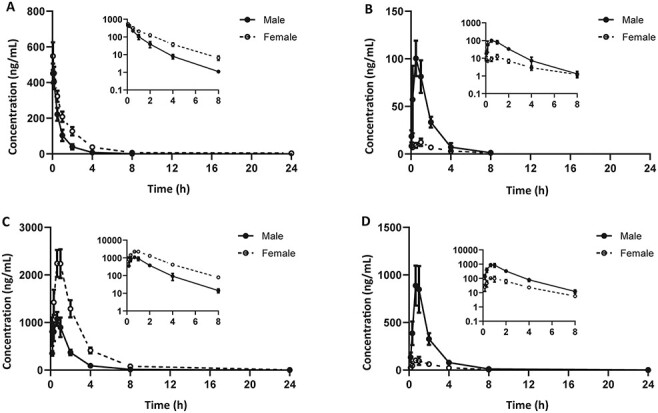
Mean plasma concentration-time curves in male and female rats (n = 5). (A) URM after 1 mg/kg intravenous administration; (B) UNO after 1 mg/kg intravenous administrations; (C) URM after 10 mg/kg oral administration (B) UNO after 10 mg/kg oral administration. The graphs are expressed as the mean ± SD.
Table III.
Pharmacokinetic Parameters of URM and UNO in Rats
| Parameters | Intravenous (1 mg/kg) | Oral (10 mg/kg) | ||||||
|---|---|---|---|---|---|---|---|---|
| URM | UNO | URM | UNO | |||||
| Male | Female | Male | Female | Male | Female | Male | Female | |
| Tmax (h) | 0.07 ± 0.05 | 0.05 ± 0.00 | 0.60 ± 0.22 | 0.81 ± 0.43 | 0.600 ± 0.149 | 0.800 ± 0.182 | 0.800 ± 0.182 | 0.867 ± 0.182 |
| Cmax (ng/mL) | 452 ± 54NS | 548 ± 78 | 102 ± 19a | 12.3 ± 3.9 | 1,089 ± 136a | 2,386 ± 297 | 943 ± 133a | 117 ± 27 |
| AUC0-t (ng/mL*h) | 363 ± 65a | 744 ± 122 | 172 ± 32a | 30.7 ± 7.4 | 1,960 ± 208a | 6,073 ± 488 | 1,637 ± 246a | 300 ± 62 |
| AUC0-∞ (ng/mL*h) | 368 ± 65a | 757 ± 123 | 177 ± 30a | 36.6 ± 6.5 | 1,985 ± 208a | 6,093 ± 491 | 1,654 ± 253a | 311 ± 59 |
| T1/2 (h) | 0.94 ± 0.29 | 1.76 ± 0.88 | 1.10 ± 0.19 | 1.95 ± 0.79 | 2.41 ± 1.84 | 3.18 ± 0.39 | 2.74 ± 1.4 | 3.65 ± 1.7 |
| MRT0-t (h) | 0.90 ± 0.14 | 1.86 ± 0.57 | – | – | 1.95 ± 0.34 | 2.85 ± 0.22 | – | – |
| MRT0-∞ (h) | 0.97 ± 0.12 | 2.08 ± 0.73 | – | – | 2.13 ± 0.46 | 2.94 ± 0.25 | – | – |
| CL (L/h/kg) | 2.77 ± 0.50a | 1.35 ± 0.19 | – | – | – | – | – | – |
| Vd (L/kg) | 2.69 ± 0.58 | 2.71 ± 0.54 | – | – | – | – | – | – |
| F (%) | – | – | – | – | 54.0 | 81.7 | – | – |
–: Not applicable.
Indicates the PK parameter is significantly difference (P < 0.05) between male and female rats.
NS: not significant.
Statistical analysis indicated significant differences (P-values < 0.05) of both AUC0-t and CL of URM in male and female rats after intravenous administration. The AUC0-t of URM in male rats (363 ± 65 ng/mL*h) was lower than females (744 ± 122 ng/mL*h), and the CL of URM in male rats (2.77 ± 0.50 L/h/kg) was higher than females (1.35 ± 0.19 L/h/kg). Regarding metabolite UNO, its Cmax and AUC0-t also showed significant differences. The higher levels of UNO in male rats indicated a higher conversion rate of URM than females.
After oral administration, the Cmax and AUC0-t of both URM and UNO also showed sex-based differences. The Cmax and AUC0-t of URM were 2.2 and 3.1 times lower in male rats than females, while the Cmax and AUC0-t of metabolite UNO were 8.1- and 5.5-fold higher in male rats than females, respectively.
Significantly higher oral bioavailability of URM was observed in female (81.7%) rats relative to males (54.0%), which could be explained by lower rate and extent of metabolism of URM in female rats than males. The UNO/URM ratios for AUC0-t after intravenous and oral administration were 47.4 and 83.5% in male rats, respectively. This indicates in addition to formation in the liver, UNO may also be formed in the intestine.
As previously reported on riddelliine (26), a retronecine-type PA, male rats showed higher levels to riddelliine N-oxide than females, while inverse relationship for riddelliine. In current study on URM, we also found the similar sex differences in rats. The metabolic patterns and DNA adduct profiles of riddelliine in liver microsomes were report by Xia et al. (27), which showed a linear relationship between the formation rates of riddelliine N-oxide and the toxic metabolite-derived DNA adducts in both rat and human. The results indicated that formation of N-oxide could partly reflect the toxic risk and the toxic mechanism in rats was relevant to humans.
It has been reported that hepatic cytochrome P450 (CYP) 3A subfamily is the major metabolic enzyme for PAs to exert toxicity in rats (27, 30, 35). It is well known that the abundance of CYP3A (especial CYP3A1 and CYP3A2) is higher in male rats than females (36, 37), which could lead to the sex-based differences in the pharmacokinetics and toxicology. However, such sex differences in rats are unlikely to occur in humans. Because the expressions of CYP3A4/5, the responsible metabolic enzymes of PAs in humans, possess considerable inter-individual variations (38). PA intoxication could exhibit greater individual differences rather than sex difference. Moreover, as the activity of CYP3A4 may be induced by many drugs (such as rifampin) (39), foods (such as St. John’s wort) (40), as well as alcohol (41), the toxic risk may increase by concurrent intake of PA-containing products and the above-mentioned drugs or foods.
Conclusion
A sensitive and rapid LC–MS-MS method for URM and UNO in rat plasma has been established and applied for the study of pharmacokinetic of URM in rats. A sex-based difference in pharmacokinetic behavior and N-oxide metabolism pathway was observed of URM and UNO after intravenous or oral administration in male and female rats.
Contributor Information
Feifei Lin, School of Chinese Materia Medica, Nanjing University of Chinese Medicine, 138 Xianlin Avenue, Qixia District, Nanjing 210023, Jiangsu, China; Chinese Academy of Sciences, Shanghai Institute of Materia Medica, 555 Zuchongzhi Road, Shanghai 201203, China.
Yan Ma, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, 536 Changle Road, Shanghai 200126, China.
Anni Pan, Chinese Academy of Sciences, Shanghai Institute of Materia Medica, 555 Zuchongzhi Road, Shanghai 201203, China.
Yang Ye, School of Chinese Materia Medica, Nanjing University of Chinese Medicine, 138 Xianlin Avenue, Qixia District, Nanjing 210023, Jiangsu, China; Chinese Academy of Sciences, Shanghai Institute of Materia Medica, 555 Zuchongzhi Road, Shanghai 201203, China.
Jia Liu, Chinese Academy of Sciences, Shanghai Institute of Materia Medica, 555 Zuchongzhi Road, Shanghai 201203, China.
Acknowledgements
This work was supported by Science and Technology Commission of Shanghai Municipality (number 19431908100).
References
- 1. Roeder E. (2000) Medicinal plants in China containing pyrrolizidine alkaloids. Pharmazie, 55, 711–726. [PubMed] [Google Scholar]
- 2. Hartmann T. (1995) Chemistry, biology and chemoecology of the pyrrolizidine alkaloids. In: Pelletier S.W. (eds.), Alkaloids: Chemical and Biological Perspectives, Volume 9, Chapter 4. Pergamon Press, Oxford, U.K., pp. 155–233. [Google Scholar]
- 3. Wiedenfeld H. (2013) Alkaloids derived from ornithine: pyrrolizidine alkaloids. In: Ramawat K.G., Mérillon J.M. (eds.), Natural Products, 1st edition, Chapter 13. Springer Press, Berlin, Heidelberg, pp. 359–379. [Google Scholar]
- 4. Edgar J.A. (2014) Food contaminants capable of causing cancer, pulmonary hypertension and cirrhosis. Medical Journal of Australia, 200, 73–74.doi: 10.5694/mja13.10227. [DOI] [PubMed] [Google Scholar]
- 5. Wiedenfeld H. (2011) Plants containing pyrrolizidine alkaloids: toxicity and problems. Food Additives & Contaminants: Part A, Chemistry, Analysis, Control, Exposure & Risk Assessment, 28, 282–292.doi: 10.1080/19440049.2010.541288. [DOI] [PubMed] [Google Scholar]
- 6. Song Z., He Y., Ma J., Fu P.P., Lin G. (2020) Pulmonary toxicity is a common phenomenon of toxic pyrrolizidine alkaloids. Journal of Environmental Science and Health, 38, 124–140.doi: 10.1080/26896583.2020.1743608. [DOI] [PubMed] [Google Scholar]
- 7. Rutz L., Gao L., Kupper J.H., Schrenk D. (2020) Structure-dependent genotoxic potencies of selected pyrrolizidine alkaloids in metabolically competent HepG2 cells. Archives of Toxicology, 94, 4159–4172.doi: 10.1007/s00204-020-02895-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gao L., Rutz L., Schrenk D. (2020) Structure-dependent hepato-cytotoxic potencies of selected pyrrolizidine alkaloids in primary rat hepatocyte culture. Food and Chemical Toxicology, 135, 110923.doi: 10.1016/j.fct.2019.110923. [DOI] [PubMed] [Google Scholar]
- 9. Pitanga B.P., Silva V.D., Souza C.S., Junqueira H.A., Fragomeni B.O., Nascimento R.P., et al. (2011) Assessment of neurotoxicity of monocrotaline, an alkaloid extracted from Crotalaria retusa in astrocyte/neuron co-culture system. Neurotoxicology, 32, 776–784.doi: 10.1016/j.neuro.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 10. Zhu L., Zhang C.Y., Li D.P., Chen H.B., Ma J., Gao H., et al. (2020) Tu-San-Qi (Gynura japonica): the culprit behind pyrrolizidine alkaloid-induced liver injury in China. Acta Pharmacologica Sinica, in press, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang X.Q., Ye J., Li X., Li Q., Song Y.H. (2019) Pyrrolizidine alkaloids-induced hepatic sinusoidal obstruction syndrome: pathogenesis, clinical manifestations, diagnosis, treatment, and outcomes. World Journal of Gastroenterology, 25, 3753–3763.doi: 10.3748/wjg.v25.i28.3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang M., Ruan J., Gao H., Li N., Ma J., Xue J., et al. (2017) First evidence of pyrrolizidine alkaloid N-oxide-induced hepatic sinusoidal obstruction syndrome in humans. Archives of Toxicology, 91, 3913–3925.doi: 10.1007/s00204-017-2013-y. [DOI] [PubMed] [Google Scholar]
- 13. Yang M., Ma J., Ruan J., Ye Y., Fu P.P., Lin G. (2019) Intestinal and hepatic biotransformation of pyrrolizidine alkaloid N-oxides to toxic pyrrolizidine alkaloids. Archives of Toxicology, 93, 2197–2209.doi: 10.1007/s00204-019-02499-2. [DOI] [PubMed] [Google Scholar]
- 14. Yang M., Ma J., Ruan J., Zhang C., Ye Y., Pi-Cheng F., et al. (2020) Absorption difference between hepatotoxic pyrrolizidine alkaloids and their N-oxides - mechanism and its potential toxic impact. Journal of Ethnopharmacology, 249, 112421.doi: 10.1016/j.jep.2019.112421. [DOI] [PubMed] [Google Scholar]
- 15. EFSA . (2017) Risks for human health related to the presence of pyrrolizidine alkaloids in honey, tea, herbal infusions and food supplements. EFSA Journal, 15, 4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mulder P.P.J., Lopez P., Castellari M., Bodi D., Ronczka S., Preiss-Weigert A., et al. (2018) Occurrence of pyrrolizidine alkaloids in animal- and plant-derived food: results of a survey across Europe. Food Additives & Contaminants: Part A, Chemistry, Analysis, Control, Exposure & Risk Assessment, 35, 118–133.doi: 10.1080/19440049.2017.1382726. [DOI] [PubMed] [Google Scholar]
- 17. Kempf M., Heil S., Hasslauer I., Schmidt L., von der Ohe K., Theuring C., et al. (2010) Pyrrolizidine alkaloids in pollen and pollen products. Molecular Nutrition & Food Research, 54, 292–300.doi: 10.1002/mnfr.200900289. [DOI] [PubMed] [Google Scholar]
- 18. Kaltner F., Kukula V., Gottschalk C. (2020) Screening of food supplements for toxic pyrrolizidine alkaloids. Journal of Consumer Protection and Food Safety, 15, 237–243.doi: 10.1007/s00003-020-01296-9. [DOI] [Google Scholar]
- 19. Mulder P.P., Sánchez P.L., These A., Preiss‐Weigert A., Castellari M. (2015) Occurrence of pyrrolizidine alkaloids in food. EFSA Supporting Publications, 12, 859E.doi: 10.2903/sp.efsa.2015.EN-859. [DOI] [Google Scholar]
- 20. Dubecke A., Beckh G., Lullmann C. (2011) Pyrrolizidine alkaloids in honey and bee pollen. Food Additives & Contaminants: Part A, Chemistry, Analysis, Control, Exposure & Risk Assessment, 28, 348–358.doi: 10.1080/19440049.2010.541594. [DOI] [PubMed] [Google Scholar]
- 21. Xu B.-Q., Zhang Y.-Q. (2017) Bioactive components of Gynura divaricata and its potential use in health, food and medicine: a mini-review. African Journal of Traditional, Complementary, and Alternative Medicines: AJTCAM, 14, 113–127.doi: 10.21010/ajtcam.v14i3.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roeder E., Eckert A., Wiedenfeld H. (1996) Pyrrolizidine alkaloids from Gynura divaricata. Planta Medica, 62, 386.doi: 10.1055/s-2006-957921. [DOI] [PubMed] [Google Scholar]
- 23. Dong X., Zhao S.X., Xu B.Q., Zhang Y.Q. (2019) Gynura divaricata ameliorates hepatic insulin resistance by modulating insulin signalling, maintaining glycolipid homeostasis and reducing inflammation in type 2 diabetic mice. Toxicology Research, 8, 928–938.doi: 10.1039/c9tx00191c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hong M.H., Jin X.J., Yoon J.J., Lee Y.J., Oh H.C., Lee H.S., et al. (2020) Antihypertensive effects of Gynura divaricata (L.) DC in rats with renovascular hypertension. Nutrients, 12, 3321–3334.doi: 10.3390/nu12113321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li J., Feng J., Wei H., Liu Q., Yang T., Hou S., et al. (2018) The aqueous extract of Gynura divaricata (L.) DC. improves glucose and lipid metabolism and ameliorates Type 2 diabetes mellitus. Evidence-Based Complementary and Alternative Medicine, 2018, 8686297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williams L., Chou M.W., Yan J., Young J.F., Chan P.C., Doerge D.R. (2002) Toxicokinetics of riddelliine, a carcinogenic pyrrolizidine alkaloid, and metabolites in rats and mice. Toxicology and Applied Pharmacology, 182, 98–104.doi: 10.1006/taap.2002.9441. [DOI] [PubMed] [Google Scholar]
- 27. Xia Q., Chou M.W., Kadlubar F.F., Chan P.C., Fu P.P. (2003) Human liver microsomal metabolism and DNA adduct formation of the tumorigenic pyrrolizidine alkaloid, riddelliine. Chemical Research in Toxicology, 16, 66–73.doi: 10.1021/tx025605i. [DOI] [PubMed] [Google Scholar]
- 28. Williams D.E., Reed R.L., Kedzierski B., Dannan G.A., Guengerich F.P., Buhler D.R. (1989) Bioactivation and detoxication of the pyrrolizidine alkaloid senecionine by cytochrome P-450 enzymes in rat liver. Drug Metabolism and Disposition: The Biological Fate of Chemicals, 17, 387–392. [PubMed] [Google Scholar]
- 29. Candrian U., Luthy J., Schlatter C. (1985) In vivo covalent binding of retronecine-labelled [3H]seneciphylline and [3H]senecionine to DNA of rat liver, lung and kidney. Chemico-Biological Interactions, 54, 57–69.doi: 10.1016/S0009-2797(85)80152-6. [DOI] [PubMed] [Google Scholar]
- 30. Chung W.G., Buhler D.R. (2004) Differential metabolism of the pyrrolizidine alkaloid, senecionine, in Fischer 344 and Sprague-Dawley rats. Archives of Pharmacal Research, 27, 547–553.doi: 10.1007/BF02980130. [DOI] [PubMed] [Google Scholar]
- 31. Lin G., Cui Y.Y., Liu X.Q. (2003) Gender differences in microsomal metabolic activation of hepatotoxic clivorine in rat. Chemical Research in Toxicology, 16, 768–774.doi: 10.1021/tx0340302. [DOI] [PubMed] [Google Scholar]
- 32. Lin G., Tang J., Liu X.Q., Jiang Y., Zheng J. (2007) Deacetylclivorine: a gender-selective metabolite of clivorine formed in female Sprague-Dawley rat liver microsomes. Drug Metabolism and Disposition: The Biological Fate of Chemicals, 35, 607–613.doi: 10.1124/dmd.106.014100. [DOI] [PubMed] [Google Scholar]
- 33.(2018) Bioanalytical Method Validation, Guidance for Industry. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Veterinary Medicine, Biopharmaceutics. https://www.fda.gov/media/70858/download (Accessed Mar 10, 2021).
- 34.National Research Council (2011) Guide for the Care and Use of Laboratory Animals. 8th edition. The National Academies Press, Washington, DC, pp. 1–200. [Google Scholar]
- 35. Miranda C.L., Reed R.L., Guengerich F.P., Buhler D.R. (1991) Role of cytochrome P450IIIA4 in the metabolism of the pyrrolizidine alkaloid senecionine in human liver. Carcinogenesis, 12, 515–519.doi: 10.1093/carcin/12.3.515. [DOI] [PubMed] [Google Scholar]
- 36. Kato R., Yamazoe Y. (1992) Sex-specific cytochrome P450 as a cause of sex- and species-related differences in drug toxicity. Toxicology Letters, 64–65, 661–667.doi: 10.1016/0378-4274(92)90245-F. [DOI] [PubMed] [Google Scholar]
- 37. Ribeiro V., Lechner M.C. (1992) Cloning and characterization of a novel CYP3A1 allelic variant: analysis of CYP3A1 and CYP3A2 sex-hormone-dependent expression reveals that the CYP3A2 gene is regulated by testosterone. Archives of Biochemistry and Biophysics, 293, 147–152.doi: 10.1016/0003-9861(92)90377-9. [DOI] [PubMed] [Google Scholar]
- 38. Eichelbaum M., Burk O. (2001) CYP3A genetics in drug metabolism. Nature Medicine, 7, 285–287.doi: 10.1038/85417. [DOI] [PubMed] [Google Scholar]
- 39. Chen J., Raymond K. (2006) Roles of rifampicin in drug-drug interactions: underlying molecular mechanisms involving the nuclear pregnane X receptor. Annals of Clinical Microbiology and Antimicrobials, 5, 3.doi: 10.1186/1476-0711-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Markowitz J.S., Donovan J.L., DeVane C.L., Taylor R.M., Ruan Y., Wang J.S., et al. (2003) Effect of St John’s wort on drug metabolism by induction of cytochrome P450 3A4 enzyme. Journal of the American Medical Association, 290, 1500–1504.doi: 10.1001/jama.290.11.1500. [DOI] [PubMed] [Google Scholar]
- 41. Feierman D.E., Melinkov Z., Nanji A.A. (2003) Induction of CYP3A by ethanol in multiple in vitro and in vivo models. Alcoholism, Clinical and Experimental Research, 27, 981–988.doi: 10.1111/j.1530-0277.2003.tb04424.x. [DOI] [PubMed] [Google Scholar]