

Abstract
The market for products containing cannabidiol (CBD) is booming globally. However, the pharmacokinetics of CBD in different oral formulations and the impact of CBD use on urine drug testing outcomes for cannabis (e.g., 11-nor-9-carboxy-Δ9-tetrahydrocannabinol (Δ9-THCCOOH)) are understudied. This study characterized the urinary pharmacokinetics of CBD (100 mg) following vaporization or oral administration (including three formulations: gelcap, pharmacy-grade syrup and or Epidiolex) as well as vaporized CBD-dominant cannabis (containing 100 mg CBD and 3.7 mg Δ9-THC) in healthy adults (n = 18). A subset of participants (n = 6) orally administered CBD syrup following overnight fasting (versus low-fat breakfast). Urine specimens were collected before and for 58 h after dosing on a residential research unit. Immunoassay (IA) screening (cutoffs: 20, 50 and 100 ng/mL) for Δ9-THCCOOH was performed, and quantitation of cannabinoids was completed via LC–MS-MS. Urinary CBD concentrations (ng/mL) were higher after oral (mean Cmax: 734; mean Tmax: 4.7 h, n = 18) versus vaporized CBD (mean Cmax: 240; mean Tmax: 1.3 h, n = 18), and oral dose formulation significantly impacted mean Cmax (Epidiolex = 1,274 ng/mL, capsule = 776 ng/mL, syrup = 151 ng/mL, n = 6/group) with little difference in Tmax. Overnight fasting had limited impact on CBD excretion in urine, and there was no evidence of CBD conversion to Δ8- or Δ9-THC in any route or formulation in which pure CBD was administered. Following acute administration of vaporized CBD-dominant cannabis, 3 of 18 participants provided a total of six urine samples in which Δ9-THCCOOH concentrations ≥15 ng/mL. All six specimens screened positive at a 20 ng/mL IA cutoff, and two of six screened positive at a 50 ng/mL cutoff. These data show that absorption/elimination of CBD is impacted by drug formulation, route of administration and gastric contents. Although pure CBD is unlikely to impact drug testing, it is possible that hemp products containing low amounts of Δ9-THC may produce a cannabis-positive urine drug test.
Introduction
Cannabis access has increased due to widespread policy changes permitting its medicinal use or decriminalization. Notably, hemp (defined in the USA as cannabis containing ≤0.3% of the psychoactive constituent Δ9-tetrahydrocannabinol (Δ9-THC)) was removed from the United States’ list of controlled substances via the Agriculture Improvement Act of 2018 (a.k.a., “The Farm Bill”). Together, sweeping hemp and cannabis policy reforms have fostered a vast retail market of cannabinoid-containing products. The growing market of hemp products is dominated by those containing cannabidiol (CBD) as the primary chemical constituent. The collective market for CBD sales (e.g., dispensary, pharmaceutical and retail sales), which was reported as $1.9 billion in 2018, is forecasted to reach $20 billion by 2024 (1). Concurrently, CBD-containing products of a variety of formulations (e.g., vaporization liquids, oral capsules and solutions and topical skin products) are marketed as nutraceuticals, dietary supplements, cosmetics and assorted other types of retail products.
The proliferation of CBD-dominant hemp and cannabis products has spurred efforts to characterize the impact of CBD use on urine drug testing. Workplace and other drug testing programs most commonly test for recent cannabis use by analyzing urine concentrations of 11-nor-9-carboxy-Δ9-THC (Δ9-THCCOOH), a metabolite of Δ9-THC (2, 3). While CBD use is not currently evaluated in most drug testing programs, there is a reason for concern that CBD-dominant cannabis or hemp product use may produce a positive result for Δ9-THCCOOH on a urine drug test. First, hemp-derived retail CBD products can legally contain up to 0.3% Δ9-THC in the USA (4), and even the FDA-approved CBD medication Epidiolex may contain trace levels (<0.1%) of Δ9-THC (U.S. Drug Enforcement Administration, 83 FR 48950). In a recent open-label, 4-week trial, participants sublingually administered a full-spectrum, high-CBD hemp extract (9.97 mg/mL CBD (1.04%) and 0.23 mg/mL of Δ9-THC (0.02%)) three times per day. Urine toxicology testing showed that 6 of 14 of participants had urinary Δ9-THCCOOH concentrations >15 ng/mL, the confirmatory cutoff concentration listed in the Mandatory Guidelines for federal workplace drug testing (5). This study demonstrated that the use of retail CBD products poses a risk of a positive drug test, but the CBD/THC dosing parameters or individual user characteristics that would likely contribute to a positive versus negative test result remain unknown. Second, retail CBD products that do not disclose the presence of Δ9-THC may still contain Δ9-THC in concentrations ranging from trace levels to concentrations capable of producing impairment. For example, an analytical study of 84 retail CBD products without labeling related to Δ9-THC content detected Δ9-THC in 18 samples with observed Δ9-THC concentrations as high as 6.43 mg/mL (6). Third, in vitro evidence indicates that CBD may degrade to Δ8- and Δ9-THC in simulated gastric fluid of pH = 1.2 (7–9), suggesting that CBD may be converted to Δ- or Δ9-THC in highly acidic human gastric fluid (pH = 1.0–2.5) (10). Fourth, in the physiological environment of the human gut, the proportion of CBD that is degraded to Δ8- and Δ9-THC may be impacted by fasting or an abstinence from food (7).
To date, few human laboratory studies have systematically evaluated the urinary pharmacokinetics of CBD or its primary metabolites (7-OH-CBD, 7-CBD-COOH) under acute dosing conditions while manipulating factors relevant to its use (e.g., route of administration, oral formulation and ingestion of food prior to use). Recently, several studies have characterized urinary CBD concentrations following acute cannabis dosing in controlled laboratory studies (11–13), including smoked CBD-dominant cannabis containing <0.2% THC (12, 13). A pilot study in our laboratory found that concentrations of CBD were higher after a single administration of an oral CBD capsule versus vaporized CBD, and the time to maximum CBD concentration was shorter in the vaporized versus oral CBD condition (14). The present study evaluated the urinary pharmacokinetics of CBD, Δ9-THC and their metabolites in healthy adults following a single dose of vaporized or orally ingested CBD (3 formulations) or vaporized CBD-dominant cannabis containing a very low amount of Δ9-THC (0.39% by dry weight), which is slightly above the threshold to be considered hemp. The influence of overnight fasting on CBD urinary pharmacokinetics, including the potential for conversion of CBD to Δ9-THC or Δ8-THC was also evaluated. Thus, this study addressed critical knowledge gaps in understanding the impact of acute CBD administration (orally ingested via multiple formulations or inhaled with a vaporizer) on urine cannabis drug testing outcomes.
Methods
Participants
Participants were healthy adults recruited via word-of-mouth and web-based advertisements. Interested individuals completed a telephone pre-screening interview, and those who appeared eligible were invited for a screening visit at the Johns Hopkins Behavioral Pharmacology Research Unit (BPRU), where written informed consent was obtained and study eligibility determined. This study was approved by the Johns Hopkins University School of Medicine’s Institutional Review Board (IRB00128331).
Inclusion criteria were as follows: age 18–45 years and body mass index (BMI) 19–36 kg/m2; good physical health per medical history and physical exam, 12-lead electrocardiogram and blood chemistry, hematology and serology analysis; self-reported no past-30-day use of cannabis or other psychoactive drugs other than alcohol, nicotine or caffeine; test negative for cannabis, other illicit drugs and alcohol per urine toxicology and breathalyzer at screen and each experimental session; prior experience inhaling cannabis and, for females, negative pregnancy test (via serum at screening and via urine test at each visit).
Key exclusion criteria were as follows: current use of prescription medication, over-the-counter medication or supplements/other drug products that could interfere with study outcomes or participant safety (e.g., drugs metabolized through CYP2D6, CYP2C9 and CYP2B10 enzymes or drugs that inhibit CYP3A4 enzymes); history or current evidence of significant medical condition (e.g., epilepsy, anemia, cardiac illness and traumatic brain injury); use of dronabinol within 6 months prior to screening or hemp seeds or oil within 3 months prior to screening and participation in another clinical trial or having received a drug as part of a research study in the 30 days prior to study participation.
A total of 18 participants provided informed consent and completed study procedures (nine men and nine women). Participants’ demographics are shown in Table S1 (Supplemental Material). Participants were predominantly white, non-Hispanic and did not smoke cigarettes. Participants (mean (SD)) were 31 (6) years of age, had a BMI of 26 (4) and had not used cannabis for 146 (251) days prior to the first drug administration session.
Study design
Eighteen participants completed four double-blind, double-dummy acute dosing sessions, each lasting at least 58 h. During each session, participants were exposed to an oral dose (either active or placebo) and then a vaporized dose (either active or placebo) exactly 1 h later. Six of the eighteen participants also completed a fifth study session in which they received an oral dose following overnight fasting (described below). A double-dummy procedure was employed to control for expectancy effects related to the route of administration.
The four dosing conditions completed by all 18 participants were as follows: (i) 100 mg oral CBD and vaporized placebo cannabis; (ii) oral placebo and 100 mg vaporized CBD; (iii) oral placebo and vaporized CBD-dominant cannabis containing 100 mg CBD and 3.7 mg Δ9-THC; (iv) oral placebo and vaporized placebo cannabis. The 100 mg oral CBD dose was delivered as one of three formulations: capsule, syrup or Epidiolex (six participants received each oral dose formulation). The four primary dosing conditions were completed in a randomized order. A fifth dose condition, always completed after the four primary dosing conditions, was completed by six of the eighteen study completers. In this fifth condition, participants fasted overnight (for at least 12 h) on a residential research unit prior to dosing and were then exposed to an oral dose of 100 mg CBD in syrup followed by vaporized placebo cannabis.
Study drug
Two batches of cannabis (CBD-dominant and placebo) were obtained for this study from the National Institute on Drug Abuse (NIDA) Drug Supply Program. The available CBD-dominant cannabis provided by the NIDA Drug Supply Program met the pre-specified criteria for “low THC (<1%) / very high CBD (>10%)” content. Specifically, the batch of CBD-dominant cannabis procured for the present study contained ∼10.5% CBD, 0.39% Δ9-THC, 0.02% Δ8-THC and 0.05% cannabinol (CBN) and was measured to yield a total CBD dose of 100 mg and a Δ9-THC dose of 3.7 mg. This was consistent with the ratio of CBD:THC found in commercial “CBD products” at the time, but note that this study was initiated before hemp was legalized in the USA and operationally defined as cannabis containing 0.3% THC or less. The placebo cannabis batch contained 0.003% CBD, 0.001% Δ9-THC, no detectable Δ8-THC and 0.005% CBN. Identical quantities of plant material (953 mg) were used under placebo and active dosing conditions. Cannabis was vaporized using the Volcano Medic® (Storz and Bickel, Tuttelingen, Germany) vaporizer at a temperature of 204°C (400°F).
Albany Molecular Research Inc. supplied pure CBD in crystalline powder form. Purity by HPLC was 100%, and independent testing confirmed the absence of Δ9-THC. CBD was prepared for dosing and dispensed by the Johns Hopkins BPRU pharmacy. For vaporization of pure CBD, the Volcano Medic® was used to heat and aerosolize the CBD powder, which was placed on a stainless-steel dosing pad accessory for the Volcano Medic®. For oral administration, three formulations were prepared.
Oral capsule
CBD powder (100 mg) was weighed and placed into a size 00, pharmacy-grade gelcap. The remaining space of the gelcap was filled with inert microcrystalline cellulose. Placebo gelcaps were filled with the same cellulose and no CBD.
Oral syrup
CBD powder (100 mg) was weighed, and ∼2 mL of ORA Plus® suspending vehicle was added directly to the CBD powder to facilitate dissolution. This mixture was suspended into a pharmacy-grade, cherry-flavored syrup to achieve a final volume of 10 mL. The placebo condition was an equal volume of cherry-flavored syrup that did not contain CBD.
Epidiolex
Epidiolex is a strawberry-flavored solution that contains CBD at a concentration of 100 mg/mL. 1 mL Epidiolex was dissolved into 9 mL of pharmacy grade, cherry-flavored syrup to obtain a final CBD dose of 100 mg at a final volume of 10 mL. The placebo condition was an equal volume of cherry-flavored syrup that did not contain Epidiolex.
The dose of 100 mg CBD was employed for two main reasons. First, there is 100 mg CBD in a single unit dose (1 mL) of Epidiolex. Second, ∼100 mg CBD is contained in a 1 g cannabis cigarette containing 10% CBD, which are common characteristics of “pre-rolled” high CBD cannabis cigarettes in legal retail markets in the USA and Canada. We maintained the 100 mg CBD dose for cannabis plant material to permit comparison with the pure CBD conditions. The use of 3.7 mg Δ9-THC yields a 25:1 CBD:THC ratio, which is common for commercially available CBD-dominant cannabis products. Moreover, the 0.39% Δ9-THC concentration in the plant material is close to what is defined as hemp in the USA (≤0.3%).
Experimental session procedures
Each 58 h dosing session consisted of an acute drug administration period, lasting 8 h, followed by a two-night, three-day inpatient stay on a closed residential unit. Sessions were scheduled so that dosing occurred at least a week apart to allow for drug washout between doses. The 8 h acute dosing period was conducted at the BPRU. Participants arrived to the BPRU the morning of dosing and first completed a urine drug test, urine pregnancy test (for females) and alcohol breathalyzer, which were all required to be negative to conduct the session. Participants were then given a standard low-fat breakfast of toast and jam (except during the fasted condition), had an intravenous catheter inserted for blood sampling and baseline blood sample collected, had baseline vital signs taken and reported the use of any medications or drugs including alcohol, cannabis and nicotine via timeline follow-back interview. Then, baseline subjective effects questionnaires and cognitive performance tasks (digit serial substitution task, divided attention task and paced serial addition task) were administered, and baseline urine and oral fluid samples were collected. Outcomes of the pharmacodynamic analyses were published previously (15).
Participants orally ingested either 100 mg CBD or a comparable placebo (see Study Drug section for descriptions of oral formulations). Exactly 1 h after oral ingestion, participants used the Volcano Medic® to administer 100 mg pure CBD, 100 mg CBD-dominant cannabis or placebo cannabis vapor by heating the drug to 204°C (400°F) and capturing the vapor generated in a balloon. Participants were given 10 minutes to inhale the contents of three fully inflated balloons ad libitum. New balloons were used for each dosing session, and they were covered with an opaque bag to obscure the appearance of the vapor in the balloon to preserve blinding.
After baseline assessments and dosing, urine specimens were collected from participants. Total volume was measured, and two 30-mL aliquots were collected and stored in polypropylene containers at −20°C. The first four urine specimens were obtained at target nominal time points post-dosing and subsequent specimens were pooled samples of all urine produced by participants over 2–10 h periods. Spot samples were collected at the end of each pooled time period and combined with the pooled sample. Given that oral drug (or placebo) administration occurred 1 h before vaporized drug (or placebo), the timelines for urine collection varied based on route of administration. The timeline for collection following oral drug administration was as follows: baseline and 1.5, 2, 3, 4, 4–6, 6–8, 8–10, 10–12, 12–22, 22–26, 26–30, 30–34, 34–46, 46–50, 50–54 and 54–58 h. The timeline for collection following vaporized drug administration was as follows: baseline and 0.5, 1, 2, 3, 3–5, 5–7, 7–9, 9–11, 11–21, 21–25, 25–29, 29–33, 33–45, 45–49, 49–53 and 53–57 h. Urine collection sometimes varied from the target time by about ±5 min across participants/sessions (e.g., if participant was unable to void immediately).
Immunoassay and creatinine
Urine specimens were analyzed with the DRI Cannabinoid Assay via the manufacturer’s procedure (Thermo Fisher Scientific, Fremont, CA) utilizing 20, 50 and 100 ng/mL cutoff concentrations. Immunoassay (IA) methods and cross-reactivity data were previously published (14). Creatinine was determined with the Siemens modified Jaffe reagent.
Hydrolysis methods for confirmatory liquid chromatography with tandem mass spectrometry (LC--MS-MS)
The following analytes were evaluated in the present study: Δ9-THC, Δ8-THC, 11-hydroxy- Δ9-THC (11-OH-Δ9-THC), Δ9-THCCOOH, Δ8-THCCOOH, tetrahydrocannabivarin (THCV), THCV carboxylic acid (THCVA), CBD, cannabinol (CBN), 7-hydroxy-CBD (7-OH-CBD) and 7-carboxy-CBD (7-CBD-COOH). For the following hydrolysis, extraction and LC–MS-MS methods, minor changes to the analytical method were employed between Study 1 and Study 2; Study 1 represents n = 6 participants (#038, 053, 054, 063, 066 and 068) who were included in the pilot study preceding this report (14). Study 2 represents the remaining n = 12 participants reported for the first time in this report. Differences in analytical methodology are described below, where applicable.
Study 1
It was anticipated that two types of conjugated metabolites would be present in urine specimens from these studies (i.e., ether-linked CBD and acid-linked THCCOOH). Because ether-linked cannabinoid conjugates are less susceptible to base-hydrolysis, a separate enzyme hydrolysis method was developed for potential ether-linked conjugates in Study 1. Base hydrolysis was conducted with 0.1 mL of 5 N KOH solution added to 0.3 mL of urine specimens, calibrators and controls and 0.1 mL of internal standard solution. Samples were incubated at 50°C for 15 min. Following incubation, 0.1 mL of 5 N formic acid and 0.4 mL of potassium phosphate buffer, pH 6.8 was added prior to extraction. Enzyme hydrolysis was conducted with 0.1 mL of BGTurbo® solution (Kura Biotec, Rancho Dominguez, CA) added to 0.3 mL of urine specimens, calibrators and controls and 0.1 mL of internal standard solution. Samples were incubated at 50°C for 30 min. Following incubation, 0.5 mL of potassium phosphate buffer, pH 6.8 was added prior to extraction.
Study 2
In Study 2, both base hydrolysis and enzyme hydrolysis were performed sequentially for all analytes. Sample volume of 0.3 mL of urine, calibrators and controls and 0.1 mL of internal standard solution were pipetted into silanized glass tubes. Enzyme hydrolysis was performed first with 0.1 mL of BGTurbo® solution (Kura Biotec, Rancho Dominguez, CA) and incubated at 50°C for 15 min. Following incubation, 0.1 mL of 5 N KOH was added and incubated at 50°C for 5 min. This was followed by 0.1 mL of 5 N formic acid and mixed with 1 mL of potassium phosphate buffer, pH 6.8 prior to extraction.
Extraction methods for confirmatory LC–MS-MS
Study 1
Clean Screen XCEL II 3 mL/130 mg SPE cartridges (UCT, Bristol, PA) were used to separately extract base or enzyme hydrolyzed samples. Samples were passed through the cartridges, and then, the extraction column was washed using 3 mL of hexane and eluted with 2 mL of solvent (49/49/2 hexane/ethyl acetate/acetic acid). Equal parts 0.1% formic acid in water and methanol totaling 0.4 M was used to evaporate and reconstitute samples. Samples were analyzed in separate runs (base hydrolyzed and enzyme hydrolyzed samples by LC–MS-MS).
Study 2
Sequentially hydrolyzed samples underwent a single extraction using Clean Screen XCEL II 3 mL/130 mg SPE cartridges. After sample passage through the cartridge, the extraction column was washed with 2.5 mL of hexane and dried. Samples were eluted with 2 mL of solvent (49/49/2 hexane/ethyl acetate/formic acid). Extracts were evaporated and reconstituted with 0.4 mL of equal parts of 0.1% formic acid in water and methanol and transferred to a 96 deep-well plate for analysis by LC–MS-MS. Samples were analyzed in a single run (since base and enzyme hydrolysis occurred sequentially rather than separately).
Please note that, in Study 1, 7-OH-CBD and 7-CBD-COOH were not included in the pilot LC–MS-MS analysis. Therefore, samples from Study 1 were re-extracted and sample extracts analyzed for 7-OH-CBD and 7-CBD-COOH using the Study 2 methods.
LC–MS-MS analyses
The cannabinoids analyzed by LC--MS-MS included: Δ9-THC, Δ8-THC, 11-OH-Δ9-THC, Δ9-THCCOOH, Δ8-THCCOOH, THCV, THCVA, CBD, CBN, 7-OH-CBD and 7-CBD-COOH. A detailed explanation of the LC–MS-MS methods is available in the Supplemental Material. Of note, the limit of detection (LOD) for all analytes was 1.0 ng/mL. The upper limit of linearity and carry over limit for all analytes was 1,000 ng/mL with the exception of Δ9-THCCOOH established at 500 ng/mL.
Data presentation and analysis
Descriptive statistics summarize participant demographics and LC–MS-MS urine results. Noncompartmental pharmacokinetic calculation of half-life (t1/2) from urine data was generated using the excretion rate method such that t1/2 = ln (2)/Kel, where Kel is equal to the elimination rate constant. Percent dose excreted of the 100 mg CBD was computed for each analyte using the following steps: first, the amount of analyte excreted ((volume of urine void, mL) * (concentration of analyte, ng)) was calculated for each time point; then, cumulative amount of analyte (ng) was generated by computing the sum of the amount of analyte (adjusted for changes in molecular weight of metabolites) excreted at each time point. Last, percent dose excreted was calculated by dividing the cumulative amount of analyte excreted by 100.
One-way ANOVA or Student’s t-test were employed as appropriate to compare within-subjects differences on pharmacokinetic parameters (maximum concentrations (Cmax), time to maximum concentrations (Tmax), t1/2, dose excreted) for CBD, 7-OH-CBD or 7-CBD-COOH by route of administration (oral, vaporized) and product type (pure CBD and high CBD cannabis). For observed main effects, post-hoc multiple comparisons were made using Tukey’s test (α = 0.05). Inferential statistics were not carried out for between-subjects comparisons of oral CBD formulation (capsule, syrup and Epidiolex) or gastric contents (low-fat breakfast and fasting) due to considerable between-subject variance and small sample size (n = 6) for these conditions. Note that, for analyses of pharmacokinetic outcomes, the midpoint time value was used for pooled specimens. For example, if Cmax was observed at “6–8” h, the Tmax value was recorded as “7 h”.
Exploratory mixed effects models (see Supplemental Material) were used, where appropriate, to compare urinary concentrations of CBD, 7-OH-CBD or 7-CBD-COOH (I) between routes of administration or (II) oral formulations with the factors of dose condition and time post-administration (baseline and 16 time points) and (III) between fasted states with the factors of dose condition (overnight fasting and standard low-fast breakfast) and time post-administration (baseline and 16 time points). For observed dose condition x time interactions, post-hoc multiple comparisons were made using Tukey’s test (α = 0.05). Statistical analyses were conducted using Prism 8 for macOS (Version 8.3.0, GraphPad Software, LLC).
Note that participants in the previously published pilot study (14) are included in this manuscript due to the fact that the present analysis includes additional metabolites and comparison of all outcomes across product formulations.
Results
Table S2 displays creatinine, IA results at 20, 50 and 100 ng/mL cutoff concentrations, and LC–MS-MS urinary results for CBD, 7-OH-CBD, 7-CBD-COOH and Δ9-THCCOOH for each participant over time. Other analytes (CBN, Δ9-THC, Δ8-THC, THC-V, THC-VA, 11-OH-THC and Δ8-THCCOOH) were detected in trace amounts (i.e., on average, below the LOD) or not at all. Thus, these analytes are not presented in the interest of parsimony. Table I contains Cmax, Tmax, t1/2, and the percent of the 100 mg CBD dose excreted for CBD, 7-OH-CBD, 7-CBD-COOH, and Δ9-THCCOOH in urine across all participants. Although some sex differences were observed in the present study, these findings were few and unsystematic. A complete report of statistical outcomes (including observed sex differences) is available as Supplemental Material.
Table I.
Summary of Urinary Pharmacokinetic Outcomes across Cannabinoid Analytes following Administration of Oral and Vaporized CBD and CBD-dominant Cannabis
| Sample information | CBD | 7-OH-CBD | 7-CBD-COOH | Δ9-THCCOOH | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Condition | Cmax (ng/mL) | Tmax (h) | Dose excreted (%) | t1/2 (h) | Cmax (ng/mL) | Tmax (h) | Dose excreted (%) | t1/2 (h) | Cmax (ng/mL) | Tmax (h) | Dose excreted (%) | t1/2 (h) | Cmax (ng/mL) | Tmax (h) | |
| Vaporized cannabis (100 mg CBD/3.7 mg THC) (n = 18) | Mean | 327.61 | 1.44 | 0.14 | 12.07 | 1,250.22 | 1.81 | 0.47 | 14.09 | 48.76 | 8.56 | 0.09 | 29.84 | 7.91 | 8.11 |
| SD | 235.53 | 1.06 | 0.10 | 11.13 | 1,479.93 | 1.13 | 0.40 | 4.88 | 40.72 | 9.82 | 0.06 | 11.65 | 8.46 | 8.51 | |
| Min | 27.10 | 0.50 | 0.01 | 0.69 | 142.00 | 0.50 | 0.08 | 2.41 | 4.76 | 1.00 | 0.01 | 11.31 | 1.20 | 1.00 | |
| Max | 808.50 | 4.00 | 0.39 | 40.66 | 5,073.04 | 4.00 | 1.56 | 24.27 | 172.30 | 31.00 | 0.18 | 51.00 | 29.90 | 31.00 | |
| 100 mg vaporized pure CBD (n = 18) | Mean | 239.95 | 1.28 | 0.11 | 7.27 | 677.40 | 1.75 | 0.32 | 11.82 | 29.39 | 7.17 | 0.06 | 29.63 | 0.43 | ND |
| SD | 240.37 | 1.13 | 0.10 | 7.73 | 596.37 | 0.91 | 0.25 | 6.30 | 19.17 | 5.95 | 0.05 | 24.49 | 1.22 | ND | |
| Min | 15.30 | 0.50 | 0.01 | 0.78 | 78.20 | 0.50 | 0.04 | 0.82 | 2.91 | 1.00 | 0.00 | 5.11 | 0.00 | ND | |
| Max | 1,007.90 | 4.00 | 0.44 | 23.69 | 2,041.80 | 3.00 | 0.92 | 23.33 | 65.80 | 16.00 | 0.16 | 107.47 | 5.00 | 4.00 | |
| 100 mg oral CBD capsule (n = 6) | Mean | 776.25 | 5.33 | 0.38 | 11.16 | 3,588.22 | 5.50 | 2.04 | 11.74 | 75.07 | 15.33 | 0.23 | 82.03 | 0.70 | ND |
| SD | 1,069.56 | 3.01 | 0.39 | 5.72 | 3,753.74 | 2.95 | 1.96 | 4.41 | 55.64 | 8.21 | 0.26 | 108.94 | 1.20 | ND | |
| Min | 210.20 | 2.00 | 0.04 | 4.29 | 373.26 | 2.00 | 0.24 | 7.72 | 25.22 | 4.00 | 0.03 | 12.64 | 0.00 | ND | |
| Max | 2,941.00 | 9.00 | 1.16 | 17.91 | 9,944.19 | 9.00 | 4.53 | 19.67 | 157.62 | 28.00 | 0.73 | 300.91 | 2.90 | 1.50 | |
| 100 mg oral CBD syrup (n = 6) | Mean | 150.57 | 4.33 | 0.12 | 11.39 | 1,075.00 | 4.00 | 0.81 | 19.06 | 79.93 | 15.17 | 0.13 | 48.96 | 0.32 | ND |
| SD | 77.81 | 1.75 | 0.08 | 7.78 | 999.20 | 1.10 | 0.45 | 15.58 | 86.79 | 12.84 | 0.05 | 58.98 | 0.78 | ND | |
| Min | 68.70 | 2.00 | 0.04 | 3.33 | 238.50 | 2.00 | 0.30 | 7.82 | 14.80 | 5.00 | 0.10 | 15.05 | 0.00 | ND | |
| Max | 228.90 | 7.00 | 0.26 | 25.94 | 2,734.80 | 5.00 | 1.49 | 49.33 | 239.20 | 40.00 | 0.24 | 167.17 | 1.90 | BL | |
| 100 mg oral Epidiolex (n = 6) | Mean | 1,273.83 | 4.33 | 0.50 | 10.78 | 5,849.10 | 4.50 | 2.53 | 13.36 | 324.75 | 13.00 | 0.62 | 24.59 | 0.20 | ND |
| SD | 1,168.15 | 0.52 | 0.37 | 2.48 | 3,588.11 | 0.55 | 0.81 | 5.52 | 113.18 | 7.77 | 0.25 | 8.97 | 0.49 | ND | |
| Min | 368.10 | 4.00 | 0.15 | 6.46 | 3,527.20 | 4.00 | 1.52 | 7.25 | 190.60 | 4.00 | 0.20 | 16.91 | 0.00 | ND | |
| Max | 3,470.20 | 5.00 | 1.08 | 13.20 | 12,898.30 | 5.00 | 3.84 | 22.29 | 520.20 | 24.00 | 0.89 | 41.79 | 1.20 | 17.00 | |
| 100 mg oral CBD (formulations combined) (n = 18) | Mean | 733.55 | 4.67 | 0.34 | 11.11 | 3,504.11 | 4.67 | 1.79 | 14.72 | 159.92 | 14.50 | 0.33 | 51.86 | 0.41 | ND |
| SD | 981.43 | 1.97 | 0.34 | 5.41 | 3,500.06 | 1.85 | 1.39 | 9.82 | 145.88 | 9.34 | 0.29 | 71.58 | 0.85 | ND | |
| Min | 68.70 | 2.00 | 0.04 | 3.33 | 238.50 | 2.00 | 0.24 | 7.25 | 14.80 | 4.00 | 0.03 | 12.64 | 0.00 | ND | |
| Max | 3,470.20 | 9.00 | 1.16 | 25.94 | 12,898.30 | 9.00 | 4.53 | 49.33 | 520.20 | 40.00 | 0.89 | 300.91 | 2.90 | 17.00 | |
| 100 mg oral CBD syrup (overnight fasting) (n = 6) | Mean | 167.88 | 4.00 | 0.07 | 35.24 | 605.38 | 4.00 | 0.28 | 49.84 | 32.58 | 16.33 | 0.05 | 105.45 | 0.00 | ND |
| SD | 212.62 | 1.10 | 0.06 | 23.69 | 578.48 | 1.10 | 0.10 | 45.43 | 22.04 | 18.60 | 0.02 | 84.64 | 0.00 | ND | |
| Min | 14.90 | 2.00 | 0.02 | 5.22 | 80.40 | 2.00 | 0.16 | 7.43 | 13.80 | 4.00 | 0.02 | 23.33 | 0.00 | ND | |
| Max | 572.50 | 5.00 | 0.15 | 65.42 | 1,358.20 | 5.00 | 0.39 | 125.38 | 60.20 | 52.00 | 0.08 | 231.47 | 0.00 | ND | |
ND = analyte was not detected for some participants/conditions, which precluded mean Tmax calculation. BL = baseline. Midpoint time value used for pooled specimens. For dose excreted (%), values are corrected for the molecular weight of the analyte.
Urinary CBD pharmacokinetics by dosing condition
Figure 1A depicts urine CBD (ng/mL) concentrations by route of administration and study time point (data points represent mean ± SEM across participants). Across oral formulations (capsule, syrup and Epidiolex conditions collapsed for this analysis), peak CBD concentrations were most commonly observed 4 h after oral dose administration (mean Cmax = 734 ng/mL, range Cmax: 69–3,470 ng/mL, mean Tmax = 4.7 h and range Tmax: 2.0–9.0 h). In the vaporized pure CBD condition, peak CBD concentrations (mean Cmax = 240 ng/mL, range Cmax: 15.3–1,008 ng/mL, mean Tmax = 1.3 h and range Tmax: 0.5–4.0 h) were most commonly observed at the first collection time point (0.5 h) and were lower than the oral CBD conditions. Similarly, in the vaporized CBD-dominant cannabis condition, peak CBD concentrations were most commonly observed 0.5 h following vaporized dose administration (mean Cmax = 328 ng/mL, range Cmax: 27.1–809 ng/mL, mean Tmax = 1.4 h and range Tmax: 0.5–4.0 h).
Figure 1.
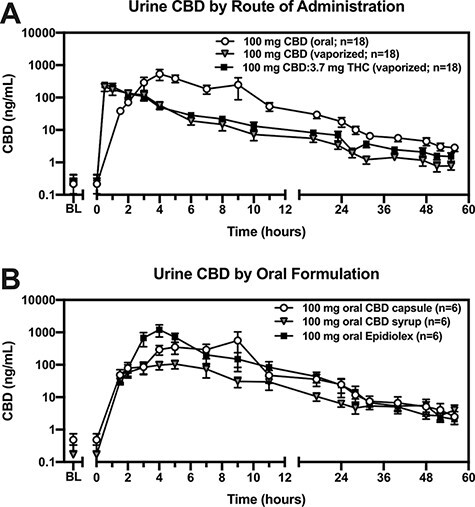
Urine CBD concentrations across dosing conditions. Urine CBD concentrations (ng/mL; y-axis; log10 scale) are plotted (mean ± SEM) by time (post-drug-administration; x-axis) for each (A) route of administration and (B) oral formulation. The 100 mg CBD (oral) condition in panel A represents each of the three oral formulations collapsed.
Tmax was significantly shorter in the 100 mg vaporized CBD (P < 0.05) and CBD-dominant cannabis (P < 0.05) conditions relative to 100 mg oral CBD. Additionally, Cmax for CBD and percent CBD dose excreted were significantly lower following 100 mg vaporized CBD (P < 0.05) but not CBD-dominant cannabis when compared to the 100 mg oral CBD condition. There was no effect of route of administration on CBD t1/2.
Figure 1B depicts urine CBD (ng/mL) concentrations across oral formulations (data points represent mean ± SEM across participants). The Epidiolex condition yielded the highest urinary CBD concentrations (mean Cmax = 1,274 ng/mL, range Cmax: 368–3,470 ng/mL) and peaked, on average, 4.3 h following administration (range Tmax: 4.0–5.0 h). The oral capsule condition peaked at 5.3 h post-administration (range Tmax: 2.0–9.0 h) and reached a lower maximum concentration (mean Cmax = 776 ng/mL, range Cmax: 210–2,941 ng/mL) than the Epidiolex conditions. The oral syrup condition also peaked at 4.3 h post-administration (range Tmax: 2.0–7.0 h) but reached a much lower maximum concentration (mean Cmax = 151 ng/mL, range Cmax: 68.7–229 ng/mL) than either the Epidiolex or capsule conditions.
Urinary 7-OH-CBD and 7-CBD-COOH pharmacokinetics by dosing condition
Figure 2 displays urine concentrations of CBD, 7-OH-CBD and 7-CBD-COOH within each dosing condition. 7-OH-CBD Cmax and percent of the dose excreted as 7-OH-CBD were greater in the oral Epidiolex versus syrup (P < 0.05) condition. Additionally, 7-CBD-COOH Cmax was greater in the oral Epidiolex versus syrup (P < 0.05) and capsule conditions (P < 0.05), and percent of the dose excreted as 7-CBD-COOH was greater in the oral Epidiolex versus syrup conditions (P < 0.05).
Figure 2.
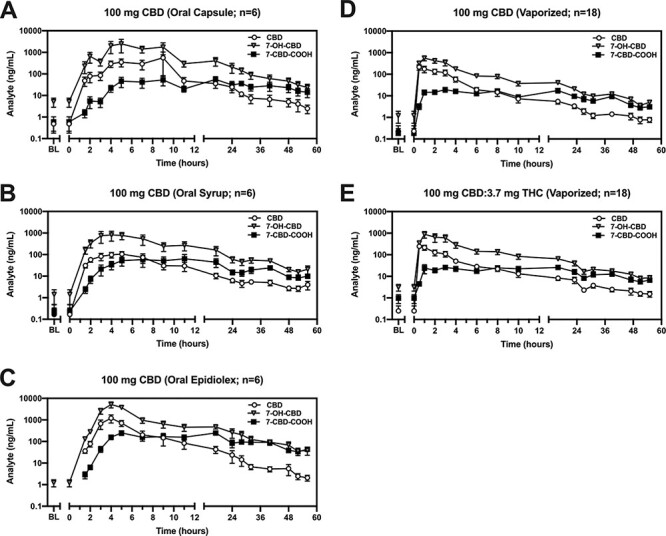
Urinary pharmacokinetic profile of CBD and its metabolites across dosing conditions. Urinary concentrations of CBD, 7-OH-CBD or 7-CBD-COOH concentrations (ng/mL; y-axis; log10 scale) are plotted (mean ± SEM) by time (post-drug-administration; x-axis) for the following, 100 mg CBD dosing conditions: (A) oral capsule, (B) oral syrup, (C) oral Epidiolex, (D) vaporized CBD and (E) vaporized CBD-dominant cannabis.
Relative to oral route, the Cmax and percent of the dose excreted for 7-OH-CBD were both significantly lower in the 100 mg vaporized CBD (Figure 2D) and CBD-dominant cannabis (Figure 2E) conditions (P < 0.05). Additionally, 7-OH-CBD Tmax was longer in the 100 mg oral CBD condition. Further, 7-CBD-COOH Cmax was significantly lower and the percent of the dose excreted as 7-CBD-COOH was significantly lower in the 100 mg vaporized CBD (Figure 2D) and CBD-dominant cannabis (Figure 2E) conditions (P < 0.05). 7-CBD-COOH Tmax was significantly shorter in the 100 mg vaporized versus oral CBD conditions (P < 0.05).
Urine cannabinoid concentrations after overnight fasting
Relative to the standard low-fat breakfast condition, CBD t1/2 was longer following overnight fasting, but there was no significant difference in CBD Cmax, Tmax, or percent of the dose excreted as CBD (Figure S1, Supplemental Material). Further, 7-OH-CBD t1/2 and Tmax were higher and the percent of the dose excreted as 7-OH-CBD was lower in the overnight fasting condition (P < 0.05 for all); there was no difference in 7-OH-CBD Cmax. Additionally, relative to the standard, low-fat breakfast condition, the percent of the dose excreted as 7-CBD-COOH was lower in the overnight fasting condition (P < 0.05). However, no differences were observed for 7-CBD-COOH Cmax, Tmax or t1/2.
Neither Δ9-THC, Δ8-THC, THC-V, THC-VA, 11-OH-THC nor Δ8-THCCOOH were detected in any of the urine voids in the overnight fasting condition. Though traces of Δ9-THCCOOH were detected in some samples, these were sporadically observed and were not different than that observed in the placebo dose condition. Thus, these trace amounts likely stem from exposure to trace amounts of THC in the placebo cannabis or reflect residual THC from prior exposure.
Urine Δ9-THCCOOH and drug testing results
Across all three 100 mg CBD oral formulation doses and the 100 mg pure vaporized CBD condition, trace amounts of Δ9-THCCOOH were detected in a subset of specimens, but these were not different than placebo. Figure 3A displays urine Δ9-THCCOOH concentrations in the vaporized CBD-dominant cannabis condition; Δ9-THCCOOH was detected in all 18 participants. Urinary Cmax for Δ9-THCCOOH ranged from 1.2 to 29.9 ng/mL (mean Cmax = 7.9 ng/mL), while Tmax values for Δ9-THCCOOH ranged from 1 to 31 h after inhalation (mean Tmax = 8 h). In the vaporized CBD-dominant cannabis condition, Δ9-THCCOOH was first detected at BL in trace amounts similar to placebo in two participants, 0.5 h in one participant, 1 h in six participants, 2 h in four participants, 3 h in three participants, 4 h in one participant and at 23 h in one participant. Δ9-THCCOOH was last detected at 8 h in two participants, 16 h in four participants, 23 h in four participants, 39 h and 51 h in one participant each and at 58 h (the final time point) in six participants.
Figure 3.
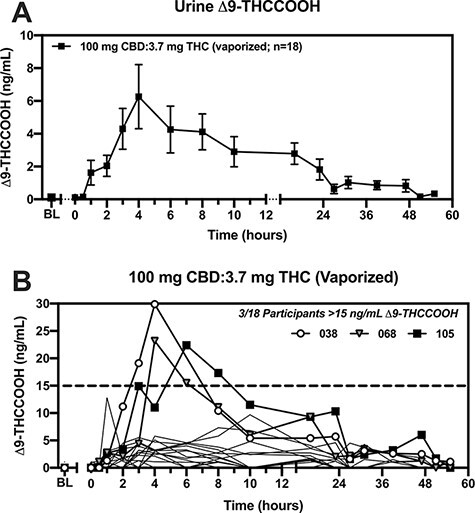
A single, vaporized administration of CBD-dominant cannabis may yield a THC-positive urine drug screen. (A) Urine Δ9-THCCOOH concentrations (ng/mL; y-axis) are plotted (mean ± SEM) by time (post drug-administration; x-axis) for the vaporized CBD-dominant cannabis condition. (B) Urine Δ9-THCCOOH concentrations (ng/mL; y-axis) are plotted by time (post-drug-administration; x-axis) for each participant in the vaporized CBD-dominant cannabis condition. Bolded lines and symbols are used to depicted the 3/18 participants who exhibited >15 ng/mL urine Δ9-THCCOOH (dashed lined), which is the common LC–MS-MS confirmatory cutoff suggested by the Mandatory Guidelines for federal workplace drug testing. The remaining 15/18 participants are indicated by faint, solid lines.
Figure 3B displays urine Δ9-THCCOOH concentrations within the vaporized CBD-dominant cannabis condition. Of note, 3/18 participants (#038, #068, and #105; all males) excreted Δ9-THCCOOH concentrations above 15 ng/mL (the confirmatory cutoff concentration listed in the Mandatory Guidelines for federal workplace drug testing). Specifically, participant #038 provided two specimens (at the 3 and 3–5 h collection points), participant #068 provided two specimens (at the 5–7 and 7–9 h collection points) and participant #105 provided two specimens (at the 5–7 and 7–9 h collection points) that exceeded 15 ng/mL. Each of these six specimens screened positive at an IA cutoff of 20 ng/mL (true positives) and two of six screened positive at a 50 ng/mL cutoff (screening cutoff suggested by the Mandatory Guidelines for federal workplace drug testing). None of these six specimens were positive at the 100 ng/mL IA screening cutoff (Table S3, Supplemental Material).
Discussion
Inhalable and ingestible CBD products are ubiquitous and widely available, but few controlled studies have evaluated the pharmacokinetics of CBD or its metabolites (7-OH-CBD, 7-CBD-COOH) under controlled acute dosing conditions while manipulating factors relevant to its use (e.g., route of administration, oral formulation and ingestion of food prior to use). The present study showed that CBD pharmacokinetics vary substantially by route of administration (vaporized, oral) and by formulation both when inhaled (CBD-dominant cannabis versus pure crystalline CBD powder) and swallowed (capsule versus syrup versus Epidiolex). The key outcomes of this study were that administration of CBD alone did not produce positive urine drug test results based on current US federal drug testing guidelines (IA cutoff of 50 ng/mL Δ9-THCCOOH; confirmation cutoff of 15 ng/mL Δ9-THCCOOH). However, vaporization of CBD-dominant cannabis (10.5% CBD, 0.39% THC) at an acute dose of 100 mg CBD and 3.7 mg Δ9-THC produced true-positive drug test results for a subset of study participants. This outcome is critically important given that the THC concentration in the cannabis used in this study is only slightly above the allowable concentration of THC in hemp products in the USA (0.3% or less). Thus, there is reasonable concern that large acute doses or frequent daily use of hemp products containing concentrations of THC ≤0.3% could result in an unexpected cannabis-positive urine drug test in some individuals. This is consistent with results of a recent clinical trial in which almost half of individuals taking a hemp extract had a positive urine toxicology test after 4 weeks of daily use (5). Moreover, hemp/CBD product users should be aware that some retail products marketed as having little or no THC may contain Δ9-THC in concentrations that are greater than those used in this study (4, 6, 16).
Another contribution of the present study was the detailed excretion profile of primary CBD metabolites in urine. Across all study conditions, the order of analyte abundance in urine was 7-OH-CBD > CBD > 7-CBD-COOH, and the 7-CBD-COOH metabolite exhibited the longest t1/2 (∼30 h across inhaled routes and ∼52 h across oral formulations). Thus, following a single administration of 100 mg CBD, 7-CBD-COOH may be excreted over the course of several days.
The present findings are important for CBD public health messaging, especially for individuals who use over-the-counter or commonly prescribed drugs in combination with CBD products (for reviews (17, 18)). For example, oral administration of Epidiolex (5–25 mg/kg/day) in combination with the anti-seizure medication clobazam elicited, on average, a 500% increase in plasma concentrations of the active metabolite norclobazam in subjects aged 4–19 years (19). Both CBD and clobazam are metabolized by cytochrome P450 (CYP) enzymes CYP2C19 and CYP3A4, which may explain this drug–drug interaction (19). Future studies are needed to evaluate the time course of potential adverse drug–drug interactions following acute use of CBD products.
In light of in vitro evidence that CBD may degrade to Δ8- and Δ9-THC in simulated gastric fluid (7–9), the present study evaluated the potential for conversion of CBD to Δ9-THC or Δ8-THC following acute CBD administration in healthy adults. However, only trace cannabinoids were detected in a few samples, which likely reflect the detection of trace cannabinoids in the placebo cannabis or residual cannabinoids from prior exposure. Between the vaporized and oral routes and across oral formulations, all 702 specimens were true negatives at the 50 and 100 ng/mL IA cutoffs for Δ9-THCCOOH after administration of CBD alone (Table S3). Thus, the present study found no evidence that CBD converted to Δ8- or Δ9-THC after inhalation or oral ingestion (three oral dose formulations).
The final key component of the present study was to evaluate fasting versus fed conditions upon oral CBD dosing, since fasting was hypothesized to create a highly acidic gastric environment, increasing the likelihood that CBD that may be degraded to Δ8- and Δ9-THC following oral ingestion (7). Few effects of overnight fasting on CBD pharmacokinetics were observed relative to the standard low-fat breakfast condition; reasons for these null results vary but may include that (I) the standard breakfast had a low fat content (II) only two of the six participants received oral syrup in both the fasted and non-fasted states, which introduced inter-subject variability and (III) the oral syrup formulation yielded, on average, the lowest CBD concentrations in urine relative to Epidiolex or the capsule. Although there was no apparent evidence that CBD converted to Δ8- or Δ9-THC following oral ingestion in a fasting state, these data are inconclusive considering that the oral syrup used in the overnight fasting experiment had the least bioavailability of the three CBD formulations in this study. Future studies are required to explicitly test the hypothesis that dietary fats impact urinary CBD pharmacokinetics and/or the likelihood that CBD that may be degraded to Δ8- and Δ9-THC following oral ingestion, perhaps by evaluating the effect of a high-fat meal and inclusion of a more optimal CBD dose formulation.
Limitations of the present study warrant discussion. First, between successive drug administration sessions, we identified several instances where low concentrations of CBD and/or Δ9-THCCOOH were detected at baseline. This suggests that the previously administered CBD/Δ9-THC dose may have failed to be completely eliminated, necessitating a longer drug wash-out period (e.g., >7 days) in future studies. Second, the cannabis used here contained 0.39% Δ9-THC by dry weight, which is narrowly above the limit mandated by the Agriculture Improvement Act of 2018 in the USA (≤0.3% THC) and was only administered via vaporization. However, the CBD-dominant cannabis used here is currently legal in many states of USA, Canada, Uruguay, and many other countries that have legalized cannabis use for medicinal and/or non-medicinal purposes. Future studies should assess CBD-dominant cannabis that meets the definition for hemp (≤0.3% THC). Third, the oral syrup used in the present study was optimized for lipophilic actives, which may have reduced solubility of CBD (a lipid-soluble compound) and impacted the CBD urinary pharmacokinetics results (20, 21). Fourth, only one batch of CBD-dominant cannabis, type of vaporizer (Volcano Medic®), and dose of CBD/THC were included in the present study, and only acute dosing sessions were employed. Other delivery methods (e.g., handheld vaporizer, gummy edible product) could alter cannabinoid delivery and warrant further exploration. Future studies employing a greater range of CBD doses and routes of administration are needed, while chronic dosing studies are required to better model the pattern of repeated CBD use that is most typical of current use of these products. Fifth, subjects did not consume a controlled amount of liquid, and thus, dilution effects were not controlled between subjects. However, dilution effects may be more ecologically valid than dilution-normalized data since this reflects “real-world” variation in urine dilution that may impact urine cannabis drug testing outcomes. Lastly, although optimization experiments in our laboratory supported the combined enzymatic and base hydrolysis procedures, the order of enzyme and base hydrolysis procedures was not evaluated in the present study.
Conclusion
The present study characterized CBD pharmacokinetics across two routes of administration (vaporized, oral) and three oral formulations (capsule, syrup and Epidiolex). Generally, urinary CBD concentrations were higher following oral versus vaporized cannabis administration, a finding that is similar to what was previously demonstrated for THC metabolites (22). Additionally, there was no evidence that CBD converted to Δ8- or Δ9-THC across oral formulations, which is discordant with in vitro studies. Importantly, these data suggest that vaporizing CBD-dominant cannabis containing ∼3.7 mg Δ9-THC (10.5% CBD, 0.39% THC) can produce a cannabis-positive drug screen at the 50 ng/mL IA/15 ng/mL LC–MS-MS cutoff suggested by the Mandatory Guidelines for federal workplace drug testing. We highlight novel directions for future controlled laboratory studies, which are of immediate importance in light of the evolving retail CBD marketplace and increasing availability of CBD-containing products.
Supplementary Material
Acknowledgments
We thank the support staff of the Johns Hopkins University Behavioral Pharmacology Research Unit for outstanding contributions to the implementation of this study. We also thank the many individuals involved with the NIDA Drug Supply Program for providing their services and cannabis for the conduct of this study. Lastly, we thank Storz & Bickel© for donating vaporization equipment for the study.
Contributor Information
Dennis J Sholler, Behavioral Pharmacology Research Unit, Johns Hopkins University School of Medicine, 5510 Nathan Shock Dr., Baltimore, MD 21224, USA.
Tory R Spindle, Behavioral Pharmacology Research Unit, Johns Hopkins University School of Medicine, 5510 Nathan Shock Dr., Baltimore, MD 21224, USA.
Edward J Cone, Behavioral Pharmacology Research Unit, Johns Hopkins University School of Medicine, 5510 Nathan Shock Dr., Baltimore, MD 21224, USA.
Elia Goffi, Behavioral Pharmacology Research Unit, Johns Hopkins University School of Medicine, 5510 Nathan Shock Dr., Baltimore, MD 21224, USA.
David Kuntz, Clinical Reference Laboratory, 8433 Quivira Rd, Lenexa, KS 66214, USA.
John M Mitchell, RTI International, 3040 East Cornwallis Road, Research Triangle Park, NC 27709, USA.
Ruth E Winecker, RTI International, 3040 East Cornwallis Road, Research Triangle Park, NC 27709, USA.
George E Bigelow, Behavioral Pharmacology Research Unit, Johns Hopkins University School of Medicine, 5510 Nathan Shock Dr., Baltimore, MD 21224, USA.
Ronald R Flegel, Division of Workplace Programs (DWP), Substance Abuse and Mental Health Services Administration (SAMHSA), 5600 Fishers Lane, Rockville, MD 20857, USA.
Ryan Vandrey, Behavioral Pharmacology Research Unit, Johns Hopkins University School of Medicine, 5510 Nathan Shock Dr., Baltimore, MD 21224, USA.
Supplementary data
Supplementary data is available at Journal of Analytical Toxicology online.
Funding
This research was supported by the Substance Abuse and Mental Health Services Administration (SAMHSA) and the National Institute on Drug Abuse (NIDA; T32DA07209).
Data Availability
The data underlying this article are available in the article and in its online supplementary material.
References
- 1. BDS Analytics . (2019) U.S. CBD Market Anticipated to Reach $20 Billion in Sales by 2024. https://bdsa.com/wp-content/uploads/2019/08/BDS-Analytics-The-Global-Cannabinoids-Market-Will-CBD-Overtake-THC.pdf (Accessed Dec 11, 2020).
- 2. Kulig K. (2017) Interpretation of workplace tests for cannabinoids. Journal of Medical Toxicology, 13, 106–110.doi: 10.1007/s13181-016-0587-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Macdonald S., Hall W., Roman P., Stockwell T., Coghlan M., Nesvaag S. (2010) Testing for cannabis in the work-place: a review of the evidence. Addiction, 105, 408–416.doi: 10.1111/j.1360-0443.2009.02808.x. [DOI] [PubMed] [Google Scholar]
- 4. Bridges M., Hanson K. (2017) Regulating hemp and cannabis-based products. National Conference of State Legislatures LegisBriefs, 25, 1–2. [PubMed] [Google Scholar]
- 5. Dahlgren M.K., Sagar K.A., Lambros A.M., Smith R.T., Gruber S.A. (2020) Urinary tetrahydrocannabinol after 4 weeks of a full-spectrum, high-cannabidiol treatment in an open-label clinical trial. JAMA Psychiatry, 78, 335–337.doi: 10.1001/jamapsychiatry.2020.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonn-Miller M.O., Loflin M.J.E., Thomas B.F., Marcy J.P., Hyke T., Vandrey R. (2017) Labeling accuracy of cannabidiol extracts sold online. JAMA, 317, 1708–1709.doi: 10.1001/jama.2017.11909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Merrick J., Lane B., Sebree T., Yaksh T., O’Neill C., Banks S.L. (2016) Identification of psychoactive degradants of cannabidiol in simulated gastric and physiological fluid. Cannabis and Cannabinoid Research, 1, 102–112.doi: 10.1089/can.2015.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nahler G., Grotenhermen F., Zuardi A.W., Crippa J.A.S. (2017) A conversion of oral cannabidiol to Delta9-Tetrahydrocannabinol seems not to occur in humans. Cannabis and Cannabinoid Research, 2, 81–86.doi: 10.1089/can.2017.0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Watanabe K., Itokawa Y., Yamaori S., Funahashi T., Kimura T., Kaji T., et al. (2007) Conversion of cannabidiol to Δ9-tetrahydrocannabinol and related cannabinoids in artificial gastric juice, and their pharmacological effects in mice. Forensic Toxicology, 25, 16–21.doi: 10.1007/s11419-007-0021-y. [DOI] [Google Scholar]
- 10. Evans D.F., Pye G., Bramley R., Clark A.G., Dyson T.J., Hardcastle J.D. (1988) Measurement of gastrointestinal pH profiles in normal ambulant human subjects. Gut, 29, 1035–1041.doi: 10.1136/gut.29.8.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Patrician A., Versic-Bratincevic M., Mijacika T., Banic I., Marendic M., Sutlovic D., et al. (2019) Examination of a new delivery approach for oral cannabidiol in healthy subjects: a randomized, double-blinded, placebo-controlled pharmacokinetics study. Advances in Therapy, 36, 3196–3210.doi: 10.1007/s12325-019-01074-6. [DOI] [PubMed] [Google Scholar]
- 12. Pacifici R., Pichini S., Pellegrini M., Rotolo M.C., Giorgetti R., Tagliabracci A., et al. (2020) THC and CBD concentrations in blood, oral fluid and urine following a single and repeated administration of “light cannabis”. Clinical Chemistry and Laboratory Medicine, 58, 682–689.doi: 10.1515/cclm-2019-0119. [DOI] [PubMed] [Google Scholar]
- 13. Pacifici R., Pichini S., Pellegrini M., Tittarelli R., Pantano F., Mannocchi G., et al. (2018) Determination of cannabinoids in oral fluid and urine of “light cannabis” consumers: a pilot study. Clinical Chemistry and Laboratory Medicine, 57, 238–243.doi: 10.1515/cclm-2018-0566. [DOI] [PubMed] [Google Scholar]
- 14. Spindle T.R., Cone E.J., Kuntz D., Mitchell J.M., Bigelow G.E., Flegel R., et al. (2020) Urinary pharmacokinetic profile of cannabinoids following administration of vaporized and oral cannabidiol and vaporized CBD-dominant cannabis. Journal of Analytical Toxicology, 44, 109–125.doi: 10.1093/jat/bkz080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Spindle T.R., Cone E.J., Goffi E., Weerts E.M., Mitchell J.M., Winecker R.E., et al. (2020) Pharmacodynamic effects of vaporized and oral cannabidiol (CBD) and vaporized CBD-dominant cannabis in infrequent cannabis users. Drug and Alcohol Dependence, 211, 107937.doi: 10.1016/j.drugalcdep.2020.107937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poklis J.L., Mulder H.A., Peace M.R. (2019) The unexpected identification of the cannabimimetic, 5F-ADB, and dextromethorphan in commercially available cannabidiol e-liquids. Forensic Science International, 294, 25–27.doi: 10.1016/j.forsciint.2018.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sholler D.J., Schoene L., Spindle T.R. (2020) Therapeutic efficacy of cannabidiol (CBD): a review of the evidence from clinical trials and human laboratory studies. Current Addiction Reports, 7, 405–412.doi: 10.1007/s40429-020-00326-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. White C.M. (2019) A review of human studies assessing cannabidiol’s (CBD) therapeutic actions and potential. Journal of Clinical Pharmacology, 59, 923–934.doi: 10.1002/jcph.1387. [DOI] [PubMed] [Google Scholar]
- 19. Geffrey A.L., Pollack S.F., Bruno P.L., Thiele E.A. (2015) Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia, 56, 1246–1251.doi: 10.1111/epi.13060. [DOI] [PubMed] [Google Scholar]
- 20. Devinsky O., Marsh E., Friedman D., Thiele E., Laux L., Sulivan J., et al. (2016) Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. The Lancet Neurology, 15, 270–278.doi: 10.1016/S1474-4422(15)00379-8. [DOI] [PubMed] [Google Scholar]
- 21. Zgair A., Wong J.C., Lee J.B., Mistry J., Sivak O., Wasan K.M., et al. (2016) Dietary fats and pharmaceutical lipid excipients increase systemic exposure to orally administered cannabis and cannabis-based medicines. American Journal of Translational Research, 8, 3448–3459. [PMC free article] [PubMed] [Google Scholar]
- 22. Huestis M.A., Sempio C., Newmeyer M.N., Andersson M., Barnes A.J., Abulseoud O.A., et al. (2020) Free and glucuronide urine cannabinoids after controlled smoked, vaporized and oral cannabis administration in frequent and occasional cannabis users. Journal of Analytical Toxicology, 44, 651–660.doi: 10.1093/jat/bkaa046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.