


Abstract
Epigenome-wide association studies often detect many differentially methylated sites, and many are located in distal regulatory regions. To further prioritize these significant sites, there is a critical need to better understand the functional impact of CpG methylation. Recent studies demonstrated that CpG methylation-dependent transcriptional regulation is a widespread phenomenon. Here, we present MethReg, an R/Bioconductor package that analyzes matched DNA methylation and gene expression data, along with external transcription factor (TF) binding information, to evaluate, prioritize and annotate CpG sites with high regulatory potential. At these CpG sites, TF–target gene associations are often only present in a subset of samples with high (or low) methylation levels, so they can be missed by analyses that use all samples. Using colorectal cancer and Alzheimer’s disease datasets, we show MethReg significantly enhances our understanding of the regulatory roles of DNA methylation in complex diseases.
INTRODUCTION
Recent epigenome-wide association studies (EWAS) have identified numerous alterations in DNA methylation (DNAm) levels that are involved in many diseases such as various cancers (1–5) and neurodegenerative diseases (6–8). Compared to genome-wide association studies (GWAS) of genetic variants, EWAS often detect a larger number of significant differences, often thousands of differentially methylated CpG sites (DMS), which are significantly associated with a disease or phenotype. Many of these DMS are located far from genes, complicating the interpretation of their functionality (9,10). Therefore, there is a critical need to better understand the functional impact of these CpG methylations and to further prioritize the significant methylation changes.
Transcription factors (TFs) are proteins that bind DNA and facilitate the transcription of DNA into RNA. Several recent studies have observed that the binding of TFs onto DNA can be affected by DNA methylation. In turn, DNA methylation can also be altered by proteins associated with TFs (11–15). Using methylation-sensitive SELEX (systematic evolution of ligands by exponential enrichment), Yin et al. (16) classified 519 TFs into several categories: TFs whose binding strength increased, decreased or was not affected by DNA methylation, as well as those not containing CpGs in their binding motifs.
Although several integrative analysis strategies (17–23) have been proposed to help assess the functional role of the DNA methylation changes in gene regulation, these methods typically integrate DNA methylation data with either gene expression (17–20) or TF binding data (21–23), but rarely both. For example, MethylMix (17) identifies CpGs predictive of transcription and then classifies the CpGs into different methylation states. Similarly, COHCAP (18) identifies a subset of CpGs within CpG islands that are most likely to regulate downstream gene expression. These methods, which test the association between DNA methylation and target gene expression, can be further improved by additionally incorporating information on TF activity.
To determine whether TF regulatory activity is enhanced or reduced by significant CpGs in EWAS, the gold standard would be to perform ChIP-seq experiments for all candidate TFs with binding sites close to the CpGs in parallel with bisulfite sequencing of DNA methylation changes and transcriptome assessment. However, performing ChIP-seq experiments on primary tissues is technically challenging because of the limited number of cells in some samples, the large number of TFs to be tested and the lack of availability for specific antibodies. Therefore, in practice, computational approaches are often used to prioritize disease-relevant TFs in DNA methylation studies. For example, LOLA (21) (locus overlap analysis) performs enrichment analysis to identify regulatory elements such as TFs with binding sites enriched in candidate genomic regions (e.g. DMRs). Alternatively, Goldmine (22) annotates individual CpGs by TFs with binding sites overlapping the CpGs. However, because the binding motifs for TFs are often nonspecific for different members of a TF family, there are often many TFs with binding sites that overlap with a given CpG. Moreover, TF binding can also occur without affecting the transcription of any gene (24,25). Therefore, methods that analyze DNA methylation and TF binding site (TFBS) data would also be greatly enhanced by additionally modeling target gene expression.
Another well-known tool is the ELMER software (26,27), which performs integrative analysis by associating DNA methylation variations with the expression of the target genes and identifying TFs that might regulate those DNA methylation loci. However, ELMER’s primary goal is not to prioritize functional CpGs. In particular, ELMER does not differentiate methylation–TF–target gene triplets in which target gene expression is regulated mainly by the TF versus those regulated by both DNA methylation and TF (Figure 1), which is crucial for tools that prioritize methylation CpGs. Supplementary Table S1 includes additional details on the key differences between ELMER and our new software MethReg.
Figure 1.

Three different scenarios in gene regulation. In (A), target gene is mainly regulated by DNA methylation (DNAm). In (B), target gene is regulated jointly by both DNA methylation and TF. Specifically, DNA methylation modulates TF activities on the target gene, so that TF–target gene association is only observed in samples with low (or high) DNA methylation, but not in all samples. In (C), target gene is regulated mainly by TF. Specifically, TF activities are associated with DNA methylation levels, and TF also regulates gene expression independently of DNA methylation. Therefore, target gene expression is mainly regulated by TF, but not DNA methylation.
To fill this critical gap in analytical methods and software for annotating and prioritizing DNA methylation changes identified in EWAS, here we present MethReg, an R/Bioconductor package that performs integrative modeling of three key components (DNA methylation, gene expression levels and TF), to provide a more comprehensive functional assessment of CpG methylation in gene regulation. In particular, MethReg leverages information from external databases on TFBS, ChIP-seq experiments and TF–target interactions, performs both promoter and distal (enhancer) analyses, implements rigorous robust regression models and can fully adjust for potential confounding effects such as copy number, age and sex that are important in DNA methylation analysis. MethReg can be used either to evaluate the regulatory potential of candidate CpG sites identified in EWAS (in supervised analysis mode) or to search for methylation-dependent TF regulatory processes in the entire genome (in unsupervised analysis mode).
Using simulated datasets, we showed that by simultaneous modeling of three key elements (DNA methylation, target gene and TF), MethReg significantly improves prioritization for true positive DNA methylation changes with regulatory roles in gene transcription compared to models that include only two key elements. In addition, we also analyzed the TCGA colorectal datasets and the ROSMAP Alzheimer’s dataset to show that MethReg was able to recover known biology and nominate novel biologically meaningful DNA methylation–TF–target associations in gene transcription.
MATERIALS AND METHODS
The MethReg analysis pipeline
To systematically search for CpG methylation with significant regulatory effects on gene expression by influencing TF activity, we developed MethReg. Figure 2 illustrates the workflow for MethReg. The input is matched DNA methylation data (methylation arrays) and gene expression data (RNA-seq); that is, the same samples in which DNA methylation is profiled by arrays and gene expression are quantified by RNA-seq. In addition, MethReg also incorporates TF binding information from the ReMap2020 (28) or the JASPAR2020 database (29), and optionally additional TF–target gene interaction databases (Supplementary Table S2), to perform both promoter and distal (enhancer) analyses. In the unsupervised mode, MethReg analyzes all CpGs on the Illumina arrays. In the supervised mode, MethReg analyzes DMS identified in EWAS. There are three main steps: (i) create a dataset with triplets of CpG, TF that binds near the CpG and the putative target gene; (ii) for each CpG–TF–target gene triplet, apply integrative statistical models to DNA methylation, target gene expression and TF activity values; and (iii) visualize and interpret results from statistical models to estimate the impact of DNA methylation on regulatory effect of TF (interaction effect of CpG methylation and TF on target gene), as well as to annotate the roles of TF and CpG methylation in regulating target gene expression. The results from the statistical models allow us to identify a list of CpGs that interact with TFs to influence target gene expressions. Here, we describe the analysis of TFs, but the method and software tool are, in principle, also applicable to other types of chromatin proteins that crosstalk with DNA methylation. MethReg is an open-source R/Bioconductor package, available at https://bioconductor.org/packages/MethReg/. There are several steps in the MethReg analysis pipeline, which we will describe next.
Figure 2.

Workflow of MethReg. Data: MethReg input datasets are (1) DNA methylation array data (HM450/EPIC) with beta values, (2) RNA-seq data with normalized counts and (3) estimated TF activities from the RNA-seq data using GSVA (gene set variation analysis) or VIPER (virtual inference by enriched regulon analysis) software. Creating triplets: There are multiple ways to create CpG–TF–target gene triplets. (A) A CpG can be mapped to TFs using TF motifs in databases such as JASPAR2020 or ReMap2020, by scanning the CpG location to identify whether it is close to a TFBS. (B) CpGs can be mapped to target genes using a distance-based approach. A CpG is linked to a gene if it is in the promoter region (< ±2 kb from the TSS). A distal CpG can be linked to either all genes within a fixed width (i.e., 500 kb) or a fixed number of genes upstream and downstream of the CpG location. (C) TF-target gene pairs can be retrieved from external databases (e.g., Cistrome Cancer and Dorothea). Using two different pairs (i.e., CpG–TF and TF–target gene), triplets can then be created. Analysis: Each triplet will be evaluated using a robust linear model, in which DNAm.group is a binary variable indicating whether a sample has high (fourth quartile) or low (first quartile) DNA methylation levels at the CpG. Results: MethReg outputs the prioritized triplets and classifies both the role of TF in the target gene expression (repressor or activator) and the role of DNA methylation on TF (enhancing or attenuating).
Step 1: Creating CpG–TF–target gene triplet dataset
MethReg first links CpGs to TFs with binding sites within a window of user-specified distance (e.g. ±250 bp) using information from the ReMap2020 (28) or the JASPAR2020 database (29). The JASPAR2020 database includes curated TF binding models, among which 637 are associated with human TFs with known DNA-binding profiles (30). Similarly, the human atlas of the ReMap2020 database contains regulatory regions for 1135 transcriptional regulators obtained using genome-wide DNA-binding experiments such as ChIP-seq. Next, in promoter analysis, CpGs located in promoter regions, defined as ±2 kb regions around the transcription start sites (TSS), are linked to target genes with promoters that overlap with the CpG. On the other hand, in distal analysis, CpGs in distal regions (i.e. >2 kb from TSS) are linked to a specific number of genes (e.g. five genes) upstream or downstream and within 1 million bp of the CpG, or to all genes within a fixed window of distance (e.g. 500 kb). The CpG–TF pairs are then combined with CpG–target gene pairs to create triplets of CpG–TF–target genes.
Alternatively, CpGs can also be linked to genes within 1 million bp in regulon-based analysis. A TF regulon consists of all the transcriptional targets of the TF. MethReg obtains TF–target pairs from curated external regulon databases (31,32) (Supplementary Table S2). Combining the CpG–TF pairs with TF–target gene pairs, we then obtain a triplet dataset where each row contains identifiers for a CpG, a TF and the target gene. Additional discussions on the parameter settings of CpG, TF and target gene linking are included in the Supplementary Text.
Step 2: Estimating regulatory effects of CpG methylation and TFs on target gene expression
Given a CpG–TF–target gene triplet, we then query the matched DNA methylation and gene expression datasets to obtain DNA methylation, target gene and TF gene expression (or activity) values and fit the following statistical model to data:
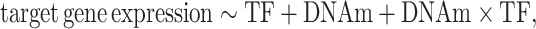 |
(Model 1) |
where the target represents log2 transformed target gene expression values, TF represents log2 transformed TF gene expression values or estimated TF activity scores (see details in the ‘Modeling TF protein activity’ section) and DNAm represents DNA methylation beta-values.
Note that Model 1 partitions the effects of DNA methylation and TF on target gene expression into three categories: the direct effect of TF (modeled by term TF), the direct effect of DNA methylation (modeled by term DNAm) and the interaction effects of TF and DNA methylation (i.e. how the effect of TF on target gene expression is modified by DNA methylation, modeled by the DNAm × TF interaction term).
For accurate statistical modeling, MethReg implements Model 1 by fitting a robust linear model. In contrast to linear regression models, which consider each sample equally, robust linear models give reduced weight to outlier gene expression values (33) to dampen their influences on the overall model fit. Note that a key feature of Model 1 is that it provides more comprehensive modeling of gene regulation by incorporating the three components (TF activity, DNA methylation and target gene expression) simultaneously. In addition to Model 1 described earlier, which included DNAm as a continuous variable, we also considered another model that modeled methylation values as a binary variable. We also propose
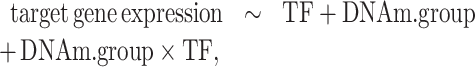 |
(Model 2) |
where DNAm.group is high or low. That is, for a given CpG, the samples with the highest DNA methylation levels (top 25%) have DNAm.group = ‘high’ and samples with lowest DNAm levels (bottom 25%) have DNAm.group = ‘low’. In this model, only samples with DNA methylation values in the first and last quartiles are considered. Note also that statistically the 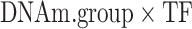 effect is estimated by comparing the magnitude of TF–target gene association in the high methylation group versus the magnitude of the TF–target gene association in the low methylation group.
effect is estimated by comparing the magnitude of TF–target gene association in the high methylation group versus the magnitude of the TF–target gene association in the low methylation group.
Step 3: Visualizing and annotating roles of CpG and TF in gene transcription
To visualize how DNA methylation interacts with TFs to influence gene expression, MethReg generates a suite of figures. Figure 3 shows an example output figure of Model 2 applied to the TCGA colorectal cancer (CRC) dataset. The first row shows figures for assessing direct pairwise TF–target and DNA methylation–target associations. In the second row are figures for assessing TF–target gene expression, stratified by high or low DNA methylation levels.
Figure 3.

Example output from MethReg analysis of the TCGA COADREAD dataset. (A) The first table shows the triplet (CpG, TF, target gene) metadata, TF role (repressor or activator) and DNA methylation effect on the TF (enhancing or attenuating). The second table shows the results from fitting robust linear model target gene expression residual ∼ DNAm group + TF + TF × DNAm group, where DNAm group is 0 if the sample has low DNA methylation levels (in the lowest quartile) at the given CpG or 1 if the sample has high methylation levels (in the highest quartile), indicating significant DNAm × TF interaction effect (P < 2 × 10–16). (B) When all samples are considered, there is no association between target gene expression and TF activity (each dot represents a sample). (C) Comparison of target gene expressions between the DNAm groups shows samples with lower DNA methylation have higher target gene expression. (D) Scatter plot of target gene expression residuals versus TF activity stratified by DNAm group (only samples in DNAm high or low groups are shown). In samples with high DNA methylation, TF represses target gene expression. In samples with low DNA methylation, target gene expression is relatively independent of the TF. Therefore, TF is predicted to be a repressor and DNAm is predicted to enhance the effect of TF on the target gene. Abbreviations: DNAm, DNA methylation; target gene expression residual, linear model residuals obtained after removing effects of copy number alteration (CNA) and tumor purity estimate from gene expression data.
Note in Figure 3, without stratifying by DNA methylation, the overall TF–target association is not significant (robust linear model P-value = 0.590). In contrast, TF–target association is highly significant in samples with high methylation levels (robust linear model P-value = 0.001). Therefore, methylation at cg00328227 might interact with TF to influence gene expression in this case. This example also demonstrates that by additionally modeling DNA methylation, we can nominate TF–target associations that might have been missed otherwise.
Figure 4 shows the different biological scenarios in which methylation and TF interact to influence target gene expression. A TF repressor decreases transcription while a TF activator increases it, and the presence of methylation can either enhance or attenuate the TF activity on the target gene. For each triplet, MethReg annotates the role of TF in the target gene (repressor, activator or dual) and how DNA methylation influences the TF (enhancing, attenuating or invert).
Figure 4.

Scenarios modeled by MethReg. (A, B) DNA methylation decreases TF activity. In (A), TF is a repressor of the target gene, while in (B) TF is an activator. (C, D) DNA methylation increases TF activity. In (C), TF is a repressor of the target gene, while in (D) TF is an activator. (E, F) DNA methylation inverts the TF role. In (E), when the DNA methylation levels are low the TF works as a repressor, while when methylation levels are high the TF acts as an activator. In (F), when the DNA methylation levels are low the TF works as an activator, while when methylation levels are high it acts as a repressor.
Modeling TF protein activity
Given that TF gene expression might not accurately reflect TF protein activity, which involves additional complex processes (e.g. post-translational modifications, protein–protein/ligand interactions and localization changes), MethReg implements an additional option to model TF activity via the VIPER (34) or GSVA (35) methods, so that the TF effects in Models 1 and 2 described earlier can also be computed by replacing TF gene expression levels with estimated TF activity. Briefly, given RNA-seq data, these methods estimate the activity of a TF by performing a rank-based gene set enrichment analysis of its target genes (i.e. its regulon). MethReg can work with different regulon databases (Supplementary Table S2), such as those described by Garcia-Alonso et al. (31), which were collected from four resources: manually curated databases, ChIP-seq binding experimental data, prediction of TF binding motifs based on gene promoter sequences or computational regulatory network analysis. The Genotype-Tissue Expression (GTEx) tissue-consensus regulons included 1 077 121 TF–target gene regulatory interactions between 1402 TFs and 26 984 target genes, and the pan-cancer regulons included 636 753 TF–target gene regulatory interactions between 1412 TFs and 26 939 target genes. Garcia-Alonso et al. (31) annotated each TF–target gene interaction with a five-level confidence score, with ‘A’ indicating most reliable, supported by multiple lines of evidence, and ‘E’ indicating least confidence, supported only by computational predictions. Benchmark experiments using three separate datasets showed that the GTEx tissue-consensus regulons performed similarly to tissue-specific regulons computed from GTEx data of specific tissue type. Notably, MethReg also provides options for users to input alternative TF regulon databases (Supplementary Table S2) and TF activity computed using alternative software such as Lisa (36).
Stage-wise method for controlling false discovery rate
MethReg implements two alternative methods for controlling false discovery rates (FDRs), using the conventional approach by the method of Benjamini and Hochberg (37) or a stage-wise approach (38). To help improve power in high-throughput experiments where multiple hypotheses are tested for each gene, Van den Berge et al. (38) proposed a stage-wise approach in the context of gene splicing analysis. First, in the screening step, a global test is applied to each gene to test the null hypothesis that there is a differential change in any of the transcripts within the gene. Second, in the confirmation step, for the genes selected in the screening step, individual transcripts are then tested while controlling the family-wise error rate (FWER). By aggregating effects from individual transcripts within a gene in the screening step, the stage-wise procedure was shown to have superior power compared with the conventional approach that tests all individual transcripts in one step.
In Models 1 and 2 described earlier, the interaction of DNA methylation and TF is estimated by the term DNAm × TF. Because the standard error of interaction effects is typically much larger than those for main effects, the conventional approach for controlling FDR often results in low power for discovering interaction effects (38). To this end, MethReg additionally implements the stage-wise procedure for testing interactions by first aggregating all CpG–TF–target gene triplets associated with the same CpG as a group. In the screening step, MethReg tests the null hypothesis that any of the individual triplets mapped to a CpG has a significant DNAm × TF effect. In the confirmation step, MethReg tests each triplet associated with the CpG selected in the screening step while controlling FWER as described in (38).
RESULTS
Figure 1 illustrates several different scenarios in which target gene expression levels are influenced by DNA methylation, TF or both. In (A), gene expression is mainly regulated by DNA methylation. In (B), target gene expression is regulated jointly by both DNA methylation and TF. Specifically, DNA methylation influences the target gene by modulating TF activity. In this case, the TF–target gene associations are only observed in samples with low (or high) DNA methylation but not in all samples. In (C), target gene expression is regulated mainly by TF activity but not DNA methylation. Specifically, TF activity is associated with DNA methylation levels, and TF also regulates gene expression independently of DNA methylation. Therefore, target gene expression is regulated by the TF and not by DNA methylation. We next discuss simulation studies comparing different methods to identify biological events described in scenarios (B) and (C).
Comparison of different models when the target gene is regulated jointly by both DNA methylation and TF
We first conducted a simulation study to compare the performance of five different approaches, including Models 1 and 2, to identify TF–target associations where TF activity is modulated by CpG methylation (Figure 1B). We considered the scenario where CpG methylation affects TF binding affinity so that TF only affects target gene expression when the methylation level is low. To assess the statistical properties of these different methods, we estimated and compared type I error rate, power and area under receiver operating characteristic (ROC) curves (AUC) for each method.
More specifically, we simulated datasets for which target gene expression levels were dependent on TF expression only in samples with low DNA methylation levels (Supplementary Figure S1). We used 38 samples of TCGA COAD matched RNA-seq and DNA methylation data on chromosome 21 included in the MethReg R package as our input dataset, from which we randomly sampled TF gene expression and methylation levels. For each simulated triplet dataset, we randomly sampled one gene from the RNA-seq dataset and one CpG site from the DNA methylation dataset to be our TF expression and DNA methylation levels. Next, target gene expression levels were simulated from negative binomial distributions as follows: the estimated medians of means and variances over all genes in our input dataset were 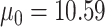 and
and 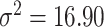 , respectively. Therefore, for target gene expression, we assumed a negative binomial distribution with parameters
, respectively. Therefore, for target gene expression, we assumed a negative binomial distribution with parameters 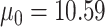 and
and 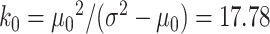 for all samples except those with the lowest DNA methylation levels in the first quartile. For the samples with low methylation levels in the first quartile, we generated target gene expression levels from negative binomial distribution (
for all samples except those with the lowest DNA methylation levels in the first quartile. For the samples with low methylation levels in the first quartile, we generated target gene expression levels from negative binomial distribution (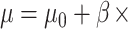 TF gene expression,
TF gene expression, 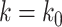 ), where
), where 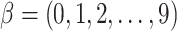 indicates different strengths of associations between TF and target gene expression, corresponding to 10 different simulation scenarios. Therefore, by the design of this simulation experiment, target gene expression was associated with TF only when methylation levels were low. For each value of
indicates different strengths of associations between TF and target gene expression, corresponding to 10 different simulation scenarios. Therefore, by the design of this simulation experiment, target gene expression was associated with TF only when methylation levels were low. For each value of 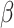 , we repeated this process 1000 times to generate 1000 triplets of TF, DNA methylation and target gene expression levels.
, we repeated this process 1000 times to generate 1000 triplets of TF, DNA methylation and target gene expression levels.
Note that when 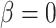 , target gene expressions were generated randomly from negative binomial distribution and did not depend on TF gene expression in the samples, so the 1000 triplet datasets simulated under this simulation scenario (null triplets) allowed us to estimate type I error rates of different models. We compared sensitivity and specificities for identifying TF–target gene associations based on P-values from five different approaches (Supplementary Figure S1):
, target gene expressions were generated randomly from negative binomial distribution and did not depend on TF gene expression in the samples, so the 1000 triplet datasets simulated under this simulation scenario (null triplets) allowed us to estimate type I error rates of different models. We compared sensitivity and specificities for identifying TF–target gene associations based on P-values from five different approaches (Supplementary Figure S1):
lm.cont: P-value for DNAm × TF term in linear model implementation of Model 1.
lm.binary: P-value for DNAm.group × TF term in linear model implementation of Model 2.
rlm.cont: P-value for DNAm × TF term in robust linear model implementation of Model 1.
rlm.binary: P-value for DNAm.group × TF term in robust linear model implementation of Model 2.
rlm.binary.en: P-value for DNAm.group × TF term in robust linear model implementation of Model 2, estimated from empirical null distribution (39) (Supplementary Text).
In the method rlm.binary.en, instead of the conventional approach, which computes P-values by comparing test statistics for DNAm.group × TF to t-distribution, this method estimates P-values for DNAm.group × TF effect using empirical null distribution (39), which is a normal distribution with empirically estimated mean 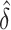 and standard deviation
and standard deviation 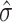 . Efron (39) showed that in large-scale simultaneous testing situations (e.g. when many triplets are tested in an analysis), serious defects in the theoretical null distribution may become obvious, while empirical Bayes methods can provide much more realistic null distributions.
. Efron (39) showed that in large-scale simultaneous testing situations (e.g. when many triplets are tested in an analysis), serious defects in the theoretical null distribution may become obvious, while empirical Bayes methods can provide much more realistic null distributions.
The results showed that all methods had type I error rates close to 5% (Supplementary Figure S2). Among them, robust linear models with binary methylation group (rlm.binary, rlm.binary.en) had the highest power (Supplementary Figure S3). In a simulation study, we designed the study so that some CpGs are truly associated with the target gene, while other CpGs are not associated with the target gene. Given the known status of CpG methylation’s role in association with target gene expression (i.e. true negative when 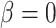 and true positive when
and true positive when 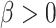 for the triplet), we next computed the AUC for each method. The ROC curves show a trade-off between sensitivity and specificity as the significance cutoff is varied. AUC assesses the overall discriminative ability of the methods to determine whether a given methylation CpG is driving target gene expression over all possible cutoffs. The best-performing models with the highest AUCs are rlm.binary.en (0.883), rlm.binary (0.874) and lm.binary (0.812), followed by models with continuous methylation levels, rlm.cont (0.755) and lm.cont (0.699) (Figure 5).
for the triplet), we next computed the AUC for each method. The ROC curves show a trade-off between sensitivity and specificity as the significance cutoff is varied. AUC assesses the overall discriminative ability of the methods to determine whether a given methylation CpG is driving target gene expression over all possible cutoffs. The best-performing models with the highest AUCs are rlm.binary.en (0.883), rlm.binary (0.874) and lm.binary (0.812), followed by models with continuous methylation levels, rlm.cont (0.755) and lm.cont (0.699) (Figure 5).
Figure 5.

ROC curves for different methods compared in the simulation study. Here, Model 1 is gene expression ∼ TF + DNAm.value + TF × DNAm.value and Model 2 is gene expression ∼ TF + DNAm.group + TF × DNAm.group. Among methods that implement Models 1 and 2, robust linear models with methylation binary group (rlm.binary, rlm.binary.en) performed best. The best performing models with the highest AUCs are rlm.binary.en (0.883), rlm.binary (0.874), and lm.binary (0.812), followed by models with continuous methylation levels, rlm.cont (0.755) and lm.cont (0.699).
Among the methods that implemented Models 1 and 2, the models that use a binary variable (low, high) to model methylation levels (rlm.binary, rlm.binary.en and lm.binary methods) performed best, probably because these models can reduce noise in data and thus can improve power. Among binary models, the robust linear models rlm.binary and rlm.binary.en performed similarly and better than regular linear model lm.binary. We also performed several additional simulation studies that evaluated the impact of different sample sizes of the simulation datasets, when methylation data were generated from beta distributions, and when the effect of TF on target gene expression (parameter 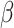 described earlier) varied in a continuum as an exponential decay function of methylation levels (Supplementary Text). In all these additional simulation scenarios, the rlm.binary model also performed best among all models (Supplementary Figures S4–S8). Thus, we selected the rlm.binary model for our subsequent analyses of real multi-omics datasets.
described earlier) varied in a continuum as an exponential decay function of methylation levels (Supplementary Text). In all these additional simulation scenarios, the rlm.binary model also performed best among all models (Supplementary Figures S4–S8). Thus, we selected the rlm.binary model for our subsequent analyses of real multi-omics datasets.
Comparison of the linear models that analyze CpG–TF–target gene triplet with methods that directly test methylation–target associations
Conventionally, DNA methylation levels are often correlated with target gene expression directly to identify those CpGs with functional effects on nearby genes, using a correlation statistic (method corr.met) or a Wilcoxon test that compares target gene expression in high versus low methylation samples (method wilcox.main.met). However, as we demonstrate below, a challenge with these simpler methods is that they cannot distinguish between the biological events in which target gene expression is regulated by both methylation and TF (Figure 1B) or only by TF (Figure 1C).
To illustrate, we next performed a simulation study to compare different methods for identifying the biological events in scenario (C), where target gene expression is mainly regulated by variations in TF but not DNA methylation. To this end, we simulated datasets in which target gene expression levels were dependent on TF expression, and DNA methylation levels were also correlated with TF expression. We used the same 38 samples of TCGA COAD matched RNA-seq and DNA methylation data described earlier as our input dataset, from which we randomly sampled TF gene expression and methylation levels.
For each simulated triplet dataset, we first randomly sampled data for one gene from the RNA-seq dataset to be our TF expression and one CpG site from the methylation dataset to be our DNA methylation levels. To obtain a dataset in which methylation levels are negatively correlated with TF expression, we ordered methylation values from largest to smallest and TF values from smallest to largest and then put the two columns of data next to each other. Therefore, by the design of the experiment, methylation values are negatively correlated with TF values.
Next, we simulated target gene expression levels in all samples from a negative binomial distribution with location parameter 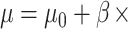 TF gene expression and scale parameter
TF gene expression and scale parameter 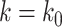 , with
, with 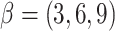 . Here, we set
. Here, we set 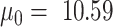 and
and 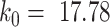 as described in the earlier section. Note that here target gene expression is dependent only on TF gene expression and not on DNA methylation levels. For each value of
as described in the earlier section. Note that here target gene expression is dependent only on TF gene expression and not on DNA methylation levels. For each value of 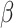 , we then repeated this process 1000 times to generate 1000 triplets of TF, DNA methylation and target gene expression levels.
, we then repeated this process 1000 times to generate 1000 triplets of TF, DNA methylation and target gene expression levels.
Figure 6 shows that methods that test methylation–target gene associations directly (corr.met, wilcox.main.met) would also identify these DNA methylation–TF–target gene triplets as significant, with the proportion of significant P-values for these methods ranging from 45% to 75%. On the other hand, methods that consider methylation, TF and target gene simultaneously (lm.binary, lm.cont, rlm.binary, rlm.cont) would not identify these triplets as significant. The proportions of significant P-values for these methods ranged from 4.3% to 8.4%. Therefore, a challenge for methods that directly test methylation–target gene association is that these methods cannot distinguish target gene expression driven mainly by the TF (Figure 1C) versus those driven by both DNA methylation and TF (Figure 1B).
Figure 6.

Performance of different models when target gene expressions are regulated only by TF expressions, and not by DNA methylation levels. Methods that test pairwise methylation–target associations (corr.met, wilcox.main.met) will identify these DNA methylation–TF–target gene triplets as significant, while methods that consider methylation, TF and target gene simultaneously (lm.binary, lm.cont, rlm.binary, rlm.cont) do not identify these triplets as significant.
Supplementary Figure S9 shows an example from the analysis of triplets in the analysis of TCGA COAD dataset, where the observed correlation between promoter DNA methylation and target gene expression is highly significant (Spearman correlation = 0.231, P-value = 4.99 × 10–5, FDR = 2.81 × 10–4). However, the result of fitting the MethReg model indicated that for this triplet, neither DNAm nor DNAm × TF terms were significant (P-values = 0.395 and 0.477, respectively), but TF was highly significant (P-value = 5.75 × 10–5). Therefore, the target gene expression is likely driven mainly by the TF EBF1, a tumor suppressor with prognostic value for CRC (40), and not by DNA methylation, even though we observed a highly significant methylation–target gene expression association.
Case study: analyses of TCGA COADREAD dataset - an unsupervised MethReg analysis
CRC is the third most commonly diagnosed cancer and the second leading cause of cancer death in the United States (41). Like many other cancers, CRC is characterized by global hypomethylation leading to oncogene activation, chromosomal instability and locus-specific hypermethylation, which leads to the silencing of tumor suppressor genes (42,43). In parallel, TFs also play instrumental roles in tumor development and metastasis (44–46). Given the strong epigenetic basis of CRC, we next applied MethReg to the TCGA COADREAD dataset, including 367 samples with matched DNA methylation, gene expression and copy number alterations (CNAs). To account for potential confounding effects, we adjusted target gene expression values by CNA and tumor purity estimates (47) first, extracted the residuals and then fitted the rlm.binary model to the residuals (Supplementary Text).
We performed an unsupervised MethReg analysis without selecting any CpG a priori. First, we divided the CpGs into those in the promoter regions (within ±2 kb regions around the TSS) or the distal regions (>2 kb from TSS). Next, we linked CpG sites in the promoter regions to genes that had promoters overlapping with the CpGs. On the other hand, CpG sites in the distal regions were linked to five genes upstream and five genes downstream within 1 million bp. Alternatively, in the regulon-based approach, we also linked CpGs in either promoter or distal regions to genes regulated by TFs with binding sites close to the CpG (Figure 2). To more accurately model TF effect, we computed TF activity scores using the VIPER algorithm (34). Stage-wise analysis using the rlm.binary model residual target gene expression ∼TF.activity + DNAm.group + DNAm.group × TF.activity was then applied to the triplet datasets to identify triplets with significant DNAm.group × TF.activity effect, in which the CpG had a significant association with the target gene expression by interacting with TF.
After multiple comparison corrections using the stage-wise approach (at a 5% FDR), the numbers of triplets with significant DNAm × TF.activity terms in the promoter, distal and regulon-based analyses were 31, 52 and 47, respectively (Table 1 and Supplementary Tables S3–S5). There was no overlap between the significant triplets obtained in these three analyses. Our results agreed well with the previous study by Wang et al., which also observed only a small number of transcriptional regulations were mediated by DNA methylation. Moreover, the TFs and target genes in the significant triplets identified by MethReg have also been previously associated with CRC. Figure 3 shows the most significant triplet among all the triplets considered in promoter and distal analyses. In this example, the TF NFATC2 represses the target gene HENMT1 in samples with high DNA methylation at cg00328227 but is relatively independent of the target gene HENMT1 expression in samples with low DNA methylation. Therefore, DNA methylation is predicted to enhance TF activity in this case. NFATC2 belongs to the NFAT family of TFs, which regulate T-cell activation and differentiation as immune cells invade the malignant tissues (48,49). In particular, NFATC2 is a critical regulator in intestinal inflammation that promotes the development of CRC, and expression levels of NFATC2 were found to be elevated in CRC patients (50,51). The MethReg prediction that NFATC2 represses HENMT1 is consistent with the recent study that demonstrated higher expression of HENMT1 is a favorable prognostic biomarker for CRC (52) and that NFATC2 is associated with tumor initiation and progression. The HENMT1 gene encodes a methyltransferase that adds a 2′-O-methyl group at the 3′-end of piRNAs and is previously shown to be dysregulated in many cancers (53). Our literature review showed the role of HENMT1 is not well understood in CRC. Therefore, this example also shows MethReg can nominate plausible TF functions and target genes that can be further studied experimentally. In contrast, without stratifying on DNA methylation levels, the direct TF–target gene association was only modest (Spearman correlation = −0.033, P-value = 0.527).
Table 1.
Results of MethReg stage-wise analysis of TCGA COADREAD dataset
| Analysis | Unique triplets | Unique CpGs | Unique TFs | Unique targets |
|---|---|---|---|---|
| Promoter analysis | ||||
| Screening | 165 | 49 | 56 | 42 |
| Confirmation | 31 | 29 | 18 | 25 |
| Distal analysis | ||||
| Screening | 2358 | 315 | 155 | 570 |
| Confirmation | 52 | 43 | 44 | 44 |
| Regulon-based analysis | ||||
| Screening | 78 | 23 | 19 | 19 |
| Confirmation | 47 | 18 | 11 | 6 |
After removing effects of copy number alteration and tumor purity estimate in gene expression data, the robust linear model target gene expression residual ∼ TF activity +DNAm.group +DNAm.group× TF activity was used to analyze CpG–TF–target gene triplets. In each analysis, TFs with binding sites within 250 bp of the CpGs were analyzed, and TF activity scores were estimated using the VIPER algorithm. Shown are significant triplets at 5% overall FDR level and the unique CpGs, TFs and targets in these triplets. The CpGs in the promoter regions (±2 kb around the TSS) were linked to genes that had promoters overlapping with them (promoter analysis), while CpGs in the distal regions (>2 kb from TSS) were linked to five genes upstream and five genes downstream within 1 million bp (distal analysis). In regulon-based analysis, CpGs were linked to TF–target gene pairs in the pan-cancer regulons described in (31). Abbreviations: DNAm, DNA methylation; TSS, transcriptional start site; TF, transcription factor
Supplementary Figure S10 shows an example in which DNA methylation is predicted to attenuate TF activity. In samples with low methylation levels at cg02816729, the TF TEAD3 downregulates target gene SMOC2. On the other hand, in samples with high methylation levels at CpG cg02816729, the expression of the target gene SMOC2 is relatively independent of TEAD3 activity (and its gene expression). TEAD3 belongs to the family of TEAD TFs, which also play critical roles in tumor initiation and progression in multiple types of malignancies, including gastric, colorectal, breast and prostate cancers (54). Higher expression of TEADs, as well as association with poor patient survival, has been observed in many cancers, including CRC (54,55). On the other hand, the target gene SMOC2 was recently shown to be a favorable prognostic biomarker for better clinical outcomes in a large cohort of CRC patients (56). Therefore, the MethReg prediction that TF TEAD3 suppresses target gene SMOC2 is consistent with the oncogenic role of TEAD3 and the favorable prognostic potential of SMOC2. Again, without stratifying on methylation levels, the TF–target gene association in all samples is only modest (Spearman correlation = 0.07, P-value = 0.183).
Interestingly, some of the TFs also exhibited different modes of regulation depending on DNA methylation levels. In Supplementary Figure S11, the expression levels of target gene BAHCC1 are increased by TF POU3F4 in low methylation samples (P-value = 6.42 × 10–4) but are decreased by POU3F4 in high methylation samples (P-value = 1.31 × 10–4). Therefore, DNA methylation may have increased the diversity of TF functions in this case. Without stratifying on DNA methylation, the TF–target gene association is not significant (Spearman correlation = 0.083, P-value = 0.112). Alternatively, the significant DNAm × TF.activity term can also be interpreted as significant DNAm–target gene association affected by the TF. Indeed, when we stratified samples by low versus high TF activity, DNAm–target gene association in samples with low TF activity (estimate = 3.06, P-value = 8.46 × 10–8) was stronger than their association in samples with high TF activity (estimate = 1.55, P-value = 7.31 × 10–5). Therefore, in these triplets where DNAm and TF interact to influence target gene expression, TF binding might also be affecting DNAm–target gene association. POU3F4 belongs to the family of POU domain TFs, which are involved in different developmental processes and were also recently found to be associated with the malignancy processes in different human tissues (57–61). In addition, expression levels of the target gene BAHCC1 were shown to be associated with survival times in different types of cancers such as melanoma (62), liver cancer (63) and pancreatic cancer (52).
Among other TFs in the top 10 triplets (Table 2), SNAI1 is a zinc finger protein involved in inducing the epithelial–mesenchymal transition process during which tumor cells become invasive with increased apoptotic resistance (64–67). The MEIS1 homeodomain protein is a tumor suppressor and was observed to be downregulated in colorectal adenomas (68). Similarly, ISL2 is also a tumor suppressor, and ISL2 loss increased cell proliferation and enhanced tumor growth in pancreatic ductal adenocarcinoma cells (69). ETV4 belongs to a subfamily of the ETS family with well-known oncogenic properties (70). In particular, ETV4 is overexpressed in colorectal tumor samples, and higher ETV4 expression is associated with a shorter patient survival time (71–77). ZNF384 is a zinc finger protein that is found to be overexpressed in several cancers, including leukemia (78,79), liver cancer (80) and CRC (81). Finally, FOXL1 is an important regulator of the Wnt/APC/β-catenin pathway, which frequently activates events in gastrointestinal carcinogenesis (82–84). Similarly, the target genes in the top 10 triplets have also been implicated previously in CRC and other cancers (Table 2). Moreover, among the significant triplets identified by MethReg, the majority of the CpGs, TFs and target genes (78%, 79% and 75%, respectively) were also differentially methylated or differentially expressed between tumor and normal samples (Supplementary Tables S3–S5). Gene ontology analysis showed many developmental biological processes (e.g. embryonic organ development and cell fate commitment) are significantly enriched with the methylation-sensitive TFs (Supplementary Table S6). Naxerova et al. showed that CRC belonged to the group of cancers exhibiting early developmental features and was undifferentiated in general compared to other types of tumors (85). Similarly, Table 2 shows the target genes in these top triplets are also highly relevant to CRC (86–93). Taken together, these results demonstrated that MethReg is able to identify biologically meaningful signals from real multi-omics datasets.
Table 2.
Top 10 most significant triplets identified by unsupervised MethReg analysis of TCGA COADREAD samples
| Triplet data | Annotations | DNAm group × TF activity | TF–target associations | Tumor versus normal comparison P-values | Literature support | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Probe ID | Position | TF | Target gene | CpG–target distance | TF.role | DNAm.effect | Estimate | P-value | Adj. P-value | Correlation | P-value | CpG | TF | Target gene | TF | Target gene |
| Promoter analysis | ||||||||||||||||
| cg00328227 | chr1: 108661703 | NFATC2 | HENMT1 | −176 | Repressor | Enhancing | −0.663 | 0 | 0 | −0.033 | 5.27E−01 | 3.19E−08 | 2.53E−01 | 8.23E−08 | (48–49,51) | (52,53) |
| cg25083481 | chr11: 1034989 | SNAI1 | MUC6 | 1727 | Activator | Attenuating | −0.413 | 1.77E−04 | 0 | 0.117 | 2.52E−02 | 8.98E−03 | 4.99E−10 | 1.97E−02 | (64,66–67) | (86) |
| cg02816729 | chr6: 168442419 | TEAD3 | SMOC2 | 1267 | Repressor | Attenuating | 0.323 | 7.77E−05 | 0 | 0.07 | 1.83E−01 | 2.96E−02 | 3.47E−02 | 7.32E−01 | (54) | (56,87) |
| cg12751565 | chr1: 206946642 | NFATC2 | PIGR | −175 | Activator | Enhancing | 0.282 | 1.09E−09 | 0 | 0.315 | 8.67E−10 | 6.49E−03 | 2.53E−01 | 1.88E−11 | (48–49,51) | (88) |
| cg05503887 | chr11: 34620378 | MEIS1 | EHF | −713 | Activator | Attenuating | −0.268 | 7.24E−05 | 0 | 0.347 | 1.10E−11 | 6.17E−01 | 1.84E−08 | 1.16E−02 | (68) | (89) |
| cg13542964 | chr4: 10457043 | ETV4 | ZNF518B | 381 | Activator | Enhancing | 0.217 | 2.08E−04 | 0 | 0.023 | 6.59E−01 | 1.31E−06 | 1.32E−14 | 1.77E−02 | (70,74) | (90) |
| Distal analysis | ||||||||||||||||
| cg14043104 | chr13: 74075149 | ISL2 | KLF12 | −80 092 | Activator | Attenuating | −0.222 | 1.03E−04 | 0 | 0.232 | 7.60E−06 | 3.89E−02 | 3.96E−02 | 1.20E−03 | (69) | (91) |
| cg09217215 | chr11: 31808034 | ZNF384 | PAX6 | 9925 | Repressor | Enhancing | −0.398 | 6.90E−07 | 1.48E−04 | −0.028 | 5.94E−01 | 7.64E−03 | 2.15E−01 | 1.68E−02 | (78,79) | (92) |
| cg11871337 | chrX: 56991382 | FOXL1 | SPIN3 | 4443 | Activator | Attenuating | −0.294 | 2.18E−07 | 2.68E−04 | 0.183 | 4.32E−04 | 7.26E−01 | 9.95E−01 | 8.21E−07 | (82,83) | (93) |
| cg04665204 | chr17: 81453398 | POU3F4 | BAHCC1 | 57 922 | Dual | Invert | −0.37 | 6.80E−07 | 2.91E−04 | 0.083 | 1.12E−01 | 2.69E−05 | 1.47E−06 | 2.38E−02 | (57,61) | (52,63) |
Triplet data included the CpGs located in promoter regions (<2 kb from TSS) or distal regions (>2 kb from TSS), its location (hg38 genome), the target genes and the TFs with binding sites within 250 bp of the CpGs. Annotations included the distance between CpG and target gene (CpG–target distance), MethReg predicted roles of the TF on the target gene and the effect of DNA methylation (DNAm) on the TF. TF activity (hereafter referred to as TF) was first estimated using the VIPER software. After removing effects of CNA and tumor purity estimate in gene expression data, the robust linear model target gene expression residual ∼ DNAm group + TF + TF × DNAm group was then fitted to data to obtain estimated TF × DNAm group effect (estimate) and its P-value; the stage-wise method was used to compute the overall FDR (adj. P-value) for the term DNAm.group × TF. TF–target association in all samples was estimated using a correlation coefficient. P-values for tumor versus normal comparisons were obtained by applying Wilcoxon rank-sum tests to compare DNAm levels, TF and target gene in tumor and normal samples separately.
Comparative analysis of MethReg (in unsupervised mode) with alternative approaches
Encouraged by the consistency of MethReg results with the recent CRC literature, we next performed a comparative analysis of MethReg with several alternative approaches. As evidence from large-scale analyses that a substantial number of TFs can interact with methylated DNA has only become available in recent years, few studies have analyzed DNA methylation, TFBS and gene expression data simultaneously using large multi-omics datasets until recently.
We considered a total of three alternative approaches: the recent studies by Wang et al. (94) and Liu et al. (95), as well as the conventional approach of directly correlating DNA methylation with gene expression. In Wang et al. (94), a rewiring score constructed based on TF–target gene correlations in high and low methylation samples was proposed to identify methylation-sensitive TFs in different cancers using TCGA data. Similarly, using a conditional mutual information (CMI)-based approach, Liu et al. (95) identified CpG–TF–target gene triplets in which the TF–target gene regulation circuit is dependent on CpG methylation levels in different cancers using TCGA data. In both of these studies, sample permutations were used to estimate P-values for test statistics based on rewiring scores or CMI.
Conceptually, compared to these previous studies, MethReg makes several distinct contributions: (i) MethReg analysis is comprehensive. While both previous works analyzed only promoter CpG methylation, MethReg analyzes methylation CpGs at both promoter and distal (enhancer) regions. The identification of distal regulatory elements is crucial as they are often not well defined. (ii) MethReg analysis is flexible. In addition to the example databases we have described here, MethReg is capable of incorporating any user-specified ChIP-seq or regulon database (Supplementary Table S2), including those that are tissue or disease specific. The power and potential of MethReg grow as more knowledge on TF regulation becomes available. (iii) MethReg analysis is rigorous. Robust linear models have been implemented to carefully model outlier samples in RNA-seq data. Further, with the permutation approaches used in the previous studies, it can be challenging to adjust for covariate information that is important for analyzing human datasets, for which confounding variables such as tumor purity are often significant contributors to both DNA methylation and gene expression. In contrast, MethReg’s regression-based approach makes it easy to adjust for potential confounding variables. (iv) Most importantly, while previous studies provided analysis results for TCGA cancer datasets, no software has been made available to the research community until now. This study provides an open-source software, MethReg, which implements comprehensive bioinformatics and statistical analysis, making it possible for researchers to analyze datasets beyond TCGA. In addition to providing estimated individual and joint regulatory effects of CpG methylation and TFs, MethReg also annotates the potential regulatory roles of TFs and CpG methylation, as well as providing a rich suite of figures for visualizing analytical results.
Next, we systematically compared MethReg analysis and other approaches empirically using the TCGA COAD dataset. Because both previous studies analyzed TF gene expression, we performed additional MethReg promoter analysis that models the TF effect based on TF gene expression instead of TF activity. More specifically, to compare with results from Wang et al. (94), which identified 3244 TF–target gene pairs, we applied the robust linear model described earlier to the corresponding triplets (average promoter DNA methylation, TF gene expression and target gene expression). Our results showed that for a majority (81.23%, n = 2613) of the significant TF–target gene pairs from (94), the corresponding triplets also had a significant DNAm × TF effect in the rlm.binary model from MethReg. Moreover, classification for promoter methylation effects on TFs in (94) and MethReg agreed very well (kappa statistic = 0.975, P-value = 0) (Supplementary Table S7).
Similarly, we also fitted our rlm.binary model described earlier to the 47 029 triplets identified in (95) for the TCGA COAD dataset. However, we observed less agreement between our significant results and those from (95), as only 5321 (11.3%) of the 47 029 triplets had significant DNAm × TF P-values by MethReg. The overlap between studies by Liu et al. (95) and Wang et al. was also very low, with only eight TF–target gene associations identified by both studies. This discrepancy might be due to the differences in methodologies. The CMI approach used by Liu et al. (95) detects any general associations that can be nonmonotonic, while the robust linear model MethReg used and the rewiring score in (94) mainly detect monotonic TF–target gene associations that are dependent on CpG methylation. Biologically, for TF–target gene pairs with nonmonotonic associations, target gene expression is higher when TF activity is either high or low than when TF activity is intermediate. Mutual information for TF and target gene expression can still be high for these TF–target gene pairs, but there would not be any monotonic TF–target association.
Finally, we compared the MethReg promoter analysis results with the conventional approach of correlating methylation–target gene directly. More specifically, Spearman correlations were computed for each promoter CpG and target gene in the COADREAD dataset. The correlation between rankings of P-values based on methylation–target gene correlation and DNAm × TF interaction effects in the MethReg model is a significant but modest association (Spearman correlation = 0.0533, P-value = 2.2 × 10–16), indicating these two approaches are identifying many different CpGs. This is not surprising, however, because the MethReg model identifies CpG methylation that can potentially influence the target gene by regulating TF activity (Figure 1B) instead of influencing the target gene directly (Figure 1A).
Comparative analysis of different approaches using methylation-sensitive TFs in MeDReaders database as the gold standard
Compared to alternative methods [e.g., the CMI approach in (95) identified 47 029 triplets in the TCGA COADREAD dataset], MethReg selected fewer significant triplets at default settings. There are several possible reasons: (i) We hypothesized that a contributing factor might be that other studies analyzed TF gene expression, while MethReg analyzed TF activity. To test this hypothesis, we re-analyzed the TCGA COADREAD dataset using the same analysis pipeline except replacing estimated TF activity with TF gene expression. Our results showed by analyzing TF gene expression, we would obtain 172 and 693 significant triplets in MethReg promoter and distal analyses (Supplementary Tables S8 and S9), compared to 31 and 52 significant triplets obtained previously by analyzing TF activity (Supplementary Tables S3 and S4). One possible reason for the reduced power in the latter approach could be the increased variance in estimated TF activity compared to the variance of TF gene expression (Supplementary Figure S12). While MethReg analyzes TF activity at default, to help users interested in analyzing TF gene expression and comparing the results obtained with the two approaches, we also implemented an additional option in MethReg for analysis using TF gene expression. (ii) MethReg’s regression-based approach allowed us to adjust for potential confounding variables, which may have also reduced the number of significant results. In contrast, adjusting for covariate variables is challenging for alternative methods that use permutation-based approaches. More specifically, in the analysis of the TCGA COADREAD dataset, we adjusted for effects from CNAs and tumor purity scores. Without adjusting for these covariate variables, MethReg promoter and distal analyses using TF gene expression would result in 354 and 810 significant triplets, compared to the 172 and 693 triplets obtained earlier after correcting for covariate variables.
To systematically compare these different MethReg approaches, we next evaluated the TFs in significant triplets identified by each approach using the MeDReaders database (96), which contains manually curated information for 731 TFs with binding activity predicted to be influenced by DNA methylation based on human or mouse studies, as the gold standard. These TFs were shown to exhibit CpG methylation-dependent DNA-binding activity using functional protein arrays (14) or SELEX (16). Because MethReg analyzed only TFs in the JASPAR database (29), we considered only TFs included in the JASPAR database in the following analysis. Specifically, for each method, we computed precision, which is the proportion of TFs identified by a method that is also in the MeDReaders database, among all TFs identified by a method, and recall, which is the proportion of TFs in the MeDReaders database identified by a method. Our results showed MethReg analysis that analyzes TF activity and adjusts for covariates (i.e. CNAs and tumor purity scores) (method A) had the highest precision (72.2%), and MethReg analysis that analyzes TF gene expression and adjusts for covariates (method B) is a close second (with a 71.2% precision). In comparison, MethReg analysis that analyzes TF gene expression without adjusting covariates (method C) performed worst with a precision of 65.9% (Supplementary Table S10). On the other hand, recall is lower, at 9.5%, 20.4% and 23.6% for methods A, B, and C, respectively. One contributing factor for these low recall rates might be the discrepancy between the cell lines used to develop the MeDReaders database and the primary tumor tissues in the TCGA dataset used for MethReg analysis. Also, the lower recall (9.5%) for method A compared to the other two methods might be due to the fewer triplets identified by MethReg analysis that analyzes TF activity and adjusts for covariates.
In addition, we also performed the analysis for the TCGA COADREAD dataset using the CMI approach as implemented in the JAMI software (97). Our results showed the CMI approach achieved a precision of 10.3% and a recall of 4.6%, significantly lower than the MethReg approaches. These results provided additional evidence for using MethReg regression models for real datasets.
Case study: analysis of ROSMAP Alzheimer’s disease dataset - a supervised MethReg analysis
In this section, we demonstrate supervised MethReg analysis using an Alzheimer’s disease (AD) dataset collected by the ROSMAP study (98). In contrast to unsupervised analysis, which tests triplets involving all CpGs, in a supervised analysis, we only test triplets involving DMS, typically obtained from EWAS. To study AD-associated DNA methylation changes in the brain, we recently performed a meta-analysis of over 1000 prefrontal cortex brain samples from four large brain studies (6,99–101) and identified 3751 significant CpGs at 5% FDR (102). To help understand the regulatory roles of these DMS, we applied MethReg to analyze matched DNA methylation and gene expression profiles measured on prefrontal cortex brain samples from 529 independent subjects in the ROSMAP dataset.
To illustrate the versatility of the MethReg analysis pipeline, we used alternative databases to analyze the ROSMAP dataset compared to the analysis pipeline for the TCGA COADREAD samples. More specifically, to map TFBS, instead of the JASPAR2020 database (29) here, we used the ReMap database (28), which contains a large collection of regulatory regions obtained using genome-wide DNA-binding experiments such as ChIP-seq. In particular, the ReMap human atlas included binding regions for 1135 transcriptional regulators. Also, to analyze these brain samples, instead of the pan-cancer regulons, we used the brain-specific TF regulons included in the ChEA3 (103) software website, along with TF activity scores estimated by GSVA (35).
More specifically, we first adjusted methylation and gene expression values separately by potential confounding effects, including age at death, sex, batch effects and markers of different cell types. Next, we computed TF activity using the GSVA method (35), which is an alternative method to VIPER (34) for computing enrichment scores of each TF by comparing enrichment in target gene expression for a TF (its regulons) with expression levels of background genes.
At a 5% FDR, MethReg identified 1, 20 and 103 triplets that included 1, 16 and 53 unique TFs that interact with DMS to influence target gene expression in the promoter, distal and regulon-based analyses, respectively. A comparison with the MeDReaders database (96) shows more than half (58.6%, 41 out of 70) of these TFs were previously shown to interact with methylated DNA sequences (Supplementary Tables S11 and S12). In Table 3, many of the TFs and target genes in the top 10 triplets were previously implicated in AD pathology. For example, in the most significant triplet (Table 3 and Supplementary Figure S13), the TF SPI1 (PU.1) is a master regulator in the AD gene network (104,105). SPI1 is critical for regulating the viability and function of microglia (106), which are resident immune cells of the brain. Microglia function as primary mediators of neuroinflammation and phagocytose amyloid-beta peptides accumulated in AD brains (107). Using transgenic mouse models for AD and comparing with gene-level variations in recent human AD GWAS meta-analysis (108), Salih et al. (109) recently showed the target gene LAPTM5 (lysosome-associated protein, transmembrane 5) belongs to an amyloid-responsive microglial gene network and predicted LAPTM5 to be one of four new risk genes for AD. Moreover, LAPTM5 was also shown to be a member of the human microglia network in AD in multiple gene expression studies (110,111). Intriguingly, SPI1 and LAPTM5 belonged to the same transcription co-expression network (109), and in mouse microglial-like BV-2 cells, results from the ChIP-seq experiment showed SPI1 binds to the regulatory region of LAPTM5 (112), consistent with the MethReg prediction that LAPTM5 is regulated by SPI1. While previous studies have implicated both SPI1 and LAPTM5 in AD pathology, how SPI1 influences LAPTM5 gene expression is less clear in the AD literature. Without stratifying on DNA methylation levels, the direct SPI1–LAPTM5 association is very low (Spearman correlation = −0.010, P-value = 0.805) (Supplementary Figure S13). To help connect the dots, MethReg analysis provided evidence that DNA methylation interacts with TF SPI1 to influence LAPTM5 gene expression jointly, which is crucial for functionally interpreting the epigenomic maps. Interestingly, SPI1 appears to have dual roles in this case, depending on DNA methylation levels at cg17418085, which is located in the gene body of LAPTM5. More specifically, SPI1 upregulates the target gene LAPTM5 in samples with low methylation at cg17418085 but downregulates the target gene when DNA methylation is high, suggesting DNA methylation and the TF might have compensatory mechanisms that control gene expression at this locus. Additional biological experiments are needed to confirm these observed cooperative interactions between DNAm and SPI1 that influence LAPTM5 gene expression.
Table 3.
Top 10 most significant triplets identified by supervised MethReg analysis of ROSMAP AD dataset
| Triplet data | Annotation | DNAm group × TF activity | TF–target | AD literature | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Probe ID | Position | TF | Target gene | CpG–target distance | TF.role | DNAm.effect | Estimate | P-value | Adj. P-value | Correlation | P-value | TF | Target gene |
| Promoter analysis | |||||||||||||
| cg17418085 | chr1: 31229122 | SPI1 | LAPTM5 | 1543 | Dual | Invert | −7.177 | 7.36E−12 | 3.06E−07 | −0.011 | 8.05E−01 | (105,106) | (109) |
| Distance-based analysis | |||||||||||||
| cg11556846 | chr2: 200468728 | ESR1 | SATB2 | −132 738 | Dual | Invert | −7.266 | 3.03E−07 | 7.80E−03 | −0.058 | 1.82E−01 | (124,126) | (157) (158) |
| cg00153919 | chr16: 88859944 | CEBPD | PIEZO1 | −8324 | Repressor | Enhancing | −8.122 | 4.92E−07 | 9.14E−03 | −0.044 | 3.10E−01 | (130,131) | (159) |
| cg08824847 | chr11: 35052388 | NR3C1 | PDHX | 115 011 | Activator | Enhancing | 4.383 | 6.13E−07 | 1.02E−02 | 0.06 | 1.66E−01 | (114,116) | (117,118) |
| cg08760493 | chr4: 109994039 | TCF12 | SEC24B | −360 887 | Dual | Invert | 8.120 | 1.26E−06 | 1.49E−02 | 0.033 | 4.44E−01 | (132,133) | (160) |
| cg05715492 | chr7: 98991138 | SRF | ARPC1B | 19 265 | Repressor | Attenuating | 11.581 | 1.34E−06 | 1.49E−02 | 0.017 | 7.00E−01 | (135) | (161) |
| cg09316954 | chr16: 67687754 | TCF12 | CARMIL2 | 8931 | Repressor | Attenuating | 8.376 | 2.28E−06 | 2.24E−02 | −0.07 | 1.08E−01 | (132,133) | (162) |
| cg21155834 | chr2: 149282209 | GABPA | ORC4 | −503 061 | Activator | Enhancing | 6.407 | 2.75E−06 | 2.41E−02 | 0.045 | 2.98E−01 | (137) | (163) |
| cg13819552 | chr9: 95799870 | TCF12 | ZNF484 | −159 565 | Activator | Enhancing | 7.989 | 2.92E−06 | 2.41E−02 | 0.039 | 3.71E−01 | (132,133) | (164) |
| cg21535772 | chr2: 171679906 | NFE2L2 | AC007405.4 | 52 282 | Dual | Invert | 7.065 | 3.10E−06 | 2.41E−02 | −0.026 | 5.47E−01 | (138,165) | (166) |
Triplet data included the CpG located in promoter regions (<2 kb from TSS) or distal regions (>2 kb from TSS), its genomic location (hg19 genome), the target gene and the TF with binding sites within 250 bp of the CpG. Annotations included the distance between CpG and target gene (CpG–target distance), MethReg predicted roles of the TF on the target gene and the effect of DNAm on the TF. TF activity (hereafter referred to as TF) was first estimated using the GSVA software. After removing age at death, sex, batch and cell type effects in gene expression data and methylation data separately, the robust linear model target gene expression residual ∼ DNAm group + TF + TF × DNAm group was then fitted to data to obtain estimated TF × DNAm group effect (estimate), its P-value and FDR (adj. P-value) for the term DNAm.group × TF. TF–target association in all samples was estimated using a correlation coefficient.
The triplet cg08824847−NR3C1−PDHX is an example in which methylation at cg08824847 is predicted to enhance activation of the target gene PDHX by NR3C1 (Table 3 and Supplementary Figure S14). NR3C1 is the glucocorticoid receptor that can act as a TF that binds glucocorticoid-responsive genes to activate their transcriptions or as a regulator for other TFs. NR3C1 regulated downstream processes such as glycolysis (113) and was observed to be dysregulated in AD (114–116). The target gene PDHX encodes the component X of the pyruvate dehydrogenase (PDH) complex, which is involved in regulating mitochondrial activity and glucose metabolism in the brain that is critical for neuron survival (117,118). Low levels of cerebral glucose metabolism often proceed with the onset of AD and have been proposed as a biomarker of AD risk (119–122). Previously, it was also shown that along with other regulators, glucocorticoids can regulate the efficacy of PDH (113,123). The MethReg prediction that methylation at cg08824847 enhances activation of PDHX by NR3C1 is consistent with these previous findings that demonstrated lower levels of PDHX in AD samples, and with results from our previous large meta-analysis of DNA methylation changes in AD, which discovered cg08824847 to be hypomethylated in AD samples across all four analyzed brain sample cohorts and has a significant negative association with AD Braak stage even after multiple comparison correction (102).
Among the other TFs in the top 10 triplets (Table 3), ESR1 is estrogen receptor alpha, one of two subtypes of the estrogen receptor. Genetic polymorphisms of ESR1 have been associated with the risk of developing cognitive impairment in older women (124–128), as well as faster cognitive decline in women AD patients (129). Induced by chronic inflammation in AD, CEBPD is associated with microglial activation and migration (130,131). TCF12 is a member of the basic helix–loop–helix E-protein family and plays important roles in developmental processes such as neurogenesis, mesoderm formation and cranial vault development. Recently, TCF12 was predicted to be affected by SNP rs10498633 (132), a top AD-associated SNP identified in the IGAP AD meta-analysis study (133). Moreover, TCF12 also belongs to a herpesvirus perturbed TF regulatory network that is implicated in AD (134). SRF is a serum response factor responsible for regulating the smooth muscle cells and blood flows in the brain, which is important for a blood vessel’s ability to remove amyloid-beta peptides accumulated in AD. Compared with healthy individuals, SRF was found to be four times higher in AD patients (135,136). GABPA belongs to the ETS family of DNA-binding factors and is a master regulator of multiple important processes, including cell cycle control, apoptosis and differentiation. Using evolutionary analysis and ChIP-seq experiments, Perdomo-Sabogal et al. (137) linked GABPA to several brain disorders, including AD, autism and Parkinson’s disease. Finally, NFE2L2/NRF2 is another master regulator and regulates genes involved in response to oxidative stress and inflammation. Motivated by the encouraging therapeutic effect of NRF2 on AD pathology in animal models and cultured human cells (138–140), modulation of the NRF2 pathway has recently been proposed as a strategy for AD drug development (138). Similarly, Table 3 shows the target genes in these top 10 triplets identified by MethReg are also highly relevant to AD. Taken together, these results demonstrated MethReg is capable of identifying biologically meaningful regulatory effects of DNA methylation in other complex diseases, such as AD, for which association signals are expected to be much weaker than those observed in cancers.
Comparative analysis of MethReg (in supervised mode) with alternative approaches
To compare the performance of MethReg in supervised mode with currently available alternative tools, we next analyzed the ROSMAP dataset using the ReMapEnrich R package (28), which identifies regulators with binding sites enriched in user-supplied regions. Several other tools, such as LOLA (21) and ChIP-Enrich (141), perform similar analyses as ReMapEnrich, but here we chose ReMapEnrich because it uses the same ReMap database as MethReg for the ROSMAP dataset analysis. More specifically, for the ReMapEnrich analysis, we also used the locations of the 3751 AD-associated CpGs (these are the DMS) from our previous AD meta-analysis (102) as the input. The results showed that ReMapEnrich identified 143 TFs with binding sites enriched with the DMS, among which a substantial number (n = 28, 20%) were also identified by MethReg (Supplementary Table S13). These 28 TFs included many well-known regulators for AD such as TCF12, SPI1, NR3C1, CEBPB, GABPA and others. On the other hand, 115 and 32 TFs were uniquely identified by ReMapEnrich or MethReg, respectively.
Although many of these significant TFs have been previously implicated in AD pathology, their specific roles in transcription regulation and the identification of their target genes in AD remain to be investigated. Notably, currently available tools such as ReMapEnrich only identify the TFs but do not consider CpGs or provide detailed information on the relevant target genes. In contrast, MethReg fills this critical gap by nominating plausible TF–target gene associations that are modulated by DNA methylation. Therefore, MethReg analysis, which leverages additional gene expression data to provide more comprehensive information on transcription regulation for the TFs, complements existing approaches.
DISCUSSION
To evaluate the role of DNA methylation in gene regulation, we developed the MethReg R package. MethReg provides a systematic approach to dissect the variations in gene transcription into three different modes of regulation: direct effects by methylation and TF individually, and the interaction effect from both DNA methylation and TF. By additionally modeling DNA methylation variations, MethReg complements existing approaches that analyze TF and target gene expression alone. In doing so, MethReg uncovers TF–target gene relations that are present only in samples with high (or low) methylation levels at the CpG that modulates TF activity. On the other hand, compared to approaches that analyze DNA methylation and TFBS data alone, MethReg analysis also reduces the noisiness in TFBS predictions by additionally modeling target gene expression data. Compared to the conventional approach of directly correlating DNA methylation with gene expression, MethReg can be useful for prioritizing DNA methylation–target gene associations driven by both DNA methylation and TFs, or mainly by DNA methylation, from those driven primarily by TFs. Computationally, MethReg is efficient. The unsupervised analysis of the TCGA COADREAD dataset, which considered all CpGs on the Illumina array, took 5, 37 and 14 min for the promoter, distal and regulon-based analyses, respectively, using a single Linux machine with 64 GB of RAM memory and Intel Xeon W-2175 (2.50 GHz) CPUs with four cores for parallel computing (Supplementary Table S14).
In addition to the conventional approach for controlling FDRs, MethReg also implements an alternative approach using the stage-wise method (38), which might help with improving power. For comparison, we re-analyzed the ROSMAP dataset using the same analysis pipeline except replacing the conventional approach for estimating FDR with the stage-wise method, and our results showed that we would get 19 and 69 significant triplets in MethReg promoter and distal analyses (Supplementary Tables S15 and S16), compared to 1 and 20 significant triplets using the conventional FDR approach (Supplementary Table S11). These results are consistent with those described in (38), which showed stage-wise method improves power over one-stage analysis in high-throughput experiments where multiple hypotheses are tested for each gene (or CpG here). Note also that because only AD-associated CpGs were considered in this supervised analysis of the ROSMAP dataset, the number of significant triplets is expected to be lower than those from unsupervised analysis, which considers all CpGs on the methylation array.
At default, after fitting statistical models, MethReg implements two additional filters to select triplets. First, to avoid triplets described in Figure 1C (target gene is mainly regulated by TF), MethReg removes a triplet if the TF is significantly associated with DNA methylation. However, this filtering step might be overly stringent since results from our simulation study (Figure 6) showed the MethReg model has very low power for identifying triplets of the scenario in Figure 1C. Next, MethReg selects only triplets in which target gene expression is significantly associated with DNA methylation, which might also be too restrictive. Note that when the direct effect of DNA methylation on the target gene and the DNAm × TF interaction effect on the target gene are in opposite directions, the overall association between DNA methylation and target gene expression might be reduced (Supplementary Figure S15). If we remove these two filters on the triplets in the analysis of the TCGA COADREAD dataset using TF gene expression, the number of significant triplets in promoter and distal analyses would be 723 and 2657, respectively. Similarly, in the stage-wise analysis of the ROSMAP dataset, without the filters, the number of significant triplets in promoter and distal analyses would increase to 70 and 401, respectively. These filters were used to prioritize the most significant results, but arguments filter.correlated.tf.exp.dnam and filter.correlated.target.exp.dnam in MethReg function interaction_model can also be used to turn these optional filters off when needed.
Because of current limitations in technology, directly measuring DNA methylation, TF binding and target gene expression in high throughput is still a difficult task, especially for a large cohort of primary tissue samples. Therefore, computational approaches are needed to prioritize regulatory elements in gene transcription. To this end, a main computational challenge is the accurate assessment of TF activity. Many integrative studies have used TF gene expression data, which are often widely available, as surrogate measurements for TF activity. However, the abundance of TF expression does not necessarily correspond to more TF binding events, which needs to be confirmed by cell type-specific ChIP-seq experiments. On the other hand, TF binding events are sometimes nonfunctional and might not lead to changes in gene expression (24,25). To this end, we implemented the option to model TF effects based on VIPER (34) or GSVA (35) estimated TF protein activity in MethReg. Both VIPER and GSVA approaches have been widely used for modeling protein activity using gene expression datasets, and these methods assume that collectively the target genes of a TF represent an optimal reporter of its activity, and they estimate TF activity based on enrichment of its target gene expression compared to background genes.
While the motivation of MethReg is to rank significant and functional DNA methylation changes identified in EWAS, a useful by-product of this analysis is the identification of enhancers, which are often located several hundreds of kb away from the target gene, where TFs bind and interact with DNA methylation to activate gene expression by looping DNA segments (142). Growing evidence indicates that in addition to promoter methylation, DNA methylation at enhancers also plays an equally or more important role in activating gene expression (143). Active cancer-specific enhancers are typically hypomethylated at CpG sites (10,144,145) in open chromatin regions free of nucleosomes (146,147). In many cases, hypermethylation of CpG sites can interfere with TF binding and lead to decreased enhancer activity in various cancers (9,148). It has been observed that TF activity often correlates with levels of demethylation at enhancer regions and subsequent target gene expression (27,148,149).
Although recently many cis-regulatory regions have been identified using genomic and epigenomic data (27,150), assigning these candidate enhancers to target genes on a genome-wide scale remains challenging and is currently an active area of research. A recent study (151) compared several published computational approaches for enhancer–gene linking using a collection of experimentally derived genomic interactions. It was shown that the best-performing method, TargetFinder (152), is only modestly better than the baseline distance-based approach, and the authors suggested further improvement in current computational methods in this area is needed. Although many of these computational methods leverage information from histone marks, chromatin accessibility and interaction, TF binding models and gene expression levels, few if any also model DNA methylation at candidate enhancer regions. As many recent studies suggested substantial crosstalk between DNA methylation and other regulatory elements such as TFs (14,16), we developed MethReg to specifically estimate and evaluate the regulatory potential of DNA methylation for interacting with candidate TFs at both promoter and distal regions. In particular, MethReg links target genes with CpGs in distal regions using two alternative approaches: linking to a fixed number of nearby genes or by using annotations in regulon databases (Figure 2). Indeed, among significant triplets identified in MethReg analysis of ROSMAP dataset, a substantial number of CpGs (6 and 22 in distal and regulon-based analyses, respectively) were located in brain-specific enhancer regions annotated in the EnhancerAtlas 2.0 database (153) (Supplementary Tables S11 and S12). Notably, the power and potential of MethReg will also grow as more knowledge on TF regulatory activity is accumulated and new ChIP-seq and TF regulon databases become available.
The aim of MethReg is to prioritize functional elements and to generate useful testable hypotheses for subsequent mechanistic studies. The significant associations identified by MethReg do not necessarily reflect causal relationships. For these DNA methylation changes that couple with TF activity, additional experimental studies are needed to determine whether the changes in methylation are causing or are caused by TF activity. Nevertheless, even if the DNA methylation changes are passive markers that accumulated as a result of TF binding (or lack of binding), they can still be useful as biomarkers. Currently, many of the large DNA methylation datasets are measured using methylation arrays because of their lower cost and the simplicity in benchwork and analysis. MethReg has been tested successfully on microarrays and can also be trivially extended to analyze large cohorts of samples measured using high-throughput sequencings such as WGBS or RRBS. In addition to CpGs identified in EWAS, MethReg can be similarly applied to evaluate the regulatory potential of candidate regions identified by other assays, including those targeting selected genomic regions. Beyond TFs, MethReg can also be applied to analyze other types of chromatin proteins, including histones that are known to crosstalk with DNA methylation (154–156). Finally, MethReg can be further extended to incorporate TF–target gene associations based on spatial enhancer–gene linking when 3D chromatin and genetic interaction data on primary cells become available.
We have presented an integrative analysis and annotation software, MethReg, which has several critical roles. First, given the large number of DMS identified from EWAS, supervised MethReg analysis can be used to analyze CpG–TF–target gene triplets involving the DMS, to identify and prioritize important CpG methylation that influences target gene expression by interacting with TFs that bind in proximity. Second, MethReg annotates CpG methylation and the TFs that bind in close proximity with their regulatory roles (i.e. activator or repressor TFs, and DNA methylation that attenuates or enhances TF effects). Third, because some TFs affect target gene expression only in samples with high methylation levels (or only in samples with low methylation levels), MethReg can help uncover TF–target gene associations that are not obvious in an analysis that uses all samples. Finally, for a particular target gene, MethReg partitions the variances in gene regulation into direct impact by methylation, direct impact by TF or joint impact of both methylation and TF, which allows MethReg to prioritize methylation–target gene associations that are likely driven by both DNA methylation and TFs, or DNA methylation alone, over those driven primarily by TFs. Using two case studies in CRC and AD, we have shown the power of MethReg to uncover biologically relevant transcriptional regulation in both diseases, which have vastly different biology. Open-source software scripts, along with extensive documentation and example data for MethReg, are freely available from the Bioconductor repository. We hope MethReg will empower researchers to gain a better understanding of the important regulatory roles of CpG methylation in many complex diseases.
DATA AVAILABILITY
The MethReg R package is available from the Bioconductor repository at https://bioconductor.org/packages/MethReg/. The scripts for the analysis performed in this study can be accessed at https://github.com/TransBioInfoLab/MethReg_supplemental. Level 3 TCGA COAD and READ cancer datasets, including genomic profiles in CNAs (gene-level copy number scores), gene expressions (HTSeq-FPKM-UQ values from RNA-seq) and DNA methylation levels (beta values from 450 000 Illumina arrays), were downloaded from the NCI’s Genomic Data Commons using TCGAbiolinks R package (version 2.17.4). DNA methylation and gene expression data for the ROSMAP project can be accessed from AMP-AD Knowledge Portal with accession numbers syn3157275 and syn3388564.
Supplementary Material
Contributor Information
Tiago C Silva, Department of Public Health Sciences, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
Juan I Young, Dr. John T. Macdonald Foundation Department of Human Genetics, University of Miami Miller School of Medicine, Miami, FL 33136, USA; John P. Hussman Institute for Human Genomics, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
Eden R Martin, Dr. John T. Macdonald Foundation Department of Human Genetics, University of Miami Miller School of Medicine, Miami, FL 33136, USA; John P. Hussman Institute for Human Genomics, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
X Steven Chen, Department of Public Health Sciences, University of Miami Miller School of Medicine, Miami, FL 33136, USA; Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
Lily Wang, Department of Public Health Sciences, University of Miami Miller School of Medicine, Miami, FL 33136, USA; Dr. John T. Macdonald Foundation Department of Human Genetics, University of Miami Miller School of Medicine, Miami, FL 33136, USA; John P. Hussman Institute for Human Genomics, University of Miami Miller School of Medicine, Miami, FL 33136, USA; Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [R21AG060459, R01AG061127 and R01AG062634 to L.W.; R01AG062634 to E.R.M; and R01CA158472 and R01CA200987 to X.S.C.]. The ROSMAP study data were collected by the Rush Alzheimer’s Disease Center, Rush University Medical Center, Chicago, IL, and supported by National Institute on Aging [P30AG10161, R01AG15819, R01AG17917, R01AG30146, R01AG36836, U01AG32984 and U01AG46152], Illinois Department of Public Health, and Translational Genomics Research Institute. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Irizarry R.A., Ladd-Acosta C., Wen B., Wu Z., Montano C., Onyango P., Cui H., Gabo K., Rongione M., Webster M.et al.. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009; 41:178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. The Cancer Genome Atlas Network Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. McInnes T., Zou D., Rao D.S., Munro F.M., Phillips V.L., McCall J.L., Black M.A., Reeve A.E., Guilford P.J.. Genome-wide methylation analysis identifies a core set of hypermethylated genes in CIMP-H colorectal cancer. BMC Cancer. 2017; 17:228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kirby M.K., Ramaker R.C., Roberts B.S., Lasseigne B.N., Gunther D.S., Burwell T.C., Davis N.S., Gulzar Z.G., Absher D.M., Cooper S.J.et al.. Genome-wide DNA methylation measurements in prostate tissues uncovers novel prostate cancer diagnostic biomarkers and transcription factor binding patterns. BMC Cancer. 2017; 17:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuang Y., Wang Y., Zhai W., Wang X., Zhang B., Xu M., Guo S., Ke M., Jia B., Liu H.. Genome-wide analysis of methylation-driven genes and identification of an eight-gene panel for prognosis prediction in breast cancer. Front. Genet. 2020; 11:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Jager P.L., Srivastava G., Lunnon K., Burgess J., Schalkwyk L.C., Yu L., Eaton M.L., Keenan B.T., Ernst J., McCabe C.et al.. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014; 17:1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Young J.I., Sivasankaran S.K., Wang L., Ali A., Mehta A., Davis D.A., Dykxhoorn D.M., Petito C.K., Beecham G.W., Martin E.R.et al.. Genome-wide brain DNA methylation analysis suggests epigenetic reprogramming in Parkinson disease. Neurol. Genet. 2019; 5:e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tarr I.S., McCann E.P., Benyamin B., Peters T.J., Twine N.A., Zhang K.Y., Zhao Q., Zhang Z.H., Rowe D.B., Nicholson G.A.et al.. Monozygotic twins and triplets discordant for amyotrophic lateral sclerosis display differential methylation and gene expression. Sci. Rep. 2019; 9:8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aran D., Sabato S., Hellman A.. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013; 14:R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Heyn H., Vidal E., Ferreira H.J., Vizoso M., Sayols S., Gomez A., Moran S., Boque-Sastre R., Guil S., Martinez-Cardus A.et al.. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol. 2016; 17:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu H., Wang G., Qian J.. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016; 17:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bonder M.J., Luijk R., Zhernakova D.V., Moed M., Deelen P., Vermaat M., van Iterson M., van Dijk F., van Galen M., Bot J.et al.. Disease variants alter transcription factor levels and methylation of their binding sites. Nat. Genet. 2017; 49:131–138. [DOI] [PubMed] [Google Scholar]
- 13. Banovich N.E., Lan X., McVicker G., van de Geijn B., Degner J.F., Blischak J.D., Roux J., Pritchard J.K., Gilad Y.. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 2014; 10:e1004663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hu S., Wan J., Su Y., Song Q., Zeng Y., Nguyen H.N., Shin J., Cox E., Rho H.S., Woodard C.et al.. DNA methylation presents distinct binding sites for human transcription factors. eLife. 2013; 2:e00726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lioznova A.V., Khamis A.M., Artemov A.V., Besedina E., Ramensky V., Bajic V.B., Kulakovskiy I.V., Medvedeva Y.A.. CpG traffic lights are markers of regulatory regions in human genome. BMC Genomics. 2019; 20:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yin Y., Morgunova E., Jolma A., Kaasinen E., Sahu B., Khund-Sayeed S., Das P.K., Kivioja T., Dave K., Zhong F.et al.. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017; 356:eaaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cedoz P.L., Prunello M., Brennan K., Gevaert O.. MethylMix 2.0: an R package for identifying DNA methylation genes. Bioinformatics. 2018; 34:3044–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Warden C.D., Lee H., Tompkins J.D., Li X., Wang C., Riggs A.D., Yu H., Jove R., Yuan Y.C.. COHCAP: an integrative genomic pipeline for single-nucleotide resolution DNA methylation analysis. Nucleic Acids Res. 2019; 47:8335–8336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saghafinia S., Mina M., Riggi N., Hanahan D., Ciriello G.. Pan-cancer landscape of aberrant DNA methylation across human tumors. Cell Rep. 2018; 25:1066–1080. [DOI] [PubMed] [Google Scholar]
- 20. Klett H., Balavarca Y., Toth R., Gigic B., Habermann N., Scherer D., Schrotz-King P., Ulrich A., Schirmacher P., Herpel E.et al.. Robust prediction of gene regulation in colorectal cancer tissues from DNA methylation profiles. Epigenetics. 2018; 13:386–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sheffield N.C., Bock C.. LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics. 2016; 32:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhasin J.M., Ting A.H.. Goldmine integrates information placing genomic ranges into meaningful biological contexts. Nucleic Acids Res. 2016; 44:5550–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lawson J.T., Tomazou E.M., Bock C., Sheffield N.C.. MIRA: an R package for DNA methylation-based inference of regulatory activity. Bioinformatics. 2018; 34:2649–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li X.Y., MacArthur S., Bourgon R., Nix D., Pollard D.A., Iyer V.N., Hechmer A., Simirenko L., Stapleton M., Luengo Hendriks C.L.et al.. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol. 2008; 6:e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fisher W.W., Li J.J., Hammonds A.S., Brown J.B., Pfeiffer B.D., Weiszmann R., MacArthur S., Thomas S., Stamatoyannopoulos J.A., Eisen M.B.et al.. DNA regions bound at low occupancy by transcription factors do not drive patterned reporter gene expression in Drosophila. Proc. Natl Acad. Sci. U.S.A. 2012; 109:21330–21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Silva T.C., Coetzee S.G., Gull N., Yao L., Hazelett D.J., Noushmehr H., Lin D.C., Berman B.P.. ELMER v.2: an R/Bioconductor package to reconstruct gene regulatory networks from DNA methylation and transcriptome profiles. Bioinformatics. 2019; 35:1974–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yao L., Shen H., Laird P.W., Farnham P.J., Berman B.P.. Inferring regulatory element landscapes and transcription factor networks from cancer methylomes. Genome Biol. 2015; 16:105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheneby J., Menetrier Z., Mestdagh M., Rosnet T., Douida A., Rhalloussi W., Bergon A., Lopez F., Ballester B.. ReMap 2020: a database of regulatory regions from an integrative analysis of human and Arabidopsis DNA-binding sequencing experiments. Nucleic Acids Res. 2020; 48:D180–D188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranašić D.et al.. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020; 48:D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 172:650–665. [DOI] [PubMed] [Google Scholar]
- 31. Garcia-Alonso L., Holland C.H., Ibrahim M.M., Turei D., Saez-Rodriguez J.. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019; 29:1363–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mei S., Meyer C.A., Zheng R., Qin Q., Wu Q., Jiang P., Li B., Shi X., Wang B., Fan J.et al.. Cistrome Cancer: a web resource for integrative gene regulation modeling in cancer. Cancer Res. 2017; 77:e19–e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Venables W.N., Ripley B.D.. Modern Applied Statistics with S. 2002; 4th edn.NY: Springer. [Google Scholar]
- 34. Alvarez M.J., Shen Y., Giorgi F.M., Lachmann A., Ding B.B., Ye B.H., Califano A.. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016; 48:838–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hanzelmann S., Castelo R., Guinney J.. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013; 14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Qin Q., Fan J., Zheng R., Wan C., Mei S., Wu Q., Sun H., Brown M., Zhang J., Meyer C.A.et al.. Lisa: inferring transcriptional regulators through integrative modeling of public chromatin accessibility and ChIP-seq data. Genome Biol. 2020; 21:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Benjamini Y., Hochberg Y.. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B. 1995; 57:289–300. [Google Scholar]
- 38. Van den Berge K., Soneson C., Robinson M.D., Clement L.. stageR: a general stage-wise method for controlling the gene-level false discovery rate in differential expression and differential transcript usage. Genome Biol. 2017; 18:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Efron B. Correlated z-values and the accuracy of large-scale statistical estimates. J. Am. Stat. Assoc. 2010; 105:1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shen Z., Chen Y., Li L., Liu L., Peng M., Chen X., Wu X., Sferra T.J., Wu M., Lin X.et al.. Transcription factor EBF1 over-expression suppresses tumor growth in vivo and in vitro via modulation of the PNO1/p53 pathway in colorectal cancer. Front. Oncol. 2020; 10:1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marley A.R., Nan H.. Epidemiology of colorectal cancer. Int. J. Mol. Epidemiol. Genet. 2016; 7:105–114. [PMC free article] [PubMed] [Google Scholar]
- 42. Berman B.P., Weisenberger D.J., Aman J.F., Hinoue T., Ramjan Z., Liu Y., Noushmehr H., Lange C.P., van Dijk C.M., Tollenaar R.A.et al.. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2011; 44:40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ehrlich M. DNA hypomethylation in cancer cells. Epigenomics. 2009; 1:239–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lee T.I., Young R.A.. Transcriptional regulation and its misregulation in disease. Cell. 2013; 152:1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ell B., Kang Y.. Transcriptional control of cancer metastasis. Trends Cell Biol. 2013; 23:603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Puisieux A., Brabletz T., Caramel J.. Oncogenic roles of EMT-inducing transcription factors. Nat. Cell Biol. 2014; 16:488–494. [DOI] [PubMed] [Google Scholar]
- 47. Aran D., Sirota M., Butte A.J.. Systematic pan-cancer analysis of tumour purity. Nat. Commun. 2015; 6:8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Daniel C., Gerlach K., Vath M., Neurath M.F., Weigmann B.. Nuclear factor of activated T cells—a transcription factor family as critical regulator in lung and colon cancer. Int. J. Cancer. 2014; 134:1767–1775. [DOI] [PubMed] [Google Scholar]
- 49. Tripathi M.K., Deane N.G., Zhu J., An H., Mima S., Wang X., Padmanabhan S., Shi Z., Prodduturi N., Ciombor K.K.et al.. Nuclear factor of activated T-cell activity is associated with metastatic capacity in colon cancer. Cancer Res. 2014; 74:6947–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gerlach K., Daniel C., Lehr H.A., Nikolaev A., Gerlach T., Atreya R., Rose-John S., Neurath M.F., Weigmann B.. Transcription factor NFATc2 controls the emergence of colon cancer associated with IL-6-dependent colitis. Cancer Res. 2012; 72:4340–4350. [DOI] [PubMed] [Google Scholar]
- 51. Lang T., Ding X., Kong L., Zhou X., Zhang Z., Ju H., Ding S.. NFATC2 is a novel therapeutic target for colorectal cancer stem cells. OncoTargets Ther. 2018; 11:6911–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A.et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 53. Begik O., Lucas M.C., Liu H., Ramirez J.M., Mattick J.S., Novoa E.M.. Integrative analyses of the RNA modification machinery reveal tissue- and cancer-specific signatures. Genome Biol. 2020; 21:97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhou Y., Huang T., Cheng A.S., Yu J., Kang W., To K.F.. The TEAD family and its oncogenic role in promoting tumorigenesis. Int. J. Mol. Sci. 2016; 17:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liu Y., Wang G., Yang Y., Mei Z., Liang Z., Cui A., Wu T., Liu C.Y., Cui L.. Increased TEAD4 expression and nuclear localization in colorectal cancer promote epithelial–mesenchymal transition and metastasis in a YAP-independent manner. Oncogene. 2016; 35:2789–2800. [DOI] [PubMed] [Google Scholar]
- 56. Jang B.G., Kim H.S., Bae J.M., Kim W.H., Kim H.U., Kang G.H.. SMOC2, an intestinal stem cell marker, is an independent prognostic marker associated with better survival in colorectal cancers. Sci. Rep. 2020; 10:14591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Purkayastha B.P., Roy J.K.. Cancer cell metabolism and developmental homeodomain/POU domain transcription factors: a connecting link. Cancer Lett. 2015; 356:315–319. [DOI] [PubMed] [Google Scholar]
- 58. Dunne J., Gascoyne D.M., Lister T.A., Brady H.J., Heidenreich O., Young B.D.. AML1/ETO proteins control POU4F1/BRN3A expression and function in t(8;21) acute myeloid leukemia. Cancer Res. 2010; 70:3985–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Diss J.K., Faulkes D.J., Walker M.M., Patel A., Foster C.S., Budhram-Mahadeo V., Djamgoz M.B., Latchman D.S.. Brn-3a neuronal transcription factor functional expression in human prostate cancer. Prostate Cancer Prostatic Dis. 2006; 9:83–91. [DOI] [PubMed] [Google Scholar]
- 60. Leblond-Francillard M., Picon A., Bertagna X., de Keyzer Y.. High expression of the POU factor Brn3a in aggressive neuroendocrine tumors. J. Clin. Endocrinol. Metab. 1997; 82:89–94. [DOI] [PubMed] [Google Scholar]
- 61. Jin T., Branch D.R., Zhang X., Qi S., Youngson B., Goss P.E.. Examination of POU homeobox gene expression in human breast cancer cells. Int. J. Cancer. 1999; 81:104–112. [DOI] [PubMed] [Google Scholar]
- 62. Gao Y., Li Y., Niu X., Wu Y., Guan X., Hong Y., Chen H., Song B.. Identification and validation of prognostically relevant gene signature in melanoma. Biomed. Res. Int. 2020; 2020:5323614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nalesnik M.A., Tseng G., Ding Y., Xiang G.S., Zheng Z.L., Yu Y., Marsh J.W., Michalopoulos G.K., Luo J.H.. Gene deletions and amplifications in human hepatocellular carcinomas: correlation with hepatocyte growth regulation. Am. J. Pathol. 2012; 180:1495–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Brzozowa M., Michalski M., Wyrobiec G., Piecuch A., Dittfeld A., Harabin-Slowinska M., Boron D., Wojnicz R.. The role of Snail1 transcription factor in colorectal cancer progression and metastasis. Contemp. Oncol. (Pozn.). 2015; 19:265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhou B.P., Deng J., Xia W., Xu J., Li Y.M., Gunduz M., Hung M.C.. Dual regulation of snail by GSK-3beta-mediated phosphorylation in control of epithelial–mesenchymal transition. Nat. Cell Biol. 2004; 6:931–940. [DOI] [PubMed] [Google Scholar]
- 66. Huang X., Xiang L., Li Y., Zhao Y., Zhu H., Xiao Y., Liu M., Wu X., Wang Z., Jiang P.et al.. Snail/FOXK1/Cyr61 signaling axis regulates the epithelial–mesenchymal transition and metastasis in colorectal cancer. Cell. Physiol. Biochem. 2018; 47:590–603. [DOI] [PubMed] [Google Scholar]
- 67. Kroepil F., Fluegen G., Vallbohmer D., Baldus S.E., Dizdar L., Raffel A.M., Hafner D., Stoecklein N.H., Knoefel W.T.. Snail1 expression in colorectal cancer and its correlation with clinical and pathological parameters. BMC Cancer. 2013; 13:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Crist R.C., Roth J.J., Waldman S.A., Buchberg A.M.. A conserved tissue-specific homeodomain-less isoform of MEIS1 is downregulated in colorectal cancer. PLoS One. 2011; 6:e23665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tufan T., Yang J., Tummala K.S., Cingoz H., Kuscu C., Adair S.J., Comertpay G., Nagdas S., Goudreau B.J., Luleyap H.U.et al.. ISL2 is an epigenetically silenced tumor suppressor and regulator of metabolism in pancreatic cancer. 2020; bioRXiv doi:26 May 2020, preprint: not peer reviewed 10.1101/2020.05.23.112839. [DOI]
- 70. Oh S., Shin S., Janknecht R.. ETV1, 4 and 5: an oncogenic subfamily of ETS transcription factors. Biochim. Biophys. Acta. 2012; 1826:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Horiuchi S., Yamamoto H., Min Y., Adachi Y., Itoh F., Imai K.. Association of Ets-related transcriptional factor E1AF expression with tumour progression and overexpression of MMP-1 and matrilysin in human colorectal cancer. J. Pathol. 2003; 200:568–576. [DOI] [PubMed] [Google Scholar]
- 72. Boedefeld W.M. 2nd, Soong R., Weiss H., Diasio R.B., Urist M.M., Bland K.I., Heslin M.J.. E1A-F is overexpressed early in human colorectal neoplasia and associated with cyclooxygenase-2 and matrix metalloproteinase-7. Mol. Carcinog. 2005; 43:13–17. [DOI] [PubMed] [Google Scholar]
- 73. Nosho K., Yoshida M., Yamamoto H., Taniguchi H., Adachi Y., Mikami M., Hinoda Y., Imai K.. Association of Ets-related transcriptional factor E1AF expression with overexpression of matrix metalloproteinases, COX-2 and iNOS in the early stage of colorectal carcinogenesis. Carcinogenesis. 2005; 26:892–899. [DOI] [PubMed] [Google Scholar]
- 74. Moss A.C., Lawlor G., Murray D., Tighe D., Madden S.F., Mulligan A.M., Keane C.O., Brady H.R., Doran P.P., MacMathuna P.. ETV4 and Myeov knockdown impairs colon cancer cell line proliferation and invasion. Biochem. Biophys. Res. Commun. 2006; 345:216–221. [DOI] [PubMed] [Google Scholar]
- 75. Liu H.Y., Zhou B., Wang L., Li Y., Zhou Z.G., Sun X.F., Xu B., Zeng Y.J., Song J.M., Luo H.Z.et al.. Association of E1AF mRNA expression with tumor progression and matrilysin in human rectal cancer. Oncology. 2007; 73:384–388. [DOI] [PubMed] [Google Scholar]
- 76. Jung Y., Lee S., Choi H.S., Kim S.N., Lee E., Shin Y., Seo J., Kim B., Jung Y., Kim W.K.et al.. Clinical validation of colorectal cancer biomarkers identified from bioinformatics analysis of public expression data. Clin. Cancer Res. 2011; 17:700–709. [DOI] [PubMed] [Google Scholar]
- 77. Deves C., Renck D., Garicochea B., da Silva V.D., Giulianni Lopes T., Fillman H., Fillman L., Lunardini S., Basso L.A., Santos D.S.et al.. Analysis of select members of the E26 (ETS) transcription factors family in colorectal cancer. Virchows Arch. 2011; 458:421–430. [DOI] [PubMed] [Google Scholar]
- 78. McClure B.J., Heatley S.L., Kok C.H., Sadras T., An J., Hughes T.P., Lock R.B., Yeung D., Sutton R., White D.L.. Pre-B acute lymphoblastic leukaemia recurrent fusion, EP300–ZNF384, is associated with a distinct gene expression. Br. J. Cancer. 2018; 118:1000–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yao L., Cen J., Pan J., Liu D., Wang Y., Chen Z., Ruan C., Chen S.. TAF15–ZNF384 fusion gene in childhood mixed phenotype acute leukemia. Cancer Genet. 2017; 211:1–4. [DOI] [PubMed] [Google Scholar]
- 80. He L., Fan X., Li Y., Chen M., Cui B., Chen G., Dai Y., Zhou D., Hu X., Lin H.. Overexpression of zinc finger protein 384 (ZNF 384), a poor prognostic predictor, promotes cell growth by upregulating the expression of cyclin D1 in hepatocellular carcinoma. Cell Death Dis. 2019; 10:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lo Sasso G., Bovenga F., Murzilli S., Salvatore L., Di Tullio G., Martelli N., D’Orazio A., Rainaldi S., Vacca M., Mangia A.et al.. Liver X receptors inhibit proliferation of human colorectal cancer cells and growth of intestinal tumors in mice. Gastroenterology. 2013; 144:1497–1507. [DOI] [PubMed] [Google Scholar]
- 82. Perreault N., Katz J.P., Sackett S.D., Kaestner K.H.. Foxl1 controls the Wnt/beta-catenin pathway by modulating the expression of proteoglycans in the gut. J. Biol. Chem. 2001; 276:43328–43333. [DOI] [PubMed] [Google Scholar]
- 83. Perreault N., Sackett S.D., Katz J.P., Furth E.E., Kaestner K.H.. Foxl1 is a mesenchymal modifier of Min in carcinogenesis of stomach and colon. Genes Dev. 2005; 19:311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kaestner K.H. The intestinal stem cell niche: a central role for Foxl1-expressing subepithelial telocytes. Cell. Mol. Gastroenterol. Hepatol. 2019; 8:111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Naxerova K., Bult C.J., Peaston A., Fancher K., Knowles B.B., Kasif S., Kohane I.S.. Analysis of gene expression in a developmental context emphasizes distinct biological leitmotifs in human cancers. Genome Biol. 2008; 9:R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Betge J., Schneider N.I., Harbaum L., Pollheimer M.J., Lindtner R.A., Kornprat P., Ebert M.P., Langner C.. MUC1, MUC2, MUC5AC, and MUC6 in colorectal cancer: expression profiles and clinical significance. Virchows Arch. 2016; 469:255–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Shvab A., Haase G., Ben-Shmuel A., Gavert N., Brabletz T., Dedhar S., Ben-Ze’ev A.. Induction of the intestinal stem cell signature gene SMOC-2 is required for L1-mediated colon cancer progression. Oncogene. 2016; 35:549–557. [DOI] [PubMed] [Google Scholar]
- 88. Traicoff J.L., De Marchis L., Ginsburg B.L., Zamora R.E., Khattar N.H., Blanch V.J., Plummer S., Bargo S.A., Templeton D.J., Casey G.et al.. Characterization of the human polymeric immunoglobulin receptor (PIGR) 3′UTR and differential expression of PIGR mRNA during colon tumorigenesis. J. Biomed. Sci. 2003; 10:792–804. [DOI] [PubMed] [Google Scholar]
- 89. Wang L., Ai M., Nie M., Zhao L., Deng G., Hu S., Han Y., Zeng W., Wang Y., Yang M.et al.. EHF promotes colorectal carcinoma progression by activating TGF-beta1 transcription and canonical TGF-beta signaling. Cancer Sci. 2020; 111:2310–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Gimeno-Valiente F., Riffo-Campos A.L., Vallet-Sanchez A., Siscar-Lewin S., Gambardella V., Tarazona N., Cervantes A., Franco L., Castillo J., Lopez-Rodas G.. ZNF518B gene up-regulation promotes dissemination of tumour cells and is governed by epigenetic mechanisms in colorectal cancer. Sci. Rep. 2019; 9:9339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Kim S.H., Park Y.Y., Cho S.N., Margalit O., Wang D., DuBois R.N.. Kruppel-like factor 12 promotes colorectal cancer growth through early growth response protein 1. PLoS One. 2016; 11:e0159899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhang X., Xu J., Zhang H., Sun J., Li N., Huang X.. MicroRNA-758 acts as a tumor inhibitor in colorectal cancer through targeting PAX6 and regulating PI3K/AKT pathway. Oncol. Lett. 2020; 19:3923–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Janecki D.M., Sajek M., Smialek M.J., Kotecki M., Ginter-Matuszewska B., Kuczynska B., Spik A., Kolanowski T., Kitazawa R., Kurpisz M.et al.. SPIN1 is a proto-oncogene and SPIN3 is a tumor suppressor in human seminoma. Oncotarget. 2018; 9:32466–32477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wang Z., Yin J., Zhou W., Bai J., Xie Y., Xu K., Zheng X., Xiao J., Zhou L., Qi X.et al.. Complex impact of DNA methylation on transcriptional dysregulation across 22 human cancer types. Nucleic Acids Res. 2020; 48:2287–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Liu Y., Liu Y., Huang R., Song W., Wang J., Xiao Z., Dong S., Yang Y., Yang X.. Dependency of the cancer-specific transcriptional regulation circuitry on the promoter DNA methylome. Cell Rep. 2019; 26:3461–3474. [DOI] [PubMed] [Google Scholar]
- 96. Wang G., Luo X., Wang J., Wan J., Xia S., Zhu H., Qian J., Wang Y.. MeDReaders: a database for transcription factors that bind to methylated DNA. Nucleic Acids Res. 2018; 46:D146–D151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hornakova A., List M., Vreeken J., Schulz M.H.. JAMI: fast computation of conditional mutual information for ceRNA network analysis. Bioinformatics. 2018; 34:3050–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Bennett D.A., Buchman A.S., Boyle P.A., Barnes L.L., Wilson R.S., Schneider J.A.. Religious orders study and rush memory and aging project. J. Alzheimers Dis. 2018; 64:S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gasparoni G., Bultmann S., Lutsik P., Kraus T.F.J., Sordon S., Vlcek J., Dietinger V., Steinmaurer M., Haider M., Mulholland C.B.et al.. DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics Chromatin. 2018; 11:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lunnon K., Smith R., Hannon E., De Jager P.L., Srivastava G., Volta M., Troakes C., Al-Sarraj S., Burrage J., Macdonald R.et al.. Methylomic profiling implicates cortical deregulation of ANK1 in Alzheimer’s disease. Nat. Neurosci. 2014; 17:1164–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Smith R.G., Hannon E., De Jager P.L., Chibnik L., Lott S.J., Condliffe D., Smith A.R., Haroutunian V., Troakes C., Al-Sarraj S.et al.. Elevated DNA methylation across a 48-kb region spanning the HOXA gene cluster is associated with Alzheimer’s disease neuropathology. Alzheimers Dement. 2018; 14:1580–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhang L., Silva T.C., Young J.I., Gomez L., Schmidt M.A., Hamilton-Nelson K.L., Kunkle B.W., Chen X., Martin E.R., Wang L.. Epigenome-wide meta-analysis of DNA methylation differences in prefrontal cortex implicates the immune processes in Alzheimer’s disease. Nat. Commun. 2020; 11:6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Keenan A.B., Torre D., Lachmann A., Leong A.K., Wojciechowicz M.L., Utti V., Jagodnik K.M., Kropiwnicki E., Wang Z., Ma’ayan A.. ChEA3: transcription factor enrichment analysis by orthogonal omics integration. Nucleic Acids Res. 2019; 47:W212–W224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Huang K.L., Marcora E., Pimenova A.A., Di Narzo A.F., Kapoor M., Jin S.C., Harari O., Bertelsen S., Fairfax B.P., Czajkowski J.et al.. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat. Neurosci. 2017; 20:1052–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Rustenhoven J., Smith A.M., Smyth L.C., Jansson D., Scotter E.L., Swanson M.E.V., Aalderink M., Coppieters N., Narayan P., Handley R.et al.. PU.1 regulates Alzheimer’s disease-associated genes in primary human microglia. Mol. Neurodegener. 2018; 13:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Smith A.M., Gibbons H.M., Oldfield R.L., Bergin P.M., Mee E.W., Faull R.L., Dragunow M.. The transcription factor PU.1 is critical for viability and function of human brain microglia. Glia. 2013; 61:929–942. [DOI] [PubMed] [Google Scholar]
- 107. Cunningham C. Microglia and neurodegeneration: the role of systemic inflammation. Glia. 2013; 61:71–90. [DOI] [PubMed] [Google Scholar]
- 108. Kunkle B.W., Grenier-Boley B., Sims R., Bis J.C., Damotte V., Naj A.C., Boland A., Vronskaya M., van der Lee S.J., Amlie-Wolf A.et al.. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 2019; 51:414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Salih D.A., Bayram S., Guelfi S., Reynolds R.H., Shoai M., Ryten M., Brenton J.W., Zhang D., Matarin M., Botia J.A.et al.. Genetic variability in response to amyloid beta deposition influences Alzheimer’s disease risk. Brain Commun. 2019; 1:fcz022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Zhang B., Gaiteri C., Bodea L.G., Wang Z., McElwee J., Podtelezhnikov A.A., Zhang C., Xie T., Tran L., Dobrin R.et al.. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013; 153:707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Forabosco P., Ramasamy A., Trabzuni D., Walker R., Smith C., Bras J., Levine A.P., Hardy J., Pocock J.M., Guerreiro R.et al.. Insights into TREM2 biology by network analysis of human brain gene expression data. Neurobiol. Aging. 2013; 34:2699–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Satoh J., Asahina N., Kitano S., Kino Y.. A comprehensive profile of ChIP-seq-based PU.1/Spi1 target genes in microglia. Gene Regul. Syst. Biol. 2014; 8:127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Landfield P.W., Blalock E.M., Chen K.C., Porter N.M.. A new glucocorticoid hypothesis of brain aging: implications for Alzheimer’s disease. Curr. Alzheimer Res. 2007; 4:205–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Canet G., Chevallier N., Zussy C., Desrumaux C., Givalois L.. Central role of glucocorticoid receptors in Alzheimer’s disease and depression. Front. Neurosci. 2018; 12:739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Dharshini S.A.P., Taguchi Y.H., Gromiha M.M.. Investigating the energy crisis in Alzheimer disease using transcriptome study. Sci. Rep. 2019; 9:18509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wetzel D.M., Bohn M.C., Kazee A.M., Hamill R.W.. Glucocorticoid receptor mRNA in Alzheimer’s diseased hippocampus. Brain Res. 1995; 679:72–81. [DOI] [PubMed] [Google Scholar]
- 117. Jha M.K., Jeon S., Suk K.. Pyruvate dehydrogenase kinases in the nervous system: their principal functions in neuronal–glial metabolic interaction and neuro-metabolic disorders. Curr. Neuropharmacol. 2012; 10:393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Vaughn A.E., Deshmukh M.. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 2008; 10:1477–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Piquet J., Toussay X., Hepp R., Lerchundi R., Le Douce J., Faivre E., Guiot E., Bonvento G., Cauli B.. Supragranular pyramidal cells exhibit early metabolic alterations in the 3xTg-AD mouse model of Alzheimer’s disease. Front. Cell. Neurosci. 2018; 12:216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Mosconi L., Mistur R., Switalski R., Brys M., Glodzik L., Rich K., Pirraglia E., Tsui W., De Santi S., de Leon M.J.. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology. 2009; 72:513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Mosconi L., Mistur R., Switalski R., Tsui W.H., Glodzik L., Li Y., Pirraglia E., De Santi S., Reisberg B., Wisniewski T.et al.. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging. 2009; 36:811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Reiman E.M., Chen K., Alexander G.E., Caselli R.J., Bandy D., Osborne D., Saunders A.M., Hardy J.. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl Acad. Sci. U.S.A. 2004; 101:284–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Huang B., Wu P., Bowker-Kinley M.M., Harris R.A.. Regulation of pyruvate dehydrogenase kinase expression by peroxisome proliferator-activated receptor-alpha ligands, glucocorticoids, and insulin. Diabetes. 2002; 51:276–283. [DOI] [PubMed] [Google Scholar]
- 124. Olsen L., Rasmussen H.B., Hansen T., Bagger Y.Z., Tanko L.B., Qin G., Christiansen C., Werge T.. Estrogen receptor alpha and risk for cognitive impairment in postmenopausal women. Psychiatr. Genet. 2006; 16:85–88. [DOI] [PubMed] [Google Scholar]
- 125. Yaffe K., Lui L.Y., Grady D., Stone K., Morin P.. Estrogen receptor 1 polymorphisms and risk of cognitive impairment in older women. Biol. Psychiatry. 2002; 51:677–682. [DOI] [PubMed] [Google Scholar]
- 126. Boada M., Antunez C., Lopez-Arrieta J., Caruz A., Moreno-Rey C., Ramirez-Lorca R., Moron F.J., Hernandez I., Mauleon A., Rosende-Roca M.et al.. Estrogen receptor alpha gene variants are associated with Alzheimer’s disease. Neurobiol. Aging. 2012; 33:198. [DOI] [PubMed] [Google Scholar]
- 127. Janicki S.C., Schupf N.. Hormonal influences on cognition and risk for Alzheimer’s disease. Curr. Neurol. Neurosci. Rep. 2010; 10:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Yaffe K., Lindquist K., Sen S., Cauley J., Ferrell R., Penninx B., Harris T., Li R., Cummings S.R.. Estrogen receptor genotype and risk of cognitive impairment in elders: findings from the health ABC study. Neurobiol. Aging. 2009; 30:607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Corbo R.M., Gambina G., Ruggeri M., Scacchi R.. Association of estrogen receptor alpha (ESR1) PvuII and XbaI polymorphisms with sporadic Alzheimer’s disease and their effect on apolipoprotein E concentrations. Dement. Geriatr. Cogn. Disord. 2006; 22:67–72. [DOI] [PubMed] [Google Scholar]
- 130. Ko C.Y., Wang W.L., Wang S.M., Chu Y.Y., Chang W.C., Wang J.M.. Glycogen synthase kinase-3beta-mediated CCAAT/enhancer-binding protein delta phosphorylation in astrocytes promotes migration and activation of microglia/macrophages. Neurobiol. Aging. 2014; 35:24–34. [DOI] [PubMed] [Google Scholar]
- 131. Ko C.Y., Chang W.C., Wang J.M.. Biological roles of CCAAT/enhancer-binding protein delta during inflammation. J. Biomed. Sci. 2015; 22:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Karch C.M., Ezerskiy L.A., Bertelsen S.Alzheimer’s Disease Genetics Consortium Alzheimer’s Disease Genetics Consortium Goate A.M.. Alzheimer’s disease risk polymorphisms regulate gene expression in the ZCWPW1 and the CELF1 loci. PLoS One. 2016; 11:e0148717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Lambert J.C., Ibrahim-Verbaas C.A., Harold D., Naj A.C., Sims R., Bellenguez C., DeStafano A.L., Bis J.C., Beecham G.W., Grenier-Boley B.et al.. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013; 45:1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Readhead B., Haure-Mirande J.V., Funk C.C., Richards M.A., Shannon P., Haroutunian V., Sano M., Liang W.S., Beckmann N.D., Price N.D.et al.. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron. 2018; 99:64–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Bell R.D., Deane R., Chow N., Long X., Sagare A., Singh I., Streb J.W., Guo H., Rubio A., Van Nostrand W.et al.. SRF and myocardin regulate LRP-mediated amyloid-beta clearance in brain vascular cells. Nat. Cell Biol. 2009; 11:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Dotti C.G., De Strooper B.. Alzheimer’s dementia by circulation disorders: when trees hide the forest. Nat. Cell Biol. 2009; 11:114–116. [DOI] [PubMed] [Google Scholar]
- 137. Perdomo-Sabogal A., Nowick K., Piccini I., Sudbrak R., Lehrach H., Yaspo M.L., Warnatz H.J., Querfurth R.. Human lineage-specific transcriptional regulation through GA-binding protein transcription factor alpha (GABPa). Mol. Biol. Evol. 2016; 33:1231–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Bahn G., Park J.S., Yun U.J., Lee Y.J., Choi Y., Park J.S., Baek S.H., Choi B.Y., Cho Y.S., Kim H.K.et al.. NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc. Natl Acad. Sci. U.S.A. 2019; 116:12516–12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Ren P., Chen J., Li B., Zhang M., Yang B., Guo X., Chen Z., Cheng H., Wang P., Wang S.et al.. Nrf2 ablation promotes Alzheimer’s disease-like pathology in APP/PS1 transgenic mice: the role of neuroinflammation and oxidative stress. Oxid. Med. Cell. Longev. 2020; 2020:3050971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Pajares M., Jimenez-Moreno N., Garcia-Yague A.J., Escoll M., de Ceballos M.L., Van Leuven F., Rabano A., Yamamoto M., Rojo A.I., Cuadrado A.. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy. 2016; 12:1902–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Welch R.P., Lee C., Imbriano P.M., Patil S., Weymouth T.E., Smith R.A., Scott L.J., Sartor M.A.. ChIP-Enrich: gene set enrichment testing for ChIP-seq data. Nucleic Acids Res. 2014; 42:e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Mukherjee S., Erickson H., Bastia D. Enhancer–origin interaction in plasmid R6K involves a DNA loop mediated by initiator protein. Cell. 1988; 52:375–383. [DOI] [PubMed] [Google Scholar]
- 143. Sur I., Taipale J.. The role of enhancers in cancer. Nat. Rev. Cancer. 2016; 16:483–493. [DOI] [PubMed] [Google Scholar]
- 144. Schubeler D. Function and information content of DNA methylation. Nature. 2015; 517:321–326. [DOI] [PubMed] [Google Scholar]
- 145. Stadler M.B., Murr R., Burger L., Ivanek R., Lienert F., Scholer A., van Nimwegen E., Wirbelauer C., Oakeley E.J., Gaidatzis D.et al.. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011; 480:490–495. [DOI] [PubMed] [Google Scholar]
- 146. Song L., Zhang Z., Grasfeder L.L., Boyle A.P., Giresi P.G., Lee B.K., Sheffield N.C., Graf S., Huss M., Keefe D.et al.. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011; 21:1757–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Thurman R.E., Rynes E., Humbert R., Vierstra J., Maurano M.T., Haugen E., Sheffield N.C., Stergachis A.B., Wang H., Vernot B.et al.. The accessible chromatin landscape of the human genome. Nature. 2012; 489:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Blattler A., Farnham P.J.. Cross-talk between site-specific transcription factors and DNA methylation states. J. Biol. Chem. 2013; 288:34287–34294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Shakya A., Callister C., Goren A., Yosef N., Garg N., Khoddami V., Nix D., Regev A., Tantin D. Pluripotency transcription factor Oct4 mediates stepwise nucleosome demethylation and depletion. Mol. Cell. Biol. 2015; 35:1014–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Encode Project Consortium Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020; 583:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Moore J.E., Pratt H.E., Purcaro M.J., Weng Z.. A curated benchmark of enhancer–gene interactions for evaluating enhancer–target gene prediction methods. Genome Biol. 2020; 21:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Whalen S., Truty R.M., Pollard K.S.. Enhancer–promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet. 2016; 48:488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Gao T., He B., Liu S., Zhu H., Tan K., Qian J.. EnhancerAtlas: a resource for enhancer annotation and analysis in 105 human cell/tissue types. Bioinformatics. 2016; 32:3543–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Schmidl C., Klug M., Boeld T.J., Andreesen R., Hoffmann P., Edinger M., Rehli M.. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 2009; 19:1165–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Reddington J.P., Perricone S.M., Nestor C.E., Reichmann J., Youngson N.A., Suzuki M., Reinhardt D., Dunican D.S., Prendergast J.G., Mjoseng H.et al.. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of polycomb target genes. Genome Biol. 2013; 14:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Brinkman A.B., Gu H., Bartels S.J., Zhang Y., Matarese F., Simmer F., Marks H., Bock C., Gnirke A., Meissner A.et al.. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res. 2012; 22:1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Cera I., Whitton L., Donohoe G., Morris D.W., Dechant G., Apostolova G.. Genes encoding SATB2-interacting proteins in adult cerebral cortex contribute to human cognitive ability. PLoS Genet. 2019; 15:e1007890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Jaitner C., Reddy C., Abentung A., Whittle N., Rieder D., Delekate A., Korte M., Jain G., Fischer A., Sananbenesi F.et al.. Satb2 determines miRNA expression and long-term memory in the adult central nervous system. eLife. 2016; 5:e17361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Velasco-Estevez M., Mampay M., Boutin H., Chaney A., Warn P., Sharp A., Burgess E., Moeendarbary E., Dev K.K., Sheridan G.K.. Infection augments expression of mechanosensing Piezo1 channels in amyloid plaque-reactive astrocytes. Front. Aging Neurosci. 2018; 10:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Fehlbaum-Beurdeley P., Jarrige-Le Prado A.C., Pallares D., Carriere J., Guihal C., Soucaille C., Rouet F., Drouin D., Sol O., Jordan H.et al.. Toward an Alzheimer’s disease diagnosis via high-resolution blood gene expression. Alzheimers Dement. 2010; 6:25–38. [DOI] [PubMed] [Google Scholar]
- 161. Wirz K.T., Bossers K., Stargardt A., Kamphuis W., Swaab D.F., Hol E.M., Verhaagen J.. Cortical beta amyloid protein triggers an immune response, but no synaptic changes in the APPswe/PS1dE9 Alzheimer’s disease mouse model. Neurobiol. Aging. 2013; 34:1328–1342. [DOI] [PubMed] [Google Scholar]
- 162. Pelucchi S., Stringhi R., Marcello E.. Dendritic spines in Alzheimer’s disease: how the actin cytoskeleton contributes to synaptic failure. Int. J. Mol. Sci. 2020; 21:908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Arendt T., Bruckner M.K.. Linking cell-cycle dysfunction in Alzheimer’s disease to a failure of synaptic plasticity. Biochim. Biophys. Acta. 2007; 1772:413–421. [DOI] [PubMed] [Google Scholar]
- 164. Xu Z., Wu C., Pan W.Alzheimer’s Disease Neuroimaging Initiative . Imaging-wide association study: integrating imaging endophenotypes in GWAS. Neuroimage. 2017; 159:159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Bahn G., Jo D.G.. Therapeutic approaches to Alzheimer’s disease through modulation of NRF2. Neuromol. Med. 2019; 21:1–11. [DOI] [PubMed] [Google Scholar]
- 166. Wang R., Reddy P.H.. Role of glutamate and NMDA receptors in Alzheimer’s disease. J. Alzheimers Dis. 2017; 57:1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MethReg R package is available from the Bioconductor repository at https://bioconductor.org/packages/MethReg/. The scripts for the analysis performed in this study can be accessed at https://github.com/TransBioInfoLab/MethReg_supplemental. Level 3 TCGA COAD and READ cancer datasets, including genomic profiles in CNAs (gene-level copy number scores), gene expressions (HTSeq-FPKM-UQ values from RNA-seq) and DNA methylation levels (beta values from 450 000 Illumina arrays), were downloaded from the NCI’s Genomic Data Commons using TCGAbiolinks R package (version 2.17.4). DNA methylation and gene expression data for the ROSMAP project can be accessed from AMP-AD Knowledge Portal with accession numbers syn3157275 and syn3388564.