


Abstract
Telomeres, the ends of linear chromosomes, are composed of repetitive DNA sequences, histones and a protein complex called shelterin. How DNA is packaged at telomeres is an outstanding question in the field with significant implications for human health and disease. Here, we studied the architecture of telomeres and their spatial association with other chromatin domains in different cell types using correlative light and electron microscopy. To this end, the shelterin protein TRF1 or TRF2 was fused in tandem to eGFP and the peroxidase APEX2, which provided a selective and electron-dense label to interrogate telomere organization by transmission electron microscopy, electron tomography and scanning electron microscopy. Together, our work reveals, for the first time, ultrastructural insight into telomere architecture. We show that telomeres are composed of a dense and highly compacted mesh of chromatin fibres. In addition, we identify marked differences in telomere size, shape and chromatin compaction between cancer and non-cancer cells and show that telomeres are in direct contact with other heterochromatin regions. Our work resolves the internal architecture of telomeres with unprecedented resolution and advances our understanding of how telomeres are organized in situ.
Graphical Abstract
Graphical Abstract.
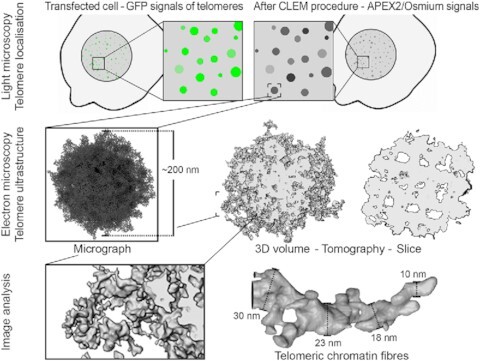
Graphical representation of a correlative light and electron microscopy workflow to visualize the ultrastructure of telomeric chromatin.
INTRODUCTION
Telomeres are large nucleoprotein complexes at the ends of linear chromosomes. Telomeres shorten with every cell division, which provides a means to regulate how many times a cell can divide. In somatic cells, telomere length maintenance is limited, while in stem cells and in ∼90% of human cancer cells telomere length and replicative potential are extended by the expression of the telomerase enzyme (1,2). Interestingly, 10% of human cancers do not express telomerase and instead use a mechanism called alternative lengthening of telomeres (ALT), which is mediated by homologous recombination (3,4). Telomeric chromatin differs from bulk chromatin in both sequence and protein composition. In mammalian cells, telomeric DNA contains thousands of double-stranded ‘TTAGGG’ repeats in tandem (5,6) followed by a short single-stranded G-overhang (7). Telomeric repeat-binding factors (TRF1 and TRF2) bind as homodimers to the double-stranded telomeric region (8,9), whereas POT1 binds to the G-overhang (10). Rap1, TPP1 and Tin2 interact with these DNA-binding factors and together form a six-protein complex called shelterin (11,12). Shelterin proteins have been shown to be critical in protecting telomeric DNA from being recognized as DNA breaks (13,14). For example, silencing of TRF2 expression leads to chromosome fusion and genome instability (15,16). In addition to shelterin, telomeres also contain nucleosomes (17,18) and RNA (TERRA) (19,20).
Structures of individual shelterin protein domains, such as the DNA-binding and dimerization domains of TRF1 and TRF2 (21,22), and a nucleosome core particle reconstituted with telomeric DNA (23) have been determined by X-ray crystallography and NMR (24,25). However, the structure of the entire shelterin complex as well as the architecture of higher order telomeric chromatin assemblies is unknown. Early electron microscopy (EM) studies of isolated telomeric DNA showed that up to 40% exhibited a lariat configuration (26,27). Based on this, a t-loop model was proposed whereby the DNA end would be protected by integration of the G-overhang into the double-stranded telomeric region (26). Although it is unclear how such a t-loop forms, both this early and a subsequent super-resolution light microscopy study (28) showed that TRF2 plays an important role in generating or stabilizing the structure. In addition, this and other super-resolution studies (28–30) demonstrated that telomeres within cells appear as compact globular foci with a diameter of ∼200 nm. However, due to the limited resolution of optical microscopy, chromatin fibres within the telomeres could not be resolved.
Here, we applied a correlative light and electron microscopy (CLEM) approach to study the structure of telomeres inside the nucleus at higher resolution. Telomeres were visualized by the expression of TRF1 or TRF2 fused in tandem to eGFP and the genetically encoded EM tag APEX2 (31,32). This allowed us to localize telomeres by confocal fluorescence microscopy and subsequently interrogate their ultrastructure and sub-nuclear organization by transmission electron microscopy (TEM), electron tomography and focused ion beam scanning electron microscopy (FIB-SEM). Using this approach, we resolve telomere ultrastructure in nuclei and demonstrate that telomeres in situ possess a fibrous and mesh-like ultrastructure. We further show that telomerase-positive and ALT cells exhibit marked differences in telomere size, shape and compaction state, and that telomeres are in direct contact with other heterochromatin domains.
MATERIALS AND METHODS
Cloning
APEX2 (31) (Addgene, Watertown, MA, USA, Cat#49385) was cloned into the Clontech eGFP-C1 vector (Clontech Laboratories/TaKaRa Bio, Mountain View, CA, USA; Cat#6084-1 of Addgene, Watertown, MA, USA) C-terminal of eGFP with a linker length of 5 aa (amino acids) between eGFP and APEX2. TRF1, TRF2 (short), TRF2 (long) (kindly provided by Daniela Rhodes; GeneBank NCBI reference sequences NM_017489.2, NM_005652.3 and NM_005652.4, respectively) or H2B (kindly provided by Gabriela Davey; NM_021058.3) was inserted C-terminal of APEX2 with linker lengths of 7 aa for TRF1, 10 aa for TRF2 short/long and 12 aa for H2B. Unless otherwise indicated, eGFP–APEX2–TRF2 long was used.
Cell culture
Immortalized mouse embryonic fibroblasts (MEFs) (kindly provided by Ben Nichols), U2OS cells (kindly provided by Siu Kwan Sze) and HT1080 cells (kindly provided by Zbynek Bozdech) were cultured in Dulbecco’s modified Eagle medium (Gibco/Thermo Fisher Scientific, Waltham, MA, USA, Cat#10569010) supplemented with 10% foetal bovine serum (FBS) (Capricorn Scientific, Ebsdorfergrund, Germany, Cat#FBS-11A) and penicillin/streptomycin (PS) (Gibco/Thermo Fisher Scientific, Waltham, MA, USA, Cat#15140122). All cell types were cultured at 37°C and 5% CO2 in humidified conditions. Coverslips or MatTek dishes (MatTek, Ashland, MA, USA, Cat#P35G-1.5-14-C) were coated with fibronectin (Sigma-Aldrich/Merck, Darmstadt, Germany, Cat#F1141) before use and cells were allowed to settle down at least overnight.
Cell transfection
Cells were transfected at a density of 20–60%. MEFs and HT1080 cells were cultivated in medium without PS for minimum 1 h prior to transfection. MEFs were transfected using a 3:1 ratio of polyethylenimine (Polysciences, Warrington, PA, USA, Cat#23966; stock: 1 mg/ml) to DNA. For a 35-mm dish, 1.25 μg of DNA was used for transfection with eGFP–APEX2–TRF1, 1 μg with eGFP–APEX2–TRF2 and 2 μg with eGFP–APEX2–H2B, and the cells were fixed after 16 h (H2B and TRF1 after hypotonic treatment, TRF2 under isotonic conditions) or 48 h (TRF1 under isotonic conditions, TRF2 under isotonic conditions, TRF2 after hypotonic treatment). HT1080 cells were transfected using calcium phosphate (2 μg of DNA, fixed after 16 h). U2OS cells were transferred to pure medium prior to transfection with a 1:1.6 ratio of Lipofectamine 2000 (Invitrogen/Thermo Fisher Scientific, Waltham, MA, USA, Cat#11668019) to DNA (1.25 μg of DNA). After 4 h, the cells were washed with phosphate-buffered saline (PBS), transferred to medium with FBS and cultured for another 16 h prior to fixation.
Sample preparation for CLEM
Sample preparation for CLEM was performed based on (31,32). Cells were grown in 35-mm MatTek dishes and transfected as described earlier. Cells under isotonic conditions were fixed with 2% glutaraldehyde (Electron Microscopy Sciences, Hatfield, PA, USA, Cat#16220) in 0.1 M sodium cacodylate (Electron Microscopy Sciences, Hatfield, PA, USA, Cat#12310) buffer (CB), pH 7.4, with 2 mM CaCl2 for 5 min at room temperature followed by 1 h on ice. For hypotonic treatment, cells were incubated in 0.3× PBS for 1 min and fixed with 2% glutaraldehyde in 0.3× PBS. All subsequent steps were performed on ice. The samples were washed 3 × 3 min with CB, blocked 6 × 5 min with 50 mM glycine in CB, rinsed 5 × 2 min in CB and subsequently mounted on a CorrSight confocal spinning disc microscope (FEI/Thermo Fisher Scientific, Waltham, MA, USA) equipped with a cooling stage (FEI/Thermo Fisher Scientific, Waltham, MA, USA and Julabo, Seelbach, Germany) and Orca R2 CCD cameras (Hamamatsu, Sunayama-cho, Naka-ku, Hamamatsu City, Japan). Confocal image stacks with 300 nm z-distance were acquired of the eGFP signals with a 40× oil objective (EC Plan-Neofluar 40×/1.30 Oil M27; Carl Zeiss, Oberkochen, Germany, Cat#420460-9900-000), 488 nm laser line and standard filter sets. Subsequently, diaminobenzidine (DAB) reaction/APEX2 labelling was carried out for 1.5–3 min using 2.5 mM (0.54 mg/ml) DAB [Sigma-Aldrich/Merck, Darmstadt, Germany, Cat#D8001; freshly prepared 10× stock: 25 mM DAB in 0.1 M HCl (5.4 mg/ml)] with 0.03% (v/v) H2O2 (ICM Pharma, Singapore) in CB filtered through a 0.2-μm filter. After washing 5 × 2 min in CB, the cells of interest were imaged with transmission light. The samples were treated with 1% osmium tetroxide (Electron Microscopy Sciences, Hatfield, PA, USA, Cat#19152) in CB with 2 mM CaCl2 for 45 min, rinsed 5 × 2 min in Milli-Q water, stained with 1% aqueous uranyl acetate (UA) (Electron Microscopy Sciences, Hatfield, PA, USA, Cat#22400; stock: 4% in water) for 1 h, washed again 5 × 2 min in Milli-Q water and dehydrated in an ascending ethanol series. The samples were transferred to room temperature, rinsed 2× in anhydrous 100% ethanol (Electron Microscopy Sciences, Hatfield, PA, USA, Cat#15055) and infiltrated with Durcupan ACM resin (Sigma-Aldrich/Merck, Darmstadt, Germany, Cat#44611/44612/44613/44614) in a stepwise procedure. After resin polymerization at 60°C for 48 h, the areas of interest were sawed out using a jeweller’s saw, mounted on dummy blocks using super glue and cut into ultrathin sections of 70–80 nm for conventional TEM imaging or 120–160 nm for electron tomography on an ultramicrotome using a diamond knife. The sections were picked up on 100 mesh copper grids or, for serial sections, on 2 mm × 0.5 mm slot copper grids coated with formvar (Electron Microscopy Sciences, Hatfield, PA, USA, Cat#1500). The samples were coated with 2–4 nm carbon from each side and, for electron tomography, glow discharged and dipped into gold fiducials [1:1 to 1.5:1 mixture of 0.1% bovine serum albumin and 10 nm gold colloids (BBI Solutions, Crumlin, UK, Cat#EM.GC10) filtered through a 0.2-μm filter].
Transmission electron microscopy
TEM imaging was carried out on a Tecnai T12 (FEI/Thermo Fisher Scientific, Waltham, MA, USA) operated at 120 kV using a 4k × 4k Eagle CCD camera (FEI/Thermo Fisher Scientific, Waltham, MA, USA). Prior to recording micrographs, sections were irradiated at low magnification (690×) for 10–20 min to prevent beam-induced shrinkage during imaging. High-magnification micrographs of telomeres were recorded at a nominal magnification of 23 000× or 30 000× (pixel size: 0.48 and 0.36 nm, respectively) and an applied defocus of −2 to −5 μm.
Electron tomography
Electron tomograms were acquired on a Tecnai Arctica (FEI/Thermo Fisher Scientific, Waltham, MA, USA) operated at room temperature at 200 kV using a Falcon3 direct electron detector (FEI/Thermo Fisher Scientific, Waltham, MA, USA). Dual-axis tilt series with manual rotation by 90° were recorded using Tomography software (FEI/Thermo Fisher Scientific, Waltham, MA, USA) applying a linear tilt scheme starting at 0° and tilt angles typically spanning ±65°. Images were acquired at every degree at a nominal magnification of 39 000× (pixel size: 0.2712/0.2787 nm), an applied defocus of −2 μm and an exposure time set to yield a dosage of 10–15 e–/Å2 per micrograph. For MEFs transfected with eGFP–APEX2–TRF2, tomograms of 49 telomeres from 17 different cells were acquired under isotonic conditions and of 15 telomeres from 4 different cells after hypotonic treatment. For MEFs transfected with eGFP–APEX2–TRF1, tomograms of 10 telomeres from 4 different cells were collected under isotonic conditions and of 8 telomeres from 2 different cells after hypotonic treatment. For MEFs transfected with eGFP–APEX2–H2B, tomograms of 9 telomeres from 4 different cells were acquired, and for U2OS cells transfected with eGPF–APEX2–TRF1 tomograms of 8 telomeres from 3 different cells were acquired. Tilt series were reconstructed by filtered backprojection into combined 3D tomograms using the Etomo IMOD software package (33,34) (version 4.9.0) and binned by a factor of 2 during the process. Further processing was performed in Fiji/ImageJ (35,36): the tomograms were filtered to 2 nm with a 3D Gaussian and again binned by a factor of 2 in all three directions (final pixel size: 1.0848/1.1148 nm). Subsequently, groups of five consecutive tomographic slices were averaged to a thickness of 5.4/5.6 nm.
Surface rendering
Surface renderings were generated in Amira (Thermo Fisher Scientific, Waltham, MA, USA; version 6.5.0). In the filtered tomograms, an ‘interactive threshold’ corresponding to the intensity of the APEX2/osmium signals was selected. Contiguous regions in 3D were identified using the ‘labelling’ function and the density corresponding to the telomere was extracted using ‘arithmetic’ operation, thereby removing all signals not connected in 3D. Subsequently, the surface of the volume was generated with a minimal smoothing of 1.5. Thin slices were generated using the ‘region of interest’ or ‘ROI’ function.
Evaluation of telomere size and chromatin measurements
Unless otherwise mentioned, measurements were performed in Fiji/ImageJ (35,36). Telomere size was evaluated from TEM images of 70 nm sections and electron tomograms selecting the average slice with the largest dimensions of the telomere. Telomere diameters were calculated as an average of the longest and shortest axes of each telomere. Surface rendering was used to facilitate measurements of telomere diameters in U2OS cells. The area covered by chromatin was evaluated from averaged tomographic slices with a thickness of 5.4/5.6 nm after thresholding. Regions corresponding to telomeres, euchromatin and heterochromatin were selected as shown in Figure 3D and Supplementary Figure S8C. Measurements of all chromatin in the entire slice, excluding cytoplasmic regions, were used to ensure similar thresholds. Chromatin fibre measurements were carried out on averaged tomographic slices with a thickness of 5.4/5.6 nm in triplets within short distances of ∼10–30 nm and subsequently averaged. APEX2 samples provided sufficient contrast for measurements of unfiltered data. Non-telomeric fibres stained with UA only were measured after filtering to 2 nm. Line scans were performed with a line width of 1 pixel across a distance of 522 nm.
Figure 3.

Electron tomography resolves the ultrastructure of telomeres in situ. (A–D) Averaged tomographic slices with a thickness of 5.4/5.6 nm of cells fixed under isotonic conditions after transfection with eGFP–APEX2–TRF1 (–TRF1) (A, C), eGFP–APEX2–TRF2 (–TRF2) (B) or eGFP–APEX2–H2B (–H2B) (D). (A, B, D) MEFs. (C) U2OS cells. Nuc = nucleolus. HC = heterochromatin. Arrowheads point at areas of telomeres of U2OS cells with comparable labelling density and compaction to telomeres of MEFs. Horizontal arrows indicate the position of the line scans shown below the images. The red line denotes the approximate half intensity between the background outside the telomere and the darkest areas. The dotted vertical lines indicate the borders of the telomeres. For H2B (D), typical examples for heterochromatin (top) and euchromatin (bottom) are shown. (E) Evaluation of the area covered by chromatin in MEFs. HC = heterochromatin. EC = euchromatin. Telom. = Telomeres. Error bars: standard deviation. Asterisks denote statistical significance based on two-sided t-tests: ***P < 0.001. Numbers of tomograms/slices analysed are 4/18 for H2B, 5/22 for TRF1 and 6/25 for TRF2. (F, G) Equivalent to panels (A)–(D), cells fixed after hypotonic treatment. (F) MEF–TRF1. (G) MEF–TRF2.
Focused ion beam scanning electron microscopy
Until complete resin polymerization, samples for FIB-SEM were prepared as described earlier for CLEM. The area of interest was sawed out using a jeweller’s saw, mounted on a standard SEM stub using silver paste and coated with 5 nm platinum. The sample was transferred to a Scios Dual Beam FIB-SEM (FEI/Thermo Fisher Scientific, Waltham, MA, USA) operated at room temperature. Images were acquired in the xz direction of the cell with a pixel size of 27 nm, a slice thickness of 40 nm, a field of view of 1536 × 1024 pixels and 30 μs acquisition time. Image registration for the correction of ‘northern drift’ of the obtained stack was performed in Fiji/ImageJ (35,36) using the MultiStackReg plugin (B. Busse; http://bradbusse.net/sciencedownloads.html) (version 1.45). The original image stack was cropped in the x dimension to a central area where the platinum layer was intact throughout the entire stack. The obtained fragment was thresholded (platinum layer to be white) and any remaining background noise was removed. The slice with the northernmost drift, i.e. with the platinum layer at the highest position, was selected and MultiStackReg was run to align the images using translation. The saved transformation file was then applied to the original image stack by running MultiStackReg again loading the transformation file, followed by final alignment through another round of MultiStackReg. Subsequently, the obtained image stack was filtered to 2.2 nm in xy (i.e. xz direction of the cell) and signal intensities were inverted.
Image processing and image correlation
Unless otherwise mentioned, images were processed in Fiji/ImageJ (35,36). For creating overlay images of the eGFP signals with TEM micrographs, low-magnification TEM images (890–1900×, covering the entire nucleus) were compared with the confocal image stacks, the corresponding optical slice identified based on cellular features and the APEX2 signal pattern, and the TEM and fluorescent images superimposed using Adobe Photoshop CS6 (Adobe Systems, San Jose, CA, USA; version 13.0.1). Typically, minor distortion and warping of the fluorescence image was necessary for proper superimposition. Overlay images of the eGFP signals with SEM images were created in a similar way, with the exception that the viewing angle of the confocal image stack was matched to the resliced FIB-SEM data set in Fiji/ImageJ using the Volume Viewer plugin (K.U. Barthel; Medieninformatik, HTW Berlin, Germany) (version 2.01). Figures were prepared using Adobe Photoshop CS6 (Adobe Systems, San Jose, CA, USA; version 13.0.1). Violin plots were generated in R (R Core Team; The R Project for Statistical Computing; R Foundation for Statistical Computing, Vienna, Austria; http://www.r-project.org/) (version 4.1.2) with the package ggplot2 (37) (version 3.3.5) part of tidyverse (38). All other graphs were generated in Excel (Microsoft, Redmond, WA, USA). Data visualization using bee swarm blots was aided by Daniel’s XL Toolbox add-in for Excel (D. Kraus; www.xltoolbox.net) (version 7.2.).
RESULTS
Telomeres can be visualized selectively by CLEM of telomeric repeat-binding factors tagged with eGFP–APEX2
To study the ultrastructure of telomeres, we selected representative telomerase-positive and telomerase-negative cell lines with long or short telomeres. Immortalized MEFs and human fibrosarcoma-derived HT1080 cells are telomerase positive (39,40), while human bone osteosarcoma cells (U2OS) are telomerase negative and belong to the group of ALT cells (41). Southern blotting confirmed that telomeres were long in MEFs and U2OS cells (19 and 25 kb, respectively) and short in HT1080 cells (∼5 kb in length) (Supplementary Figure S1A and B). In addition, western blotting confirmed that the telomerase enzyme was not expressed in the U2OS cell line (Supplementary Figure S1C).
To visualize telomeres by CLEM, we generated tandem eGFP–APEX2 constructs for the telomeric repeat-binding factors TRF1 and TRF2 (Supplementary Figure S1D). In these constructs, the eGFP tag provides fluorescence for light microscopy (LM) analysis, while the APEX2 tag generates an electron-dense stain for EM (Figure 1). Transient transfection of the TRF1 or TRF2 eGFP–APEX2 probes generated small and discrete foci in the nucleus characteristic of telomeres in all cell types analysed. We found less telomeric foci in interphase cells than estimated from chromosome spreads (Supplementary Figure S1F and G). This is consistent with previous reports (42–46) and likely due to a combination of (i) the low resolution of fluorescence microscopy, in particular along the optical axis (i.e. telomeres in close axial proximity may not be resolved), (ii) the low fluorescence intensity of short telomeres that cannot be distinguished from background and (iii) potential telomere clustering, especially in U2OS cells with ALT (44). Co-labelling with antibodies against endogenous TRF2 and quantitative line scan analyses of individual telomeres confirmed the correct targeting of the eGFP–APEX2-tagged TRF proteins to telomeres in both HT1080 and U2OS cells (Supplementary Figure S2). Telomeric levels of endogenous TRF2 were only marginally reduced upon transfection of the TRF1 eGFP–APEX2 probe, and there were no intensely stained foci in transfected nuclei that contained only one of the two TRF proteins. This indicates that both the TRF1 and TRF2 eGFP–APEX2 probes were efficiently incorporated into telomeres and that their overexpression did not interfere with the targeting of endogenous TRF2 to telomeres. Moreover, overexpression of tagged TRF1 or TRF2 did not cause any detectable increase in DNA damage compared to untransfected cells (Supplementary Figure S3), with >84% of transfected cells containing only two telomere dysfunction-induced foci or less. This indicates that overexpression of TRF1 or TRF2 did not elicit a DNA damage response at telomeres, as previously reported in cells in which TRF1 or TRF2 was downregulated (47–49). Finally, immunoprecipitations showed that eGFP–APEX2-tagged TRF2 forms a protein complex with Rap1, as expected (Supplementary Figure S1E). Taken together, these results demonstrate that the eGFP–APEX2–TRF probes are functional and correctly localized at telomeres.
Figure 1.

CLEM of telomeres. (A) Maximum intensity projection of GFP signals of MEFs transfected with eGFP–APEX2–TRF2. An untransfected cell (*) and transfected cell C (**) are magnified in the insets. Cell B is further magnified in the following panels. (B) Transmission light image of the same field of view shown in panel (A) after staining and embedding for EM. (C) Overlay images of single light optical confocal sections (GFP signals, green) and the corresponding TEM images from the physical EM section (APEX2/osmium signals, grey) for MEFs transfected with eGFP–APEX2–TRF2 (MEF–TRF2, top) or eGFP–APEX2–TRF1 (MEF–TRF1, middle) or U2OS cells transfected with eGFP–APEX2–TRF1 (U2OS–TRF1, bottom). (D, E) Corresponding TEM (D) and confocal (E) images only of MEF–TRF2 shown in panel (C). (F) Magnifications of the boxed areas in panel (C). (G) TEM images of the boxed areas in panel (F). Nuc = nucleolus. NE = nuclear envelope; position indicated by the small arrows.
For CLEM, cells transfected with the TRF1 or TRF2 probes were chemically fixed and imaged on a spinning disc confocal microscope at lateral and axial resolutions of ∼200 and ∼600 nm, respectively. Figure 1A shows a representative image of three MEF cells (labelled A–C) transfected with eGFP–APEX2–TRF2 next to an untransfected cell (labelled *). Clear eGFP foci corresponding to individual telomeres can be readily detected in the transfected but not in untransfected cells. We collected confocal z-stacks of nuclei where telomeres were clearly distinguishable and disregarded cells in which the expression level was too low (eGFP signals <2.5× above autofluorescent background signals) or too high (>10× above background levels). By co-labelling with anti-TRF2 antibodies, we estimated that the amount of TRF2 at telomeres in the selected cells was on average 4-fold higher than that in untransfected cells (Supplementary Figure S2C). After fluorescence imaging, cells were bathed in DAB and H2O2 for 1–3 min to carry out the APEX2 reaction and subsequently prepared for EM analysis in a similar way to what is generally referred to as positive stain and conventional EM (see the ‘Materials and Methods’ section). In brief, cells were post-fixed with osmium tetroxide, weakly en bloc stained with UA to introduce overall contrast to all nucleic acids and proteins, dehydrated and embedded in plastic resin. We note that in contrast to a conventionally prepared EM sample, in an APEX2 stained sample the DAB and osmium reaction creates specific contrast around the probe, allowing the protein of interest to be localized with high spatial precision. Figure 1B shows that the APEX2 reaction resulted in a dense stain at telomeres that was clearly visible by transmission light imaging. Areas of interest were then sectioned and imaged by TEM. Superimposition of eGFP fluorescence signals and the corresponding EM micrographs from eGFP–APEX2-labelled TRF2 in MEFs, TRF1 in MEFs and TRF1 in U2OS cells demonstrate that telomeres labelled by the APEX2 probes are more electron dense than other chromatin regions in the nucleus (Figure 1C–G). Figure 1G shows EM micrographs of selected areas with individual telomeres at slightly higher magnification. Non-telomeric chromatin, nucleoli and the nuclear envelope (NE) can also be recognized in these images due to the weak staining with UA. Importantly, each telomere observed in the EM images correlated precisely in space with the eGFP fluorescence signals of the corresponding optical confocal section. Given that an EM section is much thinner (70 nm) than an optical slice (600 nm), only a fraction of telomeres can be correlated in any given EM section. For example, in the first overlay image shown in Figure 1F only 6 out of 11 telomeres present in the optical slice (green eGFP dots, compare with Figure 1E) are contained within the EM section (dark EM dots, compare with Figure 1D). In summary, tagging of TRF1 or TRF2 with eGFP–APEX2 enabled us to establish a correlative imaging workflow to sequentially image telomeres by LM and EM inside the nucleus.
EM analysis reveals marked differences in telomere compaction, size and shape between telomerase-positive and ALT cells
Next, we made use of the high resolution of TEM to carry out a detailed analysis of the ultrastructure of telomeres. 2D EM images recorded from 70 nm thin sections (Figure 2 and Supplementary Figure S4) revealed darkly APEX2-stained telomeres that clearly stood out from surrounding nuclear regions. Osmium-stained cellular structures that did not correlate with eGFP fluorescence, such as peroxisomes (which inherently generate reactive oxygen species) and occasionally observed high-contrast nuclear bodies of unknown origin (Figure 6), were disregarded.
Figure 2.

Telomerase-positive and ALT cells exhibit marked differences in telomere size and shape. (A–D) TEM micrographs of cells fixed under isotonic conditions after transfection with eGFP–APEX2–TRF2 (–TRF2) (A) or eGFP–APEX2–TRF1 (–TRF1) (B–D) acquired from 70 nm thin sections. (A, B) MEFs. (C) U2OS cells. (D) HT1080 cells. The first and last images of panels (A) and (B) show the telomeres in the adjacent images at lower magnification (low mag). Arrowheads point at areas of telomeres of U2OS cells with comparable labelling density and compaction to telomeres of MEFs and HT1080 cells. Nuc = nucleolus. NE = nuclear envelope; position indicated by the small arrows. (E, F) Equivalent to panels (A)–(D), cells fixed after hypotonic treatment. (E) MEF–TRF2. (F) MEF–TRF1. (G) Evaluation of telomere size. Individual values = grey dots. Mean values = black squares. Error bars: standard deviation. Asterisks denote statistical significance based on two-sided t-tests: ***P < 0.001 and **P < 0.01. Mean values (Ø) and number of cells analysed (n) for each condition are specified at the top or bottom of the graph.
Figure 6.

FIB-SEM reveals the organization of telomeres in the 3D volume of the nucleus. (A) Xy view. Left: Overlay image of the APEX2/osmium signals (grey, average intensity projection of five SEM slices with a thickness of 135 nm) and the corresponding light optical confocal section (GFP signals, green). Middle and right: Corresponding APEX2/osmium signals (middle) and GFP signals (right) only of the overlay image. Telomere 5 is the same as in panels (B) and (C). The boxed areas are magnified below; for each of the three examples from left to right: overlay, APEX2/osmium signals alone, GFP signals alone. NB = nuclear body. (B, C) Xz view. APEX2/osmium signals. The numbered telomeres in the selected slices in panel (B) are magnified in panel (C) along with further examples; five consecutive micrographs are shown for each telomere. NE = nuclear envelope; position indicated by the small arrows. PM = plasma membrane. NB = nuclear body. Nuc = nucleolus. CC = chromocentre.
In telomerase-positive MEFs (Figure 2A and B) and HT1080 cells (Figure 2D), telomeres labelled with the TRF1 or TRF2 eGFP–APEX2 probes were round or ovoid in shape. In contrast, in U2OS ALT cells (Figure 2C) telomeres were largely heterogeneous in shape and interspersed with non-telomeric chromatin and generally appeared less compact compared to other cell types analysed. Only certain areas of the telomeres in U2OS cells (arrowheads in Figure 2C) seemed comparable to telomeres in MEFs and HT1080 cells with regards to electron density and homogeneity of the stain. Interestingly, in MEFs telomeres labelled with the TRF1 probe often exhibited patterns of low and high EM contrast (Figure 2B), while telomeres labelled with the TRF2 probe appeared to be stained more homogenously (Figure 2A). In addition, at similar transfection levels TRF1-labelled telomeres appeared slightly less electron dense than those labelled by TRF2.
We measured the average size of telomeres in the three cell types (Figure 2G). Telomeres in MEFs were ∼200 nm in diameter, which was confirmed by serial section TEM (Supplementary Figure S5). Consistent with our Southern blot analysis (Supplementary Figure S1A and B), telomeres in HT1080 cells were significantly smaller (∼160 nm), whereas telomeres in U2OS cells were much larger (∼350 nm) and generally much more heterogeneous in size than telomeres in MEFs or HT1080 cells. This likely reflects the broad range of telomere lengths found in U2OS cells and may also be due to telomere clustering as previously demonstrated to occur more frequently in this cell type (44). We conclude that telomerase-positive and ALT cells exhibit marked differences in telomere size, shape and compaction state. In addition, we did not find any difference in telomere size between MEFs in G1 phase and G2 phase of the cell cycle (Supplementary Figure S7A and C; see Supplementary Methods).
Since telomeres appeared as dense structures in both MEFs and HT1080 cells, we explored the option of gaining more detailed insights into their ultrastructure by imaging telomeres after decompaction. To this end, we exposed MEFs expressing the TRF1 or TRF2 probes to a brief hypotonic treatment. Live cells were incubated for 1 min in a low-salt buffer [0.3× PBS and 45 mM salt (hypotonic), instead of 150 mM 1× PBS (isotonic)], fixed and prepared for EM. This procedure has previously been established to decompact chromatin inside the nucleus (50,51). DNA-specific staining using osmium ammine B (Supplementary Figure S6) confirmed that MEFs under isotonic conditions contained clearly distinguishable chromocentres, regions of densely packed heterochromatin that are typical for mouse cells (52) (labelled with ‘CC’), peripheral heterochromatin (‘HC’) and large interchromatin lacunas (‘IC’), and that hypotonic treatment disrupted heterochromatin regions. As expected, hypotonic treatment increased the size of telomeres labelled with TRF1 or TRF2 probes to ∼230 and ∼260 nm, respectively (Figure 2G). Telomeres decompacted by the hypotonic treatment displayed a more open ultrastructure as compared to telomeres at isotonic conditions (Figure 2E and F). This was evident for telomeres labelled by both the TRF1 and TRF2 probes. Furthermore, after hypotonic treatment telomeres labelled with the two probes were more similar in overall appearance than observed at isotonic conditions. This shows that cations are required to stabilize the higher order structure of telomeric chromatin and that a brief hypotonic treatment provides a means to ‘open up’ telomeric chromatin inside the nucleus.
Electron tomographic analysis of the 3D structure of telomeres in MEF and U2OS nuclei
To study the ultrastructure of telomeres in 3D, we applied electron tomography. Dual-axis tomograms of ∼150 nm thick cell sections were recorded, reconstructed into a 3D volume and low-pass filtered to 2 nm (see the ‘Materials and Methods’ section). Figure 3 shows tomographic slices through the centre of representative telomeres in MEFs and U2OS cells analysed under isotonic or hypotonic conditions (see Supplementary Figure S7 for more examples of tomographic slices and Supplementary Movies S1–S5 for 3D tomograms). In agreement with our 2D analysis (Figure 2), under isotonic conditions telomeres appeared round in MEFs (Figure 3A and B) but heterogeneous in shape in U2OS cells (Figure 3C). Interestingly, line scans across the centre of the telomeres revealed alternating patterns of high and low electron densities, both in MEFs and in U2OS cells (Figure 3A–C, bottom panels). APEX2-stained fibrous densities were clearly evident in telomeres labelled with the TRF1 probe (Figure 3A and C) and were occasionally also apparent in telomeres labelled with the TRF2 probe (Figure 3B). We did not observe any difference in telomere ultrastructure between cells in G1 phase and G2 phase of the cell cycle (Supplementary Figure S7A and C; see Supplementary Methods) or between conventionally prepared samples and samples processed by high-pressure freezing and freeze substitution (Supplementary Figure S7B). Importantly, and again in line with our 2D analysis, in cells exposed to hypotonic treatment (Figure 3F and G) telomeres appeared decompacted and telomeric chromatin fibres became more evident, in particular in telomeres labelled with the TRF2 probe (Figure 3G). As a comparison to telomeric chromatin, we labelled non-telomeric chromatin in MEFs using the core histone H2B tagged with eGFP–APEX2. We found that non-telomeric chromatin also displayed a fibrous pattern (Figure 3D, Supplementary Figure S8, and Supplementary Movies S6 and S7). This shows that APEX2 can be applied to gain insight into the ultrastructure of bulk chromatin as well as specific chromatin domains such as telomeres.
Next, we analysed the organization and shape of the telomeric chromatin fibres in 3D. Figure 4 shows representative tomograms and surface rendered fibres in MEFs labelled by the TRF1 probe, under isotonic and hypotonic conditions. Thin tomographic sub-volumes (5.4 nm thick slices) through telomeres under isotonic conditions revealed an irregular 3D mesh of chromatin fibres, in which individual fibres were clearly resolved but densely packed (Figure 4A and B). As expected, under hypotonic conditions, the mesh of fibres opened up markedly (Figure 4C and D). Telomeric chromatin fibres appeared bent or looped and were often tightly opposed (centre of fibres indicated by blue lines in Figure 4B and D). In addition, they displayed a range of diameters; fibre diameters as small as 5 nm and as large as 35 nm were measured in each condition and cell type (Figure 4E–G). For comparison, the nucleosome core particle is disc shaped and has a dimension of 6 nm × 10 nm. Therefore, we interpret fibre widths in the range of 5–10 nm as ‘beads on a string’ and thicker fibres as higher order levels of chromatin compaction. It is possible, however, that some of the thicker fibres rather represent two or more individual but tightly opposed fibres, which cannot be resolved. In MEFs, the average diameter of telomeric chromatin fibres labelled with the TRF2 or TRF1 probes was 15 and 17 nm, respectively. As expected, the diameters of individual telomeric chromatin fibres were reduced under hypotonic conditions (14 nm for fibres labelled with the TRF2 probe, 13 nm for TRF1). Smaller average diameters (11 nm) were measured in U2OS cells labelled with the TRF1 probe. In MEFs, bulk chromatin fibres labelled with the eGFP–APEX2–H2B probe were on average 14 nm broad, in both euchromatin and heterochromatin regions (representative tomographic slices are shown in Figure 3D). By comparison, UA-stained fibres surrounding telomeres (within a tomographic field of view of ∼500 × 500 nm) measured 12 nm in diameter. This suggests that the APEX2-generated DAB/osmium stain contributes only minimally (∼2–3 nm) to the measured fibre width. Importantly, although the fibre width was only slightly broader in telomeric (15–17 nm) compared to non-telomeric chromatin (14 nm), telomeres under isotonic conditions appeared more compacted than other heterochromatin regions. Indeed, measurements on thresholded tomographic slices revealed that telomeric chromatin occupied ∼60–68% of the area, whereas non-telomeric chromatin labelled with the eGFP–APEX2–H2B probe occupied only 26% (euchromatin regions) to 43% (heterochromatin regions) of the nuclear space (Figure 3E).
Figure 4.

Telomeres are composed of a heterogeneous mesh of chromatin fibres. (A–E) Tomographic reconstructions of MEFs transfected with eGFP–APEX2–TRF1 fixed under isotonic conditions (A, B) or after hypotonic treatment (C–E). (A, C) From left to right: 0 degree projection image, higher magnification thereof, averaged tomographic slices of the same telomere with a thickness of 5.4 nm before (unfiltered) and after filtering to 2 nm. (B, D) Surface rendering of the same telomeres and slices shown in panels (A) and (C). Left: Full volume. Right: Slice with a thickness of 5.4 nm. Blue lines indicate the mesh of chromatin fibres. (E) Volume surface rendering of telomeric chromatin structures. Selected fibre diameters measured along the dashed lines are specified in nm. (F) Evaluation of chromatin fibre diameters of MEFs and U2OS cells transfected with eGFP–APEX2–TRF2 (TRF2), eGFP–APEX2–TRF1 (TRF1), eGFP–APEX2–H2B (H2B) or without APEX label from areas surrounding telomeres that are lightly UA stained only (UA only). Iso. = isotonic conditions. Hypo. = hypotonic treatment. Asterisks denote statistical significance based on two-sided t-tests: ***P < 0.001, **P < 0.01 and *P < 0.05. Median values (Ø) and number of cells analysed (n) for each condition are specified at the top of the graph. (G) Volume surface rendering of chromatin fibres of cells fixed under isotonic conditions. Labels equivalent to panel (F).
In conclusion, telomeric chromatin fibres display a broad range of diameters and morphologies; the fibres appear bent or looped and are closely packaged into a dense and irregular mesh-like chromatin domain. Moreover, telomere shape, fibre diameter and telomere compaction differ between MEFs and U2OS ALT cancer cells.
Association of telomeres with other heterochromatin domains
In both our 2D TEM and electron tomography data of MEFs, we regularly observed a close association of telomeres with non-telomeric heterochromatin regions, the NE and nucleoli (Figures 1–3). This was not surprising and has been described before using light microscopy (see the ‘Discussion’ section). We confirmed these observations in the cell types used here by performing 3D confocal microscopy, distance measurements and chromatin density analysis (Supplementary Figure S9). We further applied electron tomography to analyse the spatial association of telomeres with other heterochromatin regions in more detail. Data from a representative cell section are shown in Figure 5, where three telomeres (T1–T3) are located near the NE and one telomere is found close to the nucleolus (T4). Inspection of the tomographic slices showed that telomeres T1, T2 and T3 are not in direct contact with the NE (Figure 5C and D, and Supplementary Movie S8). Instead, they appeared to be attached to NE-associated heterochromatin (lamina-associated domains), separating the telomere from the NE by a thin layer of non-telomeric chromatin. Interestingly, for each telomere multiple contacts between non-telomeric and telomeric chromatin fibres were clearly discernible (indicated by arrowheads in Figure 5C and D). Similar fibrous connections were observed between telomeres and perinucleolar heterochromatin and other heterochromatin regions (Figure 5E). We conclude that telomeres are in direct physical contact with non-telomeric heterochromatin domains.
Figure 5.

Telomeres are in direct physical contact with heterochromatin domains. (A) CLEM of MEFs transfected with eGFP–APEX2–TRF2. Left: Overlay image of an optical confocal section (GFP signals, green) and the corresponding TEM image from the physical EM section (APEX2/osmium signals, grey). Right: Corresponding TEM image showing the APEX2/osmium signal only. (B) Magnification of the boxed area in panel (A). NE = nuclear envelope; position indicated by the small arrows. Nuc = nucleolus. (C) Averaged tomographic slices with a thickness of 5.4 nm of the labelled telomeres T1–T3 in panel (B) and one additional example from another cell. Arrowheads point at chromatin contacts between the telomere and non-telomeric heterochromatin. (D) Equivalent to panel (C) showing additional averaged tomographic slices of telomere T1. The position of the NE is only indicated in the first image. (E) Equivalent to panel (C) showing the labelled telomere T4 in panel (B) and additional examples from other cells associated with the nucleolus (Nuc) or other heterochromatin regions (HC).
Electron tomography provides high resolution but is limited to relatively thin cell sections and small sample areas. The technique is therefore not suited to image many telomeres simultaneously. To overcome this limitation and to better understand the localization of telomeres in the 3D space of the entire nucleus, we imaged APEX2-labelled telomeres of MEFs in whole cells using FIB-SEM. As shown in Figure 6, telomeres labelled with the APEX2–TRF2 probe exhibited strong EM contrast in SEM and appeared selectively enhanced compared to non-telomeric heterochromatin and nucleoli. Similar to our TEM images, we observed a few darkly stained nuclear structures similar to telomeres in size and appearance, which we here refer to as nuclear bodies. These structures of unknown origin could easily be discriminated from telomeres by correlating the eGFP fluorescence and the APEX2 stain (Figure 6A), demonstrating the importance of the CLEM approach. FIB-SEM analysis confirmed that a large proportion of telomeres is located close to the NE and nucleoli (Figure 6B and Supplementary Movie S9). Furthermore, it supports our observation from the tomographic images (Figure 5) that telomeres in close proximity to the NE (indicated by the small arrows) are typically separated from it by a thin layer of non-telomeric chromatin (Figure 6C). This thin layer usually exhibited slightly higher electron density in the SEM images than adjacent regions more internal in the nucleus, which indicates that it represents heterochromatin. Interestingly, telomeres frequently occurred in doublets (Figure 6C, telomeres 1, 5, 9, 13, 15, 17 and 18), as is often observed for sister loci in G2 cells, such as the one analysed here (data not shown) (53). Telomere size within some of these pairs was heterogeneous, with one telomere of the pair occasionally being smaller than the other (Figure 6C, telomeres 1, 9 and 17). This is in line with previous observations showing that telomeres from sister chromatids can vary in length (54–56). Overall, this demonstrates that APEX2 labelling can also be used for SEM volume imaging. Together, electron tomography and FIB-SEM in MEFs indicate that telomeres that are located at the very periphery of the nucleus are not in direct contact with the NE but instead are linked to lamina-associated heterochromatin.
DISCUSSION
Detailed insight into the nuclear organization of telomeric chromatin is required to understand how shelterin proteins protect chromosome ends from being recognized as DNA breaks (end-protection problem) (13,14). It is also essential for understanding cellular aging and telomere maintenance in cancer cells that escape senescence and replicate indefinitely (circumventing the end-replication problem) (1). Confocal microscopy and super-resolution microscopy have provided high-throughput information about the nuclear localization and diameter of telomeres (28–30,57). However, the resolution of current optical microscopy techniques is insufficient to resolve chromatin fibres within the telomere. EM offers much greater resolution, yet immunogold labelling to analyse telomeres in the nucleus can be compromised by poor ultrastructural preservation (58) or insufficient labelling density (59), both of which are common limitations of antibody-based approaches. New techniques for EM labelling have recently been developed, which permit the direct visualization of proteins with genetically encoded tags, similar to GFP fusion proteins for light microscopy (60). The most common labels are miniSOG (61) and APEX (32), and a modified and more sensitive version of the latter, APEX2 (31).
Fusing TRF1 or TRF2 to both eGFP and APEX2 allowed us to visualize the same cells and telomeres by fluorescence microscopy and EM, and thereby combine the strengths of the different microscopic techniques. Using this approach, we resolved ultrastructural features of telomeres in mammalian cells at a resolution of ∼5 nm, which is ∼4–5× higher in each dimension than the resolution obtained by super-resolution microscopy. We found that telomeres in MEFs and HT1080 cells were round or ovoid in shape, with an average diameter of 200 and 160 nm, respectively. This agrees with our telomere length measurements by Southern blotting and previously reported super-resolution studies of telomere size distributions in situ (28–30). In contrast, telomeres in U2OS ALT cancer cells were much larger (350 nm on average) and much more heterogeneous in shape. We note that ALT cells display a broad range of telomere lengths (4) and that exceptionally short telomeres may have escaped our analysis due to insufficient labelling by our APEX2 probes.
Previous studies have shown that telomeric chromatin has an unusually short nucleosome repeat length (157 bp) (17,18,62) and is characterized by both heterochromatic and euchromatic markers (63,64). Here, we selectively labelled and compared telomeric and non-telomeric chromatin, providing direct visual evidence that telomeres in MEFs represent highly compacted chromatin. In fact, we found that telomeres are more compact than other non-telomeric heterochromatin regions (Figure 3E). Telomeres of HT1080 cells appeared equally compact to those in MEFs. Compared to telomerase-positive MEFs (39) and HT1080 cells (40), telomeres of U2OS ALT cells (41) were clearly less compact. This is in line with the finding that telomere structure in ALT cells is more open, as determined by micrococcal nuclease digestion assays (65). In addition, telomeres in ALT cells exhibit a longer nucleosome repeat length as well as reduced levels of repressive epigenetic marks compared to telomerase-positive cells (65). Together, this indicates that different mechanisms for telomere maintenance result in marked differences in telomere ultrastructure.
How chromatin is packaged into specialized heterochromatin domains, such as telomeres, is an outstanding and largely unresolved question. Early EM studies of isolated native chromatin showed that in salt-containing buffer, linker histone (H1) compacts 10-nm nucleosomes (66) into higher order 30-nm chromatin fibres (67). Cellular EM studies have also shown that 30-nm fibres exist in the nucleus of chicken erythrocytes (68) and starfish sperm (69)—two cell types in which the entire genome is silenced—but not in mammalian cells grown in culture (70–72). In contrast to previous studies, which analysed undefined chromatin regions, we specifically imaged telomeric and non-telomeric chromatin in intact mammalian nuclei. This allowed us to visualize chromatin fibres inside telomeres that could previously not be resolved using super-resolution microscopy (28–30,57). These fibres clearly contained TRFs, as judged by the additional EM contrast introduced by the APEX2 tag and subsequent osmium staining. The telomeric fibres appeared irregular in shape and were packaged into a compact mesh. The irregular fibre diameter and structure agree with a recent report showing that the telomeric nucleosome core particle is less stable and more dynamic than nucleosome cores reconstituted with other DNA sequences (23).
Segmentation and 3D surface rendering showed that overall telomeric and non-telomeric chromatin fibres are similar in terms of dimensions and appearance. In MEFs, we measured average fibre diameters of 14–17 nm; individual fibres inside and outside of telomeres could be as thin as 5 nm and as thick as 35 nm. Our fibre measurements are consistent with the dimensions of bulk chromatin previously determined using the non-selective DNA stain DRAQ5 (73). Both APEX2 and DRAQ5 produce an osmiophilic and electron-dense stain via the local oxidation of DAB, but in contrast to APEX2, DRAQ5 staining does not permit the selective labelling of different chromatin domains and therefore cannot differentiate between telomeric and non-telomeric chromatin.
The average chromatin fibre diameters were similar in TRF1- and TRF2-labelled telomeres, under both hypo- and isotonic conditions. Under isotonic conditions, however, TRF2-labelled telomeres appeared more electron dense and more evenly stained than telomeres labelled by TRF1. This observation was unexpected since TRF1 and TRF2 are similar in sequence and structure, bind to telomeric DNA in a similar manner (21,22) and are components of the same shelterin complex (11,12). However, differences between the two proteins do exist. For instance, TRF2 (but not TRF1) interacts with the shelterin component Rap1 (74) and shelterin subcomplexes devoid of TRF1 have been suggested (12). In addition, quantitative western blot analyses suggest that the overall expression level and the chromatin-bound fraction of TRF2 are significantly higher than those of TRF1 (75). There is also evidence from FRAP (fluorescence recovery after photobleaching) experiments that TRF1 and TRF2 differ in the way they are recruited to or retained at telomeres (76). We speculate that these differences in protein abundance or recruitment to telomeric DNA might explain the differences in the staining patterns we observed in our images. An alternative explanation may be that overexpression of TRF2 induces compaction of telomeric chromatin, as previously suggested by in vitro experiments (77,78). If TRF2 did compact telomeric chromatin to a higher degree than TRF1, one would expect telomeres with elevated TRF2 levels to be smaller, which is not what we find. In fact, telomeres labelled with the TRF2 probe showed a similar size distribution to those labelled with the TRF1 probe (Figure 2G). Nevertheless, we cannot rule out that overexpression might contribute in some way to the differential appearance of TRF1- and TRF2-labelled telomeres. Finally, given the dense packing of telomeric chromatin fibres under isotonic conditions, our data neither support nor argue against the proposed t-loop model, which states that the single-stranded telomeric G-overhang loops back onto the double-stranded region to form a lasso-like structure (26). Advances in cryo-super-resolution CLEM may provide a future avenue to resolve the internal architecture of telomeres with even greater resolution.
Our tomographic analysis further revealed that telomeres are connected to non-telomeric heterochromatin via multiple fibrous densities. Such connections were observed with nucleolar heterochromatin and chromocentres as well as with NE-associated heterochromatin (lamina-associated domains). This indicates that telomeres do not form spatially isolated entities, but instead are in continuity with the surrounding heterochromatin. The close spatial association of telomeres with chromocentres (45,46,79–81), nucleoli (42,45,82,83) and the NE (42,46,83–85) has been described before, but had not been visualized with the resolution offered by electron tomography and SEM. A functional link between the NE, lamins [the main components of a 10–30 nm thick layer beneath the NE called the nuclear lamina (86)] and telomeres is well established. However, how telomeres are anchored at the nuclear periphery is still unclear. There is evidence for an association of telomeres with the nuclear matrix (58,87) as well as for interactions between lamins and shelterin proteins (88,89). On the other hand, it was shown that such interactions may be restricted to a brief time window in late mitosis/early G1 phase while being minimal or absent during the rest of interphase (84). The EM analysis presented here indicates that the majority of peripherally located telomeres are too far away from the NE to make direct contact with the nuclear membrane. If telomeres are physically anchored to the NE in interphase, this connection likely occurs through the association with NE-associated heterochromatin (lamina-associated domains) rather than direct interactions with the nuclear lamina.
In conclusion, our approach of targeting APEX2 fusion proteins to telomeres permitted us to locate and selectively visualize telomeres by EM, to resolve the organization of telomeric chromatin by tomography and to study the interaction of telomeres with non-telomeric chromatin domains inside the nucleus. This demonstrates that APEX2 labelling is a useful tool to interrogate the ultrastructure and sub-nuclear organization of different chromatin domains. Other specialized chromatin structures, such as the Barr body or centromeres, may be interesting future targets for this approach. Here, we focused our image analysis on telomere ultrastructure in three cell types (MEFs, HT1080 and ALT U2OS cells) and analysed the spatial association of telomeres with other heterochromatin regions and the NE, which was primarily conducted in MEFs. In the future, it will be interesting to explore how telomere localization and heterochromatin association compare between human and mouse cells. Analysing telomere structure in other cell types or cell states, such as in old and young primary cells, senescent cells or DNA-damaged cells, will also be of interest. Taken together, these experiments will provide a better understanding of the link between telomere ultrastructure and function.
DATA AVAILABILITY
The data that support the findings of this study are openly available in NTU research data repository DR-NTU (Data) at https://doi.org/10.21979/N9/LFBAF8.
Supplementary Material
ACKNOWLEDGEMENTS
We are indebted to Daniela Rhodes for her vision and support. We thank Andrew Wong, the NTU Institute of Structural Biology (NISB) and the Facility for Analysis, Characterisation, Testing, and Simulation (FACTS) in Singapore. We also thank Linda Sandblad and the Umeå Centre for Electron Microscopy in Sweden, Jan Karlseder and Mark Ellisman for support and discussions.
Author contributions: B.H., E.v.O. and B.A. performed all experiments, data collection and analysis, except for IP experiments that were performed by A.L., high-pressure freezing, freeze substitution and FIB-SEM by S.H., and serial section analysis by M.L.W. B.H. prepared all figures with the help of E.v.O., B.A. and M.L.W. S.S. and A.L. conceived and supervised the work. S.S., A.L. and B.H. wrote the manuscript.
Notes
Present address: Eric von Otter, Department of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge CB2 1GA, UK.
Present address: Bilal Ahsan, MRC Laboratory of Molecular Biology, Francis Crick Avenue, Cambridge Biomedical Campus, Cambridge CB2 0QH, UK.
Contributor Information
Barbara Hübner, School of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore; NTU Institute of Structural Biology, Nanyang Technological University, 59 Nanyang Drive, Singapore 636921, Singapore.
Eric von Otter, School of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore; NTU Institute of Structural Biology, Nanyang Technological University, 59 Nanyang Drive, Singapore 636921, Singapore.
Bilal Ahsan, School of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore; NTU Institute of Structural Biology, Nanyang Technological University, 59 Nanyang Drive, Singapore 636921, Singapore.
Mei Ling Wee, School of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore; NTU Institute of Structural Biology, Nanyang Technological University, 59 Nanyang Drive, Singapore 636921, Singapore.
Sara Henriksson, Umeå Centre for Electron Microscopy, Umeå University, Chemical Biological Centre (KBC) Building, Linnaeus väg 6, SE-90736 Umeå, Sweden.
Alexander Ludwig, School of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore; NTU Institute of Structural Biology, Nanyang Technological University, 59 Nanyang Drive, Singapore 636921, Singapore.
Sara Sandin, School of Biological Sciences, Nanyang Technological University, 60 Nanyang Drive, Singapore 637551, Singapore; NTU Institute of Structural Biology, Nanyang Technological University, 59 Nanyang Drive, Singapore 636921, Singapore.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Education, Singapore [MOE 2012-T3-1-001 to S.S.; MOE 2017-T1-002-067 to S.S. and A.L.]; Vetenskapsrådet, Sweden [National Microscopy Infrastructure (NMI) VR nr. 2019-00217; Molecular Infection Medicine in Sweden (MIMS) VR nr. 2016-06598]. Funding for open access charge: Ministry of Education, Singapore [MOE 2017-T1-002-067].
Conflict of interest statement. None declared.
REFERENCES
- 1. Blackburn E.H., Epel E.S., Lin J.. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science. 2015; 350:1193–1198. [DOI] [PubMed] [Google Scholar]
- 2. Nandakumar J., Cech T.R.. Finding the end: recruitment of telomerase to telomeres. Nat. Rev. Mol. Cell Biol. 2013; 14:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Apte M.S., Cooper J.P.. Life and cancer without telomerase: ALT and other strategies for making sure ends (don’t) meet. Crit. Rev. Biochem. Mol. Biol. 2017; 52:57–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Henson J.D., Neumann A.A., Yeager T.R., Reddel R.R.. Alternative lengthening of telomeres in mammalian cells. Oncogene. 2002; 21:598–610. [DOI] [PubMed] [Google Scholar]
- 5. Blackburn E.H. Structure and function of telomeres. Nature. 1991; 350:569–573. [DOI] [PubMed] [Google Scholar]
- 6. Gomes N.M., Ryder O.A., Houck M.L., Charter S.J., Walker W., Forsyth N.R., Austad S.N., Venditti C., Pagel M., Shay J.W.et al.. Comparative biology of mammalian telomeres: hypotheses on ancestral states and the roles of telomeres in longevity determination. Aging Cell. 2011; 10:761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Makarov V.L., Hirose Y., Langmore J.P.. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell. 1997; 88:657–666. [DOI] [PubMed] [Google Scholar]
- 8. Bianchi A., Smith S., Chong L., Elias P., de Lange T.. TRF1 is a dimer and bends telomeric DNA. EMBO J. 1997; 16:1785–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Broccoli D., Smogorzewska A., Chong L., de Lange T.. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat. Genet. 1997; 17:231–235. [DOI] [PubMed] [Google Scholar]
- 10. Baumann P., Cech T.R.. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001; 292:1171–1175. [DOI] [PubMed] [Google Scholar]
- 11. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005; 19:2100–2110. [DOI] [PubMed] [Google Scholar]
- 12. Lim C.J., Cech T.R.. Shaping human telomeres: from shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell Biol. 2021; 22:283–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. de Lange T. Shelterin-mediated telomere protection. Annu. Rev. Genet. 2018; 52:223–247. [DOI] [PubMed] [Google Scholar]
- 14. O’Sullivan R.J., Karlseder J.. Telomeres: protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010; 11:171–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Celli G.B., de Lange T.. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 2005; 7:712–718. [DOI] [PubMed] [Google Scholar]
- 16. van Steensel B., Smogorzewska A., de Lange T.. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998; 92:401–413. [DOI] [PubMed] [Google Scholar]
- 17. Makarov V.L., Lejnine S., Bedoyan J., Langmore J.P.. Nucleosomal organization of telomere-specific chromatin in rat. Cell. 1993; 73:775–787. [DOI] [PubMed] [Google Scholar]
- 18. Tommerup H., Dousmanis A., de Lange T.. Unusual chromatin in human telomeres. Mol. Cell. Biol. 1994; 14:5777–5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Azzalin C.M., Reichenbach P., Khoriauli L., Giulotto E., Lingner J.. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science. 2007; 318:798–801. [DOI] [PubMed] [Google Scholar]
- 20. Schoeftner S., Blasco M.A.. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat. Cell Biol. 2008; 10:228–236. [DOI] [PubMed] [Google Scholar]
- 21. Court R., Chapman L., Fairall L., Rhodes D. How the human telomeric proteins TRF1 and TRF2 recognize telomeric DNA: a view from high-resolution crystal structures. EMBO Rep. 2005; 6:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fairall L., Chapman L., Moss H., de Lange T., Rhodes D. Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Mol. Cell. 2001; 8:351–361. [DOI] [PubMed] [Google Scholar]
- 23. Soman A., Liew C.W., Teo H.L., Berezhnoy N.V., Olieric V., Korolev N., Rhodes D., Nordenskiold L.. The human telomeric nucleosome displays distinct structural and dynamic properties. Nucleic Acids Res. 2020; 48:5383–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen Y. The structural biology of the shelterin complex. Biol. Chem. 2019; 400:457–466. [DOI] [PubMed] [Google Scholar]
- 25. Smith E.M., Pendlebury D.F., Nandakumar J.. Structural biology of telomeres and telomerase. Cell. Mol. Life Sci. 2020; 77:61–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Griffith J.D., Comeau L., Rosenfield S., Stansel R.M., Bianchi A., Moss H., de Lange T.. Mammalian telomeres end in a large duplex loop. Cell. 1999; 97:503–514. [DOI] [PubMed] [Google Scholar]
- 27. Nikitina T., Woodcock C.L.. Closed chromatin loops at the ends of chromosomes. J. Cell Biol. 2004; 166:161–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Doksani Y., Wu J.Y., de Lange T., Zhuang X.. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell. 2013; 155:345–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Timashev L.A., Babcock H., Zhuang X., de Lange T. The DDR at telomeres lacking intact shelterin does not require substantial chromatin decompaction. Genes Dev. 2017; 31:578–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vancevska A., Douglass K.M., Pfeiffer V., Manley S., Lingner J.. The telomeric DNA damage response occurs in the absence of chromatin decompaction. Genes Dev. 2017; 31:567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lam S.S., Martell J.D., Kamer K.J., Deerinck T.J., Ellisman M.H., Mootha V.K., Ting A.Y.. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods. 2015; 12:51–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martell J.D., Deerinck T.J., Sancak Y., Poulos T.L., Mootha V.K., Sosinsky G.E., Ellisman M.H., Ting A.Y.. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 2012; 30:1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kremer J.R., Mastronarde D.N., McIntosh J.R.. Computer visualization of three-dimensional image data using IMOD. J. Struct. Biol. 1996; 116:71–76. [DOI] [PubMed] [Google Scholar]
- 34. Mastronarde D.N., Held S.R.. Automated tilt series alignment and tomographic reconstruction in IMOD. J. Struct. Biol. 2017; 197:102–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schindelin J., Rueden C.T., Hiner M.C., Eliceiri K.W.. The ImageJ ecosystem: an open platform for biomedical image analysis. Mol. Reprod. Dev. 2015; 82:518–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Schneider C.A., Rasband W.S., Eliceiri K.W.. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012; 9:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wickham H. ggplot2: Elegant Graphics for Data Analysis. 2016; NY: Springer-Verlag. [Google Scholar]
- 38. Wickham H., Averick M., Bryan J., Chang W., D’Agostino McGowan L., François R., Grolemund G., Hayes A., Henry L., Hester J.et al.. Welcome to the tidyverse. J. Open Source Softw. 2019; 4:1686. [Google Scholar]
- 39. Blasco M.A., Lee H.W., Hande M.P., Samper E., Lansdorp P.M., DePinho R.A., Greider C.W.. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997; 91:25–34. [DOI] [PubMed] [Google Scholar]
- 40. Kim N.W., Piatyszek M.A., Prowse K.R., Harley C.B., West M.D., Ho P.L., Coviello G.M., Wright W.E., Weinrich S.L., Shay J.W.. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994; 266:2011–2015. [DOI] [PubMed] [Google Scholar]
- 41. Bryan T.M., Englezou A., Dalla-Pozza L., Dunham M.A., Reddel R.R.. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 1997; 3:1271–1274. [DOI] [PubMed] [Google Scholar]
- 42. Billia F., de Boni U.. Localization of centromeric satellite and telomeric DNA sequences in dorsal root ganglion neurons, in vitro. J. Cell Sci. 1991; 100:219–226. [DOI] [PubMed] [Google Scholar]
- 43. Chuang T.C., Moshir S., Garini Y., Chuang A.Y., Young I.T., Vermolen B., van den Doel R., Mougey V., Perrin M., Braun M.et al.. The three-dimensional organization of telomeres in the nucleus of mammalian cells. BMC Biol. 2004; 2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Molenaar C., Wiesmeijer K., Verwoerd N.P., Khazen S., Eils R., Tanke H.J., Dirks R.W.. Visualizing telomere dynamics in living mammalian cells using PNA probes. EMBO J. 2003; 22:6631–6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ramirez M.J., Surralles J.. Laser confocal microscopy analysis of human interphase nuclei by three-dimensional FISH reveals dynamic perinucleolar clustering of telomeres. Cytogenet. Genome Res. 2008; 122:237–242. [DOI] [PubMed] [Google Scholar]
- 46. Weierich C., Brero A., Stein S., von Hase J., Cremer C., Cremer T., Solovei I.. Three-dimensional arrangements of centromeres and telomeres in nuclei of human and murine lymphocytes. Chromosome Res. 2003; 11:485–502. [DOI] [PubMed] [Google Scholar]
- 47. Martinez P., Thanasoula M., Munoz P., Liao C., Tejera A., McNees C., Flores J.M., Fernandez-Capetillo O., Tarsounas M., Blasco M.A.. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009; 23:2060–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sfeir A., Kosiyatrakul S.T., Hockemeyer D., MacRae S.L., Karlseder J., Schildkraut C.L., de Lange T.. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009; 138:90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Takai H., Smogorzewska A., de Lange T.. DNA damage foci at dysfunctional telomeres. Curr. Biol. 2003; 13:1549–1556. [DOI] [PubMed] [Google Scholar]
- 50. Cremer M., Muller S., Kohler D., Brero A., Solovei I.. Cell preparation and multicolor FISH in 3D preserved cultured mammalian cells. CSH Protoc. 2007; 2007:pdb.prot4723. [DOI] [PubMed] [Google Scholar]
- 51. Hübner B., Lomiento M., Mammoli F., Illner D., Markaki Y., Ferrari S., Cremer M., Cremer T.. Remodeling of nuclear landscapes during human myelopoietic cell differentiation maintains co-aligned active and inactive nuclear compartments. Epigenetics Chromatin. 2015; 8:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Politz J.C., Scalzo D., Groudine M.. Something silent this way forms: the functional organization of the repressive nuclear compartment. Annu. Rev. Cell Dev. Biol. 2013; 29:241–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Stanyte R., Nuebler J., Blaukopf C., Hoefler R., Stocsits R., Peters J.M., Gerlich D.W.. Dynamics of sister chromatid resolution during cell cycle progression. J. Cell Biol. 2018; 217:1985–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lansdorp P.M., Verwoerd N.P., van de Rijke F.M., Dragowska V., Little M.T., Dirks R.W., Raap A.K., Tanke H.J.. Heterogeneity in telomere length of human chromosomes. Hum. Mol. Genet. 1996; 5:685–691. [DOI] [PubMed] [Google Scholar]
- 55. Zijlmans J.M., Martens U.M., Poon S.S., Raap A.K., Tanke H.J., Ward R.K., Lansdorp P.M.. Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc. Natl Acad. Sci. U.S.A. 1997; 94:7423–7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schmutz I., Timashev L., Xie W., Patel D.J., de Lange T. TRF2 binds branched DNA to safeguard telomere integrity. Nat. Struct. Mol. Biol. 2017; 24:734–742. [DOI] [PubMed] [Google Scholar]
- 57. Bandaria J.N., Qin P., Berk V., Chu S., Yildiz A.. Shelterin protects chromosome ends by compacting telomeric chromatin. Cell. 2016; 164:735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Luderus M.E., van Steensel B., Chong L., Sibon O.C., Cremers F.F., de Lange T.. Structure, subnuclear distribution, and nuclear matrix association of the mammalian telomeric complex. J. Cell Biol. 1996; 135:867–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pierron G., Puvion-Dutilleul F.. An anchorage nuclear structure for telomeric DNA repeats in HeLa cells. Chromosome Res. 1999; 7:581–592. [DOI] [PubMed] [Google Scholar]
- 60. Ellisman M.H., Deerinck T.J., Shu X., Sosinsky G.E.. Picking faces out of a crowd: genetic labels for identification of proteins in correlated light and electron microscopy imaging. Methods Cell Biol. 2012; 111:139–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shu X., Lev-Ram V., Deerinck T.J., Qi Y., Ramko E.B., Davidson M.W., Jin Y., Ellisman M.H., Tsien R.Y.. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011; 9:e1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lejnine S., Makarov V.L., Langmore J.P.. Conserved nucleoprotein structure at the ends of vertebrate and invertebrate chromosomes. Proc. Natl Acad. Sci. U.S.A. 1995; 92:2393–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Blasco M.A. The epigenetic regulation of mammalian telomeres. Nat. Rev. Genet. 2007; 8:299–309. [DOI] [PubMed] [Google Scholar]
- 64. Galati A., Micheli E., Cacchione S.. Chromatin structure in telomere dynamics. Front. Oncol. 2013; 3:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Episkopou H., Draskovic I., Van Beneden A., Tilman G., Mattiussi M., Gobin M., Arnoult N., Londono-Vallejo A., Decottignies A.. Alternative lengthening of telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014; 42:4391–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Olins A.L., Olins D.E.. Spheroid chromatin units (ν bodies). Science. 1974; 183:330–332. [DOI] [PubMed] [Google Scholar]
- 67. Thoma F., Koller T., Klug A.. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J. Cell Biol. 1979; 83:403–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Scheffer M.P., Eltsov M., Frangakis A.S.. Evidence for short-range helical order in the 30-nm chromatin fibers of erythrocyte nuclei. Proc. Natl Acad. Sci. U.S.A. 2011; 108:16992–16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Horowitz R.A., Agard D.A., Sedat J.W., Woodcock C.L.. The three-dimensional architecture of chromatin in situ: electron tomography reveals fibers composed of a continuously variable zig-zag nucleosomal ribbon. J. Cell Biol. 1994; 125:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fussner E., Strauss M., Djuric U., Li R., Ahmed K., Hart M., Ellis J., Bazett-Jones D.P.. Open and closed domains in the mouse genome are configured as 10-nm chromatin fibres. EMBO Rep. 2012; 13:992–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hansen J.C., Connolly M., McDonald C.J., Pan A., Pryamkova A., Ray K., Seidel E., Tamura S., Rogge R., Maeshima K.. The 10-nm chromatin fiber and its relationship to interphase chromosome organization. Biochem. Soc. Trans. 2018; 46:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Maeshima K., Imai R., Tamura S., Nozaki T.. Chromatin as dynamic 10-nm fibers. Chromosoma. 2014; 123:225–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ou H.D., Phan S., Deerinck T.J., Thor A., Ellisman M.H., O’Shea C.C. ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017; 357:eaag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li B., Oestreich S., de Lange T.. Identification of human Rap1: implications for telomere evolution. Cell. 2000; 101:471–483. [DOI] [PubMed] [Google Scholar]
- 75. Takai K.K., Hooper S., Blackwood S., Gandhi R., de Lange T.. In vivo stoichiometry of shelterin components. J. Biol. Chem. 2010; 285:1457–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mattern K.A., Swiggers S.J., Nigg A.L., Lowenberg B., Houtsmuller A.B., Zijlmans J.M.. Dynamics of protein binding to telomeres in living cells: implications for telomere structure and function. Mol. Cell. Biol. 2004; 24:5587–5594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Amiard S., Doudeau M., Pinte S., Poulet A., Lenain C., Faivre-Moskalenko C., Angelov D., Hug N., Vindigni A., Bouvet P.et al.. A topological mechanism for TRF2-enhanced strand invasion. Nat. Struct. Mol. Biol. 2007; 14:147–154. [DOI] [PubMed] [Google Scholar]
- 78. Baker A.M., Fu Q., Hayward W., Victoria S., Pedroso I.M., Lindsay S.M., Fletcher T.M.. The telomere binding protein TRF2 induces chromatin compaction. PLoS One. 2011; 6:e19124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Anton T., Bultmann S., Leonhardt H., Markaki Y.. Visualization of specific DNA sequences in living mouse embryonic stem cells with a programmable fluorescent CRISPR/Cas system. Nucleus. 2014; 5:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cerda M.C., Berrios S., Fernandez-Donoso R., Garagna S., Redi C.. Organisation of complex nuclear domains in somatic mouse cells. Biol. Cell. 1999; 91:55–65. [PubMed] [Google Scholar]
- 81. Guttenbach M., Martinez-Exposito M.J., Engel W., Schmid M.. Interphase chromosome arrangement in Sertoli cells of adult mice. Biol. Reprod. 1996; 54:980–986. [DOI] [PubMed] [Google Scholar]
- 82. Dillinger S., Straub T., Nemeth A.. Nucleolus association of chromosomal domains is largely maintained in cellular senescence despite massive nuclear reorganisation. PLoS One. 2017; 12:e0178821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tam R., Smith K.P., Lawrence J.B.. The 4q subtelomere harboring the FSHD locus is specifically anchored with peripheral heterochromatin unlike most human telomeres. J. Cell Biol. 2004; 167:269–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Crabbe L., Cesare A.J., Kasuboski J.M., Fitzpatrick J.A., Karlseder J.. Human telomeres are tethered to the nuclear envelope during postmitotic nuclear assembly. Cell Rep. 2012; 2:1521–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Vourc’h C., Taruscio D., Boyle A.L., Ward D.C.. Cell cycle-dependent distribution of telomeres, centromeres, and chromosome-specific subsatellite domains in the interphase nucleus of mouse lymphocytes. Exp. Cell Res. 1993; 205:142–151. [DOI] [PubMed] [Google Scholar]
- 86. Tenga R., Medalia O.. Structure and unique mechanical aspects of nuclear lamin filaments. Curr. Opin. Struct. Biol. 2020; 64:152–159. [DOI] [PubMed] [Google Scholar]
- 87. de Lange T. Human telomeres are attached to the nuclear matrix. EMBO J. 1992; 11:717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Pennarun G., Picotto J., Etourneaud L., Redavid A.R., Certain A., Gauthier L.R., Fontanilla-Ramirez P., Busso D., Chabance-Okumura C., Theze B.et al.. Increase in lamin B1 promotes telomere instability by disrupting the shelterin complex in human cells. Nucleic Acids Res. 2021; 49:9886–9905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Travina A.O., Ilicheva N.V., Mittenberg A.G., Shabelnikov S.V., Kotova A.V., Podgornaya O.I.. The long linker region of telomere-binding protein TRF2 is responsible for interactions with lamins. Int. J. Mol. Sci. 2021; 22:3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are openly available in NTU research data repository DR-NTU (Data) at https://doi.org/10.21979/N9/LFBAF8.