

Abstract
We reported previously that the selective agonist U50,488H promoted phosphorylation of the mouse kappa opioid receptor (mKOR) in vitro at four residues in the C-terminal domain. In this study, we generated a mutant mouse line in which all the four residues were mutated to Ala (K4A) to examine the in vivo functional significance of agonist-induced KOR phosphorylation. U50,488H promoted KOR phosphorylation in brains of the wildtype (WT), but not K4A, male and female mice. Autoradiography of [3H] 69,593 binding to KOR in brain sections showed that WT and K4A mice had similar KOR distribution and expression levels in brain regions without sex differences. In K4A mice, U50,488H inhibited compound 48/80-induced scratching and attenuated novelty-induced hyperlocomotion to similar extents as in WT mice without sex differences. Interestingly, repeated pretreatment with U50,488H (80 mg/kg, s.c.) resulted in profound tolerance to the anti-scratch effects of U50,488H (5 mg/kg, s.c.) in WT mice of both sexes and female K4A mice, while in male K4A mice tolerance was attenuated. Moreover, U50,488H (2 mg/kg) induced conditioned place aversion (CPA) in WT mice of both sexes and male K4A mice, but not in female K4A mice. In contrast, U50,488H (5 mg/kg) caused CPA in male, but not female, mice, regardless of genotype. Thus, agonist-promoted KOR phosphorylation plays important roles in U50,488H-induced tolerance and CPA in a sex-dependent manner, without affecting acute U50,488H-induced anti-pruritic and hypo-locomotor effects. These results are the first to demonstrate sex differences in the effects of GPCR phosphorylation on the GPCR-mediated behaviors.
Keywords: kappa opioid receptor, phosphorylation, sex differences, tolerance, aversion
1. Introduction
The kappa opioid receptor (KOR), one of the three opioid receptors, belongs to the rhodopsin subfamily of G protein-coupled receptors (GPCRs). At the cellular level, activation of the KOR stimulates Gi/o proteins and promotes receptor phosphorylation. Phosphorylated KOR, in turn, recruits β-arrestins, which leads to desensitization and internalization of the receptor (Liu-Chen, 2004) as well as β-arrestin-mediated signaling (Bruchas and Chavkin, 2010; Brust, 2020; Cahill et al., 2021). Activation of Gi/o proteins inhibits adenylyl cyclases, reduces Ca++ channel conductance and enhances activities of K+ channels and ERK1/2 (early phase), and β-arrestins-mediated signaling includes activation of ERK1/2 (late phase) and p38 MAPK (Bruchas and Chavkin, 2010; Brust, 2020; Cahill et al., 2021). Activation of the KOR in vivo produces many effects, including analgesia, antipruritic effects, water diuresis, dysphoria / aversion, psychotomimesis, sedation, motor incoordination and hypothermia (von Voigtlander et al., 1983; Simonin et al., 1998; Cowan et al., 2015; Clark and Abi-Dargham, 2019; Cahill et al., 2021). KOR agonists produce analgesic effects without causing respiratory depression and abuse potentials associated with mu opioid receptor (MOR)-selective or preferring analgesics (Yaksh and Wallace, 2018). KOR agonists are also promising as antipruritic agents; however, their development has been hampered by dysphoric and psychotomimetic effects (Pfeiffer et al., 1986; Pande et al., 1996; Wadenberg, 2003).
Biased agonism or functional selectivity is a prevailing concept in GPCR pharmacology [reviewed in (Luttrell et al., 2015; Rankovic et al., 2016)]. Biased agonists preferentially activate G protein- or arrestin-mediated signaling and are thought to produce therapeutic effects with fewer side effects (Rankovic et al., 2016; Brust, 2020; Grim et al., 2020). It has been reported that KOR-promoted conditioned place aversion (CPA) was GRK3- and p38 MAPK-dependent (Bruchas et al., 2007; Bruchas et al., 2011; Ehrich et al., 2015; Abraham et al., 2018). However, studies performed on wildtype (WT) and β-arrestin2−/− (β-arr2 KO) mice have revealed that β-arr2 deletion attenuated the KOR agonist U69,593-induced motor incoordination, but did not affect U69,593-promoted CPA, analgesia, and hypolocomotion (White et al., 2015) or antipruritic effect induced by U50,488H, another selective KOR agonist (Morgenweck et al., 2015).
We previously determined that U50,488H promoted phosphorylation of the mouse KOR at S356, T357, T363 and S369 in the C terminal domain (Chen et al., 2016). In this study, we examined the roles of agonist-promoted KOR phosphorylation in KOR-mediated behavioral responses by generating and characterizing both male and female mice harboring S356A/T357A/T363A/S369A mutations in the KOR (K4A). K4A mutations created a KOR mutant that did not undergo agonist-promoted phosphorylation, i.e. a G protein-biased KOR. This mutant strain allowed determination of functional significance of agonist-promoted KOR phosphorylation.
2. Material and methods
2.1. Materials
Mouse N2A (N2A) cells were purchased from the A.T.C.C. (Manassas, VA, U.S.A.). (±)U50,488H, nalfurafine and nor-BNI were provided by the National Institute on Drug Abuse (Bethesda, MD, U.S.A.). Protease inhibitor cocktail was obtained from Pierce, Thermo Sci. (Rockford,IL). The rabbit anti-KOR c-tail (KC) (PA847), guinea pig anti-KC (PA5699) and rabbit anti-phospho-KOR antibodies (anti-pT363 and anti-pS369) were custom-developed by Covance and purified in our own laboratory (Chen et al., 2016). [35S]GTPᵧS (~1,250 Ci/mmol), [3H]diprenorphine (58 Ci/mmol), and [32P]orthophosphate (8,500–9,100 Ci/mmol) were purchased from PerkinElmer Life Science (Boston, MA). Pansorbin was obtained from Calbiochem, EMD Millipore (Billerica, MA). Compound 48/80 and Dynorphin A (1-17) were purchased from Sigma-Aldrich (St Louis, MO).
2.2. cDNA constructs of KOR and K4A
The cDNA constructs of wildtype mouse KOR (mKOR) and the four-alanine substitute for S356, T357, T363 and S369 (K4A) were generated as described previously (Chen et al., 2016). Briefly, the mKOR cDNA was FLAG-tagged at N-terminus by PCR and inserted into KpnI/AgeI sites of pcDNA6/6His vector with a blasticidin-resistance gene. The double tagged FLAG-mKOR-6His design was to facilitate purification of the receptor. The alanine mutants were introduced by overlap PCR to the wildtype.
2.3. Cell culture
Mouse N2A neuroblastoma cells were used in the studies because of their neuron-like nature. Cells were transfected with the KOR or K4A construct and grown in the presence of 5 μg/ml blasticidin and clonal cells stably expressing the receptor were established. Cells were maintained in 10% FBS, 1 μg/ml blasticidin and MEM at 37°C in a humidified 5% CO2 atmosphere.
2.4. Examination of phosphorylation deficiency of K4A
[32P]orthophosphate metabolic labeling of N2A-KOR or N2A-K4A cells was conducted according to a procedure described previously (Li et al., 2002). Briefly, the cells were cultured overnight in 6-well plates to 80% confluence, labeled with [32P]orthophosphate (0.25 mCi/well), treated with or without 10 μM (±)U50,488H for 30 min at 37°C. The KOR or K4A proteins were immunoprecipitated (IP) using the rabbit anti-KOR antibody (code PA847), resolved by 8% SDS-PAGE and visualized by autoradiography.
2.5. Membrane preparation from N2A-KOR and N2A-K4A cells
Membranes used for both receptor binding and [35S]GTPᵧS binding were prepared according to Li et al. (2002) with some modifications. Briefly, N2A-KOR or N2A-K4A cells were pelleted, passed through a 29G3/8 syringe needle five times and centrifuged. Pellets were resuspended in 50 mM Tris-HCl buffer (pH7.0) and the protein concentration was determined by BCA assay.
2.6. Saturation binding of [3H]Diprenorphine to KOR or K4A and competitive inhibition of [3H]Diprenorphine binding to KOR or K4A by KOR ligands
Receptor binding studies were carried out with a procedure modified from that of Huang et al. (2001). Briefly, Saturation bindings of [3H]diprenorphine to KOR or K4A were performed with at least six concentrations of [3H]diprenorphine (ranging from 25 pM to 1–2 nM) with N2A-KOR or N2A-K4A membranes (∼10 to 20 μg of protein). Binding was carried out in 50 mM Tris-HCl buffer containing 1 mM EGTA (pH 7.4) at room temperature for 1 h. Naloxone (10 μM) was used to define nonspecific binding. The Kd and Bmax values were determined. Competition binding of KOR or K4A by several KOR ligands against [3H]diprenorphine (0.4 nM) were performed with various concentrations of each ligand. Data were analyzed with Prism (GraphPad Software, San Diego, CA).
2.7. Agonist-induced [35S]GTPᵧS binding to membranes of N2A-KOR or N2A-K4A cells
Determination of [35S]GTPᵧS binding to G proteins was carried out with a procedure modified from that of Huang et al. (2001). For each experiment, ~10 μg of membrane protein was incubated with 15 μM GDP and 0.2 nM [35S]GTPᵧS in the reaction buffer (50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, 1 mM EDTA, and 0.1% bovine serum albumin, pH 7.4) in a final volume of 0.5 ml. Nonspecific binding was determined in the presence of 10 μM GTPᵧS. Seven concentrations (10 pM–10 μM) of (±)U50,488H, nalfurafine and dynorphin A (1-17) (DynA(1-17)) were used to generate dose-response curves. After 60 min of incubation at 30°C, bound and free [35S]GTPᵧS were separated by filtration with GF/B filters presoaked with water. Radioactivity on filters was determined by liquid scintillation counting. Data were analyzed with Prism (GraphPad Software, San Diego, CA).
2.8. Generation, breeding and genotyping of the K4A knockin mice
A K4A knockin mouse line was custom-generated by Cyagen Co. (Santa Clara, CA) using the homologous recombination approach in C57BL/6N genetic background. The targeting strategy is shown in Fig. 1A targeting vector was constructed in which the KOR gene (Oprk1) was modified so that a floxed neomycin resistant gene was inserted in the intron upstream of the exon 4 and S356A /T357A /T363A /S369A (K4A) mutations were introduced in the exon 4 (Fig. 1). This construct was then transfected into embryonic stem (ES) cells. A positive ES clone with proper homologous recombination was electroporated with a Cre-expressing plasmid to excise the neomycin gene and subsequently microinjected into C57BL/6N blastocysts. The resulting animals were cross-bred with C57BL/6N mice to obtain F1 heterozygous progenies. Heterozygous mice were bred to obtain wildtype and homozygous K4A mice. Age-matched male and female homozygous K4A and their wild-type (WT) littermates weighing 20–35 g (2-8 months old) were used. Breeding was also carried out among homozygous K4A or WT mice produced by K4A heterozygous parents. Animals were group-housed under standard laboratory conditions and kept on a 12 h day/night cycle (lights on at 7:00 A.M.). Mice were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All methods used were preapproved by the Institutional Animal Care and Use Committee at the Temple University.
Figure 1. Targeting strategy for generation of K4A knockin mice.
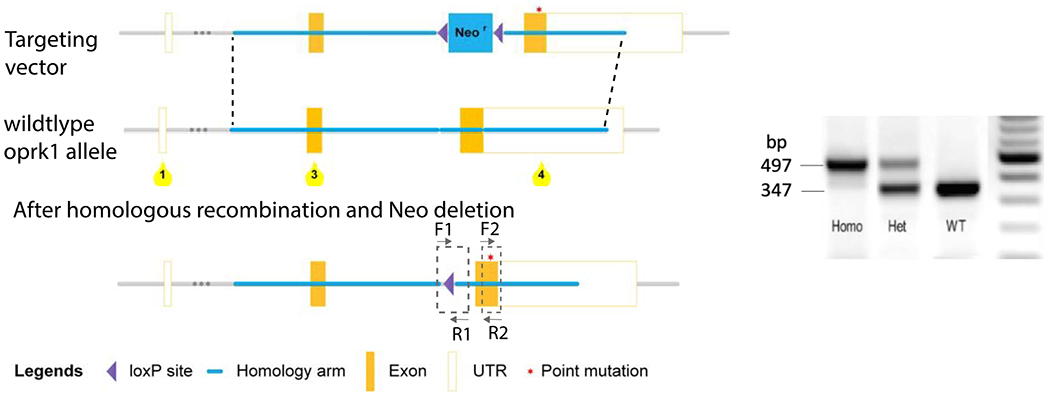
Oprk1 exons 3 and 4, and the floxed neomycin cassette are shown as orange and blue, boxes, respectively. The S356A /T357A /T363A /S369A (K4A) mutations are indicated by a red asterisk. Knockin was accomplished by homologous recombination and Neo cassette was self-deleted in germ cells. Genotyping is performed with F1 and R1 primers for detection of the remaining loxP site in the K4A mutant mice. The sizes of PCR products of K4A and KOR were 497 bp and 347 bp, respectively
Mouse genotyping was carried out by PCR with total DNA isolated from mouse ears. To detect the excision of the selection marker of knockin allele, the primer pair F1/R1 was used [5’-CTGATACAGTACAATACTGAGGACTCT-3’ (forward) / 5’-GCTCTCTTTCATTAACTGTTGTTGC-3’ (reverse)] as diagramed in Fig. 1.
2.9. Solubilization of brains, immunoprecipitation (IP) of KOR and immunoblotting (IB) of phosphorylated and total KOR
The protocol was described in detail previously (Chen et al., 2020; Liu et al., 2020). Briefly, four mouse brains were pooled as one sample, homogenized and solubilized with 12 ml buffer A. KOR or K4A in the supernatant was immunoprecipitated twice sequentially, firstly with rabbit anti-KC (PA847) and secondly with guinea pig anti-KC (PA5699) using Pansorbin as carriers. The enriched KOR or K4A proteins were resolved by SDS-PAGE (precast gel 4–12%, Invitrogen), transferred onto PVDF membranes, blotted with rabbit anti-pThr363 or anti-pSer369 (1 μg/ml) overnight followed by HRP conjugated mouse anti-rabbit IgG light chain and visualized with enhanced chemiluminescence reagents. Images were captured using LAS 1000 plus camera (Fuji Photo Film Co.) and quantified using the ImageGauge software (Version 4.1, Fuji Photo Film Co.). The blot was stripped and re-blotted with rabbit anti-KC (PA847) for 1 h for total KOR.
Brains of KOR knockout mice were used for characterization of specificity of the anti-pThr363 and anti-pSer369 antibodies. KOR knockout mice were originally generated by Dr. Brigitte Kieffer’s lab by deletion of part of the exon 1 (Simonin et al., 1998). KOR knockout mice were purchased from Jackson Labs (Bar Harbor, ME) (B6.129S2-Oprk1tm1Kff/J Stock No: 007558) and bred in house by the Animal Core of the P30 Excellence Center in our center. Wildtype C57BL/6J mice (Jackson Labs, Stock No: 000664) were used as the control.
2.10. Autoradiography of [3H]U69,593 binding to KOR in mouse brains
The experiments were performed per our published procedures (Wang et al., 2011; Chen et al., 2020). Briefly, WT or K4A mice were euthanized, brains were dissected and immediately frozen in isopentane on dry ice. Coronal sections of the brains were sliced at 20 μm, thaw-mounted onto gelatin-subbed slides and dried at room temperature for one hour, stored in −80°C until use. The slides were incubated with ~5 nM [3H]U69,593 with or without 10 μM naloxone in 50 mM Tris-HCl buffer (pH7.4) at room temperature for 1 hour, rinsed three times with ice-cold 50 mM Tris-HCl buffer and once with deionized water, dried with cold air blower. The slides were then exposed to 3H-sensitive phosphor screens for about 3 weeks and images on the screens were captured with a Cyclone Storage Phosphor Scanner (Packard Bioscience, Meriden, CT).
2.11. Inhibition of compound 48/80-induced scratching by U50,488H
The experiment was performed according to our published procedures (Liu et al., 2019a; Liu et al., 2020). On the day of the test, animals were allowed to acclimate to individual rectangular observation Plexiglas boxes for 1 h and then injected s.c. with either saline or U50,488H in volumes of 0.1 ml/10 g body weight. Twenty min later, animals were injected s.c. with 0.1 ml of compound 48/80 (0.5 mg/ml in saline; 50 μg) into the nape of the neck. Compound 48/80 causes release of histamine from mast cells and produces scratching behavior in mice that peaks within 30 min after injection and subsides within 60 min. Immediately after injection of compound 48/80, animals were observed for 36 min and the number of bouts of hindleg scratching movements directed to the neck were counted. Data were normalized to the mean values for scratching of saline groups and then plotted vs. dose of U50,488H (on a log scale). The mean A50 values were determined by nonlinear regression analysis (Prism 5, GraphPad, San Diego, CA).
2.12. U50,488H tolerance studies
Induction of U50,488H tolerance was carried out following procedures of Suzuki et al. (2004) with some modifications. Mice were subcutaneously administered with saline or 80 mg/kg (s.c.) of U50,488H five times over a 3-day period (twice a day on days1 and 2 at 10 am and 5 pm, then once a day on day 3 at 10 am). In the afternoon on day 4, mice were injected with saline or 5 mg/kg of U50,488H. Twenty min later, compound 48/80 was into the nape of the mouse necks. Scratching bouts were recorded as described in detail as in 2.11.
2.13. CPA
The 5-day CPA experiments were performed per our published procedures with some modifications using two-chamber conditioning boxes (Xu et al., 2013; Liu et al., 2019a) with some modifications. In this behavioral paradigm, the apparatus is a rectangular Plexiglas container with two distinct compartments that can be closed off by a partition. One compartment of the apparatus has checker walls and a blue light (5W) on top and the opposite one has white walls and a red light (5W) on top. Before experiments, mice were acclimated to the testing room for at least 1h on day1 and for at least 30 min on days 2-5. During the pre-test on Day 1, animals were allowed to roam freely between the two compartments for 15 min, and the time each animal spent on either side was recorded. Mice that spent more than 570 s (>63%) on one side were excluded from the study. An unbiased and counterbalanced design was used. In the 3-day conditioning phase (day 2-day 4), two sessions (AM and PM, ~4 hrs apart) per day were conducted. In the AM sessions, mice were given injections of saline s.c., and in the PM sessions, saline s.c. or U50,488H was given to mice. After injections mice were immediately placed back to their home cages for 10 min and then confined to one or the opposite compartment for 30 min for AM or PM sessions, respectively, which allowed the mice to establish a contextual basis for their responses to the drug. Mice were randomized as to which compartment they received the U50,488H injection. The PM compartments were defined as the drug-paired side. On post-test day (Day 5), the animals were once again allowed to freely roam between the two compartments for 15 minutes, and time spent in each compartment was recorded. The mouse preference score was the difference of time a mouse spent in the drug paired-side of post-test vs pre-test.
2.14. Locomotor activities
Locomotor activities will be measured using Home Cage Locomotor Activity system (Omnitech Electronics Inc., Columbus, OH). Each locomotor chamber frame can accommodate a transparent plastic cage (45cm× 20cm× 20cm) and has a pair of sensor panels with a set of 16 light beams in a horizontal direction. When a mouse interrupted light beams, the connected Fusion software recorded and determined the location of mice along X-axis. Breaking of two consecutive light beams by a mouse was interpreted as ambulatory activity. The total travel distance (cm) was the distance traveled along the horizontal direction. Adult mice were treated with U50,488H and immediately placed into the locomotor activity chambers for activity recording in 5-min bins for 1 hr. Total distance traveled (cm) and ambulatory activity were recorded with the Fusion software.
2.15. Statistics
Data were graphed and analyzed with Graph Pad Prism5 (San Diego, CA). One-way ANOVA was used to analyze one-factor experiment, followed by Tukey post-hoc test. Two-way ANOVA was used to analyze two-factor experiments followed by Bonferroni post-hoc test.
3. Results
3.1. S356, T357, T363 and S369 are the only phosphorylation sites in the mouse KOR following U50,488H stimulation
Prior to generation of a K4A knockin mouse line, we examined if there were other phosphorylation sites, in addition to the four sites identified (Chen et al., 2016) because in the LC-MS/MS analyses, only the C-terminal domain Glu-C proteolytic fragments were large enough for analyses. We constructed a mouse KOR mutant cDNA with all the four residues mutated to alanines (K4A) and stably expressed the mutant and the wildtype KOR in neuro2A mouse neuroblastoma cells (N2A-K4A and N2A-KOR, respectively). We investigated whether the mouse KOR-4A underwent U50,488H-induced phosphorylation by metabolic labeling of N2A-K4A and N2A-KOR cells with [32P]orthophosphate. Following incubation of cells with vehicle or 10 μM U50,488H for 30 minutes. KOR was immunoprecipitated, the precipitated mixture was resolved with SDS-PAGE. By autoradiography, we found that U50,488H induced robust phosphorylation of the wildtype KOR, but no phosphorylation of K4A, indicating that there are no phosphorylation sites other than the four sites identified (Supplemental Fig.1).
3.2. K4A mutations did not affect binding affinity of KOR ligands or potency and efficacy of KOR agonists in [35S]GTPᵧS binding
We then examined the effect of K4A mutations on binding affinity of KOR ligands. Saturation binding of [3H]diprenorphine was conducted with membranes of N2A-K4A and N2A-KOR cells. K4A did not appreciably affect Kd value of [3H]diprenorphine (Supplemental Table 1). We identified a clonal N2A-K4A cell line with a similar Bmax value as a N2A-KOR cell line (1.14 ± 0.02 and 1.08 ± 0.02 pmole/mg protein, respectively) to perform the subsequent binding experiments. Binding affinity of KOR ligands was determined by competitive inhibition of [3H]diprenorphine binding to KOR or K4A. As shown in Supplemental Table 1, K4A mutations did not affect Ki values of U50,488H, nalfurafine, dynorphin A (1-17) or norbinaltorphimine (norBNI).
We next investigated effects of K4A mutations on agonist-induced [35S]GTPᵧS binding. K4A mutations did not affect EC50 values and maximal effects of U50,488H, nalfurafine and dynorphin A (1-17) (Supplemental Table 2). Thus, K4A mutations did not affect KOR-G protein coupling.
3.3. Generation and genotyping of the K4A knockin mice
To investigate the in vivo functional significance of agonist-induced KOR phosphorylation, we custom-generated a K4A knockin mouse line. The targeting strategy is shown in Fig. 1. The mutations are indicated with an asterisk within the exon 4. After homologous recombination and deletion of the Neo cassette, the knockin allele has a loxP site, which was used for genotyping using the F1 and R1 primers (Fig. 1). The sizes of PCR products of K4A and KOR were 497 bp and 347 bp, respectively (Fig. 1). The K4A mice were fertile and appeared to develop and breed normally and their overall appearance and behaviors are indistinguishable from those of the WT.
3.4. K4A mutations abolished U50,488H-induced KOR phosphorylation
We demonstrated previously that T363 and S369 were primary phosphorylation sites and phosphorylation of T363 and S369 is required for S356/T357 phosphorylation (Chen et al., 2016). Therefore, our phosphorylation studies were focused on T363 and S369. We first examined whether our previously developed phospho-KOR antibodies (anti-pT363 and anti-pS369) (Chen et al., 2016) were specific for immunoblotting (IB) of phospho-KOR in mouse brains. Wildtype (WT) and KOR−/− (KO) C57BL/6 adult mice were injected with saline or U50,488H (5 mg/kg, s.c.) and euthanized 30 min later and brains were immediately removed and frozen. Four brains were pooled as one sample because of the low expression level of KOR. Phosphorylated KOR had to be enriched by IP before IB because of the low level of KOR and high complexity of brain proteins. KOR in solubilized membranes was immunoprecipitated twice and IB was performed with the rabbit anti-pT363 or anti-pS369 for detection of phosphorylated KOR or rabbit antibodies against KOR(368-380) for total KOR.
In wildtype C57BL/6N mice, U50,488H treatment greatly increased the intensity of anti-pT363 and anti-pS369 staining in brains (Fig. 2A). In contrast, in the brains of KOR knockout mice, there was no staining by either antibody following saline or U50,488H pretreatment (Chen et al., 2021), indicating specificity for the phosphorylated KOR.
Figure 2. Immunoblotting of phosphorylated KOR in mouse brains.

(A) Wildtype (WT), KOR−/− (KO) or K4A adult mice were injected with saline or U50,488H (5 mg/kg, s.c.) and euthanized 30 min later and brains were immediately removed and frozen. Four brains were pooled as one sample because of the low expression level of KOR. Brains were solubilized. KOR in the cleared supernatant was enriched by two-step immunoprecipitation as described in Material and Methods. The enriched KOR was dissolved with Laemmli buffer. The mixture was resolved with SDS-PAGE followed by IB with rabbit anti-pT363 and anti-pS369 antibodies.
(A)(B) U50,488H greatly enhanced anti-pT363 and anti-pS369 staining in the wildtype mice, but not in (A) KOR−/− mice (Chen et al., 2021) or (B) K4A mice. In the wildtype, the p-KOR band appeared as a broad diffuse band with a median Mr of 60 ~kDa. (A) KOR−/− mice did not show any staining in saline- or U50,488H-treated group, indicating that anti-pT363 and anti-pS369 are specific for the phosphorylated KOR in the mouse brain (Chen et al., 2021). (B) In male and female K4A mice, there was no anti-pT363 and anti-pS369 staining in saline- or U50,488H-treated group, indicating that K4A mutations abolished U50,488H-induced KOR phosphorylation.
(A)(B) Following staining with pT363 and pS369 antibodies, the blot was stripped and re-blotted for total KOR with purified rabbit antibodies against the KOR(371-380) peptide (PA847, customed generated, antigen affinity purified). The experiment was performed twice with similar results.
We then conducted similar experiments with WT and K4A mice. U50,488H treatment enhanced the intensity of anti-pT363 and anti-pS369 staining in WT brains of both male and female mice, but it had no effect in the brains of K4A mice of both sexes (Fig. 2B). Thus, K4A mutations abolished U50,488H-promoted KOR phosphorylation at T363 and S369 in both male and female mouse brains.
3.5. K4A mutations did not affect KOR distribution and level of the KOR in male or female mouse brains
We next examined whether K4A mutations affect distribution and expression levels of the KOR in mouse brain by autoradiography of [3H]U69,593 (~5 nM) binding to the KOR in coronal brain sections. Naloxone (10 μM) was used to define non-specific binding, which was negligible (not shown). In brains of male and female WT and K4A mice, KOR had similar distributions (Fig. 3). The KOR levels were highest in the claustrum-endopiriform nucleus, and moderate levels were observed in the amygdala, nucleus accumbens, hypothalamus, periaqueductal gray, substantia nigra and deep layers of cortex. In each of these brain regions, there was no significant difference (Student’s t test, Prism 5) in KOR levels between WT and K4A (Fig. 3).
Figure 3. Autoradiograms of [3H]U69593 (~5 nM) binding to the KOR in coronal sections of the male WT and K4A mouse brains at 4 anatomical levels.

Sections were incubated with ~5 nM [3H]U69,593 for 1 hour at 25°C, washed, dried and exposed to tritium-sensitive phosphor screens. Exposure time was 3 weeks. Quantitation was done from both left and right sides of each section. Data are the mean ± S.E.M. of three animals, 2-10 sections from each brain depending on brain regions. Abbreviations(labels): Acb(3), nucleus accumbens; Amg(4), amygdala; Cl(1), claustrum; En(2), endopiriform nucleus; Hypo(5), hypothalamus; PAG(6), periaqueductal gray; SN(7), substantia nigra.
In addition, two-way ANOVA analyses showed that there were no differences in KOR expression in brain regions between male and female mice (K4A or WT), except for claustrum. For claustrum, KOR expression levels in females were significantly higher than in males [F(1,8)=7.23, p=0.028], while there were no main effects in genotype (K4A vs WT) [F(1,8)=0.0076, p=0.93] and interaction between genotype and sex [F(1,8)=0.00068, p=0.98].
3.6. U50,488H inhibited compound 48/80-induced scratching similarly in WT and K4A mice with no sex differences
Mice were injected with vehicle or U50,488H (1, 2.5, 5 mg/kg, sc) and 20 min later with compound 48/80 into the nape. The number of scratching bouts were counted for 30 min. As shown in Figure 4, U50,488H caused dose-dependent inhibition of scratching bouts in both WT and K4 Amice similarly. The A50 values were determined to be: male WT, 3.11 mg/kg; male K4A, 3.32 mg/kg; female WT, 2.84 mg/kg; female K4A, 2.40 mg/kg. Two-way ANOVA analysis revealed that, in male or female mice, no main effects were found in genotype [for males, F(1,57)=0.074, p=0.78; for females, F(1,64)=1.22, p=0.27] and interaction [for males, F(3,57)=0.93, p=0.43; for females, F(3,64)=0.25, p=0.85] while there were main effects in U50,488 treatment [for males, F(3,57)=22.6, p<0.0001; for females, F(3,64)=21.8, p<0.0001]. Thus, there are no significant differences in anti-scratching effects of U50,488H between the wildtype and K4A mice in either male or female mice (Fig. 4).
Figure 4. Effects of K4A mutations on the inhibitory effect of U50,488H on compound 48/80-induced scratching.

Mice were acclimated in test cages for at least 1 h and injected with various doses of U50,488H or saline (s.c.). Twenty min later the pruritogen compound 48/80 was subcutaneously injected into the nape and starting immediately the number of scratching bouts was counted for 36 min. Data were normalized to the mean value of each saline group and represents the mean ± S.E.M (n = 8–14). Results were analyzed by two-way ANOVA followed by Bonferroni posttests (Prism 5). In WT and K4A mice, the dose-dependent anti-pruritic effects of U50,488H were not different significantly, which was independent of sex.
3.7. K4A mutations attenuated U50,488H tolerance in males but not in females
We used the treatment paradigm of Suzuki et al. (2004) to induce tolerance to U50,488H. Mice were treated with saline or U50,488H (80 mg/kg, s.c.) twice a day on days 1 and 2, and once in the morning of day 3 and tested on day 4 with U50,488H (5 mg/kg, s.c.). As shown in Fig. 5, the treatment paradigm caused robust and complete tolerance in female WT and K4A mice and male WT mice, as no significant differences were found in scratching bouts between the “U50 80 mg/kg 5x-U50 5 mg/kg” group and the “saline 5x-saline” group for each type of mice by one-way ANOVA analysis. However, male 4A mice displayed significant differences between the two treatment groups ($$p<0.01) and also between the “U50 80 g/kg 5x-U50 5 mg/kg” group and the “saline 5x- U50 5 mg/kg” group (*p<005), and thus exhibited only partial tolerance. Therefore, K4A mutations attenuated U50,488H tolerance in male, but not in female, mice (Fig. 5).
Figure 5. Effect of K4A mutations on U50,488H-induced tolerance in the anti-scratch test.

WT and K4A mice were treated with saline or U50,488H (80 mg/kg, s.c.) five times in 2 ½ days and tested on the fourth day for anti-scratch effect of U50,488H (5 mg/kg, s.c.) with saline as the control. Data of scratching bouts were normalized to the mean value of each “saline 5x-saline” group and shown as mean ± sem (n= 8-10) and analyzed by one-way ANOVA followed by Tukey’s post-hoc test (Prism 5). $$p<0.01, n.s.not significant, for comparison of “U50 80 mg/kg 5x-U50 5 mg/kg” vs “saline 5x-saline” group; ***p<0.001, **p<0.01, *p<0.05, for comparison of “U50 80 mg/kg 5x-U50 5 mg/kg” vs “saline 5x-U50 5 mg/kg” group; ###p<0.001, #p<0.05, for comparison of “saline 5x-U50 5 mg/kg” vs “saline 5x-saline” group.
3.8. K4A mutations abolished U50,488H (2 mg/kg)-induced CPA in female, but not in male, mice
As shown in Fig. 6, two-way ANOVA analysis revealed that, in male mice, main effects were found in U50,488H treatment [F(2,70)=18.1, p<0.0001], but not in interaction [F(2,70)=0.0022, p=0.10] or genotype [F(1,70)=3.80, p=0.060]. Among male mice, WT and K4A mice displayed similar degrees of U50,488H (2 or 5 mg/kg, s.c.)-induced CPA (p<0.001 or p<0.01 vs each saline group by Bonferroni post-tests) (Fig. 6). In female mice, two-way ANOVA analysis revealed main effects in interaction [F(1,37)=7.66, p=0.0087] and U50,488 (2 mg/kg) treatment [F(1,37)=5.41, p=0.026], but not in genotype [F(1,37)=2.25, p=0.14]. While female WT exhibited strong CPA to 2 mg/kg U50,488H (p<0.001 vs saline group by two-way ANOVA followed by Bonferroni post-tests), female K4A mice showed no U50,488H (2 mg/kg, s.c.)-promoted CPA (Fig. 6). Thus, K4A mutations eliminated U50,488H (2 mg/kg)-induced CPA in female mice, but not in male mice. The mechanisms underlying the sex differences remain to be investigated.
Figure 6. Effects of K4A mutations on U50,488H-induced CPA.

Male and female WT and K4A were conditioned using a two-chamber setup as previously described (Liu et al., 2019a). On Day 1 (pretest), mice were allowed to roam in either chamber for 15 min and those that spent 570 sec or more in either chamber was excluded. On Day 2 to Day 4, mice were conditioned once in the morning with saline and once in the afternoon with U50 or saline subcutaneously. Mice were injected with saline or U50,488H 10 min before each 30-min conditioning session (2 sessions/day) for 3 days. On Day 5 (posttest), the length of time the mouse spent on the U50,488H-paired side was measured. Preference score = posttest time – pretest time. Each value is mean ± sem (n = 14-19 for male K4A mice, 13 and 15 for female WT mice treated with U50,488H at 2 and 5 mg/kg, respectively, and 8-10 for the other seven mouse groups). **p <0.01, ***p<0.001 compared with the saline group, by two-way ANOVA followed by Bonferroni posttests (Prism 5).
Unexpectedly, U50,488H (5 mg/kg, s.c.) did not induce CPA in female mice of either genotype (WT or K4A), in contrast to the results in male mice. Two-way ANOVA analysis demonstrated no main effects in U50,488H (5 mg/kg) treatment [F(1,41)=1.53, p=0.22], genotype [F(1,41)=0.27, p=0.60] and interaction [F(1,41)=0.75, p=0.39]. It has been shown that U50,488H may induce biphasic CPA responses in females of certain mouse strains (discussed in section 4.5).
3.9. U50,488H inhibited novelty-induced locomotor activities similarly in WT and K4A mice without sex differences
Male mice: As shown in Fig. 7, for data of total distance in male mice, two-way ANOVA analysis demonstrated main effects in U50,488H treatment [F(1,43)=37.2, p<0.0001] and genotype (K4A vs WT) [F(1,43)=5.38, p=0.025], but not in interaction [F(1,43)=0.83, p=0.37]. As determined by Bonferroni posttests, U50,488H similarly decreased total distance traveled in both WT and K4A mice (p<0.001). For data of ambulatory activity in male mice, two-way ANOVA analysis demonstrated main effects in U50,488H treatment [F(1,43)=22.6, p<0.0001], but not in genotype (K4A vs WT) [F(1,43)=2.42, p=0.13] and interaction [F(1,43)=0.16, p=0.69]. As determined by Bonferroni posttests, U50,488H similarly compromised mouse ambulation in both WT and K4A male mice (p<0.01).
Figure 7. Effect of K4A mutations on inhibitory effect of U50,488H on novelty-induced locomotor activity.

Experiments were performed as we described previously (Liu et al., 2019a). Mice were treated with vehicle or U50,488H (5 mg/kg, sc) and immediately placed in locomotor activity chambers. Locomotor activities (total distance and ambulatory activity) of the mice were recorded for 60 minutes in 5-min bins. Cumulative data are shown as mean ± sem (n = 9-14). *p<0.05, **p <0.01, ***p<0.001, compared with the saline group, by two-way ANOVA followed by Bonferroni posttests (Prism 5).
Female mice: For data of total distance in female mice, two-way ANOVA analysis demonstrated main effects in U50,488H treatment [F(1,33)=21.5, p<0.0001], but not in genotype (K4A vs WT) [F(1,33)=0.42, p=0.52] and interaction [F(1,33)=0.088, p=0.77]. As determined by Bonferroni posttests, U50,488H similarly decreased total distance traveled in both WT and K4A mice (p<0.01). For data of ambulatory activity in female mice, two-way ANOVA analysis demonstrated main effects in U50,488H treatment [F(1,33)=16.9, p=0.0002], but not in genotype (K4A vs WT) [F(1,33)=0.0043, p=0.95] and interaction [F(1,33)=0.0008, p=0.98]. As determined by Bonferroni posttests, U50,488H similarly compromised mouse ambulation in both WT and K4A female mice (p<0.05). Thus, U50,488H significantly reduced total distance traveled and ambulatory activities in 60 minutes in WT and K4A mice to similar extents (Fig. 7) and similar results were obtained for male and female mice.
4. Discussion
The salient findings of this study are that lack of agonist-induced KOR phosphorylation, due to the K4A mutations, decreased KOR agonist U50,488H-induced tolerance in male, but not female, mice and abolished U50,488H (2 mg/kg)-induced CPA in female, but not male, mice. These results are the first to demonstrate sex differences in the effects of GPCR phosphorylation deficiency on the GPCR-mediated behaviors. These sex differences were unexpected, and the underlying mechanisms remain to be investigated. In contrast, K4A mutations did not affect U50,488H-induced anti-pruritic effect or hypolocomotion in male or female mice.
4.1. K4A and tolerance
The findings that U50,488H (5mg/kg)-induced KOR phosphorylation in both male and female mice, while phosphorylation-deficient KOR (K4A) attenuated U50,488H-induced tolerance in male mice, but not in female mice, indicates that tolerance is related to KOR phosphorylation in male mice only.
Observations on two other phosphorylation-deficient GPCRs in male mice appeared also corroborative. In male mice harboring a phosphorylation-deficient mu opioid receptor (MOR) mutant, analgesic tolerance to fentanyl and morphine was significantly diminished (Kliewer et al., 2019). Similarly, mutation of phosphorylation sites in the cannabinoid receptor 1 (CB1R) confers resistance to tolerance induced by delta-9-tetrahydrocannabinol (Δ9-THC) in male mice (Morgan et al., 2014). In contrast, in female mice KOR phosphorylation deficiency did not affect U50,488H-induced tolerance in the current study. This result was unexpected. It is unknown whether MOR or CB1R phosphorylation deficiency would have sex-specific effects on opioids or cannabinoids tolerance, since only male mice were utilized in previous studies (Morgan et al., 2014; Kliewer et al., 2019). In addition, β-arr2 deletion has been shown to abolish morphine tolerance in mice with sex not specified (Bohn et al., 2000).
4.2. Tolerance and KOR phosphorylation
We used the treatment regimen of Suzuki et al. (2004) to produce U50,488H tolerance. In this regimen, mice were injected with a very high dose of U50,488H (80 mg/kg, s.c.) twice a day for two days and once on the 3rd day and tolerance was observed on the 4th day. Prior to using this regimen, we injected wildtype mice with 5 mg/kg U50,488H (s.c.) twice a day for 5 days; however, mice did not develop tolerance when tested on the 6th day (data not shown). U50,488H at 5 mg/kg promoted robust KOR phosphorylation at 30 min after drug injection (Fig. 2); therefore, KOR phosphorylation alone is insufficient to cause tolerance. McLaughlin et al. (2004) used an escalating daily dose regimen (10, 25, 50, 70 mg/kg U50,488H) to induce tolerance and showed that prolonged KOR phosphorylation was required for tolerance to develop. This suggests that tolerance to KOR agonists may not be of clinical significance when they are used at the dose ranges effective for analgesic or anti-pruritic effects.
4.3. K4A and CPA
The observations that K4A mutations reduced U50,488H (2 mg/kg)-induced CPA in female, but not in male mice, whereas U50,488H caused similar extent of KOR phosphorylation suggest that CPA is related to KOR phosphorylation in female mice only. This is not simply because repeated U50,488H administration using the CPA paradigm induced less tolerance in K4A males rather than K4A females, compared with the WT mice of each sex, because, as mentioned above in section 4.2., mice repeatedly treated with U50,488H at dose of 5 mg/kg did not develop any tolerance.
In male mice, K4A mutations did not affect U50,488H-indued CPA. This finding is consistent with the study of White et al. (2015) performed on wildtype and β-arr2 KO mice of mixed sexes (an equal number of male and female subjects were age-matched). They showed that β-arr2 deletion had no effects on CPA induced by KOR agonists including U69,593, salvinorin A and RB-64. However, Bruchas et al. reported that, in male mice, KOR agonist-induced CPA was GRK3- and p38 MAPK-dependent, which was proposed to involve a β-arrestins-dependent signaling pathway (Bruchas et al., 2007; Bruchas et al., 2011; Ehrich et al., 2015). Recently, using a large-scale mass spectrometry-based phosphoproteomics of U50,488H-mediated signaling in the male mouse brain, changes in multiple signaling pathways were observed to be induced by acute U50,488H treatment. The changes involved PKC-dependent modulation of GRK5/6 and Wnt pathways at 5 min post-U50,488H; mTOR signaling, stress signaling, cytoskeleton and CB1R phosphorylation at 30 min post-U50,488H (Liu et al., 2019a; Liu et al., 2020). Furthermore, the mTOR and/or CB1R signaling pathways have been found to play roles in KOR agonists-induced CPA in male mice (Liu et al., 2019a; Liu et al., 2020). PKC also appears to be involved in U50,488H-induced KOR phosphorylation at Ser369, but not at Thr363, in the male mouse brain (Liu et al., 2020). However, significant modulation of the p38 MAPK pathway was not detected in male mouse brains at 5 or 30 min after U50,488H treatment by the non-hypothesis driven phosphoproteomic approach (Liu et al., 2019a).
In contrast, in female mice, phosphorylation-deficient KOR (K4A) abolished U50,488H-indued CPA. Corroborative with this finding, U50,488H-induced CPA in female mice was also proposed to be mediated by activation of β-arrestin and p38 MAPK similarly as in male mice by Chavkin and colleagues (Abraham et al., 2018). It will be intriguing to adopt the novel large-scale phosphoproteomic approach to further pinpoint the mechanisms underlying the sex differences observed.
It is noteworthy that estrogen regulation of GRK2 has been shown by Chavkin’s group to inactivate KOR-mediating analgesia, but not CPA (Abraham et al., 2018). Estradiol diminished G protein-mediated signaling of KOR through increased phosphorylation of GRK2 and possible sequestration of Gβγ by GRK2 (Abraham et al., 2018). In addition, Valentino’s group (Curtis et al., 2006) reported that locus coeruleus (LC) neurons in female rats were 10x-30x more sensitive to corticotropin-releasing factor (CRF) than in males. The effect was independent of circulating sex hormones (Curtis et al., 2006) and may be related to the greater amount of Gαs protein and the lower amount of β-arr2, associated with the CRF1 receptor in females than in males (Bangasser et al., 2010).
4.4. K4A mutations did not affect U50,488H-induced antipruritic or hypo-locomotor effects
The phosphorylation-deficient, presumably G-protein-biased, KOR (K4A) did not affect binding affinity of KOR ligands and agonist-stimulated G protein activation in N2A cells, nor did it change the distribution and expression levels of KOR in either male or female mouse brains. These findings were in accord with that, in K4A mice, U50,488H inhibited compound 48/80-induced scratching and attenuated novelty-induced hyperlocomotion, similar to wildtype mice in both males and females. Consistent with these observations, β-arrestin2 deletion did not affect antipruritic or hypo-locomotor effects of acute administration of KOR agonists (U69,593 or U50,488H) in male mice (Morgenweck et al., 2015; White et al., 2015). In contrast, the phosphorylation-deficient G-protein-biased mu opioid receptor (MOR) or β-arrestin2 deletion increased morphine or fentanyl-induced analgesia in male mice (Kliewer et al., 2019) or mice with sex unspecified (Bohn et al., 1999). In addition, male mice expressing phosphorylation-deficient CB1R were more sensitive to acutely administered Δ9-THC. Thus, GPCRs are diverse regarding whether lack of phosphorylation or β-arrestin2 deletion enhances acutely administrated GPCR agonist-mediated behavioral effects.
4.5. U50,488H-induced CPA in wildtype rodents: dose dependence?
It should be noted that results have been inconsistent regarding whether certain dose(s) of U50,488H induces CPA in wildtype mice or rats and whether there are sex differences in U50,488H-induced CPA. Our data to date have consistently shown that in male CD-1 mice U50,488H (0.25-10 mg/kg) induced significant CPA independent of the dose used (Liu et al., 2019a; Liu et al., 2020). In this study, 2 and 5 mg/kg of U50,488H caused similar degrees of CPA in male WT (C57BL/6N) mice, and 2 mg/kg induced similar extents of CPA in both male and female WT (C57BL/6N) mice. These results are in accord with data from Chavkin’s group (Abraham et al., 2018) that 2.5 mg/kg U50,488H induced CPA similarly in both male and female C57BL/6N mice. Furthermore, U50,488 (1.25-5 mg/kg) caused CPA in male and female C57BL/6J mice (Kaski et al., 2019). However, in female WT (C57BL/6N) mice, 5 mg/kg of U50,488H caused no CPA, which was surprising because a lower dose (2 mg/kg) did induce CPA, in the current study. This is reminiscent of one previous report demonstrating that, among three doses of U50,488H tested (2.5, 5 and 10 mg/kg) in female California mice, U50,488H induced CPA only at 2.5 mg/kg, but not at the two higher doses, while in male mice U50,488H only at 10 mg/kg caused CPA (Robles et al., 2014). Taken together, U50,488H may induce biphasic CPA responses only in females of certain mouse strains. Another report showed that U50,488 induced CPA in male C57BL/6J mice only at 10 mg/kg, but not at lower doses (1 and 5 mg/kg), while none of the doses induced CPA in female C57BL/6J mice (Liu et al., 2019b). In addition, U50,488H was shown to induce biphasic CPA responses in male Sprague Dawley rats (Bals-Kubik et al., 1989; Bals-Kubik et al., 1993). Thus, the discrepancies in U50,488H-induced CPA responses in wildtype rodents have been considerable, which may be ascribed to differences in sex, experimental designs, CPA setups, mouse strains and/or species.
5. Conclusions
KOR agonist-induced phosphorylation has effects on the KOR agonist-mediated behaviors in a behavioral endpoint-dependent and sex-specific way. The mechanisms underlying the observed sex differences remain to be investigated.
Supplementary Material
Acknowledgements:
The study was funded by NIH grants R01 DA041359, R21 DA045274 and P30DA013429.
Abbreviations:
- β-arr
β-arrestin
- Acb
nucleus accumbens
- Amg
amygdala
- Cl
claustrum
- CPA
conditioned place aversion
- DynA(1-17)
dynorphin A (1-17)
- En
endopiriform nucleus
- GPCRs
G protein-coupled receptors
- Hypo
hypothalamus
- IB
immunoblotting
- IP
immunoprecipitation
- K4A
a mouse KOR mutant cDNA with all the four residues mutated to alanine
- KC
KOR c-tail
- KOR
kappa opioid receptor
- mKOR
mouse kappa opioid receptor
- MOR
mu opioid receptor
- CB1R
cannabinoid receptor 1
- N2A-K4A and N2A-KOR
neuro2A mouse neuroblastoma cells which express K4A and wildtype mKOR, respectively
- nor-BNI
norbinaltorphimine
- PAG
periaqueductal gray
- SN
substantial nigra
- Δ9-THC
delta-9-tetrahydrocannabinol
- WT
wildtype
Footnotes
Disclosure: The authors declare that they have no competing interests.
References
- Abraham AD, Schattauer SS, Reichard KL, Cohen JH, Fontaine HM, Song AJ, Johnson SD, Land BB, Chavkin C, 2018. Estrogen Regulation of GRK2 Inactivates Kappa Opioid Receptor Signaling Mediating Analgesia, But Not Aversion. J Neurosci 38, 8031–8043, 10.1523/JNEUROSCI.0653-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bals-Kubik R, Ableitner A, Herz A, Shippenberg TS, 1993. Neuroanatomical sites mediating the motivational effects of opioids as mapped by the conditioned place preference paradigm in rats. J Pharmacol Exp Ther 264, 489–495. [PubMed] [Google Scholar]
- Bals-Kubik R, Herz A, Shippenberg TS, 1989. Evidence that the aversive effects of opioid antagonists and kappa-agonists are centrally mediated. Psychopharmacology (Berl) 98, 203–206, 10.1007/BF00444692. [DOI] [PubMed] [Google Scholar]
- Bangasser DA, Curtis A, Reyes BA, Bethea TT, Parastatidis I, Ischiropoulos H, Van Bockstaele EJ, Valentino RJ, 2010. Sex differences in corticotropin-releasing factor receptor signaling and trafficking: potential role in female vulnerability to stress-related psychopathology. Mol Psychiatry 15, 877, 896–904, 10.1038/mp.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG, 2000. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 408, 720–723, 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT, 1999. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 286, 2495–2498, 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bruchas MR, Chavkin C, 2010. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology (Berl) 210, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C, 2007. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J. Neurosci. 27, 11614–11623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, Palmiter RD, Chavkin C, 2011. Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron 71, 498–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brust TF, 2020. Biased Ligands at the Kappa Opioid Receptor: Fine-Tuning Receptor Pharmacology. Handb Exp Pharmacol, 10.1007/164_2020_395. [DOI] [PubMed] [Google Scholar]
- Cahill C, Tejeda HA, Spetea M, Chen C, Liu-Chen LY, 2021. Fundamentals of the Dynorphins/Kappa Opioid Receptor System: From Distribution to Signaling and Function. Handb Exp Pharmacol, 10.1007/164_2021_433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Chiu YT, Wu W, Huang P, Mann A, Schulz S, Liu-Chen LY, 2016. Determination of sites of U50,488H-promoted phosphorylation of the mouse kappa opioid receptor (KOPR): Disconnect between KOPR phosphorylation and internalization. Biochem J 473 497–508, 10.1042/BJ20141471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Widmann M, Schwarzer C, Liu-Chen LY, 2021. Considerations on Using Antibodies for Studying the Dynorphins/Kappa Opioid Receptor System. Handb Exp Pharmacol, 10.1007/164_2021_467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Willhouse AH, Huang P, Ko N, Wang Y, Xu B, Huang LHM, Kieffer B, Barbe MF, Liu-Chen LY, 2020. Characterization of a Knock-In Mouse Line Expressing a Fusion Protein of kappa Opioid Receptor Conjugated with tdTomato: 3-Dimensional Brain Imaging via CLARITY. eNeuro 7, 10.1523/ENEURO.0028-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SD, Abi-Dargham A, 2019. The Role of Dynorphin and the Kappa Opioid Receptor in the Symptomatology of Schizophrenia: A Review of the Evidence. Biol Psychiatry 86, 502–511, 10.1016/j.biopsych.2019.05.012. [DOI] [PubMed] [Google Scholar]
- Cowan A, Kehner GB, Inan S, 2015. Targeting itch with ligands selective for kappa opioid receptors. Handb. Exp. Pharmacol. 226, 291–314, 10.1007/978-3-662-44605-8_16. [DOI] [PubMed] [Google Scholar]
- Curtis AL, Bethea T, Valentino RJ, 2006. Sexually dimorphic responses of the brain norepinephrine system to stress and corticotropin-releasing factor. Neuropsychopharmacology 31, 544–554, 10.1038/sj.npp.1300875. [DOI] [PubMed] [Google Scholar]
- Ehrich JM, Messinger DI, Knakal CR, Kuhar JR, Schattauer SS, Bruchas MR, Zweifel LS, Kieffer BL, Phillips PE, Chavkin C, 2015. Kappa opioid receptor-induced aversion requires p38 MAPK Aactivation in VTA dopamine neurons. J. Neurosci. 35, 12917–12931, 10.1523/JNEUROSCI.2444-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grim TW, Acevedo-Canabal A, Bohn LM, 2020. Toward Directing Opioid Receptor Signaling to Refine Opioid Therapeutics. Biol Psychiatry 87, 15–21, 10.1016/j.biopsych.2019.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang P, Kehner GB, Cowan A, Liu-Chen LY, 2001. Comparison of pharmacological activities of buprenorphine and norbuprenorphine: norbuprenorphine is a potent opioid agonist. J Pharmacol Exp Ther 297, 688–695. [PubMed] [Google Scholar]
- Kaski SW, White AN, Gross JD, Trexler KR, Wix K, Harland AA, Prisinzano TE, Aube J, Kinsey SG, Kenakin T, Siderovski DP, Setola V, 2019. Preclinical Testing of Nalfurafine as an Opioid-sparing Adjuvant that Potentiates Analgesia by the Mu Opioid Receptor-targeting Agonist Morphine. J Pharmacol Exp Ther 371, 487–499, 10.1124/jpet.118.255661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer A, Schmiedel F, Sianati S, Bailey A, Bateman JT, Levitt ES, Williams JT, Christie MJ, Schulz S, 2019. Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat Commun 10, 367, 10.1038/s41467-018-08162-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Li JG, Chen C, Zhang F, Liu-Chen LY, 2002. Molecular basis of differences in (−)(trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidiny)-cyclohexyl]benzeneacetamide -induced desensitization and phosphorylation between human and rat kappa-opioid receptors expressed in Chinese hamster ovary cells. Mol. Pharmacol. 61, 73–84. [DOI] [PubMed] [Google Scholar]
- Liu-Chen L-Y, 2004. Agonist-induced regulation and trafficking of kappa opioid receptors. Life Sci. 75, 511–536. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Chiu YT, Chen C, Huang P, Mann M, Liu-Chen LY, 2020. Pharmacological and phosphoproteomic approaches to roles of protein kinase C in kappa opioid receptor-mediated effects in mice. Neuropharmacology 181, 108324, 10.1016/j.neuropharm.2020.108324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Chiu YT, DiMattio KM, Chen C, Huang P, Gentile TA, Muschamp JW, Cowan A, Mann M, Liu-Chen LY, 2019a. Phosphoproteomic approach for agonist-specific signaling in mouse brains: mTOR pathway is involved in kappa opioid aversion. Neuropsychopharmacology 44, 939–949, 10.1038/s41386-018-0155-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SS, Pickens S, Burma NE, Ibarra-Lecue I, Yang H, Xue L, Cook C, Hakimian JK, Severino AL, Lueptow L, Komarek K, Taylor AMW, Olmstead MC, Carroll FI, Bass CE, Andrews AM, Walwyn W, Trang T, Evans CJ, Leslie FM, Cahill CM, 2019b. Kappa Opioid Receptors Drive a Tonic Aversive Component of Chronic Pain. J Neurosci 39, 4162–4178, 10.1523/JNEUROSCI.0274-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, Maudsley S, Bohn LM, 2015. Fulfilling the promise of “biased” G protein-coupled receptor agonism. Mol Pharmacol 88, 579–588, 10.1124/mol.115.099630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Myers LC, Zarek PE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Chavkin C, 2004. Prolonged kappa opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance. J. Biol. Chem. 279, 1810–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DJ, Davis BJ, Kearn CS, Marcus D, Cook AJ, Wager-Miller J, Straiker A, Myoga MH, Karduck J, Leishman E, Sim-Selley LJ, Czyzyk TA, Bradshaw HB, Selley DE, Mackie K, 2014. Mutation of putative GRK phosphorylation sites in the cannabinoid receptor 1 (CB1R) confers resistance to cannabinoid tolerance and hypersensitivity to cannabinoids in mice. J Neurosci 34, 5152–5163, 10.1523/JNEUROSCI.3445-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenweck J, Frankowski KJ, Prisinzano TE, Aube J, Bohn LM, 2015. Investigation of the role of betaarrestin2 in kappa opioid receptor modulation in a mouse model of pruritus. Neuropharmacology 99, 600–609, https://doi.org/S0028-3908(15)30072-1 [pii]; 10.1016/j.neuropharm.2015.08.027 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande AC, Pyke RE, Greiner M, Wideman GL, Benjamin R, Pierce MW, 1996. Analgesic efficacy of enadoline versus placebo or morphine in postsurgical pain. Clin Neuropharmacol 19, 451–456, 10.1097/00002826-199619050-00009. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM, 1986. Psychotomimesis mediated by kappa opiate receptors. Science 233, 774–776, 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- Rankovic Z, Brust TF, Bohn LM, 2016. Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett. 26, 241–250, 10.1016/j.bmcl.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles CF, McMackin MZ, Campi KL, Doig IE, Takahashi EY, Pride MC, Trainor BC, 2014. Effects of kappa opioid receptors on conditioned place aversion and social interaction in males and females. Behav Brain Res 262, 84–93, 10.1016/j.bbr.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonin F, Valverde O, Smadja C, Slowe S, Kitchen I, Dierich A, Le Meur M, Roques BP, Maldonado R, Kieffer BL, 1998. Disruption of the kappa-opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa-agonist U-50,488H and attenuates morphine withdrawal. EMBO J 17, 886–897, 10.1093/emboj/17.4.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Izumimoto N, Takezawa Y, Fujimura M, Togashi Y, Nagase H, Tanaka T, Endoh T, 2004. Effect of repeated administration of TRK-820, a kappa-opioid receptor agonist, on tolerance to its antinociceptive and sedative actions. Brain Res 995, 167–175, 10.1016/j.brainres.2003.09.057. [DOI] [PubMed] [Google Scholar]
- von Voigtlander PF, Lahti RA, Ludens JH, 1983. U-50,488: a selective and structurally novel non-Mu (kappa) opioid agonist. J. Pharmacol. Exp. Ther. 224, 7–12. [PubMed] [Google Scholar]
- Wadenberg ML, 2003. A review of the properties of spiradoline: a potent and selective kappa-opioid receptor agonist. CNS Drug Rev 9, 187–198, 10.1111/j.1527-3458.2003.tb00248.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Rasakham K, Huang P, Chudnovskaya D, Cowan A, Liu-Chen LY, 2011. Sex difference in kappa-opioid receptor (KOPR)-mediated behaviors, brain region KOPR level and KOPR-mediated guanosine 5’-O-(3-[35S]thiotriphosphate) binding in the guinea pig. J Pharmacol Exp Ther 339, 438–450, 10.1124/jpet.111.183905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, Roth BL, 2015. The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J Pharmacol Exp Ther 352, 98–109, 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Wang Y, Ma Z, Chiu YT, Huang P, Rasakham K, Unterwald E, Lee DY, Liu-Chen LY, 2013. L-isocorypalmine reduces behavioral sensitization and rewarding effects of cocaine in mice by acting on dopamine receptors. Drug Alcohol Depend 133, 693–703, 10.1016/j.drugalcdep.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Wallace MS, 2018. Opioids, Analgesia, and Pain Management. In: Brunton LL, Hilal-Dandan R, Knollmann BC, (Eds), Goodman & Gilman’s The Pharmacological Basis of Therapeutics. McGraw-Hill Co., Inc., New York, NY. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.