

Summary
The pore-forming protein Gasdermin D (GSDMD) executes lytic cell death called pyroptosis to eliminate the replicative niche of intracellular pathogens. Evolution favors pathogens that circumvent this host defense mechanism. Here we show that the Shigella ubiquitin ligase IpaH7.8 functions as an inhibitor of GSDMD. Shigella is an enteroinvasive bacterium that causes hemorrhagic gastroenteritis in primates, but not rodents. IpaH7.8 contributes to species specificity by ubiquitinating human, but not mouse, GSDMD and targeting it for proteasomal degradation. Accordingly, infection of human epithelial cells with IpaH7.8-deficient Shigella flexneri results in increased GSDMD-dependent cell death compared to wild-type. Consistent with pyroptosis contributing to murine disease resistance, eliminating GSDMD from NLRC4-deficient mice, which are already sensitized to oral infection with Shigella flexneri, leads to further enhanced bacterial replication and increased disease severity. This work highlights a species-specific pathogen arms race focused on maintenance of host cell viability.
Keywords: GSDMD, inflammasome, proteasome, pyroptosis, Shigella, ubiquitin ligase, virulence
eTOC blurb
Luchetti et al. identify Shigella IpaH7.8, a bacterial ubiquitin ligase, as a species-specific inhibitor of inflammatory cell death termed pyroptosis. IpaH7.8 prevents pyroptosis by targeting the pore-forming protein gasdermin D for degradation. This activity contributes to Shigella causing severe gastroenteritis in humans, but not rodents.
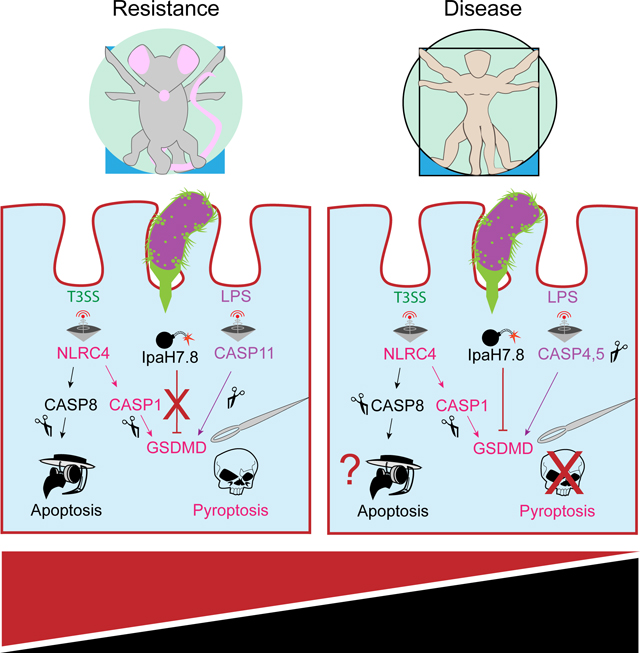
Introduction
A fundamental question in innate immunity is how intracellular bacterial pathogens have evolved to evade cell-intrinsic clearance mechanisms. Pathogen-associated molecular patterns (PAMPs), including lipopolysaccharide (LPS), are sensed by host inflammasome pattern recognition receptors (PRRs) triggering lytic cell death termed pyroptosis (Jorgenson et al., 2017; Broz and Dixit, 2016). The inflammasome sensors assemble complexes that activate human caspases 1, 4, and 5 (or mouse caspases 1 and 11) leading to cleavage of gasdermin D (GSDMD). Release of the N-terminal pore-forming domain (PFD) of GSDMD promotes cell lysis (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015). This cell death mechanism is crucial to host defense because inflammasome-deficient mice exhibit increased susceptibility to opportunistic microorganisms such as C. violaceum and B. thailandensis. Although wild-type mice eliminate up to 20,000,000 colony forming units (CFUs), inflammasome-deficient mice succumb to as few as 100 CFUs (Aachoui et al., 2015; Maltez et al., 2015).
Other pathogens have acquired virulence mechanisms to evade or disable pyroptosis. Salmonella and Listeria evade pyroptosis by repressing expression of PrgJ and flagellin, two of the PAMPs that are detected by the PRR NLRC4 (NLR family, CARD-containing 4) (Miao et al., 2010; Sauer et al., 2011; Warren et al., 2011). Yersinia and enteropathogenic Escherichia coli secrete YopM and NleF that inhibit caspases 1 and 4 (LaRock and Cookson, 2012; Pallett et al., 2017). Enterovirus 71 inactivates GSDMD through the proteolytic activity of virally encoded 3C protease (Lei et al., 2017). The relative competitive fitness between these pathogens and their hosts derives from their natural environmental co-evolutionary history.
Humans and non-human primates are the natural reservoir for bacteria of the genus Shigella. Infections cause severe gastrointestinal disease referred to as shigellosis (Trofa et al., 1999; Mulder, 1971). In severe infections, hemorrhagic dysentery can be life-threatening, especially for children under 5 years of age (Khalil et al., 2018). Rising antibiotic resistance complicates treatment, and currently, there is no clinically approved Shigella vaccine (Baker and The, 2018). Shigella is highly infectious, with oral ingestion of as few as 10–100 microbes eliciting disease (DuPont et al., 1969; Dupont et al., 1989). Mice, however, are non-natural hosts and resist millionfold higher doses (McGuire et al., 1958). Shigella virulence depends on a Type III secretion system (T3SS) that injects 30 effectors of largely uncharacterized activity (Parsot, 2009). Murine resistance stems from host-detection of the T3SS and cytosolic LPS (Mitchell et al., 2020). These PAMPs trigger assembly of canonical NAIP-NLRC4-caspase-1 (Kofoed and Vance, 2011; Zhao et al., 2011) and non-canonical caspase-11 (Kayagaki et al., 2013; Shi et al., 2014) inflammasome complexes. It is unclear why humans are vulnerable to Shigella despite expressing homologous inflammasome components.
To address how Shigella evades pyroptosis in humans, we screened a collection of Shigella virulence factors for their ability to suppress LPS-induced pyroptosis. We identified the bacterial ubiquitin ligase IpaH7.8 as an inhibitor of human, but not mouse pyroptosis. Mechanistically, IpaH7.8 targeted human GSDMD for proteasomal degradation.
Results
IpaH7.8 ubiquitin ligase activity blocks pyroptosis in human cells
Human Ea.hy926 endothelial cells expressing a library of doxycycline (dox)-inducible Shigella effectors were electroporated with LPS to trigger caspase-4- and GSDMD-dependent pyroptosis (Figures 1A and 1B; Table S1). Two factors suppressed pyroptosis: OspC3, a known inhibitor of caspase-4 (Kobayashi et al., 2013), and IpaH7.8, an E3 ubiquitin ligase (Rhode et al., 2007) (Figure 1C). Paradoxically, IpaH7.8 has been shown to activate NLRP3 and NLRP1B inflammasomes in mouse macrophages (Sandstrom et al., 2019; Suzuki et al., 2014).
Figure 1. IpaH7.8 ubiquitin ligase activity blocks pyroptosis in human cells.

(A) Shigella effector positive selection screen strategy (NGS, next generation sequencing).
(B) The non-canonical inflammasome pathway.
(C) Volcano plot depicting changes in effector abundance in the LPS- and dox-treated sample relative to the dox-only control. The x-axis corresponds to the Log2 fold change in gene expression, and the y-axis indicates the adjusted p-value. Wald test, FDR adjustment by Benjamini-Hochberg method (n = 3 per group).
(D, E, G and H) Graphs indicate the percentage of LDH released from Ea.hy926 cells (D, G, and H) or iMacs (E) after LPS electroporation (D, E and G) or treatment with 25 μM Valboro-Pro (H, VbP).
(F) Domain architecture of IpaH7.8.
Where indicated, doxycycline (dox) was used to induce expression of wild-type (WT) IpaH7.8 or mutant IpaH7.8(C357A). Bars represent the mean of 3–4 biological replicates, each plotted as a single data point.
See also figure S1.
We confirmed the species-specific anti-pyroptotic role of IpaH7.8 in Ea.hy926 cells and Hoxb8-immortalized mouse myeloid progenitor cells expressing doxycycline-inducible IpaH7.8 (Figure S1A). As expected, in the absence of doxycycline, cytoplasmic LPS caused both cell types to release lactate dehydrogenase (LDH), a hallmark of lytic cell death (Figures 1D and 1E). Doxycycline-induced expression of IpaH7.8 blocked LPS-induced LDH release from human Ea.hy926 cells (Figure 1D), but not mouse immortalized macrophages (iMacs) (Figure 1E). Thus, IpaH7.8 interferes with the non-canonical pyroptosis pathway specifically in human cells.
Members of the IpaH family of ubiquitin ligases in Shigella share a common domain architecture. They have a less conserved N-terminal Leucine-Rich Repeat (LRR) followed by a highly conserved novel E3 ubiquitin ligase domain (NEL) bearing an essential catalytic cysteine residue (Rohde et al., 2007) (Figure 1F). Catalytically inactive IpaH7.8(C357A) could be expressed to the same extent as wild-type IpaH7.8 in Ea.hy926 cells (Figure S1B), but only the latter blocked LPS-induced LDH release (Figure 1G). Wild-type, but not catalytically inactive IpaH7.8 also prevented caspase-1-mediated LDH release from Ea.hy926 cells stimulated with Val-boroPro (VbP), an agonist of the NLRP1/CARD8 inflammasomes (Johnson et al., 2018) (Figure 1H). We determined the kinetics of cell death by live cell imaging of pyroptotic cells taking up the cell impermeable dye YOYO-1. This assay confirmed that wild-type, but not catalytically inactive IpaH7.8 prevented the death of Ea.hy926 cells either transfected with LPS (Figures S1C and S1D) or treated with VbP (Figures S1E and S1F) up to 24 h. Thus, the ubiquitin ligase activity of IpaH7.8 is crucial to inhibition of either caspase-1- or caspase-4-dependent pyroptosis.
IpaH7.8 targets GSDMD for proteasomal degradation
To identify the substrate(s) of IpaH7.8, we used mass spectrometry to measure changes in protein abundance and ubiquitination following induction of IpaH7.8 (Figure 2A). Out of 7,995 quantified proteins (Data S1), GSDMD showed the greatest decline in abundance (Figure 2B). Consistent with GSDMD being a direct substrate of IpaH7.8, ubiquitination of three unique GSDMD peptides increased prior to the disappearance of GSDMD (Figure 2C). In addition, the loss of GSDMD protein did not coincide with a reduction in GSDMD transcription (Figures S2A and S2B). Previously, mouse Glomulin (GLMN) and NLRP1B were identified as pro-pyroptotic substrates of IpaH7.8 (Sandstrom et al., 2019; Suzuki et al., 2014), but neither of their human counterparts appeared affected by IpaH7.8 in Ea.hy926 cells (Figure 2D and 2E). Accordingly, expression of IpaH7.8 alone did not trigger marked pyroptosis in Ea.hy926 cells (Figure 1D). Thus, it appears IpaH7.8 targets GSDMD to prevent pyroptosis in human cells.
Figure 2. IpaH7.8 targets GSDMD for proteasomal degradation.

(A) Proteomics strategy to identify IpaH7.8 substrate(s) in Ea.hy926 cells.
(B) Scatter plot depicting doxycycline (dox)-induced IpaH7.8-dependent changes in global protein abundance. Results represent 2 biological replicates. The x-axis corresponds to the Log2 ratio of 6 h dox / 0 h no dox. The Log2 ratio of 6 h dox / 6 h no dox is on the y-axis. P-values determined by ANOVA.
(C, D and E) GSDMD (C), GLMN (D), and NLRP1, (E) (red circles) and associated ubiquitinated lysines (blue circles) are quantified at different times in Ea.hy926 cells after doxycycline (dox)-induced expression of IpaH7.8. Each circle represents the Log2 ratio of dox treated / no dox (2 biological replicates each).
(F) Immunoblots of 293T cells co-transfected with myc-tagged gasdermins and Flag-IpaH7.8 (WT, wild-type or CA, mutant C357A). Results representative of 3 independent experiments.
(G) The mammalian ubiquitin pathway. Specific inhibitors of UAE/E1 (MLN-7243), NAE1 (MLN-4924), and the proteasome (Btz) are annotated at the relevant steps. Human cullin-RING family E3 ligases require the co-factor NEDD8 (NED) for activity.
(H) Immunoblots of Ea.hy926 cells with dox-inducible Flag-IpaH7.8 after treatment for 4 h with DMSO vehicle or 1 μM Btz, MLN-7243 (UAE/E1), or MLN-4924 (NAE1).
(I) Immunoblots of in vitro ubiquitination reactions using GSDMD and IpaH7.8 (WT or C357A). Results representative of 3 independent experiments.
Another human Gasdermin, GSDMB, also appeared susceptible to degradation in 293T cells co-expressing IpaH7.8, but not IpaH7.8(C357A) (Figure 2F). Note that Ea.hy926 cells only express GSDMD and GSDME, and the latter resists degradation by IpaH7.8 (Figures 2F and S2C). These observations explain why GSDMB and GSDME were not identified in our earlier proteomics analysis. The primary sequences of the different Gasdermins do not provide obvious clues as to why GSDMB and GSDMD are uniquely susceptible to IpaH7.8 (Figure S2D). Although cleavage of GSDMD is mediated largely by caspases 1, 4, and 5 (He et al., 2015; Kayagaki et al., 2015; Shi et al., 2015), cleavage of GSDMB, which has no murine ortholog, is mediated by granzyme A from cytotoxic T-lymphocytes and natural killer cells (Zhou et al., 2020). Since cell-intrinsic pyroptosis is probably the initial immune barrier for Shigella to overcome, we focused on the mechanism by which IpaH7.8 targets GSDMD for degradation.
Other IpaH E3 ligases require the host ubiquitin pathway (Figure 2G) to target their substrates for proteasomal degradation (Rohde et al., 2007). The proteasome inhibitor Bortezomib (Btz) or the ubiquitin-activating enzyme E1 (UAE/E1) inhibitor MLN-7243 blocked IpaH7.8-induced degradation of GSDMD (Figure 2H), indicating that IpaH7.8 utilizes the host proteasome to eliminate GSDMD. Previously, IpaH7.8 was shown to target mouse GLMN, leading to activation of host cullin-RING ubiquitin ligases (Suzuki et al., 2014.). IpaH7.8 induced degradation of GSDMD in human Ea.hy926 cells even when host cullin-RING ubiquitin ligases were inhibited with the NEDD8-activating enzyme E1 subunit 1 (NAE1) inhibitor MLN-4924 (Brownell et al., 2010) (Figure 2H). As a positive control for NAE1 inhibition by MLN-4924, a Cul1 immunoblot confirmed loss of NEDD8 (Cul1-NED) (Figure 2H). Thus, IpaH7.8 does not promote the degradation of human GSDMD indirectly by targeting GLMN for degradation.
Consistent with human GSDMD being a direct substrate of IpaH7.8, we could reconstitute its ubiquitination by IpaH7.8 in vitro using purified components (Figures 2I and S2E). K48-linked polyubiquitin, and to a lesser extent, K63-linked polyubiquitin were conjugated to human GSDMD (Figure S2F). Interestingly, ubiquitination of GSDMD by IpaH7.8 in vitro did not prevent its cleavage by caspase-4; the ubiquitinated pore-forming domain (PFD) released by caspase-4 was detected using an antibody that recognizes cleaved, but not full-length GSDMD (Figures S2G and S2H). Moreover, ubiquitination of the GSDMD PFD did not prevent it from associating with artificial membranes in a membrane flotation assay (Figures S2I-S2L). Finally, when we blocked proteasomal degradation of IpaH7.8-ubiquitinated GSDMD in Ea.hy926 cells with Btz, LPS still triggered pyroptosis (Figures S2M and S2O). Thus, ubiquitination of GSDMD by IpaH7.8 is necessary but not sufficient for pyroptosis inhibition; there must also be proteasomal degradation of the ubiquitinated GSDMD. As a positive control for proteasome inhibition by Btz, VbP-induced pyroptosis, which relies on proteasomal activation of NLRP1/CARD8, was blocked (Figure S2N).
IpaH7.8 is a molecular determinant of Shigella species-specificity
In keeping with IpaH7.8 not preventing pyroptosis in mouse cells (Figure 1E), mouse GSDMD was not ubiquitinated by IpaH7.8 in vitro (Figure 3A), nor was it degraded in cells expressing IpaH7.8 (Figure 3B). To gain insights into why IpaH7.8 targets human but not mouse GSDMD, we tested which features of human GSDMD allow it to be modified by IpaH7.8 and then degraded. Three lysines in human GSDMD exhibited clear IpaH7.8-dependent ubiquitination (K55, K62, and K204; Figure 2C). Mutating each of these lysines to arginine (mutant 3KR) did not prevent IpaH7.8-induced degradation of GSDMD, whereas mutation of all 15 lysines in GSDMD (mutant 15KR) did (Figure 3C). Six of these 15 lysines were not conserved in mouse GSDMD (Figure S3A), but neither single nor tandem introduction of these lysines into mouse GSDMD conferred susceptibility to IpaH7.8 (Figures 3D and S3B).
Figure 3. IpaH7.8 targets human GSDMD, but not mouse GSDMD.

(A) Immunoblots of in vitro ubiquitination reactions using human (hs) or mouse (mm) GSDMD and IpaH7.8. Results representative of 3 independent experiments.
(B) Immunoblots of 293T cells transfected with the GSDMD and IpaH7.8 constructs indicated. Results representative of 3 independent experiments.
(C and D) Immunoblots of 293T cells co-transfected with IpaH7.8 and GSDMD (wild-type, WT, or the mutants indicated). 3KR, K55R/K62R/K205R. 15KR, pan KR mutant. mm, mouse. hs, human. mm-K, introduces the 6 non-conserved lysine residues from human GSDMD into mouse GSDMD.
(E) Immunoblots showing co-immunoprecipitation of IpaH7.8(C357A) and GSDMD from co-transfected 293T cells.
(F) Binding isotherms of hsGSDMD (black circles) and mmGSDMD (turquoise circles) with IpaH7.8 measured by microscale thermophoresis (MST). The y-axis indicates the fraction bound, the x-axis is the Log10 of the IpaH7.8 concentration in molar. Each circle represents the mean ± SD of 3 technical replicates. Results representative of 3 independent experiments.
See also figure S3.
We considered that IpaH7.8 might not ubiquitinate mouse GSDMD because the two proteins fail to interact, but mouse GSDMD co-immunoprecipitated with IpaH7.8(C357A) to the same extent as, or better than, human GSDMD (Figure 3E). In reciprocal pull-downs, IpaH7.8(C357A) also co-immunoprecipitated with either mouse or human GSDMD (Figure S3C). By microscale thermophoresis, IpaH7.8 displayed 4-fold higher affinity for mouse GSDMD (Kd = 47 nM) than for human GSDMD (Kd = 194 nM) (Figure 3F). We conclude that more subtle differences must explain why human GSDMD is ubiquitinated by IpaH7.8, while mouse GSDMD is not.
The pore-forming domain of GSDMD is the target of IpaH7.8
During our initial characterization of GSDMD as an IpaH7.8 substrate, we observed N-terminal tagging of human GSDMD with a FLAG epitope prevented its degradation in cells co-expressing IpaH7.8 (Figure S4A). Analyses of chimeric GSDMD proteins composed of mouse and human sequences indicated that the N-terminal PFD of human GSDMD was required for IpaH7.8-induced degradation (Figures 4A and 4B). Alanine-scanning mutagenesis of human GSDMD also identified a contiguous 15 amino acid sequence (aa 11–25) near the N-terminus as crucial for degradation (Figures 4C and 4D). Analyses of chimeric GSDMD proteins composed of human (hs) residues 16–20 and mouse (mm) residues 16–21 confirmed this minimal region is required for degradation by IpaH7.8 (Figure 4E). This region contains features unique to human GSDMD, including positively or negatively charged residues (hs17D vs. mm17S, hs19G vs. mm20R, and hs18H vs mm19S), and a missing glycine insertion (between hs17D and 18H, mm18G). When mapped onto the structures of human and mouse GSDMD, this region coincides with a solvent-exposed loop that follows an N-terminal alpha helix (Figure 4F). Interestingly, the mouse loop is extended further, and substituted by amino acid side-chains that are absent from the human loop (mm19S and mm20R). Future structural studies may indicate why this region facilitates IpaH7.8 driven degradation of GSDMD.
Figure 4. The GSDMD Pore-forming domain is the target of IpaH7.8.

(A) Domain swapping strategy depicts PFD-boundaries for human and mouse GSDMD.
(B, C and E) Immunoblots of 293T cells co-transfected with IpaH7.8 (wild-type, WT, unless mutant C357A, CA is indicated) and GSDMD (wild-type, WT, or the mutants indicated). mm, mouse. hs, human. 5XA, the five residues indicated were replaced with five alanines. PFD, pore-forming domain; CTD, C-terminal domain. hs↔mm aa swap, human-mouse GSDMD chimera with indicated residues swapped in.
(D) Alignment of mouse (mm) and human (hs) GSDMD N-terminal protein sequences highlighting human residues 11–25 (red bar).
(F) Structural overlay of human (hs) GSDMD (6N9O, magenta, aa 11–25) and mouse (mm) GSDMD (6N9N, cyan, aa 11–26). N, N-terminal. C, C-terminal. mm18G is colored red.
(G) Truncation mutants of IpaH7.8(C357A) used in (H) and (I).
(H and I) Immunoblots showing co-immunoprecipitation of the IpaH7.8 LRR and GSDMD from co-transfected 293T cells. Results representative of 3 independent experiments.
See also figure S4.
Of the 6 IpaH family members tested, only IpaH7.8 eliminated expression of human GSDMD (Figure S4B). Substrate specificity in IpaH family members derives from sequence divergence of their N-terminal LRR domains (Ashida et al., 2016; Ji et al., 2019) (Figures S4C and S4D). Accordingly, the N-terminal LRR of IpaH7.8 was sufficient to co-immunoprecipitate either human or mouse GSDMD (Figures 4G–4I). Of the 24 complete Shigella flexneri genomes available containing a 200+ kb virulence plasmid (Table S2), 22 (92%) of these encode IpaH7.8. Strain G1663 appears to have two identical copies of IpaH7.8. Sequence identity amongst the IpaH7.8 variants detected was greater than 90% (Figures S4E and S4F). Notably, the LRR domain was entirely conserved (Figure S4E). The prevalence and conservation of IpaH7.8 among disease-causing clinical isolates of Shigella supports the notion that targeting of GSDMD by IpaH7.8 is an important virulence mechanism.
GSDMD-dependent death during S. flexneri infection is reduced by IpaH7.8
Next, we examined the impact of IpaH7.8 on GSDMD during Shigella flexneri infection. Human epithelial HeLa cells infected with GFP-expressing S. flexneri contained less GSDMD than uninfected cells after 1 hour. After 2 hours, GSDMD was barely detected in the infected population (Figure 5A). The disappearance of GSDMD was dependent on IpaH7.8 because GSDMD persisted in HeLa cells infected with a GFP-expressing mutant strain of S. flexneri lacking IpaH7.8 (ΔipaH7.8; Figure 5A). Importantly, both strains showed equivalent invasion and intracellular replication between 1–3 hours post-infection, and outside the ipaH7.8 locus, shared genome-wide consensus sequences (Figures S5A and S5B). Inhibiting the host ubiquitin-proteasome system with either MLN-7243 or Btz prevented the disappearance of GSDMD during S. flexneri infection (Figure 5B), consistent with proteasomal degradation of GSDMD. As expected, mouse GSDMD did not decline in mouse iMacs infected with S. flexneri (Figure 5C).
Figure 5. GSDMD restricts S. flexneri replication in Nlrc4−/− mice.

(A) Immunoblots of HeLa cells infected by S. flexneri 5a strain M90T wild-type (WT) or ΔipaH7.8. h.p.i, hours post-infection. Results representative of 3 independent experiments.
(B) Immunoblots of HeLa cells infected with S. flexneri 5a strain M90T for 2 h in the presence of DMSO vehicle or 1 μM Bortezomib (Btz), or MLN-7243 (UAE1). Results representative of 3 independent experiments.
(C) Immunoblots of iMacs infected by S. flexneri 5a strain M90T WT or ΔipaH7.8. Results representative of 3 independent experiments.
(D) Percentage of infected Caco-2 cells killed by GFP-expressing S. flexneri 5a strain M90T wild-type (WT, grey circles), ipaH7.8 deficient (ΔipaH7.8, green circles), or un-infected control (black circles) based on their uptake of propidium iodide (GFP/PI++ cells over total GFP+ cells). Circles represent the mean ± SD calculated from 6 infected wells. Data representative of 3 independent experiments.
(E to L) Characterization of control (black circles, n = 6), Gsdmd−/− (grey circles, n = 5), Nlrc4−/− (green circles, n = 9) and Nlrc4−/−Gsdmd−/−(dKO, white circles, n = 6) female mice at 48 h.p.i. challenged with 107 CFU (colony forming units) of S. flexneri 2a strain 2457T. Endpoint harvests were performed 48 h.p.i. (E) Representative images of cecum and colon. Note the cecum tissue thickening (size reduction), macroscopic edema, and lack of stool pellets (blue arrows) in dKO tissue. (F) Percentage change in body weight. (G) Feces weights before and after dehydration. (H) Fecal blood scores (1 = occult blood, 2 = macroscopic blood). (I) Representative images of H&E stained cecum and colon tissue from infected mice (50 μm scale bar). (J) Blinded quantification of histology score (cumulative) for tissues in I. (K) Tissue KC levels measured by ELISA. (L) CFUs in the IEC-enriched fraction of gentamicin-treated cecum and colon (combined). (G and H, J to L) Each symbol represents one mouse, lines represent mean ± SD. (F) Each symbol represents the mean of all animals within the indicated group ± SD. Mann-Whitney test, * p < 0.05, ** p < 0.01, *** p < 0.001, ns = not significant (p > 0.05).
See also figure S5.
Infected cells lacking GSDMD should be less susceptible to Shigella-induced pyroptosis. We monitored the death of S. flexneri-infected GFP+ HeLa and Ea.hy926 cells by their uptake of the membrane impermeable dye propidium iodide (PI). Consistent with IpaH7.8 suppressing GSDMD-dependent pyroptosis, cell death was accelerated in cultures infected with S. flexneri ΔipaH7.8 when compared to cultures infected with wild-type S. flexneri (Figures S5C and S5D). In contrast, S. flexneri ΔipaH7.8 did not accelerate death in GSDMD-deficient HeLa and Ea.hy926 cells (Figures S5E-S5H). As expected, mouse iMacs died at the same rate whether they were infected with wild-type or ΔipaH7.8 S. flexneri (Figure S5I). Finally, we confirmed that the more physiologically relevant human enteric Caco-2 cell line also died faster when infected with S. flexneri ΔipaH7.8 than when infected with wild-type S. flexneri (Figure 5D).
Mice lacking both GSDMD and NLRC4 are hyper susceptible to oral S. flexneri infection
We used GSDMD-deficient mice (Kayagaki et al., 2015) to investigate whether failure of IpaH7.8 to target murine GSDMD for proteasomal degradation contributes to the resistance of mice to shigellosis. Mice lacking NLRC4, which activates caspase-1 in response to the T3SS apparatus, are sensitized to oral Shigella infection (Mitchell et al., 2020). Therefore, we compared disease severity after infection in controls (Nlrc4+/− n=2, Gsdmd+/− n=3, wild-type n=1), Gsdmd−/− (Gsdmd−/− Nlrc4+/+ n=4, Gsdmd−/− Nlrc4+/− n=1), Nlrc4−/− (Nlrc4−/− Gsdmd+/+ n=4, Nlrc4−/− Gsdmd+/− n=5), and Gsdmd−/− Nlrc4−/− (n=6) mice. As expected, due to the presence of the compensatory protective NLRC4-caspase-8 death pathway (Rauch et al., 2017), Gsdmd−/− mice were as resistant as control mice to Shigella infection. However, Gsdmd−/− Nlrc4−/− mice exhibited more severe hallmarks of disease than Nlrc4−/− mice, including bloody diarrhea (Figures 5E, 5G, and 5H), more pronounced weight loss (Figure 5F), elevated bacterial burdens in intestinal epithelial cells (IECs; Figure 5L), increased inflammation (Figures 5K and S5L-S5N), and greater macroscopic tissue abnormalities (Figures 5I-5J and S5J-K). Unexpectedly, resistant Gsdmd−/− mice and sensitized Nlrc4−/− mice had similar numbers of bacteria in their IECs, while Gsdmd−/− Nlrc4−/− IECs had approximately 10-fold more bacteria than the individual knockouts (Figure 5L). We speculate that GSDMD provides an innate immune barrier to intracellular bacterial replication in mice that is eliminated during human infection by IpaH7.8.
Discussion
Identification of IpaH7.8 as a virulence factor that specifically targets human GSDMD highlights the evolutionary importance of GSDMD-dependent pyroptosis to host defense. This virulence strategy is one mechanism by which Shigella resists cell-intrinsic immunity while simultaneously fortifying its intracellular replicative niche. By blocking pyroptosis and the release of intracellular DAMPs (damage associated molecular patterns) that can activate neutrophils and the complement system, IpaH7.8 may also dampen cell-extrinsic immunity (Jogensen et. al., 2016). The ability of IpaH7.8 to target GSDMB (Figure 2F; Hansen et al., 2021) may protect infected cells from attack by cytotoxic lymphocytes or natural killer cells (Zhou et al., 2020).
Previous in vitro studies have led to the suggestion that GSDMD may exert direct bactericidal activity (Ding et al., 2016 and Liu et al., 2019). IpaH7.8 appears to target the entire intracellular pool of GSDMD, so it would likely counter all GSDMD-dependent mechanisms. We suspect little, if any, GSDMD PFD is released from the infected cell.
While the pore-forming activity of human and mouse GSDMD is conserved within the PFD, only the human PFD is susceptible to IpaH7.8-mediated degradation. The IpaH7.8-degron maps to an N-terminal 5–6 amino acid window in GSDMD, suggesting subtle differences confer IpaH7.8 sensitivity. This region forms a solvent-exposed loop, the relative positioning of which is influenced by a murine-specific glycine residue (Figure 4F). Interestingly, substrate recognition by the Cullin-RING ligase 4 (CRL4) substrate adapter protein cereblon (CRBN) is mediated through its interaction with an analogous surface turn motif that also contains a critical glycine residue (Matyskiela et al., 2016 and Petzold et al., 2016). Identification of this degron was achieved by solving structures of CRL4CRBN in a complex with different neo-substrates. Accordingly, structural insights will be needed to reconcile why IpaH7.8 binds to, but does not ubiquitinate mouse GSDMD. Currently available structural data on IpaH-family enzymes suggests that ligase activity is regulated by substrate binding (Keszei et al., 2017). For example, the LRR of IpaH9.8 auto-inhibits the NEL domain until the substrate human guanylate binding protein 1 (hGBP1) binds to and releases it (Ye et al., 2020). Consistent with this model, the LRR of IpaH7.8 is sufficient to mediate interactions with human and mouse GSDMD, but the consequence of these interactions may be distinguished by their ability to promote subsequent conformational changes necessary for regulating enzymatic activity.
Mitchell et al (2020) identified the NLRC4 inflammasome as an important murine barrier against Shigella infection. Using the 129 mouse strain, which lacks caspase-11, they also demonstrated the importance of LPS-mediated pyroptosis in restricting Shigella pathogenesis. Both of these pathways converge on GSDMD to eliminate the infected cell, but the consequence of GSDMD-deficiency remained unexplored. Our finding that GSDMD-deficient mice are resistant to Shigella infection (Figure 5) is consistent with the ability of NLRC4 to also trigger caspase-8-dependent apoptosis as a protective measure against Shigella (Rauch et al., 2017). Thus, flexible usage of different pathways to cell death probably explains the observed patterns of murine susceptibility and disease severity. Activation of caspase-11 does not fully compensate for NLRC4 deficiency, but it limits disease severity to an intermediate level. However, when both the NLRC4 and caspase-11 pathways are eliminated, as in Gsdmd−/− Nlrc4−/− mice, disease severity increases, and infected mice exhibit clinical features more consistent with human disease.
In the natural context of human pathogenesis, Shigella must overcome these or related pathways to establish a successful infection. We show that Shigella deploys IpaH7.8 to eliminate GSDMD-dependent pyroptosis triggered by the NLRC4 and caspase-4,5 (equivalent to murine caspase-11) inflammasomes. The prevalence and high conservation of IpaH7.8 among disease causing clinical isolates of S. flexneri highlights the importance of its virulence activity against GSDMD. Whether an auxiliary NLRC4-caspase-8 pathway to apoptosis can restrict human infections, and if so, to what extent, is unknown. Shigella may encode additional factors to combat such a pathway. For example, Shigella disables cytosolic LPS detection with two additional T3SS effectors, IpaH9.8 and OspC3 (Li et al., 2017; Wandel et al., 2017; Kobayashi et al., 2013).
This work epitomizes Van Valen’s Red Queen hypothesis that pathogens and their hosts must co-evolve increasingly sophisticated virulence and immune mechanisms to ensure mutual survival (Lacey and Miao, 2020). A robust network of interconnected host cell death pathways is met by an array of Shigella effector molecules, each competing for the decisive victory over cell viability. In humans, IpaH7.8 shifts this balance in favor of Shigella, and when phenotypically translated to mice, furnishes an improved disease model that more faithfully recapitulates human shigellosis. This model provides an opportunity for developing vaccine and therapeutic strategies aimed at eliminating this prominent cause of childhood morbidity and mortality in developing nations.
Limitations of the Study
Genetic studies indicate how mice employ cell death pathways to combat Shigella infection, but it remains unclear which cell death pathways pose the greatest threat to productive Shigella infection in humans. Although Gsdmd−/− Nlrc4−/− mice provide an improved model of shigellosis, they may not recapitulate all aspects of the human disease.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Lead Contact
Further information should be requested from the lead contact, Vishva M. Dixit (dixit@gene.com).
Materials Availability
Mouse and bacterial strains generated in this study may be requested through Genentech’s MTA program.
Data and Code Availability
Mass spectrometry raw data is available as a supplementary data table (Data S1). Bacterial sequencing data has been deposited in Genbank, accession number WP_010921637.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Nlrc4−/− Gsdmd−/− mice were generated by crossing Nlrc4−/− mice (Mariathasan et al., 2004) with Gsdmd−/− mice (Kayagaki et al., 2015). Both alleles were maintained on C57BL6/N genetic background and were bred in compliance with the Genentech institutional animal care and use committee. Mice were maintained in a specific pathogen free colony until three weeks prior to infection, maintained under a 12 hr light-dark cycle (7 am to 7 pm), and given a standard chow diet (Harlan irradiated laboratory animal diet) ad libitum. Females used in infection experiments were aged 12–16 weeks and were generated by intercrossing Nlrc4+/− Gsdmd+/− mice or their offspring. Females that were not littermates were co-housed for at least three weeks prior to infection in an ABSL2 facility. All animal experiments complied with the regulatory standards of, and were approved by, the University of California, Berkeley Animal Care and Use Committee.
Mouse infections were performed as described (Mitchell et al., 2020). Briefly, mice deprived of food and water for 4–6 h were dosed with 100 μL of 250 mg/mL streptomycin sulfate dissolved in water by oral gavage and placed in a cage with fresh bedding. After 24 h, mice were again deprived of food and water for 4–6 h before dosing with 107 CFU of log-phase, streptomycin resistant Shigella flexneri 2457T suspended in 100 μL of PBS by oral gavage. Infection inputs were determined by serially diluting a fraction of the initial inoculum and plating on TSB plates containing 0.01% CR and 100 μg/mL streptomycin. Mouse weights and fecal pellets were recorded or collected daily from 1 day prior to infection to the day of euthanasia (2 days post-infection). MPO ELISAs were performed as described (Mitchell et al., 2020). Upon euthanasia, mouse ceca and colons were removed and a section fixed in methacarn. The remaining organ was cleaned of fecal matter and incubated in gentamicin for 1 h to remove luminal Shigella. To determine intestinal epithelial cell CFU, the intestinal epithelial cell (IEC) fraction of this section was isolated, lysed, and plated as described (Mitchell et al., 2020). To determine levels of IL-18, IL-1β, and KC, the remaining tissue was homogenized in 1% PBSA + protease inhibitors and supernatants were used for ELISAs as described (Mitchell et al., 2020).
Bacterial strains
Shigella flexneri M90T was obtained from John Rohde (Dalhouse, Canada). The S. flexneri M90T ipaH7.8 open reading frame was replaced with a kanamycin resistance cassette from the pACYC177 vector and using lambda red recombinase expressed by pKD46 (Datsenko et al., 2000). Gene replacement was verified using whole-genome sequencing. Illumina paired-end reads were mapped onto the Shigella flexneri M90T 5a reference genome (GenBank accession number CP037923.1 and CP037924.1) using GSNAP version 2013–10-10 (Wu et al., 2010). Single nucleotide variant detection was performed using VariantTools R package with bqual threshold set to 30. Wild-type S. flexneri M90T and ΔipaH7.8 S. flexneri M90T were transformed by electroporation with the pBR322-AmpR-dasher GFP plasmid to visualize bacteria. For in vivo infections (see below), S. flexneri serovar 2a was used.
Cell culture
Ea.hy926 human endothelial, 293T human embryonic kidney, and HeLa human cervical epithelial cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) high glucose containing media supplemented with 10 mM HEPES pH 7.4, 1X Glutamax (gibco), 1X Penicillin-Streptomycin (gibco), 1X non-essential amino acids (gibco), 1 mM sodium pyruvate (gibco), and 10% (v/v) fetal bovine serum (FBS, VWR) at 37°C with 5% CO2. ER-Hoxb8-immortalized murine myeloid progenitor cells (Wang et al., 2006) were cultured in myeloid medium comprised of RPMI 1640 medium supplemented with 10% (v/v) FBS, 20 ng/mL murine granulocyte-macrophage colony-stimulating factor (GM-CSF, eBioscience), and 1 μM β-estradiol (MilliporeSigma). Immortalized Hoxb8 myeloid progenitor cells were differentiated into macrophages (iMacs) in DMEM supplemented with 10% (v/v) FBS, and 20% (v/v) L929-conditioned medium at 37°C with 5% CO2 and were harvested on day 5 for experiments.
Method Details
Reagents and antibodies
Ultra-pure LPS (E. coli 0111:B4, InvivoGen), Val-boroPro (MilliporeSigma), Doxycycline (Clontech), 3XFLAG peptide (MilliporeSigma), 1D4 peptide (TETSQVAPA, Genscript), MLN-7243 (ChemieTek), MLN-4924 (ChemieTek), Bortezomib (ChemieTek), Propidium iodide (Millipore Sigma), Congo red (Sigma-Aldrich), Poly-L-lysine (Millipore Sigma), Lipofectamine 2000 (ThermoFisher), Egg-phosphatidylcholine (Avanti Polar Lipids), Egg-phosphatidylethanolamine (Avanti Polar Lipids), Egg lissamine rhodamine phosphatidylethanolamine (Avanti Polar Lipids). Antibodies used include: Actin (clone C4, MPbio), β-Tubulin (ab6046, abcam RRID:AB_2210370), IpaH9.8 (A55475, EpiGentek ), Cul1 (2H4C9, ThermoFisher RRID:AB_86218), Myc-tag (9B11, Cell Signaling Technology RRID:AB_331783), FLAG-epitope (M2 and M2-HRP, SigmaAldrich RRID:AB_259529 and RRID:AB_439702), GSDMD (clone 17G2G9, Genentech (Aglietti et al., 2016)), GSDMD (E8G3F, Cell Signaling Technology RRID:AB_2864253), GSDMD-Asp275 (E7H9G, Cell Signaling Technology RRID:AB_2799099), Caspase-4 (4B9, Enzo Life Sciences RRID:AB_10621609), Rho-1D4 (University of British Columbia), Ubiquitin (VU-1, LifeSensors RRID:AB_2716558), K11-ubiquitin (Genentech (Matsumoto et al., 2010)), K48-ubiquitin (Genentech (Newton et al., 2008)), K63-ubiquitin (Genentech (Newton et al., 2008)), mouse-HRP (Jackson), rabbit-HRP (Jackson), Rat-HRP (SouthernBiotech), human-HRP (Jackson).
CRISPR-cas9 gene-editing
GSDMD was deleted from HeLa and Ea.hy926 cells using the Alt-R CRISPR-Cas9 system (IDT). A single-guide gRNA (sgRNA, IDT) targeting human GSDMD (5’-TTCCACTTCTACGATGCCA-3’) was used for generating ribonucleoprotein (RNP) complexes. First, 10 nmole of sgRNA was re-suspended in duplex buffer (2 μg/mL, IDT) for 10 min. 4 μg of sgRNA (2 μL) was combined with 4 μg Alt-R Cas9 (0.4 μL), and 3.6 uL duplex buffer (IDT). HeLa and Ea.hy926 cells were washed twice with PBS, and 5.6 × 106 cells were each diluted in 100 μL of Buffer R (Neon, ThermoFisher). For electroporation, 5 uL of RNP complex was combined with 10 uL of cells (5 × 105 cells), and 5 uL of Buffer R. Cells were electroporated using the Neon™ Transfection System 10 μL Kit (Voltage = 1,005, Pulse Width = 35, Pulse # = 2). Cells were recovered in 6-well plates containing DMEM, and GSDMD deletion was confirmed in pooled cells.
Cell assays
For Ea.hy926 cell death assays, dox-inducible cells were seeded into 96-well plates at a density of 8,000 cells per well. After 1 d, cells were treated with 250 ng/mL doxycycline for 24 h. On the day of stimulation, LPS (0.5 μg/well, InvivoGen) was transfected using Lipofectamine LTX transfection reagent (0.2 μL/well, ThermoFisher), or treated with 25 μM Val-boroPro (Millipore), and cultured for 24 h. For Hoxb8 cell death assays, myeloid progenitor cells were first differentiated into iMacs, and seeded into 96-well plates at a density of 1 × 105 cells per well. After 1 d, cells were treated with 250 ng/mL doxycycline for 24 h. On day 3, LPS (1 μg/well, Invivogen) was transfected using FugeneHD (0.5 μL/well, Promega). In both cell-lines, doxycycline was maintained throughout as needed. On day 4, LDH-release into culture medium was quantified using CytoTox 96 Non-radioactive Cytotoxicity Assay (Promega). Uptake of YOYO-1 was conducted in similar format as for LDH-release, only cells were stimulated in the presence of 0.2 μM YOYO-1. Plates were imaged (phase contrast and green channel) on the IncuCyte S3 using the 10X objective every 30 min up to 24 h. At the endpoint, cells were stained with Nuclight Rapid Red dye (essen bioscience) and imaged one final time (red channel) to measure the total number of cells. Image quantitation was performed using Incucyte®Base Analysis software to generate segmented images used for quantification. Data are presented as the number of YOYO-1+ cells over the total number of cells. The mRNA levels of GSDMD and ipah7.8 were measured in Ea.hy926 cells after 24 h induction of IpaH7.8 with doxycycline using Power SYBR Green CellsTo-CT kit (ThermoFisher). The primers used for GSDMD (forward primer: 5’- GTGTGTCAACCTGTCTATCAAGG-3’ and reverse primer: 5’-CATGGCATCGTAGAAGTGGAAG-3’), ipaH7.8 (forward primer: 5’-CCCTGATCGTGGAGAACAATAA-3’ and reverse primer: 5’-CACGGACAGGCGATTGTTA3’), and GAPDH (forward primer: 5’- GTCTCCTCTGACTTCAACAGCG-3’ and reverse primer: 5’- ACCACCCTGTTGCTGTAGCCAA-3’). Transcript levels relative to GAPDH were calculated using the ΔCt method.
Plasmids, lentiviral, and transient expression
The genetically-barcoded Shigella effector library was synthesized and cloned into pMIN-ducer, a custom synthesized lentiviral vector that provides doxycycline-inducible control of transgene expression (Genscript). cDNAs encoding N-terminal FLAG IpaH7.8 WT, C357A mutant, LRR domain (aa 2–273), NEL domain (aa 274–565), and 5-Alanine-scanning mutants were synthesized and sub-cloned into pMIN-ducer or pCDNA3.1Zeo(+) (Genscript). cDNAs encoding untagged IpaH, IpaH1.4, IpaH2.5, IpaH4.5, IpaH7.8, and IpaH9.8 were obtained from the Shigella effector library used for screens. cDNAs encoding C-terminal FLAG, Myc, nano-Luciferase (nLuc) or Rho-1D4 (1D4) GSDMD (human and mouse), A-, B-, C-, E-, F-, single or tandem lysine to arginine mutants (K/R, 3K/R, and 15K/R), Pore-forming domain swap chimeras (hsPFD, mmPFD, hs16–21, and mm16–20), and 5-Alanine-scanning mutants were synthesized and sub-cloned into pCDNA3.1Zeo(+) (Genscript). For transient expression in 293T cells, 3 × 106 cells were plated in 10 cm plates 1 d prior to transfection. On the day of transfection, 3 μg of pCDNA3.1Zeo(+) total plasmid DNA was transfected using Lipofectamine 2000 (ThermoFisher) following the manufacturer’s instructions. For lentiviral packaging, 293T cells plated in 10 cm plates at a density of 2.5 × 106 cells were transfected with 5 μg pMIN-ducer, 10 μg pCMV-Δ8.9, and 0.5 μg Pcmv-VSVG (1:2.3:0.2 mole ratio). After 72 h, viral-containing culture supernatants were passed through 0.45 um syringe filters, and used immediately for infecting recipient cells. The day prior to infection, Ea.hy926 cells were seeded into 6-well plates at a density of 2 × 105 cells per well. Lentiviral-containing medium was supplemented with polybrene (Millipore) to a final concentration of 10 μg/mL, and used to infect recipient cells. After 48 h, selection of transduced cells was initiated with 4 μg/mL puromycin (Takara). Mock infected cells were used to judge selection duration and efficiency. For Hoxb8 lentiviral infections, 2.5 × 105 cells were seeded into 24-well plates in 0.5 mL of myeloid media, and spinfected with lentiviral supernatant supplemented with 10 μg/mL polybrene (Millipore) at 1,500 × g for 1 h at room temp. Cells received 3 mL of additional media and were allowed 48 h for recovery prior to selection with 10 μg/mL puromycin (Takara).
Immunoblotting and immunoprecipitation
Cell lysates were prepared after washing cells once with phosphate buffered saline, and lysing cells in radioimmunoprecipitation assay buffer (RIPA) supplemented with protease inhibitors (Roche) and 1% NP-40 (Pierce). Lysates were clarified by centrifugation at 20,000 × g for 30 min, and protein concentrations were measured using the BCA assay (Pierce). For IPs, lysates were incubated with 20 μL Flag-M2 sepharose (MilliporeSigma) or Rho-1D4 sepharose (Rho-1D4 Ab was coupled to CNBr-activated sepharose beads, GE Healthcare) slurry (10 μL packed resin) for 1 h at 4°C. Beads were washed with lysis buffer, and captured proteins were eluted with 100 μg/mL 3XFLAG or 250 μM Rho-1D4 peptide O/N at 4°C. Samples were reduced after boiling with 4X NuPAGE sample buffer, and resolved using NuPAGE Bis-Tris gels in MES running buffer (ThermoFisher). Proteins were transferred using wet-transfer boxes (Bio-Rad) onto nitrocellulose membranes using NuPAGE transfer buffer (ThermoFisher) supplemented with 10% (v/v) methanol. Membranes were blocked with 5% milk TBS-T for 30 min, and probed with primary Abs dissolved in blocking buffer O/N at 4°C. The following day, membranes were washed with TBS-T, and incubated with secondary HRP-conjugated Abs diluted in blocking buffer for 1 h at room temperature. Membranes were washed with TBS-T, incubated for 5 min with ECL substrate (ThermoFisher), and visualized by exposing and developing films (GE healthcare).
Positive selection screens
The lentiviral library was packaged in twelve 15 cm cell culture plates of 293T cells. Each plate (2.7 × 107 cells) was transfected with 50.8 μg of DNA mix comprising the library plasmid, pCMV-Δ8.9 and pCMV-VSVG plasmids at a molar ratio of 1:2:0.2 using Lipofectamine 2000 reagent (ThermoFisher). At 6 h post-transfection, the medium containing the transfection mix was aspirated and replaced with fresh growth medium supplemented with 1 U/mL DNase I, 5 mM MgCl2 and 20 mM HEPES pH 7.2. After overnight incubation at 37°C, the medium was aspirated and replaced with fresh growth medium. After another 24 hours incubation, the lentiviruscontaining medium from each plate was collected, pooled, clarified by filtering through a 0.45 μm bottle-top filter, and concentrated using the Lenti-X Concentrator (Clontech). Concentrated lentivirus was re-suspended in PBS/1% BSA, aliquoted, frozen in dry ice/ethanol and stored at 80°C. Ea.hy926 human endothelial cells were infected with pooled lentivirus at an MOI of 0.3 to ensure a single integrant frequency of 97% with 1000X fold-coverage. On day 1, Ea.hy926 cells were seeded into 2 × 10 cm plates (1.2 × 106 each) and cultured for 24 h. The next day, cells were infected with lentivirus diluted in DMEM supplemented with 10 μg/mL polybrene (Millipore) for 24 h. On day 3, virus-containing media was replaced with fresh DMEM, and cells were cultured for another 24 h. On day 4, cells were expanded into a 15 cm plate and cultured further for 24 h. On day 5, antibiotic selection was initiated by replacing the culture media with DMEM containing 2 μg/mL puromycin (Takara). After 5 d, cells were expanded into 4 × 15 cm plates with antibiotic-free DMEM, and cultured for 2 d.
For LPS screens, 6 × 15 cm plates of Ea.hy926-Shigella-effector-library cells were seeded at a density of 1.5 × 106 cells per plate (3 biological replicates each, ±LPS electroporation). Shigella effectors were dox-induced (250 ng/mL) for 48 h (6 plates). On the day of LPS-electroporation, cells were lifted into suspension with TrpLE Express (ThermoFisher), washed once with PBS, and re-suspended in 110 μL Buffer R each (Neon, ThermoFisher). Cells were electroporated ±7 μg of LPS (InvivoGen) using the Neon™ Transfection System 100 μL Kit. Cells were washed once with PBS to remove LPS, and plated for recovery in 6-well plates containing fresh DMEM medium. Mock electroporated control cells were passaged until LPS-electroporated cells recovered. After 11 d, LPS-resistant and mock-electroporated cells were collected for genomic DNA (gDNA) extraction using the Gentra Puregene Cell Kit (Qiagen) following the manufacturer’s instructions. Using the resulting gDNA as templates for PCR amplification, genetically-barcoded regions were amplified within the linear range using custom primers. PCR-cleanup was conducted on an automated liquid-handler (Beckman), DNA concentration was determined fluorometrically using the Qubit (ThermoFisher), and the samples were submitted for next generation sequencing.
Next generation sequencing analysis
A sample ORF matrix was generated by counting exact matches of ORF barcodes in the sample FASTQ files. Count matrix was normalized by library size-based factors. Differentially enriched ORFs were identified using DESeq2 (Love et al., 2014) by comparing LPS treated sample to the untreated sample.
Bioinformatics analysis of S. flexneri genomes
The ipaH7.8 Shigella protein sequence (Genbank accession: WP_010921637) was compared to 24 complete Shigella flexneri genomes (Table S2) using TBLASTN 2.7.1+ (Camacho et al., 2009) with default parameter settings. ipaH7.8 was determined to be present in a genome if BLAST searches yielded at least one high-scoring segment pair satisfying expect value < 0.001, query coverage > 80% and sequence identity > 90%.
Multiple sequence alignment
Alignments were generated using Clustal Omega at EMBL-EBI, and results were displayed using Jalview.
IpaH7.8 substrate identification
Ea.hy926 Flag-IpaH7.8(dox)-inducible cells were used for identifying IpaH7.8 substrate(s) using multiplexed quantitative proteomics. Timed expression of IpaH7.8 was carried out as follows: no dox 6 h (n = 2), plus dox 6 h (n = 2), plus dox 4 h (n = 2), plus dox 2 h (n = 2), plus dox 1 h (n = 1), and no dox 0 h (n = 2) for a total of 11 samples (each encoded within a single TMT11-plex experiment). A yield of 40 mg of protein per treatment group necessitated 143 × 15 cm plates of confluent cells. All treatment groups were harvested at the same endpoint by scraping into MS buffer containing 50 mM HEPES pH 8.5, 9 M Urea, 150 mM NaCl, and protease inhibitors (Roche). Samples were rotated end-over-end at room temperature for 1 h, centrifuged at 15,000 × g for 20 min, and the clarified lysates were transferred to fresh tubes. The total yield per sample was determined by BCA assay (Pierce).
Multiplexed quantitative proteomics sample preparation
20 mg of protein lysate in denaturing buffer (8 M Urea, 20 mM HEPES, pH 8.0) was reduced (5 mM dithiothreitol (DTT), 45 min at 37°C), alkylated (15 mM iodoacetamide (IAA), 20 min at room temperature in the dark), and quenched (5 mM DTT, 15 min at room temperature in the dark). Proteins were pelleted by chloroform-methanol precipitation. The resulting pellet was resuspended in denaturing buffer, diluted to 4M Urea, and digested for 4 hours at 37°C with lysylendopeptidase (Wako) at an enzyme to protein ratio of 1:100. The sample was further diluted to 1.3 M Urea and subjected to overnight enzymatic digestion at 37°C with sequencing grade trypsin (Promega, enzyme : protein ratio = 1:50). Resultant peptides were acidified with 20% Trifluoroacetic acid (TFA, 1% final concentration), centrifuged at 18,000 × g for 15 min, and desalted using a Sep-Pak C18 column (Waters). ~500 μg of eluted peptides from each treatment group was lyophilized and reserved for global proteome abundance. The remaining eluted peptides were lyophilized and used for di-glycine (KGG) analysis. For global proteome samples, 100 μg of peptides from each sample was dissolved in 20 mM HEPES pH 8.0 (1 mg/mL). Isobaric labeling was performed using TMT11-plex reagents (ThermoFisher). Each unit (0.8 mg) of TMT reagent was allowed to reach room temperature immediately before use, spun down on a benchtop centrifuge, and re-suspended with occasional vortexing in 41 μL anhydrous acetonitrile (ACN) prior to mixing with peptides (29% final ACN concentration). After incubation at room temperature for 1 h, the reaction was quenched for 15 min with 20 μL of 5% hydroxylamine. Labeled peptides were combined in equimolar ratios and dried. The TMT-labeled sample was re-dissolved in 80 μL 0.1% TFA, centrifuged at 16,000 × g, and the supernatant was processed further. Offline high pH reversed-phase fractionation was performed on a 1100 HPLC system (Agilent) using an ammonium formate based buffer system. Peptides (400 μg) were loaded onto a 2.1 × 150 mm 3.5 μm 300 Extend-C18 Zorbax column (Agilent) and separated over a 75-min gradient from 5% to 85% ACN into 96 fractions (flowrate = 200 μL/min). The fractions were concatenated into 24 fractions, mixing different parts of the gradient to produce samples that would be orthogonal to downstream low pH reversed phase LC-MS/MS. Fractions were dried and desalted using C18 stage-tips as previously described (Rappsilber et al., 2007). Peptides were lyophilized and resuspended in 10 μL Buffer A (2% ACN, 0.1% formic acid) for LC-MS/MS analysis.
For ubiquitylome quantitation of KGG peptides, lyophilized peptides were reconstituted in 1X detergent containing IAP buffer (Cell Signaling Technology) for immunoaffinity enrichment. KGG peptide enrichment was performed at 4°C on a MEA2 automated purification system (Phynexus) using 1 mL Phytips (Phynexus) packed with 20 μL ProPlus resin coupled to 200 μg of anti-KGG (Cell Signaling Technology) antibody. Phytip columns were equilibrated for 2 cycles (1 cycle = aspiration and dispensing, 0.9 mL, 0.5 mL/min) with 1 mL 1X IAP buffer prior to contact with peptides. Phytip columns were incubated with peptides for 16 cycles of capture, followed by 6 cycles of wash, 2X with 1 mL 1X IAP buffer and 4X with 1 mL water. Captured peptides were eluted with 60 μL 0.15% TFA in 8 cycles where the volume aspirated/dispensed was adjusted to 60 μL. Enriched ubiquitinated peptides were prepared as previously described (Rose et al., 2016). Labeled peptides were combined, dried, and re-solubilized in 0.15% TFA for high pH reversed-phase fractionation using a commercially available kit (ThermoFisher). Fractionation was performed according to the manufacturer’s protocol with a modified elution scheme where 11 fractions were collected (F1: 13.5% ACN, F2: 15% ACN, F3: 16.25% ACN, F4: 17.5 ACN,F5: 20% ACN, F6: 21.5% ACN, F7: 22.5% ACN, F8: 23.75% ACN, F9: 25% ACN, F10: 27.5% ACN and F11: 30% ACN) and then combined into 6 fractions (F1+F6, F2+F7, F8, F3+F9, F4+F10, F5+F11). Peptides were lyophilized and re-suspended in 10 μL Buffer A for LCMS/MS analysis.
Mass spectrometry instrumental analysis
For global proteome and ubiquitylome quantitation of KGG peptides, LC-MS/MS analysis was performed by injecting 5 μL of each fraction on an Orbitrap Lumos mass spectrometer (ThermoFisher) coupled to a Dionex Ultimate 3000 RSLC (ThermoFisher) employing a 25 cm IonOpticks Aurora Series column (IonOpticks, Parkville, Australia) with a gradient of 2% to 30% buffer B (98% ACN, 2% H2O with 0.1% FA, flow rate = 300 nL/min). All samples were analyzed with a total run time of 180 min. The Orbitrap Lumos collected FTMS1 scans at 120,000 resolution with an AGC target of 1 × 106 and a maximum injection time of 50 ms. FTMS2 scans on precursors with charge states of 3–6 were collected at 15,000 resolution with CID fragmentation at a normalized collision energy of 35%, an AGC target of 2 × 104 (proteome) or 2 × 105 (KGG), and a max injection time of 100 ms (proteome) or 200 ms (KGG). Synchronous-precursor-selection (SPS) MS3 scans were analyzed in the Orbitrap at 50,000 resolution with the top 8 most intense ions in the MS2 spectrum subjected to HCD fragmentation at a normalized collision energy of 55%, an AGC target of 2 × 105, and a max injection time of 100 ms (proteome) or 350 ms (KGG).
Mass spectrometry data analysis
For both global proteome and ubiquitylome quantitation of KGG peptides, MS data were searched using Mascot against a concatenated target-decoy human database (downloaded August, 2017) containing common contaminant sequences, and the protein sequence of Shigella IpaH7.8 ligase with a precursor mass tolerance of 50 ppm, 0.8 Da fragment ion tolerance, and tryptic specificity up to 2 (proteome) or 3 missed cleavages (KGG). For global proteome analysis, the following modifications were considered: carbamidomethyl cysteine (+57.0214), TMT labeled N-terminus (+229.1629), and TMT labeled lysine (+229.1629) as static modifications, and oxidized methionine (+15.9949) and TMT labeled tyrosine (+229.1629) as variable modifications. For analysis of KGG peptides, TMT labeled di-glycine modified lysine (+343.2059) was also included as a variable modification. Peptide spectral matches for each run were filtered using linear discriminant analysis (LDA) to a false discovery rate (FDR) of 2% and subsequently in aggregate to a protein level FDR of 2%. TMT-MS3 quantification was performed using Mojave, with only those PSMs possessing isolation specificities greater than or equal to 0.5 considered for the final dataset. The abundance of ubiquitination on each peptide or each identified protein was estimated by using a model fitted with Tukey Median Polish summarization with imputation of missing values below a censoring threshold of 28. For each pairwise comparison, the change in abundance (log2 ‘fold’ values) and the results of an ANOVA test were reported. We used the implementation of these methods in MSstats 3.16.0 (Choi et al., 2014). Data were further processed in R to produce figures.
Bacterial infection of cultured cells
Bacteria used for infection studies were freshly streaked from glycerol stocks onto trypticase soy agar (TSA) supplemented with Congo red (Sigma-Aldrich), and used for experiments within 1 week. For biochemical experiments, HeLa cells were plated in 6-well plates at a density of 3.5 × 105 cells per well in antibiotic-free DMEM. On the same day, a single red colony of the desired strain was used to inoculate a tube containing 5 mL of terrific broth (TB), and cultured overnight at 37°C. On the morning of infection, overnight cultures were diluted 1:50 (v/v) in TB and cultured until bacteria reached an OD600 of 0.6–0.8. Bacteria were pelleted at 3,000 × g and washed once with PBS. Bacteria were pelleted again, and re-suspended in PBS supplemented with 5 μg/mL poly-L-lysine (Millipore Sigma), and incubated at room temperature for 15 min. Bacteria were pelleted and washed with PBS once more, and re-suspended in DMEM without supplements. Infections were carried out at a multiplicity of infection (MOI) of 25 (1 OD600 = 8 × 108 bacteria per mL) by spinfecting bacteria onto HeLa cells at 1,000 × g for 10 min. Bacterial invasion occurred at 37°C for 30 min, after which, cells were washed twice with PBS, once with DMEM containing 50 ug/mL gentamicin, and overlaid with the same antibiotic medium until sample processing steps. Lysates used for immunoblot were generated by gently washing cells once with PBS, and lysing cells with 0.5 mL of RIPA buffer supplemented with 1% NP-40 (Pierce) and protease inhibitors (Roche) at 4°C for 1 h. Viable bacteria from these lysates were serially diluted in PBS supplemented with 0.1% Tween-20 (Pierce), and cultured on TSA plates overnight at 37°C for CFU determination. For propidium iodide (PI) uptake studies, HeLa, Ea.hy926, or Caco-2 cells were plated in black-wall 96-well plates (Corning) at a density of 20,000 cells per well in antibiotic-free DMEM. Bacteria were prepared identically as for biochemical experiments, but infections were carried out at an MOI of 1. Cells were spinfected as before, but the final overlay also contained 50 μg/mL propidium iodide (Sigma-Aldrich). Cells were imaged every 30 minutes for 24 h using the IncuCyte S3 (essen bioscience) to monitor cells by phase contrast (cell confluency), GFP fluorescence (bacteria infected cells), and red fluorescence (PI+ dying cells) using a 10X objective. Image quantitation was performed using Incucyte®Base Analysis software to generate segmented images used for quantification. The y-axis indicates the percentage of infected cells that are killed by the indicated strains of S. flexneri. Calculated as:
Protein expression and purification
Shigella IpaH7.8 2–565 WT/C357A constructs were expressed as N-terminal His fusion constructs in E. coli Rosetta 2 (Millipore). Human GSDMD 1–484 was expressed as a His-SUMO fusion construct in E. coli Rosetta 2 (Millipore). Cell pellets were frozen and stored at −80°C and re-suspended prior to purification in Lysis Buffer: 50 mM Tris pH 8.0, 200 mM NaCl, 5 % glycerol, 5 mM MgCl2, 1 mM TCEP or 2 mM β-mercaptoethanol (BME) supplemented with protease inhibitors (Roche), DNAse and Lysozyme. Cells were lysed via microfluidization, the lysate was clarified by centrifugation at 18,000 rpm for 1 h, and filtered (2 μm pore). NTA Superflow resin (QIAGEN) or Talon® Superflow™ (GE Healthcare) were used for affinity purification. The resin was incubated with the clarified lysate for 1 h at 4°C and washed with 1–2 L of Wash Buffer: 50 mM Tris pH 8.0, 200 mM NaCl, 5 % glycerol, 1 mM TCEP or 2 mM BME. Proteins were eluted in Wash Buffer supplemented with 250 mM Imidazole pH 8.0 (Sigma). Eluted fractions were analyzed by SDS-PAGE gel and protein containing fractions were pooled. IpaH7.8 proteins were concentrated and further purified by size exclusion chromatography (SEC) using a Superdex 200 column (GE Healthcare) pre-equilibrated with 50 mM Tris pH 8.0, 300 mM NaCl, 5% glycerol, and 1 mM TCEP. His-tagged SENP1 was used for cleaving the remaining His-SUMO tag from purified GSDMD. Cleavage reactions proceeded in dialysis membranes O/N against Wash Buffer at 4°C. The His-SUMO tag was captured by NTA Superflow resin (QIAGEN) and the flowthrough containing untagged GSDMD was collected. Cleavage was confirmed by LC/MS. GSDMD was concentrated further and purified by SEC using a Superdex 200 column (GE Healthcare) pre-equilibrated with 50 mM Tris pH 8.0, 300 mM NaCl, 5% glycerol, 1 mM TCEP. SEC fractions were analyzed by SDS-PAGE gel, and pure protein containing fractions were pooled, concentrated, snap-frozen in liquid nitrogen, and stored at −80°C.
in vitro ubiquitination assays
Reactions were set up with with 2.5 ng/μL human E1 (Boston Biochem), 0.125 μg/μL Ub (Boston Biochem), 0.01 μg/μL UBE2D3 (Boston Biochem), 0.01 μg/μL IpaH7.8 (WT or C357A, CA), and 0.1 μg/μL human GSDMD in reaction buffer: 50 mM Tris pH 8.0, 50 mM NaCl, 5 mm MgCl2, and 0.1 mm DTT. Reactions were initiated with 5 mM ATP, incubated at 37°C, and quenched with LDS sample loading buffer (ThermoFisher) at the indicated time points. Equimolar amounts of K11-, K48-, and K63-linked Ub4 chains (Boston Biochem) were loaded as controls for the ubiquitin linkage detection assay.
in vitro cleavage and membrane flotation assays
Cleavage assays were carried out under identical reaction conditions as described for in vitro ubiquitination. Prior to the addition of ATP to initiate ubiquitination, input samples containing an equal volume were removed for un-cleaved control and cleavage reactions (Pre-Ub). Ubiquitination was initiated by ATP addition, and allowed to proceed for 60 min at 37°C. Reactions were quenched with DTT, and equal volumes were removed for un-cleaved control and cleavage reactions (post-Ub). Recombinant ΔCARD-Caspase-4 was added (2 μM final concentration), and incubated at 37°C for 15 min. LDS sample buffer was added to a subset of the pre-Ub and post-Ub cleavage samples, and the remainder was used for membrane flotation assay. Large unilamellar vesicles (LUVs) containing phosphatidylcholine (PC, Avanti), phosphatidylethanolamine (PE, Avanti), and Rhodamine-phosphatidylethanolamine (Rhod-PE, Avanti) (1:1 mole ratio PC:PE doped with 0.1 mole % of total lipids with Rhod-PE) were prepared by extrusion. Briefly, lipids were dispensed from organic solutions into as conical glass container. The lipids were concentrated under reduced pressure using a rotary evaporator, and dried further on a hi-vacuum line until no residual solvent remained. The lipid cake was hydrated in 50 mM Hepes pH 7.5, 150 mM NaCl, and 1 mM TCEP. The solution was vortexed to generate a suspension, and briefly sonicated in a bath sonicator. A miniature extruder (Avanti) was used to extrude the lipid suspension first through a 0.4 μm membrane (15 passes), and then through a 0.1 μm membrane (15 passes). Equal volumes of pre-Ub and post-Ub cleavage reactions (20 μL ea.) were mixed with LUVs (20 uL), and allowed to equilibrate at rt for 30 min. The samples were mixed with an equal volume of 80% (w/v) Histobenz (40 μL, 40% final), and loaded into an ultracentrifuge tube. A layer of 20% (w/v) Histobenz (250 μL) was placed above the samples, followed by a final layer of liposome buffer (40 μL). Samples were centrifuged in a TLS-55 swinging bucket rotor at 200,000 × g in a table-top ultracentrifuge for 4 h. The gradients were fractionated, and the fluorescence signal in each fraction was measured (Ex-560/Em-583). LDS sample loading buffer was added to each fraction, and resolved by SDS-PAGE. Plots were generated with GraphPad Prism 8, with the fluorescence signal being normalized to the signal obtained from the top of the gradient.
Microscale thermophoresis (MST)
Proteins used for fluorescent labeling and binding reactions were expressed and purified from 293T cells. Briefly, hsGSDMD-1D4 and mmGSDMD-1D4 constructs were transfected in 293T cells using the procedures outlined in the methods under transient expression. After 48 h, cells were lysed in buffer that contained 50 mM Hepes pH 7.5, 150 mM NaCl, 1% NP-40, and 1X Roche cOmplete protease inhibitor cocktail at 4°C for 1 h. Lysates were clarified by centrifugation at 20,000 × g for 30 min and transferred to fresh tubes. Rho-1D4 resin was incubated with the lysates at 4°C for 1 h, followed by 3 consecutive washes in lysis buffer. Proteins were eluted with 250 μM 1D4-peptide dissolved in 50 mM Hepes pH 7.5, 150 mM NaCl at 4°C overnight. Eluted proteins were labeled using the 2nd generation red-maleimide protein labeling kit from Nanotemper following the manufacturer’s instructions. Briefly, a 10:1 molar ratio of dye to protein was used for labeling reactions. Excess, unreacted dye was removed using the de-salting columns supplied in the kit. Binding reactions were set up in 50 mM Hepes pH 7.5, 150 mM NaCl, and 0.1% Tween-20 using each of the dye-labeled GSDMD proteins. IpaH7.8 was purified from bacteria as described before, and titrated against GSDMD in a two-fold dilution series with a final volume of 20 μL. Reactions were allowed to equilibrate for 1 h prior to recording the MST-signal. Data were collected on the Monolith NT.115 adjusted to 5% LED and medium MST power. Plots were generated using GraphPad Prism 8, signals (Fnorm) were normalized to 1, and curves were fitted using non-linear regression to estimate the Kd s for each reaction.
Histology
Hematoxylin and eosin stained sections of cecum and colon were scored based on previously established scoring systems (Mitchell et al., 2020). In brief, histologic sections were scored by a qualified veterinary pathologist in a randomized and blinded manner for the severity of edema, inflammation, epithelial injury, and hyperplasia. Edema severity was scored as: 0) no edema; 1) mild, focal to segmental edema with less than 2-fold expansion of the submucosa and mucosa; 2) moderate, focal to segmental edema with 2 to 3-fold expansion of the submucosa and mucosa; 3) severe, multifocal to segmental edema with at least 1 region of > 3-fold expansion of the submucosa and mucosa; 4) diffuse, > 3-fold expansion of the submucosa and mucosa. Inflammatory infiltrates were scored as: 0) no inflammation; 1) minimal, focal to multifocal (usually less than 3 foci) inflammation with minimal disruption of the mucosal architecture; 2) multifocal inflammation associated with mild to moderate separation of crypts and/or crypt elevation; 3) severe, multifocal to coalescing inflammatory infiltrates that expand and disrupt the mucosal architecture; 4) extensive, confluent inflammatory infiltrates distributed throughout the mucosa. Epithelial damage was scored as: 0) no epithelial injury observed; 1) rare superficial epithelial erosions or crypt degeneration/atrophy; 2) multifocal, superficial epithelial attenuation, erosions or cobblestoning and/or crypt degeneration/ atrophy; 3) multifocal ulcers and/or regional crypt degeneration/ atrophy; 4) locally extensive to diffuse ulceration and/or crypt loss. Crypt hyperplasia was scored as: 0) no hyperplasia identified; 1) mild, hyperplasia with less than 2-fold crypt expansion associated with inflammation; 2) multifocal, hyperplasia with less than 2-fold crypt expansion associated with inflammation; 3) moderate, focal to multifocal crypt hyperplasia with > 2-fold crypt expansion. 4) extensive crypt hyperplasia of > 2-fold crypt expansion occurring in greater than 50% of the mucosa. Individual scores were summed to derive final scores of the colonic and cecal lesions.
Statistics
Statistical analysis of results was performed by unpaired, two-tailed t-test or Mann-Whitney test. P-value calculations for mass-spectrometry analysis in Fig. 2b were performed as described above. All other statistical analyses were performed using GraphPad Prism 8.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti Actin | MP Biomedicals | SKU: 0869100-CF, Clone C4 |
| Rabbit polyclonal anti beta-tubulin | Abcam | Cat#: Ab6046, RRID:AB_2210370 |
| Rabbit polyclonal anti IpaH9.8 | EpiGentek | Cat#: A55475 |
| Mouse monoclonal anti Cul1 | ThermoFisher | Cat#: 32–2400, 2H4C9, RRID:AB_86218 |
| Mouse monoclonal anti Myc | Cell Signaling | Cat#: 2276S, RRID:AB_331783 |
| Mouse monoclonal anti FLAG M2-Peroxidase | Millipore Sigma | SKU: A8592, RRID:AB_439702 |
| Mouse monoclonal anti FLAG M2 | Millipore Sigma | SKU: F3165, RRID:AB_259529 |
| Rat monoclonal anti GSDMD | Aglietti et al., 2016 | Clone 17G2G9, Genentech |
| Mouse monoclonal anti Rho-1D4 | University of British Colombia | https://ubc.flintbox.com/technologies/0f1ef64b-fa5d-4a58-9003-3e01f6f672a6 |
| Mouse monoclonal anti-Ubiquitin | LifeSensors | SKU: VU-0101, clone VU-1, RRID:AB_2716558 |
| Human monoclonal anti K11-Ubiquitin | Matsumoto et al., 2010 | Genentech |
| Human monoclonal anti K48-Ubiquitin | Newton et al., 2008 | Genentech |
| Human monoclonal anti K63-Ubiquitin | Newton et al., 2008 | Genentech |
| Rabbit monoclonal anti GSDMD | Cell Signaling Technology | Cat#: 97558S, E8G3F, RRID:AB_2864253 |
| Rabbit monoclonal anti GSDMD(Asp275) | Cell Signaling Technology | Cat#: 36425S, E7H9G, RRID:AB_2799099 |
| Mouse monoclonal caspase-4 | Enzo | Cat#: ADI-AAM-114 RRID:AB_10621609 |
| Bacterial and virus strains | ||
| Shigella flexneri M90T wild-type (WT) | John Rohde, Dalhousie University | N/A |
| Shigella flexneri M90T wild-type (WT) GFP | This study | Genentech |
| Shigella flexneri M90T ΔIpaH7.8 (WT) GFP | This study | Genentech |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| Ultra-pure LPS E. coli 0111:B4 | InvivoGen | Cat#: 0111:B4 |
| Egg phosphatidylcholine (Egg PC) | Avanti Polar Lipids | Cat#: 840051 |
| Egg phosphatidylethanolamine (Egg PE) | Avanti Polar Lipids | Cat#: 840021 |
| Egg Liss Rhod phosphatidylethanolamine | Avanti Polar Lipids | Cat#: 810146 |
| Val-boroPro | Millipore Sigma | Cat#: 531465 |
| Doxycycline | Fisher scientific (Clontech) | Cat#: NC042034 |
| 3X FLAG® Peptide | Millipore Sigma | SKU: F4799 |
| 1D4 peptide (TETSQVAPA) | Genscript | Custom peptide |
| MLN-7243 | ChemieTek | Cat#: CT-M7243 |
| MLN-4924 (Pevonedistat) | ChemieTek | Cat#: CT-M4924 |
| Bortezomib | ChemieTek | Cat#: CT-BZ001 |
| Propidium iodide | Millipore Sigma | SKU: P4170 |
| YOYO-1 Iodide | ThermoFisher | Cat#: Y3601 |
| Nuclight Rapid Red Dye | essenbioscience | Cat#: 4717 |
| Congo Red | Millipore Sigma | SKU: C6277 |
| Poly-L-lysine | Millipore Sigma | SKU: P4707 |
| Alt-R CRISPR-Cas9 | IDT | Cat#: 1081060 |
| IpaH7.8 wild-type (recombinant) | This study | Genentech |
| IpaH7.8 C357A (recombinant) | This study | Genentech |
| Human GSDMD (recombinant) | This study | Genentech |
| Mouse GSDMD (recombinant) | This study | Genentech |
| Human E1 | R&D systems | Cat#: E-304–050 |
| Human UBE2D3 | R&D systems | Cat#: E2–627-100 |
| Human Ubiquitin | R&D systems | Cat#: U-100H-10M |
| K11-linked tetra-Ub chains | R&D systems | Cat#: UC-45–025 |
| K48-linked tetra-Ub chains | R&D systems | Cat#: UC-210B-025 |
| K63-linked tetra-Ub chains | R&D systems | Cat#: UC-310B-025 |
| Critical commercial assays | ||
| CytoTox 96® Non-radioactive Cytotoxicity Assay | Promega | Cat#: G1780 |
| Power SYBR™ Green Cells-to-CT™ Kit | ThermoFisher | Cat#: 4402955 |
| Protein labeling kit RED-MALEIMIDE 2nd Generation | NanoTemper | SKU: MO-L014 |
| Deposited data | ||
| Shigella flexneri whole-genome sequencing | This study | SRA: PRJNA697993 |
| Experimental models: cell lines | ||
| Ea.hy926 human endothelial cells | Genentech gCell facility | N/A |
| Ea.hy926 GSDMD−/− CRISPR-KO cells | This study | N/A |
| 293T human embryonic kidney cells | Genentech gCell facility | N/A |
| HeLa human cervical epithelial cells | Genentech gCell facility | N/A |
| Caco-2 human intestinal epithelial cells | Genentech gCell facility | N/A |
| HeLa GSDMD−/− CRISPR-KO cells | This study | N/A |
| ER-Hoxb8-immortalized murine myeloid progenitor cells | Wang et al., 2006 | Genentech |
| Experimental models: organisms/strains | ||
| Nlrc4−/− mice (C57BL6/N) | Mariathasan et al., 2004 | Genentech |
| Gsdmd−/− mice (C57BL6/N) | Kayagaki et al., 2015 | Genentech |
| Nlrc4−/− Gsdmd−/− mice (C57BL6/N) | This study | Genentech |
| Oligonucleotides | ||
| Human GSDMD gRNA | IDT | TTCCACTTCTACGATGCCA |
| GSDMD RT-qPCR forward primer | IDT | GTGTGTCAACCTGTCTATCAAGG |
| GSDMD RT-qPCR reverse primer | IDT | CATGGCATCGTAGAAGTGGAAG |
| ipaH7.8 RT-qPCR forward primer | IDT | CCCTGATCGTGGAGAACAATAA |
| ipaH7.8 RT-qPCR reverse primer | IDT | CACGGACAGGCGATTGTTA |
| GAPDH RT-qPCR forward primer | IDT | GTCTCCTCTGACTTCAACAGCG |
| GAPDH RT-qPCR reverse primer | IDT | ACCACCCTGTTGCTGTAGCCAA |
| Recombinant DNA | ||
| pMIN-ducer (dox-inducible, lentiviral expression) | Genscript, this study | N/A |
| pMIN-ducer Shigella effector library | Genscript, Table S1, this study | N/A |
| pMIN-ducer FLAG-IpaH7.8 | Genscript, this study | N/A |
| pMIN-ducer FLAG-IpaH7.8 C357A | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo (mammalian transient overexpression) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo FLAG-IpaH7.8 | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo FLAG-IpaH7.8 C357A | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo FLAG-IpaH7.8 LRR | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo FLAG-IpaH7.8 NEL | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMA-myc (human) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMB-myc (human) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMC-myc (human) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-myc (human) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-myc (mouse) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDME-myc (human) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMF-myc (human) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-1D4 (human) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-1D4 (mouse) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-1D4 (human) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-3KR-1D4 (human) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-15KR-1D4 (human) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo mmPFD-1D4 (chimera) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo hsPFD-1D4 (chimera) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-nLuc-1D4 (human) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-nLuc-1D4 (mouse) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-nLuc-1D4 (human 5X Ala mutant, aa 11–15) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-nLuc-1D4 (human 5X Ala mutant, aa 16–20) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo GSDMD-nLuc-1D4 (human 5X Ala mutant, aa 21–25) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo hsGSDMD-mmGSDMD-16–21-1D4 (chimera) | Genscript, this study | N/A |
| pCDNA3.1 (+) zeo mmGSDMD-hsGSDMD-16–20-1D4 (chimera) | Genscript, this study | N/A |
| pCMV-Δ8.9 (lentiviral packaging) | Genentech | N/A |
| pCMV-VSVG (lentiviral packaging) | Genentech | N/A |
| pACYC177 (bacterial kanamycin resistance cassette) | Genentech | N/A |
| pKD46 (lambda red recombinase) | Genentech | N/A |
| pBR322-AmpR-dasher GFP | Genentech | N/A |
| Software and algorithms | ||
| Prism 8 | GraphPad software, LLC | https://www.graphpad.com/, RRID:SCR_002798 |
| DESeq2 | Bioconductor | https://bioconductor.org/packages/release/bioc/html/DESeq2.html, RRID:SCR_015687 |
| MSstats 3.16.0 | Choi et al., 2014 | https://github.com/MeenaChoi/UW2019-MSstats/blob/master/uw2019-msstats.Rmd, RRID:SCR_014353 |
| The R-project | The R Foundation | https://www.r-project.org/ |
| Pymol 2.3.2 | Schrodinger LLC. | https://pymol.org, RRID:SCR_000305 |
| Jalview 2.11.1.4 | The Barton Group, University of Dundee, Scotland, UK | http://www.jalview.org/, RRID:SCR_006459 |
| Incucyte®Base Analysis software | Essenbioscience | https://www.essenbioscience.com/en/products/software/incucyte-base-software/ |
| Other | ||
Highlights.
The Shigella ubiquitin ligase effector IpaH7.8 blocks pyroptosis in human cells
IpaH7.8 targets human, but not mouse, gasdermin D for proteasomal degradation
IpaH7.8 targets the pore-forming domain of gasdermin D to suppress pyroptosis
Mice lacking both GSDMD and NLRC4 are hyper susceptible to S. flexneri infection
Acknowledgments
This work was funded by Genentech and NIH grants AI075039, AI063302, and AI155634 (REV). We thank Susan Haller for technical assistance, Erin Dueber for advice on membrane flotation assays, and members of the Dixit and Vance laboratories for discussions and reagents.
Footnotes
Declaration of interests
All authors except JR, RAC, and REV are employees of Genentech. REV is an investigator of the Howard Hughes Medical Institute and a consultant for Ventus Therapeutics and Tempest Therapeutics.
Supplemental Data Sets
Data S1. Quantitative proteomics data gathered from IpaH7.8 time-course. Related to Figures 2A and 2B.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JPY, Coers J, Aderem A, Buxbaum JD, Miao EA (2015). Canonical inflammasomes drive IFN-Y to prime caspase-11 in defense against a cytosol-invasive bacterium. Cell Host Microbe 18, 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aglietti RA, Estevez A, Gupta A, Gonzalez M, Liu PS, Kayagaki N, Ciferri C, Dixit VM, Dueber EC (2016). GSDMD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc. Natl. Acad. Sci. USA 113, 7858–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashida H, Sasakawa C (2016). Shigella IpaH family effectors as a versatile model for studying pathogenic bacteria. Front. Cell Infect. Microbiol. 5, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker S, The HC (2018). Recent insights into Shigella. Curr. Opin. Infect. Dis. 31, 449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt AL, Ma J, Loke HK., Lingaraj T, Wu D, Hamman KB, Spelman JL, Cullis CA, Langston SP, Vyskocil S, Sells TB, Mallender WD, Visiers I, Li P, Claiborne CF, Rolfe M, Bolen JB, Rick LR (2010) Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the nedd8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol. Cell 37, 102–111. [DOI] [PubMed] [Google Scholar]
- Broz P, Dixit VM (2016). Inflammasomes: mechanism of assembly, regulation and signaling. Nat. Rev. Immunol. 16, 407–420. [DOI] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL (2009). BLAST+: architecture and applications. BMC Bioinformatics 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Chang CY., Clough T, Broudy D, Killeen T, MacLean B, Vitek O (2014). MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC., Shao F (2016). Poreforming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. [DOI] [PubMed] [Google Scholar]
- DuPont HL, Hornick RB, Dawkins AT, Snyder MJ, Formal SB (1969). The response of man to virulent Shigella flexneri 2a. J. Infect. Dis. 119, 296–299. [DOI] [PubMed] [Google Scholar]
- DuPont HL, Levine MM, Hornick RB, Formal SB (1989). Inoculum size in shigellosis and implications for extended mode of transmission. J. Infect. Dis. 159, 1126–1128. [DOI] [PubMed] [Google Scholar]
- Hansen JM, de Jong MF, Wu Q, Zhang L, Heisler DB, Alto LT, Alto NM (2021). Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions. Cell S0092–8674(21)00571–7. doi: 10.1016/j.cell.2021.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang Z, Zhong C, Han J (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 12, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji C, Du S, Li P, Zhu Q, Yang X, Long C, Yu J, Shao F, Xiao J (2019). Structural mechanism for guanylate-binding proteins (GBPs) targeting by the Shigella E3 ligase IpaH9.8. PLoS Pathog. 15, e1007876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Taabazuing CY, Okondo MC, Chui AJ, Rao SD, Brown FC, Reed C, Peguero E, de Stanchina E, Kentsis A, Bachovchin DA (2018). DPP8/DPP9 inhibitor-induced pyroptosis for treatment of acute myeloid-leukemia. Nat. Med. 24, 1151–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Rayamajhi M, Miao EA (2017). Programmed cell death as a defence against infection. Nat. Rev. Immunol. 17, 151–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Zhang Y, Krantz BA, Miao EA (2016). Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 213, 2113–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Roarke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signaling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, Forsberg LS, Carlson RW, Dixit VM (2013). Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 341, 1246–1249. [DOI] [PubMed] [Google Scholar]
- Keszei AFA, Sicheri F (2017). Mechanism of catalysis, E2 recognition, and autoinhibition for the IpaH family of bacterial E3 ubiquitin ligases. Proc. Natl. Acad. Sci. USA 114, 1311–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil IA, Troeger C, Blacker BF, Rao PC, Brown A, Atherly DE, Brewer TG, Engmann CN, Houpt ER, Kang G, Kotloff KL, Levine MM, Luby SP, MacLennan CA, Pan WK, Pavlinac PV, Platts-Mills JA, Qadri F, Riddle MS, Ryan ET, Shoultz A, Steele AD, Walson JL, Sanders JW, Mokdad AH, Murray CJL, Hay SI, Reiner RC (2018). Morbidity and mortality due to shigella and enterotoxigenic Esherichia coli diarrhea: global burden of disease study 1990–2016. Lancet Infect. Dis. 18, 1229–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, Akakura R, Yoshida M, Kawalec M, Reichhart JM., Mizushima T, Sasakawa C (2013). The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe 13, 570–583. [DOI] [PubMed] [Google Scholar]
- Kofoed EM, Vance RE (2011). Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey CA, Miao EA (2020). Programmed cell death in the evolutionary race against bacterial virulence factors. Cold Spring Harb. Perspect. Biol. 12(2):a036459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRock C, Cookson BT (2012). The yersenia virulence effector YopM binds caspase-1 to arrest inflammasome assembly and processing. Cell Host Microbe 12, 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X, Zhang Z, Xiao X, Qi J, He B, Wang J (2017). Enterovirus 71 inhibits pyroptosis through cleavage of GSDMD. J Virol., 91, e01069–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Jiang W, Yu Q, Liu W, Zhou P, Li J, Xu J, Xu B, Wang F, Shao F (2017). Ubiquitination and degradation of GBPs by a Shigella effector to suppress host defence. Nature 551, 378–383. [DOI] [PubMed] [Google Scholar]
- Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, Abbott DW, Xiao TS (2019) Crystal structures of the full-length murine and human gasdermin D reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity 51, 23–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014). Moderated estimation of fold change and dispersion forRNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maltez VI, Tubbs AL, Cook KD, Aachoui Y, Falcone EL, Holland SM, Whitmire JK, Miao EA (2015). Inflammasomes coordinate pyroptosis and natural killer cell cytotoxicity to clear infection by a ubiquitous environmental bacterium. Immunity 43, 987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S, Newton K, Monack DM, Vucic D, French DM, Lee WP, Roose-Girma M, Erickson S, Dixit VM (2004). Differential activation of the inflammasome by caspase-1 adapters ASC and Ipaf. Nature 430, 213–218. [DOI] [PubMed] [Google Scholar]
- Matsumoto ML, Wickliffe KE, Dong KC, Yu C, Bosanac I, Bustos D, Phu L, Kirkpatrick DS, Hymowitz SG, Rape M, Kelley RF, Dixit VM (2010). K11-linked polyubiquitin in cell cycle control revealed by K11 linkage-specific antibody. Cell 39, 477–484. [DOI] [PubMed] [Google Scholar]
- Matyskiela ME, Lu G, Ito T, Pagarigan B, Lu CC, Miller K, Fang W, Wang NY, Nguyen D, Houston J, Carmel G, Tran T, Riley M, Nosaka L, Lander GC, Gaidarova S, Xu S, Ruchelman AL, Handa H, Carmichael J, Daniel TO, Cathers BE, LopezGirona A, Chamberlain PP (2016) A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature 535, 252–257. [DOI] [PubMed] [Google Scholar]
- McGuire CD, Floyd TM (1958). Studies on experimental shigellosis. J. Exp. Med. 108, 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A (2010). Caspase-1 induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell PS, Roncaioli JL, Turcotte EA, Goers L, Chavez RA, Lee AY, Lesser CF, Rauch I, Vance RE (2020). NAIP-NLRC4-deficient mice are susceptible to shigellosis. eLife 2020;9:e59022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder JB (1971). Shigellosis in nonhuman primates: a review. Lab Anim. Sci. 21, 734–738. [PubMed] [Google Scholar]
- Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, Komuves L, French DM, Ferrando RE, Lam C, Compaan D, Yu C, Bosanac I, Hymowitz SG, Kelley RF, Dixit VM (2008). Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell 134, 668–678. [DOI] [PubMed] [Google Scholar]
- Pallet MA, Crepin VF, Serafini N, Habibzay M, Kotik O, Sanchez-Garrido J, Santo JPD, Shenoy AR, Berger CN, Frankel G (2017). Bacterial virulence factor inhibits caspase-4/11 activation in intestinal epithelial cells. Mucosal Immunol. 10, 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsot C (2009). Shigella type III secretion effectors: how, where, when, and for what purpose? Curr. Opin. Microbiol. 12, 110–116. [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Mann M, Ishihama Y (2007). Protocol for micro-purification, enrichment, prefrationation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906. [DOI] [PubMed] [Google Scholar]
- Petzold G, Fischer ES, Thomä NH (2016) Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature 532, 127–130. [DOI] [PubMed] [Google Scholar]
- Rauch I, Deets KA, Ji DX, Moltke JV, Tenthorey JL, Lee AY, Philip NH, Ayres JS, Brodsky IE, Gronert K, Vance RE (2017). NAIP-NLRC4 inflammasomes coordinate intestinal epithelial cell expulsion with eicosanoid and IL-18 release via activation of Caspase-1 and −8. Immunity 46, 649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde JR, Breitkreutz A, Chenal A, Sansonetti PJ, Parsot C (2007). Type III secretion effectors of the IpaH family are E3 ubiquitin ligases. Cell Host Microbe. 1, 77–83. [DOI] [PubMed] [Google Scholar]
- Rose CM, Isasa M, Ordureau A, Prado MA, Beausoleil SA, Jedrychowski MP, Finley DJ, Harper JW, Gygi SP (2016). Highly multiplexed quantitative mass spectrometry analysis of ubiquitylomes. Cell Systems 3, 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE (2019). Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 364, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer JD., Pereyre S, Archer KA, Burke TP, Hanson B, Lauer P, Portnoy DA (2011). Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc. Natl. Acad. Sci. USA 108, 12419–12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Mimuro H, Kim M, Ogawa M, Ashida H, Toyotome T, Franchi L, Suzuki M, Sanada T, Suzuki T, Tsutsui H, Nunez G, Sasakawa C (2014). Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc. Natl. Acad. Sci. USA 111, E4254–E4263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trofa AF, Ueno-Olsen H, Oiwa R, Yoshikawa M (1999). Dr. Kiyoshi Shiga: discoverer of the dysentery bacillus. Clin. Infect. Dis. 29, 1303–1306. [DOI] [PubMed] [Google Scholar]
- Wandel MP, Pathe C, Werner EI, Ellison CJ, Boyle KB, Malsburg AVD, Rohde J, Randow F (2017). GBPs inhibit motility of Shigella flexneri but are targeted for degradation by the bacterial ubiquitin ligase IpaH9.8. Cell Host Microbe 22, 432–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GG, Calvo KR, Pasillas MP, Sykes DB, Hacker H, Kamps MP (2006). Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat. Methods 3, 287–293. [DOI] [PubMed] [Google Scholar]
- Warren SE, Duong H, Mao DP, Armstrong A, Rajan J, Miao EA, Aderem A (2011). Generation of a listeria vaccine strain by enhanced caspase-1 activation. Eur. J. Immunol. 41, 1934–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TD, Nacu S (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Xiong Y, Huang H (2020). Substrate-binding destabilizes the hydrophobic cluster to relieve the autoinhibition of bacterial ubiquitin ligase IpaH9.8. Commun. Biol. 3, 752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Shi J, Gong Y, Lu Q, Xu H, Liu L, Shao F (2011). The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600. [DOI] [PubMed] [Google Scholar]
- Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, Wang Y, Li D, Liu W, Zhang Y, Shen L, Han W, Shen L, Ding J, Shao F (2020). Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368, 1–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry raw data is available as a supplementary data table (Data S1). Bacterial sequencing data has been deposited in Genbank, accession number WP_010921637.