

Abstract
Niche concept is a core tenet of ecology that has recently been applied in marine microbial research to describe the partitioning of taxa based either on adaptations to specific conditions across environments or on adaptations to specialised substrates. In this study, we combine spatiotemporal dynamics and predicted substrate utilisation to describe species-level niche partitioning within the NS5 Marine Group. Despite NS5 representing one of the most abundant marine flavobacterial clades from across the world’s oceans, our knowledge on their phylogenetic diversity and ecological functions is limited. Using novel and database-derived 16S rRNA gene and ribosomal protein sequences, we delineate the NS5 into 35 distinct species-level clusters, contained within four novel candidate genera. One candidate species, “Arcticimaribacter forsetii AHE01FL”, includes a novel cultured isolate, for which we provide a complete genome sequence—the first of an NS5—along with morphological insights using transmission electron microscopy. Assessing species’ spatial distribution dynamics across the Tara Oceans dataset, we identify depth as a key influencing factor, with 32 species preferring surface waters, as well as distinct patterns in relation to temperature, oxygen and salinity. Each species harbours a unique substrate-degradation potential along with predicted substrates conserved at the genus-level, e.g. alginate in NS5_F. Successional dynamics were observed for three species in a time-series dataset, likely driven by specialised substrate adaptations. We propose that the ecological niche partitioning of NS5 species is mainly based on specific abiotic factors, which define the niche space, and substrate availability that drive the species-specific temporal dynamics.
Subject terms: Microbial ecology, Marine microbiology, Water microbiology
Introduction
An ecological niche is defined as a specific set of conditions (environmental and biotic interactions) that allow a population to perform its evolutionarily adapted function and as a result, persist or grow [1, 2]. Although a long-standing concept in ecology, niche theory has only recently been incorporated in the study of marine microbial populations [3–6]. Such studies have either focused on adaptations to specific conditions across environments [4] or on specific functional adaptations within an environment, e.g. specialised substrate utilisation [7]. However, to obtain a more detailed understanding on microbial populations’ niches, an in-depth analysis on the adaptation to conditions across different spatial and temporal scales in combination with an assessment of ecological function is needed.
Microbial populations exhibit distinct distribution patterns across the world’s oceans, which are most influenced by depth [8] and changes in temperature [9, 10] and salinity [11]. However, the effect these have on microbial populations varies. Although some appear to be ubiquitously distributed, such as the SAR11 or Prochlorococcus Clade, further analysis has shown that distinct genetic variations exist, resulting in ecotypes that are driven by environmentally mediated selection processes [9, 12]. Within specific environments, microbial populations also exhibit distinct dynamics that are driven by temporally derived shifts, such as seasons [6]. This is particularly evident with heterotrophic microbes in the Bacteroidetes phylum, that show recurrent and potentially predictable, seasonal dynamics driven by substrate availability [7, 13, 14]. From these studies, it is clear that conditions and resources influence microbial populations, however, to what extent do these determine niches?
In this study, we phylogenetically and ecologically characterise members of the NS5 marine group (referred to as NS5 from hereon) and subsequently identify the key niche-determining factors over spatial and temporal scales. The NS5 was selected as it represents a ubiquitous and abundant group of the Flavobacteriia class for which our knowledge on phylogeny and function is limited. Since the name was introduced 14 years ago, from a study describing high local and temporal diversity of Flavobacteriia in the North Sea [15], NS5-classified sequences have been recovered from across the world’s marine water masses, ranging from semi-enclosed seas in tropical regions [16, 17] to the Antarctic peninsula [18] and North Pacific oxygen minimum zone [19]. They are frequently reported as one of the most abundant groups of Flavobacteriia from studies using 16S rRNA gene analysis [17, 20] and fluorescence in situ hybridisation (FISH) cell counts [21] (referred to as VIS1 in that study). The VIS1 clade, which represents only a fraction of the NS5, was reported to reach 29 ± 3 × 103 cells ml−1 in the Arctic province of the North Atlantic. Members of the NS5 have been shown to associate with spring phytoplankton blooms [13, 22, 23] and increasing chlorophyll α concentrations [21, 24], however they are typically more prominent in early bloom stages or are more tightly coupled to the fluctuations in flagellate abundance [13]. A study conducted in the South Sea of Korea concluded that NS5 was a good indicator species for coastal waters [25] whilst they were also shown to be linked to eutrophication in coastal bays of Vietnam [22]. In contrast, other findings have indicated NS5 sequence abundance maxima to occur in winter [26] and a dominance of NS5 affiliated sequences in open ocean Arctic waters [20, 21], which motivated us to here provide novel data from the Fram Strait region. From these studies, it is clear that the NS5 may represent a diverse group of bacteria with different ecological niches.
With a global perspective, we here characterise the NS5 based on (1) phylogenetic analysis using 16S rRNA gene and ribosomal protein tree reconstructions, (2) functional descriptions using MAGs and a complete genome of a cultured isolate, (3) spatiotemporal distribution patterns of species-level cluster representatives and (4) visual identification of cells in culture using transmission electron microscopy (TEM) and in the environment using catalysed reporter deposition-fluorescence in situ hybridisation (CARD-FISH). As a result, we identify and characterise 35 species assigned to four novel candidate genera, which we have named Candidatus Marisimplicoccus (NS5_A), Candidatus Marivariicella (NS5_B), Candidatus Maricapacicella (NS5_D) and Candidatus Arcticimaribacter (NS5_F), for each of which, we propose a candidate type species based on a genome voucher.
Materials and methods
Fram Strait sampling and sequencing
Seawater samples were collected at the deep chlorophyll maximum (DCM) layer from 11 stations across the Fram Strait region in July and August 2018 during the PS114 Polarstern cruise, as described previously [27]. Seawater was fractionated using filtration and DNA extracted using a modified SDS-based extraction method after Zhou et al. [28]. Metagenomes were generated from the 0.2–3 µm fraction using the HiSeq 3000 (Illumina) and Sequel II (PacBio) platforms, as described previously [27] (Supplementary Material 1).
Generation of MAG dataset
The metagenomic reads from the Fram Strait samples were assembled (using Megahit [29] for Illumina reads and MetaFlye [30] for PacBio reads), binned (using Concoct [31], Metabat2 [32], Maxbin2 [33] and DAStool [34]) and manually refined as described previously [27], resulting in MAGs identified in this study by the prefix “FRAM18_“. Estimation of genome completion and contamination was determined using CheckM v1.1.2 [35]. MAGs belonging to the NS5 were identified by 16S rRNA gene phylogeny (Supplementary Material 1) and additionally assigned to taxonomic groups in the Genome Taxonomy Database (GTDB) (Release 89) using the classify_wf pipeline of GTDB-tk v1.0.2 [36, 37]. The dataset was expanded by retrieving all species-representative assemblies within the assigned GTDB taxa along with MAGs from two additional 0.2–3 µm marine microbial metagenomic datasets (Bioproject accessions: PRJEB28156 [38, 39], PRJEB43746) that had been assigned the same GTDB taxonomy (Supplementary Tables S1 and S2). The resulting dataset was de-replicated using FastANI v1.9 [40] with a cut-off threshold of 95%.
Helgoland NS5 isolate AHE01FL: sampling, isolation and genome sequencing
Seawater from the long-term ecological research station Helgoland Roads (54°11′03″N, 7°54′00″E) was sampled on the 28th April, 2016 and serially diluted with artificial seawater [41]. An inoculum of 2.6 nl, statistically containing three cells, was grown in an oligotrophic HaHa medium with the addition of vitamins [42] in the dark at 12 °C. After several transfers and another dilution to extinction series, purity controls confirmed that the culture contained a pure strain. It was maintained by transfers every 3 months. Growth in HaHa100V medium [42] yielded a turbid, orange-coloured culture and provided biomass for DNA extraction that was performed according to Zhou et al. [28]. Genome sequencing was performed by the Max Planck-Genome-centre Cologne, Germany (https://mpgc.mpipz.mpg.de/home/) using Sequel I (PacBio) and HiSeq 2500 (Illumina) platforms. Circular long read sequences from PacBio were assembled using Canu v2.1 [43] whilst short Illumina reads were assembled using Spades v3.13.2 (parameters: -isolate) [44]. The contigs from both datasets were aligned and assembled together in Geneious Prime v2019.1.3 (https://www.geneious.com) before a final round of error-correction using the Illumina reads as a reference. The assembled genome was submitted to EMBL-EBI and assigned the name “Flavobacteriaceae bacterium AHE01FL” with the taxid 2820661 and in this study, is named “Iso_AHE01FL”.
Helgoland NS5 isolate AHE01FL: cell visualisation
To accurately determine cell morphology of Iso_AHE01FL, TEM was used. An aliquot of the liquid culture was retrieved and fixed with 25% glutaraldehyde (EM grade Science Services) for 1 h at room temperature followed by centrifugation (5 min at 21,100 × g) and resuspension in the growth media (HaHa 100 V medium). An aliquot of this resuspension was pipetted onto a Formvar coated 400-mesh copper grid and stained with 1% uranylacetate for 5 min before being air dried overnight.
NS5 MAG phylogenetic tree reconstruction
The reconstruction of a phylogenetic tree for species-representative MAGs was performed using a concatenated alignment of 16 ribosomal proteins (L2, L3, L4, L5, L6, L14, L16, L18, L22, L24, S3, S8, S10, S17, S19), following the procedure described by Hug et al. [45]. In brief, Muscle v3.8.15 [46] was used to align amino acid sequences that were subsequently trimmed using TrimAI v1.4.1 [47] and concatenated into a single alignment that was provided as an input to FastTree v2.1.10 [48] (Supplementary Material S1). This workflow was then repeated with the addition of 1275 Flavobacteriaceae assemblies from the RefSeq database (Supplementary Material S1). To corroborate the inferred phylogenetic separation of MAGs, average nucleotide identity (ANI) and average amino acid identity (AAI) were calculated using FastANI and CompareM v0.1.1 (https://github.com/dparks1134/CompareM), respectively.
16S rRNA phylogenetic tree construction
16S rRNA gene sequences, longer than 1 kbp in length, were extracted from species-representative MAGs using Barrnap [49]. The sequences were imported to the ARB programme [50], aligned using the SINA aligner [51] and phylogenetically placed into the SILVA 138.1 SSU Ref NR99 reference tree using the parsimony algorithm. The MAG-derived sequences along with 100 of the highest quality NS5 sequences in the SILVA database were used for phylogenetic tree reconstruction. Three tree algorithms were used, RaxML v8.2.8 maximum likelihood (GTR-Gamma rate distribution model, rapid bootstrap algorithm, 100 repetitions) [52], neighbour-joining (Jukes-Cantor’s substitution model, 1000 bootstrap repetitions) and Parsimony v3.6, each with two different positional variability conservation filters, a 30% for all Flavobacteriia and the “termini” filter provided with the ARB SILVA database. A consensus tree was constructed from these six input trees and groups that remained stable throughout all tree methods were designated.
Probe design and environmental cell visualisation
CARD-FISH [53, 54] probes could be designed in ARB for two genus-level clades, NS5_A and NS5_F (Supplementary Table S2). Optimal hybridisation conditions were determined by testing on filtered pelagic water samples from the Fram Strait region [27], the same samples used to generate the “FRAM18_” MAGs. The probes were subsequently applied to five samples from that dataset to obtain information on morphology and cell count data. More detailed information is provided in Supplementary Material S1.
Global distribution of NS5 subgroups, MAGs and their correlation to physical parameters
The distribution of NS5 members was determined by recruiting metagenomic reads from the Tara Oceans dataset (ENA study accessions: PRJEB1787, PRJEB9740) [55] to species-representative MAGs using BBMap v38.73 [56], with a 99% identity threshold (minid = 99, idfilter = 99). In total, 122 surface water, 95 DCM and 47 mesopelagic metagenomes were used. To provide comparability between samples, the number of mapped reads was converted to reads per kilobase per million (RPKM) [57]. The generated data were imported into RStudio [58] and visualised using the packages rnaturalearth [59], sf [60] and ggplot2 [61]. To check the accuracy and provide support for the RPKM values, comparisons were made to genome coverage of mapped reads, cell counts (see “Probe design and environmental cell visualisation”) and another, more robust metric, the truncated average depth (TAD) [62], detailed information is provided in Supplementary Material S1.
To determine the effect of abiotic characteristics on NS5 species distribution, physical parameter measurements (depth, chlorophyll a, nitrite, nitrate + nitrite, oxygen, phosphate, salinity and silicate) of Tara Oceans samples were obtained from ENA-EBI. Scatter plots of RPKM values across physical parameters were produced using ggplot2 and Pearson’s correlation analysis performed using log transformed parameter values.
Seasonal dynamics of species-representative MAGs
Temporal dynamics of species-representative MAGs was determined by read recruitment of oligotypes from a multiyear time-series dataset [63] sampled at Helgoland Roads, North Sea (Supplementary Material S1). Recruitment was performed by BBMap with a 100% identity threshold (minid = 100, idfilter = 100). The distribution dynamics, based on relative abundance of oligotypes taken from the original manuscript, were visualised using the vegan [64] and ggplot2 packages in RStudio.
Functional characterisation
The presence of major metabolic pathways was determined using KofamKoala [65] and RAST v2.0 [66]. For each MAG, initial gene prediction was performed by Prokka v1.14.6 [67]. Carbohydrate-active enzymes (CAZymes) were predicted using a combination of HMMscan against the dbCAN v9 database [68] (E-value threshold: 1E−5) and Diamond blastp v0.9.14 [69] against the CAZy database (release 07312020) [70] (E-value threshold: 1E−20, parameters: -more-sensitive –query-cover 40 –id 30 –k 15). Sulfatases were annotated by blastp search against the SulfAtlas v1.3 database [71] (E-value threshold: 1E−4) and HMMscan against the Pfam sulfatase family PF00884 (E-value threshold: 1E−5). Peptidases were identified by blastp search against the MEROPS database [72] (E-value threshold: 1E−4). TonB-dependent transporters (TBDTs) were predicted by HMMscan against TIGRFAM profiles TIGR01352, TIGR01776, TIGR01778, TIGR01779, TIGR01782, TIGR01783, TIGR01785, TIGR01786, TIGR02796, TIGR02797, TIGR02803, TIGR02804, TIGR02805, TIGR04056 and TIGR04057 (E-value threshold: 1E−10). SusD genes were identified by HMMscan against the Pfam profiles PF12741, PF12771, PF14322, PF07980. Annotations of carbohydrate esterases, carbohydrate binding modules, glycoside hydrolases (GH) and polysaccharide lyases (PL) were designated correct only if both the dbCAN and CAZy annotations agreed. Annotations were combined into a single “gene_table” for each MAG. To identify potential polysaccharide utilisation loci (PULs), text searches were performed in the “gene_table” for regions on contigs that contained either a SusC/SusD gene pair with two or more degradative CAZymes or contained at least three substrate utilisation genes in close proximity (maximum of 6 genes in between each). PULs were manually inspected and visualised using the gggenes [73] and ggplot2 packages in RStudio.
The composition of CAZyme, sulfatase, peptidase and TBDT gene families for all MAGs was subsequently converted to a Bray-Curtis dissimilarity matrix and used as an input for hierarchical clustering and a non-metric multi-dimensional scaling analysis, using the hclust and metaMDS functions of the vegan package in RStudio. The visualisation of the analyses was carried out using the ggplot2 and ggdendro [74] packages.
SusC/SusD protein trees
Amino acid sequences of SusC/SusD genes identified in PULs were extracted and used for tree calculation. Additional SusC/SusD sequences were included from previously published marine flavobacteria MAGs [38] and cultured isolates [75]. Multiple sequence alignments were calculated using MAFFT v7.310 [76] with L-INS-I and trees calculated using FastTree. Trees were visualised and annotated in iTOL v4 [77].
Results
Seven species-representative MAGs retrieved from Fram Strait metagenomes were identified as members of the NS5 marine group through 16S rRNA gene analysis and assigned to four different genera within the GTDB database (MED-G11, GCA-002723295, MS024-2A, UBA7428). The GTDB species-representatives within these groups along with MAGs from two other metagenome datasets were acquired (Supplementary Tables S1 and S2). In addition, we sequenced and assembled a complete genome of an isolate retrieved from surface seawater at Helgoland Roads, North Sea in 2016. The derived dataset of assembled genomes was de-replicated at a 95% ANI threshold, resulting in 35 species-level clusters that provided the foundation for a detailed phylogenetic and ecological characterisation of the NS5 marine group (Supplementary Table S1).
Phylogenetic analysis of the NS5 marine group
Phylogenetic tree reconstruction using 16S rRNA genes from NS5 species-representatives and sequences from the SILVA 138 database resulted in six distinct clusters being formed (NS5_A–NS5_F) (Fig. 1a). However, the MAG sequences were positioned only within five of the ribosomal protein-based clusters (not NS5_E), indicating that part of NS5’s diversity is not yet captured by MAGs. Minimum intra-group 16S rRNA gene sequence similarity varied from 93% in NS5_D to 97.0% in NS5_F whilst the median values ranged from 94.5% in NS5_B to 98.9% in NS5_F. The lower values observed were typically a result of only a few sequences, with the majority of minimum values being >94.5% and median values >96.4% and therefore, in agreement with genus-level thresholds [78].
Fig. 1. Phylogenetic tree reconstruction of the NS5 Marine Group.

a 16S rRNA gene tree constructed using MAG sequences, from this and previous studies and the GTDB database, and 100 sequences classified as NS5 Marine Group in the SILVA 138 database. The tree represents a consensus from six input trees, constructed using three different algorithms, RaxML, Neighbour-joining and parsimony, with two different positional variability filters. b Ribosomal protein tree generated from a concatenated alignment of 16 proteins identified within NS5 species-representatives MAGs and genomes of Polaribacter and Tenacibaculum retrieved from the NCBI RefSeq database. The cultured isolate, Iso_AHE01FL, is highlighted in bold and with ***.
Reconstruction of a MAG-based ribosomal protein tree (Fig. 1b) revealed five distinct groups that corresponded to clusters in the 16S rRNA gene tree. The number of species-representative MAGs in each cluster ranged from 17 in NS5_D to 1 in NS5_C. Genomic comparisons between MAGs revealed intra-cluster average AAI values of >65% and inter-cluster values of <65%, further supporting the delineation of groups at the genus-level [79]. The coherence and stability of the clusters was additionally confirmed by phylogenetic tree reconstruction at the family level (Supplementary Fig. S1). Due to the genetic coherence, the defined clusters will now be referred to as genera. The cultured isolate, Iso_AHE01FL, belonged to the NS5_F genus. In order to provide an indication on genetic conservation, the species-representative genomes from NS5_F were aligned to the complete isolate genome and a visualisation provided on the conserved syntenic gene blocks identified (Supplementary Fig. S2).
Clear distinctions between genera were evident with respect to genome size and GC content (Supplementary Fig. S3). The average genome size of the three most complete MAGs from each genus were 2.05 Mbp for NS5_F, 2.02 Mbp for NS5_D, 1.82 Mbp for NS5_B and 1.17 Mbp for NS5_A, whilst the GC content of NS5_A and _B representatives was ~30% compared to 36 – 37% in NS5_D and _F.
Cell visualisation
Following the design and optimisation of CARD-FISH probes (Supplementary Material S1) for the NS5_A and NS5_F genera (Supplementary Table S3), cells were visualised on filtered seawater samples from the Fram Strait region [27] (Fig. 2). Probe design for the other genera was unsuccessful due to sequence similarities with neighbouring taxa. Hybridised cells visualised using the NS5_A probe were of a small coccoid shape with a diameter of ~0.5 µm. Those identified with the NS5_F probe were rod-shaped cells with a length of 0.5–1.5 µm and width of ~0.5 µm. Enumeration of FISH signals that overlapped with a nucleic acid stain (DAPI) revealed similar peak counts for both NS5_A, 1.70 × 104 cells ml−1, and NS5_F, 1.76 × 104 cells ml−1 (Supplementary Fig. S4).
Fig. 2. Visualisation of cells from NS5_A and NS5_F.
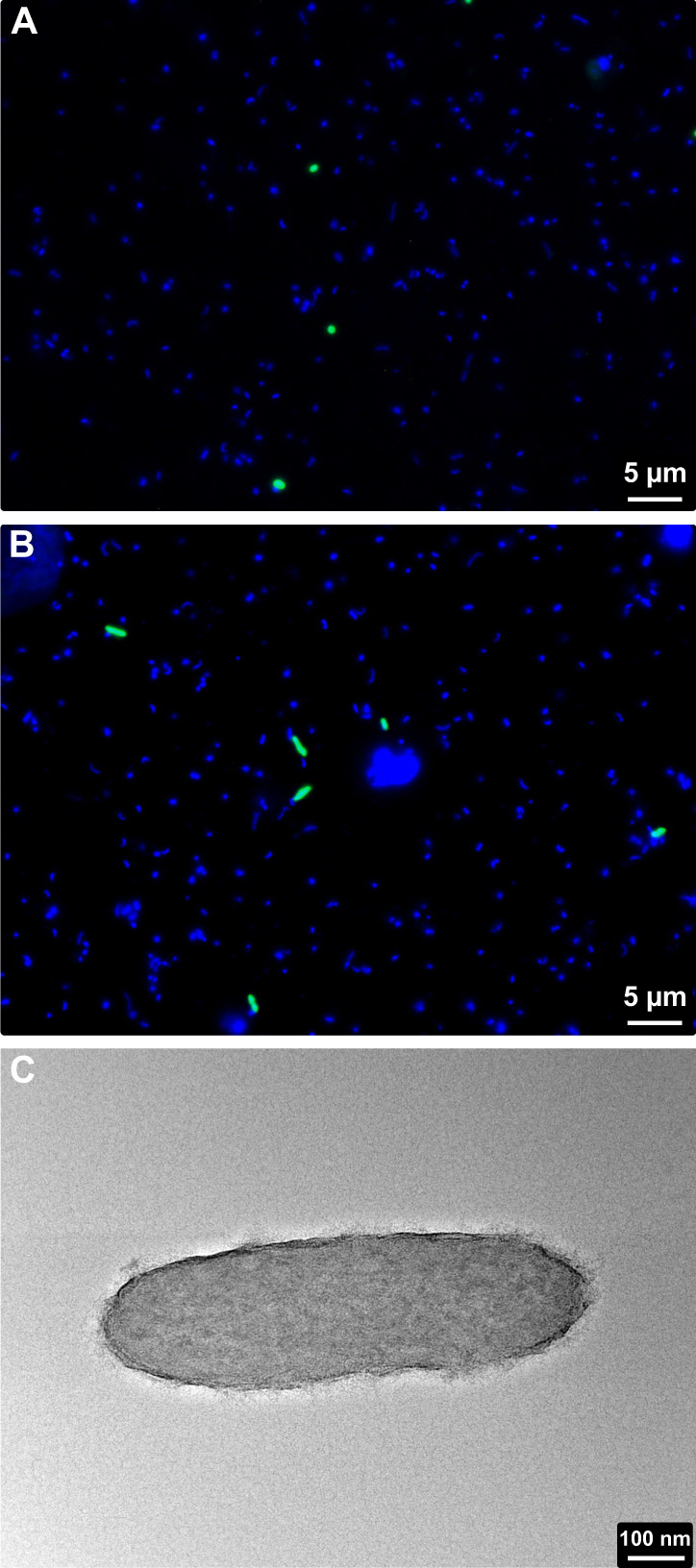
Environmental cells hybridised using CARD-FISH probes targeting the NS5_A (A) and NS5_F (B). FISH probe signals are shown in green and DNA stain in blue. C Transmission electron microscopy image of the isolate, Iso_AHE01FL, in the NS5_F.
TEM on the Iso_AHE01FL revealed rod-shaped cells with a length of 0.5–1 µm and width of <0.5 µm (Fig. 2), in agreement with the observations on environmental samples, based on FISH.
Global distribution of NS5 genera, MAGs and their correlation to physical parameters
The distribution of NS5 genera was determined by read recruitment from Tara Oceans metagenomes to each individual species and subsequently summing the RPKM values (Supplementary Fig. S5). To provide additional support, RPKM values were compared to genome coverage of mapped reads, CARD-FISH cell counts and another, more robust sequence-based metric, the TAD [62] (Supplementary Material S1 and Supplementary Figs. S4, S6 and S7). Based on this, a cut-off threshold of 0.25 RPKM was applied for inclusion in further analysis, which ensured a coverage of >40%. The four genera each exhibited a ubiquitous presence across all oceanic regions in the surface and DCM layers, although the NS5_F genus was less widespread in the DCM than surface. All genera showed lower RPKM values in the mesopelagic than DCM layer. The magnitude of RPKM values observed for NS5_B was six-fold lower than for the other genera. Variations in distribution patterns were evident, with the NS5_D and NS5_F reaching higher RPKM values in Arctic and geographically connected areas whilst NS5_A appeared more prevalent in specific locations, such as the North Atlantic and Chilean upwelling system. These patterns were further confirmed by grouping samples into oceanic regions (Supplementary Fig. S8).
On a species-level, an almost universal distribution pattern with depth was identified, with all but two species exhibiting highest RPKM values in surface waters (<30 m) (Supplementary Fig. S9). The two contrasting species were MS024-2A_sp7 (NS5_D), which peaked between 100–200 m, and MED-G11_sp2 (NS5_A), which peaked at ~300 m depth. Additionally, several species exhibited a bimodal peak, with highest values in surface waters but an additional, smaller peak in RPKM observed in mesopelagic depths, such as FRAM18_bin185 (NS5_B). Asides from depth, the geographical distribution patterns of species within and between genera varied (Supplementary Figs. S10–S14) which typically reflected distinct dynamics in relation to temperature (Supplementary Fig. S15), salinity (Supplementary Fig. S16) and oxygen (Supplementary Fig. S17). However, no clear patterns were evident with respect to nitrate + nitrite, phosphate, silicate or chlorophyll a. The geographical distribution patterns of species could be categorised into three types.
The first, encompasses species-representatives with higher RPKM values in a specific geographical region, e.g. the Mediterranean and Red Sea for MS024-2A_sp5 (Fig. 3). As a result, representatives of this type exhibited narrow peaks in RPKM values in relation to abiotic conditions, e.g. for MS024-2A_sp5 at ~15 °C and ~38 psu. The second pattern is represented by changes in RPKM values with latitude, e.g. higher values in the Arctic for species UBA7428_sp2 (Fig. 3) and all species in NS5_F or in temperate regions for GCA-002723295_sp2 in NS5_B (Supplementary Fig. S11) and MS024-2A_sp7 in NS5_D (Supplementary Fig. S13). These species typically exhibited peak RPKM values within a defined range of each abiotic condition. For example, the Arctic-preference distribution of NS5_F species was related to peaks in RPKM values at temperatures <5 °C, oxygen concentrations >300 µM and salinities <33 psu whereas the temperate-preference distribution of UltraPac_E_bin_84_1 was related to peaks across a wide range of temperatures, 12–30 °C, and at salinity values of 33–38 psu. Lastly, the remaining species exhibited an unclear distribution pattern, either due to below-threshold RPKM values in most samples or comparable RPKM values in samples without a clear pattern, e.g. GCA-002723295_sp1 in NS5_B (Fig. 3). Representatives of this last distribution type, as could be expected, showed a lack of or an inconsistent pattern with shifts in abiotic conditions. Species RPKM values across all Tara Oceans samples are provided in Supplementary Table S4.
Fig. 3. Three select species that represent different distribution types observed across the NS5 and their dynamics in relation to abiotic conditions.

RPKM values were calculated based on read recruitment from Tara Oceans metagenomes to species-representative MAGs using BBMap with a 99% identity threshold. A minimum threshold of 0.25 RPKM was applied which ensured a minimum genome coverage of 40%. a Global distribution and b dynamics in species RPKM values across abiotic conditions. Within the scatter plots, each point represents a Tara Oceans sample where the species RPKM value was >0.25. Alongside each scatterplot is a density diagram showing the distribution of points.
Seasonal dynamics of NS5 species
By performing read recruitment analysis of 16S rRNA gene oligotypes from a previously published time-series dataset, we were able to visualise the temporal dynamics of six NS5 species-representatives at Helgoland Roads, German Bight. Each of the identified oligotypes exhibited distinct and recurrent temporal dynamics over three consecutive years (Fig. 4), with three also showing a successional pattern from spring to summer. This succession began with 20100330_Bin_64_1 (NS5_D) that peaked from early to late spring (up to 4.5% of the community), followed by FRAM18_bin181 (NS5_F) in late spring and FRAM18_bin161 (NS5_D) that also peaked in late spring but persisted throughout summer (up to 3.5% of the community). Although the isolate, Iso_AHE01FL, was recovered from Helgoland Roads, the respective oligotype was present in low relative read abundance throughout the annual cycle (<0.1% of the community).
Fig. 4. Temporal dynamics of NS5 species-representatives in surface waters at Helgoland Roads, North Sea.

Distributions were obtained by recruiting oligotype representatives from a previously published dataset (62) to each species-representative MAG using BBMap with a 100% identity threshold. Next to each species-representative name, the original oligotype number is provided for direct comparison with previous dataset. Only recruitments successful with a 100% identity threshold are included. The relative abundances for each oligotype were taken from the original publication.
Functional characterisation
In order to assess functional differences across the NS5 genera, the gene annotations that were consistent across the three most complete MAGs in each genus were compared (referred to as genus-level values from hereon). The necessary genes for glycolysis, gluconeogenesis, the pentose phosphate pathway, the tricarboxylic acid cycle and for the major components of the electron transport chain were identified in all genera, confirming an aerobic heterotrophic metabolism. An additional unifying feature was the presence of a green-light proteorhodopsin (PR), which is not found in low light conditions. Mechanisms for nitrogen and phosphorous metabolism were conserved across all groups and restricted to an ammonium transporter (Amt family) and nitrogen response regulatory proteins (e.g. NtrC) along with the ability to build and hydrolyse long chain polyphosphates with a polyphosphate kinase (ppk) and an exopolyphosphatase. In addition, all species contained a glycogen synthase gene, indicating the capacity to use glycogen as a storage molecule. In contrast, genes related to sulfur metabolism were not conserved across genera, with only the NS5_B harbouring the capacity for assimilatory sulfate reduction. The ability to synthesise riboflavin was conserved whilst all genera lacked the genes required for biotin, thiamine and vitamin B12 synthesis. In contrast, significant differences were evident with respect to substrate acquisition and degradation potential between NS5 genera.
The annotation of CAZymes varied considerably across genera and species (Supplementary Tables S5 and S6). The number of glycoside hydrolase (GH) genes across all species-representatives ranged from 0–12 per Mbp (Fig. 5 and Supplementary Table S5), whilst genus-level values ranged from 9 per Mbp in NS5_D and _B to 5 per Mbp in NS5_A. There were also clear distinctions in the composition of conserved and non-conserved GH gene families within each genus (Supplementary Fig. S18). The NS5_D harboured eight conserved gene family annotations compared to four in NS5_F, three in NS5_B and one in NS5_A. There were no universally conserved gene families. However, two conserved gene families were shared between the NS5_B, _D and _F, including a GH16_3 (β-1,3-glucanase) and a GH3. Conserved gene families specific to a single genus included GH29 and GH95 (both known as α-fucosidases) in NS5_D and GH113 (β-mannanase) in NS5_F. The large range in non-conserved GH gene family annotations across genera indicated a varying degree of substrate-metabolic diversity on the species-level (Supplementary Fig. S18). Most notable was the diversity within NS5_B, with 26 different GH gene families or sub-families. This also provided evidence that potential substrates, not conserved at the genus-level, are shared between species of different genera. For example, annotations for α-fucosidases were not restricted to NS5_D, but also found in some species of NS5_B (GH151) and NS5_F (GH107). The presence of GH16_3, GH18 and GH20 genes across species from all genera indicated a shared potential to degrade β-1,3-glucans, such as laminarin, and β-hexosamines, such as peptidoglycan. In addition, a number of annotations were unique to some species within a single genus, including GH43_1 (β-xylosidase/α-L-arabinofuranosidase) and GH142 (β-L-arabinofuranosidase) in NS5_B, GH13_31 (α-glucosidase), GH28 (α-L-arabinofuranosidase) and GH28 (poly-/rhamno-galacturonase) in NS5_D and GH144 (β-1,2-glucosidase) in NS5_F. Further comparisons on GH gene family annotations revealed that each species’ composition is unique (Supplementary Table S6).
Fig. 5. Summary of substrate utilisation genes annotated in species-representative MAGs.

a Number of glycoside hydrolase genes based on agreeing annotations from HMMscan against dbCAN database and Diamond blastp search against the CAZy database. b Number of peptidases annotated using blastp search against the MEROPS database. c Number of sulfatase genes based on HMMscan against the Pfam sulfatase profile and blastp search against the SulfAtlas database. d Number of TonB-dependent transporters based on HMMscan against TIGRFAM TonB profiles.
A major process in carbohydrate catabolism in heterotrophic microbes involves glycan transport into the cell, a process mediated by, among others, TBDTs. The number of annotated TBDTs at the genus-level ranged from 7–9 per Mbp (Table 1) and at a species-level, from 4–13 per Mbp (Fig. 5 and Supplementary Table S5). In addition to TBDTs, the composition of all transporters was compared between genera (Supplementary Fig. S19). There were 23 universal transporters, including for vitamins and metals (Vitamin B12, zinc and magnesium), peptides (Di-tripeptide and D-serine) and carbohydrates (sodium/glucose, L-idonate, high-affinity gluconate and sugar SemiSWEET). In addition, 11 transporters were shared between more than one genus whilst 23 were unique to a single genus. The NS5_B and NS5_D both shared transporters indicative of more versatile metabolisms, including fatty acid and C4-dicarboxylate TRAP transporters.
Table 1.
Genomic statistics and carbohydrate and peptide-degradation gene repertoire of NS5 genera.
| Genome completeness (%) | Genome contamination (%) | Genome size (Mbp) | GC content (%) | GHs/Mbp | CAZymes/Mbp | Peptidases/Mbp | Sulfatases/Mbp | TBDTs/Mbp | SusCs/Mbp | SusDs/Mbp | PULs | GH: peptidase | GH: sulfatase | GH: TBDT | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NS5_A | 81.4 | 0.3 | 1.17 | 30 | 5 | 7 | 8 | 8 | 7 | 2 | 3 | 0 | 1:1.6 | 1:1.6 | 1:1.4 |
| NS5_B | 97.3 | 0.5 | 1.82 | 30 | 9 | 12 | 8 | 13 | 8 | 2 | 2 | 2 | 1:0.9 | 1:1.4 | 1:0.9 |
| NS5_D | 99.1 | 1.4 | 2.02 | 37 | 9 | 12 | 7 | 9 | 9 | 4 | 4 | 3 | 1:0.8 | 1:1 | 1:1 |
| NS5_F | 97.0 | 0.1 | 2.05 | 36 | 7 | 10 | 7 | 4 | 9 | 2 | 3 | 2 | 1:1 | 1:0.6 | 1:1.3 |
Values are derived from the average of the three most complete metagenome-assembled genomes in each genus. Completeness and contamination were estimated using CheckM v1.1.2. Gene groups are shown as per Mbp values.
TBDT TonB-dependent transporters.
Distinct differences were also observed for sulfatase and peptidase gene annotations. The number of sulfatases ranged considerably, from 2 to 24 per Mbp across species (Fig. 5) and on the genus-level, from 4 per Mbp in NS5_F to 13 per Mbp in NS5_B (Table 1). In comparison, peptidases were more consistent at the genus-level, ranging from 7 to 8 per Mbp (Table 1) and exhibited a narrow range across species, 6–11 per Mbp (Fig. 5).
To provide an additional perspective on substrate preferences, the ratio of GH genes to other substrate utilisation genes was calculated (Table 1), a metric that has previously been employed for other flavobacterial groups [7]. The NS5_A consistently had the lowest ratios of GH genes and was the only genus to harbour less GH genes than peptidases, 1:1.6. The ratio of CAZymes:sulfatases also varied across genera, with NS5_F being the only genera to contain more GH genes than sulfatases (Table 1).
Comparing the gene repertoire of CAZymes and peptidases across all species-representatives through a dissimilarity distance matrix approach, resulted in a clustering of species based on phylogeny (Fig. 6). This suggests that the substrate utilisation potential is primarily determined through evolution, and not an adaptation to habitats based on lateral gene transfer. However, refining the dataset to only contain specific sets of genes, e.g. only CAZymes, resulted in less coherent phylogeny-based clustering, although the effects across genera varied depending upon the chosen gene set (Supplementary Fig. S20).
Fig. 6. Comparison of the substrate utilisation gene composition between species-representatives.

A dissimilarity matrix was generated from the CAZyme, peptidase, sulfatase and TonB-dependent transporter gene compositions and subsequently used for a hierarchical clustering analysis and b non-metric multi-dimensional scaling ordination. *indicates the isolate, Iso_AHE01FL.
Polysaccharide utilisation loci (PULs) and SusC/SusD protein trees
PULs are genetic clusters of functionally related genes that are involved in the binding and cleavage of polysaccharides and subsequent uptake of oligosaccharides into the cell [80]. Canonical PULs are those containing degradative CAZymes and a SusC/SusD gene pair [80], which provides the transport mechanism for large oligosaccharides across the outer membrane. However, atypical or non-canonical PULs, lacking the SusC/SusD gene pair but still consisting of numerous degradative CAZymes, have also been described [81]. PULs are typically specific towards certain polysaccharides and can thus provide valuable information on variations in substrate metabolism across species, even if the SusC/SusD gene pair is absent. The total number of PULs across NS5 species ranged from 0 to 6, with an almost complete absence in NS5_A (Table 1 and Supplementary Table S7).
PUL structures were highly diverse across species, but four examples of conserved gene colocalisations were identified at the genus-level, two in NS5_D and two in NS5_F (Fig. 7). In NS5_D, one conserved colocalisation consisted of several GH29 genes (α-fucosidases), a single GH33 (sialidase) and/or GH3 gene and at least two sulfatases. Additionally, species within NS5_D harboured a PUL containing a solute-binding protein, C4-dicarboxylate TRAP transporter, 2,3-diketo-L-gulonate TRAP transporter, uronate isomerase and mannonate dehydratase which was accompanied by a GH95 and fructokinase gene in many of the representatives. Such a structure was also identified in one MAG from NS5_B, UltraPac_3515_G_bin_18, and indicates an ability to uptake sugar acids and C4 carbon compounds. In the NS5_F, one conserved colocalisation consisted of several GH16_3 (β-1,3-glucosidase) genes with a GH3 gene, with all but one MAG also encoding for a GH109 (N-acetylhexosaminidase) and galactokinase gene in the same region. In Iso_AHE01FL, this was further supplemented by a sodium/glucose cotransporter, a GH65 and β-phosphoglucomutase gene, providing additional machinery for β-glucan degradation. The second conserved colocalisation in NS5_F consisted of a double SusC/SusD gene pair along with at least two polysaccharide lyase (PL) genes from the PL6, PL7 and PL17 families, suggesting alginate as a potential substrate target (Fig. 7).
Fig. 7. Polysaccharide utilisation loci containing gene colocalisations that are conserved within candidate genera.

The structures presented include two each from the candidate species of NS5_D, 20100330_Bin_64_1, and NS5_F, Iso_AHE01FL. Although the exact PUL structures vary across species, the colocalisation of CAZymes are conserved within all species of the genus. The predicted substrate targets for NS5_D PULs are, PUL_1 = α-fucan and PUL_2 = unclear, and for NS5_F, PUL_3 = laminarin and PUL_4 = alginate.
The reconstruction of protein trees using SusC and SusD genes additionally confirmed the predicted substrate targets of PULs (Supplementary Fig. S21). For example, the SusC and SusD genes derived from potential alginate-targeting PULs identified in NS5_F species, clustered with those from alginate-targeting PULs of previously recovered MAGs and cultured isolates of Flavobacteriia.
Novel candidate genera
Based on the phylogenetic and ecological partitioning of NS5 species, sufficient information has been collected to formally describe four candidate species and genera within the Flavobacteriaceae family, Candidatus Marisimplicoccus (NS5_A), Candidatus Marivariicella (NS5_B), Candidatus Maricapacicella (NS5_D) and Candidatus Arcticimaribacter (NS5_F). The etymology and metabolic descriptions of these are provided in Supplementary Material S1 and Supplementary Figs. S22–S25.
Discussion
The NS5 marine group represent one of the most prevalent groups of marine flavobacteria across the world’s oceans yet our knowledge on their phylogenetic diversity and ecological functions is limited. Here, we phylogenetically and ecologically characterise four novel candidate genera within the NS5, each with a candidate representative species. The genera encapsulate 35 distinct species that are distinguishable by genomic characteristics, spatiotemporal distribution patterns and predicted functional potentials, from which we can hypothesise ecological niche partitioning. Furthermore, we present the first complete genome sequence and morphological description of an NS5 isolate, “Arcticimaribacter forsetii AHE01FL”.
Phylogeny and genomic characteristics
Phylogenetic tree reconstructions revealed four distinct, coherent genera in the NS5, each with multiple species-representatives, which formed a novel branch within the Flavobacteriaceae family. It is clear from tree topologies that two additional genera likely also exist, but a lack of representative genomes hinders further investigation.
NS5 species-representatives across genera are distinguishable by their genomic characteristics. The larger, average genome size of NS5_F and NS5_D (2.05 ± 0.19 and 2.02 ± 0.38 Mbp, respectively) are above the median size reported for a dataset of >1200 marine Bacteroidetes MAGs with comparable completeness values (1.96 Mbp) [38] whilst NS5_A is within the smallest 10% of genome sizes from that dataset. In general, the genome size of NS5 representatives are smaller than those of related cultured marine Flavobacteriia, such as Polaribacter (3.1–4.0 Mbp), Tenacibaculum (3.2–5.5 Mbp) and Formosa sp. B (2.7 Mbp). Genome sizes of cultured isolates and MAGs has previously been shown to vary by up to 1 Mbp in Polaribacter [7], however, the genome sizes of NS5_F MAGs were comparable to “Arcticimaribacter forsetii AHE01FL” (2.03 Mbp) in the same genus. Furthermore, within NS5_D, one of the species-representatives, MS024-2A_sp8, is a high quality draft single cell genome [16] which also has a comparable genome size to MAGs within the same genus. Therefore, the smaller genome sizes are likely not a methodological artefact but reflect differences in the life strategy and ecological role of NS5 compared to the other well described Flavobacteriia.
Life strategy and metabolism
The major metabolic pathways and cellular functions were largely conserved across all four newly described candidate genera and indicative of an aerobic photoheterotrophic lifestyle with supplemental energy acquisition through a proteorhodopsin (PR). PRs are light-driven proton pumps that can generate ATP through the proton motive force [82]. PR-mediated photoheterotrophy is widely distributed among marine Archaea and Bacteria inhabiting the photic zone [83], and it has been shown that PR-containing marine flavobacteria have smaller genomes than PR-lacking flavobacteria [84]. Such findings are in-line with the genome sizes reported here. Another key finding of this study is that NS5 species exhibit free-living lifestyles, evidenced by a lack of flagella machineries and gliding motility and a distinct separation of visualised cells from particles. This is in agreement with previous studies that reported an enrichment of NS5 marine group in the free-living fraction (<3 µm) in the North Sea [85] and Southern Ocean [18].
For free-living aerobic heterotrophs, the main source of carbon and nutrients for growth is dissolved organic matter (DOM). As is known for other groups of marine Flavobacteriia [84, 86], NS5 species are shown to encode a suite of degradative CAZymes, indicating a specialised capacity for high molecular-weight DOM degradation. The number of GH genes in NS5_B, _D and _F representatives, 7–9 per Mbp, is similar to values reported for MAGs classified in the Polaribacter 1-b (9 per Mbp) and 2-a clusters (9 per Mbp) as well as some cultured isolates such as Formosa B (7.7 per Mbp) [86] and Gramella forsetii KT0803T (10.5 per Mbp) [87]. These organisms are known as specialist degraders of algal-derived carbohydrates and are key members of microbial communities following spring phytoplankton blooms [86, 88]. In contrast, the number of peptidases present in NS5 species is considerably lower, 7–8 per Mbp, than in Gramella forsetii KT0803T, 30.5 per Mbp, and Formosa B, 25 per Mbp, indicating a reduced capacity for protein hydrolysis. Furthermore, the number of canonical PULs in NS5 species is lower than the average recently reported for a large dataset of marine Bacteroidetes MAGs [38]. Such features may be evidence of narrow substrate niches for NS5 species, which has also been suggested previously [16].
Unique substrate-degradation potentials
NS5 species harbour distinct substrate utilisation capacities, with evidence of genus-wide conserved substrate targets also evident in NS5_D and _F. In NS5_F, these consist of laminarin and alginate. Laminarin is a major storage polysaccharide in marine diatoms and a common substrate of marine flavobacteria, based on the widespread presence of PULs [38] and rapid hydrolysis rates in incubations [89]. The NS5_F laminarin-targeting PULs resemble those previously described for Gramella forsetii KT0803T [75], Gramella sp. MAR_2010_147 and Gillisia spp. Hel1_29 and Hel1_33_132 [75], shown to be upregulated in the presence of laminarin [88]. Alginate, also a widely available polysaccharide, constitutes a key component in brown algal cell walls, representing up to 45% of their dry weight biomass. Alginate-targeting PULs have been identified in a number of marine Flavobacteriia [75], however NS5 representatives dominated the alginate PUL cluster in the Helgoland Roads time-series dataset [38]. These PULs contain the Aly (PL6 and PL7) and Oal families (PL15 and PL17) that together, provide the capacity for complete alginate degradation [90, 91]. Additional substrate targets, shared by a minority of, or unique to a single species, also included algal-derived polysaccharides, such as α-fucan (GH107) for “Arcticimaribacter forsetii AHE01FL”.
A conserved PUL structure identified in species of NS5_D, containing α-fucosidases and sulfatases, suggests a potential to utilise fucose-containing sulphated polysaccharides (FCSP). However, previous FCSP-targeting PULs identified in fourteen isolates of marine Flavobacteriia, encoded a more extensive CAZyme gene repertoire, reflecting the complexity of FCSP [92, 93]. It is thus unlikely that NS5_D species utilise FCSPs but instead, cleave fucose groups bound to other carbohydrate structures or hydrolyse less complex fucose-containing oligosaccharides in a scavenging-like mechanism. Additionally conserved within NS5_D species, is a genetic loci containing numerous transporters for amino acids, D-xylose, acid sugars and C4 compounds without any degradative CAZymes. Such a structure may be evidence of genome rearrangement to increase the efficiency of gene regulation for substrate acquisition under certain conditions.
A lack of intra-genus conserved gene colocalisations were found in the NS5_B, however, all species contained a PUL targeting bacterial-derived polysaccharides, such as glycogen. In addition, unique PUL structures targeting algal-derived polysaccharides were identified in some species, such as an α-mannose-targeting PUL in GCA-002723295_sp2 (GH95). In NS5_A species, PULs were either absent or low in number and the comparable number of CAZyme to peptidase genes, which was unique to this genus, suggests that proteins are a more important substrate for growth.
Ecological niche partitioning of species
Niche concept has been applied in marine microbial ecology to describe the partitioning of taxa either based on adaptations to specific conditions across environments [4, 6, 94] or adaptations to specialised substrates within an environment [5, 14]. Using time-series data from a coastal ecosystem, it has been shown that populations of Bacteroidetes occupy distinct substrate specific niches that drive recurrent temporal dynamics [7, 13, 14]. For the NS5 representatives identified in that dataset, several specific substrate targets were reported, including β-glucan, α-glucan, α-mannan and alginate [38]. We show in this study that these substrates are indicative of different genera. Furthermore, by using an oligotype dataset from the same time-series, we identified successional-like dynamics for some NS5 species. Those dynamics were also likely driven by substrate utilisation capacity, with the early spring responder, 20100330_Bin_64_1 (NS5_D), encoding for twice as many GH genes and sulfatases as well as a broader diversity of predicted substrate targets than the late spring responder, FRAM18_bin161 (NS5_D). This suggests that substrate may be a major factor in the niche partitioning of these species in that environment. It is important to note however, that other factors, not assessed in this study, likely also contribute to these temporal dynamics, such as grazing by microeukaryotes and viral-induced mortality.
Although substrate may act as a key niche-determining factor for NS5 species in a given environment, we show that species’ spatial distribution dynamics across environments and throughout the water column are influenced by distinct shifts in abiotic conditions. Depth, and the associated changes in light and temperature, is well evidenced to structure the vertical distribution of microbial taxa [8]. Such a pattern is also clear for NS5, with nearly all species showing a preference for the upper euphotic zone (<100 m). Adaptations to this environment are evident within the genomes and predicted metabolisms of NS5 species, e.g. the presence of PR and utilisation of HMW-DOM as a substrate that is primarily produced by, or a result of lysis of phytoplankton in the euphotic zone. On geographical spatial scales, studies on microbial biogeography have reported that temperature and oxygen are the strongest correlates to changes in taxonomic and functional composition [95, 96]. The distribution dynamics of NS5 species are in agreement with this, although distinct patterns could also be identified in relation to salinity. It is clear that the niche-determining conditions vary considerably across species, with adaptations to narrow and broad ranges of conditions observed.
The widespread presence of many NS5 species but with distinct preferences for specific environmental conditions are in support of previous theories such as “everything is everywhere but the environment selects” [97] and the microbial seed bank hypothesis [98, 99]. The capacity to survive “everywhere” likely reflects an evolutionary adaptation that resulted in small genomes and cell sizes with advantageous features such as a PR and a potential to utilise widely available substrates. However, it is clear that each species has adapted to a specific set of conditions under which it can proliferate within its defined ecological niche. The factors that determine the partitioning of niches for NS5 species is a combination of abiotic conditions, such as temperature, and substrate utilisation. We propose that abiotic conditions influence spatial and temporal niche space across environments for each species, whereas substrate availability most strongly influences temporal niche dynamics within an environment.
We recognise that additional factors, particularly biotic interactions, can play an important role in determining a species’ niche, but we were unable to address these in the scope of this study. Thus, further work would be required to understand the influence these factors have.
We present evidence here that NS5 genera are distinguishable by phylogeny, cell shape and size, genomic characteristics, spatiotemporal distribution patterns and predicted substrate metabolism. Based on this, we formally describe four novel candidate genera and type species within the Flavobacteriaceae family—etymology and metabolic descriptions are provided in Supplementary Material S1 and Supplementary Figs. S22–S25.
Supplementary information
Acknowledgements
We thank Sabine Kühn for her technical support on isolation and cultivation. We thank Monike Oggerin for providing the metagenome-assembled genomes from the South Pacific Gyre dataset. We thank Bruno Huettel and the entire team at the Max-Planck-Genome-centre Cologne (http://mpgc.mpipz.mpg.de/home/) for their efforts with genome and metagenome sequencing. We thank Aharon Oren for his advice on etymology. We thank Susanne Erdmann for her assistance with the transmission electron microscopy. TP is a member of the International Max Planck Research School of Marine Microbiology (MarMic). This study was funded by the Max Planck Society.
Author contributions
TP, BMF and RA conceived and designed the study. TP performed all bioinformatic and molecular analyses. AH and JH performed the isolation and cultivation of the isolate. TP wrote the manuscript with contributions and input from all coauthors. All authors read and approved the final version of the manuscript.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Data availability
Metagenomes and MAGs used in this study were either previously deposited or deposited for this study in the European Nucleotide Archive, with all accession numbers provided in Supplementary Table S2. Those deposited for this study include the cultured isolate genome, PRJEB45371, and the MAGs derived from the South Pacific gyre metagenomes, PRJEB43746. The 16S rRNA gene amplicon time-series dataset used was previously published [63] and stored by JGI in the GOLD database under the project ID Gp0056779 as part of the community sequencing project COGITO. The ARB database containing the 16S rRNA gene NS5 phylogenetic tree is provided as Supplementary File S1. All data tables and the R script required to recreate the main body figures are available at https://github.com/tpriest0/NS5_marine_group_manuscript_figures.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41396-022-01209-8.
References
- 1.Hutchinson GE. Concluding remarks. Cold Spring Harb Symp Quant Biol. 1957;22:415–27. [Google Scholar]
- 2.Hutchinson GE. An introduction to population biology. New Haven, CT: Yale University Press; 1978.
- 3.Larkin AA, Martiny AC. Microdiversity shapes the traits, niche space, and biogeography of microbial taxa. Environ Microbiol Rep. 2017;9:55–70. doi: 10.1111/1758-2229.12523. [DOI] [PubMed] [Google Scholar]
- 4.Mena C, Reglero P, Balbín R, Martín M, Santiago R, Sintes E. Seasonal niche partitioning of surface temperate open ocean prokaryotic communities. Front Microbiol. 2020;11:1749. doi: 10.3389/fmicb.2020.01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sarmento H, Morana C, Gasol JM. Bacterioplankton niche partitioning in the use of phytoplankton-derived dissolved organic carbon: quantity is more important than quality. ISME J. 2016;10:2582–92. doi: 10.1038/ismej.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Auladell A, Barberán A, Logares R, Garcés E, Gasol JM, Ferrera I. Seasonal niche differentiation among closely related marine bacteria. ISME J. 2022;16:178–89. doi: 10.1038/s41396-021-01053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Avcı B, Krüger K, Fuchs BM, Teeling H, Amann RI. Polysaccharide niche partitioning of distinct Polaribacter clades during North Sea spring algal blooms. ISME J. 2020;14:1369–83. doi: 10.1038/s41396-020-0601-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghiglione J-F, Galand PE, Pommier T, Pedrós-Alió C, Maas EW, Bakker K, et al. Pole-to-pole biogeography of surface and deep marine bacterial communities. Proc Natl Acad Sci USA. 2012;109:17633–8. doi: 10.1073/pnas.1208160109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson ZI, Zinser ER, Coe A, McNulty NP, Woodward EMS, Chisholm SW. Niche partitioning among Prochlorococcus ecotypes along ocean-scale environmental gradients. Science. 2006;311:1737–40. doi: 10.1126/science.1118052. [DOI] [PubMed] [Google Scholar]
- 10.Wang Z, Juarez DL, Pan J-F, Blinebry SK, Gronniger J, Clark JS, et al. Microbial communities across nearshore to offshore coastal transects are primarily shaped by distance and temperature. Environ Microbiol. 2019;21:3862–72. doi: 10.1111/1462-2920.14734. [DOI] [PubMed] [Google Scholar]
- 11.Herlemann DP, Labrenz M, Jürgens K, Bertilsson S, Waniek JJ, Andersson AF. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011;5:1571–9. doi: 10.1038/ismej.2011.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Delmont TO, Kiefl E, Kilinc O, Esen OC, Uysal I, Rappé MS, et al. Single-amino acid variants reveal evolutionary processes that shape the biogeography of a global SAR11 subclade. eLife. 2019;8:e46497. doi: 10.7554/eLife.46497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teeling H, Fuchs BM, Bennke CM, Krüger K, Chafee M, Kappelmann L, et al. Recurring patterns in bacterioplankton dynamics during coastal spring algae blooms. eLife. 2016;5:e11888. doi: 10.7554/eLife.11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science. 2012;336:608–11. doi: 10.1126/science.1218344. [DOI] [PubMed] [Google Scholar]
- 15.Alonso C, Warnecke F, Amann R, Pernthaler J. High local and global diversity of flavobacteria in marine plankton. Environ Microbiol. 2007;9:1253–66. doi: 10.1111/j.1462-2920.2007.01244.x. [DOI] [PubMed] [Google Scholar]
- 16.Ngugi DK, Stingl U. High-quality draft single-cell genome sequence of the NS5 Marine Group from the Coastal Red Sea. Genome Announc. 2018;26:e00565-18. doi: 10.1128/genomeA.00565-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meziti A, Kormas KA, Moustaka-Gouni M, Karayanni H. Spatially uniform but temporally variable bacterioplankton in a semi-enclosed coastal area. Syst Appl Microbiol. 2015;38:358–67. doi: 10.1016/j.syapm.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 18.Milici M, Vital M, Tomasch J, Badewien TH, Giebel H-A, Plumeier I, et al. Diversity and community composition of particle-associated and free-living bacteria in mesopelagic and bathypelagic Southern Ocean water masses: evidence of dispersal limitation in the Bransfield Strait. Limnol Oceanogr. 2017;62:1080–95. [Google Scholar]
- 19.Beman JM, Vargas SM, Vazquez S, Wilson JM, Yu A, Cairo A, et al. Biogeochemistry and hydrography shape microbial community assembly and activity in the eastern tropical North Pacific Ocean oxygen minimum zone. Environ Microbiol. 2020;23:2765–81. doi: 10.1111/1462-2920.15215. [DOI] [PubMed] [Google Scholar]
- 20.Rapp JZ, Fernández-Méndez M, Bienhold C, Boetius A. Effects of ice-algal aggregate export on the connectivity of bacterial communities in the Central Arctic Ocean. Front Microbiol. 2018;9:01035. doi: 10.3389/fmicb.2018.01035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gómez-Pereira PR, Fuchs BM, Alonso C, Oliver MJ, van Beusekom JEE, Amann R. Distinct flavobacterial communities in contrasting water masses of the North Atlantic Ocean. ISME J. 2010;4:472–87. doi: 10.1038/ismej.2009.142. [DOI] [PubMed] [Google Scholar]
- 22.Choi DH, An SM, Yang EC, Lee H, Shim J, Jeong J, et al. Daily variation in the prokaryotic community during a spring bloom in shelf waters of the East China Sea. FEMS Microbiol Ecol. 2018;94:fiy134. doi: 10.1093/femsec/fiy134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang C, Li Y, Zhou B, Zhou Y, Zheng W, Tian Y, et al. Illumina sequencing-based analysis of free-living bacterial community dynamics during an Akashiwo sanguine bloom in Xiamen sea, China. Sci Rep. 2015;5:8476. doi: 10.1038/srep08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Díez‐Vives C, Nielsen S, Sánchez P, Palenzuela O, Ferrera I, Sebastián M, et al. Delineation of ecologically distinct units of marine Bacteroidetes in the Northwestern Mediterranean Sea. Mol Ecol. 2019;28:2846–59. doi: 10.1111/mec.15068. [DOI] [PubMed] [Google Scholar]
- 25.Seo J-H, Kang I, Yang S-J, Cho J-C. Characterization of spatial distribution of the bacterial community in the South Sea of Korea. PLoS ONE. 2017;12:e0174159. doi: 10.1371/journal.pone.0174159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alonso‐Sáez L, Díaz‐Pérez L, Morán XAG. The hidden seasonality of the rare biosphere in coastal marine bacterioplankton. Environ Microbiol. 2015;17:3766–80. doi: 10.1111/1462-2920.12801. [DOI] [PubMed] [Google Scholar]
- 27.Priest T, Orellana LH, Huettel B, Fuchs BM, Amann R. Microbial metagenome-assembled genomes of the Fram Strait from short and long read sequencing platforms. PeerJ. 2021;9:e11721. doi: 10.7717/peerj.11721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou J, Bruns MA, Tiedje JM. DNA recovery from soils of diverse composition. Appl Environ Microbiol. 1996;62:316–22. doi: 10.1128/aem.62.2.316-322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li D, Luo R, Liu C-M, Leung C-M, Ting H-F, Sadakane K, et al. MEGAHIT v1.0: a fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods. 2016;102:3–11. doi: 10.1016/j.ymeth.2016.02.020. [DOI] [PubMed] [Google Scholar]
- 30.Kolmogorov M, Bickhart DM, Behsaz B, Gurevich A, Rayko M, Shin SB, et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat Methods. 2020;17:1103–10. doi: 10.1038/s41592-020-00971-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, et al. Binning metagenomic contigs by coverage and composition. Nat Methods. 2014;11:1144–6. doi: 10.1038/nmeth.3103. [DOI] [PubMed] [Google Scholar]
- 32.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7:e7359. doi: 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu Y-W, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32:605–7. doi: 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 34.Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol. 2018;3:836–43. doi: 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–55. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 2020;36:1925–7. doi: 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parks DH, Chuvochina M, Chaumeil P-A, Rinke C, Mussig AJ, Hugenholtz P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat Biotechnol. 2020;38:1079–86. doi: 10.1038/s41587-020-0501-8. [DOI] [PubMed] [Google Scholar]
- 38.Krüger K, Chafee M, Ben Francis T, Glavina del Rio T, Becher D, Schweder T, et al. In marine Bacteroidetes the bulk of glycan degradation during algae blooms is mediated by few clades using a restricted set of genes. ISME J. 2019;13:2800–16. doi: 10.1038/s41396-019-0476-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Francis TB, Bartosik D, Sura T, Sichert A, Hehemann J-H, Markert S, et al. Changing expression patterns of TonB-dependent transporters suggest shifts in polysaccharide consumption over the course of a spring phytoplankton bloom. ISME J. 2021;15:2336–50. doi: 10.1038/s41396-021-00928-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Winkelmann N, Harder J. An improved isolation method for attached-living Planctomycetes of the genus Rhodopirellula. J Microbiol Methods. 2009;77:276–84. doi: 10.1016/j.mimet.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 42.Hahnke RL, Bennke CM, Fuchs BM, Mann AJ, Rhiel E, Teeling H, et al. Dilution cultivation of marine heterotrophic bacteria abundant after a spring phytoplankton bloom in the North Sea. Environ Microbiol. 2015;17:3515–26. doi: 10.1111/1462-2920.12479. [DOI] [PubMed] [Google Scholar]
- 43.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–36. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, et al. A new view of the tree of life. Nat Microbiol. 2016;1:1–6. doi: 10.1038/nmicrobiol.2016.48. [DOI] [PubMed] [Google Scholar]
- 46.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3. doi: 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Price MN, Dehal PS, Arkin AP. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seeman T. Barrnap 0.9 (version 3): rapid ribosomal RNA prediction. 2017. https://github.com/tseemann/barrnap.
- 50.Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–71. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pruesse E, Peplies J, Glöckner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–9. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amann RI, Krumholz L, Stahl DA. Fluorescent-oligonucleotide probing of whole cells for determinative, phylogenetic, and environmental studies in microbiology. J Bacteriol. 1990;172:762–70. doi: 10.1128/jb.172.2.762-770.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pernthaler A, Pernthaler J, Amann R. Fluorescence in situ hybridization and catalyzed reporter deposition for the identification of marine bacteria. Appl Environ Microbiol. 2002;68:3094–101. doi: 10.1128/AEM.68.6.3094-3101.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pesant S, Not F, Picheral M, Kandels-Lewis S, Le Bescot N, Gorsky G, et al. Open science resources for the discovery and analysis of Tara Oceans data. Sci Data. 2015;2:150023. doi: 10.1038/sdata.2015.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bushnell B. BBTools software package. 2017. https://sourceforge.net/projects/bbmap/.
- 57.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 58.RStudio Team. RStudio: integrated development of R. Boston, MA: RStudio Inc.; 2015.
- 59.South A. rnaturalearth: World map data from Natural Earth. R packag version 0.1.0; 2017.
- 60.Pebesma E. Simple features for R: standardized support for spatial vector data. R J. 2018;10:439–46. [Google Scholar]
- 61.Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2016.
- 62.Orellana LH, Francis TB, Ferraro M, Hehemann J-H, Fuchs BM, Amann RI. Verrucomicrobiota are specialist consumers of sulfated methyl pentoses during diatom blooms. ISME J. 2021. [DOI] [PMC free article] [PubMed]
- 63.Chafee M, Fernàndez-Guerra A, Buttigieg PL, Gerdts G, Eren AM, Teeling H, et al. Recurrent patterns of microdiversity in a temperate coastal marine environment. ISME J. 2018;12:237–52. doi: 10.1038/ismej.2017.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al. Vegan community ecology package version 2.5, 7 November. 2020.
- 65.Aramaki T, Blanc-Mathieu R, Endo H, Ohkubo K, Kanehisa M, Goto S, et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics. 2020;36:2251–2. doi: 10.1093/bioinformatics/btz859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST) Nucleic Acids Res. 2014;42:D206–14. doi: 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 68.Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, et al. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018;46:95–101. doi: 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 70.Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014;42:490–5. doi: 10.1093/nar/gkt1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barbeyron T, Brillet-Guéguen L, Carré W, Carrière C, Caron C, Czjzek M, et al. Matching the diversity of sulfated biomolecules: creation of a classification database for sulfatases reflecting their substrate specificity. PLoS ONE. 2016;11:e0164846. doi: 10.1371/journal.pone.0164846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, Finn RD. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018;46:624–32. doi: 10.1093/nar/gkx1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilkins D. gggenes: Draw Gene Arrow Maps in ‘ggplot2’. R package version 0.4.1; 2020.
- 74.de Vries A, Ripley BD. ggdendro: create dendrograms and tree diagrams using ‘ggplot2’. 2020.
- 75.Kappelmann L, Krüger K, Hehemann J-H, Harder J, Markert S, Unfried F, et al. Polysaccharide utilization loci of North Sea Flavobacteriia as basis for using SusC/D-protein expression for predicting major phytoplankton glycans. ISME J. 2019;13:76–91. doi: 10.1038/s41396-018-0242-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–66. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Letunic I, Bork P. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019;47:256–9. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer K-H, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12:635–45. doi: 10.1038/nrmicro3330. [DOI] [PubMed] [Google Scholar]
- 79.Konstantinidis KT, Rosselló-Móra R, Amann R. Uncultivated microbes in need of their own taxonomy. ISME J. 2017;11:2399–406. doi: 10.1038/ismej.2017.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bjursell MK, Martens EC, Gordon JI. Functional genomic and metabolic studies of the adaptations of a prominent adult human gut symbiont, Bacteroides thetaiotaomicron, to the suckling period. J Biol Chem. 2006;281:36269–79. doi: 10.1074/jbc.M606509200. [DOI] [PubMed] [Google Scholar]
- 81.Ficko-Blean E, Préchoux A, Thomas F, Rochat T, Larocque R, Zhu Y, et al. Carrageenan catabolism is encoded by a complex regulon in marine heterotrophic bacteria. Nat Commun. 2017;8:1685. doi: 10.1038/s41467-017-01832-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Johnson ET, Baron DB, Naranjo B, Bond DR, Schmidt-Dannert C, Gralnick JA. Enhancement of survival and electricity production in an engineered bacterium by light-driven proton pumping. Appl Environ Microbiol. 2010;76:4123–9. doi: 10.1128/AEM.02425-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dubinsky V, Haber M, Burgsdorf I, Saurav K, Lehahn Y, Malik A, et al. Metagenomic analysis reveals unusually high incidence of proteorhodopsin genes in the ultraoligotrophic Eastern Mediterranean Sea. Environ Microbiol. 2017;19:1077–90. doi: 10.1111/1462-2920.13624. [DOI] [PubMed] [Google Scholar]
- 84.Fernández-Gómez B, Richter M, Schüler M, Pinhassi J, Acinas SG, González JM, et al. Ecology of marine Bacteroidetes: a comparative genomics approach. ISME J. 2013;7:1026–37. doi: 10.1038/ismej.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heins A, Reintjes G, Amann RI, Harder J. Particle collection in Imhoff sedimentation cones enriches both motile chemotactic and particle-attached bacteria. Front Microbiol. 2021;12:643730. doi: 10.3389/fmicb.2021.643730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Unfried F, Becker S, Robb CS, Hehemann J-H, Markert S, Heiden SE, et al. Adaptive mechanisms that provide competitive advantages to marine Bacteroidetes during microalgal blooms. ISME J. 2018;12:2894–906. doi: 10.1038/s41396-018-0243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bauer M, Kube M, Teeling H, Richter M, Lombardot T, Allers E, et al. Whole genome analysis of the marine Bacteroidetes ‘Gramella forsetii’ reveals adaptations to degradation of polymeric organic matter. Environ Microbiol. 2006;8:2201–13. doi: 10.1111/j.1462-2920.2006.01152.x. [DOI] [PubMed] [Google Scholar]
- 88.Kabisch A, Otto A, König S, Becher D, Albrecht D, Schüler M, et al. Functional characterization of polysaccharide utilization loci in the marine Bacteroidetes ‘Gramella forsetii’ KT0803. ISME J. 2014;8:1492–502. doi: 10.1038/ismej.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reintjes G, Arnosti C, Fuchs B, Amann R. Selfish, sharing and scavenging bacteria in the Atlantic Ocean: a biogeographical study of bacterial substrate utilisation. ISME J. 2019;13:1119–32. doi: 10.1038/s41396-018-0326-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thomas F, Barbeyron T, Tonon T, Génicot S, Czjzek M, Michel G. Characterization of the first alginolytic operons in a marine bacterium: from their emergence in marine Flavobacteriia to their independent transfers to marine Proteobacteria and human gut Bacteroides. Environ Microbiol. 2012;14:2379–94. doi: 10.1111/j.1462-2920.2012.02751.x. [DOI] [PubMed] [Google Scholar]
- 91.Hehemann J-H, Arevalo P, Datta MS, Yu X, Corzett CH, Henschel A, et al. Adaptive radiation by waves of gene transfer leads to fine-scale resource partitioning in marine microbes. Nat Commun. 2016;7:12860. doi: 10.1038/ncomms12860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deniaud-Bouët E, Hardouin K, Potin P, Kloareg B, Hervé C. A review about brown algal cell walls and fucose-containing sulfated polysaccharides: cell wall context, biomedical properties and key research challenges. Carbohydr Polym. 2017;175:395–408. doi: 10.1016/j.carbpol.2017.07.082. [DOI] [PubMed] [Google Scholar]
- 93.Sichert A, Corzett CH, Schechter MS, Unfried F, Markert S, Becher D, et al. Verrucomicrobia use hundreds of enzymes to digest the algal polysaccharide fucoidan. Nat Microbiol. 2020;5:1026–39. doi: 10.1038/s41564-020-0720-2. [DOI] [PubMed] [Google Scholar]
- 94.Duerschlag J, Mohr W, Ferdelman TG, LaRoche Desai D, Croot PL, Desai D, Croot PL. Niche partitioning by photosynthetic plankton as a driver of CO2-fixation across the oligotrophic South Pacific Subtropical Ocean. ISME J. 2022;15:465–76. doi: 10.1038/s41396-021-01072-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, et al. Structure and function of the global ocean microbiome. Science. 2015;348:1261359. doi: 10.1126/science.1261359. [DOI] [PubMed] [Google Scholar]
- 96.Raes J, Letunic I, Yamada T, Jensen LJ, Bork P. Toward molecular trait-based ecology through integration of biogeochemical, geographical and metagenomic data. Mol Syst Biol. 2011;7:473. doi: 10.1038/msb.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baas Becking L. G. M.. Geobiologie of inleiding tot de milieukunde. WP Van Stock Zoon, Den Haag; 1934.
- 98.Gibbons SM, Caporaso JG, Pirrung M, Field D, Knight R, Gilbert JA. Evidence for a persistent microbial seed bank throughout the global ocean. Proc Natl Acad Sci USA. 2013;110:4651–5. doi: 10.1073/pnas.1217767110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lennon JT, Jones SE. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol. 2011;9:119–30. doi: 10.1038/nrmicro2504. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Metagenomes and MAGs used in this study were either previously deposited or deposited for this study in the European Nucleotide Archive, with all accession numbers provided in Supplementary Table S2. Those deposited for this study include the cultured isolate genome, PRJEB45371, and the MAGs derived from the South Pacific gyre metagenomes, PRJEB43746. The 16S rRNA gene amplicon time-series dataset used was previously published [63] and stored by JGI in the GOLD database under the project ID Gp0056779 as part of the community sequencing project COGITO. The ARB database containing the 16S rRNA gene NS5 phylogenetic tree is provided as Supplementary File S1. All data tables and the R script required to recreate the main body figures are available at https://github.com/tpriest0/NS5_marine_group_manuscript_figures.