

Abstract
The naturally occurring imino acid azetidine-2-carboxylic acid (Aze) is consumed by humans and can be misincorporated in place of proline in myelin basic protein (MBP) in vitro. To determine Aze effects on the mammalian CNS in vivo, adult CD1 mice were given Aze orally or intraperitoneally. Clinical signs reminiscent of MBP-mutant mice occurred with 600 mg/kg Aze exposure. Aze induced oligodendrocyte (OL) nucleomegaly and nucleoplasm clearing, dilated endoplasmic reticulum, cytoplasmic vacuolation, abnormal mitochondria, and Aze dose-dependent apoptosis. Immunohistochemistry demonstrated myelin blistering and nuclear translocation of unfolded protein response (UPR)/proinflammatory molecules (ATF3, ATF4, ATF6, eIF2α, GADD153, NFκB, PERK, XBP1), MHC I expression, and MBP cytoplasmic aggregation in OL. There were scattered microglial nodules in CNS white matter (WM); other CNS cells appeared unaffected. Mice given Aze in utero and postnatally showed more marked effects than their dams. These OL, myelin, and microglial alterations are found in normal-appearing WM (NAWM) in multiple sclerosis (MS) patients. Thus, Aze induces a distinct oligodendrogliopathy in mice that recapitulates MS NAWM pathology without leukocyte infiltration. Because myelin proteins are relatively stable throughout life, we hypothesize that Aze misincorporation in myelin proteins during myelinogenesis in humans results in a progressive UPR that may be a primary process in MS pathogenesis.
Keywords: Autoimmunity, Azetidine-2-carboxylic acid, Multiple sclerosis, Myelin basic protein, Oligodendrocyte, Proline, Unfolded protein response
INTRODUCTION
Edward Rubenstein hypothesized in 2008 that early life dietary exposure to the nonprotein imino acid azetidine-2-carboxylic acid (Aze), and its substitution for proline (Pro) in CNS myelin proteins, particularly the proline-rich myelin basic protein (MBP), contribute to the pathogenesis of multiple sclerosis (MS) (1). This novel unifying hypothesis suggests a biochemical mechanism for some of the distinct aspects of MS history, epidemiology, clinical heterogeneity, progression, and neuropathology (1, 2).
Aze is abundant in sugar beets and garden beets (3); sugar beets continue to be among the 10 most important agricultural products in more than 30 nonequatorial countries throughout the world, including the United States (4). Thus, the extent of sugar beet agriculture correlates with the known latitudinal gradient of MS prevalence (5). However, the amounts of Aze consumed by humans from dietary sources including beets, beet byproducts, meat, dairy products, and other derivative foods remain unknown (1). Moreover, the utilization of Aze as a carbon and nitrogen source by microorganisms raises the possibility that the gut or maternal microbiome could be an additional source of Aze exposure in humans (6, 7).
CNS myelin proteins are largely stable throughout life (8, 9). Therefore, Aze misincorporation in CNS myelin in humans would necessarily occur predominantly during myelinogenesis in fetal and early postnatal life. Aze can be preferentially misincorporated in place of Pro in MBP in vitro resulting in its misfolding (10, 11). The in vitro and in vivo effects of Aze and other nonprotein amino acids have been documented (12–16), but the neuropathological effects of Aze on the mammalian CNS have not been investigated.
We assessed clinical and pathological effects of Aze exposure in adult and neonatal mice. High doses of Aze alone induced a clinical phenotype in adult mice similar to that seen in MBP mutant (shiverer) mice. Pathological analyses demonstrated dose-dependent effects on oligodendrocytes (OL) including nucleomegaly and nuclear clearing (i.e. “watery swelling”), apoptosis, alterations indicating endoplasmic reticulum (ER) stress/unfolded protein response (UPR), features of mitophagy, and a proinflammatory phenotype and subtle myelin degeneration. Activated microglia and microglial nodules were observed in CNS white matter (WM). These pathological findings are present in MS patient normal-appearing (i.e. noninflamed), WM (NAWM), and adjacent to inflammatory and demyelinating MS lesions (17–22). Our results indicate that Aze induces a novel oligodendrogliopathy relevant to both inflammatory and degenerative aspects of MS pathophysiology and progression. They provide support for the concept that misincorporation of noncanonical amino acids may contribute to autoimmune mechanisms in human diseases.
MATERIALS AND METHODS
Adult Mice
Male CD1 mice (ages 6–8 weeks) (Charles River, Wilmington, MA) were exposed to 2 different doses of Aze (Sigma, St. Louis, MO), or normal saline. In experiment A1, 5 groups of 5 mice each were treated daily with either 300 or 600 mg/kg by intraperitoneal injection (IP), or oral gavage PO or saline IP. Clinical disease was observed in the high dose groups (see Results for details); 2 of these were found dead and 4 were killed due to declining clinical condition after 2–3 weeks of treatment. The remaining mice showed no clinical signs and were treated over 4 weeks. They were then killed by CO2 inhalation. At the time of death, half brain tissue samples were frozen in OCT. Remaining samples of brain and of heart, liver, spleen, kidney, and skin were snap frozen and stored at −80°. Portions of these tissues, spinal cord, and pylorus/duodenum/pancreas were also fixed in 10% neutral buffered formalin and subsequently processed routinely for paraffin sections, histology, and immunohistochemistry (IHC) (Table 1).
TABLE 1.
Summary of Azetidine Treatment Groups and Analyses Performed
| Experiment | Aze Dose (mg/kg) | Treatment |
Treatment Duration | Analyses Performed | Non-CNS Tissues | |
|---|---|---|---|---|---|---|
| IP | PO | |||||
| A1 (adult mice) | 0, 300, 600 | QD (5 mice/group) | QD (n = 5 mice/group) | Up to 4 weeks* | + | |
| A2 (adult mice) | 0, 300, 600 | 5×/week (3–4 mice/group) | — | Up to 4 weeks |
|
+ |
| N1 pups | 0, 350 | — | 3×/week (n = 37 total) | D23 (postweaning) to D44 | LM† | + |
| N1 dams | 0, 350 | — | 3×/week (n = 3/group) | 5 weeks (postbirth) | LM† | + |
| N2 pups | 0, 300 | — | 3×/week (n = 5/group) | E14 to D22 (weaning) to 6 weeks | LM†, TUNEL, Caspase 3 IHC | +§ |
| N2 dams | 0, 300 | — | 3×/week (n = 2/group) | E14 to 5 weeks postbirth | LM†, TUNEL, Caspase 3 IHC | +§ |
LM, light microscopy; EM, electron microscopy; IHC immunohistochemistry; TUNEL, terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling; +, tissues sampled.
Excludes mice that died or were euthanized in the 600 mg/kg group.
LM, Light microscopy: H&E, Luxol fast blue, Bielschowsky.
IHC, immunostained with antibodies listed in Table 2.
Samples include eyes and optic nerves.
Iba-1 IHC only.
In experiment A2, mice were treated with saline (n = 4), 300 mg/kg Aze (n = 3), or Aze 600 mg/kg (n = 4) IP, 5×/week for 4 weeks. After 4 weeks of treatment, mice were killed by isoflurane inhalation and perfused with 1% paraformaldehyde/2% glutaraldehyde (electron microscopy [EM] grade) in 0.1 M PBS. Three mice were killed at 2–3 weeks after treatment onset due to the development of clinical disease; a paired mouse in the saline control group was killed on the same day. The remaining control and Aze-treated mice were killed after 4 weeks of treatment. For all perfused animals, the skulls were opened and the skull and vertebral column were placed in the fixative along with a separate small piece of spinal cord. Perfusion quality was considered satisfactory (tail writhe, blanched liver), for 8 of the 11 mice; only samples from these mice were included in the analyses. Samples for EM were stored at 2–8°C. Non-CNS tissues were fixed in formalin and processed for routine histology as in experiment A1 (Table 1).
Neonatal Mice
In experiment N1, timed pregnant CD-1 females (Charles River) were obtained at E17; their pups were born on E20 and were weaned on postnatal day 23. Aze was administered PO, 350 mg/kg 3×/week, to nursing dams and weaned pups. Controls received sterile water PO. Three pups each in the Aze and water treatment groups were killed by CO2 inhalation at postnatal days 9, 16, 24, 30, and 38; 4 Aze-treated and 3 water-treated pups were killed on day 44. The 3 dams in each treatment group were killed at 5 weeks postbirth. Frozen, OCT, and formalin-fixed samples of CNS and non-CNS tissues were obtained from all groups at the time of death (Table 1).
In experiment N2, Aze exposure and observation of weaned pups were extended. Four timed pregnant CD-1 dams were obtained at E14 and dosed 3×/week with Aze 300 mg/kg PO or saline, and continued to be dosed through the nursing period. After weaning on day 22 postbirth, 5 males and 5 females per group were separated and dosing continued with 300 mg/kg Aze or saline for 6 weeks. The pups were killed 3 months from weaning and the dams were killed at 5 weeks postbirth. Frozen and formalin-fixed samples of CNS and non-CNS tissues including eyes were collected (Table 1).
All mice were maintained under controlled conditions in a facility accredited by the AAALAC International. All studies were approved by the Institutional Animal Care and Use Committee of Stanford University.
Tissue Pathology and Immunohistochemistry
Formalin-fixed paraffin-embedded CNS and non-CNS tissue sections 8- to 10-μm thick were stained routinely with hematoxylin and eosin (H&E) CNS tissue sections were also stained with Luxol fast blue (LFB)-PAS or LFB-H&E, Bielschowsky impregnation, and by IHC using the antibodies listed in Table 2. Immunostaining following microwave antigen retrieval and the appropriate species second antibodies was performed as previously described (23). Normal mouse spleen samples were used for IHC controls. TUNEL staining was performed using ApopTag peroxidase in situ apoptosis detection kits (S7100, Sigma) with hematoxylin counterstain according to the manufacturer’s instructions. Each CNS tissue slide contained representative cross-sections of brain (6–8 coronal sections) and cross- and longitudinally oriented spinal cord tissues (10–20 sections) for each mouse.
TABLE 2.
Antibodies
| Antigen/Cell Type | Antibody/Clone Name | Species/Isotype | Immunostain Concentration | Source |
|---|---|---|---|---|
| ATF3 (C-19) | SC-188 | Rb P | 1:200 | Santa Cruz Biotechnology, Dallas, TX |
| ATF4 (pSer245) | NB100-81802 | Rb P IgG | 1:100 | Novus Biologicals, Littleton, CO |
| ATF6 | LS-B2516 | Rb P | 1:200/1:500 | LifeSpan Biosciences, Seattle, WA |
| β-2 microglobulin (FL-199) | SC-15366 | Rb P | 1:50 | Santa Cruz Biotechnology |
| Calnexin | ab2595 | Rb P | 1:500 | Abcam, Cambridge, MA |
| Caspase-3, active (cleaved form) | AB3623 | Rb P | 1:50 | Millipore, Burlington, MA |
| Caspase-12 | ab62484 | Rb P | 1:500 | Abcam |
| CNPase (OGC) | C9743 | Rb P | 1:150 | Sigma-Aldrich, St. Louis, MO |
| EIF2S1 (phosphor S51) [E90] | ab32157 | Rb mAb IgG | 1:100 | Abcam |
| GADD 153 (R-20) | SC-793 | Rb P | 1:400 | Santa Cruz Biotechnology |
| Iba-1 (Macrophages/Microglia) | 09-19741 | Rb P | 1:250 | Wako Diagnostics USA, Mountain View, CA |
| MBP 82-87 | MAB 386 | Ra mAb IgG2a | 1:1000 | Sigma |
| MBP 119-131 | MCA1845 | M mAb IgG1 | 1:100* | Serotec, Oxford, UK |
| MHC Class I H2Db (28–14-8) | ab25228 | M mAb IgG2aκ | 1:200* | Abcam |
| MHC Class II (M5/114.15.2) | NBP1-43312 | Ra mAb IgG2b | 1:100 | Novus Biologicals |
| NFκB p65 (A) | SC-109 | Rb P | 1:200 | Santa Cruz Biotechnology |
| PERK (H-300)† | SC-13073 | Rb P | 1:200 | Santa Cruz Biotechnology |
| XBP1 | LS-B188 | Rb P | 1:200 | LifeSpan Biosciences |
CNPase, 2′,3′-cyclic-nucleotide 3′-phosphodiesterase; mAb, monoclonal antibody; M, mouse; P, polyclonal antibody; Ra, rat; Rb, rabbit.
Using mouse-on-mouse kit (Vector).
This antibody has been discontinued by the supplier.
OL Nuclear Diameters
For measurements of OL nuclear diameters, 5 random fields of uniformly processed H&E-stained paraffin section slides containing spinal cord WM, cerebellar WM, and corpus callosum of 3 mice each from the saline IP, 300 mg/kg Aze IP, and the 600 mg Aze IP and PO groups (Experiment A1), were photographed with a 60× magnification lens using a Nikon E1000M Eclipse microscope with an attached digital camera (Nikon, Melville, NY) and imaging software program (SPOT Imaging Solutions, Sterling Heights, MI). Fields of the spinal cord posterior columns consisting only of intact WM tracts in cross-section were photographed with 50-µm calibration bars merged into the fields. Fields of the cerebellar WM were selected adjacent to but did not include the dentate nucleus; fields with cerebellar nuclei and granular layer neurons were excluded. JPG files were modified to optimize color balance, brightness, and contrast using Adobe Photoshop CS6 (Adobe Systems, Inc., San Jose, CA) without altering the appearance of the original materials. Each of the 45 (3 mice/group×5 images/mouse×3 conditions) files was then printed in color on 8″ X 11″ paper; field areas were 153×264 µm.
After an initial training period, blinded observers measured the cross-sectional diameter of each round cell nucleus in each of the printed fields. For noncircular nuclei, the largest diameter was used as the measurement. Nuclei that had angulated shapes and cells that were closely proximal to microvessels were excluded. The observer labeled each measured nucleus on the paper and identified any for which there was a question as to the appropriateness of inclusion in the counts (∼10%–20% in each field). The images and measured nuclei were then reviewed by the neuropathologist as to whether they were morphologically consistent with OL nuclei and either included or excluded from the counts. The diameters of each OL nucleus were then calculated using the scale bars in the images.
OL Apoptosis
To assess the frequency of OL apoptosis in tissue samples, fields centered on TUNEL- and caspase-3-positive apoptotic cells in spinal cord WM were photographed as described above (Experiment A1). A blinded observer then counted the total positive apoptotic cells in the field. In experiment N2, sections of Aze-treated and control mouse eyes were immunostained for caspase-3 and numbers of positively stained cells in optic nerves were determined for each mouse.
Immunohistochemistry Analyses
To assess MHC 1 expression on apoptotic OL, 10 high power (60×) fields in WM were centered on an immunopositive cell with a pyknotic or apoptotic nucleus in spinal cord and brain WM samples. The fields were photographed from 5 mice that had been treated with 600 mg/kg Aze and 5 mice that had received 300 mg/kg Aze (Experiment A1). Numbers of immunopositive apoptotic cells were determined for each mouse. This analysis could not be performed on samples from mice treated with saline because of the near total absence of apoptotic cells in those mice. To assess nuclear translocation of UPR-related transcription factors, immunopositive nuclei were counted in 10 similarly selected high-power fields of spinal cord WM samples from 3 to 5 mice in each treatment group and controls.
Microglial nodules consisting of aggregates of closely apposed Iba-1-positive dendritic cells with increased amounts of cytoplasm and thickened processes were counted in spinal cord WM in Iba-1-immunostained slides from 4 adult mice that had been treated with 600 mg/kg Aze, 5 mice that had been treated with 300 mg/kg Aze, and 5 saline controls (Experiment A1).
To ascertain the frequency of OLs with cytoplasmic aggregation of myelin in mouse pups, 10 high-power fields of WM bundles in caudoputamen immunostained for MBP were centered on individual cells with immunopositive coarse cytoplasmic vacuoles and photographed. Samples from 4 pups that had been treated with 300 mg/kg Aze and 3 saline controls were analyzed (Experiment N2). The caudoputamen was selected for this analysis because individual OL are identifiable in this anatomic region in which there is a background of sparsely immunostained gray matter (GM). Numbers of cells with MBP-positive cytoplasmic aggregates in each field were determined for each mouse.
Electron Microscopy
The CNS samples in experiment A2 were sectioned into ∼1 mm3 pieces and further fixed in 150 mM phosphate buffer pH 7.4, 2.5% glutaraldehyde, 1% paraformaldehyde, 2 mM CaCl2 for 1 hour at 4°C, washed with 4 changes in 100 mM sodium cacodylate buffer, pH 7.4 with 2 mM CaCl2, secondarily fixed in cacodylate buffer with 1% OsO4 for 1 hour at room temperature (RT), washed with 3 changes with 50 mM sodium maleate, pH 6.0 1 hour at RT, and stained en bloc with 0.5% uranyl acetate in 50 mM sodium maleate for 1 hour at RT, then washed, and stored in 50 mM sodium maleate. The samples were then dehydrated in graded alcohols to 100% propylene oxide and embedded in BEEM size 00 embedding capsules with EMBed-812 Prep (Electron Microscopy Sciences, Hatfield, PA), according to the manufacturer’s protocol. After curing for 24 hours at 60°C, resin block sections were cut at 1-μm thickness, mounted on glass slides, heated, and stained with 1% toluidine blue, 2% sodium borate solution then washed with distilled water. Fields with optimally preserved OL were identified and photographed.
Statistical Analyses
Histologic data were analyzed using t-tests and 1-way ANOVA with Welch correction as appropriate using GraphPad Prism 5 (GraphPad, Inc., La Jolla, CA).
RESULTS
Clinical Disease
In experiment A1, clinical signs (body weight loss, tail paresis, ruffled fur, diarrhea, dyspnea) were observed in 6 of the 10 mice in the 600 mg/kg-treated groups from 2 to 3 weeks after onset of treatment; 2 of these mice were found dead (1 in the IP-treated group, 1 in the PO-treated group) and 4 were killed prior to the planned experimental endpoint due to declining clinical condition. In experiment A2, 3 mice in the 600 mg/kg group appeared thin, hunched, and dyspneic, and showed hind limb ataxia and tremors with movement. Videos demonstrate the tremulous phenotype of an Aze-treated mouse and a normal control littermate from this experiment (Supplementary Data Videos 1 and 2). No clinical abnormalities were observed in the 300 mg/kg- or saline-control-treated mice in experiments A1 and A2 or in any of the pups or dams in experiments N1 and N2.
Morphologic Alterations of OL
Routine brain and spinal cord tissue sections of Aze-treated adult and neonatal mice demonstrated clearing of nucleoplasm and nucleomegaly (watery swelling) of WM OL as compared to controls (Fig. 1A). OL nuclear diameter increases were Aze dose-dependent in both adult and neonatal mice (Fig. 1B; Supplementary Data Fig. S1). Pyknotic nuclei suggesting individual apoptotic OL were also identified in these sections and in sections immunostained for CNPase (Fig. 1C). No alterations of macroglia were identified in routine sections or by glial fibrillary acidic protein IHC (not shown). In particular, astrocyte nuclear swelling characteristic of hepatic and other metabolic encephalopathies was not observed in either GM or WM in any mice. Neuron cell bodies also appeared unaffected by Aze doses that altered OLs (Fig. 1A, C). Immunostains for T cells (CD3g) were negative in the CNS of Aze-treated as well as control mice (not shown).
FIGURE 1.

OL nucleomegaly and apoptosis in white matter of Aze-treated adult mice (Experiment A1). (A) Diffuse enlargement and clearing of nucleoplasm in OL in cerebellar white matter in Aze-treated mice as compared to those in a saline-treated control. Arrows indicate areas shown at higher power in the insets. H&E, original magnification: 240×. (B) OL nuclear diameters in Aze-treated and control mice. Numbers of OL nuclear measurements are indicated. *p<0.001, One-way ANOVA. (C) Immunostains for the OL-specific marker CNPase highlight nuclear swelling (black arrows) and pyknotic (possibly apoptotic), nuclei (red arrows) in the Aze-treated but not the control-treated mouse spinal cord white matter (WM). OL nuclear swelling is not evident in the gray matter (GM). No effects of Aze on GM neurons are evident. Original magnification: 240×.
EM confirmed the watery swelling of WM OL nuclei in Aze-treated versus control mice (Fig. 2). The WM OL in Aze-treated mice showed dilated ER and vacuoles in the cytoplasm and mitochondria (Fig. 2). The mitochondria in the OL of Aze-treated mice also showed abnormal shapes and vesicular autophagic profiles suggestive of mitophagy (Fig. 3; Supplementary Data Fig. S2).
FIGURE 2.
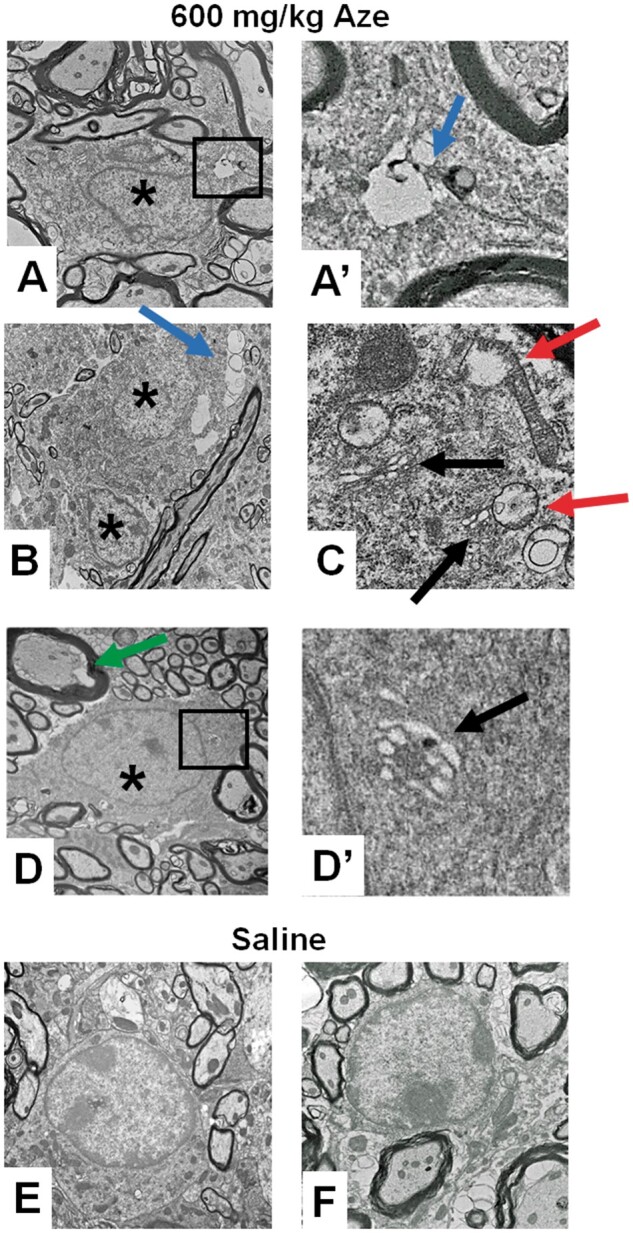
Ultrastructural analysis of Aze effects on mouse spinal cord OL (Experiment A2). (A–D) OL of Aze-treated mice show watery nuclei (* in A, B, D), clear cytoplasmic vacuoles (blue arrows in A, A′, B), vacuolated mitochondria (red arrows in C), dilated ER (black arrows in C, D′), and enlarged inner tongues of myelin (green arrow). (E, F) Normal OL morphology in saline control mice. Boxed areas in (A) and (D) are shown at higher magnifications in (A′) and (D′), respectively. Original magnifications: A, D, E, 9300×; B, 6800×; F, 13 000×; A′, C, D′, 59 300×.
FIGURE 3.

Mitochondrial abnormalities suggestive of mitophagy in OL of adult mice that received 600 mg/kg Aze (Experiment A2). (A–D) There are membrane-bound vesicles and degenerative profiles within mitochondria; some show abnormal shapes and loss of cristae. They are in close proximity to ER (arrows). Enlarged inner tongues of myelin are also seen in (A) (green arrow). Boxed areas in (A) are shown at higher magnification in (B–D). Original magnification: 13 000×.
Myelin and Axons
By light microscopy, CNS compact myelin in Aze-treated adult mice appeared to be largely intact in routine H&E (Fig. 1A) and Luxol fast blue (not shown) stains. Anti-MBP IHC also did not detect frank demyelination. There were, however, blister-like swellings in the inner myelin layers in the WM of Aze-treated mice (Fig. 4; Supplementary Data Fig. S3) and enlarged inner tongues of myelin were observed in Aze-treated mice by EM (Figs. 2D and 3A). Extensive abnormalities also could not be identified in compact myelin in the Aze-treated mouse pups (Experiment N2). However, there were small numbers of OL with coarse MBP-positive cytoplasmic aggregates that could be counted in the caudoputamen and were more numerous in Aze-treated versus control mice (Fig. 5).
FIGURE 4.

Compact myelin in sections of cerebellum from adult mice immunostained for MBP show watery swelling of OL nuclei in the Aze-treated (600 mg/kg) mouse but not the control mouse (Experiment A1). Black arrows indicate fields magnified in panels to the right of the larger fields. There are blister-like areas of immunoreactivity on the periphery of some of the OL in the treated mouse (red arrows). Original magnification: 160×.
FIGURE 5.

Cytoplasmic aggregation of MBP in individual caudoputamen oligodendrocytes in Aze-exposed neonatal mice (Experiment N2). (A, B) Examples of coarse granular immunostaining for MBP in individual OL. Black arrows indicate individual cells magnified in black-bordered insets. Red arrows indicate WM bundles with myelin blistering magnified in red-bordered insets. (C) The OL in a saline control mouse shows normal nuclear staining without MBP aggregation. Green arrow indicates a WM bundle from the saline control magnified in the green-bordered inset. (D) Counts of cells with MBP aggregates in Aze-treated versus saline control mice. *p = 0.0032. Data are from 40 fields of Aze-treated and 30 fields of control mice. (A–C), original magnification: 160×.
Bielschowsky preparations did not demonstrate widespread abnormalities of WM axons but there were rare dystrophic axonal swellings in the WM of Aze-treated mice that were not seen in the control mice (Fig. 6). No abnormalities of neuron cell bodies or of other cells were detected in Aze-treated mice by either light microscopy or EM.
FIGURE 6.

Bielschowsky preparations demonstrate rare axonal swellings in cerebellar white matter of Aze-treated (600 mg/kg) but not in saline-treated adult mice (Experiment A1). Upper and lower panels are from 2 different mice from each treatment group. Arrows indicate swollen axons shown in magnified images to the right. Original magnification: 240×.
OL Apoptosis in CNS White Matter and Optic Nerves
In TUNEL- and caspase-3-immunostained sections of brain and spinal cord of Aze-treated adult mice, there were individual TUNEL- and caspase-3-positive cells in different stages of apoptosis in cerebellar and spinal cord WM and corpus callosum. The apoptotic OL appeared to be randomly distributed through the WM without alterations of adjacent cells or structures (Fig. 7). The extent of OL apoptosis was Aze dose-dependent (Fig. 7). There were similar caspase-12-positive apoptotic OL in WM of Aze-treated adult mice (Supplementary Data Fig. S4). In Aze-treated mouse pup optic nerves, there were more apoptotic cells than in controls (Experiment N2) (Fig. 8). The Aze-treated pups also showed more numerous apoptotic cells in the spinal cord than their dams, which had received the maximal cumulative amount of Aze before they were killed (Fig. 9A, B).
FIGURE 7.

Aze induces OL apoptosis in spinal cord white matter of adult mice (Experiment A1). Counts of apoptotic cells in the spinal cord white matter of each mouse in the 3 treatment groups are shown in the graphs. (A) TUNEL stains demonstrate individual OL undergoing apoptosis more frequent in the Aze-treated than in a saline control mouse. Areas in boxes are shown at higher power in the insets. (B) Caspase-3 immunostained section of spinal cord of an Aze-treated mouse. The graphs demonstrate that there are more numerous immunopositive cells in the Aze-treated mice versus controls. A, p = 0.0019; B, p = 0.0002, by ordinary ANOVA. Original magnification: 240×.
FIGURE 8.

Effects of in utero and neonatal Aze on mouse pup optic nerves (Experiment N2). (A) Optic nerves of Aze-treated mouse pups showed scattered individual cells apparently undergoing apoptosis (arrows). Higher magnification of these cells is shown in (A′) and (A″). (B) No apoptotic cells were identified in the saline-treated controls. (C–F) Caspase-3 immunostained sections demonstrate multiple immunopositive cells and apoptosomes (arrows) in the Aze-treated (D–F) but not the control mice (C). Higher magnifications of the indicated cells are shown in (D′), (E′), and (F′). (G) There were more numerous caspase-3-positive cells in the Aze-treated versus control optic nerves (p = 0.0037, 2-tailed t-test). A, B, H, and E; C–F, caspase-3 IHC. All original magnifications: 240×.
FIGURE 9.

Newborn mice with intrauterine and neonatal exposure to low dose Aze (Experiment N2), show greater apoptosis in spinal cord white matter than their equally exposed dams and versus water-treated controls. (A) TUNEL. p = 0.045; (B) Caspase-3 IHC, p < 0.0001, ANOVA.
MHC 1 Expression on Apoptotic OL
Immunostaining for MHC I demonstrated individual immunopositive apoptotic WM OL in Aze-treated but not control adult mice; the extent of the MHC I labeling of the apoptotic OL was Aze dose-dependent (Fig. 10). There was similar immunostaining for β-2 microglobulin of apoptotic OL in Aze-treated mice (Supplementary Data Fig. S5). Expression of MHC II was not detected in the CNS of any mice; there was positive immunostaining of normal mouse spleen control tissue (not shown).
FIGURE 10.

Apoptotic OL in white matter of Aze-treated mice are immunostained for MHC I (Experiment A1). (A) Saline control shows no MHC 1 immunostaining of OL. Asterisk indicates positive control staining of endothelial cells in a microvessel. (B–D) Positive staining of individual cells with pyknotic or apoptotic nuclei from samples at the indicated levels of Aze exposure. Cells in boxes are shown at higher power in insets and adjacent. Original magnification: 240×. (E) Counts of MHC 1-positive cells in Aze-treated mice. 300 mg/kg versus 600 mg/kg, p = 0.0146, t-test. ND, not done.
Reactive Microglia and Microglial Nodules
Microglia in Aze-treated adult mice identified by Iba-1 IHC showed thickened long processes, amoeboid shapes, and more abundant cytoplasm as compared to those in saline-treated controls (Fig. 11A–D). There were also scattered Iba-1-positive microglial nodules in the WM of Aze-treated mice (Fig. 11E–H; Supplementary Data Fig. S6). Microglial nodules were more numerous in mice that had received 600 mg/kg than those given 300 mg/kg; no microglial nodules were identified in controls (Fig. 11I). No abnormal Iba-1 immunostaining was identified in GM or spinal nerve roots.
FIGURE 11.

Reactive microglia and microglial nodules in WM of Aze-treated adult mice (Experiment A1). (A, B) Saline controls show quiescent microglial features including thin elongated processes (red arrows) in cerebellar (A) and anterior spinal cord (B) WM. (C–F) Microglia in cerebellar (C, E, F), and anterior spinal cord (D, G, H) WM in Aze-treated mice show shorter, thicker processes, and more abundant cytoplasm. There are aggregates of reactive microglia with overlapping cytoplasm, i.e. microglial nodules (black arrows in [E] and examples in [G] and [H]). The numbers of microglial nodules increased with increasing doses (I). *p = 0.0019, one-way ANOVA. All are Iba-1 IHC. Original magnification: 160×.
Nuclear Translocation of ER Stress/UPR-Related Molecules in OL
To understand the intracellular mechanisms in OL apoptosis and MHC I expression, we analyzed components of the UPR using IHC. In immunostains for each of the ER stress/UPR-related molecules ATF3, ATF4, ATF6, eIF2α, GADD153, NFκB, PERK, and XBP1, OL with nuclear staining were more numerous in Aze-treated mice versus controls in a dose-dependent pattern (Figs. 12–15). By contrast, there were no comparable alterations in the OL subcellular localization of the ER chaperone calnexin (Supplementary Data Fig. S7).
FIGURE 12.

Aze induces nuclear translocation of ATF3, ATF4, and ATF6 in OL in adult mice (Experiment A1). (A–I) Representative fields of spinal cord WM from mice with the indicated treatments show increased proportions of OL with nuclear immunostaining for ATF3 (A, B), ATF4 (D, E), and ATF6 (G, H) (arrows). Boxes in (B), (E), and (H) indicate OL shown at higher magnifications in adjacent panels. Red arrow and box in (B) indicate an OL apparently undergoing apoptosis. (C, F, I) Graphs demonstrate the effect of Aze dose. *p < 0.0001, ordinary ANOVA (C, I). *p = 0.0007, Welch ANOVA (F). Original magnification: 360×.
FIGURE 13.

Aze induces nuclear translocation of EIF2α, GADD153, and PERK in OL in adult mice (Experiment A1). (A–I) Representative fields of spinal cord WM from mice with the indicated treatments show increased proportions of OL with nuclear immunostaining for EIF2α (A, B), GADD153 (D, E), and PERK (G, H) (arrows). Boxes in (B), (E), and (H) show OL at higher magnifications in adjacent panels. C, F, I: Graphs demonstrate the effect of Aze dose. *p < 0.0001, ordinary and Welch ANOVA. Original magnification: 360×.
FIGURE 14.

Aze induces nuclear translocation of NFκB in OL in adult mice (Experiment A1). (A–C) Representative fields of spinal cord WM from mice with the indicated treatments show increased proportions of OL with nuclear immunostaining for NFκB (red arrows). Boxes indicate OL shown at higher magnifications in insets. Original magnification: 240×. (D) Graph demonstrates the effect of Aze dose. *p < 0.0001, ordinary ANOVA.
FIGURE 15.

Aze induces nuclear translocation of XBP1 in OL in adult mice (Experiment A1). (A–D) Representative fields of spinal cord WM from mice with the indicated treatments show increased proportions of OL with nuclear immunostaining for XBP1 (red arrows). A preparation without hematoxylin (C) highlights increased immunoreactivity of OL nuclei. Boxes in (D) indicate OLs shown in higher mag. in insets. Original magnification: A–D, 360. (E) Graph demonstrates the effect of Aze dose. *p = 0.0338, ordinary ANOVA.
Non-CNS Tissues
In experiment A1, 8 out of the 10 mice treated with 600 mg/kg had gastric distension; their livers were grossly abnormal with pale and/or mottled areas in 5 of these mice (Supplementary Data Fig. S8A). Similar macroscopic lesions were observed in 2 of the 4 high dose-treated mice in experiment A2. Histologic examination of 5 of 10 of the livers in the 600 mg/kg group in experiment A1 and 3 of 4 similarly treated mice in experiment A2 demonstrated variable extents of hepatocyte vacuolation and nuclear pyknosis (Supplementary Data Fig. S8B–D). One mouse in the latter group in experiment A2 also showed foci of hepatocyte necrosis and inflammation. These abnormalities were not observed in any of the livers of the lower dose- or saline-treated mice (Supplementary Data Fig. S8E, F). No macroscopic or microscopic abnormalities in other non-CNS tissues were identified in the other Aze-treated dose or control mice.
In experiments N1 and N2, there were no macroscopic abnormalities observed in any organs. In experiment N1, the livers of 9 of 18 Aze-treated pups showed mild histopathologic abnormalities consisting of hepatocyte vacuolation, nucleomegaly, and focal inflammation (Supplementary Data Fig. S9). The frequency of these abnormalities increased with the duration of postnatal Aze exposure prior to sacrifice. The livers of the 3 Aze-treated dams also showed these changes. In experiment N2, 9 of 10 Aze-treated mouse pups also showed these liver abnormalities. The livers of all 15 control pups and 3 dams in experiment N1 and the livers of all 10 control-treated pups in experiment N2 were histologically normal. No microscopic abnormalities were observed in any other non-CNS organs in experiments N1 and N2. Table 3 summarizes the major effects of Aze treatment in the 4 experiments.
TABLE 3.
Summary of Effects of Azetidine Exposure in Adult and Neonatal Mice
| Experiment | Clinical Disease (600 mg/kg group only) | OL Nucleomegaly and Nuclear Clearing (LM, EM) | OL Apoptosis (TUNEL, Caspase-3 IHC) | WM Microglial Reaction/Nodules (Iba-1 IHC) | OL ER stress/UPR |
↑MHC 1 on Apoptotic OL (IHC) | WM Myelin Blistering and Myelinosomes (MBP IHC) | Dystrophic WM Axons (LM, Biel) | Liver Abnormalities (LM) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Nuclear translocation of UPR proteins (IHC) | Dilated ER, autophagy/mitophagy (EM) | |||||||||
| A1 (adult mice) | +† | +† | +† | +† | +† | +† | + | +† | +† | |
| A2 (adult mice) | − | + | ND | + | ND | + | ND | ND | ND | +† |
| N1 pups* | − | + | ND | ND | ND | ND | ND | ND | ND | +‡ |
| N1 dams* | − | + | ND | ND | ND | ND | ND | ND | ND | + |
| N2 pups | − | + | +§ | ND | ND | ND | + | ND | ND | + |
| N2 dams | − | + | + | ND | ND | ND | ND | − | ND | + |
Biel, Bielschowsky silver impregnation; EM, electron microscopy; ER, endoplasmic reticulum; IHC, immunohistochemistry; LM, light microscopy; MBP, myelin basic protein; ND, not done; OL, oligodendrocyte; UPR, unfolded protein response; WM, white matter; +, present; −, analysis performed with negative results.
Treatment PO-initiated postweaning with 350 mg/kg Aze resulted in no definite effects on the CNS by routine LM analysis.
Aze dose-dependent effects.
Increased frequency in pups with longer exposures to Aze.
Greater incidence of OL apoptosis in Aze-treated pups versus dams.
DISCUSSION
Clinical Effects of Aze
Adult mice that had been treated with high doses of Aze developed clinical abnormalities including shivering (Supplemental Data Videos 1 and 2). Although this phenotype may be seen in multiple myelin mutants, it is the phenotype of genetically MBP-deficient shiverer mice (24). However, Aze had been administered systemically and consistent abnormalities were also observed in their livers (Supplementary Data Fig. S8). These were particularly common in the livers of Aze-treated mouse pups (Supplementary Data Fig. S9; Table 3), and it is possible that metabolic derangements resulting from the liver abnormalities contributed to the clinical phenotype of Aze-treated mice. Conversely, CNS dysfunction could have led to impaired feeding and failure to thrive that affected the liver as well as the functions of other non-CNS organs despite their normal histology. Therefore, the Aze-associated clinical signs suggest but do not prove that CNS MBP was specifically targeted or that they can be entirely attributed to Aze effects on the CNS. Clinical disease was not observed in the neonatal mice or their dams likely because they had received lesser amounts of Aze.
OL Alterations Induced by Systemic Aze
By routine light microscopy, the most prominent CNS abnormalities induced by Aze were in WM OL with apparent overall preservation of compact myelin (Fig. 1). Watery swelling of nuclei and apoptosis of individual OL with relative preservation of myelin are not typical features of shiverer mice or of other MBP mutants (25, 26) but, since abnormalities of other CNS cells were not observed, the distinct Aze-induced pathology implies cell-specific metabolic effects on OL. Similar OL nucleomegaly has been documented in MS NAWM by Prineas and Parratt and by earlier authors (21).
Ultrastructural analyses concentrated on WM OL confirmed the nuclear swelling and identified dilated ER and vacuolation of OL cytoplasm in Aze-treated mice (Figs. 2 and 3); these findings are characteristic of ER stress in rodent OL (27). OL mitochondria also had morphological abnormalities suggestive of autophagy and mitophagy (Fig. 3; Supplementary Data Fig. S2), which are also characteristic of ER stress (28–30), hypoxic injury (31), and mitochondriopathies (32). They are also seen in differentiating OL (33), and suggest involvement of multiple intersecting cell stress pathways.
OL Apoptosis
Routine H&E, TUNEL, and caspase-3 and -12 immunostains demonstrated dose-dependent apoptosis of WM OL in Aze-treated adult and neonatal mice (Figs. 6 and 7; Supplementary Data Fig. S4). Identification of individual randomly distributed apoptotic cells required scanning of the entire CNS tissue sections at high magnification. Apoptosis of OL was not noted in GM but detection may have been precluded by the overall lower density of OL in GM versus WM. The greater extent of apoptosis in the neonatal pups as compared to their equally dosed dams (Fig. 9), indicates particular susceptibility to Aze-induced effects during myelinogenesis and myelination (34). OL apoptosis and depletion have also been documented in MS NAWM as well as adjacent to active inflammatory/demyelinating lesions by multiple investigators and may determine the extent of subsequent myelin loss (17, 18, 35–37).
Effects of Aze on Myelin and Axons
Aze did not induce frank demyelinating lesions or overt abnormalities of compact myelin but blister-like swellings suggestive of myelin detachment from axons were observed in Aze-treated adult mice (Fig. 4) and neonatal mice (Supplementary Data Fig. S3) by IHC for MBP. Myelin blistering has recently been reported in MS NAWM by Luchicchi et al who suggest that the accompanying biochemical alterations may precede an autoimmune inflammatory attack (38). Recent studies employing novel imaging and spectroscopic techniques have also identified alterations in MS NAWM not readily identifiable by conventional microscopy that may precede frank inflammatory lesions (39, 40).
At the EM level, myelin lamellae and major dense lines were not clearly different between Aze-treated versus saline-treated control mice in Experiment A2 (Fig. 2). The enlarged inner tongues of myelin in Aze-treated mice identified by EM (Figs. 2D and 3A), likely represent the ultrastructural counterpart of the blister-like areas seen at the light microscopic level. Similar findings are reported in some myelin mutants and inflammatory and myelin toxicity models; they suggest incipient myelin degeneration and disturbance of the function of the axon-myelin unit (41, 42). The inner tongues adjacent to axons are the location of incorporation of new myelin membranes (43). The enlargements imply that these regions are particularly susceptible to effects of Aze and might account for significant functional effects on axons despite apparent overall integrity of the compact myelin. This occurrence in adult mice also raises the possibility that exogenous Aze might have effects on myelin postcompaction, for example, as part of a focal adaptive myelination process (44) or remyelination following injury or demyelination.
Intracytoplasmic MBP-positive aggregates in OL in Aze-treated mice were not seen in controls and were particularly numerous in Aze-treated mouse pups versus adults, suggesting greater effects of Aze on newly forming as opposed to intact compact myelin (Fig. 5). Romanelli et al reported similar cytoplasmic aggregates termed, “myelinosomes” in active MS lesions, in MS NAWM, and in the WM of mice with experimental autoimmune encephalomyelitis (EAE) (45). In the present study there was no leukocyte infiltration in the CNS indicating that the MBP aggregation in OL arose autonomously.
Bielschowsky preparations also demonstrated subtle WM axonal abnormalities in Aze-treated mice (Fig. 6), which are similar to the dystrophic and degenerating axons reported in MS NAWM (46–48).
ER Stress/Unfolded Protein Response and Proinflammatory OL Phenotype
Because the light and electron microscopic findings suggested ER stress and UPR activation, we investigated the immunolocalization of key UPR components in OL. We consistently identified dose-dependent nuclear immunostaining consistent with activation of multiple cell stress pathways in Aze-treated mice (Figs. 12–15). The ER marker calnexin did not show the same nuclear translocation pattern, indicating the specificity of observations on the UPR components (Supplementary Data Fig. S7).
The UPR is a complex, dynamic cytoprotective response that preserves cell viability and function under proteostatic stress but triggers apoptosis when the ER becomes overwhelmed with misfolded proteins (49, 50). Multiple studies implicate OL ER stress/UPR in the pathogenesis of myelin disorders including MS as well as numerous other CNS disorders (19, 20, 51–54). UPR activation can result in the induction or enhancement of inflammation and autoimmunity (55–57). In this regard, the nuclear translocation of NFκb and XBP1 are particularly relevant to the induction of a proinflammatory OL phenotype as manifested by increased expression of MHC 1 and β-2 microglobulin (Fig 10; Supplementary Data Fig. S5). NFκb is a central regulator of protein quality control in response to Aze-induced protein aggregation (13), but it could also indicate a protective mechanism for OL under inflammatory conditions (58). MHC I expression indicates the potential for OL cytotoxicity and presentation to CD8-positive T cells of neuroantigens released as a consequence of the OL apoptosis (59). Thus, a predisposition to OL activation resulting from ER stress and the UPR could enhance or prolong a cellular immune reaction within the CNS initiated by a second event such as a common viral infection (60–62). Moreover, MHC I expression by OL may inhibit remyelination and promote neurodegeneration (59, 63).
Microglial Activation and Microglial Nodules
Aze-induced microglial activation and scattered microglial nodules indicate focal responses to OL apoptotic cell death, focal myelin degeneration, and axonal injury in otherwise intact WM. Similar findings are well recognized in MS NAWM (64, 65). Microglial activation as a consequence of Aze exposure might result in priming and increased susceptibility to subsequent immune challenge (66, 67). The complex morphological patterns and the diverse functional roles for microglia in MS-affected CNS tissues have recently been documented in detail by Prineas and Parratt (22).
Comparison to Noninflammatory and Inflammatory Animal Models of MS
Unlike extensively studied inflammatory MS models induced by immunization with self-antigens such as EAE, or by viruses (68), Aze induces noninflammatory, OL-specific injury in the CNS associated with UPR activation. The induction of ER stress/UPR in EAE and MS has been attributed to an inciting inflammatory response, which is by definition minimal or absent in MS NAWM. Cuprizone toxicity is a widely employed as model of OL injury, demyelination, and remyelination (69–71); it is also hepatotoxic (72). However, both the pathology and the molecular mechanisms of cuprizone and other toxic oligodendrogliopathies, such as that induced with ethidium bromide (73) or LPS (74) differ considerably from those of Aze-induced OL toxicity. Importantly, unlike Aze, cuprizone, and other myelinotoxins are not consumed by humans; they do not as closely mimic MS NAWM; and they are not directly implicated in MS pathogenesis. There are also features of OL stress in the genetically engineered OBiden mouse model (75), but Aze-induced oligodendrogliopathy is acquired and is mechanistically distinct.
Relevance to MS Pathogenesis
Despite normal macroscopic appearances and overall histopathologic appearance of CNS compact myelin, subtle abnormalities in MS NAWM have long been suggested to indicate an increased susceptibility to injury (76). The present observations indicate that despite the absence of inflammation and overt demyelinating lesions, Aze induces alterations in WM OL, including the UPR and proinflammatory phenotype, and on compact myelin, axons, and microglia that closely replicate findings in MS NAWM and adjacent to active MS lesions.
Myelin breakdown as a consequence of progressive MBP misfolding might induce an expanded autoantigen repertoire (77–79). The misincorporation of Aze during myelinogenesis in the fetal and early postnatal period would necessarily be a primary event, as in the “inside-out” hypothesis of MS etiology (80). The more severe effects of Aze in mice exposed during intrauterine and early postnatal life are consistent with effects on myelinogenesis. A “second hit,” such as infection, trauma, necrosis, ADEM, or other injury that otherwise might be less severe or more short-lived would be enhanced in a proinflammatory WM milieu in which there has been Aze misincorporation in major myelin proteins. The multiplicity of such potential second hits might account for the difficulties in identifying a single infectious agent or autoimmune target antigen molecule in MS patients. Moreover, instability of proteins in formed compact myelin resulting in increased breakdown over many years would impair the capacity for OL regeneration and remyelination (81, 82). These processes would likely result in progressive WM atrophy and associated clinical progression in MS patients. How Aze may initiate these hypothesized mechanisms in humans is summarized in Figure 16.
FIGURE 16.

Hypothesized pathogenetic processes in the human CNS resulting from Aze misincorporation into MBP based on results of this study. The MBP misincorporation portion of this figure is adapted from Figure 6 in reference (10) with permission from Elsevier. Broken arrows indicate progressive degenerative processes that may develop over many years.
Limitations of This Study
Because no definite abnormalities were observed in the CNS of pups in experiment N1 (up to postnatal D44), the time of exposure was begun earlier, (i.e. in utero), and the periods of observation (up to 3 months postweaning) were extended in experiment N2 (Table 1). The earlier exposure and longer observation period resulted in more marked effects in those mice (Table 3). It is possible that observation periods longer than 3 months would have resulted in spontaneous clinical disease and more pronounced histopathologic effects in the neonatally treated mice. Moreover, because the neonatal mice were only studied by light microscopy, ultrastructural alterations, such as of the integrity of compact myelin or possible major dense line splitting, could not be assessed in this study.
The pathophysiological mechanisms leading to the clinical phenotype in 600 mg/kg Aze-treated mice, particularly with the apparent relative preservation of compact myelin, are not clear. Although suggested by the ultrastructural findings of enlarged inner mesaxonal tongues in OL, there could have been undetected effects on neuroaxonal function that contributed to the Aze-induced clinical phenotype. Analysis of potential physiologic disturbances of neurons was beyond the scope of the present study.
As is the case for most rodent MS model studies, we report the effects of a severe acute injury and analyses over relatively short time periods in both adult and neonatal mice. This is analogous to the often-minimized differences in time course (as well as in species differences), that prevent precise extrapolations from rodent EAE (particularly acute EAE), models to MS. Low doses of Aze administered at critical developmental periods and analyses over longer time periods may demonstrate a progressive UPR and more closely mimic chronic effects in humans in future studies.
Because of the key structural roles that Pro and polyproline sequences have in the human proteome (e.g. zinc finger proteins, which control DNA expression [83]), there are multiple potential aberrant CNS and non-CNS proteins that might result from systemic administration of Aze. Indeed, although in the CNS the predominant effects identified were on WM OL, the induction of the liver pathology indicates significant non-CNS effects of systemic administration (Table 3). Because myelin proteins are stable throughout life, the potential for a slow degenerative process resulting from misincorporation during myelinogenesis may be greater over decades than effects on proteins that turn over more rapidly, such as those in the liver. MBP is the second most abundant CNS myelin protein and has many more Pro residues than the more abundant PLP (84). Although the in vitro experiments in the literature as well as the present findings strongly suggest that MBP is a likely target for misincorporation of Aze, formal proof of the presence of Aze and its misincorporation in myelin proteins in either mouse or human CNS tissues is lacking at this time. Such proof is difficult to obtain with current methodologies (85), and remains a challenge for future investigations.
Summary
In summary, we provide a model of noninflammatory OL injury and proof-of-concept that exogenous Aze has specific deleterious effects on OL in the mammalian CNS in vivo. The clinical and morphological findings imply but do not prove that Aze misincorporation in MBP during myelinogenesis results in protein misfolding with associated downstream consequences. Nevertheless, the present study supports and extends the Aze hypothesis. In view of the many shared morphologic and mechanistic processes of inflammation and degeneration in MS NAWM and Aze oligodendrogliopathy, the results are potentially highly relevant to the pathogenesis of chronic human demyelinating diseases. Moreover, recognition of OL ER stress/UPR as a primary event in MS pathogenesis raises possibilities for new translational therapies (86–91).
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Wing Fei Wong and Shawn Lee for technical assistance. This study received the Weil Award for Best Paper on Experimental Neuropathology at the Annual Meeting of the American Association of Neuropathologists, Monterey, CA, June 2020.
This study was supported by Progressive MS Alliance PA 0082, SPARK, Stanford University Department of Pathology Gift Fund and Palo Alto Veterans Institute for Research.
The authors have no duality or conflicts of interest to declare.
Supplementary Data can be found at academic.oup.com/jnen.
REFERENCES
- 1. Rubenstein E. Misincorporation of the proline analog azetidine-2-carboxylic acid in the pathogenesis of multiple sclerosis: A hypothesis. J Neuropathol Exp Neurol 2008;67:1035–40 [DOI] [PubMed] [Google Scholar]
- 2. Sobel RA. A novel unifying hypothesis of multiple sclerosis. J Neuropathol Exp Neurol 2008;67:1032–4 [DOI] [PubMed] [Google Scholar]
- 3. Rubenstein E, Zhou H, Krasinska KM, et al. Azetidine-2-carboxylic acid in garden beets (Beta vulgaris). Phytochemistry 2006;67:898–903 [DOI] [PubMed] [Google Scholar]
- 4.Central Intelligence Agency. The World Factbook, Field Listing, Agriculture Products. Washington, DC: Central Intelligence Agency; 2021. [Google Scholar]
- 5. Simpson S Jr, Wang W, Otahal P, et al. Latitude continues to be significantly associated with the prevalence of multiple sclerosis: An updated meta-analysis. J Neurol Neurosurg Psychiatry 2019;90:1193–200. [DOI] [PubMed] [Google Scholar]
- 6. Bach TM, Takagi H.. Properties, metabolisms, and applications of (L)-proline analogues. Appl Microbiol Biotechnol 2013;97:6623–34 [DOI] [PubMed] [Google Scholar]
- 7. Biratsi A, Athanasopoulos A, Kouvelis VN, et al. A highly conserved mechanism for the detoxification and assimilation of the toxic phytoproduct L-azetidine-2-carboxylic acid in Aspergillus nidulans. Sci Rep 2021;11:7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Toyama BH, Savas JN, Park SK, et al. Identification of long-lived proteins reveals exceptional stability of essential cellular structures. Cell 2013;154:971–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Truscott RJW, Friedrich MG, Review. Can the fact that myelin proteins are old and break down explain the origin of multiple sclerosis in some people? J Clin Med 2018;7:281–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bessonov K, Bamm VV, Harauz G.. Misincorporation of the proline homologue Aze (azetidine-2-carboxylic acid) into recombinant myelin basic protein. Phytochemistry 2010;71:502–7 [DOI] [PubMed] [Google Scholar]
- 11. Song Y, Zhou H, My-Nuong V, et al. Double mimicry evades tRNA synthetase editing by toxic vegetable-sourced non-proteinogenic amino acid. Nat Commun 2017;8:2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nunn PB, Bell EA, Watson AA, et al. Toxicity of non-protein amino acids to humans and domestic animals. Nat Prod Commun 2010;5:485–504 [PubMed] [Google Scholar]
- 13. Nivon M, Fort L, Muller P, et al. NFκB is a central regulator of protein quality control in response to protein aggregation stresses via autophagy modulation. Mol Biol Cell 2016;27:1712–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Onselen R, Downing S, Kemp G, et al. Investigating β-N-methylamino-L-alanine misincorporation in human cell cultures: A comparative study with known amino acid analogues. Toxins 2017;9:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roest G, Hesemans E, Welkenhuyzen K, et al. The ER stress inducer L-azetidine-2-carboxylic acid elevates the levels of phospho-eIF2α and of LC3-II in a Ca2+-dependent manner. Cells 2018;7:239–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Samardzic K, Rodgers KJ.. Cell death and mitochondrial dysfunction induced by the dietary non-proteinogenic amino acid L-azetidine-2-carboxylic acid (Aze). Amino Acids 2019;51:1221–32 [DOI] [PubMed] [Google Scholar]
- 17. Raine CS. The Norton Lecture: A review of the oligodendrocyte in the multiple sclerosis lesion. J Neuroimmunol 1997;77:135–52 [DOI] [PubMed] [Google Scholar]
- 18. Bonetti B, Stegagno C, Cannella B, et al. Activation of NF-κB and c-jun transcription factors in multiple sclerosis lesions—Implications for oligodendrocyte. Pathology. Am J Pathol 1999;155:1433–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ní Mháille A, McQuaid S, Windebank A, et al. Increased expression of endoplasmic reticulum stress-related signaling pathway molecules in multiple sclerosis lesions. J Neuropathol Exp Neurol 2008;67:200–11 [DOI] [PubMed] [Google Scholar]
- 20. Cunnea P, Ní Mháille A, McQuaid S, et al. Expression profiles of endoplasmic reticulum stress-related molecules in demyelinating lesions and multiple sclerosis. Mult Scler 2011;17:808–18 [DOI] [PubMed] [Google Scholar]
- 21. Prineas JW, Parratt JD.. Oligodendrocytes and the early multiple sclerosis lesion. Ann Neurol 2012;72:18–31 [DOI] [PubMed] [Google Scholar]
- 22. Prineas JW, Parratt JDE.. Multiple Sclerosis: Microglia, Monocytes, and Macrophage-Mediated Demyelination. Journal of Neuropathology & Experimental Neurology 2021;80:975–96 10.1093/jnen/nlab083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sobel RA, Eaton MJ, Jaju PD, et al. Anti-myelin proteolipid protein peptide monoclonal antibodies specifically bind multiple cell surface proteins on developing neurons and inhibit their differentiation. J Neuropathol Exp Neurol 2019;78:819–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Popko B, Puckett C, Lai E, et al. Myelin deficient mice: Expression of myelin basic protein and generation of mice with varying levels of myelin. Cell 1987;48:713–21 [DOI] [PubMed] [Google Scholar]
- 25. Rosenbluth J. Central myelin in the mouse mutant shiverer. J Comp Neurol 1980;194:639–48 [DOI] [PubMed] [Google Scholar]
- 26. Duncan ID, Radcliff AB.. Inherited and acquired disorders of myelin: The underlying myelin pathology. Exp Neurol 2016;283:452–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bauer J, Bradl M, Klein M, et al. Endoplasmic reticulum stress in PLP overexpressing transgenic rats: Gray matter oligodendrocytes are more vulnerable than white matter oligodendrocytes. J Neuropathol Exp Neurol 2002;61:12–22 [DOI] [PubMed] [Google Scholar]
- 28. Manoli I, Alesci S, Blackman MR, et al. Mitochondria as key components of the stress response. Trends Endocrinol Metab 2007;18:190–8 [DOI] [PubMed] [Google Scholar]
- 29. Senft D, Ronai ZA.. UPR, autophagy and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 2015;40:141–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lebeau J, Saunders JM, Moraes VWR, et al. The PERK arm of the unfolded protein response regulates mitochondrial morphology during acute endoplasmic reticulum stress. Cell Rep 2018;22:2827–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rathnasamy G, Murugan M, Ling EA, et al. Hypoxia-induced iron accumulation in oligodendrocytes mediates apoptosis by eliciting endoplasmic reticulum stress. Mol Neurobiol 2016;53:4713–27 [DOI] [PubMed] [Google Scholar]
- 32. Szalardy L, Molnar M, Torok R, et al. Histopathological comparison of Kearns-Sayre syndrome and PGC-1a-deficient mice suggests a novel concept for vacuole formation in mitochondrial encephalopathy. Folia Neuropathol 2016;54:9–22 [DOI] [PubMed] [Google Scholar]
- 33. Yazdankhah M, Ghosh S, Shang P, et al. BNIP3L-mediated mitophagy is required for mitochondrial remodeling during the differentiation of optic nerve oligodendrocytes. Autophagy 2021;17:3140–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sturrock RR. Myelination of the mouse corpus callosum. Neuropathol Appl Neurobiol 1980;6:415–20 [DOI] [PubMed] [Google Scholar]
- 35. Ozawa K, Suchanek G, Breitschopf H, et al. Patterns of oligodendroglia pathology in multiple sclerosis. Brain 1994;117:1311–22 [DOI] [PubMed] [Google Scholar]
- 36. Lucchinetti C, Brück W, Parisi J, et al. Heterogeneity of multiple sclerosis lesions: Implications for the pathogenesis of demyelination. Ann Neurol 2000;47:707–17 [DOI] [PubMed] [Google Scholar]
- 37. Lassmann H. Pathology of inflammatory diseases of the nervous system: Human disease versus animal models. Glia 2020;68:830–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luchicchi A, Hart B, Frigerio I, et al. Axon-myelin unit blistering as early event in MS normal appearing white matter. Ann Neurol 2021;89:711–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Teo W, Caprariello AV, Morgan ML, et al. Nile Red fluorescence spectroscopy reports early physicochemical changes in myelin with high sensitivity. Proc Natl Acad Sci U S A 2021;118:e2016897118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Elliot C, Momayyezsiahkal P, Arnold DL, et al. Abnormalities in normal-appearing white matter from which multiple sclerosis lesions arise. Brain Commun 2021;3:fcab176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bando Y, Nomura T, Bochimoto H, et al. Abnormal morphology of myelin and axon pathology in murine models of multiple sclerosis. Neurochem Int 2015;81:16–27 [DOI] [PubMed] [Google Scholar]
- 42. Stassart RM, Möbius W, Nave K-A, et al. The axon-myelin unit in development and degenerative disease. Front Neurosci 2018;12:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Snaidero N, Möbius W, Czopka T, et al. Myelin membrane wrapping of CNS axons by PI(3,4,5)P3-dependent polarized growth at the inner tongue. Cell 2014;156:277–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baraban M, Mensch S, Lyons DA.. Adaptive myelination from fish to man. Brain Res 2016;1641:149–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Romanelli E, Merkler D, Mezydlo A, et al. Myelinosome formation represents an early stage of oligodendrocyte damage in multiple sclerosis and its animal model. Nat Commun 2016;7:13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sobel RA. Ephrin A receptors and ligands in lesions and normal-appearing white matter in multiple sclerosis. Brain Pathol 2005;15:35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Howell OW, Rundle JL, Garg A, et al. Activated microglia mediate axo-glial disruption that contributes to axonal injury in multiple sclerosis. J Neuropathol Exp Neurol 2010;69:1017–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Moll NM, Rietsch AM, Thomas S, et al. Multiple sclerosis normal-appearing white matter: Pathology-imaging correlations. Ann Neurol 2011;70:764–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hiramatsu N, Chiang W-C, Kurt TD, et al. Multiple mechanisms of unfolded protein response-induced cell death. Am J Pathol 2015;185:1800–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Costa-Mattioli M, Walter P.. Proteostasis. The integrated stress response: From mechanism to disease. Science 2020;368:eaat5314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ohri SS, Maddie MA, Zhao Y, et al. Attenuating the endoplasmic reticulum stress response improves functional recovery after spinal cord injury. Glia 2011;59:1489–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Volpi VG, Touvier T, D’Antonio M.. Endoplasmic reticulum protein quality control failure in myelin disorders. Front Mol Neurosci 2016;9:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lin W, Stone S.. Unfolded protein response in myelin disorders. Neural Regen Res 2020;15:636–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Shi M, Chai Y, Zhang J, Chen X.. Endoplasmic reticulum stress-associated neuronal death and innate immune response in neurological diseases. Front Immunol 2021;12:794580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bettigole SE, Glimcher LH.. Endoplasmic reticulum stress in immunity. Annu Rev Immunol 2015;33:107–38 [DOI] [PubMed] [Google Scholar]
- 56. Smith JA. Regulation of cytokine production by the unfolded protein response; implications for infection and autoimmunity. Front Immunol 2018;422:1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. García-González P, Cabral-Miranda F, Hetz C, et al. Interplay between the unfolded protein response and immune function in the development of neurodegenerative diseases. Front Immunol 2018;9:2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lei Z, Yue Y, Stone S, et al. NF-κB Activation accounts for the cytoprotective effects of PERK activation on oligodendrocytes during EAE. J Neurosci 2020;40:6444–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kirby L, Jin J, Cardona JG, et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat Commun 2019;10:3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Li Y, Jiang W, Niu Q, et al. eIF2α-CHOP-BCl-2/JNK and IRE1α-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of Newcastle disease virus. Cell Death Dis 2019;10:891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Meier U-C, Cipian RC, Karimi A, et al. Cumulative roles for Epstein-Barr virus, human endogenous retroviruses, and human herpes virus-6 in driving an inflammatory cascade underlying MS pathogenesis. Front Immunol 2021;12:757302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bjornevik K, Cortese M, Healy BC, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022;375:296–301 [DOI] [PubMed] [Google Scholar]
- 63. Psenicka MW, Smith BC, Tinkey RA, et al. Connecting neuroinflammation and neurodegeneration in multiple sclerosis: Are oligodendrocyte precursor cells a nexus of disease? Front. Front Cell Neurosci 2021;15:654284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Maeda A, Sobel RA.. Matrix metalloproteinases in the normal human central nervous system, microglial nodules and multiple sclerosis lesions. J Neuropathol Exp Neurol 1996;55:300–9 [DOI] [PubMed] [Google Scholar]
- 65. Singh S, Metz I, Amor S, et al. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol 2013;125:595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ramaglia V, Hughes TR, Donev RM, et al. C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc Natl Acad Sci U S A 2012;109:965–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Neher JJ, Cunningham C.. Priming microglia for innate immune memory in the brain. Trends Immunol 2019;40:358–74 [DOI] [PubMed] [Google Scholar]
- 68. Kim BS. Review: Excessive innate immunity steers pathogenic adaptive immunity in the development of Theiler’s virus-induced demyelinating disease. Int J Mol Sci 2021;22:5254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ludwin SK, Johnson ES.. Evidence for a “dying-back” gliopathy in demyelinating disease. Ann Neurol 1981;9:301–5 [DOI] [PubMed] [Google Scholar]
- 70. Matsushima GK, Morell P.. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol 2001;11:107–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Praet J, Guglielmetti C, Berneman Z, et al. Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neurosci Biobehav Rev 2014;47:485–505 [DOI] [PubMed] [Google Scholar]
- 72. Suzuki K, Kikkawa Y.. Status spongiosus of CNS and hepatic changes induced by cuprizone (biscyclohexanone oxalyldihydrazone). Am J Pathol 1969;54:307–25 [PMC free article] [PubMed] [Google Scholar]
- 73. Yajima K, Suzuki K.. Ultrastructural changes of oligodendroglia and myelin sheaths induced by ethidium bromide. Neuropathol Appl Neurobiol 1979;5:49–62 [DOI] [PubMed] [Google Scholar]
- 74. Zhang F, Yao S-Y, Whetsell WO Jr, et al. Astrogliopathy and oligodendrogliopathy are early events in CNS demyelination. Glia 2013;61:1261–73 [DOI] [PubMed] [Google Scholar]
- 75. Radecki DZ, Johnson EL, Brown AK, et al. Corticohippocampal dysfunction in the OBiden mouse model of primary oligodendrogliopathy. Sci Rep 2018;8:16116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Allen IV, McKeown SR.. A histological, histochemical and biochemical study of the macroscopically normal white matter in multiple sclerosis. J Neurol Sci 1979;41:81–91 [DOI] [PubMed] [Google Scholar]
- 77. Matsuo A, Lee GC, Takami K, et al. Unmasking of an unusual myelin basic protein epitope during the process of myelin degeneration in humans: A potential mechanism for the generation of autoantigens. Am J Pathol 1997;150:1253–66 [PMC free article] [PubMed] [Google Scholar]
- 78. Winger RC, Zamvil SS.. Antibodies in multiple sclerosis oligoclonal bands target debris. Proc Natl Acad Sci U S A 2016;113:7696–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Caprariello AV, Rogers JA, Morgan ML, et al. Biochemically altered myelin triggers autoimmune demyelination. Proc Natl Acad Sci U S A 2018;115:5528–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sen MK, Almuslehi MSM, Shortland PJ, et al. Revisiting the pathoetiology of multiple sclerosis: Has the tail been wagging the mouse? Front Immunol 2020;11:572186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. de la Fuente AG, Queiroz RML, Ghosh T, et al. Changes in the oligodendrocyte progenitor cell proteome with aging. Mol Cell Proteomics 2020;19:1281–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nicaise AM, Wagstaff LJ, Willis CM, et al. Cellular senescence in progenitor cells contributes to diminished remyelination potential in progressive multiple sclerosis. Proc Natl Acad Sci U S A 2019;116:9030–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Morgan AA, Rubenstein E.. Proline: The distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS One 2013;8:e53785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Jahn O, Tenzer S, Werner HB.. Myelin proteomics: Molecular anatomy of an insulating sheath. Mol Neurobiol 2009;40:55–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Steele JR, Italiano CJ, Phillips CR, et al. Misincorporation proteomics technologies: A review. Proteomes 2021;9:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rivas A, Vidal RL, Hetz C.. Targeting the unfolded protein response for disease intervention. Expert Opin Ther Targets 2015;19:1203–18 [DOI] [PubMed] [Google Scholar]
- 87. Morris G, Puri BK, Walder K, et al. The endoplasmic reticulum stress response in neuroprogressive diseases: Emerging pathophysiological role and translational implications. Mol Neurobiol 2018;55:8765–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Almanza A, Carlesso A, Chintha C, et al. Endoplasmic reticulum stress signalling – From basic mechanisms to clinical applications. FEBS J 2019;286:241–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Titus HE, Chen Y, Podojil JR, et al. Pre-clinical and clinical implications of “inside-out” vs. “outside-in” paradigms in multiple sclerosis etiopathogenesis. Front Cell Neurosci 2020;14:599717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Nutma E, Marzin MC, Cillessen SAGM, et al. Autophagy in white matter disorders of the CNS: Mechanisms and therapeutic opportunities. J Pathol 2021;253:133–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chen Y, Kunjamma RB, Weiner M, et al. Prolonging the integrated stress response enhances CNS remyelination in an inflammatory environment. eLife 2021;10:e65469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.