

Graphical abstract

Keywords: Fentanyl, µ-Opioid receptor, Molecular dynamics simulations, Metadynamics, Structure–activity relationship
Abstract
Fentanyl and its analogs are selective agonists of the µ-opioid receptor (MOR). Among novel synthetic opioids (NSOs), they dominate the recreational drug market and are the main culprits for the opioid crisis, which has been exacerbated by the COVID-19 pandemic. By taking advantage of the crystal structures of the MOR, several groups have investigated the binding mechanism of fentanyl, but have not reached a consensus, in terms of both the binding orientation and the fentanyl conformation. Thus, the binding mechanism of fentanyl at the MOR remains an unsolved and challenging question. Here, we carried out a systematic computational study to investigate the preferred fentanyl conformations, and how these conformations are being accommodated in the MOR binding pocket. We characterized the free energy landscape of fentanyl conformations with metadynamics simulations, and compared and evaluated several possible fentanyl binding conditions in the MOR with long-timescale molecular dynamics simulations. Our results indicate that the most preferred binding pose in the MOR binding pocket corresponds well with the global minimum on the energy landscape of fentanyl in the absence of the receptor, while the energy landscape can be reconfigured by modifying the fentanyl scaffold. The interactions with the receptor may stabilize a slightly unfavored fentanyl conformation in an alternative binding pose. By extending similar investigations to fentanyl analogs, our findings establish a structure–activity relationship of fentanyl binding at the MOR. In addition to providing a structural basis to understand the potential toxicity of the emerging NSOs, such insights will contribute to developing new, safer analgesics.
1. Introduction
Selective agonists of the µ-opioid receptor (MOR), such as morphine, codeine, and meperidine, have a long history of being used as analgesics to relieve pains [1]. Based on meperidine, which shares the piperidine core with morphine, Janssen and his colleagues discovered fentanyl in 1960 [2]. Fentanyl is easy to synthesize, and much more potent in analgesic effect, but has higher addiction liability than morphine [1], [2], [3]. Fentanyl and its analogs dominate the current recreational drug market [4] and are the main culprits for the ongoing and growing opioid crisis [5], which has been exacerbated by the COVID-19 pandemic [6].
MOR, a class A G-protein coupled receptor (GPCR), couples with both G-protein and β-arrestin to regulate downstream signaling pathways. The mechanisms of action of analgesics at the MOR have been extensively studied for decades (reviewed in [1]). However, whether and how the functional selectivity is involved in inducing the side effects of fentanyl and analgesics in general, such as respiratory suppression, is still being actively studied and debated [7], [8], [9], [10]. In addition, fentanyl was found to access the binding pocket of the MOR by partitioning into the membrane first, and thus has an anomalous pharmacology compared to morphine [11].
Early computational studies, based on homology modeling, shed light on the potential binding pose of fentanyl at the MOR [12], [13]. In recent years, high-resolution structures of the MOR have been acquired by both x-ray crystallography [14], [15] and cryo-electron microscopy (cryo-EM) [16], for an inactive conformation of the receptor bound with a morphinan antagonist, β-FNA [15], and active conformations bound with agonists BU72 and DAMGO [14], [16]. These structures have been leveraged to investigate the binding mechanisms, and related MOR conformations, of the opioids having therapeutic implications, such as methadone, morphine, and TRV130 [17], [18].
By taking advantage of these structures, several groups have computationally investigated the binding mechanism of fentanyl and its analogs at the MOR, but they have not reached a consensus [19], [20], [21], [22], [23], [24], [25]. While these studies commonly found that the positively charged nitrogen on the piperidine ring forms a salt bridge with Asp3.32 (superscripts denote Ballesteros-Weinstein numbering [26]) in the binding site of the MOR, they proposed different binding modes of fentanyl and its analogs. Specifically, based on the crystal structure of the MOR in an active conformation, Vo et al. carried out extensive molecular dynamics (MD) simulations recently and proposed that, in addition to forming a salt-bridge with Asp3.32 in the orthosteric binding site (OBS), fentanyl can move deeper toward the intracellular side with its positively charged nitrogen forming a hydrogen bond to His6.52 [24]. They found that the carbonyl and aniline groups of fentanyl point towards the extracellular portions of transmembrane segments (TMs) 2 and 7, an orientation of which is similar to that proposed previously by Lipinski et al. and Eshleman et al. [20], [24], [25] (defined in this study as the phenyl-piperidine-amide (FPA) orientation in Fig. 1C). However, using long unbiased MD simulations, Ricarte and colleagues identified an opposite orientation of fentanyl in the binding site of the MOR, with the amide and aniline groups located near the middle portions of TMs 3 and 6 [23] (defined in this study as the amide-piperidine-phenyl (APF) orientation in Fig. 1B). The Ricarte pose is similar to that previously proposed by de Waal et al., but in a deeper position in the binding pocket [21], [23]. In addition, the conformations of the bound fentanyl are also different in these studies. The amide bond in the propanamide moiety of fentanyl was in the cis-amide configuration in the Ricarte, de Waal, Lipinski, and Eshleman poses [20], [21], [23], [25], and trans-amide in the Vo and Podlewska poses [22], [24] (see the cis- and trans-amide configurations in Fig. 1D). Together, these studies proposed varied binding poses of fentanyl at the MOR, not only in opposite orientations and different positions in the binding site, but also the divergent fentanyl conformations. Thus, the binding mechanism of fentanyl at the MOR remains an unsolved and challenging question.
Fig. 1.

Two possible binding orientations of fentanyl in the orthosteric binding site of μ-opioid receptor. (A) An overview of a fentanyl bound MOR-Gi complex. The binding pocket is highlighted with a dashed box. (B) and (C) are schematic orientations of how a bound fentanyl can be oriented in the MOR binding pocket. F, P, and A stand for phenyl, piperidine, and amide moieties of fentanyl, respectively. Two binding orientations, APF (B) and FPA (C), have been proposed in previous studies (see text). In addition, the rotation of the amide bond results in cis-amide and trans-amide configurations of fentanyl, as shown in panel D.
The 3- and 4-positions on the piperidine ring of the fentanyl scaffold have been exploited to develop more potent fentanyl analogs, including carfentanil, 3-methylfentanyl, and lofentanil [2], [27] (Fig. S1). In particular, carfentanil (4-carbomethoxy fentanyl) has evolved from being used as an animal anesthetic to a deadly abused drug [28], and even has the potential of being used as a chemical weapon [29]. Intriguingly, with the same addition of a 3-methyl moiety, 3-methylfentanyl has much higher potency (as high as ∼200-fold, (3R,4S)-isomer) than fentanyl [30], [31], while lofentanil (i.e., 3-methyl carfentanil) only has slightly better affinity at the MOR than carfentanil [32], [33].
Lofentanil was originally referred to as the compound having the (3R,4S) chirality, which has three other possible stereoisomers, (3S,4R), (3S,4S), and (3R,4R) [34], [35] (Fig. S1). The two cis ((3R,4S)- and (3S,4R)-) isomers have been well characterized pharmacologically. While the (−)-cis-isomer has much higher binding affinity at the MOR than the (+)-cis-isomer, especially in the presence of NaCl (>100-fold), the affinity of the racemic mixture of (+)-trans and (−)-trans-isomers is in between those of the two cis isomers [35], [36], [37], [38] (Note the cis and trans configurations referred to here are that of the 3- and 4-modifications but not the amide bond mentioned above). In addition, the (−)-cis-isomer of lofentanil has been found to be the most active and long-acting in both pharmacological and in vivo analgesic tests, but the (+)-cis-isomer was reported to be the first fentanyl derivative having short-acting antagonistic properties [39]. However, the absolute configurations of (−)-cis- and (+)-cis-isomers, i.e., which one is (3R,4S)- and which one is (3S,4R)-configuration, have never been definitively determined by X-ray crystallography.
For 3-methylfentanyl, it has been determined by crystallography that (+)-cis-isomer corresponds to the (3R,4S)-configuration [40], while (−)-cis-(3S,4R)-3-methylfentanyl has drastically lower affinity (>1000 fold) than its active (+)-cis-(3R,4S)-isomer at the MOR [31], [40]. In addition, a close analog of 3-methylfentanyl, ohmefentanyl (Fig. S1), was found to have a similar pharmacological profile, with its (3S,4R)-isomers having lower affinity (>200-fold) than its (3R,4S)-isomers [41].
In this study, we first carried out systematic characterizations of the possible conformations of fentanyl and a few selected analogs with modifications at the 3- and/or 4-positions on the piperidine ring. In the context of the revealed energy landscapes of these ligand conformations in the absence of the receptor, based on the cryo-EM structure of the MOR in complex with the Gi protein [16], we performed long-timescale MD simulations to identify the most favored binding poses of fentanyl and its analogs at the MOR, by comparing the stability and energetics of their possible binding orientations and conformations. We found potential impacts of modifying the fentanyl scaffold on both conformational rigidity and binding potency. We further compared MOR conformations, stabilized by either fentanyl or by DAMGO, to reveal the initial clues about the structural basis of their potentially divergent signaling preferences.
2. Materials and methods
2.1. Ligand preparation
The 3D models of fentanyl and its analogs were prepared using Ligprep of Schrodinger (version 2020-3). The protonation state of each ligand was determined by Epik of Schrodinger (version 2020-3) in pH 7.0 ± 2.0 condition, which protonates the nitrogen in the piperidine ring of the fentanyl scaffold to the positively charged state. We protonate the nitrogen in the direction observed in the crystal structure of 3-methylfentanyl [40].
The accurate force field parameters of opioid drugs are critical for their modeling studies [42], [43]. In this study, we used both OPLS3 and OPLS4 force fields, which have been extensively parametrized for drug-like small compounds and cover much larger chemical space than other available force fields [44], [45]. Specifically, the force field parameters for fentanyl and its analogs were first assigned by atom typing, then the parameters requiring further optimization were identified by the Force Field Builder (see Fig. S1). These parameters are optimized quantum mechanically through the Jaguar program of Schrodinger [46].
2.2. Metadynamics simulations to characterize the free energy landscape
For fentanyl and each of its analogs, we immersed the compound in a water (or octanol) box with a dimension of 30 × 30 × 30 Å3, which also included 0.15 M NaCl. The total number of atoms of each system is ∼2,300.
Two dihedral angles of the fentanyl scaffold were selected as collective variables (CV) (Fig. 2C). The dihedral angle between amide carbonyl and aniline was defined as CV1, and that between aniline and piperidine as CV2. In the metadynamics simulations, the height of the biased Gaussian potential was 0.01 kcal/mol, and the window width was 5 degrees for the CVs. Metadynamics simulations were performed with Desmond [47] (version 2020-3 with the OPLS3e force field [44] and version 2021-2 with the OPLS4 force field [45], Schrodinger LLC, New York, NY, USA). For each compound or isomer, 10 replicate runs were performed – 5 runs were started from each of the CV1 trans and cis configurations. The length of each run for the fentanyl and carfentanil systems was 100 ns. In order to reach convergence, the runs for the (3S,4R)-lofentanil and (3R,4S)-lofentanil systems were prolonged to 400 ns each. The free energy surface (FES), defined by CV1 and CV2, was rebuilt with the Metadynamics Analysis utility of Schrodinger, with in-house modifications to flexibly increase the number of bins. To integrate the results for each compound or isomer, we carried out 100 bootstrap samplings of the FES from the pool of 10 replicate runs. The averages are shown in Figs. 2, 3, S2, S3 and S4, while the standard deviations are shown in Fig. S5. The FES was normalized by defining the lowest energy point on the FES as 0.
Fig. 2.

Metadynamics simulations of fentanyl and carfentanil reveal their possible conformations. The free energy surfaces (FESs) reconstructed from the metadynamics results are shown in panels A-B for the indicated ligands. For each FES, we identified four minima, labelled as TL, TH, CL, and CH for fentanyl, TL1, TL2, CL1, and CL2 for carfentanil. The dihedral angle of the amide bond is defined as CV1, and that between aniline and piperidine as CV2, as shown in panel C. The stable states of fentanyl and carfentanil in the MOR binding pocket are CL and CL1, respectively (Fig. 6). The minimum free energy paths (MFEP) between the TL and CL states for fentanyl and the TL1 and CL1 states for carfentanil are plotted with dotted curves on each FES. The conformations corresponding to the energy minima are shown in panel C for fentanyl, and panel D for carfentanil.
Fig. 3.

Metadynamics simulations of 3-methylfentanyl and lofentanil demonstrate the impact of 3-methyl modification. The FESs reconstructed from the metadynamics results are shown in panels A-B for the ligands in the indicated configurations. For each FES, we identified four minima, labelled as TL, TH, CL, and CH for 3-methylfentanyl, TL1, TL2, CL1, and CL2 for lofentanil. The stable state of the cis-isomers of lofentanil in the MOR binding pocket is CL1 (Fig. 6). The MFEP between the TL1 and CL1 states for the cis-isomers of lofentanil are plotted with dotted curves on each FES. The conformations corresponding to the energy minima are shown in panels C and D for representative 3-methylfentanyl and lofentanil isomers, respectively.
We implemented Dijkstra’s algorithms [48] to identify the minimum free energy path (MFEP) between each pair of local minima. Briefly, we choose one of the local minima as the starting vertex and identify the neighboring vertex that has the lowest energy as the next step on the path and repeat this process. To lead the path towards the targeted minimum, the path is not allowed to revisit the same vertex. The process can generate multiple paths, when more than one neighboring vertex have the same lowest energy. Energy barrier is defined as the difference between the starting vertex and the vertex with the highest energy on the path. The MFEP is identified by finding the path with the lowest energy barrier between two minima. While the MFEP between two minima is unique, the value of energy barrier depends on the starting vertex (Figs. 2 and S2, Tables S1 and S2).
2.3. Energy minimization of the ligand models closest to the minima
By scanning all frames of metadynamics simulation trajectories, the structures with the shortest Euclidean distance (of CV1 and CV2) to the local minima were extracted. Ligands were separated from the extracted frames and energy minimized through Prime of Schrodinger (Version 2021-1) with the VSGB implicit solvation model to mimic either water environment (dielectric constant = 80) or octanol solvent (dielectric constant = 10.3) [49]. We used the default options except for increasing the maximum number of iterations to 100.
2.4. Molecular modeling of the human MOR in an active conformation
From the two available MOR structures in active states, as the structure 5C1M was stabilized by “nanobody 39”, which might introduce some small artifacts, we chose the cryo-EM structure of the mouse MOR (mMOR) in complex with the Gi protein (PDB code: 6DDF) as the main template to build our human MOR (hMOR)-Gi models. In the structure 6DDF, some MOR residues are missing in the N terminus and at H8. We added 8 missing N terminal residues (residues 59–66 in hMOR numbering), present in another MOR structure in an active conformation (PDB code: 5CM1), and the missing H8 residues (residues 348–354 in hMOR numbering), from the MOR structure (PDB code: 4DKL), to the main template using homology modelling with Modeller (version 9.24) [50]. In this modeling process, the structure 5CM1 also provided some missing sidechains in the structure 6DDF, which has relatively low resolution. The added N-terminal residues are necessary to prevent the entry of lipid molecule to the binding pocket in the following molecular dynamics (MD) simulations. In addition, the divergent residues between the mMOR and hMOR in the main template are converted to the aligned human residues (V681.30I, V1894.45I, I308EL3V). No change or addition was made on the Gi protein template. The hMOR model with the lowest DOPE score was selected for following studies.
2.5. Establish the defined binding modes with molecular docking and manual adjustments
The selected hMOR model was further processed through the Protein Preparation Wizard in Maestro of Schrodinger. Hydrogen bond assignment was optimized with PROPKA [51] at pH 7.0. Energy minimization of the structure was conducted with the default constraint of 0.3 Å heavy atoms root-mean-square deviation (RMSD). Using the induced-fit docking protocol implemented in Schrodinger [52], we first docked fentanyl in the binding site of the prepared hMOR model. The center of docking box was determined by the center of mass of the ligand bound in the structure 6DDF, DAMGO. We applied a restraint to filter the poses that have the protonated N atom on the piperidine ring forming an ionic interaction with Asp3.32. We found both APF and FPA (Fig. 1B, C) poses in the docking results, and selected or manually adjusted the CV1 dihedral angle, so to acquire both the trans-amide and cis-amide conformations of each pose. Based on these hMOR models bound with fentanyl in different poses and conformations, we manually modified the fentanyl scaffold to add the extra moieties with the 3D builder in Maestro of Schrodinger (version 2020-3), and established the corresponding hMOR models bound with carfentanil, (3S,4R)-lofentanil, or (3R,4S)-lofentanil.
2.6. Molecular dynamics simulation protocol
The hMOR models with the defined binding poses of fentanyl and its analogs were further processed to build the simulation systems with the Desmond System Builder of Schrodinger suites (version 2020-3 with OPLS3e force field and version 2021-2 with OPLS4 force field). Briefly, the MOR-Gi complex models were immersed in explicit 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine lipid bilayer (POPC). The simple point charge (SPC) water model was used to solvate the system, the net charge of the system was neutralized by Cl- ions, and then 0.15 M NaCl was added. Residues Asp1162.50 and Asp1663.49 are protonated to their neutral forms as assumed in the active state of rhodopsin-like GPCRs [53], and we manually adjusted the His2996.52 protonation form to either HIE or HID as well (see Results). We additionally protonated Asp3427.57, which is positioned in between TM7 and H8 and was predicted to be in a neutral protonated state in all MOR structures (6DDF, 5C1M, and 4DKL) by PROPKA. Our test run with Asp3427.57 deprotonated shows that the negative charge at this location likely destabilized the local interaction network with Gi. The process resulted in a system with a dimension of 106x117x151 Å3 and total number of atoms of ∼190,000. The initial parameters for carfentanil and lofentanil were further optimized by the force field builder of the Schrodinger Suites (version 2020-3 with OPLS3e force field and version 2021-2 with OPLS4 force field).
Desmond MD systems (D. E. Shaw Research, New York, NY) was used for the MD simulations. Similar to our previous simulation protocols used for GPCRs [54], the system was initially minimized and equilibrated with restraints on the ligand heavy atoms and protein backbone atoms. The NPγT ensemble was used with constant temperature maintained with Langevin dynamics. Specifically, 1 atm constant pressure was achieved with the hybrid Nose-Hoover Langevin piston method on an anisotropic flexible periodic cell with a constant surface tension (x-y plane). In the production runs at 310 K, all restraints on the hMOR were released; however, to retain the integrity of the Gi protein, while allowing adequate flexibility to interact with the receptor, the heavy atoms of residues 46–55, 182–189, and 230–242 of Gα, and the entire Gβ and Gγ subunits, were restrained with a force constant of 1 kcal/mol/Å.
For each condition, we collected at least three trajectories starting from different random number seeds. Overall, more than 200 trajectories, with an aggregated simulated time of more than 300 μs, were collected (Table S3).
2.7. Conformational analysis
Dihedral angles and distances were calculated with VMD-python (version 3.0.6) [55]. We used the Protein Interaction Analyzer [56], [57] in analyzing the MD simulation results of the MOR. For analysis of coarse-grained interaction network of the hMOR, we defined the following structural elements: TM1e (the extracellular section (e) of TM1, residues 68–74), TM1m (the middle section (m) of TM1, residues 75–84),TM1i (the intracellular section (i) of TM1, residues 85–97), TM2i (residues 105–117), TM2m (residues 118–126), TM2e (residues 127–131), TM3e (residues 140–148), TM3m (residues 149–157), TM3i (residues 158–172), TM4i (residues 183–193), TM4m (residues 194–200), TM4e (residues 201–207), TM5e (residues 229–240), TM5m (residues 241–248), TM5i (residues 249–259), TM6i (residues 275–293), TM6m (residues 294–301), TM6e (residues 302–307), TM7e (residues 314–323), TM7m (residues 324–331), and TM7i (residues 332–341).
We assembled the representative ensembles of frames for analysis by randomly selecting 5,000 frames with replacement (bootstrapping) for each condition from all the trajectories of that condition, which was repeated 10 times. The same datasets were used for all the geometric calculations and analyses. The presented results are the average of the 10 bootstrap samplings.
Pairwise Root Mean Square Deviations (RMSDs) can avoid the bias of a single reference. To evaluate the stability of ligand binding, we aligned all possible pairs of the representative MD frames for a given condition to each other according to the Cα atoms of the binding site residues of hMOR: Thr1222.56, Phe1252.59, Gln1262.60, Asn1292.63, Trp135EL1.50, Val1453.28, Ile1463.29, Asp1493.32, Tyr1503.33, Met1533.36, Asp218EL2.49, Cys219EL2.50, Trp2956.48, Ile2986.51, His2996.52, Trp3207.35, His3217.36, Ile3247.39, Gly3277.42, and Tyr3287.43, then calculated the RMSD based on the ligand heavy atoms. The values reported in Fig. 3, Fig. 4 are the averages and standard deviations of each condition.
Fig. 4.

APFDC and FPADT are the more stable poses in each orientation. The average pairwise ligand RMSDs for each indicated condition are shown for the APF and FPA orientations in panels A and B, respectively. The darker colored bars are OPLS3e results, and those of the lighter colored bars are OPLS4 results. The APFDC and FPADT are the more stable conditions for APF and FPA orientations, respectively, and the fentanyl binding poses in these two conditions are demonstrated in panels C and D.
2.8. MM/GBSA calculation
Binding free energies between the bound ligands and the hMOR were estimated with the Molecular mechanics/generalized Born surface area calculations (MM/GBSA) method, using the same force field in the MD simulations for the proteins and ligands, but with VSGB2.1 solvation model [49]. We extracted frames every 3 ns from the production runs to carry out the MM/GBSA calculations using the thermal_mmgbsa.py script from the Schrodinger suite. The binding free energies for each condition were the averages of the selected frames.
3. Results
Previous MD simulation studies showed that the amide bond of fentanyl could be stable in either the trans or cis configuration (Fig. 1D) in the MOR binding pocket, without any reported transition between these two conformations [20], [21], [22], [23], [24]. These results suggest the transition between the trans- and cis-amide configurations may have a relatively high energy barrier to overcome in the MD simulations, but these studies did not evaluate which conformation has lower energy. While the trans configuration of the amide bond in proteins (peptide bond) usually has lower energy, interestingly in a crystal structure of a computationally designed fentanyl binding protein, the amide bond of the bound fentanyl molecule is in the cis-amide configuration [58].
Thus, to thoroughly understand the energy landscape of the fentanyl conformation and to provide reasonable starting conformations of these ligands in the unbiased MD simulations of ligand-receptor complexes, we first carried out metadynamics simulations with a fentanyl (or its analogs) molecule immersed in a water box (see Methods).
3.1. Fentanyl and its analogs favor the cis-amide configuration
Metadynamics simulation is an enhanced sampling method that can efficiently overcome energy barriers by adding time-dependent bias potential acting on selected collective variables (CVs) [59]. In the metadynamics simulations of this study, we defined two CVs, CV1 is the dihedral angle of the amide bond, while CV2 is the dihedral angle between aniline and piperidine moieties of the fentanyl scaffold (Fig. 2C).
As expected, in our metadynamics simulation results of fentanyl plotted on a 2D free energy surface (FES) (Fig. 2A), we observed that CV1 has two energy minima, near 0° (trans-amide, referred to as the “T” state below) and 180° (cis-amide, the “C” state). Interestingly, CV2 has two minima as well, near −80° (denoted as the “L” state) and 105° (the “H” state). The examination of the FES indicates that the C state has more than 2 kcal/mol lower energy than the T state, while the L state has lower energy and a wider distribution than the H state (Fig. 2A and Table S1). Thus, on the FES determined by CV1 and CV2, the C state of CV1, in combination with L state of CV2 (referred to as CL), has the globally lowest energy (Fig. 2A and Table S1). This preference for the cis-amide configuration was also found previously in fentanyl following a quantum mechanical approach [43].
Using the Dijkstra’s algorithm, we identified the minimum free energy paths (MFEP) [60] between the energy minima and calculated the height of the energy barrier along each of such paths on the FES (see Methods). The results show that the transition between the CL and CH states has a significantly lower energy barrier than other transitions, while the C to T transitions (both of the CH to TH and CL to TL MFEPs) have energy barriers of more than 10 kcal/mol (Fig. 2A and Table S2). While this manuscript was being prepared, an updated OPLS force field (OPLS4) became available [45]. Using OPLS4, we carried out the same set of metadynamics simulations and analyses and found similar results and conclusions as those described above using OPLS3e (Fig. S2, Tables S1 and S2).
An interesting finding by Kelly et al. indicates that fentanyl can enter the MOR binding pocket from the lipid membrane phase [11]. We carried out the metadynamics simulations of fentanyl immersed in an octanol box using OPLS4, which mimics the membrane environment due to its amphiphilic nature [61]. The resulting energy landscape is very similar to that derived from simulations in a water box (Fig. S3 Tables S1 and S2).
3.2. Fentanyl can be stable in two opposite binding orientations in the MOR binding pocket
To comprehensively evaluate the possible binding poses of fentanyl in the active conformation of the MOR, we first docked fentanyl in the cryo-EM structure of the MOR-Gi complex (PDB code: 6DDF) [16]. By selecting and then manually adjusting the docked poses, we then established fentanyl poses in both the APF and FPA orientations and in both trans- and cis-amide configurations, as proposed by previous studies [20], [21], [22], [23], [24] (Fig. 1B-D). In addition, Vo et al. showed that the protonation state of the His2996.52 might have a significant impact on the fentanyl binding pose [24], which prompted us to consider two neutral protonation states (HIE and HID) of His2996.52 in our study as well. Thus, we built the fentanyl bound MOR-Gi complex models in 8 different conditions (Table S3). For example, the APFDT condition has the bound fentanyl molecule in the APF orientation and the trans-amide (subscript “T”) configuration, with His6.52 of the MOR in the HID state (subscript “D”). For each condition, we immersed the resulting models in the explicit lipid bilayer simulation environment, and collected multiple prolonged MD simulation trajectories, using either or both OPLS3e and OPLS4 force fields (Table S3). For the condition that we have both OPLS3e and OPLS4 data, we did not find any noticeable difference resulting from different force fields. We analyzed the data separately but drew the conclusions by integrating all the results.
To evaluate the stability of ligand binding, we calculated the pairwise ligand RMSDs for each condition (see Methods). Our results show that fentanyl in the APF cis-amide conditions (both APFEC and APFDC, “E” and “D” in the subscripts represent the His2996.52 in its HIE and HID forms, respectively) are very stable with the pairwise ligand RMSDs of ∼1.0 Å (Fig. 4A). Interestingly, the bound fentanyl in the APFET condition has a tendency to transition to the cis-amide configuration (Fig. S6), therefore this condition is not considered to be stable for further analysis. We then carried out MM/GBSA binding energy calculations for the same set of representative MD frame ensembles of each condition. The results show that the APFDC condition has the most favored binding energy, and the APFEC and APFDT conditions have 2.5 and 8.1 kcal/mol higher energies (ΔΔG), respectively. Thus, both the pairwise ligand RMSD and MM/GBSA results are consistent with the findings from our metadynamics simulations (Fig. 2A and Table S1) in which we found that the cis-amide configuration (C state) has lower energy. When comparing the APFEC and APFDC conditions, we found the bottom of the binding pocket near His2996.52 is less dynamic in APFDC than in APFEC. In particular, the distribution of His2996.52 χ1 rotamer in APFDC is tighter than in APFEC, which also results in a slightly different orientation of Trp2956.48 χ2 (Fig. S7).
In contrast to the APF conditions, among the FPA conditions of fentanyl, only FPADT appears to be a reasonably stable condition with a pairwise ligand RMSD of 1.8 Å, while the other three FPA conditions have drastically higher RMSDs (Fig. 4B). This trend is reflected on the MM/GBSA calculation results in which the FPAEC, FPADC, and FPAET conditions have more than 6 kcal/mol higher energies than FPADT. However, the relative stability of fentanyl in FPADT is surprising given our findings from the metadynamics simulations that show the trans-amide configuration (T state) has higher energy (Fig. 2A). From our inspection of the FPADT condition, we found that the bound fentanyl molecule in the trans-amide configuration forms a hydrogen bond (H-bond) interaction with the sidechain of either Gln1262.60 or Asn1292.63 (Fig. 4D and S6), which compensates the less favored trans-amide configuration. Similar to what we observed between the APFEC and APFDC conditions, the bottom of the binding pocket in FPAET is more dynamic than in FPADT, with the His2996.52 χ1 of the former in wider distribution (Fig. S7).
Interestingly, even though APF is a more stable pose than FPA, the carbonyl of the amide moiety does not directly interact with any MOR residue. In our analysis of the waters in the binding pocket, we found that an average of 3 water molecules are within 5 Å of the carbonyl oxygen in both of APFEC and APFDC conditions, mediating an H-bond network between the carbonyl and the sidechain of Tyr1503.33 (Fig. S8A-C). In the comparison of the FPAET and FPADT conditions, we found that FPAET on average has more than one water molecule within 5 Å of the carbonyl oxygen than FPADT (Fig. S8D-F), suggesting that fentanyl is more exposed to water in the FPAET condition.
In summary, we found that fentanyl can be stable in both APF and FPA orientations, with the APFC (combining APFDC and APFEC, which are similarly stable) and FPADT conditions being more favored than the other conditions in each orientation. Our results also show that APFC is more stable than FPADT. The detailed contact frequency analysis (see Method) indicates that the bound fentanyl molecules in both APFC and FPADT conditions form the ionic interaction with Asp1493.32, and commonly contact many other residues (Table 1). From previously reported mutagenesis work relevant to fentanyl binding at the MOR (Table S4), we found mutations of the residues that are not directly involved in interacting with either APF or FPA pose, Ile4.56, Val4.60, and Lys6.58 have no effect on fentanyl binding [62], [63]. However, the substitutions of His6.52 to Asn or Gln have no effect as well, indicating a polar residue can be tolerated at this position, while the aromatic property of His is not critical [64].
Table 1.
The contact frequencies of the residues interacting with fentanyl and its analogs in the hMOR. In a molecular dynamics (MD) simulation frame, if the shortest heavy-atom distance between the ligand and any given residue of the MOR was within 5 Å, we defined that the ligand forms an interaction with this residue. The residues that have at least one contact frequency >0.6 in any indicated condition are included in this table. For a specific condition, the residue having frequency >0.6 are shaded in green (APF) and blue (FPA). The results shown here are based on the simulations with the OPLS4 force field. For the APF poses, we combined the results of APFEC and APFDC into APFC for each indicated ligand. The differences between the analogs and fentanyl in their APFC conditions are shown in the “- fentanyl” columns.
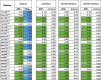 |
Interestingly, fentanyl contacts Asn1523.35 only in APFC, and His3217.36 only in FPADT (Table 1). Specifically in APFC, the N-phenyl moiety of fentanyl interacts with the amide sidechain of Asn1523.35 and renders its χ1 rotamer to switch from gauche- observed in the active MOR structure 6DDF to trans found in the inactive structure 4DKL (Fig. S9A-C). However, such an aromatic-amide interaction is relatively weak and crowded. Indeed, the binding affinity of fentanyl at the MOR was drastically improved for ∼20 fold in the presence of the Asn3.35Ala mutation [62] (Table S4), which may substitute with a more favored aromatic-hydrophobic interaction with more space. However, as Asn1523.35 contributes to the Na binding site and the associated interaction network of the MOR [65], [66], we cannot exclude the possibility of indirect effects of the mutation.
The Ala mutation of residue His3217.36, which interacts with the bound fentanyl in the FPADT condition but has no chance of interacting in APFC in our simulations (Table 1), resulted in significant disruption of the ohmefentanyl binding at the MOR [41] (Table S4). To understand the structural basis of this disruption, we docked ohmefentanyl in the MOR binding site, in either the APF or FPA orientations. We found that the potential interaction with His3217.36 could not be from the extra modifications of ohmefentanyl on the fentanyl scaffold, i.e., the 3-methyl of the piperidine ring or the 2-hydroxy on the linker between phenyl and piperidine (Fig. S9E-F). Similar to fentanyl, the alkyl tail of the propanamide of ohmefentanyl interacts with His3217.36 in the FPA orientation (Fig. S9F). In the fentanyl FPADT simulations, such an interaction results in a more defined His3217.36 conformation reflected in its tighter χ2 rotamer distribution, compared to that in APFC (Fig. S9D). Thus, the FPA pose may explain the His7.36Ala mutagenesis data better; however, we cannot exclude the possibility of indirect effect of the mutation.
3.3. 3-methyl and 4-carbomethoxy modifications change the energy landscape
To understand the impact of the modifications to the fentanyl scaffold on the FES, we carried out metadynamics simulations of three fentanyl analogs with either or both of 3-methyl and 4-carbomethoxy modifications similar to those of fentanyl (section 3.1). Carfentanil has a carbomethoxy group at the 4-position of the piperidine ring (Fig. S1). Compared to the FES of fentanyl, carfentanil has a similar CV1 profile, i.e., the T and C states are similarly located, while the C states always have lower energy than the T states (Fig. 2B, Table S1). However, carfentanil has a drastically different CV2 profile from that of fentanyl, suggesting significant impacts of the modifications on both the conformation and the flexibility of the scaffold. Specifically, the FES regions corresponding to the H states of fentanyl are high-energy forbidden regions for carfentanil (Fig. 2B), due to a steric clash between the 4-carbomethoxy and the N-phenyl groups (Fig. S10).
Further, an energy barrier arises in the middle of the L state and splits the state into two, which we termed L1 and L2 states. The L1 and L2 states are located near −10° and −110°, respectively. Given the same CV1 state (either the T or C state), the L1 and L2 states of carfentanil have comparable energies (Table S1). On the FES of carfentanil, the identifications of MFEPs and calculations of energy barriers show that the transition from the TL1 to CL1 state, which is favored in the MOR binding site (see below), has more than 2 kcal/mol lower energy barrier than the TL to CL transition of fentanyl (Table S2), suggesting the carbomethoxy modification may facilitate this transition.
In addition to the 4-carbomethoxy modification, lofentanil has an extra methyl group at the 3-position of the piperidine ring, the chirality of which can result in the (3S,4R)-, (3R,4S)-, (3S,4S)-, and (3R,4R)-isomers of the compound (Fig. S1). Overall, these lofentanil isomers have similar FESs as that of carfentanil, especially the two trans ((3S,4S)- and (3R,4R)-) isomers (Fig. 3B). However, given the same CV1 state (either the T or C state), the L1 state of (3S,4R)-lofentanil has ∼6 kcal/mol higher energy than its L2 state, while (3R,4S)-lofentanil has a reversed trend. In contrast, the L1 and L2 states of the trans isomers have only minor differences, demonstrating less impact of the 3-methyl modification when it is at the equatorial position of the piperidine ring. For all four isomers, their energetic differences between the most favored C states (e.g., CL2 for (3S,4R)-lofentanil and CL1 for (3R,4S)-lofentanil) and the T states are larger than that of fentanyl (Table S1). On their FESs, the identifications of MFEPs and calculations of energy barriers show that the additional methyl group on the piperidine ring of (3S,4R)-lofentanil further lowered the energy barrier of the TL1 to CL1 state transition to ∼7 kcal/mol, while that for (3R,4S)-lofentanil is similar to carfentanil (Table S2).
To further dissect the roles of 3-methyl and 4-carbomethoxy in altering the FES, we performed metadynamics simulations of ((3S,4R)-, (3R,4S)-, (3S,4S)-, and (3R,4R)-isomers of 3-methylfentanyl. For the two cis ((3S,4R)- and (3R,4S)-) isomers, their FESs are similar to that of fentanyl. However, the positions of their L and H states of CV2 are altered, with those of (3S,4R) moved slighted downwards and those of (3R,4S) moved upwards. In addition, the TL states of 3-methylfentanyl have slightly narrower distributions than that of fentanyl (Fig. 3A). Interestingly, the minima of the L states (both TL and CL) on the FES of (3R,4S)-3-methylfentanyl are pushed to similar minima positions of the L1 states of carfentanil and lofentanil, while those of (3S,4R)-3-methylfentanyl are pushed to those of the L2 states (Fig. 3 and Table S1). These different impacts are consistent with L1 and L2 states having lower energies for (3R,4S)- and (3S,4R)-lofentanil, respectively. For the two trans isomers of 3-methylfentanyl, while some shifts of the L and H states are observed, they are relatively small and correspond to similar profiles of the trans isomers of lofentanil.
Taken together, the C state is the most favored state for fentanyl and all its analogs investigated in this study, while the modifications on the piperidine ring in carfentanil and lofentanil make this trend even stronger and lower the energy barrier for the transition from the T to C state. Using OPLS4, we carried out the same set of metadynamics simulations and analyses and found similar results and conclusions as those described above using OPLS3e (Fig. S4, Tables S1 and S2).
3.4. The APF cis-amide binding poses of carfentanil and lofentanil are more stable
To characterize the impact of modifications to the fentanyl scaffold on the binding pose preferences at the MOR, we then carried out MD simulations of the MOR in complex with carfentanil, (3S,4R)- or (3R,4S)-lofentanil, each in various conditions as those for the MOR-fentanyl complex (Table S3).
In the APF trans-amide conditions (both APFET and APFDT), both carfentanil and (3S,4R)-lofentanil could not stay in the trans-amide configuration and transitioned to cis-amide in all the trajectories within 3 µs, while the transitions in the APFET condition were noticeably faster than in APFDT (Fig. S6). This is consistent with the results of our metadynamics simulations that show the differences between cis-amide and trans-amide configurations are higher for carfentanil and lofentanil than for fentanyl (Table S1), while the transitions from the T to C state are easier, i.e., having lower energy barriers than those of fentanyl (Table S2).
In the FPA trans-amide conditions, FPAET is not stable for carfentanil and both isomers of lofentanil, and the bound compounds in the majority of the FPAET trajectories transitioned to the cis-amide configuration. In the FPADT condition, however, while (3S,4R)-lofentanil transitioned to cis-amide in all trajectories and (3R,4S)-lofentanil has high pairwise ligand RMSDs (Fig. 5), carfentanil could form a similar H-bond interaction with either Gln1262.60 or Asn1292.63 as fentanyl (Fig. S6) but has higher RMSDs than fentanyl in this condition. An analysis of the dihedral angle between aniline and piperidine (i.e., the CV2 in the metadynamics simulations) of the FPADT conditions of both fentanyl and carfentanil revealed that these bound ligands form the H-bond with either Gln1262.60 or Asn1292.63 only in the TL1 state (Fig. 6), which can be significantly populated in the metadynamics simulations (Fig. 2). In contrast, the TL1 state of (3S,4R)-lofentanil has at least more than 6 kcal/mol higher energy than the other energy minima on the FES, and is an unstable state (Fig. 3, and Table S1). Thus, the potential H-bonds to Gln1262.60 or Asn1292.63 could not retain (3S,4R)-lofentanil in this state.
Fig. 5.

Carfentanil and lofentanil prefer the APF orientation. The average pairwise ligand RMSDs for each indicated ligand and condition are in panels A-C. APFDC conditions for each analog are shown in panels D-F. As a reference, the fentanyl binding pose (green) is superimposed in each panel. The carbomethoxy moiety of these analogs form a hydrophobic-aromatic interaction with Trp3207.35 (large circles in panels D-F), which is likely responsible for the higher potencies of these analogs compared to fentanyl. In addition, (3R,4S)-lofentanil has a hydrophobic interaction between its 3-methyl to Tyr1503.33 (small circle in panel F). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 6.

The distributions of the CV1 and CV2 dihedral angles of fentanyl and its analogs bound in the MOR. Only the relatively stable conditions in the MD simulations, APFEC, APFDC, FPAET, and FPADT, are shown for each indicated ligands. Darker dots are the OPLS3e results and lighter ones are the OPLS4 results. See Fig. 2, Fig. 3 for the CV1 and CV2 definitions.
Comparing the APF and FPA cis-amide conditions, FPAEC and FPADC of carfentanil and (3S,4R)-lofentanil have significantly higher pairwise ligand RMSDs than APFEC and APFDC, indicating these FPA conditions are likely not stable (Fig. 5). Interestingly, in the MM/GBSA calculations, we found the APFEC condition is slightly more favored than APFDC for carfentanil, and lofentanil (APFEC has 1.9, 2.6, and 1.7 kcal/mol lower energy than APFDC for carfentanil, (3S,4R)-lofentanil, and (3R,4S)-lofentanil, respectively), which is different from the situation in fentanyl.
For these two preferred APF cis-amide conditions, the analysis of the MD simulation results of the MOR complexes shows that carfentanil and both isomers of lofentanil stay in their CL1 states and never get into the CL2 state, while the CL state of fentanyl is in a much wider distribution (Fig. 6). From the FES derived from the metadynamics simulations of these compounds alone in the absence of the receptor, we note that the CL1 state is the most preferred state for (3R,4S)-lofentanil, while the CL1 state has only slightly higher energy than the CL2 state for carfentanil. However, the CL1 state of (3S,4R)-lofentanil has more than 6 kcal/mol higher than its CL2 state (Fig. 3 and Table S1). Thus, the less favored CL1 state of (3S,4R)-lofentanil bound in the MOR binding pocket is likely stabilized by the interactions with the receptor residues.
In summary, carfentanil prefers the APFC conditions and can be relatively stable in FPADT, but both isoforms of lofentanil can only be in APFC due to the conformational restraints rendered by the extra methyl group on the piperidine ring.
Carfentanil has been reported to have at least 6-fold [3], [33] or even ∼50-fold [32] higher binding affinity than fentanyl at the MOR, while lofentanil was found to have an even slightly higher affinity than carfentanil [33]. By comparing the contact frequencies in the APFDC conditions of carfentanil and lofentanil versus that of fentanyl, we found the 4-carbomethoxy group of carfentanil and the cis-isomers of lofentanil forms a strong hydrophobic-aromatic interaction with Trp3207.35 (Fig. 5 and Table 1), which is likely the structural basis for their enhanced affinities at the MOR. In the APFDC conditions of (3R,4S)- and (3S,4R)-lofentanil, the 3-methyl group on the piperidine ring of the fentanyl scaffold is located to different sides of the 4-carbomethoxy group in the binding pocket of the MOR. The 3-methyl of (3R,4S)-lofentanil forms a weak hydrophobic interaction with Tyr1503.33, which may further stabilize the APFDC condition compared to those of carfentanil, though only marginally (see Discussion).
3.5. Conformational changes of the MOR when binding to fentanyl
DAMGO, which is bound in the cryo-EM structure of the MOR-Gi complex used as the main template for this study, has been found to be a full agonist [67], while fentanyl has been proposed to be a β-arrestin-biased agonist at the MOR [7]. In order to fully understand the functional consequence of fentanyl binding, it is critical to evaluate whether fentanyl binding would result in a different receptor conformation compared to that stabilized by DAMGO. Thus, we compared the conformations of the MOR-fentanyl and MOR-DAMGO models. For the former, we chose the more favored MOR-fentanyl APFDC condition; for the latter, based on the original cryo-EM structure, we immersed a refined MOR-DAMGO model in a lipid bilayer and carried out extensive MD simulations using the same simulation protocols as those for the MOR-fentanyl models (Table S3). In these control simulations of the MOR-DAMGO model, we found only limited changes from the original conformation revealed by the cryo-EM structure, as demonstrated by the low and quickly plateaued RMSD evolutions along the MOR-DAMGO MD trajectories (Fig. S11).
We adapted a previously developed in-house tool, the Protein Interaction Analyzer (see Methods), to compare the representative MD frame ensembles of the MOR-fentanyl and MOR-DAMGO models, with a focus on the regions in or near the binding pocket (see Methods). We found the conformations of the OBS in two models are very similar (the region enclosed by the dotted box in Fig. 7D); however, there are significant changes in a secondary binding pocket (SBP) near the extracellular portion of TM1 (TM1e, see the definition in Methods), TM2e, TM3e, and TM7e. Our inspections of the models indicated that these changes likely resulted from an outward rearrangement of Trp135EL1.50 due to its interaction with the phenyl moiety of fentanyl, while Tyr1302.64 rotates inward, compared to their configurations in the presence of the bound DAMGO (Fig. 7B). These coordinated changes are reflected in the shortened distance between Tyr1302.64 and Trp135EL1.50 in the presence of fentanyl (Fig. 7C). We further quantified the differential effects of fentanyl and DAMGO binding by plotting the distances of TM1e-TM3e versus TM2e-TM7e in two conditions. We found fentanyl reduced the TM1e-TM3e distance, elongated the TM2e-TM7e distance, and therefore changed both the size and shape of the SBP (Fig. 7E,F).
Fig. 7.

Fentanyl induced divergent conformation near the binding pocket. (A) An overview the superimposed DAMGO and fentanyl binding poses in the MOR. (B) A zoom-in view demonstrating the diverged positions and orientations of Tyr1302.64 and Trp135EL1.50 when the receptor is bound with either DAMGO or fentanyl (APFDC). Panel C shows the center-of-mass distances between the phenyl ring of the indole moiety of Trp135EL1.50 and the sidechain of Tyr1302.64 in the fentanyl and DAMGO bound conditions. In the analysis with the Protein Interaction Analyzer, we calculated the center-of-mass distances among the extracellular and middle subsegments of the MOR. The differences of these distances between the DAMGO and fentanyl bound conditions (i.e., distanceDAMGO - distancefentanyl) are plotted on a heatmap shown in panel D. Specifically, the differences in the orthosteric binding site enclosed by TMs 3, 5, 6, and 7 (dotted box) are small, however, significant differences are detected in a secondary binding pocket encircled by TMs 1, 2, 3, and 7. In particular, the TM1e-TM3e distance is larger in the presence of the bound DAMGO, while TM2e-TM7e is larger in the fentanyl-bound condition. These two distances are indicated by the dotted lines in panel A, and their distributions are shown in panels E and F for DAMGO- and fentanyl-bound conditions, respectively.
4. Discussion
The mechanistic understanding of the binding of fentanyl and its analogs at the MOR is the foundation to reveal how the impact of these abused synthetic opioids is propagated toward the intracellular side of the receptor to initiate downstream signaling cascades, which will then elicit both beneficial analgesic and a variety of undesired side effects. However, in the absence of high-resolution structural information of the MOR bound with fentanyl, previous molecular modeling and simulation studies have proposed drastically different fentanyl binding modes, both in orientations within the binding pocket and ligand conformations [20], [21], [22], [23], [24]. To this end, we carried out a systematic computational study to investigate the preferred conformations of fentanyl and how these conformations are being accommodated in the MOR binding pocket, by characterizing the free energy landscape of fentanyl conformations with metadynamics simulations, as well as performing long-timescale MD simulations to compare 8 possible binding conditions. Our results indicate that the APFC binding condition is preferred, while the FPADT condition is also possible, with the slightly unfavored trans-amide configuration of fentanyl stabilized by forming a H-bond with Gln1262.60 or Asn1292.63. Indeed, a large-scale energetic analysis of the ligand–protein complexes in the Protein Data Bank (PDB) indicates that the bound ligands in many cases are not at its global minimum [68]. Thus, if there is a significant ligand conformational strain energy, it must be compensated by favored ligand–protein interactions. While the majority of the available mutagenesis data of the MOR in literature cannot differentiate these two poses, the improved fentanyl affinity with the Asn3.35Ala mutation is consistent with an interaction to Asn3.35 that is present only in APF, while the His7.36Ala mutagenesis results support FPA (Table S4). Due to the alterations of the energy landscape by the modifications to the fentanyl scaffold, we found that carfentanil has a weaker tendency to be in FPADT, and lofentanil essentially cannot be in this alternative pose.
Our APF cis-amide poses of fentanyl are largely consistent with the fentanyl pose reported by Ricarte et al., which was based on the modeling and the simulations with the structure 5C1M [23], and we found that the same set of hMOR residues interact with fentanyl (Table 1). Our FPADT is in a similar orientation as the pose proposed by Vo et al., whom, however, found that when His6.52 was in the HID form, fentanyl could move deeper and form a hydrogen bond between the protonated piperidine amine to the Nε atom of His6.52, instead of forming a salt bridge with Asp3.32 [24]. In our simulations, the bound fentanyl in the FPADT condition retains the interaction with Asp3.32 persistently. Our results show that the protonation state of His6.52, had only limited impact on the APF pose of fentanyl and carfentanil (Fig. 4, Fig. 5); nevertheless, the HID form facilitated stabilizing the FPA pose of fentanyl (Fig. 4).
The drastic improved affinity of 3-methylfentanyl over fentanyl, which is not observed in lofentanil over carfentanil, indicate that modifications at the 3- and 4-positions may have overlapping roles in improving the potency; at least they do not have additive effects. If (3R,4S)-lofentanil has higher affinity than (3S,4R)-lofentanil, i.e., like the situation of 3-methylfentanyl, our results may provide the structural basis for why lofentanil prefers the (3R,4S)- but not (3S,4R)- configuration in binding to the MOR. Specifically, carfentanil and both (3S,4R)- and (3R,4S)-isomers of lofentanil all stay in the CL1 state in the APF poses when bound in the MOR binding pocket (Fig. 6). However, the CL1 state of (3R,4S)-lofentanil on the FES has lower energy than the CL2 state, while its (3S,4R)-isomer has the reversed trend (Fig. 3B). Thus, if the (3S,4R)-lofentanil has to be in the CL1 state in the MOR binding pocket, it is not in its preferred state and may result in lower binding affinity. In addition, the stronger interaction with Tyr3.33, when the ligand is in the (3R,4S)-configuration (Table 1), is expected to slightly improve the affinity. However, our results do not provide a clear clue about the lack of additive effects between 3-methyl and 4-carbomethoxy. It is possible that both 3-methyl and 4-carbomethoxy have significant positive impacts on the binding affinity, but they are somewhat in conflict. Thus, it is not necessarily a safe assumption that (3R,4S)-lofentanil has higher affinity than (3S,4R)-lofentanil like the situation of 3-methylfentanyl. Notably, these two cis-isomers of lofentanil are confused in recent literature [19], [21], [31], [32], [69], as well as in wikipedia (https://en.wikipedia.org/wiki/Lofentanil, (3S,4R)-isomer) and PubChem (https://pubchem.ncbi.nlm.nih.gov/compound/Lofentanil, (3R,4S)-isomer).
In this study, we compared the results from metadynamics and MD simulations of fentanyl in the absence and presence of the receptor, respectively, and combined them to establish the structure–activity relationship (SAR) of the fentanyl scaffold at the MOR. The FES deduced from metadynamics simulations lay the foundation for our understanding of how fentanyl and its analogs would behave in the binding pocket of the MOR. The most favored poses of fentanyl and its analogs are consistent with the global minima on the FES, the FES guided us to seek the specific ligand-receptor interactions that may compensate the unfavored ligand conformations. Such an approach to thoroughly characterize the energy landscape of the ligand conformation has not been well appreciated. While long-timescale MD simulations is powerful in revealing ligand-induced receptor conformation changes to accommodate specific ligand scaffolds, it is not trivial to identify the proper binding pose with MD simulations in the first place, such as our previous work in identifying the binding modes of paroxetine at the serotonin transporter [70]. Indeed, even at relatively long timescales, we have not observed any transition between APF and FPA orientations. Thus, this combined approach provides a framework not only for the current fentanyl study but can also be applied to other SAR studies at other GPCRs.
In a companion manuscript, by combining the experimental and computational approaches, we found that the alkyl modifications of the amide moiety of the fentanyl scaffold affect the efficacy at the MOR (Xie et al., manuscript in preparation). Together, our findings start to establish a SAR of fentanyl binding at the MOR, which will facilitate our prediction and understanding of the potential toxicity of emerging novel synthetic opioids based on the fentanyl scaffold. Such insights will also contribute to developing new, safer analgesics with desired pharmacological properties.
Author Contributions
B.X. and L.S. designed the study. B.X. and A.G. carried out computational modeling, simulations, and analysis. All authors took part in interpreting the results. B.X. and L.S. wrote the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We thank Michael Baumann, Istvan Ujvary, Kenner Rice, and Arthur Jacobson for insightful discussions. Support for this research was provided by the National Institute on Drug Abuse–Intramural Research Program, Z1A DA000606 (L.S.). This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.05.013.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Pasternak G.W., Pan Y.X. Mu opioids and their receptors: evolution of a concept. Pharmacol Rev. 2013;65(4):1257–1317. doi: 10.1124/pr.112.007138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stanley T.H. The fentanyl story. J Pain. 2014;15(12):1215–1226. doi: 10.1016/j.jpain.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Baumann M.H., Kopajtic T.A., Madras B.K. Pharmacological research as a key component in mitigating the opioid overdose crisis. Trends Pharmacol Sci. 2018;39(12):995–998. doi: 10.1016/j.tips.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 4.The Drug Enforcement Administration's (DEA) special testing and research laboratory’s emerging trends program. DEA Emerging Threat Reports. 2020, May 1; Available from: https://ndews.umd.edu/resources/dea-emerging-threat-reports.
- 5.Comer S.D., Cahill C.M. Fentanyl: Receptor pharmacology, abuse potential, and implications for treatment. Neurosci Biobehav Rev. 2019;106:49–57. doi: 10.1016/j.neubiorev.2018.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haley D.F., Saitz R. The opioid epidemic during the COVID-19 pandemic. JAMA. 2020;324(16):1615–1617. doi: 10.1001/jama.2020.18543. [DOI] [PubMed] [Google Scholar]
- 7.Schmid C.L., Kennedy N.M., Ross N.C., Lovell K.M., Yue Z., et al. Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell. 2017;171(5):1165–1175 e13. doi: 10.1016/j.cell.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ehrlich A.T., Semache M., Gross F., Da Fonte D.F., Runtz L., et al. Biased signaling of the mu opioid receptor revealed in native neurons. iScience. 2019;14:47–57. doi: 10.1016/j.isci.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gillis A., Gondin A.B., Kliewer A., Sanchez J., Lim H.D., et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci Signal. 2020;13(625) doi: 10.1126/scisignal.aaz3140. [DOI] [PubMed] [Google Scholar]
- 10.Stahl E.L., Bohn L.M. Low intrinsic efficacy alone cannot explain the improved side effect profiles of new opioid agonists. Biochemistry. 2021 doi: 10.1021/acs.biochem.1c00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kelly E., Sutcliffe K., Cavallo D., Ramos-Gonzalez N., Alhosan N., et al. The anomalous pharmacology of fentanyl. Br J Pharmacol. 2021 doi: 10.1111/bph.15573. [DOI] [PubMed] [Google Scholar]
- 12.Subramanian G., Paterlini M.G., Portoghese P.S., Ferguson D.M. Molecular docking reveals a novel binding site model for fentanyl at the mu-opioid receptor. J Med Chem. 2000;43(3):381–391. doi: 10.1021/jm9903702. [DOI] [PubMed] [Google Scholar]
- 13.Dosen-Micovic L., Ivanovic M., Micovic V. Steric interactions and the activity of fentanyl analogs at the mu-opioid receptor. Bioorg Med Chem. 2006;14(9):2887–2895. doi: 10.1016/j.bmc.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 14.Huang W., Manglik A., Venkatakrishnan A.J., Laeremans T., Feinberg E.N., et al. Structural insights into micro-opioid receptor activation. Nature. 2015;524(7565):315–321. doi: 10.1038/nature14886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manglik A., Kruse A.C., Kobilka T.S., Thian F.S., Mathiesen J.M., et al. Crystal structure of the micro-opioid receptor bound to a morphinan antagonist. Nature. 2012 doi: 10.1038/nature10954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koehl A., Hu H., Maeda S., Zhang Y., Qu Q., et al. Structure of the micro-opioid receptor-Gi protein complex. Nature. 2018;558(7711):547–552. doi: 10.1038/s41586-018-0219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapoor A., Provasi D., Filizola M. Atomic-level characterization of the methadone-stabilized active conformation of µ-opioid receptor. Mol Pharmacol. 2020;98(4):475–486. doi: 10.1124/mol.119.119339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mafi A., Kim S.K., Goddard W.A., 3rd Mechanism of β-arrestin recruitment by the μ-opioid G protein-coupled receptor. Proc Natl Acad Sci U S A. 2020;117(28):16346–16355. doi: 10.1073/pnas.1918264117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellis C.R., Kruhlak N.L., Kim M.T., Hawkins E.G., Stavitskaya L. Predicting opioid receptor binding affinity of pharmacologically unclassified designer substances using molecular docking. PLoS ONE. 2018;13(5):e0197734. doi: 10.1371/journal.pone.0197734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipinski P.F.J., Jaronczyk M., Dobrowolski J.C., Sadlej J. Molecular dynamics of fentanyl bound to mu-opioid receptor. J Mol Model. 2019;25(5):144. doi: 10.1007/s00894-019-3999-2. [DOI] [PubMed] [Google Scholar]
- 21.de Waal P.W., Shi J., You E., Wang X., Melcher K., et al. Molecular mechanisms of fentanyl mediated beta-arrestin biased signaling. PLoS Comput Biol. 2020;16(4):e1007394. doi: 10.1371/journal.pcbi.1007394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Podlewska S., Bugno R., Kudla L., Bojarski A.J., Przewlocki R. Molecular modeling of µ opioid receptor ligands with various functional properties: PZM21, SR-17018, morphine, and fentanyl-simulated interaction patterns confronted with experimental data. Molecules. 2020;25(20) doi: 10.3390/molecules25204636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ricarte A., Dalton J.A.R., Giraldo J. Structural assessment of agonist efficacy in the μ-opioid receptor: morphine and fentanyl elicit different activation patterns. J Chem Inf Model. 2021;61(3):1251–1274. doi: 10.1021/acs.jcim.0c00890. [DOI] [PubMed] [Google Scholar]
- 24.Vo Q.N., Mahinthichaichan P., Shen J., Ellis C.R. How mu-opioid receptor recognizes fentanyl. Nat Commun. 2021;12(1):984. doi: 10.1038/s41467-021-21262-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eshleman A.J., Nagarajan S., Wolfrum K.M., Reed J.F., Nilsen A., et al. Affinity, potency, efficacy, selectivity, and molecular modeling of substituted fentanyls at opioid receptors. Biochem Pharmacol. 2020;182 doi: 10.1016/j.bcp.2020.114293. [DOI] [PubMed] [Google Scholar]
- 26.Ballesteros JA, Weinstein H. Modeling transmembrane helix contacts in GPCR. In: Biophysical Society Meeting. 1995. San Francisco, CA.
- 27.Van Bever W.F., Niemegeers C.J., Janssen P.A. Synthetic analgesics. Synthesis and pharmacology of the diastereoisomers of N-(3-methyl-1-(2-phenylethyl)-4-piperidyl)-N-phenylpropanamide and N-(3-methyl-1-(1-methyl-2-phenylethyl)-4-piperidyl)-N-phenylpropanamide. J Med Chem. 1974;17(10):1047–1051. doi: 10.1021/jm00256a003. [DOI] [PubMed] [Google Scholar]
- 28.Zawilska J.B., Kuczynska K., Kosmal W., Markiewicz K., Adamowicz P. Carfentanil - from an animal anesthetic to a deadly illicit drug. Forensic Sci Int. 2021;320 doi: 10.1016/j.forsciint.2021.110715. [DOI] [PubMed] [Google Scholar]
- 29.Wax P.M., Becker C.E., Curry S.C. Unexpected “gas” casualties in Moscow: a medical toxicology perspective. Ann Emerg Med. 2003;41(5):700–705. doi: 10.1067/mem.2003.148. [DOI] [PubMed] [Google Scholar]
- 30.Jin W.Q., Xu H., Zhu Y.C., Fang S.N., Xia X.L., et al. Studies on synthesis and relationship between analgesic activity and receptor affinity for 3-methyl fentanyl derivatives. Sci Sin. 1981;24(5):710–720. [PubMed] [Google Scholar]
- 31.Xu H., Kim C.H., Zhu Y.C., Weber R.J., Jacobson A.E., et al. (+)-cis-3-methylfentanyl and its analogs bind pseudoirreversibly to the mu opioid binding site: evidence for pseudoallosteric modulation. Neuropharmacology. 1991;30(5):455–462. doi: 10.1016/0028-3908(91)90006-w. [DOI] [PubMed] [Google Scholar]
- 32.Maguire P., Tsai N., Kamal J., Cometta-Morini C., Upton C., et al. Pharmacological profiles of fentanyl analogs at mu, delta and kappa opiate receptors. Eur J Pharmacol. 1992;213(2):219–225. doi: 10.1016/0014-2999(92)90685-w. [DOI] [PubMed] [Google Scholar]
- 33.Yeadon M., Kitchen I. Differences in the characteristics of opioid receptor binding in the rat and marmoset. J Pharm Pharmacol. 1988;40(10):736–739. doi: 10.1111/j.2042-7158.1988.tb07008.x. [DOI] [PubMed] [Google Scholar]
- 34.Janssen P.A. Potent, new analgesics, tailor-made for different purposes. Acta Anaesthesiol Scand. 1982;26(3):262–268. doi: 10.1111/j.1399-6576.1982.tb01765.x. [DOI] [PubMed] [Google Scholar]
- 35.Leysen J.E., Gommeren W., Niemegeers C.J. [3H]Sufentanil, a superior ligand for mu-opiate receptors: binding properties and regional distribution in rat brain and spinal cord. Eur J Pharmacol. 1983;87(2–3):209–225. doi: 10.1016/0014-2999(83)90331-x. [DOI] [PubMed] [Google Scholar]
- 36.Tollenaere J.P., Moereels H., Van Loon M. On conformation analysis, molecular graphics, fentanyl and its derivatives. Prog Drug Res. 1986;30:91–126. doi: 10.1007/978-3-0348-9311-4_3. [DOI] [PubMed] [Google Scholar]
- 37.Leysen J.E., Laduron P.M. Receptor binding properties in vitro and in vivo of some long-acting opiates. Arch Int Pharmacodyn Ther. 1978;232(2):243–246. [PubMed] [Google Scholar]
- 38.Stahl K.D., van Bever W., Janssen P., Simon E.J. Receptor affinity and pharmacological potency of a series of narcotic analgesic, anti-diarrheal and neuroleptic drugs. Eur J Pharmacol. 1977;46(3):199–205. doi: 10.1016/0014-2999(77)90334-x. [DOI] [PubMed] [Google Scholar]
- 39.Leysen, J, Laduron, P, and Niemegeers, C, Receptor binding properties in vitro and in vivo of new long acting narcotic analgesics. In: Characteristics and function of opioids. 1978, Elsevier North-Holland Biomedical Press Amsterdam. p. 479-482.
- 40.Flippenanderson J.L., George C., Bertha C.M., Rice K.C. X-ray crystal-structures of potent opioid receptor ligands - etonitazene, cis-(+)-3-methylfentanyl, etorphine, diprenorphine, and buprenorphine. Heterocycles. 1994;39(2):751–766. [Google Scholar]
- 41.Xu H., Lu Y.F., Partilla J.S., Zheng Q.X., Wang J.B., et al. Opioid peptide receptor studies, 11: involvement of Tyr148, Trp318 and His319 of the rat mu-opioid receptor in binding of mu-selective ligands. Synapse. 1999;32(1):23–28. doi: 10.1002/(SICI)1098-2396(199904)32:1<23::AID-SYN3>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 42.Lesnik S., Hodoscek M., Bren U., Stein C., Bondar A.N. Potential Energy Function for Fentanyl-Based Opioid Pain Killers. J Chem Inf Model. 2020;60(7):3566–3576. doi: 10.1021/acs.jcim.0c00185. [DOI] [PubMed] [Google Scholar]
- 43.Giannos T., Lesnik S., Bren U., Hodoscek M., Domratcheva T., et al. CHARMM force-field parameters for morphine, heroin, and oliceridine, and conformational dynamics of opioid drugs. J Chem Inf Model. 2021;61(8):3964–3977. doi: 10.1021/acs.jcim.1c00667. [DOI] [PubMed] [Google Scholar]
- 44.Roos K., Wu C., Damm W., Reboul M., Stevenson J.M., et al. OPLS3e: extending force field coverage for drug-like small molecules. J Chem Theory Comput. 2019;15(3):1863–1874. doi: 10.1021/acs.jctc.8b01026. [DOI] [PubMed] [Google Scholar]
- 45.Lu C., Wu C., Ghoreishi D., Chen W., Wang L., et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput. 2021 doi: 10.1021/acs.jctc.1c00302. [DOI] [PubMed] [Google Scholar]
- 46.Bochevarov A.D., Harder E., Hughes T.F., Greenwood J.R., Braden D.A., et al. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int J Quantum Chem. 2013;113(18):2110–2142. [Google Scholar]
- 47.Bowers K.J., Chow E., Xu H., Dror R.O., Eastwood M.P., et al. ACM/IEEE SC 2006 conference (SC'06) 2006. Scalable algorithms for molecular dynamics simulations on commodity clusters. [Google Scholar]
- 48.Dijkstra E.W. A note on two problems in connexion with graphs. Numer Math. 1959;1(1):269–271. [Google Scholar]
- 49.Li J., Abel R., Zhu K., Cao Y., Zhao S., et al. The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins. 2011;79(10):2794–2812. doi: 10.1002/prot.23106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.John B., Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003;31(14):3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Olsson M.H., Sondergaard C.R., Rostkowski M., Jensen J.H. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J Chem Theory Comput. 2011;7(2):525–537. doi: 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- 52.Sherman W., Day T., Jacobson M.P., Friesner R.A., Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49(2):534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 53.Dror R.O., Arlow D.H., Maragakis P., Mildorf T.J., Pan A.C., et al. Activation mechanism of the beta2-adrenergic receptor. Proc Natl Acad Sci U S A. 2011;108(46):18684–18689. doi: 10.1073/pnas.1110499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lane J.R., Abramyan A.M., Adhikari P., Keen A.C., Lee K.H., et al. Distinct inactive conformations of the dopamine D2 and D3 receptors correspond to different extents of inverse agonism. Elife. 2020;9 doi: 10.7554/eLife.52189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph, 1996; 14(1): 33-8, 27-8. [DOI] [PubMed]
- 56.Stolzenberg, S, Michino, M, LeVine, MV, Weinstein, H, and Shi, L, Computational approaches to detect allosteric pathways in transmembrane molecular machines. Biochim Biophys Acta, 2016; 1858(7 Pt B): 1652-62. [DOI] [PMC free article] [PubMed]
- 57.Michino M., Boateng C.A., Donthamsetti P., Yano H., Bakare O.M., et al. Toward understanding the structural basis of partial agonism at the dopamine D3 receptor. J Med Chem. 2017;60(2):580–593. doi: 10.1021/acs.jmedchem.6b01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bick M.J., Greisen P.J., Morey K.J., Antunes M.S., La D., et al. Computational design of environmental sensors for the potent opioid fentanyl. Elife. 2017;6 doi: 10.7554/eLife.28909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laio A., Parrinello M. Escaping free-energy minima. Proc Natl Acad Sci U S A. 2002;99(20):12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Branduardi D., Faraldo-Gomez J.D. String method for calculation of minimum free-energy paths in Cartesian space in freely-tumbling systems. J Chem Theory Comput. 2013;9(9):4140–4154. doi: 10.1021/ct400469w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu X., Testa B., Fahr A. Lipophilicity and its relationship with passive drug permeation. Pharm Res. 2011;28(5):962–977. doi: 10.1007/s11095-010-0303-7. [DOI] [PubMed] [Google Scholar]
- 62.Mansour A., Taylor L.P., Fine J.L., Thompson R.C., Hoversten M.T., et al. Key residues defining the mu-opioid receptor binding pocket: a site-directed mutagenesis study. J Neurochem. 1997;68(1):344–353. doi: 10.1046/j.1471-4159.1997.68010344.x. [DOI] [PubMed] [Google Scholar]
- 63.Bonner G., Meng F., Akil H. Selectivity of mu-opioid receptor determined by interfacial residues near third extracellular loop. Eur J Pharmacol. 2000;403(1–2):37–44. doi: 10.1016/s0014-2999(00)00578-1. [DOI] [PubMed] [Google Scholar]
- 64.Spivak C.E., Beglan C.L., Seidleck B.K., Hirshbein L.D., Blaschak C.J., et al. Naloxone activation of mu-opioid receptors mutated at a histidine residue lining the opioid binding cavity. Mol Pharmacol. 1997;52(6):983–992. doi: 10.1124/mol.52.6.983. [DOI] [PubMed] [Google Scholar]
- 65.Katritch V., Fenalti G., Abola E.E., Roth B.L., Cherezov V., et al. Allosteric sodium in class A GPCR signaling. Trends Biochem Sci. 2014;39(5):233–244. doi: 10.1016/j.tibs.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lesnik S., Bertalan E., Bren U., Bondar A.N. Opioid receptors and protonation-coupled binding of opioid drugs. Int J Mol Sci. 2021;22(24) doi: 10.3390/ijms222413353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kelly E. Efficacy and ligand bias at the mu-opioid receptor. Br J Pharmacol. 2013;169(7):1430–1446. doi: 10.1111/bph.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tong J., Zhao S. Large-scale analysis of bioactive ligand conformational strain energy by ab initio calculation. J Chem Inf Model. 2021;61(3):1180–1192. doi: 10.1021/acs.jcim.0c01197. [DOI] [PubMed] [Google Scholar]
- 69.Lalinde N., Moliterni J., Wright D., Spencer H.K., Ossipov M.H., et al. Synthesis and pharmacological evaluation of a series of new 3-methyl-1,4-disubstituted-piperidine analgesics. J Med Chem. 1990;33(10):2876–2882. doi: 10.1021/jm00172a032. [DOI] [PubMed] [Google Scholar]
- 70.Abramyan A.M., Slack R.D., Meena S., Davis B.A., Newman A.H., et al. Computation-guided analysis of paroxetine binding to hSERT reveals functionally important structural elements and dynamics. Neuropharmacology. 2019;161 doi: 10.1016/j.neuropharm.2018.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.