

Abstract
Triple-negative breast cancer (TNBC) presented as high heterogeneous immunogenicity that lacks useful clinical signatures to risk-stratify immune-benefit subtypes. We hypothesized that molecular-based phenotypic characterization of TNBC tumors and their immunity may overcome these challenges. We enrolled 1,145 patients with TNBC for analysis. Through combining algorithm integration analysis and TNBC datasets, a tumor immune risk score (TIRS) panel consisting of 8 potential biomarkers was identified. The TIRS panel represented excellent effectiveness as an independent predictor. High- and low risk stratification of patients was further achieved by TIRS, and significant survival and immune-infiltration pattern differences were found in each cohort, both at the transcriptome and protein levels. Non-negative matrix factorization clustering further identified four different tumor immune microenvironment types (TIMTs), among which TIMT-II was associated with the best prognosis and immune status, whereas TIMT-IV had the opposite effect, TIMT-III was associated with highly unstable genomes, and TIMT-I displayed stem-cell-related characteristics along with high stromal scores and may have extensive enrichment of tumor-associated fibroblasts and vascular cells. In conclusion, our TIRS panel could serve as a robust prognostic signature and provide therapeutic benefits for immunotherapy. Additionally, coordinating four TIMTs may be helpful for clinical decision-making in TNBC patients.
Keywords: triple-negative breast cancer, immune risk signature, risk stratification, tumor immune microenvironment types, treatment strategy
Graphical abstract
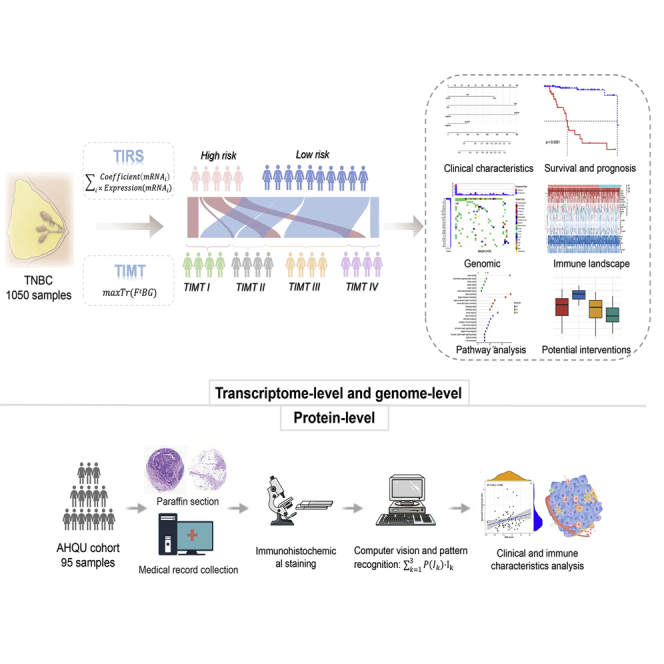
The immunotherapy of TNBC lacks clinical guidance based on precise stratification. Sun and colleagues established a TIRS multi-molecular panel and systematically identified four tumor immune microenvironment types. The findings highlight the potential clinical implications of more appropriate choices and improved individualized treatment strategies for TNBC patients.
Introduction
Triple-negative breast cancer (TNBC) is the most aggressive type of breast cancer, with higher rates of distant recurrence and poorer prognosis than other subtypes.1,2 Owing to the lack of expression of the estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), therapeutic targeting is difficult. The TNBC subgroup is traditionally considered a single entity based on immunohistochemistry (IHC), which demonstrates that TNBC has an unexpected level of heterogeneity but encompasses the characteristics of histopathological and molecular profiling, and an immune state with significant differences.3 Nonetheless, chemotherapy has been the standard treatment of choice for TNBC, but successful TNBC management remains an elusive subject in medicine. Individualized strategies are urgently needed to improve this situation.
The heterogeneous immunogenicity of TNBC has attracted the attention of clinicians and scientists. Compared with other breast cancer subtypes, TNBC has higher PD-L1 and tumor mutational burden (TMB) expression levels and tumor-infiltrating lymphocyte (TIL) density, which has the potential to change the chemotherapy-dominated treatment model of TNBC for half a century. IMPASSION 130, a phase III randomized controlled trial, identified the immune checkpoint inhibitor atezolizumab combined with nanoparticle albumin-bound (nab) paclitaxel as the standard first-line therapy for PD-L1-positive peritumoral immune-infiltrate patients with metastatic TNBC, which opened up possibilities for TNBC clinical immunotherapy.4 We have witnessed a rapid increase in the number of trials investigating immunotherapy in recent years3,5; however, low benefits of single immunotherapy have also been reported. Although the treatment population is already screened for PD-L1 and TMB expression levels, only a subset of patients with TNBC have benefited from immunotherapy in clinical practice.6 This is largely because of the poor sensitivity and specificity of these methodologies (limiting the beneficiary population by PD-L1 or TMB). The performance of current clinicopathological variables and prognostic factors is limited; none are sufficiently robust to guide therapy, although some should be considered as stratification factors for future trials.7 An imperative need exists to identify a new signature to stratify TNBC and immune characteristics and personalized survival risk of patients through a better understanding of the immune environment and molecular characteristics of TNBC.
The tumor immune microenvironment is a complex system that has been characterized at the cellular level for a long time. The development and application of high-throughput data has facilitated the identification of previously unrecognized molecular biomarkers, and technological advances in biological computing algorithms and bioinformatics have enabled innovative analysis of complex molecular profiling in malignancies and are strongly warranted to pave the way for individualized anti-cancer treatment.8 In particular, technologies based on DNA microarray and next-generation sequencing have led to the development of big data in molecular biology, on the basis of which many genetic features have emerged to predict clinical outcome as well as molecular subclassification.9 For example, the classic 50-gene subtype predictor (PAM50) has been widely used as a basis for breast cancer classification.10 Omics technology is of great significance to characterize the heterogeneity of tumor immune status and to predict responses to drugs and the clinical course of the disease. However, it should be noted that although a large number of genomic studies allow us to understand the input codes (genes) of cancer, their output codes (proteins) are needed to fully capture the state of the tumor and provide a complete and accurate map to understand and treat potential molecular pathology. This is because proteins and their post-translational modifications are important biological features and processes, which cannot be analyzed using genomics and are involved in most therapeutic interventions.11 Further verification at the protein level is essential to narrow the gap between cancer genotypes and cancer phenotypes.
In this study, we systematically identified highly reliable immune markers of TNBC, established a tumor immune risk score (TIRS) multi-molecular panel, and analyzed its potency at the immune, risk-benefit, and patient-prognosis levels. Furthermore, a non-negative matrix factorization (NMF) clustering based on immune genes was performed, and four subtypes with distinct molecular signatures, clinical characteristics, and immune infiltration were identified. Multi-dimensional cohort data at both gene and protein levels were used for independent verification and further showed that TIRS subtypes were superior to clinicopathological variables in terms of immune status and survival benefits in patients with TNBC.
Results
Analytic pipeline
A total of 1,096 breast cancer samples were collected from The Cancer Genome Atlas (TCGA) cohort, and 156 TNBC breast cancer samples were screened based on the inclusion criteria. The survival and expression data of the samples were called training sets to construct the TIRS models. Clinicopathological parameters, including age, tumor node metastasis (TNM) stage, and grade, are presented in Table S1. A total of 27 transcriptome data series with survival information for patients with TNBC were sorted out in the Gene Expression Omnibus (GEO) database (Table S2). Duplicated samples from different datasets were removed, and 659 TNBC sample data were obtained. After further screening of their survival information, patients with the following three outcome states were distinguished: overall survival (OS; n = 431), disease-free survival (DFS; n = 206), and recurrence-free survival (RFS; n = 132). The de-batching effect across multiple platforms was harmonized and eliminated using SangerBox. In the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) database, after excluding patients without complete information, 235 samples were obtained finally and used together with the GEO data as the verification cohort to verify the robustness of the model. Basic common clinical information of patients with TNBC is presented in Table S3, while detailed patient information obtained from each database is summarized in Table S1.
In the Affiliated Hospital of Qingdao University (AHQU) cohort, all samples were obtained before patients underwent therapy, and 95 patients were available for analysis after omitting samples that failed quality checks. All patients underwent surgery at the AHQU. The age of patients ranged from 23 to 80 years (median: 52 years), while the duration of follow up ranged from 160 to 2,801 days (mean: 1,146 days; median: 1,065 days), with recurrence and death occurring in 9 and 11 of these patients, respectively. Clinicopathological parameters, including patient age, tumor size, grade, lymphovascular invasion, and axillary lymph node status, are presented in Table S1.
TIRS signature construction based on a stable 8-gene panel
The immune gene sets from three databases (GSEA, Immport, and InnateDB) were included and integrated with the collated gene expression of patients with TNBC to obtain specific immune-related genes of TNBC (Figure 1A). A set of 1,471 targets was finally obtained and was further used to integrate the expression matrix and match clinical information. Based on this, a stable 8-gene (ARTN, GBP1, DLL4, PDK1, BCL2A1, MAP2K6, TOR2A, EIF4EBP1) panel was identified and printed as a TIRS signature (Figures 1B and 1C) using least absolute shrinkage and selection operator (LASSO) Cox regression analysis. The risk score was calculated according to the genetic coefficient, and the TIRS formula was obtained after allocating weights as follows: TIRS = 0.0274 × ARTN - 0.1770 × GBP1 + 0.0918 × DLL4 - 0.1025 × PDK1 - 0.0580 × BCL2A1 - 0.2023 × MAP2K6 + 0.4355 × TOR2A + 0.0920 × EIF4EBP1. We calculated the risk score for each training cohort of patients with TNBC using the TIRS formula and stratified them into high- (n = 45) and low-risk (n = 111) TIRS subtypes according to the optimal cutoff generated by X-tile.
Figure 1.

Construction and validation of the TIRS
(A) Venn diagram of TNBC-associated immune genes.
(B) Partial likelihood deviance revealed by the LASSO regression model. The red dots represent the partial likelihood values, the gray lines represent the standard error (SE), and the vertical dotted lines are drawn at the optimal values by 1-SE criteria.
(C) LASSO coefficient profiles.
(D) Survival differences between high- (red) and low- (blue) risk groups.
(E) ROC curve analysis of the TIRS model, age, stage, and TNM stage.
(F) Time-dependent ROC curves of TIRS prediction on the 1-, 3-, and 5-year survival rates.
(G–J) Between high- (red) and low- (blue) risk groups, OS (G), RFS (H), and DFS (I) in the GEO database and OS (J) in the METABRIC database were included in the analysis and displayed as a Kaplan-Meier plot. Statistical significance was set at p < 0.05.
The resulting model was subjected to Kaplan-Meier analysis in the training cohort to evaluate the relationship between the risk score and survival, as shown in Figure 1D. The OS of patients in the high-risk group was significantly shorter than that in the low-risk group (p < 0.0001). Meanwhile, receiver operating characteristic (ROC) curve results (Figure 1E) showed that TIRS had the highest prognostic performance for risk and survival status compared with other clinical models (area under the curve [AUC] = 0.932). Furthermore, the classification efficiency of prognosis prediction at 1, 3, and 5 years was analyzed and is shown in Figure 1F. It can be seen that the TIRS model has a high AUC offline region and is stable above 0.9. Subsequently, the Cox proportional hazards were calculated, and the training set displayed a good outcome, with a hazard ratio of 19.49 (95% confidence interval [CI]: 7.77–48.88), p < 0.001 in univariate Cox regression, and 16.06 (95% CI: 5.03–51.31), p < 0.001 in multivariate Cox regression.
Validation of TIRS model
The net reclassification improvement (NRI) and integrated discrimination improvement (IDI) were used to further qualify the improvement in TIRS.12 Specifically, the Akaike information criterion (AIC), which measures the goodness of fit in statistical models, was used as a quantitative criterion. A lower AIC value represents a better model prediction capability. The results of NRI showed that compared with stage (AIC = 121.36), the TIRS (AIC = 98.31) versus stage was 0.78 (95% CI: 0.37–0.92, p < 0.01), while the result of IDI was 0.53 (95% CI: 0.21–0.74, p < 0.01). We then calculated the NRI and IDI of the TIRS model versus TIRS combined with the stage model (AIC = 95.22) and found them to be 0.19 (95% CI: −0.34–0.58, p = 0.40) and -0.01 (95% CI: −0.05–0.10, p = 0.85), respectively. Obviously, the prognostic effect of the TIRS model was better than that of the stage standard, and this advantage was not strengthened by the combination of factors such as tumor stage, which reflects the independent-risk prognostic value of TIRS.
To further verify the robustness of TIRS, the effectiveness of TIRS in predicting the risk of survival recurrence and progression was confirmed in the validation cohort. After the batch effect was removed, patients were divided into high/low TIRS subtypes using the same procedures as in the training cohort. Consistent with the results of the training cohort, survival analysis varied substantially across the low- and high-risk groups (Figures 1G–1J).
Clinical characteristics of TIRS subtypes
To further describe and understand the biological and clinical differences between the two TIRS subtypes, we performed a stratified analysis of patients with TNBC. The expression level of the risk score for each sample in the training cohort was calculated, and the risk-score distribution was plotted (Figure 2A). A higher number of deaths in the high-risk group was displayed, and the same results were observed in the GEO and METABRIC cohorts (Figures 2B and 2C).
Figure 2.

Correlation between TIRS and clinic characteristics
(A–C) Corresponding distribution of risk score and survival status of patients in different groups in the training cohort (A), as well as in the GEO OS (B) and METABRIC (C) cohorts. Top: patient risk score for each sample (green, low risk; red, high risk). Bottom: patient outcomes for each sample (green, survival; red, death) plotted with vertical jitter along the sample’s model prediction (x axis).
(D–G) Analysis of patient survival status after further stratification of clinical status. Kaplan-Meier analysis was used for two TIRS groups in no lymph node metastasis (N0)(D) and non-N0 (E) patients, as well as stages I–II (F) and stages III–IV (G) patients.
(H) Difference display of risk score in different stage, tumor, and node through violin plots.
(I) The specific difference distribution of characteristics between two TIRS groups in the TCGA cohort.
As mentioned above, the low- and high-risk subtypes displayed significant survival differences with good prediction accuracy. Further in-depth analysis revealed that this difference was significant regardless of lymph node status and stage (Figures 2D–2G). In addition, it was found that the risk score was closely related to stage (Figure 2H), especially tumor and lymph node status. The stack diagram shows this feature (Figure 2I). Specifically, low-risk groups tended to be staged earlier, and most of them did not have lymph node metastasis, with the age distribution concentrated in the vicinity of 45–59 (peri-menopausal period).
To provide better clinical assistance, we constructed a nomogram based on risk score, age, staging, and other factors (Figures 3A–3C) to intuitively understand the 3- and 5-year survival probabilities of patients. A good degree of fit between the calibration curve and the ideal curve was observed in each cohort (Figure S1). In addition, decision curve analysis (DCA) was plotted to assess clinical benefits. Compared with other models, the TIRS model had a higher net benefit in predicting immune benefits (Figure 3D).
Figure 3.

Construction analysis of clinical nomogram
(A–C) The nomogram was constructed in the training cohort, with the TIRS, age, and stage incorporated. Calibration plot of the nomogram in terms of agreement between the predicted and observed (B) 3- and (C) 5-year outcomes. The 95% confidence intervals are represented by the close-ended vertical lines. The ideal performance of a nomogram is represented by the dashed lines along the 45-degree line.
(D) Decision curve analysis of the nomogram for different predicting model.
Immune cells and molecules differences
Further research found that immune subtypes could be distinguished by TIRS and are closely related to specific immune states. We quantified the differences in the distributions of 22 immune cell types between the high- and low-TIRS groups using CIBERSORT and found that the infiltration of immune-associated cells with high and low risk presented two completely different states. Significant differences were observed in the enrichment of a series of immune cells, such as naive B cells, plasma cells, CD4 memory T cells, CD4 memory activated T cells, follicular helper T cells, regulatory T cells (Tregs), and M1-type macrophages (Figure 4A). In short, the expression of immune-activated cells (such as M1-type macrophages and CD4 memory resting cells) was significantly reduced in high-risk subtypes, while the expression of immune-suppressive cells (such as Tregs) was increased.
Figure 4.

Immune landscape of TIRS subtypes
(A and B) Difference of immune cell infiltration and immune-target expression between two TIRS groups through CIBERSORT (A) and ESTIMATE (B). Wilcoxon test was used for statistical analysis, with ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05.
(C and D) Gene mutation analysis in high- (C) and low-risk (D) groups.
(E) GSEA showing significant differences in biological processes between high- and low-risk groups, which were the T cell receptor signaling pathway, the JAK/STAT signaling pathway, and natural-killer-cell-mediated cytotoxicity. Statistical significance was set at p < 0.05. NES, normalized enrichment score.
(F and G) GO and KEGG analysis of the differentially expressed genes.
The ESTIMATE analysis corroborated this result. Based on the single-sample gene set enrichment analysis (ssGSEA) algorithm, the stroma and immune cell score were estimated using transcriptome expression data (Figure S2A). The low-risk group showed good immune activity, which was characterized by a higher immune score and more obvious expression of immune cells compared with the high-risk group (Figure 4B).
The key immune target genes are closely related to the immune status and treatment of TNBC, and their correlation with TIRS characteristics needs to be further studied. First, as endogenous peptides are delivered to T lymphocytes via the major histocompatibility complex (MHC) class I pathway, we evaluated the RNA expression of HLA class I genes (Figure S2B) in high- and low-TIRS subtypes. Notably, the HLA family genes were highly expressed and statistically significant in the low-risk group. A more diverse library of HLA-I classes will lead to a wider array of antigens, increasing the odds of presenting more immunogenic antigens and increasing the likelihood of benefiting from immune checkpoint inhibitor (ICI) therapy.13
In addition, the correlation between subtypes and key immune checkpoint molecules (PD-L1, CTLA4) was studied (Figure 4B). As protective effector molecules of the human immune system, immune checkpoint molecules may be overexpressed in the tumor microenvironment and inhibit the immune response of the host, resulting in immune escape. ICIs can block the transmission of immunosuppressive signals, restore the killing activity of T cells, and reverse the immunosuppressive tumor microenvironment.14 Higher expression of immune checkpoint molecules could mean more benefits for immunotherapy and a longer tumor control period, which is significant in the low-risk group.
Furthermore, chemokines, as regulators of directed chemotaxis of immune cells, were enriched in the low-risk group, which was also reflected in chemokine receptors (Figure S2C). Finally, several immune-process targets, as well as oncogene families that are closely related to the development of breast cancer, were also included in the analysis, and the differences between TIRS subgroups were described (Figure S2D).
The mutation status was calculated and characterized based on somatic mutation sites in the MAF file. Specifically, both groups had high mutations in the TP53, PIK3CA, and MUC families, but the mutation frequency was higher in the high-risk group (Figure 4C) than in the low-risk group (Figure 4D). There were 259 genes with a mutation frequency of more than 5% in the high-risk group and 43 in the low-risk group. Higher frequency mutations lead to more genetic instability, which may be associated with poor clinical outcomes in patients in the high-risk group.
Immune processes
To better understand the biological basis of immune score and TNBC survival, the potential mechanisms of the molecules and pathways were further explored. First, we analyzed the differentially expressed genes from the TCGA cohort. The low-risk group had elevated expression of genes related to immune infiltration and a high apoptosis rate. GSEA was performed to clarify which signals were enriched in the tumor microenvironment (Figure 4E). The chemokine signaling pathway (p = 0.02, normalized enrichment score [NES] = 1.56), the JAK/STAT signaling pathway (p = 0.02, NES = 1.40), natural killer (NK)-cell-mediated cytotoxicity (p = 0.04, NES = 1.58), and the T cell receptor signaling pathway (p = 0.01, NES = 1.72) were activated in low-risk patients.
The results of KEGG and GO pathway enrichment analyses were consistent with the above description (Figures 4F and 4G). Cytokine receptor interactions, chemokine signaling pathways, and NK-cell-mediated cytotoxicity pathways were significantly enriched in KEGG. Meanwhile, GO analysis suggested that in the low-risk group, the molecular function was mainly reflected in immune response, signal transduction, inflammatory response, cell-surface-receptor signaling pathway, and innate and adaptive immune response, while biological processes were mainly concentrated for chemokines, transmembrane signal receptors, and cytokines. These results suggest that patients with lower risk scores may have higher levels of immune activity.
Protein-level validation based on AHQU cohorts
Transcriptome-level outcomes were extended to the protein level to explore the clinical potential of TIRS. The technical route is illustrated in Figure 5A. In this study, formalin fixation and paraffin embedding (FFPE) from 95 patients with TNBC was obtained and used to prepare tumor-associated macrophages (TAMs), and IHC based on eight key targets was performed. A total of 2,280 patches were finally sampled to create the test dataset. Intensity conversion was performed on the RGB 3D data of the image to obtain the staining-intensity value of the patch. Then, the fitting histogram of each patch area was calculated to clarify its probability distribution in the image. Finally, we calculated the risk score based on the first three parts of the captured probability values and used X-tile to obtain truncation values to divide the high- (n = 37) and low-risk (n = 58) groups.
Figure 5.

Protein-level verification of TIRS based on the AHQU cohort
(A) Workflow for training value matrix. A pathologist reviewed the slides and marked any regions with tumor, lymphocytes, and normal tissues. The tumor regions were then separated from the IHC images to create patches, which were input into computer vision and pattern recognition for training values. The top 3 probability sets were obtained.
(B) Kaplan-Meier survival analysis of patients with TNBC grouped by TIRS.
(C) ROC curves showing the prognostic prediction efficiency for the TIRS, age, ki67, NLR, and P53 index in AHQU cohort.
(D) Time-dependent ROC curve of TIRS prediction on the 1- and 5-year survival rates.
(E) Linear correlation between TIRS and neutrophil to lymphocyte ratio.
(F) Group-based distribution of risk scores (top) and survival status of patients in different groups (bottom) in the AHQU cohort.
(G) Image representing the pathological H&E staining between the high- and low-TIRS groups, with low lymphocyte infiltration in the high-risk group.
(H) Immunohistochemistry detects the expression of PDK1, GBP1, MAP2K6, EIF4EBP1, TOR2A, BCL2A, ARTN, and DLL4 in the AHQU cohort.
Survival analysis was further performed using TIRS obtained from the risk model in the AHQU cohort by separating patients into high- and low-risk groups. The survival curve (Figure 5B) showed that the survival time of the low-risk group was significantly higher than that of the high TIRS group (p = 0.014). In addition, compared with other risk factors such as age, ki67, and p53, TIRS had the highest AUC value (AUC = 0.810) (Figures 5C and 5D), and a higher neutrophil-to-lymphocyte ratio tended to be associated with a higher TIRS (Figure 5E). Patient death was more likely to occur in high-risk groups (Figure 5F). Meanwhile, the histopathological section confirmed that the low-TIRS group showed higher infiltration of lymphocytes (Figure 5G), indicating that TIRS was negatively correlated with immune infiltration and prognosis. The protein-level validation (Figure 5H) suggested that the risk-scoring model has a good predictive power for patient prognosis, which highlights the potential significance of these findings for risk assessment and survival in patients with TNBC.
Immune landscape of four tumor immune microenvironment types (TIMTs)
Considering the limited clinical benefit of patients with TNBC receiving immunotherapy, it is necessary to further identify different patterns of TNBC immune infiltration in detail. We scored 1,471 immune expression signatures and used NMF algorithm cluster analysis to identify modules of immune signature sets. The iteration number was set to 50, and the optimal clustering number was obtained as four based on the analysis of the relevant features (Figure 6A). The detailed grouping is shown in Table S4.
Figure 6.

Identification of the four distinct TIMTs
(A) The consensus map of NMF clustering.
(B) The schematic diagram of immune cell infiltration microenvironment of four TIMTs. The most significant immunoreactive subtype was reflected in TIMT-II, while the opposite was true for TIMT-IV. TIMT-I was enriched in neovascularization, and TIMT-III had the presence of a highly unstable genome.
(C) Overall survival of four TIMTs.
(D) Violin plot showing riskscore between four subtypes.
(E) Heatmap showing the immune-infiltration landscape of four subtypes.
(F) Differential distribution of key immune infiltrating cells in four TIMTs. The asterisk indicates the p value, with ∗∗∗p < 0.001.
(G) KEGG and GO analyses of the DEGs between four TIMTs.
Four TIMTs were identified by reclassification of TNBC based on immune characteristics (Figure 6B). Survival and immune characteristics were further described for different TIMTs. TIMT-II exhibited the best prognosis with statistically significant differences among the four subtypes (p = 0.035), while TIMT-IV showed the contrary (Figure 6C). According to the Wilcoxon test, a significant correlation between TIRS and the four TIMTs was found (Figure 6D). TIMT-IV patients with the worst survival had the highest risk score, while TIMT-II had the lowest risk score.
The immune cell infiltration and the scoring of key immune targets of the four TIMTs were further analyzed in detail (Figure S3A, S3B, and S4). These results suggest that TIMT-IV may have a higher immune risk and poorer immune infiltration. To test this hypothesis, heatmaps based on the results of the ESTIMATE and CIBERSORT analyses for the expression of immune cell infiltration state were developed in patients with TNBC (Figure 6E). Proinflammatory response, CD8+ T cells, cytotoxicity, checkpoints, B cells, and TILs were all enriched in the TIMT-II type (Figure 6F), suggesting an immune-activated tumor microenvironment. However, the expression of these cells and pathways was exhausted in the TIMT-IV type.
Moreover, in the KEGG and GO pathway analysis (Figures 6G, S5A, and S5B), inflammatory response, innate immunity, and adaptive immunity were prominent in the TIMT-II type, further confirming its better immune cell infiltration and immunotherapy response. Specifically, TIMT-II reflected the most important immunoreactivity subtype, which displayed the highest immune score, best survival, and smallest mutation frequency (Figures 7A and 7B). It is more likely to be in an immune-activated tumor microenvironment, which is characterized by high infiltration of immune cells (CD8+ T cells), high expression of immune cytokines, and effective enrichment of immune cell signals. In addition, the elevation of CD8a, GZMA, and PRF1 mRNA levels suggested that TIMT-II met the “hot tumor” criteria (Figure 7C), further suggesting the possible benefit of immunotherapy.
Figure 7.

Characteristic analysis of four TIMTs
(A) Waterfall diagram showing the mutation status of the four TIMTs.(B and C) Expression level of immune score and markers (CD8A, GZMA, PRF1) between four TIMTs. ∗∗∗p < 0.001.(D) The gene-expression alterations of characteristic pathways in four TIMTs. The heatmap shows the fold changes, with red representing upregulated genes and blue representing downregulated genes.(E) Boxplot showing the differential expression of HRD-related indicators. ∗∗p < 0.01, ∗∗∗p < 0.001.(F) Drug-sensitivity analysis of a single TIMT.
Conversely, TIMT-IV showed obvious immune-rejection characteristics. The significant downregulation of immune-related genes and more frequent mutations made it more prone to “cold tumor” features. The low expression of immunoregulatory factors (IDO1, ICOs, CD27, CTLA4, and CD274) and low infiltration of TILs (markers as CD8a, GZMA, PRF1) further indicated a lower immunotherapy response rate (Figures 7C and S4).
TMIT-I patients had more non-synonymous mutations (Figure 7A) and the highest estimated stromal score (Figure S3C), suggesting that they might comprise the distribution of interstitial cells, such as tumor-associated fibroblasts. Moreover, despite the lack of unique genomic changes, they showed features associated with cancer stem cells (CSCs). To further understand the alterations in this subtype, the JAK/STAT3 signaling pathway (Figure 7D), which plays a crucial role in the maintenance of breast CSCs, was investigated. The high expression of JAK1, interleukin-6 (IL-6), and STAT3, suggesting that the TIMT-I subtype may be a potential beneficiary of STAT3 inhibitors. In response to these characteristics, we tested TIMT-I samples for drug sensitivity to provide a possible medication strategy.15 We found that, except for being sensitive to JAK inhibitors (CEP.701), these patients may also benefit from vascular endothelial growth factor (VEGF) inhibitors (Axitinib, AZ628), TKI, SRC inhibitors (Bosutinib), and PI3K and mTOR inhibitors (NVP BEZ235) (Figure 7F).
For TIMT-III, the main difference was the highly unstable genome. The number of mutations in this subtype was equal to the sum of the mutations of other subtypes, and most of them were non-synonymous mutations (Figure 7A). Most of the TP53 mutations in TIMT-III were frameshift insertions (ins) or frameshift deletions (dels), while most of the others were missense mutations. The high level of gene instability may be one of the factors contributing to the higher immune risk score of this subtype. In addition, higher homologous recombination deficiency (HRD) (Figure 7E) is present in TIMT-III as a matter of course and is a common molecular feature of genomic instability. Interestingly, we also found that the expression of CDK4 and CDK6 (Figure 7D) was preserved in TIMT-III tumors, while the expression of RB1 mRNA was significantly reduced, suggesting that this type may be a potential candidate for CDK4/6 inhibitors. Based on these characteristics, we conducted drug-sensitivity tests (Figure 7F) on the samples and found that cisplatin, which acts on DNA function and microtubule-associated inhibitors (Epothilone.b), may produce better clinical benefits.
Discussion
When immunotherapy has changed the pattern of tumor management, the analysis and identification of immune benefits of patients with TNBC will be more important than ever before. Because of limited performance and inconsistent predictive values of clinicopathologic variables for survival, patients with TNBC, as a heterogeneous group, urgently need reliable classification criteria to identify subgroups that might benefit more from the addition of immunotherapy. We have adopted a systematic and comprehensive biomarker detection and verification method and successfully identified a novel multi-molecular panel that can effectively identify the benefits of immunotherapy and clinical prognosis of TNBC patients. These findings were verified in several independent clinical cohorts, which further confirmed the effectiveness of TIRS in depicting the immune landscape of TNBC. We further collected FFPE samples and confirmed the clinical potential of this feature at protein level, highlighting the potential significance of these findings for their clinical translation for improved risk assessment and survival in patients with TNBC. Moreover, based on the gene-expression signals derived from intratumoral immune infiltrates, we distinguished four immune clusters with significantly different molecular landscapes, ultimately providing a comprehensive description of the immune environment in TNBC.
We established an 8-gene panel to distinguish between high and low risk of the sample and successfully stratified prognosis in all cohorts. However, because the AUC focuses on discrimination rather than risk prediction, and the significance of its increment cannot be explained intuitively from a clinical perspective, the degree of improvement in the AUC alone is not enough to explain the contribution of newly added indicators to the clinical model degree. The model utility quantitative evaluation indicators IDI and NRI were applied to quantify the contribution of TIRS. The results showed that TIRS has a better risk prediction than the traditional TNBC staging assessment, and this advantage was not optimized after the joint TNM staging. It is suggested that TIRS can be used as a good index to independently assess prognosis. In addition, the risk assessment and survival probability were further quantified according to the nomogram containing TIRS with clinical information, and the DCA model also showed that TIRSs all exhibited better clinical net income at different threshold probabilities. This has overcome its unsatisfactory efficiency, in contrast to some clinicopathological variables. Therefore, the panel provides a potential supplemental prognostic tool for TNBC in the clinic.
In the past few years, immunotherapy has been recognized as an important clinical option in cancer therapy,16 and TNBC with stronger immunogenicity is no exception. Extensive clinical trials have shown that immunotherapy raises great hope for the treatment of TNBC, especially in PD-L1-positive subgroups. However, we must be aware that PD-L1 does not seem to be an ideal predictor of immune benefits. The phase III Keynote-522 trial described that the proportion of PCR among patients in the pembrolizumab arm (64.8%) was significantly higher than that in patients without anti-PD-1 treatment (51.2%).17 However, this benefit was observed regardless of PD-L1 status. The same numerical improvement results in pathological complete response were observed in the IMpassion031 and GeparNuevo study, where atezolizumab and durvalumab were beneficial in early TNBC regardless of PD-L1 status.18,19 The response observed in patients with PD-L1-negative tumors underscores the need to study more powerful biomarkers. In this context, precise immune signatures are essential for defining refined TNBC immune characteristics to better predict the response to immunotherapy.
The superiority of TIRS was reflected in the immune landscape of TNBC. The immune characteristics of the low-risk group were as follows: (1) more immune cell infiltration and higher estimate immune scores; (2) higher expression of immune checkpoints; (3) higher expression of immunocyte-associated chemokines and their receptors; and (4) higher levels of MHC genes involved in immune regulation. In addition, in terms of mutation frequency, both groups contained high mutations of TP53, PIK3CA, and MUC families, but the high-risk group had more frequent mutations (gene number with mutation frequency >5%: 259 versus 43), specifically reflected in oncogenes such as MED12, BAP1, and FGD5, which are associated with poor survival in patients with breast cancer.20, 21, 22, 23 Then, we attempted to analyze the potential pathway mechanism. Pathways such as the T cell receptor signaling pathway, NK-cell-mediated cytotoxicity, and the chemokine signaling pathway were significantly enriched in the low-risk group by both GSEA and KEGG. In addition, GO analysis also showed the immune characteristics of the low-risk group, such as immune response, adaptive immune response, and chemokine activity. TIRS not only supports the fact that high-risk patients have a poor prognosis but also predicts the immune characteristics of the TNBC cohort well, suggesting that ICI administration for subgroups may directly guide the curative effect of these patients.
Previous studies have shown that in addition to the classification of PAM50, the genome-wide map seems to have identified several TNBC subgroups, namely immunomodulatory (IM), luminal androgen receptor (LAR), basal-like 1 (BL1), BL2, mesenchymal (MES), and mesenchymal stem-like (MSL).24,25 Jiang further overlapped it into four subtypes: basal-like/immune suppressed (BLIS), basal-like/immune activated (BLIA), LAR, and mesenchymal (MES),26 which is consistent with the research of Burstein.27 Based on these data and considering the limited clinical benefits of immunotherapy in unselected patients with TNBC, research toward optimizing treatment through molecular subtyping needs to be continuously promoted. Therefore, in addition to constructing the immune panel TIRS, we performed NMF clustering to further subdivide TNBC subgroups specifically based on immune characteristics. Four TIMTs with different characteristics were identified. The most significant immunoreactive subtype was reflected in TIMT-II (16%), which showed the highest estimated immune score and the best survival. Moreover, it had effective enrichment of immune cell signals and a better expression of immune cytokines such as immunocyte-associated chemokines and MHC genes, especially the significant overexpression of PD-L1, CTLA4, and IDO1, providing a rationale for the use of immune checkpoint blockade as a therapeutic approach. In addition, TIMT-II also conforms to the criteria of hot tumors according to the criteria of Chen,28 specifically manifested as higher levels of CD8A, GZMA, and PRF1 mRNA, which further suggests the possible benefits of immunotherapy. In contrast, TIMT-IV showed immune rejection, with the worst survival and the highest TIRS, and was significantly associated with the downregulation of immune genes. Genetic mutations in ATM, ARTX, and ERCC2 suggest that the TIMT-IV subtype is related to an imbalance in the DNA damage repair mechanism.
Despite the lack of distinctive genomic alterations, tumors in the TIMT-I subtype (23.7%) displayed characteristics associated with CSCs. Meanwhile, this subtype displayed the highest estimated stromal score, suggesting a possible correlation with the widespread distribution of cancer-related fibroblasts and tumor-associated vascular cells.29 The significant enrichment of the JAK/STAT3 signaling pathway confirmed this feature because it not only plays a crucial role in the maintenance of breast CSCs but is also a key driver in regulating the interaction between fibroblasts and tumors.30,31 This subtype exhibited a higher expression of JAK1, IL-6, and STAT3, which are important drivers of JAK/STAT3 activation.32 Despite the recent evidence of the limited activity of the JAK1/2 inhibitor ruxolitinib in metastatic TNBC,33,34 our data suggest that the identification of TIMT-I subtypes in JAK/STAT-activated populations may be a potential beneficiary of STAT3 inhibitors. In addition, despite being genetically more stable, TIMT-I tumors showed overexpression of “inducing angiogenesis” hallmarks such as VEGF, PDGF, and PDGFR. It may also be affected by the activation of JAK/STAT3,35 but this feature promoted the occurrence of TIMT-I tumors and supported the benefits of anti-angiogenic therapy in this group. The outcome of drug-susceptibility prediction confirmed this view.
Differentiated TIMT-III (44.3%) to other TNBC subtypes was the presence of a highly unstable genome, with which the number of mutations in this subtype is equal to the sum of others, and we demonstrated that TIMT-III tumors retained characteristics of DNA damage while showing significantly lower half maximal inhibitory concentration (IC50) of cisplatin and epothilone B. This suggests that patients diagnosed with TIMT-III tumors may be potential candidates for chemotherapy. Meanwhile, they may also benefit from MEK1/2 inhibitors because of the mutation in MAP2K1/2 and overexpression of KRAS, NRAS, and BRAF mRNA. Based on the different immune states observed in these four subgroups, we further explored the applicability of our TIRS signature. Specifically, the patients with high TIRS risk were significantly distributed in the TIMT-IV subtype with immune-escape characteristics, while the opposite was true for the immune-benefit subtype TIMT-II, which is completely consistent with the characteristic analysis of the above subtypes.
Although this study provides important evidence for the application of immune models for the benefit of immunotherapy and prognosis of TNBC, it still has some shortcomings. The retrospective nature of our study is an inevitable limitation. We must recognize that sampling biases caused by tumor genetic heterogeneity and cross-platform integration can only be reduced, not completely eliminated. To minimize this bias, we included as many datasets (including our own cohort) as possible for rigorous verification and combined different functions, such as Combat, to reduce batch effects. Second, the incomplete clinicopathological information of most breast cancer cohorts used in our study led to the instability of TIRS in the construction of a clinical nomogram, and the adjustment for more clinicopathological information in multivariate Cox regression analysis may affect the efficiency of TIRS as an independent prognostic factor. However, compared with TNM staging, which is already excellent in clinical practice, IDI and NRI both showed better benefits of TIRS. Compared with previous studies, we strengthened the connection between mRNA and proteome and conducted clinical cohort verification at protein level. In contrast to relying solely on pathologists to judge the intensity of staining, as has been done in the past, we also performed machine analysis on immunohistochemical images to quantify the pathological information and more intuitively present the actual expression status of different markers. In addition, IHC can be widely used as a routine test in subsequent studies at hospitals. Therefore, our current study provides convincing evidence that the proposed TIRS model is valuable for predicting prognosis and immune-invasion characteristics. Nevertheless, future prospective studies with larger patient cohorts are still necessary before considering these biomarkers in clinical settings.
In conclusion, we have identified and developed a novel TIRS signature that is a robust tool for survival prediction and treatment guidance for TNBC. Four TIMT immune-landscape classifications of TNBC were obtained. These outcomes need to be validated in future prospective studies, but our findings highlight the potential clinical implications of more appropriate patient choices and improved individualized treatment strategies for TNBC patients.
Materials and methods
TNBC cohort dataset acquisition and preprocessing
To obtain the mRNA expression profiles of TNBC patients, we conducted a systematic search using TCGA (https://portal.gdc.cancer.gov/), GEO (https://www.ncbi.nlm.nih.gov/geo/), and METABRIC (http://www.cbioportal.org/) databases. Detailed steps are provided in the supplemental methods.
The specimens for the validation cohort at the protein level were collected independently from the AHQU cohort. Based on the WHO criteria, FFPE samples from patients in the cohort were screened by pathologists to further clarify the sample quality and molecular subtypes.
TIRS panel identification
To determine the differential expression signature with the diagnostic value of patients with TNBC, the Limma package in R was used for patients with and without TNBC. The differentially expressed genes were integrated with immune-related genes for further analysis. The LASSO Cox model was used to identify the most predictive immune risk markers in TNBC and output gene coefficient. The regression coefficient was used to calculate the risk score and construct the TIRS. The details are shown in the supplemental methods.
Robustness verification of the TIRS model
There are many shortcomings in the application of AUCs in clinical models. It focuses more on differentiation, and the improvement degree of the AUC alone is not enough to explain the contribution degree of newly added indicators to clinical models. Therefore, the improvement in TIRS was measured by calculating the NRI and IDI.12 Specific methods and formulas are shown in the supplemental methods.
Clinical feature analysis
The significance of the AUC increment usually cannot be directly explained from a clinical perspective. Therefore, we adopted the DCA curve to obtain the net benefit of this scoring model. Riskplot and risk heatmaps were used to reflect the specific distribution state of patients related to the risk score. In addition, multivariate Cox regression analysis was used to assess whether the risk score was independent of other clinical features, and a visual risk prediction was provided with a nomogram score for each clinical feature. Nomograms and calibration plots were drawn using the “rms” package of R software. The performance of the Nomo diagram was assessed using a calibration curve, time-dependent ROC analysis, and DCA curve analysis.
Immune cell characteristics and mutational landscape
CIBERSORT deconvolution analysis36 and ssGSEA were performed to quantify the immune infiltrating cells in TNBC specimens. Both estimated the proportions of cell types in a mixed cell population based on normalized data and matched more than 20 immune cells. Furthermore, the ratio of immune stroma of each TNBC sample was expressed in the estimate score, stromal and immune scores, and tumor purity, calculated by the ESTIMATE package with default parameters.37 These scores were calculated by decomposing the immune components from the expression profiles.
The somatic mutation and copy-number-variation profiles obtained from TCGA were transformed. Using “Maftools” and “Genvisr” in R software, a waterfall diagram was drawn to visualize the mutation frequency.In addition, HRD scores38 were defined as the sum of loss of heterozygosity (LOH), Large-scale state Transitions (LST), and Telomeric Allelic imbalance (TAI), that is, HRD=ScoreLOH+ScoreLST+ScoreTAI.
Pathway enrichment analysis
The analysis of immune landscapes among different subtypes was further characterized based on transcriptome and mutation maps. After identifying differentially expressed genes in high- and low-risk groups, GO and KEGG pathway enrichment analyses were performed, and p values were adjusted using the Benjamini and Hochberg methods. GSEA was performed between different subtypes using the R package clusterProfiler.39 The NES was defined as the degree of each immune infiltration characteristic.
Preparation and IHC of tissue microarray
For each TNBC tissue specimen, the typical tissue was first identified on the H&E section, and then the tissue was transferred to a wax mold to make a tissue microarray (TMA) module. Further H&E staining and IHC of key targets were implemented based on TMA, and detailed steps are provided in the supplemental methods.
Fully automatic image analysis and design
The image-data-information-processing method based on MATLAB software was chosen for image recognition of immunohistochemical outcomes. As the whole immunohistochemical image contains redundant information and artifacts such as folding, missing, and broken tissue, three representative image patches were selected for each complete TNBC image by two pathologists selecting tumor regions. This procedure eliminated artifacts and low-quality regions and was performed on 8-indicator staining immunohistochemical images.
Next, we extracted all representative regions of each index of each patient from its original index image and gathered them into a rectangular image. Detailed calculation methods are provided in the supplemental methods. Then, we matched the dyeing intensity value with the risk weight of each index to obtain the risk score. X-tile software was used to calculate the best truncation value, and then that was divided into high- and low-risk groups to verify the survival difference.
Construction of four TIMTs
To further reveal the distribution of features in patients with TNBC and identify robust clusters, unsupervised NMF was used to process the transcriptome expression data in the training set. The cluster number K was set to 2–10 and 50 iterations. The optimal clustering number was determined using the cophenetic, dispersion, and silhouette indicators. NMF clustering was combined with ssGSEA to reveal immune-related expression patterns in different subtypes and was set as differential TIMTs.
Based on NMF clustering, Kaplan-Meier analysis was used to evaluate the survival results of each group. Subsequently, we compared the expression levels of various immune cells and immune factors, as well as molecules or pathways associated with immune invasion and immune escape enriched in each group. The prediction of the drug-sensitivity response based on the sorted expression profiles for the different subtypes was executed to obtain a better clinical-benefit scheme.
Statistics analysis
All statistical analyses were performed using R software (v.4.0.3). The differences in OS times between different subtypes can be determined by constructing a survival curve and estimating by the bilateral log rank test. The Spearman method was used to calculate the p value in the correlation analysis. Kruskal-Wallis test was performed for more than two groups, and that of two groups was compared by the Wilcoxon test. All statistical tests were two-sided unless otherwise stated, and statistical significance was set at p <0.05.
Acknowledgments
This work was supported by funds from the National Natural Science Foundation of China (grant numbers 82174222 and 81973677) and the Shandong Provincial Natural Science Foundation (grant number ZR2019MD011).
Author contributions
Research design, C.L., Y.L., X.X., and C.S.; data collection, Y.L., J.W., XX., Y.Y., and L.Z.; data analysis, C.L., L.Z., C.W., F.F., and J.L.; manuscript preparation, C.L., Y.L., C.G., H.L., and J.Z.; chart preparation, Y.L., C.W., C.G., and L.L.; revisions, C.L., J.Z., X.X., and C.S. All authors confirm that they contributed to manuscript reviews, critical revision for important intellectual content, and read and approved the final draft for submission. All authors are also responsible for the manuscript content.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.04.034.
Supplemental information
References
- 1.Cao L., Niu Y. Triple negative breast cancer: special histological types and emerging therapeutic methods. Cancer. Biol. Med. 2020;17:293–306. doi: 10.20892/j.issn.2095-3941.2019.0465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaudhary L.N. Early stage triple negative breast cancer: management and future directions. Semin. Oncol. 2020;47:201–208. doi: 10.1053/j.seminoncol.2020.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marra A., Trapani D., Viale G., Criscitiello C., Curigliano G. Practical classification of triple-negative breast cancer: intratumoral heterogeneity, mechanisms of drug resistance, and novel therapies. NPJ. Breast. Cancer. 2020;6:54. doi: 10.1038/s41523-020-00197-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmid P., Adams S., Rugo H.S., Schneeweiss A., Barrios C.H., Iwata H., Diéras V., Hegg R., Im S.A., Shaw Wright G., et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N. Engl. J. Med. 2018;379:2108–2121. doi: 10.1056/nejmoa1809615. [DOI] [PubMed] [Google Scholar]
- 5.Mediratta K., El-Sahli S., D'Costa V., Wang L. Current progresses and challenges of immunotherapy in triple-negative breast cancer. Cancers (Basel) 2020;12:3529. [Google Scholar]
- 6.de Melo Gagliato D., Buzaid A.C., Perez-Garcia J., Cortes J. Immunotherapy in breast cancer: current practice and clinical challenges. BioDrugs. 2020;34:611–623. doi: 10.1007/s40259-020-00436-9. [DOI] [PubMed] [Google Scholar]
- 7.Hudeček J., Voorwerk L., van Seijen M., Nederlof I., de Maaker M., van den Berg J., van de Vijver K.K., Sikorska K., Adams S., Demaria S., et al. Application of a risk-management framework for integration of stromal tumor-infiltrating lymphocytes in clinical trials. NPJ Breast Cancer. 2020;6:15. doi: 10.1038/s41523-020-0155-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Judes G., Rifaï K., Daures M., Dubois L., Bignon Y.J., Penault-Llorca F., Bernard-Gallon D. High-throughput «Omics» technologies: new tools for the study of triple-negative breast cancer. Cancer Lett. 2016;382:77–85. doi: 10.1016/j.canlet.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 9.Ma C.X., Luo J., Ellis M.J. Molecular profiling of triple negative breast cancer. Breast. Dis. 2011;32:73–84. doi: 10.3233/bd-2010-0309. [DOI] [Google Scholar]
- 10.Parker J.S., Mullins M., Cheang M.C., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z., et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009;27:1160–1167. doi: 10.1200/jco.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez H., Zenklusen J.C., Staudt L.M., Doroshow J.H., Lowy D.R. The next horizon in precision oncology: proteogenomics to inform cancer diagnosis and treatment. Cell. 2021;184:1661–1670. doi: 10.1016/j.cell.2021.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pencina M.J., D'Agostino R.B., D' Agostino R.B., D'Agostino R.B., Jr., Vasan R.S. Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat. Med. 2008;27:157–172. doi: 10.1002/sim.2929. discussion 207-12. [DOI] [PubMed] [Google Scholar]
- 13.Chowell D., Morris L.G.T., Grigg C.M., Weber J.K., Samstein R.M., Makarov V., Kuo F., Kendall S.M., Requena D., Riaz N., et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science. 2018;359:582–587. doi: 10.1126/science.aao4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia H., Truica C.I., Wang B., Wang Y., Ren X., Harvey H.A., Song J., Yang J.M. Immunotherapy for triple-negative breast cancer: existing challenges and exciting prospects. Drug Resist. Updat. 2017;32:1–15. doi: 10.1016/j.drup.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Yang C., Huang X., Li Y., Chen J., Lv Y., Dai S. Prognosis and personalized treatment prediction in TP53-mutant hepatocellular carcinoma: an in silico strategy towards precision oncology. Brief. Bioinform. 2021;22:bbaa164. doi: 10.1093/bib/bbaa164. [DOI] [PubMed] [Google Scholar]
- 16.Wolchok J. Putting the immunologic brakes on cancer. Cell. 2018;175:1452–1454. doi: 10.1016/j.cell.2018.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Schmid P., Cortes J., Pusztai L., McArthur H., Kümmel S., Bergh J., Denkert C., Park Y.H., Hui R., Harbeck N., et al. Pembrolizumab for early triple-negative breast cancer. N. Engl. J. Med. 2020;382:810–821. doi: 10.1056/nejmoa1910549. [DOI] [PubMed] [Google Scholar]
- 18.Mittendorf E.A., Zhang H., Barrios C.H., Saji S., Jung K.H., Hegg R., Koehler A., Sohn J., Iwata H., Telli M.L., et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): a randomised, double-blind, phase 3 trial. Lancet. 2020;396:1090–1100. doi: 10.1016/s0140-6736(20)31953-x. [DOI] [PubMed] [Google Scholar]
- 19.Sinn B.V., Loibl S., Hanusch C.A., Zahm D.M., Sinn H.P., Untch M., Weber K., Karn T., Becker C., Marmé F., et al. Immune-related gene expression predicts response to neoadjuvant chemotherapy but not additional benefit from PD-L1 inhibition in women with early triple-negative breast cancer. Clin. Cancer Res. 2021;27:2584–2591. doi: 10.1158/1078-0432.ccr-20-3113. [DOI] [PubMed] [Google Scholar]
- 20.Li K., Zhang T.T., Zhao C.X., Wang F., Cui B., Yang Z.N., Lv X.X., Yeerjiang Z., Yuan Y.F., Yu J.M., et al. Faciogenital Dysplasia 5 supports cancer stem cell traits in basal-like breast cancer by enhancing EGFR stability. Sci. Transl. Med. 2021;13:eabb2914. doi: 10.1126/scitranslmed.abb2914. [DOI] [PubMed] [Google Scholar]
- 21.Yoshida M., Ogawa R., Yoshida H., Maeshima A., Kanai Y., Kinoshita T., Hiraoka N., Sekine S. TERT promoter mutations are frequent and show association with MED12 mutations in phyllodes tumors of the breast. Br. J. Cancer. 2015;113:1244–1248. doi: 10.1038/bjc.2015.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mishima C., Kagara N., Tanei T., Naoi Y., Shimoda M., Shimomura A., Shimazu K., Kim S.J., Noguchi S. Mutational analysis of MED12 in fibroadenomas and phyllodes tumors of the breast by means of targeted next-generation sequencing. Breast. Cancer. Res. Treat. 2015;152:305–312. doi: 10.1007/s10549-015-3469-1. [DOI] [PubMed] [Google Scholar]
- 23.Masclef L., Ahmed O., Estavoyer B., Larrivée B., Labrecque N., Nijnik A., Affar E.B. Roles and mechanisms of BAP1 deubiquitinase in tumor suppression. Cell Death Differ. 2021;28:606–625. doi: 10.1038/s41418-020-00709-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lehmann B.D., Bauer J.A., Chen X., Sanders M.E., Chakravarthy A.B., Shyr Y., Pietenpol J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011;121:2750–2767. doi: 10.1172/jci45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bareche Y., Venet D., Ignatiadis M., Aftimos P., Piccart M., Rothe F., Sotiriou C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann. Oncol. 2018;29:895–902. doi: 10.1093/annonc/mdy024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang Y.Z., Ma D., Suo C., Shi J., Xue M., Hu X., Xiao Y., Yu K.D., Liu Y.R., Yu Y., et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell. 2019;35:428–440. doi: 10.1016/j.ccell.2019.02.001. e5. [DOI] [PubMed] [Google Scholar]
- 27.Burstein M.D., Tsimelzon A., Poage G.M., Covington K.R., Contreras A., Fuqua S.A., Savage M.I., Osborne C.K., Hilsenbeck S.G., Chang J.C., et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015;21:1688–1698. doi: 10.1158/1078-0432.ccr-14-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y.P., Zhang Y., Lv J.W., Li Y.Q., Wang Y.Q., He Q.M., Yang X.J., Sun Y., Mao Y.P., Yun J.P., et al. Genomic analysis of tumor microenvironment immune types across 14 solid cancer types: immunotherapeutic implications. Theranostics. 2017;7:3585–3594. doi: 10.7150/thno.21471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walter S.G., Scheidt S., Nißler R., Gaisendrees C., Zarghooni K., Schildberg F.A. Depth characterization of stromal cells within the tumor microenvironment yields novel therapeutic targets. Cancers (Basel) 2021;13:1466. doi: 10.3390/cancers13061466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu H., Lee H., Herrmann A., Buettner R., Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer. 2014;14:736–746. doi: 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 31.Kuzet S.E., Gaggioli C. Fibroblast activation in cancer: when seed fertilizes soil. Cell. Tissue. Res. 2016;365:607–619. doi: 10.1007/s00441-016-2467-x. [DOI] [PubMed] [Google Scholar]
- 32.Taher M.Y., Davies D.M., Maher J. The role of the interleukin (IL)-6/IL-6 receptor axis in cancer. Biochem. Soc. Trans. 2018;46:1449–1462. doi: 10.1042/bst20180136. [DOI] [PubMed] [Google Scholar]
- 33.Lynce F., Williams J.T., Regan M.M., Bunnell C.A., Freedman R.A., Tolaney S.M., Chen W.Y., Mayer E.L., Partridge A.H., Winer E.P., Overmoyer B. Phase I study of JAK1/2 inhibitor ruxolitinib with weekly paclitaxel for the treatment of HER2-negative metastatic breast cancer. Cancer Chemother. Pharmacol. 2021;87:673–679. doi: 10.1007/s00280-021-04245-x. [DOI] [PubMed] [Google Scholar]
- 34.Stover D.G., Gil Del Alcazar C.R., Brock J., Guo H., Overmoyer B., Balko J., Xu Q., Bardia A., Tolaney S.M., Gelman R., et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ. Breast. Cancer. 2018;4:10. doi: 10.1038/s41523-018-0060-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garg M., Shanmugam M.K., Bhardwaj V., Goel A., Gupta R., Sharma A., Baligar P., Kumar A.P., Goh B.C., Wang L., Sethi G. The pleiotropic role of transcription factor STAT3 in oncogenesis and its targeting through natural products for cancer prevention and therapy. Med. Res. Rev. 2020;41:1291–1336. doi: 10.1002/med.21761. [DOI] [Google Scholar]
- 36.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoshihara K., Shahmoradgoli M., Martínez E., Vegesna R., Kim H., Torres-Garcia W., Treviño V., Shen H., Laird P.W., Levine D.A., et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi Z., Shen J., Qiu J., Zhao Q., Hua K., Wang H. CXCL10 potentiates immune checkpoint blockade therapy in homologous recombination-deficient tumors. Theranostics. 2021;11:7175–7187. doi: 10.7150/thno.59056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.