

Abstract
A maltogenic amylase gene was cloned in Escherichia coli from a gram-negative thermophilic bacterium, Thermus strain IM6501. The gene encoded an enzyme (ThMA) with a molecular mass of 68 kDa which was expressed by the expression vector p6xHis119. The optimal temperature of ThMA was 60°C, which was higher than those of other maltogenic amylases reported so far. Thermal inactivation kinetic analysis of ThMA indicated that it was stabilized in the presence of 10 mM EDTA. ThMA harbored both hydrolysis and transglycosylation activities. It hydrolyzed β-cyclodextrin and starch mainly to maltose and pullulan to panose. ThMA not only hydrolyzed acarbose, an amylase inhibitor, to glucose and pseudotrisaccharide (PTS) but also transferred PTS to 17 sugar acceptors, including glucose, fructose, maltose, cellobiose, etc. Structural analysis of acarbose transfer products by using methylation, thin-layer chromatography, high-performance ion chromatography, and nuclear magnetic resonance indicated that PTS was transferred primarily to the C-6 of the acceptors and at lower degrees to the C-3 and/or C-4. The transglycosylation of sugar to methyl-α-d-glucopyranoside by forming an α-(1,3)-glycosidic linkage was demonstrated for the first time by using acarbose and ThMA. Kinetic analysis of the acarbose transfer products showed that the C-4 transfer product formed most rapidly but readily hydrolyzed, while the C-6 transfer product was stable and accumulated in the reaction mixture as the main product.
Several maltogenic amylases (EC 3.2.1.-) and closely related enzymes were cloned from gram-positive bacteria, including Bacillus species (4, 13). The enzymes were different from typical amylases in that they (i) were not secreted outside the cell, (ii) preferred cyclodextrins to starch or pullulan as a substrate, and (iii) exhibited both transglycosylation and hydrolysis activities on various substrates. They hydrolyzed starch and β-cyclodextrin mainly to maltose and pullulan to panose. Many of these properties, if not all, were shared by some amylolytic enzymes, including neopullulanases (EC 3.2.1.135) and cyclomaltodextrinases (EC 3.2.1.54; CDases) (7, 10, 17, 20, 26).
The action modes of two maltogenic amylases (4, 13) and a CDase (17) isolated from three different Bacillus species were investigated by time course experiments with soluble starch or maltotriose as a substrate. The enzymes transferred a sugar molecule (donor) released after the hydrolysis of an α-(1,4)-glycosidic linkage to a reducing end of another sugar molecule (acceptor) by forming an α-(1,6)-glycosidic linkage. The coupled transglycosylation and hydrolysis activities of these enzymes were used for the production of branched oligosaccharides (BOS) from liquefied starch (15, 23), giving a more efficient process than the traditional one (31).
The maltogenic amylases from Bacillus licheniformis (BLMA [13]), Bacillus stearothermophilus (BSMA, [4]), and B. subtilis (unpublished data) could hydrolyze acarbose, an amylase inhibitor, at different levels of efficiency. Acarbose is a pseudotetrasaccharide that has a pseudosugar ring at the nonreducing end [4,5,6-trihydroxy-3-(hydroxymethyl)-2-cyclohexene-1-yl] linked to the nitrogen of 4-amino-4,6-dideoxy-d-glucopyranose (4-amino-4-deoxy-d-quinovo-pyranose), which is linked via an α-(1,4)-glycosidic linkage to maltose. The pseudotrisaccharide (PTS) resulting from the hydrolysis of acarbose by these enzymes was transferred to the C-6 of glucose forming isoacarbose. This indicated that the catalytic properties unique to maltogenic amylases are probably due to differences in the tertiary structures of the proteins. The primary structures of maltogenic amylases in four regions were well conserved, and their secondary structure was likely to constitute a (β/α)8-barrel domain as with other amylolytic enzymes (11, 12). The characterization of amylolytic enzymes that exhibit transglycosylation and/or cyclodextrin hydrolyzing activity at the level of protein structure and enzymatic properties would be quite useful for understanding catalytic activities and substrate binding patterns more precisely.
In this paper, we report on the cloning and physicochemical properties of another maltogenic amylase of a Thermus strain (ThMA) that was capable of hydrolyzing acarbose and transferring PTS to various acceptors. The enzyme was isolated from a thermophilic gram-negative bacterium, Thermus strain IM6501, and was more stable at high temperatures than other maltogenic amylases. Studies of the transferring activity of the thermostable enzyme by using acarbose and methylation of the resulting transfer products revealed additional modes of transglycosylation. Transglycosylation of a donor sugar molecule (PTS) to an acceptor molecule by forming an α-(1,3)-glycosidic linkage was demonstrated for the first time by using acarbose and ThMA.
MATERIALS AND METHODS
Bacterial strains and cultivation.
Thermus strain IM6501 was isolated from compost (16). Escherichia coli MC1061 [F− araD139 recA13 Δ(araABC-leu)7696 galU galK ΔlacX74 rpsL thi hsdR2 mcrB] and HB101 [F− supE44 ara-14 proA2 galK2 lacY1 rpsL20 xyl-5 mtl-1 hsdS20(rB− mB−)] were used as hosts for DNA manipulation and transformation. Thermus strain IM6501 and E. coli strains were grown in Luria-Bertani medium (1% Bacto Tryptone, 0.5% yeast extract, 0.5% NaCl) at 65 and 37°C, respectively. E. coli transformants were grown in Luria-Bertani medium containing ampicillin (100 μg/ml). pBR322, pUC119, and pBluescript II SK (Stratagene) were used as cloning vectors.
Gene cloning.
Chromosomal DNA of Thermus IM6501 isolated by the spool method (8) was digested with PstI, SalI, or SacI and ligated into pUC119 at the corresponding restriction enzyme sites. Each genomic DNA library was transformed into E. coli, and the resulting transformants were screened for starch hydrolyzing activity by the iodine test after treating the colonies with d-cycloserine as described previously (13).
Nucleotide sequence analysis.
Plasmid DNA sequencing was carried out by the chain termination method (29) with two automatic DNA sequencers, ALFexpress (Pharmacia Biotechnology) and ABI377 PRISM (Perkin-Elmer). Sequencing reactions were carried out by using the Cy5 AutoRead sequencing kit for ALFexpress and the ABI Prism BigDye terminator cycle sequencing kit for ABI377 PRISM as the manufacturers recommended. A PE9600 thermal cycler (Perkin-Elmer) was used for thermal cycling sequencing. Both DNA strands were sequenced.
Overexpression of ThMA.
An expression vector, p6xHis119, was constructed by using the promoter for the maltogenic amylase gene of B. licheniformis for stable overexpression and six histidine residues for the easy purification of foreign proteins. pUCIJ119 containing the BLMA gene (6) was used as the frame for the construct. At first, an NcoI site was introduced at the putative translation initiation site of the BLMA structural gene by site-directed mutagenesis, and it was designated pUCIJm1. Two complementary oligonucleotides (HisTag5 and HisTag3) encoding a six-His tag were designed and inserted at the NcoI site of pUCIJm1, and it was designated p6xHBLMA. The DNA fragment of p6xHBLMA digested with EcoRI and BamHI was isolated and ligated into pUC119 at the corresponding restriction sites to introduce various cloning sites. The ThMA gene with proper restriction enzyme sites was amplified by using two primers (TMNdeI and TMHdIII) from pThMA119. The resulting PCR product was digested with NdeI and HindIII and ligated into p6xHis119 digested with the two restriction enzymes. The p6xHThMA insert was then replaced with the SalI-HindIII fragment of pThMA119 to minimize possible errors that might have been introduced during PCR.
Enzyme purification.
The extract of the E. coli transformant harboring the ThMA gene on pThMA119 was applied to fast protein liquid chromatography (Pharmacia, Uppsala, Sweden) as follows. First, it was loaded on a DEAE-TOYOPEARL 650 column (3.0 by 15 cm) equilibrated with 20 mM Tris-HCl (pH 7.5) and eluted with a linear gradient of NaCl from 0.15 to 0.4 M in the same buffer at a flow rate of 7 ml/min. The fractions with enzyme activity were concentrated by ultrafiltration with a PM-10 membrane (Amicon Co.) and then dialyzed to remove salts. They were applied to a Mono-Q HR 5/5 column, and elution was done with a linear gradient of NaCl from 0.15 to 0.3 M in 20 mM Tris-HCl (pH 7.5) at a flow rate of 1 ml/min. Active fractions were concentrated to a one-third volume and dialyzed again as described above.
ThMA with the six-His tag was purified from E. coli harboring p6xHThMA by using a nickel-nitrilotriacetic acid (Ni-NTA) column. A 50% Ni-NTA slurry was added to the cell lysate and mixed gently at 4°C for 1 h. The lysate-Ni-NTA mixture was loaded onto a column with the bottom outlet capped. After the bottom cap was removed, it was washed twice with 4 ml of a wash buffer (50 mM NaH2PO4 [pH 7.0], 300 mM NaCl, 20 mM imidazole). ThMA was eluted twice with 2.5 ml of an elution buffer consisting of 50 mM NaH2PO4 (pH 7.0), 300 mM NaCl, and 250 mM imidazole. The molecular mass of purified ThMA was determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) with 4% (wt/vol) stacking and 12% (wt/vol) resolving gels as described by Laemmli (22). Matrix-assisted laser desorption-ionization time-of-flight mass spectrometry was performed with a Lasermat 2000 (Finnigan). All the results presented in this study were obtained by using purified ThMA.
Enzyme assay.
ThMA activity was assayed at 60°C in a 50 mM sodium-acetate buffer (pH 6.0) with 3,5-dinitrosalicylic acid according to the method described by Miller (24). β-Cyclodextrin (Miwon Inc., Inchon, Korea) was used as the substrate. One unit of β-cyclodextrin hydrolyzing activity (CU) was defined as the amount of enzyme that forms reducing sugars to give an increase in absorbance at 575 nm of 1.0 as described previously (13). The protein concentration was measured by the Bradford method (2) with bovine serum albumin (Sigma Co., St. Louis, Mo.) as the standard.
Hydrolytic activity of ThMA.
Purified ThMA (5 CU) was incubated with 0.5 ml of 0.5% (wt/vol) β-cyclodextrin, soluble starch (Showa Chemicals Inc., Tokyo, Japan), pullulan (Sigma Co.), or acarbose (see below) in a 50 mM sodium-acetate buffer (pH 6.0) at 60°C for 12 h to determine its hydrolytic action mode. The reaction was stopped by boiling the mixture for 5 min. The resulting products were analyzed by thin-layer chromatography (TLC) and high-performance ion chromatography (HPIC) as described previously (23).
Formation and isolation of acarbose transfer products.
Acarbose {O-4,6-dideoxy-4-[(4,5,6-trihydroxy-3-hydroxymethyl-2-cyclohexene-1-yl)amino]-α-d-glucopyranosyl-(1→4)-O-α-d-gluco-pyranosyl-(1→4)-d-glucose}, a pseudotetrasaccharide amylase inhibitor, was a generous gift from Bayer Korea Ltd. (Seoul, Korea). ThMA (10 CU per gram of acarbose) was incubated in a reaction mixture consisting of 5% acarbose and 10% carbohydrate acceptor in 5 ml of a 50 mM Na-acetate buffer (pH 6.0) at 60°C for 48 h. The reaction mixture was loaded onto a Bio-Gel P-2 (Bio-Rad) column (2 by 90 cm; Pharmacia Biotech) equilibrated with a 50 mM NaCl solution and eluted with the same solution. Each fraction was analyzed by TLC to confirm the removal of glucose, PTS, and acceptors by gel permeation chromatography. The fractions containing transfer products were desalted by using the gel column equilibrated with deionized water. The desalted solutions were freeze-dried and resuspended in deionized and distilled water. The mixture of transfer products was spotted on a Whatman K6F TLC plate, which was irrigated with 2 ascents of isopropyl alcohol-ethyl acetate-water (3:1:1 [vol/vol/vol]) at room temperature. The plate was thoroughly dried between each ascent.
The spots representing each transfer product on the TLC plate were cut out, and the moisturized silica powder scratched off the plate was collected into a Falcon tube separately for each step. Each sample was extracted with water shaking at room temperature for 2 h. The extract was centrifuged at 15,000 × g for 15 min, and the supernatant was filtered through a 0.2-μm-pore-size membrane filter (Micro Filtration System). Each product was freeze-dried and resuspended in methanol (about 10 mg/ml) for methylation analysis.
Structure analysis of transfer products.
The structure of each transfer product was analyzed by TLC, HPIC, and nuclear magnetic resonance (NMR). TLC analysis was carried out as described above. For HPIC analysis, the sample was mixed with an equal volume of acetonitrile and boiled for 5 min. The pellet collected by centrifugation (12,000 × g for 5 min) was filtered (0.2-μm-diameter filter; Gelman Science). Twenty microliters of the sample was applied to a Carbopak PA1 column (0.4 by 25 cm; diameter, 10 μm; Dionex), and elution was done with a 0 to 30% (vol/vol) gradient of 600 mM Na-acetate in 150 mM NaOH at a flow rate of 1.0 ml/min. For NMR (JEOL JNM-LA-400; Fourier transform NMR at 400 MHz) analysis, samples (1 to 2 mg/100 μl) were dissolved in D2O and analyzed at 90°C.
Methylation analysis of transfer products.
Methylation of acarbose transfer products was carried out according to the method described by Mukerjea et al. (25). Aliquots (1 to 3 μl) were applied onto a TLC (Whatman K6F) plate and irrigated by using a solvent system made of ethylacetate-isopropyl alcohol-water (1:3:1 [vol/vol/vol]). After two irrigations, the plate was dried and visualized by dipping it in 0.3% (wt/vol) N-(1-naphthyl)-ethylenediamine–5% (vol/vol) H2SO4 in methanol and heating it at 110°C for 10 min. Methylation of β-cyclodextrin and alternan by the same method gave rise to 2,3,6-tri-O-methyl-d-glucose and 2,3,4-tri-O-methyl-d-glucose and 2,4,6-tri-O-methyl-d-glucose, respectively, and the mixture of these was used as the standard.
Nucleotide sequence accession number.
The nucleotide sequence of the ThMA gene was deposited in the EMBL and GenBank databases under accession no. AF060204.
RESULTS
Cloning and nucleotide sequence analysis of the ThMA gene.
Thermus strain IM6501 is a thermophilic, gram-negative, aerobic bacterium that does not produce endospores under any circumstances. Previous analysis of Thermus strain IM6501 for starch-hydrolyzing activity indicated that the bacterial strain contained multiple amylolytic enzymes, including an α-glucosidase and a pullulanase (unpublished data). The presence of the maltogenic amylase gene was detected by PCR amplification of the Thermus genomic DNA with primers whose sequences were deduced from multiple alignment of the conserved sequences of several maltogenic amylase genes (16). A nucleotide sequence of 300 bp amplified as a major PCR product showed 59 to 82% homology to those of maltogenic amylases (4, 13, 17, 19, 26, 27, 32). Southern blot analysis of the Thermus genomic DNA with the insert as a probe indicated that the gene was located either on a 5-kb PstI, 4.3-kb SalI, or 2.9-kb SacI fragment. A putative SacI maltogenic amylase clone selected because of its starch-hydrolyzing activity had an insert of 2.9 kb (Fig. 1A). Southern blot analysis of the Thermus strain IM6501 genomic DNA with the insert as a probe confirmed that the DNA fragment originated from the bacterial strain. The clone was designated pThMA119.
FIG. 1.

Restriction maps of the ThMA gene clones. (A) The box represents the 2.9-kb SacI DNA fragment cloned in pThMA119. The black box indicates the location of the ThMA structural gene, directing from left to right, and the lines represent the vector. (B) For overexpression and easy purification of ThMA, the structural gene on the NdeI*-HindIII DNA fragment was fused to six histidines in frame under the control of the BLMA promoter (PBLMA) by subcloning it into p6xHis119 at the NdeI and HindIII sites. The NdeI* site was introduced into the ThMA gene for convenience in subcloning.
An open reading frame of 1,761 nucleotides encoded 587 amino acids of a protein with a molecular mass of 68,207 Da. The primary structure of ThMA was compared to those of various closely related enzymes, including maltogenic amylases, neopullulanases, and CDases. ThMA had 55.3% homology with BLMA, 55.9% with CDase I-5, 69.6% with BSMA, 86.1% with neopullulanase, and 45.9% with amylase II of Thermoactinomyces vulgaris R-47 (TVA-II) at the predicted amino acid sequence level. The homology level was even higher in the four conserved regions. The sequences of the four conserved regions in ThMA were identical to those of BSMA and neopullulanase from Bacillus stearothermophilus strains and differed by only one amino acid residue from those of BLMA and CDase I-5. The spacings between the conserved regions are also known to be similar among maltogenic amylases, CDases, and neopullulanases.
Physicochemical properties of ThMA.
The purification of ThMA from recombinant E. coli harboring pThMA119 by anion-exchange chromatography resulted in a 3.5-fold purification with 27.5% recovery. The procedure, including several steps, was laborious and inefficient. Therefore, six-His-tagged ThMA was subcloned into an expression vector, which contains the BLMA promoter for stable overexpression and six histidine residues for the easy purification of the foreign proteins (Fig. 1B). ThMA was overproduced from the recombinant E. coli harboring p6xHThMA and purified by using an Ni-NTA column. By this one-step procedure, 60% of the total enzyme activity was recovered. The apparent molecular mass of ThMA purified by either method was estimated to be about 64,000 Da by SDS-PAGE (Fig. 2). However, matrix-assisted laser desorption-ionization time-of-flight mass spectrometry spectra indicated that the enzyme had a molecular mass of 68,238 Da, close to the deduced molecular mass (68,207 Da).
FIG. 2.

SDS-PAGE of purified ThMA. The apparent molecular mass of ThMA purified by a traditional chromatographic procedure (panel A, lane 1) and that of six-His-ThMA purified by using an Ni-NTA column (panel B, lane 2) from the E. coli lysate (panel B, lane 1) were approximately 64,000 Da. Standard size markers (lanes M) were myosin (205 kDa), β-galactosidase (116 kDa), phosphorylase b (97.4 kDa), bovine serum albumin (66 kDa), ovalbumin (45 kDa), and carbonic anhydrase (29 kDa).
The optimum reaction temperature of ThMA was 60°C when β-cyclodextrin was used as a substrate in a 50 mM sodium-acetate buffer (pH 6.0). The thermal inactivation of ThMA in the temperature range between 70 and 80°C showed that the enzyme had higher thermal stability than other maltogenic amylases reported up to date. Maltogenic amylases from Bacillus subtilis, B. licheniformis, and B. stearothermophilus had optimal temperatures at 45, 50, and 55°C, respectively (4, 13). The addition of 10 mM EDTA increased the thermal stability of ThMA, while the addition of CaCl2 decreased it. Thermal inactivation analysis of ThMA followed first-order kinetics, and the enthalpy change of activation (ΔH‡) of ThMA was greatest in the presence of 10 mM EDTA and smallest in the presence of 10 mM CaCl2 (Table 1).
TABLE 1.
Thermodynamic parameters for the thermal inactivation of ThMA
| ThMA in the presence of: | Temp (°C) | k (s−1) | ΔG‡ (kJ/mol) | ΔS‡ (J/mol · temp [K]) | ΔH‡ (kJ/mol) |
|---|---|---|---|---|---|
| Nothing | 70 | (4.18 ± 0.46) × 105 | 113.0 ± 0.3 | ||
| 75 | (3.84 ± 0.15) × 104 | 108.7 ± 0.1 | 844.6 ± 38.7 | 402.6 ± 13.6 | |
| 80 | (2.33 ± 0.07) × 103 | 104.5 ± 0.1 | |||
| EDTA (10 mM) | 75 | (1.85 ± 0.03) × 104 | 110.5 ± 0.0 | 1,112.1 ± 15.4 | 497.5 ± 5.4 |
| 80 | (2.14 ± 0.07) × 103 | 104.9 ± 0.1 | |||
| CaCl2 (10 mM) | 75 | (6.26 ± 0.13) × 104 | 107.0 ± 0.1 | 760.4 ± 17.2 | 371.6 ± 6.0 |
| 80 | (3.91 ± 0.09) × 103 | 103.2 ± 0.1 |
The enzyme activity was strongly inhibited by most of the metal ions tested, especially by Cu2+, Ag2+, Mn2+, and Zn2+ but not by Ba2+. ThMA was strongly inhibited by EGTA, contrary to EDTA. These results were somewhat different from those obtained with CDase I-5, which showed a two-fold higher enzyme activity in the presence of EDTA or EGTA (17). ThMA was stable in a broad range of pH values (5.5 to 10) with an optimum of 6.0 in the 50 mM sodium-acetate buffer.
Hydrolysis and transglycosylation activity of ThMA.
In order to determine the hydrolysis pattern of the enzyme, β-cyclodextrin, soluble starch, pullulan, or acarbose was reacted with purified ThMA. TLC analysis of the reaction products indicated that ThMA hydrolyzed β-cyclodextrin, pullulan, and soluble starch readily, with relative hydrolysis activities of 1,322, 100, and 80, respectively. ThMA hydrolyzed β-cyclodextrin and soluble starch to maltose and glucose (Fig. 3, lanes A and B). Pullulan was hydrolyzed mainly to panose by ThMA, but small amounts of maltose and glucose were also detected (Fig. 3, lane C). The hydrolysis of panose to maltose and glucose by ThMA was not efficient (Fig. 3, lane D). Maltotriose was hydrolyzed to glucose and maltose by the enzyme with the formation of panose and/or maltotetraose (Fig. 3, lane E). ThMA also hydrolyzed acarbose, an effective amylase inhibitor, to glucose and acarviosine-glucose (PTS; Fig. 3, lane F). Most amylases could not hydrolyze acarbose but were strongly inhibited by the compound. In an inhibition study of ThMA with acarbose and β-cyclodextrin, the enzyme hydrolyzed acarbose to glucose and PTS, but the β-cyclodextrin hydrolyzing activity of ThMA was partially inhibited by acarbose (data not shown). No apparent degradation of substrates was observed by TLC when they were incubated in the absence of ThMA.
FIG. 3.

TLC analysis of ThMA hydrolysis activity. ThMA was reacted with various substrates (0.5% [wt/vol]), including β-cyclodextrin (lane A), soluble starch (lane B), pullulan (lane C), panose (lane D), maltotriose (lane E), and acarbose (lane F). The reactions were carried out at 60°C for 12 h. Maltodextrins (MD) of G1 to G7, PTS, acarbose (Acb), and isoacarbose (IAcb) were used as standards (Std).
The transferring activity of ThMA was investigated by using acarbose as a donor and 17 different carbohydrates as acceptors. As shown in Fig. 4, mostly two or three acarbose transfer products were produced and detected by TLC in every reaction done with each carbohydrate acceptor. This indicated that PTS resulting from the hydrolysis of acarbose (the donor molecule) was transferred to the acceptors in various modes by ThMA. The transferring activity of ThMA was also tested with 30% (wt/vol) liquefied corn starch solution as a substrate. ThMA produced various BOS with panose as the major product (data not shown). Therefore, ThMA was likely to have a transferring activity like other maltogenic amylases.
FIG. 4.

TLC analysis of acarbose transfer products. Purified ThMA transferred PTS resulting from the hydrolysis of acarbose to various acceptors: glucose (A), α-MG (B), galactose (C), fructose (D), maltose (E), cellobiose (F), lactose (G), sucrose (H), and gentiobiose (I). Maltodextrins (MD) G1 to G7, PTS, acarbose (Acb), and isoacarbose (IAcb) were used as standards (Std). The resulting transfer products were numbered (1, 2, and 3) in the order of traveling distance.
Mode of transglycosylation reaction catalyzed by ThMA.
To analyze the structures of the acarbose transfer products, several of them formed in large quantities were isolated and analyzed by TLC, HPIC, NMR, and methylation. The results of TLC and HPIC chromatograms complemented each other in their abilities to separate the various compounds. The mobility of compounds having α-(1,4)- or α-(1,3)-linkages was faster than those of compounds having α-(1,6)-linkages on TLC plates, while the opposite was observed for HPIC.
The transglycosylation of PTS to the acceptor molecule catalyzed by ThMA was observed with all the carbohydrate acceptors used. However, the results of the transfer reaction, carried out with methyl-α-d-glucopyranoside (α-MG) as the acceptor, are presented in detail in this paper. Two transfer products, 1 and 2 (Fig. 4, lane B), were detected by TLC analysis as the result of the reaction. On the other hand, HPIC of the reaction mixture indicated that at least three transfer products were formed during the reaction that were linked by α-(1,6)- (Fig. 5, peak 3), α-(1,4)- (Fig. 5, peak 4), or α-(1,3)- (Fig. 5, peak 5) linkage. The fractions of peaks 3 to 5 were isolated and subjected to methylation. Methylation analysis of the transfer product in peak 3 yielded 2,3,6- and 2,3,4-tri-O-methyl-d-glucose, indicating the presence of α-(1,6)-linkage (Fig. 6, lane 1); that in peak 5 yielded 2,3,6- and 2,4,6-tri-O-methyl-d-glucose, indicating that the linkage was α-(1,3) (Fig. 6, lane 2); and that in peak 4 yielded 2,3,6-tri-O-methyl-d-glucose, indicating that the linkage was α-(1,4) (Fig. 6, lane 3).
FIG. 5.

Results from HPIC of acarbose transfer products. Acarbose and α-MG were reacted in the presence of ThMA, and the resulting reaction mixture was subjected to HPIC. Peak 1 represents α-MG; peak 2, glucose; peak 3, α-(1,6)-linked transfer product; peak 4, α-(1,4)-linked transfer product; peak 5, α-(1,3)-linked transfer product; peak 6, PTS; peak 7, isoacarbose; and peak 8, acarbose.
FIG. 6.

Methylation analysis of acarbose transfer products formed with α-MG. Lane M was loaded with a mixture of methylated alternan and β-cyclodextrin. Lane 1, methylated transfer product of peak 3 in Fig. 5; lane 2, methylated transfer product of peak 5 in Fig. 5; lane 3, methylated transfer product of peak 4 in Fig. 5.
Based on these results, an NMR spectrum of these transfer products was obtained (Fig. 7). Transfer products purified by HPIC were subjected to 1H-NMR spectroscopy, a method useful in estimating the degree of branching of glycogens and amylopectin β-limit dextrins, to distinguish between anomeric protons involved in glycosidic linkages (9). The α-(1,6)-linked transfer product (from Fig. 6, lane 1) showed a unique peak at 5.45 ppm as in panose and isoacarbose (Fig. 7A). A peak representing α-(1,3)-linkage (Fig. 6, lane 2) was detected at 5.85 ppm (Fig. 7C), which exhibited a slight chemical shift from that of α-(1,4)-linkage detected at 5.80 ppm (Fig. 7B).
FIG. 7.

1H-NMR spectroscopy of α-(1,6)-acarbose transfer product (A), α-(1,4)-acarbose transfer product (B), α-(1,3)-acarbose transfer product (C), and α-MG (D). Peaks at 5.45, 5.82, and 5.85 ppm were assigned to H1 of α-(1,6)-, α-(1,4)-, and α-(1,3)-linked units between glucose and α-MG, respectively.
In order to understand the mode of transfer more precisely, the transfer of PTS to α-MG was monitored for 48 h (Fig. 8) and reaction rate constants (k) were calculated for each hydrolysis and transglycosylation reaction as follows:
FIG. 8.

Time course analysis of acarbose transfer product formation. ThMA was reacted with acarbose (5% [wt/vol]) and α-MG (10% [wt/vol]) at 60°C for 48 h. The graph shows the hydrolysis of acarbose (black circles), formation of α-(1,4)-acarbose transfer product (squares), formation of α-(1,6)-acarbose transfer product (white circles), and formation of α-(1,3)-acarbose transfer product (triangles).
 |
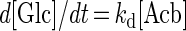 |
 |
 |
 |
 |
Under the assumption of first-order kinetics, the reaction rate constants obtained for the period of 48 h were evaluated by nonlinear regression by using the BMDP statistical analysis method (1a, 5). Therefore, the reaction rate constant, k, was given in dimension per seond.
At the beginning of the reaction, the formation of the α-(1,4)-linkage was predominant, 6.3- and 4.5-fold faster than α-(1,3)-linkage and α-(1,6)-linkage formation, respectively, but the resulting molecules kept being hydrolyzed rapidly during the rest of the reaction (Table 2). The rate constant for α-(1,4)-linkage hydrolysis was higher than that for formation of the linkage. The formation of the α-(1,6)-linkage increased during the first 5 h of the reaction, and the resulting molecules were stable throughout the reaction, with a rate constant of nearly 0. The α-(1,3)-linkage was formed at the lowest rate at the beginning, but the resulting transfer product was relatively stable for the rest of the time. Therefore, the transfer products containing an α-(1,6)- or α-(1,3)-linkage accumulated, while those with an α-(1,4)-linkage were readily hydrolyzed and transferred mainly to the C-6 of α-methylglucoside by ThMA during the reaction.
TABLE 2.
Reaction rate constants of acarbose hydrolysis and transglycosylation by ThMA
| Total (kd) | Rate constant (104 s−1)a for:
|
|||||
|---|---|---|---|---|---|---|
| Hydrolysis
|
Transglycosylation
|
|||||
| k1 | k5 | k6 | k2 | k3 | k4 | |
| 4.06 ± 0.44 | 1.28 ± 0.01 | 2.70 ± 0.16 | 0.23 ± 0.04 | 2.02 ± 0.09 | 0.32 ± 0.02 | 0.45 ± 0.02 |
kd to k6, as described in the text.
The products derived from the transfer acceptor reactions with various sugars are summarized in Table 3. The results indicated that many different carbohydrates, including mono-, di-, and trisaccharides, could play the role of acceptors for the transfer of PTS from acarbose by ThMA and that this enzyme favored an acceptor containing a pyranose ring structure. Among the acceptors tested, glucose, maltose, and cellobiose were the most efficient ones. Furthermore, the transfer was primarily to a C-6, primary alcohol moiety, to form an α-(1,6)-glycosidic linkage, and secondly to a C-3 and/or C-4, which was similar to the transfer acceptor reactions described previously for a dextransucrase (28). PTS was transferred primarily to a C-5 when the acceptor contained a furanose ring structure, as in fructose, or to a C-4 in the case of xylose.
TABLE 3.
Acarbose transfer products formed by the action of ThMA with acarbose and various carbohydrate acceptors
| Acceptor | PTS acceptor products containing linkage
|
|||
|---|---|---|---|---|
| α-(1,6) | α-(1,5) | α-(1,4) | α-(1,3) | |
| Glucose | +++++a | NSb | + | |
| Mannose | +++ | ++ | ||
| Galactose | ++++ | + | ++ | |
| Fructose | +++ | + | ||
| Xylose | ++++ | ++ | ||
| α-MG | +++ | + | ++ | |
| Maltose | ++++++ | NS | + | |
| Cellobiose | ++++++ | + | ||
| Lactose | ++++ | + | ||
| Gentiobiose | +++++ | + | + | |
| Sucrose | ++ | +++ | ||
| Trehalose | + | + | ||
| Raffinose | ++ | + | ||
| Maltotriose | ++ | ++ | ||
| Sorbitol | + | |||
| Xylitol | ++ | + | + | |
| Maltitol | ++ | +++ | ||
Relative efficacy of each linkage formation was represented in the number of +.
NS, not separated.
DISCUSSION
Maltogenic amylases are likely to constitute a subfamily of amylolytic enzymes with CDases, neopullulanases, and TVA-II. This subfamily shares the characteristics of hydrolyzing starch, pullulan, and cyclodextrin and transfers the hydrolyzed sugar moiety to another sugar molecule. ThMA was the first maltogenic amylase isolated from the gram-negative bacterium Thermus. This thermophilic bacterium shares many common physiological characteristics with B. stearothermophilus, a gram-positive thermophilic bacterium. The primary structure of ThMA was very homologous to that of a neopullulanase (86% identity [20]) and that of a maltogenic amylase (70% identity [4]), both of which had originated from B. stearothermophilus. The sequences and the spacing pattern of four conserved regions in these three enzymes were identical, while those of other enzymes in the subfamily were homologous but not identical. B. licheniformis thermostable α-amylase (14), a typical α-amylase, and cyclodextrin glucanotransferase (CGTase [18]) showed spacing patterns different from that of maltogenic amylases. CGTase was remarkably different in that its C-terminal region, which is related to its cyclization activity, was longer than the C termini of other amylolytic enzymes. All maltogenic amylases, neopullulanases, and CDases had longer N-terminal regions than those of CGTases and α-amylases. Therefore, the enzymes in the subfamily including maltogenic amylases should have tertiary structures different from those of α-amylases and CGTases.
Among the enzymes in the subfamily, however, BSMA (4), CDase I-5 (17), and ThMA were not inhibited by acarbose but could hydrolyze the amylase inhibitor to glucose and PTS. ThMA exhibited the highest acarbose-hydrolyzing activity among the enzymes. Previous studies of BSMA indicated that the enzyme cleaved the first glycosidic bond of acarbose to produce glucose and PTS, which was then transferred to the C-6 of the glucose to give an α-(1,6)-glycosidic linkage, resulting in the formation of isoacarbose (4). Crystallographic studies of the amylolytic enzyme (α-amylase, CGTase, or glucoamylase)-acarbose complex have been reported (1, 3, 30), but none of these enzymes hydrolyzed acarbose or produced acarbose transfer products. A time course study of BOS formation with liquefied starch and maltogenic amylases suggested that coupled hydrolysis and transglycosylation reactions on maltooligosaccharides and transfer products proceeded until the reaction had reached equilibrium with products that were not hydrolyzable by the enzymes.
The transglycosylation modes of TVA-II and neopullulanase have been investigated (21, 33). TVA-II transferred a glucosyl residue from the donor (pullulan) to the acceptor molecules with the formation of both α-(1,4)- and α-(1,6)-linkages (33). Similar observations have been reported for the neopullulanase (21). In either case, an α-(1,3)-linked transfer product was not detected. The transglycosylation of sugar to a donor molecule by forming an α-(1,3)-glycosidic linkage was observed only when acarbose was used as a substrate in this study.
By methylation of the various acarbose transfer products, the formation of α-(1,4)-, α-(1,3)-, α-(1,5)-, and with an α-(1,6)-linkages was observed during transglycosylation reactions with ThMA, and the last three products accumulated in significant amounts. Binding of acarbose to the active site of ThMA was likely, such that the first α-(1,4)-glycosidic linkage of acarbose, not the second one, was favorably positioned for the catalytic groups to carry out hydrolysis and transglycosylation. However, the acarbose transfer product either with an α-(1,3)- or with an α-(1,6)-linkage might be unable to bind to the active site of ThMA in an appropriate position for cleavage. The results indicated that many different carbohydrates could play the role of acceptor for the transfer of PTS from acarbose by ThMA and that this enzyme favored an acceptor containing a pyranose ring structure. Furthermore, the transfer is primarily to a C-6, primary alcohol to form an α-(1,6)-glycosidic linkage, but not exclusively so, similarly to the transfer acceptor reactions of dextransucrase (28).
A proposed action pattern for the transfer of PTS from acarbose by ThMA is suggested (Fig. 9). In this scheme, acarbose is hydrolyzed to PTS and glucose when water acts as an acceptor. However, as the concentration of glucose builds up in the reaction mixture, it serves as the acceptor and PTS is transferred mostly to the C-4 of the glucose, forming acarbose again. When other acceptors are added, ThMA cleaves the first α-(1,4)-glycosidic linkage of acarbose and transfers PTS to the acceptors, primarily forming α-(1,6)-linkages between PTS and the acceptors, although an α-(1,3)-linkage is also formed. The acarbose transfer product with an α-(1,6)-linkage is not hydrolyzed further by the enzyme, thereby increasing gradually in concentration. The acarbose transfer product with an α-(1,3)-linkage is hydrolyzed by the enzyme at a rate low enough to keep the concentration almost constant. With some acceptors, such as d-fructose and d-xylose, α-(1,5)- and α-(1,4)-linkages were formed. The main linkage was to a primary alcohol, if it was present on the acceptor. Preliminary studies on the action of the acarbose transfer products formed using acarbose and various mono- and disaccharides indicated that they would act as inhibitors of amylolytic enzymes (data will be published elsewhere).
FIG. 9.

Proposed action modes of ThMA in the transfer reaction with acarbose and water or methyl-α-d-glucopyranoside as acceptors. If water acts as an acceptor, acarbose is hydrolyzed to PTS and glucose. If methyl-α-d-glucopyranoside (α-MG) acts as an acceptor, α-(1,4)-, α-(1,3)-, and α-(1,6)-acarbose transfer products are formed. α-(1,4)- and α-(1,3)-acarbose transfer products are hydrolyzed further to PTS and α-MG, while α-(1,6)-acarbose transfer products are accumulated without further hydrolysis.
To understand the action pattern of ThMA and the structure-function relationship of amylolytic enzymes more precisely, mutagenesis studies and crystallographic analysis of the enzymes are under investigation in the laboratory.
ACKNOWLEDGMENTS
This study was supported by the Korea Science and Engineering Foundation (KoSEF) through a grant to the Research Center for New Bio-Materials in Agriculture and a grant to J.-W. Kim (971-0604-031-2).
REFERENCES
- 1.Aleshin A E, Firsov L M, Honzatke R B. Refined structure for the complex of acarbose with glucoamylase from Aspergillus awamori var. X100 to 2.4-Å resolution. J Biol Chem. 1994;269:15631–15639. [PubMed] [Google Scholar]
- 1a.BMDP statistical software. 1981. California State University.
- 2.Bradford M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 3.Brzozowski A M, Davies M J. Structure of the Aspergillus oryzae α-amylase complexed with the inhibitor acarbose at 2.0 Å. Biochemistry. 1997;36:10837–10845. doi: 10.1021/bi970539i. [DOI] [PubMed] [Google Scholar]
- 4.Cha H J, Yoon H G, Kim Y W, Lee H S, Kim J W, Kweon K S, Oh B H, Park K H. Molecular and enzymatic characterization of novel maltogenic amylase that hydrolyzes and transglycosylates acarbose. Eur J Biochem. 1998;253:251–262. doi: 10.1046/j.1432-1327.1998.2530251.x. [DOI] [PubMed] [Google Scholar]
- 5.Chang B S, Park K H, Lund D B. Thermal inactivation kinetics of horseradish peroxidase. J Food Sci. 1988;53:920–923. [Google Scholar]
- 6.Cheong T K. Modulation of Bacillus licheniformis amylases by site-directed and deletion mutagenesis. Ph.D. thesis. Seoul, Korea: Seoul National University; 1995. [Google Scholar]
- 7.DePinto J A, Campbell L L. Purification and properties of the cyclodextrinase of Bacillus macerans. Biochemistry. 1968;7:121–123. doi: 10.1021/bi00841a016. [DOI] [PubMed] [Google Scholar]
- 8.Dubnau D, Abenson R D. Fate of transforming DNA following uptake by competent Bacillus subtilis. J Mol Biol. 1971;56:209–221. doi: 10.1016/0022-2836(71)90460-8. [DOI] [PubMed] [Google Scholar]
- 9.Gridley M J. Quantification of the structural features of starch polysaccharides by N. M R Spectroscopy Carbohydr Res. 1985;139:85–93. [Google Scholar]
- 10.Igarashi K, Ara K, Saeki K, Ozaki K, Ozaki S, Ito S. Nucleotide sequence of the gene that encodes a neopullulanase from an alkalophilic Bacillus. Biosci Biotechnol Biochem. 1992;56:514–516. doi: 10.1271/bbb.56.514. [DOI] [PubMed] [Google Scholar]
- 11.Janecek S, Svensson B, Henrissat B. Domain evolution in the alpha-amylase family. J Mol Evol. 1997;45:322–331. doi: 10.1007/pl00006236. [DOI] [PubMed] [Google Scholar]
- 12.Jespersen H M, MacGregor E A, Henrissat B, Sierks M R, Svensson B. Starch- and glycogen-debranching and branching enzymes: prediction of structural features of the catalytic (β/α)8-barrel domain and evolutionary relationship to other amylolytic enzymes. J Prot Chem. 1993;12:791–805. doi: 10.1007/BF01024938. [DOI] [PubMed] [Google Scholar]
- 13.Kim I C, Cha J H, Kim J R, Jang S Y, Seo B C, Cheong T K, Lee D S, Choi Y D, Park K H. Catalytic properties of the cloned amylase from Bacillus licheniformis. J Biol Chem. 1992;267:22108–22114. [PubMed] [Google Scholar]
- 14.Kim I C, Jang S Y, Cha J H, Ko Y H, Park K H, Rho H M. Cloning and expression of thermostable alpha-amylase gene in E. coli from Bacillus licheniformis ATCC27811. Korean J Appl Microbiol Bioeng. 1988;16:369–373. [Google Scholar]
- 15.Kim I C, Yoo S H, Lee S J, Oh B H, Kim J W, Park K H. Synthesis of branched oligosaccharides from starch by two amylases cloned from Bacillus licheniformis. Biosci Biotechnol Biochem. 1994;58:416–418. [Google Scholar]
- 16.Kim T J. Molecular cloning and characterization of thermostable maltogenic amylase and pullulanase from Thermus strain IM6501. Ph.D. thesis. Seoul, Korea: Seoul National University; 1998. [Google Scholar]
- 17.Kim T J, Shin J H, Oh J H, Kim M J, Lee S B, Ryu S, Kwon K, Kim J W, Choi E H, Robyt J F, Park K H. Analysis of the gene encoding cyclomaltodextrinase from alkalophilic Bacillus sp. I-5 and characterization of enzymatic properties. Arch Biochem Biophys. 1998;535:221–227. doi: 10.1006/abbi.1998.0639. [DOI] [PubMed] [Google Scholar]
- 18.Kimura K, Kataoka S, Nakamura A, Takano T, Kovayashi S, Yamane K. Functions of the COOH-terminal regional of cyclodextrin glucanotransferase of alkalophilic Bacillus sp. #1011: relation to catalyzing activity and pH stability. Biochem Biophys Res Commun. 1989;161:1273–1279. doi: 10.1016/0006-291x(89)91380-6. [DOI] [PubMed] [Google Scholar]
- 19.Kuriki T, Imanaka T. Nucleotide sequence of the neopullulanase gene from Bacillus stearothermophilus. J Gen Microbiol. 1993;135:1521–1528. doi: 10.1099/00221287-135-6-1521. [DOI] [PubMed] [Google Scholar]
- 20.Kuriki T, Park J-H, Okada S, Imanaka T. Purification and characterization of thermostable pullulanase from Bacillus stearothermophilus and molecular cloning and expression of the gene in Bacillus subtilis. Appl Environ Microbiol. 1988;54:2881–2883. doi: 10.1128/aem.54.11.2881-2883.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuriki T, Yanase M, Takata H, Takesada Y, Imanaka T, Okada S. A new way of producing isomalto-oligosaccharide syrup by using the transglycosylation reaction of neopullulanase. Appl Environ Microbiol. 1993;59:953–959. doi: 10.1128/aem.59.4.953-959.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 23.Lee S J, Yoo S H, Kim M J, Kim J W, Seok H M, Park K H. Production and characterization of branched oligosaccharides from liquefied starch by the action of Bacillus licheniformis amylase. Starch. 1995;47:127–134. [Google Scholar]
- 24.Miller C L. Use of dinitrosalicylic acid reagent for determination reducing sugar. Anal Chem. 1959;31:426–428. [Google Scholar]
- 25.Mukerjea R, Kim D, Robyt J F. Simplified and improved methylation analysis of saccharides, using a modified procedure and thin layer chromatography. Carbohydr Res. 1996;292:11–20. [Google Scholar]
- 26.Oguma T, Matsuyama A, Kikuchi M, Nakano E. Cloning and sequence analysis of the cyclomaltodextrinase gene from Bacillus sphaericus and expression in Escherichia coli cells. Appl Microbiol Biotechnol. 1993;39:197–203. doi: 10.1007/BF00228606. [DOI] [PubMed] [Google Scholar]
- 27.Podkovyrov S M, Zeikus J G. Structure of the gene encoding cyclomaltodextrinase from Clostridium thermohydrosulfuricum 39E and characterization of the enzyme purified from Escherichia coli. J Bacteriol. 1992;174:5400–5405. doi: 10.1128/jb.174.16.5400-5405.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robyt J F. Mechanisms in the glucansucrase synthesis of polysaccharides and oligosaccharides from sucrose. Adv Carbohydr Chem Biochem. 1995;51:151–157. doi: 10.1016/s0065-2318(08)60193-6. [DOI] [PubMed] [Google Scholar]
- 29.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strokopytov B, Penninga D, Rozeboom H J, Kalk K H, Diikhyizen L, Dijstra B W. X-ray structure of cyclodextrin glycosyltransferase complexed with acarbose: implications for the catalytic mechanism of glycosidases. Biochemistry. 1995;34:2234–2240. doi: 10.1021/bi00007a018. [DOI] [PubMed] [Google Scholar]
- 31.Takaku H. Handbook of amylases and related enzymes: their sources, isolation methods, properties and applications. New York, N.Y: Pergamon Press; 1988. pp. 215–217. [Google Scholar]
- 32.Tonozuka T, Ohtsuka M, Mogi S, Sakai H, Ohta T, Sakano T. A neopullulanase-type alpha-amylase gene from Thermoactinomyces vulgaris R-47. Biosci Biotechnol Biochem. 1993;57:395–401. doi: 10.1271/bbb.57.395. [DOI] [PubMed] [Google Scholar]
- 33.Tonozuka T, Sakai H, Ohta T, Sakano Y. A convenient enzymatic synthesis of 4(2)-alpha-isomaltosylisomaltose using Thermoactinomyces vulgaris R-47 alpha-amylase II (TVAII) Carbohydr Res. 1994;261:157–162. doi: 10.1016/0008-6215(94)80014-6. [DOI] [PubMed] [Google Scholar]